Похідні індолу як активатори nurr-1 та їх застосування для лікування хвороби паркінсона
Номер патенту: 102397
Опубліковано: 10.07.2013
Автори: Таллондьє Мірей, Маккрірі Ендрю, ван Вліт Бернард Йоханнес, Бубіа Бенаїсса, ден Хартог Якобус Антоніус Йозеф, Пупарден-Олів'є Олівія, ван Донген Марія Йоханна Петронелла











Формула / Реферат
1. Похідне індолу, що має терапевтичну активність, вибране із
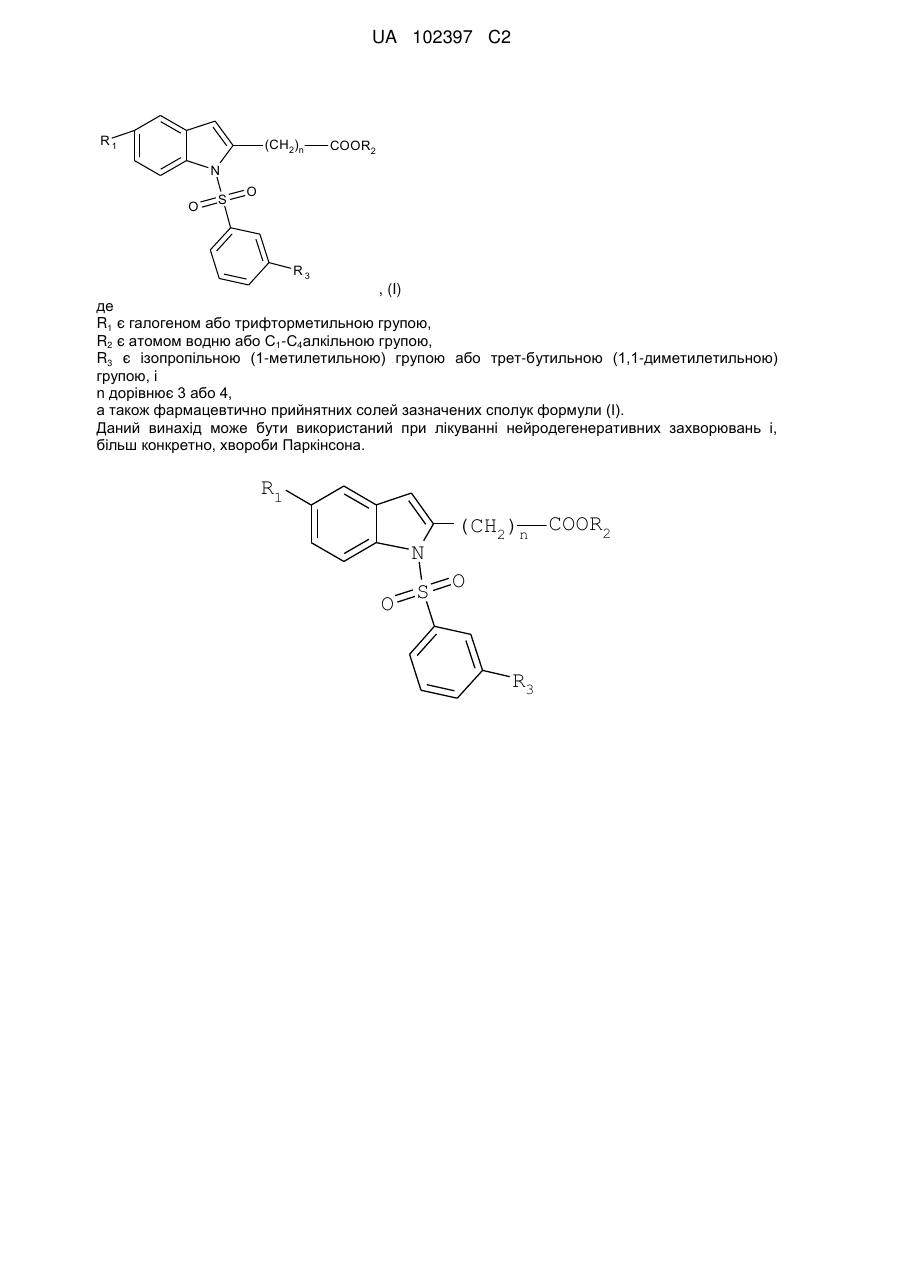
і) сполук формули (І)
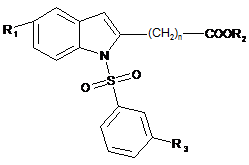 , (I)
, (I)
де
R1 є галогеном або трифторметильною групою,
R2 є атомом водню або С1-С4алкільною групою,
R3 є ізопропільною (1-метилетильною) групою або трет-бутильною (1,1-диметилетильною) групою, і
n дорівнює 3 або 4,
і
іі) фармацевтично прийнятних солей зазначених сполук формули (І).
2. Сполука згідно з п. 1, де в наведеній вище формулі (І) R3 є ізопропільною групою.
3. Сполука згідно з п. 1, де в наведеній вище формулі (І) R3 є тpeт-бутильною групою.
4. Сполука за будь-яким з пп. 1-3, де в наведеній вище формулі (І) R2 є атомом водню.
5. Фармацевтична композиція, яка містить щонайменше одну сполуку за будь-яким з пп. 1-4 як активну речовину й щонайменше один фармацевтично прийнятний ексципієнт.
6. Застосування похідної індолу за будь-яким з пп. 1-4 для виготовлення ліків, призначених для лікування або профілактики захворювань, в які залучений рецептор NURR-1.
7. Застосування згідно з п. 6 для виготовлення ліків, призначених для лікування і профілактики нейродегенеративних захворювань.
8. Застосування згідно з п. 7, де вищезгадане захворювання є хворобою Паркінсона.
Текст
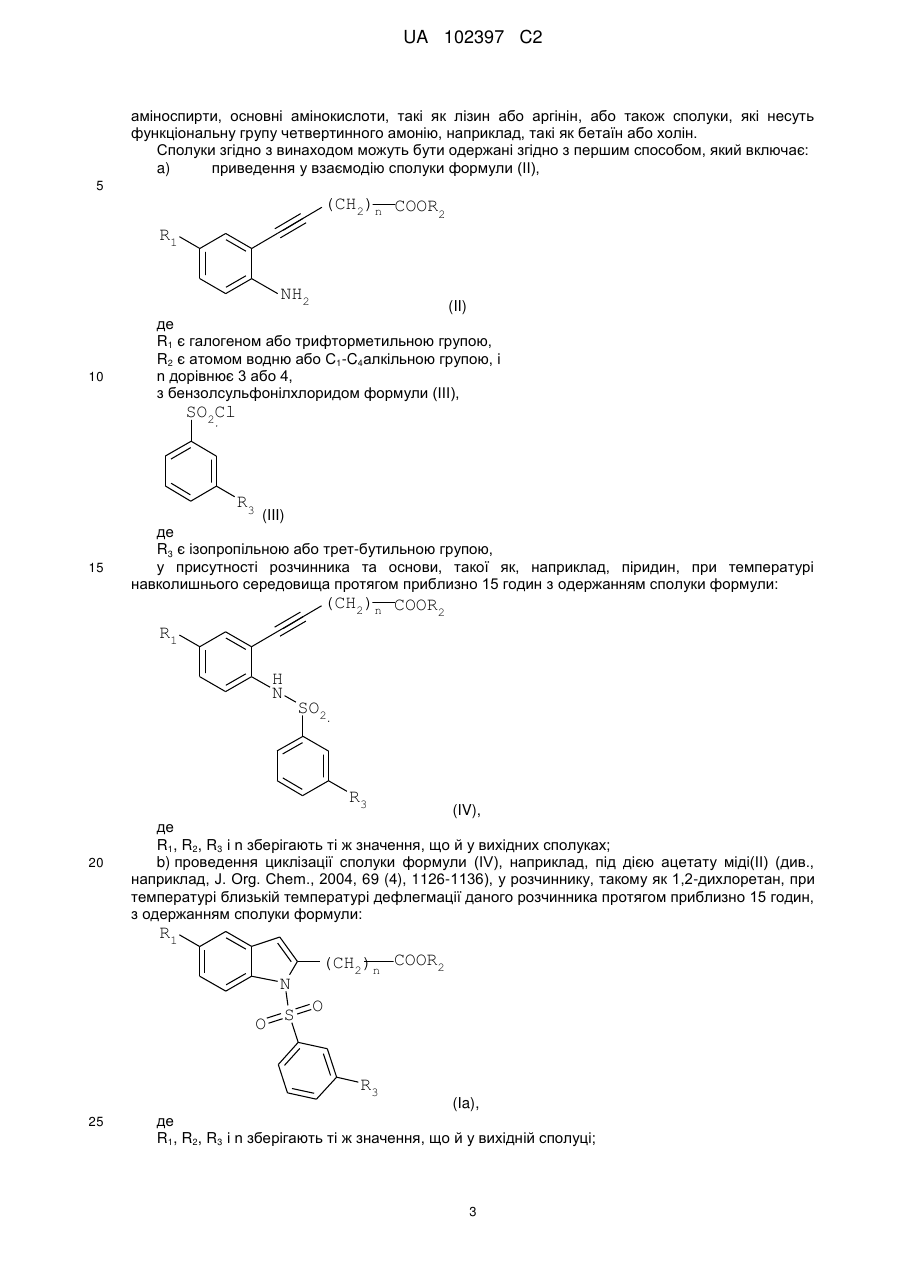
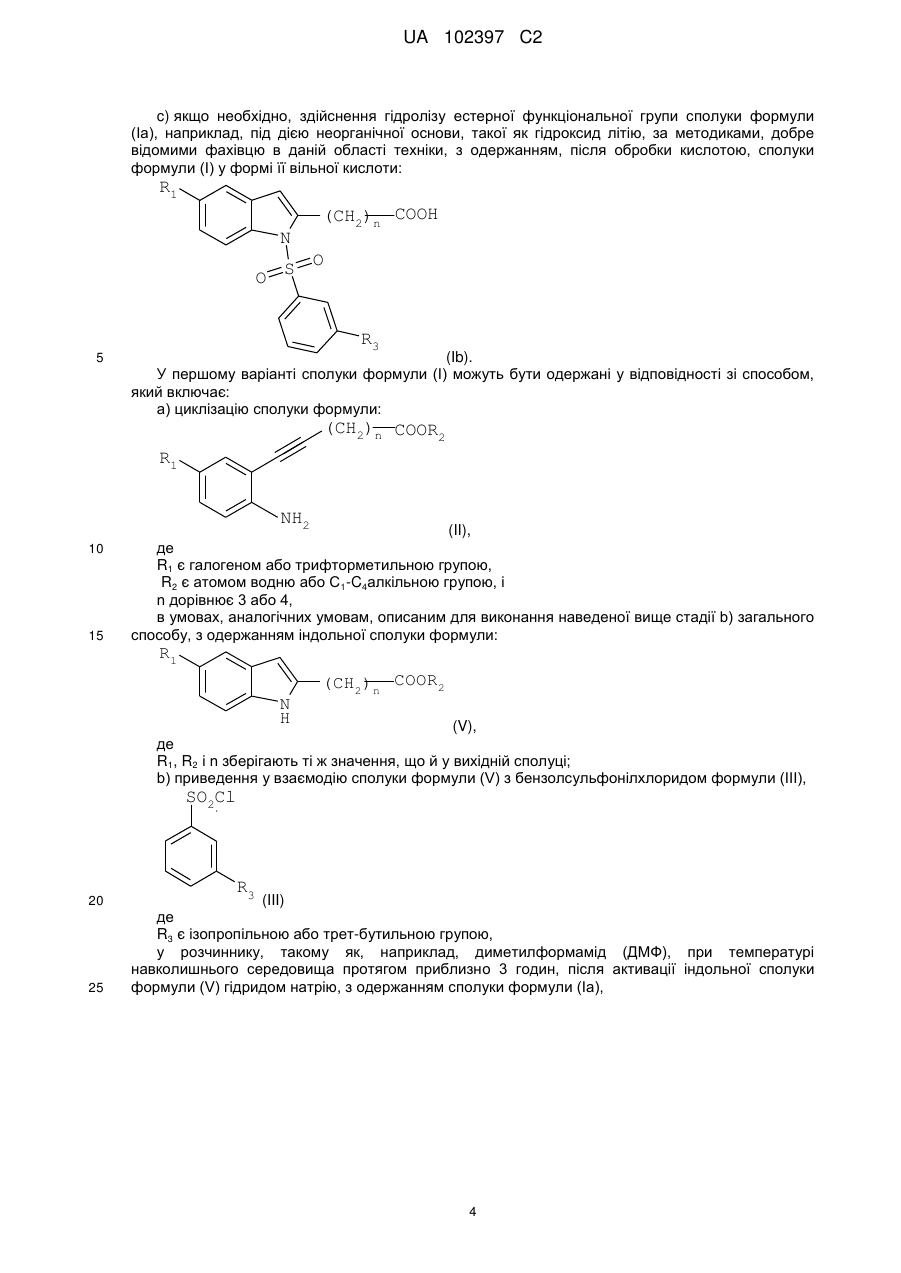
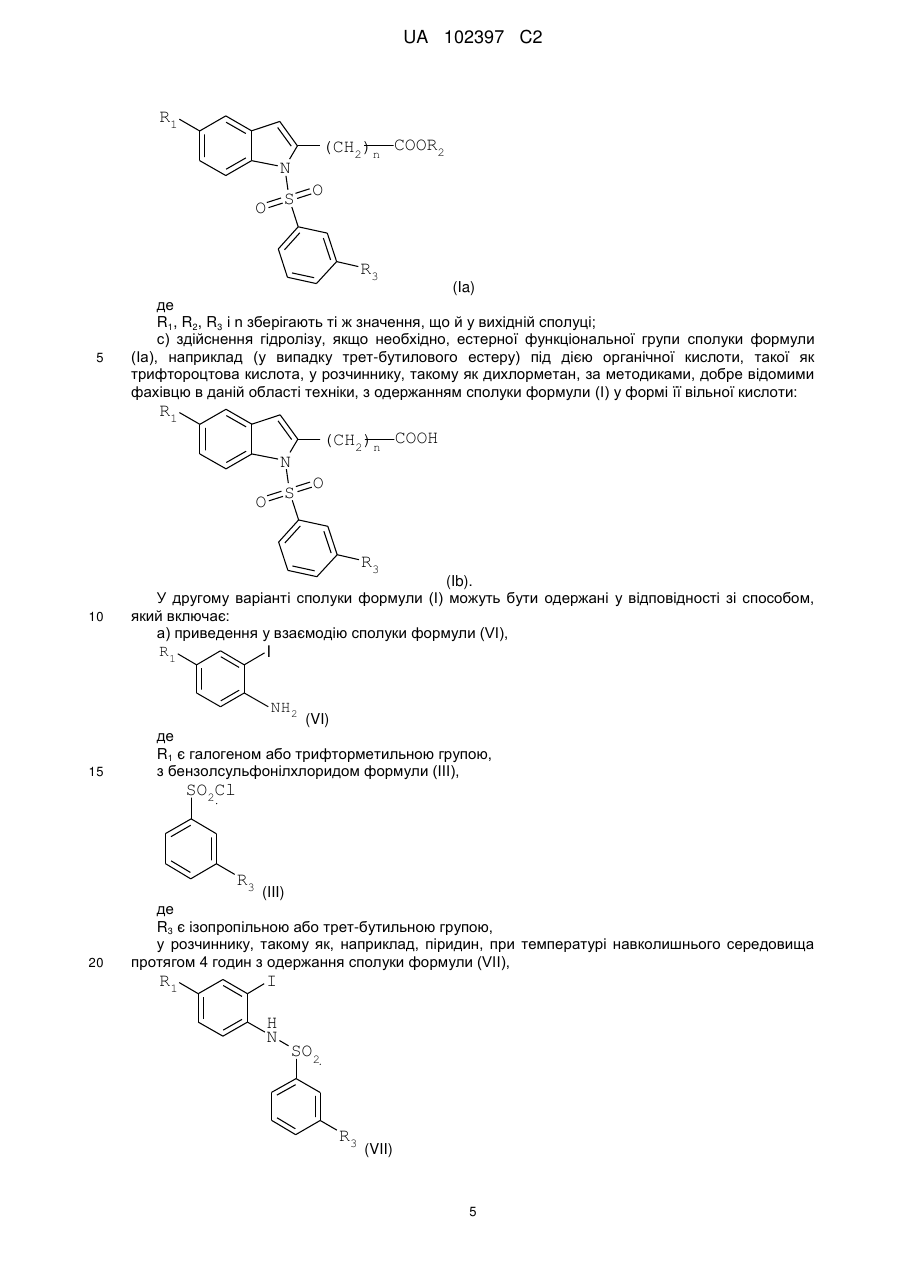
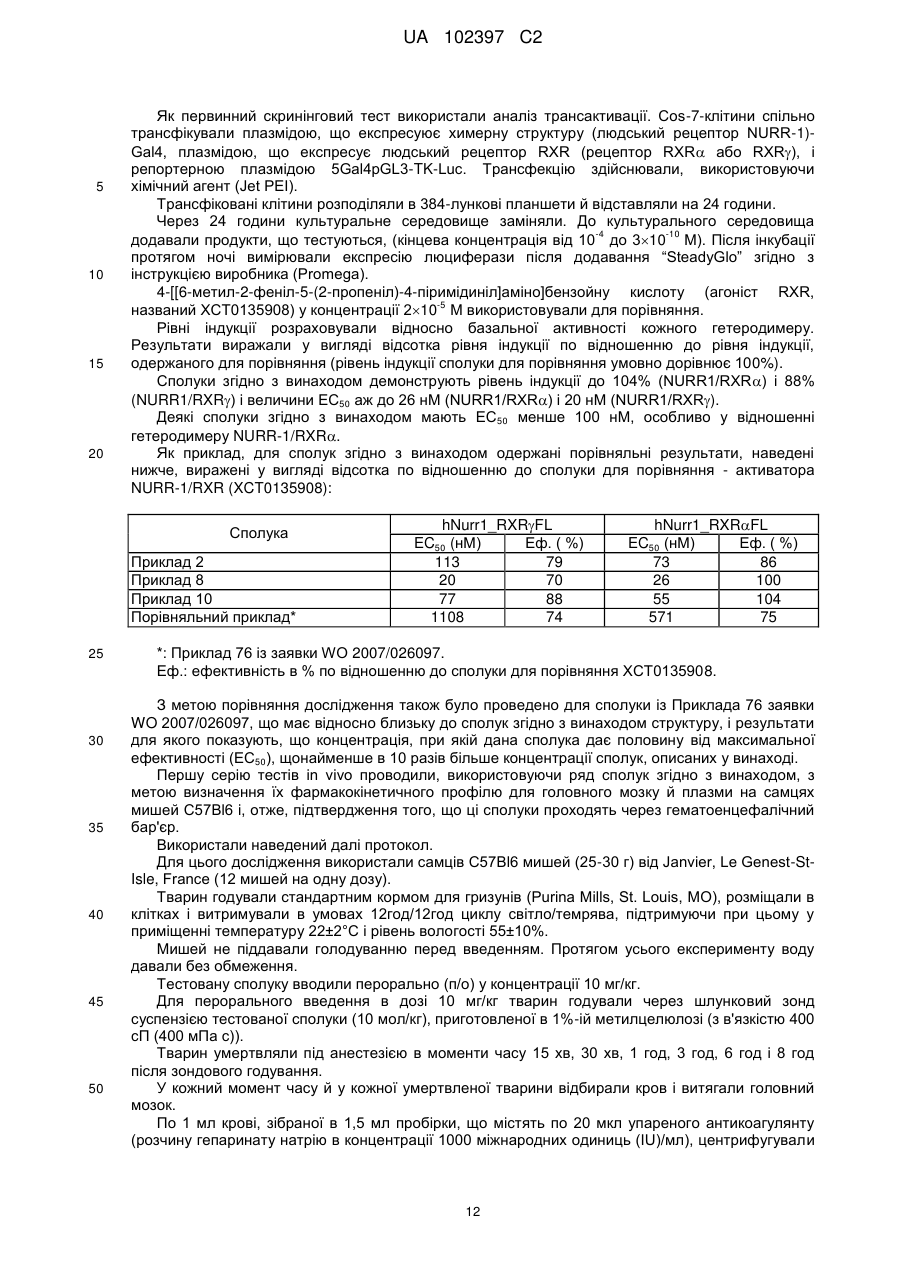
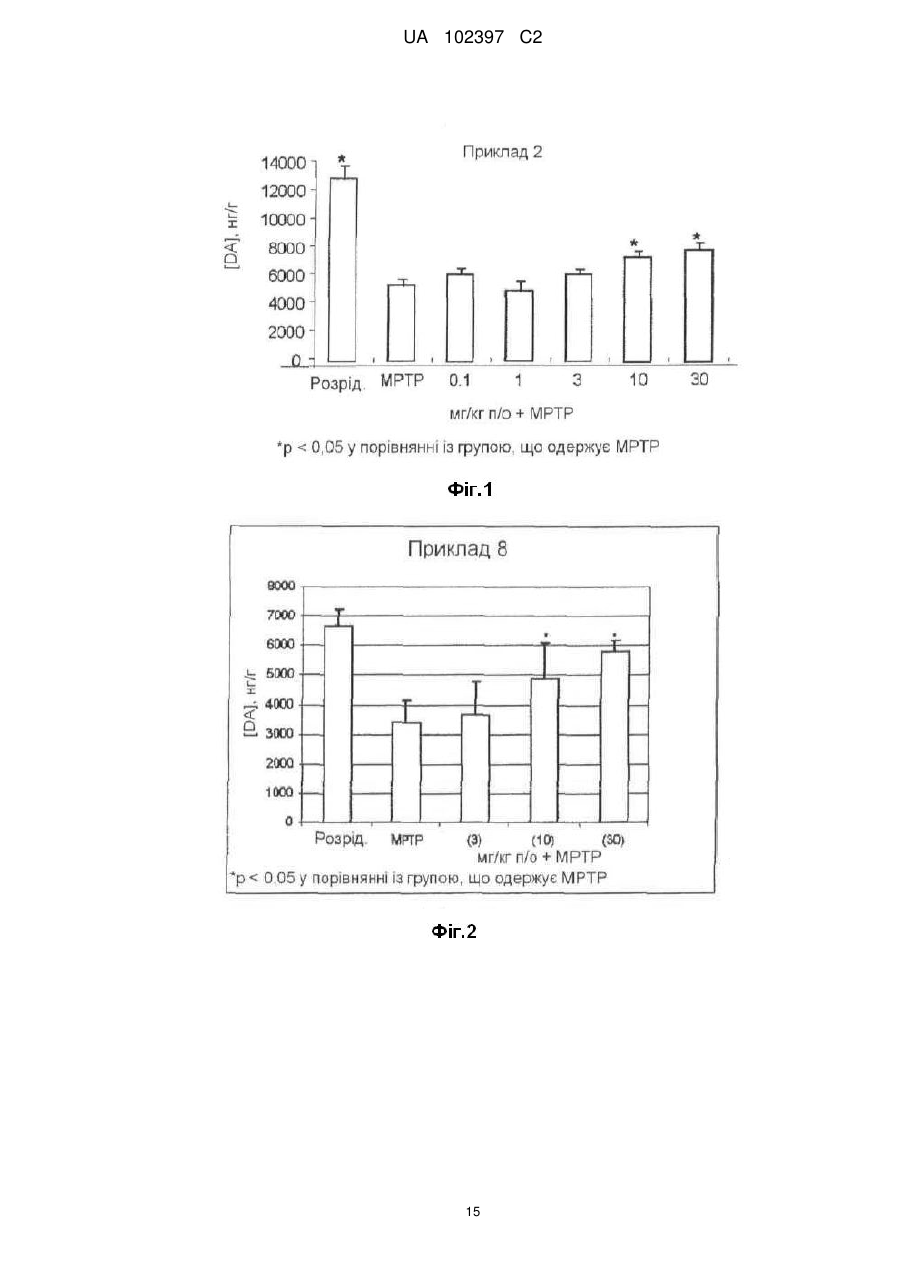
Реферат: Даний винахід стосується похідного індолу, що має терапевтичну активність, вибраного із сполук формули (І) UA 102397 C2 (12) UA 102397 C2 R1 (CH2)n COOR2 N O S O R3 , (I) де R1 є галогеном або трифторметильною групою, R2 є атомом водню або С1-С4алкільною групою, R3 є ізопропільною (1-метилетильною) групою або трет-бутильною (1,1-диметилетильною) групою, і n дорівнює 3 або 4, а також фармацевтично прийнятних солей зазначених сполук формули (І). Даний винахід може бути використаний при лікуванні нейродегенеративних захворювань і, більш конкретно, хвороби Паркінсона. R1 N O S (CH2)n COOR2 O R3 UA 102397 C2 5 10 15 20 25 30 35 40 45 50 55 Даний винахід стосується нового терапевтичного застосування деяких похідних індолу в лікуванні і/або профілактиці захворювань, які залучають ядерні рецептори NURR-1. Більш конкретно, даний винахід стосується застосування цих сполук для виготовлення ліків для лікування і/або профілактики хвороби Паркінсона. Попередній рівень техніки Нейродегенеративні захворювання визначають як захворювання, які характеризуються прогресуючою дисфункцією нервової системи. Найчастіше їх асоціюють із атрофією структур центральної або периферійної нервової системи. Вони включають, серед іншого, такі захворювання, як хвороба Альцгеймера, хвороба Крейцфельдта-Якоба, хвороба Гентінгтона, хвороба Паркінсона, “лізосомальні хвороби”, прогресуючий супрануклеарний параліч, множинний склероз і аміотрофічний бічний склероз. Серед нейродегенеративних захворювань хвороба Паркінсона є захворюванням, яке уражає приблизно чотири мільйона людей по всьому світі. Хоча ураженню піддаються індивідууми будь-якого віку, у більшості випадків вона зустрічається у людей похилого віку (причому цією хворобою уражені 2% популяції людей старіше 65 років). Вона характеризується дегенерацією дофамінергічних нейронів чорної субстанції. Дофамін є нейротрансмітером, який відіграє центральну роль у контролі довільних рухів, у когнітивних функціях і у прояві поведінки, асоційованої з емоціями. Сучасна терапевтична стратегія лікування хвороби Паркінсона полягає в ослабленні симптомів за допомогою компенсації дефіциту дофаміну завдяки введенню метаболічного попередника, такого як L-DOPA (L-3,4-дигідроксифенілаланін). У даний час збільшення частоти виникнення цієї патології зробило необхідною розробку нових терапевтичних агентів, які відіграють істотну роль у диференціюванні й застосуванні нейронів. Дана розробка привела до ідентифікації сполук, здатних активувати ядерні рецептори, залучені у патогенез хвороби Паркінсона. Високо експресований у головному мозку транскрипційний фактор NURR-1, член суперродини орфанових ядерних рецепторів, ідентифікований як такий, що відіграє істотну роль у розвитку й збереженні дофамінергічних нейронів середнього мозку (Zetterstrom, Solomin and al. 1997, Science. 1997 Apr 11; 276(5310): 248-50). Ядерний рецептор NURR-1 бере участь у збереженні дофамінергічного фенотипу шляхом регуляції специфічних генів дофамінергічних (DA) нейронів. Він також сприяє виживаності DA нейронів за допомогою захисту їх від впливу токсичних речовин. Таким чином, ядерний рецептор NURR-1 діє як специфічний транскрипційний фактор дофамінергічних нейронів, активність яких можна регулювати шляхом модулювання дофамінергічної нейропередачі у хворобі Паркінсона. Цей рецептор зв'язується із ДНК у формі мономерів, гомодимерів або гетеродимерів з RXR (ретиноїдним X рецептором), ядерним рецептором, що є гетеропартнером для багатьох інших членів сімейства ядерних рецепторів. RXR бере участь у багатьох фізіологічних процесах, таких як метаболізм ліпідів, метаболізм глюкози, розвиток і диференціювання. Так, NURR-1 взаємодіє з ізоформами і RXR. Експресія RXR здійснюється повсюдно, тоді як експресія RXR зосереджена в першу чергу у головному мозку й більш конкретно в смугастому тілі, гіпоталамусі й гіпофізі. Утворені комплекси NURR-1/RXR і NURR-1/RXR мають здатність регулювати транскрипцію у відповідь на ліганд RXR. Таким чином, RXR позитивно модулює здатність до активації транскрипції NURR-1. Отже, ідентифікація сполук, здатних індукувати активність комплексів NURR-1/RXR і NURR-1/RXR, з імовірністю дозволити зробити доступними нові напрямки для лікування хвороби Паркінсона. У документі WO2003/015780 описуються гетероциклічні сполуки, які є дієвими щодо лікування хвороби Паркінсона. Крім того, в документах WO2004/072050, FR 2 903 105, FR 2 903 106 і FR 2 903 107 описуються сполуки, які представляють собою активатори рецептора NURR-1, а застосування гетероциклічних сполук, які модулюють активність рецепторів сімейства NGFI-B (членом якого є NURR-1), описано в документі WO2005/047268. І зрештою, у документі WO2005/056522 описуються похідні індолу, які є активаторами ядерних рецепторів PPAR (рецепторів, що активуються проліфератором пероксисом) і знаходять застосування як активний початок ліків для лікування деяких захворювання серцевосудинної системи. 1 UA 102397 C2 5 10 У цьому контексті виявлено, - і це є тим, що становить основу даного винаходу, - що деякі сполуки, які є похідними індолу й охоплюються загальною формулою, наведеною в документі WO2005/056522, є селективними агоністами NURR-1/RXR і NURR-1/RXR, здатними інгібувати дегенерацію нейронів, що спостерігається при хворобі Паркінсона. Таким чином, несподівано виявлено, що сполуки згідно з винаходом, на додаток до їх ефективності як активатора PPAR, демонструють дуже високий потенціал до активації гетеродимерів NURR-1/RXR і NURR-1/RXR. Тому дані сполуки, завдяки своїм унікальним властивостям, становлять особливий інтерес з точки зору їх застосування у лікуванні або профілактиці захворювань, що залучають рецептор NURR-1, особливо нейродегенеративних захворювань і, більш конкретно, хвороби Паркінсона. По-перше, згідно з даним винаходом як нові продукти запропоновані сполуки, що представляють собою похідні індолу, вибрані з 1) сполук формули (I), R1 N O S (CH2)n COOR2 O R3 15 20 25 30 35 40 45 50 (I) де R1 є галогеном або трифторметильною групою, R2 є атомом водню або C1-C4алкільною групою, R3 є ізопропільною (1-метилетильною) групою або трет-бутильною (1,1-диметилетильною) групою, і n дорівнює 3 або 4 і 2) фармацевтично прийнятних солей зазначених сполук формули (I). Виявлено (і це є внеском у рівень техніки), що одночасна присутність: - ізопропільного замісника або трет-бутильного замісника у мета-положенні на бензолсульфонільній групі; і - галогену або трифторметильної групи у положенні 5 індолу наділяє сполуки згідно з винаходом чудовою і зовсім неочікуваною активністю відносно рецепторів NURR-1. Таким чином, сполуки згідно з винаходом, мають хімічну структуру, яка, незважаючи на те, що в більшості випадків охоплюється загальною формулою, описаною в документі WO 2005/056522, є результатом вибору, який фахівець у даній області техніки не зміг би здійснити в процесі дослідження сполук, призначених для лікування хвороби Паркінсона. По-друге, згідно з даним винаходом, запропоновані вищезгадані сполуки для їх застосування як фармакологічно активних речовин, а також фармацевтичні композиції, що містять їх. У третіх, згідно з даним винаходом, запропоноване застосування щонайменше однієї сполуки формули (I) або однієї з його фармацевтично прийнятних солей як активного початку для виготовлення ліків, призначених для лікування захворювань, що залучують рецептор NURR-1, особливо нейродегенеративних захворювань, таких як, більш конкретно, хвороба Паркінсона. Докладний опис У даному описі C1-C4алкільна група є лінійним або розгалуженим насиченим вуглецевим ланцюгом, що має 1-4 атоми вуглецю, і більш конкретно групу метил, етил, пропіл, 1-метилетил, бутил, 1-метилпропіл, 2-метилпропіл або 1,1-диметилетил. Галоген є атомом фтору або хлору. Сполуки формули (I), в яких R2 є атомом водню, є карбоновими кислотами, які можуть бути використані у формі вільної кислоти або у формі солей, причому зазначені солі одержують шляхом об'єднання даної кислоти з нетоксичною органічною або неорганічною основою, яка переважно є фармацевтично прийнятною. Неорганічні основи, які можуть бути використані, включають, наприклад, гідроксид натрію, гідроксид калію, гідроксид магнію або гідроксид кальцію. Органічні основи, які можуть бути використані, включають, наприклад, аміни, 2 UA 102397 C2 аміноспирти, основні амінокислоти, такі як лізин або аргінін, або також сполуки, які несуть функціональну групу четвертинного амонію, наприклад, такі як бетаїн або холін. Сполуки згідно з винаходом можуть бути одержані згідно з першим способом, який включає: a) приведення у взаємодію сполуки формули (II), 5 (CH2)n COOR2 R1 NH2 10 (II) де R1 є галогеном або трифторметильною групою, R2 є атомом водню або C1-C4алкільною групою, і n дорівнює 3 або 4, з бензолсульфонілхлоридом формули (III), SO2Cl R3 15 (III) де R3 є ізопропільною або трет-бутильною групою, у присутності розчинника та основи, такої як, наприклад, піридин, при температурі навколишнього середовища протягом приблизно 15 годин з одержанням сполуки формули: (CH2)n COOR2 R1 H N SO2 R3 20 (IV), де R1, R2, R3 і n зберігають ті ж значення, що й у вихідних сполуках; b) проведення циклізації сполуки формули (IV), наприклад, під дією ацетату міді(II) (див., наприклад, J. Org. Chem., 2004, 69 (4), 1126-1136), у розчиннику, такому як 1,2-дихлоретан, при температурі близькій температурі дефлегмації даного розчинника протягом приблизно 15 годин, з одержанням сполуки формули: R1 N O S (CH2)n COOR2 O R3 25 (Ia), де R1, R2, R3 і n зберігають ті ж значення, що й у вихідній сполуці; 3 UA 102397 C2 c) якщо необхідно, здійснення гідролізу естерної функціональної групи сполуки формули (Ia), наприклад, під дією неорганічної основи, такої як гідроксид літію, за методиками, добре відомими фахівцю в даній області техніки, з одержанням, після обробки кислотою, сполуки формули (I) у формі її вільної кислоти: R1 N O S (CH2)n COOH O R3 5 (Ib). У першому варіанті сполуки формули (I) можуть бути одержані у відповідності зі способом, який включає: a) циклізацію сполуки формули: (CH2)n COOR2 R1 NH2 10 15 (II), де R1 є галогеном або трифторметильною групою, R2 є атомом водню або C1-C4алкільною групою, і n дорівнює 3 або 4, в умовах, аналогічних умовам, описаним для виконання наведеної вище стадії b) загального способу, з одержанням індольної сполуки формули: R1 N H (CH2)n COOR2 (V), де R1, R2 і n зберігають ті ж значення, що й у вихідній сполуці; b) приведення у взаємодію сполуки формули (V) з бензолсульфонілхлоридом формули (III), SO2Cl 20 25 R3 (III) де R3 є ізопропільною або трет-бутильною групою, у розчиннику, такому як, наприклад, диметилформамід (ДМФ), при температурі навколишнього середовища протягом приблизно 3 годин, після активації індольної сполуки формули (V) гідридом натрію, з одержанням сполуки формули (Ia), 4 UA 102397 C2 R1 (CH2)n COOR2 N S O O R3 5 (Ia) де R1, R2, R3 і n зберігають ті ж значення, що й у вихідній сполуці; c) здійснення гідролізу, якщо необхідно, естерної функціональної групи сполуки формули (Ia), наприклад (у випадку трет-бутилового естеру) під дією органічної кислоти, такої як трифтороцтова кислота, у розчиннику, такому як дихлорметан, за методиками, добре відомими фахівцю в даній області техніки, з одержанням сполуки формули (I) у формі її вільної кислоти: R1 (CH2)n COOH N S O O R3 10 (Ib). У другому варіанті сполуки формули (I) можуть бути одержані у відповідності зі способом, який включає: a) приведення у взаємодію сполуки формули (VI), R1 I NH2 15 (VI) де R1 є галогеном або трифторметильною групою, з бензолсульфонілхлоридом формули (III), SO2Cl R3 20 (III) де R3 є ізопропільною або трет-бутильною групою, у розчиннику, такому як, наприклад, піридин, при температурі навколишнього середовища протягом 4 годин з одержання сполуки формули (VII), R1 I H N SO2 R3 (VII) 5 UA 102397 C2 де R1 і R3 зберігають ті ж значення, що й у вихідних сполуках; b) приведення у взаємодію сполуки формули (VII) з похідною ацетилену формули: (CH2)n COOR2 5 10 (VIII), де R2 є атомом водню або C1-C4алкільною групою, n дорівнює 3 або 4; у присутності йодиду одновалентної міді, каталізатора на основі паладію, такого як, наприклад, біс(трифенілфосфін)паладію хлорид, і органічної основи, такої як, наприклад, триетиламін, у розчиннику, такому як, наприклад, диметилформамід (ДМФ), при температурі від температури навколишнього середовища до 80°C протягом 12 годин з одержанням сполуки формули: (CH2)n COOR2 R1 H N SO2 R3 15 (IV), де R1, R2, R3 і n зберігають ті ж значення, що й у вихідних сполуках; c) циклізацію наведеної вище сполуки формули (IV) в умовах, аналогічних умовам, описаним для виконання наведеної вище стадії (b) загального способу, з одержанням індольної сполуки формули: R1 N O S (CH2)n COOR2 O R3 20 (Ia), де R1, R2, R3 і n зберігають ті ж значення, що й у вихідній сполуці; d) здійснення гідролізу, якщо необхідно, естерної функціональної групи сполуки формули Ia, наприклад (у випадку трет-бутилового естеру) під дією органічної кислоти, такої як трифтороцтова кислота, у розчиннику, такому як дихлорметан, за методиками, добре відомими фахівцю в даній області техніки, з одержанням сполуки формули I у формі її вільної кислоти: R1 N O S (CH2)n COOH O R3 25 (Ib), де R1, R2, R3 і n зберігають ті ж значення, що й у вихідній сполуці. 6 UA 102397 C2 5 Необхідно відзначити, що в деяких умовах стадії b) і c) цього способу переважно можуть бути виконані за одну операцію (так званий спосіб в одному реакторі). Сполука формули (II), в якій R1 є галогеном або трифторметильную групою, R2 є C1C4алкільною групою, а n дорівнює 3 або 4, може бути одержана в результаті взаємодії ортойоданіліну формули: R1 I NH2 (VI) зі естером алкінової кислоти формули: (CH2)n COOR2 10 15 (VIII), де R2 є C1-C4алкільною групою, і n дорівнює 3 або 4; у присутності йодиду одновалентної міді, каталізатора на основі паладію, такого як, наприклад, біс(трифенілфосфін)паладію хлорид, і органічної основи, такої як, наприклад, триетиламін, у розчиннику, такому як, наприклад, диметилформамід (ДМФ), при температурі від температури навколишнього середовища до 80°C протягом 1-12 годин. Естер алкінової кислоти формули: (CH2)n COOR2 20 25 30 35 40 45 50 (VIII), де R2 є C1-C4алкільною групою, і n дорівнює 3 або 4, може бути одержаний, починаючи з відповідної алкінової кислоти, в результаті послідовної дії оксалілхлориду і потім алкілату металу формули R 2OM, в якому M є лужний метал, такий як, наприклад, натрій або калій. Сполуки згідно з винаходом у формі солей кислоти формули (Ib) з органічною або неорганічною основою можна одержати традиційним способом, використовуючи методи, добре відомі фахівцю в даній області техніки, наприклад шляхом змішування стехіометричних кількостей кислоти формули (Ib) і основи в розчиннику, такому як, наприклад, вода або водноспиртова суміш, і потім за допомогою ліофілізації одержаного розчину. На деяких реакційних стадіях, описаних вище, можливо й переважно замінити традиційні методи нагрівання нагріванням мікрохвильовим опроміненням, використовуючи реактори, які підходять для цього типу реакції. У цьому випадку фахівцю в даній області техніки буде зрозуміло, що тривалість нагрівання буде істотно знижена у порівнянні із тривалістю, необхідною у випадку загальноприйнятого нагрівання. Наведені далі приклади одержання сполук, які відповідають формулі (I), забезпечать краще розуміння винаходу. У цих прикладах, які не обмежують об’єм винаходу, приклади, які мають назву “підготовчий приклад”, є прикладами, що описують синтез проміжних сполук, а приклади, що мають назву “приклади”, описують синтез сполук формули (I) згідно з винаходом. Використані такі скорочення: - мМ: мілімоль(і), - ТГФ: тетрагідрофуран, - ДМФ: диметилформамід, - DCM: дихлорметан. Температури плавлення вимірюють на стенді Кофлера, а спектральні величини ядерного магнітного резонансу охарактеризовують хімічним зрушенням, розрахованим відносно TMS (тетраметилсилану), за кількістю протонів, асоційованих із сигналом, і за формою сигналу (с для синглету, д для дублету, т для триплету, к для квадруплету, м для мультиплету). Для кожної сполуки вказують використовувані робочу частоту і розчинник. Температура навколишнього середовища становить 20°C + 5°C. ПІДГОТОВЧИЙ ПРИКЛАД 1 6-[2-(((3-(1-метилетил)феніл)сульфоніл)аміно]-5-(трифтор-метил)феніл]-5-гексинової 7 UA 102397 C2 5 10 15 20 25 30 35 40 45 50 55 60 кислоти метиловий естер Одержували розчин 42,90 г (150,39 мМ) метилового естеру 6-[2-аміно-5(трифторметил)феніл]-5-гексинової кислоти в 500 мл піридину і додавали 37,90 г (173,29 мМ) 3(1-метилетил)бензолсульфонілхлориду. Суміш перемішували при температурі навколишнього середовища протягом 15 годин і потім виливали в суміш льоду й соляної кислоти. Одержану кислотну суміш тричі екстрагували етилацетатом. Об'єднані органічні фази сушили над сульфатом магнію й концентрували при зниженому тиску. Масло, що залишилося, очищували хроматографією на силікагелі, елююючи сумішшю циклогексан/етилацетат (9/1; об./об.). Це дозволило одержати 29,09 г цільової сполуки у вигляді блідо-жовтої маслянистої рідини (вихід = 41%). 1 H ЯМР (ДМСОd6, 250 МГц) δ =1,12 (д, J=6,9, 6H), 1,76 (к, J=7,0, 2H), 2,40 (т, J=7,0, 2H), 2,44 (т, J=7,0, 2H), 2,92 (к, J=6,9, 1H), 3,62 (с, 3H), 7,47-7,51 (м, 4H), 7,62-7,66 (м, 3H), 9,68 (с, 1H). ПРИКЛАД 1 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-бутанова кислоти метиловий естер Одержували розчин 28,12 г (60,15 мМ) естеру, одержаного згідно з Підготовчим прикладом 1, в 250 мл 1,2-дихлоретану й додавали 12,49 г (62,55 мМ) ацетату (двовалентної) міді моногідрату. Суміш поміщали в атмосферу азоту й піддавали кип'ятінню зі зворотним холодильником при перемішуванні протягом приблизно 15 годин. Реакційну суміш фільтрували й твердий залишок після фільтрації промивали на фільтрі, використовуючи DCM. Об'єднані фільтрати концентрували при зниженому тиску. Це дозволило одержати 27,70 г цільової сполуки у вигляді бежевих кристалів (вихід = 99%). т.пл. = 115°C. ПРИКЛАД 2 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-бутанова кислота 27,50 г (58,82 мМ) естеру, одержаного згідно з Прикладом 1, змішували з 450 мл ТГФ і додавали 4,23 г (176,47 мМ) гідроксиду літію в 100 мл води. Суміш перемішували протягом приблизно 15 годин при температурі навколишнього середовища й потім охолоджували до 0°C. Потім поступово додавали 180 мл 1 н. соляної кислоти при ретельному перемішуванні. Органічну фазу відокремлювали й половину розчинника випарювали без нагрівання, при зниженому тиску. Залишок після випарювання тричі екстрагували дихлорметаном. Об'єднані органічні фази сушили над сульфатом магнію, фільтрували й концентрували при зниженому тиску. Це дозволило одержати 26,22 г цільового продукту у вигляді білого порошку (вихід = 98%). т.пл. = 160°C. ПРИКЛАД 2a 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-бутанової кислотиі натрієва сіль 68 мг (0,15 мМ) кислоти, одержаної згідно з Прикладом 2, у розчині в 4 мл тетрагідрофурану змішували з 6 мг (0,15 мМ) гідроксиду натрію в розчині в 3 мл води. Суміш перемішували при температурі навколишнього середовища протягом 6 годин і потім концентрували при зниженому тиску. Це дозволило одержати 65 мг цільової солі у вигляді білого кристалічного порошку (вихід = 91%). т.пл. = 231°C. ПРИКЛАД 2b 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-бутанова кислоти сіль піперазину 400 мг (0,88 мМ) кислоти, одержаної згідно з Прикладом 2, розчиняли в 10 мл тетрагідрофурану й додавали 76 мг (0,88 мМ) піперазину. Реакційну суміш перемішували протягом ночі при температурі навколишнього середовища й потім концентрували при зниженому тиску. Це дозволило одержати 400 мг цільової солі у вигляді білого кристалічного порошку (вихід = 46%). т.пл. = 147°C. ПРИКЛАД 2c 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-бутанової кислоти сіль тріс(гідроксиметил)амінометану 400 мг (0,88 мМ) кислоти, одержаної згідно з Прикладом 2, розчиняли в 10 мл тетрагідрофурану й додавали 106,85 мг (0,88 мМ) тріс(гідроксиметил)-амінометану. Додавали 3 мл води, одержуючи розчин. Реакційну суміш перемішували протягом ночі при температурі навколишнього середовища й потім концентрували при зниженому тиску. Залишок тричі 8 UA 102397 C2 5 10 15 20 25 30 35 40 45 50 55 60 переносили у метанол, щораз випарюючи розчинник при зниженому тиску. Це дозволило одержати 480 мг цільової солі у вигляді білого кристалічного порошку (вихід = 95%). т.пл. = 126°C. ПІДГОТОВЧИЙ ПРИКЛАД 2 6-[5-хлор-2-[[[3-(1-метилетил)феніл]сульфоніл]аміно]феніл]-5-гексинової кислоти метиловий естер Процедуру виконували тим же способом, як і в Підготовчому прикладі 1, починаючи зі метилового естеру 6-(2-аміно-5-хлорфеніл)-5-гексинової кислоти, цільову сполуку одержували у вигляді коричневої маслянистої рідини (вихід = 96%). 1 H ЯМР (ДМСОd6, 300 МГц) δ =1,13 (д, J=6,9, 6H) 1,71 (к, J=7,1, 2H), 2,33 (т, J=7,1, 2H), 2,42 (т, J=7,4, 2H), 2,91 (к, J=6,9, 1H), 3,61 (с, 3H), 7,26 (д, J=7,3, 1H), 7,34-7,40 (м, 3H), 7,49-7,57 (м, 2H), 7,76-7,78 (м, 1H), 9,68 (с, 1H). ПРИКЛАД 3 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-хлор-1H-індол-2-бутанової кислоти метиловий естер Готували розчин 0,3 г (0,69 мМ) естеру, одержаного згідно з Підготовчим прикладом 2, в 13 мл 1,2-дихлоретану й додавали 0,21 г (1,05 мМ) ацетату двовалентної міді моногідрату. Проводили опромінення реакційної суміші в мікрохвильовій печі при 120°C протягом 15 хвилин, потім охолоджували й фільтрували. Залишок на фільтрі промивали DCM і потім фільтрат концентрували при зниженому тиску. Неочищений продукт очищали хроматографією на силікагелі, елююючи сумішшю циклогексан/етилацетат (9/1; об./об.). Це дозволило одержати 0,23 г цільової сполуки у вигляді бежевої твердої речовини (вихід = 77%). т.пл. = 94-97°C. 1 H ЯМР (ДМСОd6, 250 МГц) δ = 1,11 (д, J=6,9, 6H), 1,95 (к, J=7,4, 2H), 2,42 (т, J=7,4, 2H), 2,94 (к, J=7,4, 1H), 3,02 (т, J=7,4, 2H), 3,59 (с, 3H), 6,61 (с, 1H), 7,32 (дд, J=2,2 і 8,9, 1H), 7,47 (т, J=7,9, 1H), 7,56-7,63 (м, 4H), 8,06 (д, J=8,9, 1H). ПРИКЛАД 4 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-хлор-1H-індол-2-бутанова кислота Процедуру виконували способом, аналогічним такому Приклада 2, починаючи із сполуки, одержаної згідно з Прикладом 3, з одержанням цільового продукту у вигляді темно-бежевої твердої речовини (вихід = 93%). т.пл. = 128°C. ПІДГОТОВЧИЙ ПРИКЛАД 3 6-гептинової кислоти 1,1-диметилетиловий естер 8,00 г (63,41 мМ) 6-гептинової кислоти розчиняли в суміші 137 мл безводного дихлорметану й 0,70 мл безводного диметилформаміду. Краплями додавали 16,10 г (126,83 мМ) оксалілхлориду. Реакційну суміш перемішували при температурі навколишнього середовища протягом 1 години в атмосфері азоту й потім упарювали в атмосфері азоту. Продукт, що залишився, переносили в 137 мл тетрагідрофурану. Суміш охолоджували до 0°C і змішували порціями з 14,23 г (126,83 мМ) трет-бутилату калію. Реакційну суміш витримували при температурі навколишнього середовища при перемішуванні протягом години. Потім додавали 200 г льоду й 200 мл води. Суміш екстрагували 3 рази, використовуючи 200 мл ефіру, потім об'єднані органічні фази сушили над сульфатом магнію й концентрували при зниженому тиску. Це дозволило одержати 7,46 г цільової сполуки у вигляді коричневої маслянистої рідини (вихід = 65%). 1 H ЯМР (ДМСОd6, 250 МГц) δ = 1,40 (с, 9H), 1,40-1,45 (м, 4H), 2,13-2,22 (м, 4H), 2,75 (т, J=2,7, 1H). ПІДГОТОВЧИЙ ПРИКЛАД 4 7-[2-аміно-5-(трифторметил)феніл]-6-гептинової кислоти 1,1-диметилетиловий естер Одержували розчин 9,78 г (34,07 мМ) 2-йод-4-(трифторметил)аніліну й 7,45 г (40,89 мМ) естеру 6-гептинової кислоти, одержаного згідно з Підготовчим прикладом 3, в 136 мл триетиламіну. Додавали 1,20 г (1,70 мМ) дихлорбіс(трифенілфосфін)паладію й 0,3 г (1,70 мМ) йодиду одновалентної міді. Реакційну суміш перемішували, нагрівали при температурі дефлегмації в атмосфері азоту протягом 3 годин і потім концентрували при зниженому тиску. Залишок після випарювання переносили в етилацетат і промивали розчином гідрокарбонату натрію (приблизно 1 М у воді), потім 1 н. соляною кислотою й остаточно дистильованою водою. Органічну фазу сушили над сульфатом магнію, фільтрували й концентрували при зниженому тиску. Це дозволило одержати 12,38 г цільової сполуки у вигляді коричневої маслянистої рідини (вихід = 71%). 1 H ЯМР (ДМСОd6, 250 МГц) δ = 1,40 (с, 9H), 1,53-1,68 (м, 4H), 2,24 (т, J=8,4, 2H), 2,48 (т, J=8,1, 2H), 5,93 (с, 2H), 6,78 (д, J=10,2, 1H), 7,28-7,33 (м, 2H). 9 UA 102397 C2 5 10 15 20 25 30 35 40 45 50 55 ПІДГОТОВЧИЙ ПРИКЛАД 5 5-трифторметил-1H-індол-2-пентанової кислоти 1,1-диметилетиловий естер Одержували розчин 7,63 г (22,35 мМ) трет-бутилового естеру 7-[2-аміно-5(трифторметил)феніл]-6-гептинової кислоти в 44,70 мл 1,2-дихлоретану й додавали 6,69 г (33,52 мМ) ацетату двовалентної міді моногідрату. Суміш піддавали кип'ятінню зі зворотним холодильником при перемішуванні протягом 48 годин. Реакційну суміш фільтрували через поліамідний фільтр і потім фільтрат концентрували при зниженому тиску. Неочищений продукт очищали хроматографією на силікагелі, елююючи сумішшю циклогексан/етилацетат (9/1; об./об.). Це дозволило одержати 3,42 г цільової сполуки у вигляді жовтого порошку (вихід = 45%). 1 H ЯМР (ДМСОd6, 250 МГц) δ = 1,38 (с, 9H), 1,51-1,57 (м, 2H), 1,67-1,73 (м, 2H), 2,23 (т, J=8,4, 2H), 2,75 (т, J=8,7, 2H), 6,31 (с, 1H), 7,28 (дд, J=2,1 і 10,2, 1H), 7,44 (д, J=10,2, 1H), 7,79 (с, 1H). ПРИКЛАД 5 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-пентанової кислоти 1,1диметилетиловий естер 46,87 мг (1,17 мМ) гідриду натрію (60%-го в маслянистій рідині) додавали до розчину 200,00 мг (0,59 мМ) естеру, одержаного згідно з Підготовчим прикладом 5, в 0,5 мл ДМФ при 0°C. Цю суміш перемішували протягом 5 хвилин і як і раніше при 0°C додавали розчин 192,20 мг (0,88 мМ) 3-(1-метилетил)бензолсульфонілхлориду в 0,5 мл ДМФ. Суміш перемішували при температурі навколишнього середовища протягом 3 годин і потім додавали розчин хлориду амонію, щоб нейтралізувати слідові кількості гідриду натрію. Суміш екстрагували дихлорметаном. Органічну фазу концентрували при зниженому тиску й потім одержану реакційну суміш використали в реакції на наступній стадії без очищення. ПРИКЛАД 6 1-[[3-(1-Метилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-пентанова кислота Одержували розчин 200,00 мг (0,38 мМ) естеру, одержаного згідно з Прикладом 5, в 1 мл DCM і додавали 1 мл трифтороцтової кислоти. Реакційну суміш перемішували при температурі навколишнього середовища протягом 3 годин, потім переносили у DCM і концентрували при зниженому тиску. Неочищений продукт очищали хроматографією на силікагелі, елююючи сумішшю циклогексан/етилацетат (6/4; об./об.). Це дозволило одержати 50,00 мг цільової сполуки у вигляді білуватого порошку (вихід = 26%). т.пл. = 119°C. ПІДГОТОВЧИЙ ПРИКЛАД 6 3-(1,1-Диметилетил)-N-[2-йод-4-(трифторметил)феніл]бензолсульфонамід Одержували розчин 1,03 г (3,59 мМ) 2-йод-4-(трифторметил)аніліни в 5 мл піридину й додавали 1,00 г (4,31 мМ) 3-(1,1-диметилетил)бензолсульфонілхлориду. Після цього реакційну суміш перемішували при температурі навколишнього середовища протягом 4 годин. Реакційну суміш промивали 1 н. соляною кислотою й двічі екстрагували етилацетатом. Органічну фазу сушили над сульфатом магнію й потім фільтрували й концентрували при зниженому тиску. Неочищений продукт очищали хроматографією на силікагелі, елююючи сумішшю циклогексан/етилацетат (градієнт від 100/0 до 90/10; об./об.). Це дозволило одержати 730 мг цільової сполуки у вигляді білого кристалічного порошку (вихід = 42%). т.пл. = 111°C. ПРИКЛАД 7 1-[[3-(1,1-Диметилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-бутанової кислоти метиловий естер В атмосфері азоту готували суміш 250 мг (0,52 мМ) сполуки, одержаної згідно з Підготовчим прикладом 6, 4,93 мг (0,03 мМ) йодиду одновалентної міді, 9,08 мг (0,01 мМ) біс(трифенілфосфін)дихлорпаладію й 3 мл триетиламіну. Реакційну суміш перемішували при температурі навколишнього середовища протягом 10 хвилин. Додавали 120,31 мг (0,95 мМ) метилового естеру 5-гексинової кислоти в розчині в 3 мл диметилформаміду. Реакційну суміш нагрівали при температурі дефлегмації протягом 3 годин, потім промивали водою й екстрагували етилацетатом. Органічну фазу сушили над сульфатом магнію й концентрували при зниженому тиску. Неочищений продукт очищали хроматографією на силікагелі, елююючи сумішшю циклогексан/етилацетат (95/5; об./об.). Це дозволило одержати 115 мг цільового продукту у вигляді бежевого кристалічного порошку (вихід = 46%). т.пл. = 84°C. ПРИКЛАД 8 1-[[3-(1,1-Диметилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-бутанова кислота 10 UA 102397 C2 5 1015 20 25 Процедуру виконували тим же способом, як і для Приклада 2, починаючи із сполуки, одержаної згідно з Прикладом 7, з одержанням цільового продукту у вигляді білого порошку (вихід = 27%). т.пл. = 135-141°C. ПРИКЛАД 9 1-[[3-(1,1-Диметилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-пентанової кислоти метиловий естер В атмосфері азоту готували суміш 57,93 г (119,87 мМ) сполуки, одержаної згідно з Підготовчим прикладом 6, і 350 мл диметилформаміду й перемішували до повного розчинення продукту. Потім послідовно додавали 21,84 г (155,83 мМ) метилового естеру 4-пентинової кислоти, 1,14 г (5,99 мМ) йодиду одновалентної міді й 1,68 г (2,40 мМ) біс(трифенілфосфін)дихлорпаладію. Цю суміш перемішували при температурі навколишнього середовища протягом 15 хвилин і потім змішували, додаючи краплями 174 мл триетиламіну. Реакційну суміш нагрівали протягом 14 годин при 80°C, охолоджували, потім проводили гідроліз, використовуючи 1 л води, і екстрагували етилацетатом. Органічну фазу сушили над сульфатом магнію, фільтрували й концентрували при зниженому тиску. Одержаний маслянистий продукт розчиняли при 40°C в ізопропіловому ефірі. Одержаний розчин фільтрували й концентрували при зниженому тиску. Одержаний продукт перекристалізовували із суміші 140 мл ізопропанолу й 60 мл води. Це дозволило одержати 46,51 г цільового продукту у вигляді білуватої твердої речовини (вихід = 78%). т.пл. = 77°C. ПРИКЛАД 10 1-[[3-(1,1-Диметилетил)феніл]сульфоніл]-5-(трифторметил)-1H-індол-2-пентанова кислота Процедуру виконували тим же способом, як і для Прикладу 2, починаючи із сполуки, одержаної згідно з Прикладом 9, з одержанням цільового продукту у вигляді білуватої твердої речовини (вихід = 94%). т.пл. = 135°C. Описані вище сполуки згідно з винаходом представлені у наведеній нижче таблиці. Таблиця I R1 (CH2)n COOR2 N O O S R3 Ін. 1 2 3 4 5 6 7 8 9 10 R1 5-CF3 5-CF3 5-Cl 5-Cl 5-CF3 5-CF3 5-CF3 5-CF3 5-CF3 5-CF3 n 3 3 3 3 4 4 3 3 4 4 R2 CH3 H CH3 H C(CH3)3 H CH3 H CH3 H R3 CH(CH3)2 CH(CH3)2 CH(CH3)2 CH(CH3)2 CH(CH3)2 CH(CH3)2 C(CH3)3 C(CH3)3 C(CH3)3 C(CH3)3 30 35 Фармакологічна активність Сполуки згідно з винаходом піддавали біологічним тестам з метою оцінки їх потенціалу на предмет лікування або профілактики деяких нейродегенеративних патологій. Почнемо з того, що з використанням аналізу in vitro вимірювали здатність сполук згідно з винаходом діяти як активатор гетеродимерів, утворених ядерним рецептором NURR-1 і ядерними рецепторами RXR. 11 UA 102397 C2 5 10 15 20 Як первинний скринінговий тест використали аналіз трансактивації. Cos-7-клітини спільно трансфікували плазмідою, що експресуює химерну структуру (людський рецептор NURR-1)Gal4, плазмідою, що експресує людський рецептор RXR (рецептор RXR або RXR), і репортерною плазмідою 5Gal4pGL3-TK-Luc. Трансфекцію здійснювали, використовуючи хімічний агент (Jet PEI). Трансфіковані клітини розподіляли в 384-лункові планшети й відставляли на 24 години. Через 24 години культуральне середовище заміняли. До культурального середовища -4 -10 додавали продукти, що тестуються, (кінцева концентрація від 10 до 310 M). Після інкубації протягом ночі вимірювали експресію люциферази після додавання “SteadyGlo” згідно з інструкцією виробника (Promega). 4-[[6-метил-2-феніл-5-(2-пропеніл)-4-піримідиніл]аміно]бензойну кислоту (агоніст RXR, -5 названий XCT0135908) у концентрації 210 M використовували для порівняння. Рівні індукції розраховували відносно базальної активності кожного гетеродимеру. Результати виражали у вигляді відсотка рівня індукції по відношенню до рівня індукції, одержаного для порівняння (рівень індукції сполуки для порівняння умовно дорівнює 100%). Сполуки згідно з винаходом демонструють рівень індукції до 104% (NURR1/RXR) і 88% (NURR1/RXR) і величини EC50 аж до 26 нМ (NURR1/RXR) і 20 нМ (NURR1/RXR). Деякі сполуки згідно з винаходом мають EC50 менше 100 нМ, особливо у відношенні гетеродимеру NURR-1/RXR. Як приклад, для сполук згідно з винаходом одержані порівняльні результати, наведені нижче, виражені у вигляді відсотка по відношенню до сполуки для порівняння - активатора NURR-1/RXR (XCT0135908): Сполука Приклад 2 Приклад 8 Приклад 10 Порівняльний приклад* 25 30 35 40 45 50 hNurr1_RXRFL EC50 (нМ) Еф. ( %) 113 79 20 70 77 88 1108 74 hNurr1_RXRFL EC50 (нМ) Еф. ( %) 73 86 26 100 55 104 571 75 *: Приклад 76 із заявки WO 2007/026097. Еф.: ефективність в % по відношенню до сполуки для порівняння XCT0135908. З метою порівняння дослідження також було проведено для сполуки із Приклада 76 заявки WO 2007/026097, що має відносно близьку до сполук згідно з винаходом структуру, і результати для якого показують, що концентрація, при якій дана сполука дає половину від максимальної ефективності (EC50), щонайменше в 10 разів більше концентрації сполук, описаних у винаході. Першу серію тестів in vivo проводили, використовуючи ряд сполук згідно з винаходом, з метою визначення їх фармакокінетичного профілю для головного мозку й плазми на самцях мишей C57Bl6 і, отже, підтвердження того, що ці сполуки проходять через гематоенцефалічний бар'єр. Використали наведений далі протокол. Для цього дослідження використали самців C57Bl6 мишей (25-30 г) від Janvier, Le Genest-StIsle, France (12 мишей на одну дозу). Тварин годували стандартним кормом для гризунів (Purina Mills, St. Louis, MO), розміщали в клітках і витримували в умовах 12год/12год циклу світло/темрява, підтримуючи при цьому у приміщенні температуру 22±2°C і рівень вологості 55±10%. Мишей не піддавали голодуванню перед введенням. Протягом усього експерименту воду давали без обмеження. Тестовану сполуку вводили перорально (п/о) у концентрації 10 мг/кг. Для перорального введення в дозі 10 мг/кг тварин годували через шлунковий зонд суспензією тестованої сполуки (10 мол/кг), приготовленої в 1%-ій метилцелюлозі (з в'язкістю 400 сП (400 мПа с)). Тварин умертвляли під анестезією в моменти часу 15 хв, 30 хв, 1 год, 3 год, 6 год і 8 год після зондового годування. У кожний момент часу й у кожної умертвленої тварини відбирали кров і витягали головний мозок. По 1 мл крові, зібраної в 1,5 мл пробірки, що містять по 20 мкл упареного антикоагулянту (розчину гепаринату натрію в концентрації 1000 міжнародних одиниць (IU)/мл), центрифугували 12 UA 102397 C2 5 10 при 4500 g протягом 3 хв, одержуючи приблизно 400 мкл плазми. Плазму розділяли на дві аліквоти по 200 мкл, які зберігали при -20°C до моменту проведення екстракції шляхом осадження білків з подальшим аналізом рідинною хроматографією у сполученні з тандемною мас-спектрометрією (LC-MS/MS) для кількісного визначення тестованої сполуки. Відразу після вилучення зразки головного мозку занурювали в рідкий азот і потім зберігали при -20°C для аналізу. Після цього зразки головного мозку подрібнювали у присутності суміші водного/органічного розчинника з одержанням гомогенату. Потім ці гомогенати центрифугували, тестовану сполуку екстрагували з одержаного супернатанту рідина-рідинною екстракцією й далі проводили кількісне визначення за допомогою LC-MS/MS. Фармакокінетичні (ФК) параметри визначали, ґрунтуючись на некомпартментальному підході в Excel. Площу під кривою (AUC0-t) визначали методом трапецій з лінійною апроксимацією. Як приклад представлені такі результати, одержані з використанням сполук Прикладів 2, 8 і 10: 15 Сполука Приклад 2 Приклад 8 Приклад 10 20 25 30 35 40 45 50 55 ФК дані після перорального введення мишам: 10 мг/кг AUCмозок Відношення AUCмозок/AUCплазма 3318 0,67 2371 0,87 1689 0,80 Другу серію тестів in vivo проводили, використовуючи сполуку згідно з винаходом, з метою підтвердження того, що дані молекули мають очікуваний нейропротективний ефект. Сполуку з Приклада 2 тестували в моделі мишей, оброблених 1-метил-4-феніл-1,2,3,6тетрагідропіридином (MPTP), щоб підтвердити її потенційну активність. MPTP є нейротоксином, який підсилює необоротні симптоми хвороби Паркінсона, руйнуючи деякі нейрони у чорній субстанції головного мозку. Нижче наведений використаний протокол. Самців C57BL6/J мишей у віці 10-12 тижнів на початку досліджень, розділяли на групи по 8 тварин. Сполуку вводили перорально і два рази на добу в цілому протягом 11 діб. Введення починали за 3 доби до обробки токсином MPTP у концентрації 20 або 25 мг/кг. MPTP уводили один раз на добу шляхом внутрішньочеревинної ін'єкції протягом 5 діб. Введення тестованої сполуки продовжували протягом 3 діб після обробки MPTP. Одна група мишей одержувала тільки розріджувач (0,5%-ий розчин метилцелюлози). Тварин піддавали евтаназії після закінчення останнього зондового годування й витягали смугасте тіло. Зі смугастого тіла екстрагували дофамін і кількість дофаміну (DA), виражена в нг на один г смугастого тіла (середнє ± SEM (стандартна помилка середнього)), вимірювали високоефективною рідинною хроматографією (HPLC) із застосуванням електрохімічної детекції. Одержані результати наведені на доданих Фіг. 1-3. Ці результати показують, що введення MPTP підсилює характерне зниження рівня дофаміну в смугастому тілі, і що сполуки, які відповідають Прикладам 2, 8 і 10, послабляють дозозалежним чином дію MPTP, токсину, який підсилює синдром паркінсонізму. Таким чином, значний ефект спостерігається в дозах 10 і 30 мг/кг: сполуки згідно з винаходом, уведені перорально, мають здатність відновлення дофамінергічної активності, інгібованої MPTP, у головному мозку. Сполуки цього типу, які проходять через гематоенцефалічний бар'єр і сприятливо діють на комунікацію між нейронами, можуть бути переважно використані як активний початок у ліках, призначених для лікування хвороби Паркінсона. Ці результати in vitro і in vivo показують, що сполуки згідно з винаходом здатні модифікувати механізми даного захворювання в деяких тваринних і клітинних моделях, і припиняти дегенеративний процес шляхом збільшення кількості нейропротекторних агентів, які протидіють клітинній смерті дофамінергічних нейронів. Таким чином, дані результати підтверджують інтерес до цих сполук із точки зору їх застосування як активний початок у ліках, призначених для профілактики або лікування нейродегенеративних захворювань і, більш конкретно, хвороби Паркінсона. Крім того, згідно з винаходом запропонована фармацевтична композиція, яка містить як активний початок щонайменше одну сполуку формули (I) або одну з її фармацевтично прийнятних солей. В іншому аспекті дана заявка ставить задачу застосування фармацевтичної композиції такого типу для профілактики або лікування захворювань, що залучають рецептор NURR-1, особливо нейродегенеративних захворювань і, більш конкретно, хвороби Паркінсона. 13 UA 102397 C2 5 10 15 20 25 Ці фармацевтичні композиції можна виготовити традиційним способом, використовуючи фармацевтично прийнятні ексціпієнти, з одержанням форм, які можна вводити парентерально або, переважно, перорально, таких як, наприклад, таблетки або капсули. У випадку ін'єкційних форм сполуки формули (I) варто переважно застосовувати у вигляді солей, які розчинні у водному середовищі. Як зазначено вище, такі солі переважно утворюються між сполукам формули (Ib) (кислотою) і фармакологічно прийнятною нетоксичною основою. Композиція може бути або розчином сполуки в ізотонічному водному середовищу в присутності розчинних ексціпієнтів, або ліофілізат сполуки, до якого екстемпорально додають розчинник. Ці сполуки з Підготовчих прикладів можна вводити в перфузійній формі або у вигляді болюсної ін’єкції, залежно від потреб пацієнта. Із практичної точки зору у випадку парентерального введення сполуки добова доза для людини переважно складає 2-250 мг. Сполуки з Підготовчих прикладів, які можуть бути уведені перорально, переважно будуть представлені у вигляді капсули або таблетки, що містить тонко здрібнене або, ще краще, мікронізовану сполуку згідно з винаходом, змішану з ексціпієнтами, добре відомими фахівцю в даній області техніки, такими як, наприклад, лактоза, прежелатинізований крохмаль і стеарат магнію. Наприклад, суміш, яка складається з 500 г сполуки із Приклада 2, тонко подрібненої, 500 г прежелатинізованого крохмалю, 1250 г лактози, 15 г лаурилсульфату натрію й 235 г полівінілпіролідону, піддавали грануляції. Потім цю гранульовану суміш додавали до 20 г стеарату магнію й 80 г мікрокристалічної целюлози й одержану суміш розподіляли, після розмелювання й просіювання, по 260 мг у капсули. Це дозволило виготовити капсули, кожна з яких містить 50 мг активного початку. Із практичної точки зору у випадку перорального введення сполуки добова доза для людини переважно складає 5-500 мг. ФОРМУЛА ВИНАХОДУ 30 1. Похідне індолу, що має терапевтичну активність, вибране із і) сполук формули (І) R1 (CH2)n COOR2 N O S O R3 35 40 45 , (I) де R1 є галогеном або трифторметильною групою, R2 є атомом водню або С1-С4алкільною групою, R3 є ізопропільною (1-метилетильною) групою або трет-бутильною (1,1-диметилетильною) групою, і n дорівнює 3 або 4, і іі) фармацевтично прийнятних солей зазначених сполук формули (І). 2. Сполука згідно з п. 1, де в наведеній вище формулі (І) R3 є ізопропільною групою. 3. Сполука згідно з п. 1, де в наведеній вище формулі (І) R3 є тpeт-бутильною групою. 4. Сполука за будь-яким з пп. 1-3, де в наведеній вище формулі (І) R2 є атомом водню. 5. Фармацевтична композиція, яка містить щонайменше одну сполуку за будь-яким з пп. 1-4 як активну речовину й щонайменше один фармацевтично прийнятний ексципієнт. 6. Застосування похідної індолу за будь-яким з пп. 1-4 для виготовлення ліків, призначених для лікування або профілактики захворювань, в які залучений рецептор NURR-1. 7. Застосування згідно з п. 6 для виготовлення ліків, призначених для лікування і профілактики нейродегенеративних захворювань. 8. Застосування згідно з п. 7, де вищезгадане захворювання є хворобою Паркінсона. 14 UA 102397 C2 15 UA 102397 C2 Комп’ютерна верстка А. Крулевський Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 16
ДивитисяДодаткова інформація
Назва патенту англійськоюIndole derivatives as nurr-1 activators and use thereof fro the treatment of parkinson's disease
Автори англійськоюBoubia, Benaissa, van Vliet, Bernard, Johannes, den Hartog, Jacobus, Antonius, Joseph, McCheary, Andrew, Tallandier, Mireille, van Dongen, Maria, Johanna, Petronella, Poupardin-Olivier, Olivia
Назва патенту російськоюПроизводные индола как активаторы nurr-1 и их применение для лечения болезни паркинсона
Автори російськоюБубиа Бенаисса, ван Влит Бернард Йоханнес, ден Хартог Якобус Антониус Йозеф, МакКрири Эндрю, Таллондье Мирей, ван Донген Мария Йоханна Петронелла, Пупарден-Оливье Оливия
МПК / Мітки
МПК: A61P 25/16, C07D 209/18, C07D 209/12, A61K 31/405, A61P 25/28
Мітки: індолу, хвороби, похідні, застосування, лікування, активаторі, nurr-1, паркінсона
Код посилання
<a href="https://ua.patents.su/18-102397-pokhidni-indolu-yak-aktivatori-nurr-1-ta-kh-zastosuvannya-dlya-likuvannya-khvorobi-parkinsona.html" target="_blank" rel="follow" title="База патентів України">Похідні індолу як активатори nurr-1 та їх застосування для лікування хвороби паркінсона</a>
Катехоламінові похідні, придатні для лікування хвороби паркінсона

Номер патенту: 97989
Опубліковано: 10.04.2012
Автори: Ларсен Дженіфер, Банґ-Андерсен Бенні, Йорґенсен Мортен, Мьорк Нільс, Пюшл Аск
МПК: A61P 25/00, A61K 31/4741, C07D 491/04
Мітки: паркінсона, лікування, катехоламінові, придатні, похідні, хвороби
Формула / Реферат:
1. Сполуки структури І, де n дорівнює 0 або 1; де R1 і R2 об'єднані і утворюють метиленову (СН2) групу, та де R3 вибраний з групи, що складається з водню, метилу, етилу, н-пропілу, циклопропілу, циклобутилу, алілу, пропаргілу, гідроксіетилу, 3-фторпропілу і 2-фторетилу,та їх фармацевтично...
Застосування заміщених 2-амінотетралінів для випереджувального лікування хвороби паркінсона

Номер патенту: 83691
Опубліковано: 11.08.2008
Автори: Шеллер Дітер, Дрессен Франк
МПК: A61P 25/16, A61K 31/381
Мітки: заміщених, застосування, 2-амінотетралінів, лікування, випереджувального, паркінсона, хвороби
Формула / Реферат:
1. Застосування сполуки загальної формули І,де:n = 1-5;R1 вибрано з групи: , ,де X - S, О або NH;R2 означає ОА; з А, вибраним з групи: Н, C1-12 алкіл, алкоксиметил або група
Похідні індолу, композиція на їх основі (варіанти) та їх застосування

Номер патенту: 73507
Опубліковано: 15.08.2005
Автори: Зісапель Нава, Лаудон Моше
МПК: A61P 15/18, A61P 11/06, A61P 25/00, A61P 25/18, A61P 25/28, A61K 31/427, A61K 31/4045, C07D 417/12, A61P 29/00, A61P 25/04, A61P 25/22, A61P 25/16, C07D 209/16, A61P 5/06, A61P 7/00, A61K 8/49, A61P 27/02, A61P 25/20, A61P 9/12, A61P 7/02, A61P 35/00, C07D 405/12, A61K 31/422, A61P 19/02, C07D 413/12, A61Q 17/00, A61P 25/08, A61P 9/10, A61P 3/04, A61P 25/06, A61Q 19/00, C07D 209/14, A61P 31/18
Мітки: індолу, похідні, варіанти, основі, застосування, композиція
Формула / Реферат:
1. Сполуки за формулою (IIА): (IIА)і їх кислотні адитивні солі, де дані сполуки є основними, які відрізняються тим, що:A є C 1-4 алкіленом;Z1 є -NH-X-Y або дигідроксицинамоїлокси,Х є >CH2 або >С=О;Y є тетрагідрофурилом, фурилом, ацетоксифенілом або стирилом, заміщеним одним або більше гідрокси і метокси, за винятком...
Спосіб лікування хвороби паркінсона, adhd та мікроаденом за допомогою піридопіразинів та фармацевтична композиція

Номер патенту: 72471
Опубліковано: 15.03.2005
Автори: Джексон Еліза Роуз, Зорн Стевін Говард, Маклін Стеффорд
МПК: A61P 25/16, A61K 31/505
Мітки: лікування, паркінсона, спосіб, хвороби, мікроаденом, композиція, фармацевтична, допомогою, піридопіразинів
Формула / Реферат:
1. Спосіб лікування розладів, вибраних з хвороби Паркінсона, гіперактивного розладу дефіциту уваги (ADHD) та мікроаденом у ссавця, який полягає у введенні ссавцеві, що потребує такого лікування, сполуки формулиабо її фармацевтично прийнятної солі приєднання кислоти, де Х є N або СН;Y є, ,або ,Z є, SCH2, OCH2, Y1(СH2)n або Y1(СН2)n, заміщений на вуглеці метильними групами у кількості до...
Активне начало лікарського засобу, призначеного для лікування хвороби паркінсона і синдромів паркінсона та спосіб одержання вказаного лікарського засобу

Номер патенту: 29464
Опубліковано: 15.11.2000
Автори: ЛУВЕЛЬ Ерік, Добль Адам, ШТУТЦМАНН Жан-Марі, Міке Жан-Марі, Дюбеда П'єр, Буаро Ален, Меньє Міріель
МПК: A61K 31/425
Мітки: активне, призначеного, одержання, лікування, синдромів, вказаного, засобу, лікарського, паркінсона, спосіб, хвороби, начало
Текст:
...могут использоваться орально или парентерально. Примером твердых композиций для орального введения могут служить таблетки, пилюли, порошки (желатиновые капсулы, облатки) или гранулы. В этих композициях основное действующее вещество, предлагаемое в настоящем изобретении, смешано с одним или несколькими инертными разбавителями, например, крахмалом, целлюлозой, сахарозой, лактозой или двуокисью кремния в потоке аргона. Эти композиции...
Попередній патент: Спосіб вимірювання часових інтервалів та пристрій для його здійснення
Наступний патент: Підвищення пропускної здатності бездротового зв’язку
Випадковий патент: Елемент для захисту напою від підробки