Похідні 5,7-дизаміщених[1,3]тіоазоло[4,5-d]піримідин-2(3н)аміну та їх застосування у терапії
Номер патенту: 95966
Опубліковано: 26.09.2011
Автори: Йоганссон Рольф, Карлстрьом Софія, Сліво Кен, Нордволл Ґуннар, Рейн Тобіас, Керс Анніка





Формула / Реферат
1. Сполука формули (І)
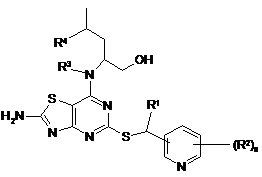 , (І)
, (І)
де:
R1 - CH3 aбo CF3;
R2 - галоген, CN або С1-6алкіл;
R3 - H абоСН3;
R4 - Н або СН3;
n = 0, 1 або 2;
як вільна основа або її фармацевтично прийнятна сіль, сольват або сольват солі.
2. Сполука за п. 1, де n = 1.
3. Сполука за п. 1, де R1 - СН3.
4. Сполука за п. 1, де R2 - галоген або CN.
5. Сполука за п. 1, де R2 - F або Сl.
6. Сполука за п. 1, де R2 - CN.
7. Сполука за п. 1, де n = 1; R1 - СН3; та R2 - F, Сl або CN.
8. Сполука за п. 1, де R3 - Н.
9. Сполука за п. 1, де R4 - СН3.
10. Сполука, яку вибрано з групи:
(2R)-2-[(2-аміно-5-{[1-(5-флуорпіридин-2-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]-4-метилпентан-1-ол;
(2R)-2-[(2-аміно-5-{[(1S)-1-(5-хлорпіридин-2-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]-4-метилпентан-1-ол;
(2R)-2-[(2-аміно-5-{[(1S)-1-(5-флуорпіридин-2-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]-4-метилпентан-1-ол;
(2R)-2-[(2-аміно-5-{[1-(3-хлорпіридин-4-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]-4-метилпентан-1-ол;
(2R)-2-[(2-аміно-5-{[(1S)-1-(3-хлорпіридин-4-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]-4-метилпентан-1-ол;
(2R)-2-[(2-аміно-5-{[(1R)-1-(3-хлорпіридин-4-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]-4-метилпентан-1-ол;
(2R)-2-[(2-аміно-5-{[(1S)-1-(3-флуорпіридин-2-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]-4-метилпентан-1-ол;
(2R)-2-[(2-аміно-5-{[(1S)-1-(6-хлорпіридин-3-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)(метил)аміно]пентан-1-ол;
(2R)-2-[(2-аміно-5-{[(1S)-1-(6-хлорпіридин-3-іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]пентан-1-ол та
2-{(1S)-1-[(2-аміно-7-{[(1R)-1-(гідроксиметил)-3-метилбутил]аміно}[1,3]тіоазоло[4,5-d]піримідин-5-іл)тіо]етил}ізонікотинонітрил;
як вільна основа або її фармацевтично прийнятна сіль, сольват або сольват солі.
11. Сполука за будь-яким з пп. 1-10 або її фармацевтично прийнятна сіль для застосування як медикаменту.
12. Сполука за будь-яким з пп. 1-10 або її фармацевтично прийнятна сіль у суміші з фармацевтично прийнятним розріджувачем або носієм.
13. Застосування сполуки формули (І) за будь-яким з пп. 1-10 або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики нейродегенеративних розладів, демієлінізаційної хвороби, серцево- та церебрально-васкулярних атеросклеротичних розладів, хвороби периферійних артерій, ревматоїдного артриту, хвороб легень, як-то COPD, астма або біль.
14. Застосування сполуки формули (І) за будь-яким з пп. 1-10 або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики розсіяного склерозу.
15. Застосування сполуки формули (І) за будь-яким з пп. 1-10 або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики атеросклерозу зміною складу бляшок для зменшення ризику руйнування бляшок та випадків атеротромбозу.
Текст
1. Сполука формули (І) C2 2 (19) 1 3 95966 4 як вільна основа або її фармацевтично прийнятна сіль, сольват або сольват солі. 11. Сполука за будь-яким з пп. 1-10 або її фармацевтично прийнятна сіль для застосування як медикаменту. 12. Сполука за будь-яким з пп. 1-10 або її фармацевтично прийнятна сіль у суміші з фармацевтично прийнятним розріджувачем або носієм. 13. Застосування сполуки формули (І) за будьяким з пп. 1-10 або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики нейродегенеративних розладів, демієлінізаційної хвороби, серцево- та церебрально васкулярних атеросклеротичних розладів, хвороби периферійних артерій, ревматоїдного артриту, хвороб легень, як-то COPD, астма або біль. 14. Застосування сполуки формули (І) за будьяким з пп. 1-10 або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики розсіяного склерозу. 15. Застосування сполуки формули (І) за будьяким з пп. 1-10 або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики атеросклерозу зміною складу бляшок для зменшення ризику руйнування бляшок та випадків атеротромбозу. Заявлений винахід розкриває нові похідні 5заміщених 7-аміно-[1,3]тіоазоло[4,5-d]піримідинів разом зі способами їх отримання, фармацевтичні композиції, що їх містять та їх застосування у терапії. Хемокіни грають важливу роль в імунній та запальній реакціях у різних хворобах та розладах, як-то астма, атеросклероз та алергічні хвороби, а також автоімунні патології, як-то ревматоїдний артрит та розсіяний склероз. Ці малі секретовані молекули є зростаючою надродиною білків по 8-14 кДа, що характеризуються збереженим цистеїновим мотифом. Зараз надродина хемокінів має чотири групи, що виявляють характеристичні структурні мотифи, родини С-Х-С, С-С та С-Х3-С та ХС. Родини С-Х-С та С-С мають подібність послідовностей та відрізняються одна від одної на основі одиночної амінокислотної вставки між NHпроксимальною парою цистеїнових залишків. Родина С-Х3-С відрізняється від інших двох родин на основі того, що має потрійну амінокислотну вставку між NH-проксимальною парою цистеїнових залишків. На відміну, члени родини ХС втратили один з перших двох цистеїнових залишків. Хемокіни С-Х-С охоплюють кілька потужних хемоатрактантів та активаторів нейтрофілів, як-то інтерлейкін-8 (IL-8) та нейтрофіл-активувальний пептид 2 (NAP-2). Хемокіни С-С охоплюють потужні хемоатрактанти моноцитів, лімфоцитів та нейтрофілів. Приклади охоплюють тимотаксини моноцитів людини 1-3 (МСР-1, МСР-2 та МСР-3), RANTES (Регульовані на активацію, експресовані та секретовані нормальними Т-клітинами), еотаксин та запальні білки макрофагів 1 та 1 (ΜΙΡ-1 та ΜΙΡ-1). Хемокін С-Х3-С (також відомий як фракталкін) є потужним хемоатрактантом та активатором мікроглій у центральній нервовій системі (ЦНС) а також моноцитів, Т-клітин, NK-клітин та мастоцитів. Дослідження продемонстрували, що дія хемокінів опосередкована підродинами сполучених з Gбілком рецепторів, серед котрих є рецептори, позначені як CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 та CCR11 (для родини С-С); CXCR1, CXCR2, CXCR3, CXCR4 та CXCR5 (для родини С-Х-С) та CX3CR1 для родини С-Х3-С. Ці рецептори є гарними цілями для розробки ліків, оскільки засоби, що модулюють ці рецептори, могли б бути корисними у лікуванні вищезгаданих розладів та хвороб. WO 01/58907 розкриває деякі 2-заміщені похідні 4-аміно-тіоазолопіримідинів, що є корисними як антагоністи рецепторів, зв'язаних з родинами хемокінів С-Х-С та С-С, зокрема як антагоністи рецептору CX3CR2. Заявлений винахід розкриває групу сполук, що є спорідненими зі сполуками, розкритими у WO 01/58907, але мають тип структури, що не представлений там конкретно. При порівнянні з прикладами, розкритими у WO 01/58907, сполуки заявленого винаходу виявляють неочікувано корисні властивості як антагоністи рецептору CX3CR1. Заявлений винахід розкриває сполуку формули (І) де 1 R - CH3 або CF3; 2 R - галоген, CN або С1-6алкіл; 3 R - H або CH3; 4 R - H абоСН3; n=0, 1 або 2; як вільну основу або її фармацевтично прийнятну сіль, сольват або сольват солі. В одному втіленні винаходу розкрито сполуки формули (І), де n=1. У ще одному втіленні винаходу розкрито спо1 луки формули (І), де R - СН3. У ще одному втіленні винаходу розкрито спо2 луки формули (І), де R - галоген або CN. 5 У ще одному втіленні винаходу розкрито спо2 луки формули (І), де R - F або СІ. У ще одному втіленні винаходу розкрито спо2 луки формули (І), де R - CN. У ще одному втіленні винаходу розкрито спо1 2 луки формули (І), де n=1; R - СН3; та R - F, СІ або CN. У ще одному втіленні винаходу розкрито сполуки формули (І), де піридин є приєднаним через свою 5-позицію та має СІ у 2-позиції. У ще одному втіленні винаходу розкрито сполуки формули (І), де піридин є приєднаним через свою 2-позицію та має CN у 4-позиції. У ще одному втіленні винаходу розкрито сполуки формули (І), де піридин є приєднаним через свою 2-позицію та має F у 5-позиції. У ще одному втіленні винаходу розкрито сполуки формули (І), де піридин є приєднаним через свою 2-позицію та має СІ у 5-позиції. У ще одному втіленні винаходу розкрито сполуки формули (І), де піридин є приєднаним через свою 2-позицію та має F у 3-позиції. У ще одному втіленні винаходу розкрито сполуки формули (І), де піридин є приєднаним через свою 4-позицію та має СІ у 3-позиції. У ще одному втіленні винаходу розкрито спо3 луки формули (І), де R - Н. У ще одному втіленні винаходу розкрито спо4 луки формули (І), де R - СН3. У ще одному втіленні винаходу розкрито сполуки формули (І), вибрані з групи: (2R)-2-[(2-аміно-5-{[1-(5-флуорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол; (2R)-2-[(2-аміно-5-{[(1S)-1-(5-хлорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол; (2R)-2-[(2-аміно-5-{[(1S)-1-(5-флуорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол; (2R)-2-[(2-аміно-5-{[1-(3-хлорпіридин-4іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол; (2R)-2-[(2-аміно-5-{[(1S)-1-(3-хлорпіридин-4іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол; (2R)-2-[(2-аміно-5-{[(1R)-1-(3-хлорпіридин-4іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол; (2R)-2-[(2-аміно-5-{[(1S)-1-(3-флуорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин~7іл)аміно]-4-метилпентан-1-ол; (2R)-2-[(2-аміно-5-{[(1S)-1-(6-хлорпіридин-3іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7іл)(метил)аміно]пентан-1-ол; (2R)-2-[(2-аміно-5-{[(1S)-1-(6-хлорпіридин-3іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7іл)аміно]пентан-1-ол;та 2-{(1S)-1-[(2-аміно-7-{[(1R)-1-(гідроксиметил)-3метилбутил]аміно}[1,3]тіоазоло[4,5-d]піримідин-5іл)тіо]етил}ізонікотинонітрил; як вільну основу або її фармацевтично прийнятну сіль, сольват або сольват солі. Сполуки формули (І) можуть існувати у стереоізомерних та/або таутомерних формах. Слід 95966 6 розуміти, що усі енантіомери, діастереомери, рацемати, таутомери та їх суміші є охопленими рамками винаходу. При порівнянні зі сполуками, розкритими у WO 01/58907, сполуки заявленого винаходу характеризуються наявністю розгалуженого тіоалкілпіридилу у 5-позиції тіоазолопіримідинової кільцевої системи. Тобто, сполуки заявленого винаходу ма1 ють R , що не є гідрогеном. Згідно з винаходом ми крім того розкриваємо спосіб отримання сполуки формули (І), або її фармацевтично прийнятної солі, котрий полягає у наступному: a) реакція сполуки формули (II): 3 4 де R та R визначені у формулі (І); зі сполукою формули (III): 1 2 1 1 2 де R , R , та n визначені у формулі (І) та L відщеплювана група; або b) реакція сполуки формули (IV) 2 де R , R та n визначені у формулі (І) та L - відщеплювана група; зі сполукою формули (V) 3 4 де R та R визначені у формулі (І); 7 та де необхідно, перетворення утвореної сполуки формули (І), або ще одної її солі в її фармацевтично прийнятну сіль; або перетворення утвореної сполуки формули (І) в іншу сполуку формули (І); та, де потрібно, перетворення утвореної сполуки формули (І) в її оптичний ізомер. У способі (а), реагенти (II) та (III) сполучають разом у придатному органічному розчиннику, як-то диметилсульфоксид (ДМСО), ацетонітрил або 1метил-2~піролідинон (NMP). Реакцію необов'язково проводять при наявності доданої органічної або неорганічної основи, як-то триетиламін, N,Nдіізопропілетиламін (DIPEA) або натрій гідрид. Реакцію необов'язково проводять при наявності помірного відновнику, як-то натрій борогідрид. Реакцію проводять при придатній температурі, звичайно між кімнатною температурою та температурою кипіння розчиннику. Реакція загалом продовжується протягом приблизно одної години одного тижня, або доки аналіз не показує, що утворення потрібного продукту завершене. У способі (b), реагенти (IV) та (V) сполучають разом у придатному органічному розчиннику, як-то тетрагідрофуран, ацетонітрил, диметилсульфоксид або 1-метил-2-піролідинон. Реакцію необов'язково проводять при наявності доданої основи. Ця основа може бути органічною основою, як-то триетиламін або N,N-діізопропілетиламін, або неорганічною основою, як-то калій карбонат. Реакцію проводять при придатній температурі, звичайно між кімнатною температурою та температурою кипіння розчиннику, але необов'язково при вищих температурах, якщо застосовують герметизовану реакційну посудину. Реакція загалом продовжується протягом приблизно одної години - одного тижня, або доки аналіз не показує, що утворення потрібного продукту завершене. 1 2 Придатні відщеплювані групи L та L - галоген, 1 зокрема хлор або бром. В одному втіленні, L та 2 L , кожний, - хлор. Спеціалісту треба розуміти, що у вищенаведених способах може бути потрібно або необхідно захищати амін, гідроксил або інші потенційно реа 95966 8 ктивні групи. Придатні захисні групи та способи додавання та видалення таких груп є, загалом, добре відомими у рівні техніки. Дивись, наприклад, "Protective Groups in Organic Synthesis", 3rd Edition (1999) by Greene та Wuts. Заявлений винахід розкриває сполуки формули (І) у формі солей. Придатні солі охоплюють солі, утворені з органічними або неорганічними кислотами або органічними або неорганічними основами. Такі солі повинні звичайно бути фармацевтично прийнятними, хоча солі фармацевтично неприйнятних кислот або основ можна використовувати при отриманні та очистці сполук. Солі сполук формули (І) можна утворювати реакцією вільної сполуки, або її солі, енантіомеру або рацемату з одним або більше еквівалентами прийнятної кислоти або основи. Реакцію можна проводити у розчиннику або середовищі, у котрих сіль є нерозчинною або у розчиннику, у котрому сіль є розчинною, як-то, вода, діоксан, етанол, тетрагідрофуран або діетил-етер, або суміші розчинників, котрі можна видаляти у вакуумі або сушкою сублімацією. Реакція може також бути обмінною або її можна проводити на іонообмінній смолі. Сполуки формули (II) є відомими, наприклад, з WO 01/58907, WO01/25242, або WO 02/76990, або можуть бути отриманими відомими способами, що спеціалісту треба розуміти. Сполуки формули (IV) можуть бути отриманими способами, аналогічними способам, розкритим, наприклад, у WO 00/09511, або іншими відомими способами, що спеціалісту треба розуміти. Сполуки формул (III) та (V) є у продажу, або відомі у літературі, або можуть бути отриманими відомими способами, що спеціалісту треба розуміти. Придатні конкретні способи отримання сполук формул (II), (III), (IV) та (V) наведені далі у прикладах, такі способи представляють конкретні втілення способів винаходу. Наприклад, сполуки формули (II), та тим самим сполуки формули (І), можуть бути отриманими, як показано у схемі 1: 9 Проміжні сполуки можна застосовувати як такі або у захищеній формі. Придатні захисні групи та способи додавання та видалення таких груп є, загалом, добре відомими у рівні техніки. Дивись, наприклад, "Protective Groups in Organic Synthesis", 3rd Edition (1999) by Greene та Wuts. Сполуки винаходу та проміжні сполуки можна виділяти з їх реакційних сумішей та, якщо необхідно, далі очищали звичайними способами. Сполуки формули (І) можуть існувати у стереоізомерних формах. Тому, усі енантіомери, діастереомери, рацемати та їх суміші є охопленими 95966 10 рамками винаходу. Різні оптичні ізомери можна виділяти розділенням стереоізомерної суміші сполук звичайними способами, наприклад, фракційною кристалізацією або ВЕРХ. Альтернативно, різні оптичні ізомери можуть бути отриманими безпосередньо з оптично активних вихідних матеріалів. Сполуки формули (І) містять два стереогенні центри та можуть тому існувати у чотирьох окремих стереоізомерних формах, як показано у формулах (la)-(Id) 11 Усі такі чотири стереоізомери та будь-які їх суміші є охопленими рамками винаходу. В одному втіленні, сполуки формули (І) мають стереохімію, показану у формулі (Іа). У ще одному втіленні, сполуки формули (І) мають стереохімію, показану у формулі (Іb). Проміжні сполуки можуть також існувати у стереоізомерних формах та їх можна застосовувати як очищені енантіомери, діастереомери, рацемати або суміші. У цьому описі термін "С1-6алкіл" охоплює як нерозгалужений, так і розгалужений ланцюг а також циклічні алкіли. С1-6алкіл, що має 1-6 атомів карбону та може означати, але без обмеження, метил, етил, н-пропіл, i-пропіл, н-бутил, i-бутил, втор-бутил, m-бутил, н-пентил, i-пeнтил, m-пентил, нео-пентил, н-гексил, i-гексил або циклогексил. У цьому описі термін "галоген" стосується флуору, хлору, брому та йоду. Сполуки формули (І), та їх фармацевтично прийнятні солі є корисними, оскільки вони виявляють фармакологічну активність як антагоністи рецептору CX3CR1. Зокрема, при порівнянні зі сполуками, конкретно представленими у WO 01/58907, сполуки формули (І) заявленого винаходу виявляють значно поліпшену ефективність стосовно інгібування рецептору CX3CR1 та /або зменшену ефективність стосовно інгібування рецептору CX3CR2. Кращі сполуки заявленого винаходу виявляють поліпшену ефективність стосовно інгібування CX3CR1 та зменшену ефективність стосовно інгібування CXCR2. В одному аспекті заявлений винахід розкриває сполуку формули (І) або її фармацевтично прийнятну сіль для застосування як медикаменту. У ще одному аспекті заявлений винахід розкриває застосування сполуки формули (І) або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики хвороб або станів, у котрих антагонізм рецептору CX3CR1 є цілющим. У ще одному аспекті заявлений винахід розкриває застосування сполуки формули (І) або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики нейродегенеративних розладів, демієлінілувальної хвороби, серцево- та церебрально-васкулярних атеросклеротичних розладів, хвороби периферійних артерій, ревматоїдного артриту, хвороб легень, як-то COPD, астма або біль. У ще одному аспекті заявлений винахід розкриває застосування сполуки формули (І) або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики розсіяного склерозу (MS). У ще одному аспекті заявлений винахід розкриває застосування сполуки формули (І) або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики атеросклерозу попередженням та/або зменшенням утворення нових атеросклеротичних уражень або бляшок та/або попередженням або уповільненням прогресування існуючих уражень та бляшок. 95966 12 У ще одному аспекті заявлений винахід розкриває застосування сполуки формули (І) або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики атеросклерозу зміною складу бляшок для зменшення ризику руйнування бляшок та випадків атеротромбозу. У ще одному аспекті заявлений винахід розкриває застосування сполуки формули (І) або її фармацевтично прийнятної солі у виробництві медикаменту для лікування або профілактики інсульту або тимчасового пошкодження мозку (ТВІ). Згідно з винаходом також розкрито спосіб лікування або зменшення ризику хвороб або станів, у котрих антагонізм рецептору CX3CR1 є цілющим, котрий полягає у застосуванні до особи, яка потерпає від вказаної хвороби або стану або при її ризику, терапевтично ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі. Також розкрито спосіб лікування або зменшення ризику нейродегенеративних розладів, демієлінілувальної хвороби, серцево- та церебрально-васкулярних атеросклеротичних розладів, хвороби периферійних артерій, ревматоїдного артриту, хвороб легень, як-то COPD, астма або біль, у особи яка потерпає від вказаної хвороби або стану або при її ризику, де спосіб полягає у застосуванні до особи терапевтично ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі. Також розкрито спосіб лікування або зменшення ризику розсіяного склерозу (MS) у особи, яка потерпає від вказаної хвороби або стану або при її ризику, де спосіб полягає у застосуванні до особи терапевтично ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі. Також розкрито спосіб лікування або зменшення ризику атеросклерозу попередженням та/або зменшенням утворення нових атеросклеротичних уражень або бляшок та /або попередженням або уповільненням прогресування існуючих уражень та бляшок у особи, яка потерпає від вказаної хвороби або стану або при її ризику, де спосіб полягає у застосуванні до особи терапевтично ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі. Також розкрито спосіб лікування або зменшення ризику атеросклерозу зміною складу бляшок, щоб зменшити ризик руйнування бляшок та випадків атеротромбозу у особи, яка потерпає від вказаної хвороби або стану або при її ризику, де спосіб полягає у застосуванні до особи терапевтично ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі. Також розкрито спосіб лікування або зменшення ризику інсульту або тимчасового пошкодження мозку (ТВІ) у особи, яка потерпає від вказаної хвороби або стану або при її ризику, де спосіб полягає у застосуванні до особи терапевтично ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі. Сполуки можна застосовувати як монотерапію або у комбінації, як профілактичне або терапевтичне лікування запальних хвороб або станів 13 центральної нервової системи, як-то інсульт та тимчасове пошкодження мозку (ТВІ). (Soriano et al. J. Neuroimmunology 2002, 125, 59-65.). У ще одному аспекті заявлений винахід розкриває фармацевтичну композицію, що містить терапевтично ефективну кількість сполуки формули (І), або її фармацевтично прийнятної солі у суміші з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм, для застосування у лікуванні або профілактиці хвороб або станів, у котрих антагонізм рецептору CX3CR1 є цілющим. У ще одному аспекті заявлений винахід розкриває фармацевтичну композицію, що містить терапевтично ефективну кількість сполуки формули (І), або її фармацевтично прийнятної солі у суміші з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм, для застосування у лікуванні або профілактиці нейродегенеративних розладів, демієлінілувальної хвороби, серцево- та церебрально-васкулярних атеросклеротичних розладів, хвороби периферійних артерій, ревматоїдного артриту, COPD, астма або біль. У ще одному аспекті заявлений винахід розкриває фармацевтичну композицію, що містить терапевтично ефективну кількість сполуки формули (І), або її фармацевтично прийнятної солі у суміші з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм, для застосування у лікуванні або профілактиці розсіяного склерозу. У ще одному аспекті заявлений винахід розкриває фармацевтичну композицію, що містить терапевтично ефективну кількість сполуки формули (І), або її фармацевтично прийнятної солі у суміші з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм, для застосування у лікуванні або профілактиці атеросклерозу попередженням та зменшення утворення нових атеросклеротичних уражень та/або бляшок та/або попередженням або уповільненням прогресування існуючих уражень та бляшок. У ще одному аспекті заявлений винахід розкриває фармацевтичну композицію, що містить терапевтично ефективну кількість сполуки формули (І), або її фармацевтично прийнятної солі у суміші з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм, для застосування у лікуванні або профілактиці атеросклерозу зміною складу бляшок, щоб зменшити ризик руйнування бляшок та випадків атеротромбозу. Сполуки формули (І) та їх фармацевтично прийнятні солі показані для застосування у лікуванні або профілактиці хвороб або станів, у котрих модуляція активності стосовно рецептору CX3CR1 є потрібною. Зокрема, сполуки показані для застосування у лікуванні нейродегенеративних розладів або демієлінілувальної хвороби у ссавців, у тому числі людини. Більш конкретно, сполуки показані для застосування у лікуванні розсіяного склерозу. Сполуки також показані як корисні у лікуванні болю, ревматоїдного артриту, остеоартриту, серцево- та церебрально-васкулярних атеросклеротичних розладів, хвороби периферійних артерій та легеневої артеріальної гіпертензії. Станами, що можна конкретно згадати, є: нейродегенеративні хвороби та розлади з деменцією, 95966 14 наприклад, хвороба Альцгеймера, бічний аміотрофічний склероз та інші хвороби моторних нейронів, хвороба Крейтцфельда-Якоба та інші пріонні хвороби, ВІЛ-енцефалопатія, хвороба Хантингтона, лобно-скронева деменція, деменція з тільцями Леві та мультіінфарктна деменція; поліневропатії, наприклад, мієлополірадикулоневрит, хронічна запальна демієлінілувальна полірадикулоневропатія, багатофокальна моторна невропатія та плексопатії; демієлінізація центральної нервової системи, наприклад, гострий розсіяний/геморагічний енцефаломієліт та підгострий склерозувальний паненцефаліт; нейром'язові розлади, наприклад, бульбоспінальний параліч та синдром Лабберта-Ітона; спінальні розлади, наприклад, тропічний спастичний парапарез та синдром тугорухливості чоловіків; паранеопластичні синдроми, наприклад, мозочкова дегенерація та енцефаломієліт; травматичне пошкодження мозку, мігрень; рак; відторгнення алотрансплантату; системний склероз; вірусні інфекції; передавані паразитами хвороби, наприклад, малярія; періодонтальна хвороба; інфаркт міокарду; інсульт; коронарна серцева хвороба; ішемічна серцева хвороба; рестеноз; ревматоїдний артрит; хвороби легень, як-то COPD; астма або біль. Сполуки винаходу також показані для застосування у лікуванні атеросклерозу попередженням та/або зменшенням утворення нових атеросклеротичних уражень або бляшок та/або попередженням або уповільненням прогресування існуючих уражень та бляшок. Сполуки винаходу також показані для застосування у лікуванні атеросклерозу зміною складу бляшок, щоб зменшити ризик руйнування бляшок та випадків атеротромбозу. Сполуки винаходу також показані для застосування у лікуванні запальної хвороби кишечнику (IBD), як-то хвороба Крона та виразковий коліт, індукуванням ремісії та/або підтримуванням ремісії IBD. Профілактика, як очікують, буде особливо релевантною стосовно лікування осіб, які раніше потерпали від такої хвороби або стану або мають збільшений ризик захворіти. Особи з ризиком розвитку конкретної хвороби або стану загалом охоплюють осіб, які мають родинну історію хвороби або стану, або осіб, які ідентифіковані генетичним тестуванням або скринінгом як особливо схильні до розвитку хвороби або стану. Для вищезгаданих терапевтичних показань застосовуване дозування повинне, безумовно, варіювати залежно від застосовуваної сполуки, режиму застосування та потрібного лікування. Однак, загалом, задовільні результати отримують, коли сполуки застосовують при дозуванні твердої форми у межах 1 мг - 2000 мг на добу. Сполуки формули (І) та їх фармацевтично прийнятні похідні можна застосовувати самі по собі або у формі прийнятної фармацевтичної композиції, у котрій сполука або похідне ε у суміші з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм. Застосування може бути, але без обмеження, ентеральним (у тому числі пероральним, сублінгвальним або ректальним), 15 інтраназальним, внутрішньовенним, місцевим або іншим парентеральним. Звичайні способи відбору та отримання придатних фармацевтичних композицій описані, наприклад, у "Pharmaceuticals - The Science of Dosage Form Designs", Μ. Ε. Aulton, Churchill Livingstone, 1988. Фармацевтична композиція переважно містить менше 80% та більш переважно менше 50% сполуки формули (І), або її фармацевтично прийнятної солі. Також розкрито спосіб отримання такої фармацевтичної композиції, спосіб полягає у змішуванні складових. Заявлений винахід крім того розкриває комбінаційну терапію, де сполуку формули (І) або її фармацевтично прийнятну сіль чи фармацевтичну композицію або композицію, що містить сполуку формули (І), застосовують одночасно або послідовно з терапією та/або засобом для лікування будьчого з серцево- та церебрально-васкулярних атеросклеротичних розладів та хвороб периферійних артерій. Зокрема, сполуку формули (І) або її фармацевтично прийнятну сіль можна застосовувати в асоціації зі сполуками з одної або більше наступних груп: 1) антизапальні засоби, наприклад, a) NSAID (наприклад, ацетилсаліцилова кислота, ібупрофен, напроксен, флурбіпрофен, диклофенак, індометацин); b) інгібітори синтезу лейкотриєну (інгібітори 5LO, наприклад, AZD4407, зилейтон, лікофелон, CJ13610, CJ13454; інгібітори FLAP, наприклад, BAY-Y-1015, DG-031, MK591, МК886, А81834; інгібітори гідролази LTA4, наприклад, SC56938, SC57461A); c) антагоністи рецептору лейкотриєну; (наприклад, СР195543, амелубант, LY293111, аколат, МК571); 2) антигіпертензивні засоби, наприклад, a) бета-блокатори (наприклад, метопролол, атенолол, соталол); b) інгібітори ферменту перетворення ангіотензину (наприклад, каптоприл, раміприл, квінаприл, еналаприл); c) блокатори каналу кальцію (наприклад, верапаміл, дилтіазем, фелодипін, амлодипін); d) антагоністи рецептору ангіотензину II (наприклад, ірбесартан, кандесартан, телмісартан, лосартан); 3) антикоагулянти, наприклад, a) інгібітори тромбіну (наприклад, ксимелагатран), гепарини, інгібітори фактору Ха; b) інгібітори агрегації тромбоцитів (наприклад, клопідогрель, тиклопідин, прасугель, AZ4160); 4) модулятори метаболізму ліпідів, наприклад, a) сенсибілізатори інсуліну, як-то агоністи PPAR (наприклад, піоглітазон, розиглітазон, Галіда, мураглітазаар, гефемрозил, фенофібрат); b) Інгібітори редуктази HMG-CoA, статини (наприклад, симвастатин, правастатин, аторвастатин, розувастатин, флувастатин, пітавастатин); c) інгібітори поглинання холестерину (наприклад, езетиміб); 95966 16 d) інгібітори ІВАТ (наприклад, AZD-7806); е)агоністи LXR (наприклад, GW-683965A, Т0901317); f) модулятори рецептору FXR; g) інгібітори фосфоліпази; 5) засоби проти стенокардії, наприклад, нітрати та нітрити; 6) модулятори окиснювального стресу, наприклад, антиоксиданти, (пробукол), інгібітори мієлопероксидази. Винахід ілюстровано, але без обмеження, наступними прикладами: Загальні способи Усі застосовувані розчинники були аналітичного гатунку та наявні у продажу безводні розчинники застосовували для реакції. Реакції звичайно перебігали в інертній атмосфері азоту або аргону. 1 13 Спектри Н та С ЯМР реєстрували при 400 МГц для протона та 100 МГц для карбона-13 на спектрометрі Varian Unity+ 400 ЯМР із зондом ВВО 5 мм із Z-градієнтами, або спектрометрі Bruker Avance 400 ЯМР із дуальним зондом 60 мкл інверсного потоку із Z-градієнтами, або спектрометрі Bruker DPX400 ЯМР з 4-центровим зондом із Z1 градієнтами. Спектри 600 МГц Н ЯМР реєстрували на спектрометрі Bruker av600 ЯМР із зондом 1 ВВО 5 мм із Z-градієнтами. Спектри 300 МГц Н ЯМР реєстрували на спектрометрі Varian Gemini 300 ЯМР із зондом ВВО 5 мм. Якщо не показане інше у прикладах, спектри реєстрували при 400 МГц дляпротона та 100 МГц для карбона-13. Наступні стандартні сигнали застосовували: середня 1 13 лінія ДМСО-d6 2,50 ( Н), 39,51 ( С); середня 1 13 лінія CD3OD 3,31 ( Н) або 49,15 ( С); ацетон-d6 1 13 1 2,04 ( Н), 206,5 ( С); та CDCI3 7,26 ( Н), середня 13 лінія CDCI3 77,16 ( С) (якщо не показане інше). Енантіомерний надлишок (eн) визначали за допомогою газової хроматографії на колонці Cyclodex В (ізотермічне елювання 100°С) або на колонці Cyclosil В (градієнт температури 110130°С). Діастереомерний надлишок (дн) визначали за допомогою ВЕРХ. Мас-спектри реєстрували на РХ-МС Waters, що складався з Alliance 2795 (РХ) та одиночного квадрупольного мас-спектрометра ZQ. Масспектрометр мав джерело електророзпилення іонів (ІЕР) у режимі позитивних чи негативних іонів. Напруга на капілярі була 3 кВ та масспектрометр сканував при m/z 100-700 з часом сканування 0,3 або 0,8 s. Розділення проводили на колонках Waters X-Terra MC, C8, (3,5 мкм, 50 або 100 мм x 2,1 мм в.д.), колонці ScantecLab's АСЕ 3 AQ (100 мм х 2,1 мм в.д.). Температуру колонки доводили до 40°С. Лінійний градієнт застосовували, застосовуючи систему нейтральних або кислотних мобільних фаз, при 0%-100% органічної фази у 4-5 хвилин, швидкість потоку 0,3 мл/хвил. Система нейтральної або мобільної фази: ацетонітрил/[10 мМ NH4OAc (водн.)/MeCN (95:5)], або [10 мМ NH4OAc (водн.)/MeCN (1/9)]/[10 мМ NH4OAc (водн.)/MeCN (9/1)]. Система кислотної мобільної фази: [133 мМ НСООН (водн.)/MeCN (5/95)]/[8 мМ НСООН (водн.)/MeCN (98/2)]. Альтернативно, мас-спектри реєстрували за допомогою газової хроматографії-мас 17 спектроскопії (GC 6890, 5973N MSD, Agilent Technologies) на колонці VF-5 МС (ID 0,25 мм x 30 м, 0,25 мкм (Varian Inc.)). Лінійний градієнт температури застосовували (40°С-300°С), 25°С/хвилин. МС був з джерелом іонів XI та газом-реагентом був метан. МС сканував між m/z 50-500 та швидкість сканування доводили до 3,25 сканувань/с. ВЕРХ-аналізи проводили на системі Agilent HP1000, що складалася з мікровакуумного дегазатора G1379A, бінарного насоса G1312A, автовідбірника G1367A Wellplate, термостатованої колонки G1316A та детектора з діодною матрицею G1315B. Колонка: Х-Terra МС, Waters, 4,6x50 мм, 3,5 мкм. Температуру колонки доводили до 40°С та швидкість потоку до 1,5 мл/хвил. Детектор з діодною матрицею сканував при 210-300 нм, крок та ширину піку доводили до 2 нм та 0,05 хвил, відповідно. Лінійний градієнт застосовували від 0% до 100% ацетонітрилу, у 4 хвил. Мобільна фаза: ацетонітрил/10 мМ амоній ацетат у 5% ацетонітрилі у воді MilliQ. Звичайний спосіб обробки після реакції складався з екстракції продукту розчинником, як-то етилацетат, промивання водою, а потім сушки органічної фази MgSO4 або Na2SO4, та концентрації розчину у вакуумі. Тонко-шарову хроматографію (ТШХ) проводили на ТШХ-планшетах Merck (Silica gel 60 F254) та УФ застосовували для візуалізації плям. Флешхроматографію проводили на Combi Flash Companion на нормально-фазових флешколонках RediSep або на Merck Silica gel 60 (0,040-0,063 мм). Звичайними розчинниками, застосовуваними для флеш-хроматографії, були суміші хлороформ/метанол, толуєн/етилацетат та етилацетат/гексани. Препаративну хроматографію проводили на автопрепаративній ВЕРХ Gilson з детектором з діодною матрицею на колонці ХTerra МС (С8, 19x300 мм, 7 мкм), та градієнт із ацетонітрил/0,1М амоній ацетат у 5% ацетонітрилу у воді MilliQ, від 20% до 60% ацетонітрилу, протягом 13 хвил, та швидкість потоку 20 мл/хвил., якщо не показане інше у прикладах. Альтернативно, очистка була на напівпрепаративній ВЕРХ Shimadzu РХ-8А з УФдетектором Shimadzu SPD-10A з колонкою Waters Symmetry (C18, 5 мм, 100 мм x 19 мм). Градієнт ацетонітрил/0,1% трифлуороцтової кислоти у воді MilliQ, при від 35% до 60% ацетонітрилу у 20 хвил. Швидкість потоку: 10 мл/хвил. Перекристалізацію звичайно проводили у розчинниках або суміші розчинників, як-то етер, етилацетат/гептани та метанол/вода. Застосовано такі скорочення: ДХМ = дихлорметан; дн = діастереомерний надлишок; DIPCI = хлордіізопінокамфенілборан (DIP-Хлорид); DIPEA = Ν,Ν-діізопропілетиламін; ДМФ = N,Nдиметилформамід; ДМСО = диметилсульфоксид; eн = енантіомерний надлишок; NCS = Nхлорсукцинімід; ΝΜΡ = 1-метил-2-піролідинон; ТГФ = тетрагідрофуран; конц. = концентрований. Вихідні матеріали були з комерційних джерел або отриманими літературними способами. Приклади вихідних матеріалів, що були отриманими: 95966 18 (2R)-2-[(2-аміно-5-меркапто[1,3]тіоазоло[4,5d]піримідин-7-іл)аміно]-4-метилпентан-1-ол: WO 02/076990 (Приклади 1-5 та 8); 5-(бензилтіо)-7-хлор[1,3]тіоазоло[4,5фіримідин-2-амін: WO 00/09511 (Приклад 8); 5-флуор-2-формілпіридин: WO 2005/066155 (Приклад 1); 5-Флуор-піридин-2-карбонітрил WO 2005/066155 (Приклад 3); 1-(3-Хлорпіридин-4-іл)етанон: Marsais, F. et al. J. Organometal. Chem. 1981, 216, 139-147 (Приклад 4); 2-ацетил-ізонікотинонітрил: Citterio et al. J. Chem. Res. Synopses 1982, 10, 272-273 (Приклад 8); та 1-(6-Хлорпіридин-3-іл)етанон: Lee, С. et al. J. Med. Chem. 2001, 44, 2133 (Приклади 6 та 7). У нижченаведених загальних способах Ру необов'язково заміщений піридил. Загальний спосіб А Натрій борогідрид (0,1 еквів.), DIPEA (1,5 еквів.) та сполуку загальної формули (III) (1,2 еквів.) додавали до сполуки загальної формули (II) (1,0 еквів.) у ДМСО в атмосфері азоту. Утворену реакційну суміш перемішували при 40°C, доки реакція не була завершеною (за РХ-МС, ВЕРХ або ТШХ). Суміш виливали у льодяну воду та продукт екстрагували ДХМ або EtOAc. Комбіновані органічні фази сушили та концентрували у вакуумі. Сирий продукт, якщо необхідно, очищали препаративною ВЕРХ або колонковою флеш-хроматографією. Загальний спосіб В1 (VI) (1,0 еквів.) у ТГФ додавали при 0°C до (+)DIPCI (для отримання (VII)) або (-)-DIPCI (для отримання (VIII)) (1,5 еквів.) у ТГФ в атмосфері аргону. Реакційній суміші давали повільно досягти кімнатної температури протягом ночі. Розчинник випарювали з наступним додаванням Et2O та діетаноламіну (2,2 еквів.). Суміш перемішували, доки реакція не була завершеною (за РХ-МС, ВЕРХ або ТШХ). Осад, що утворився відфільтровували, промивали Et2O та фільтрат концентрували у вакуумі. Сирий продукт, якщо необхідно, очищали препаративною ВЕРХ або колонковою флешхроматографією. Загальний спосіб В2 (R)-(+)-метил-СВS-оксазаборолідин (1М у толуєні, 0,1-1 еквів.) розчиняли у ТГФ та охолоджували до 0°C. Комплекс боран-метилсульфід (2М у ТГФ, 1 еквів.) додавали краплями та реакційну суміш перемішували протягом 1 години. Реакційну суміш охолоджували до -10°C та (VI) (1 еквів.), 19 95966 20 розчиняли у ТГФ і додавали краплями протягом 0,5 годин. Утворену суміш перемішували протягом 1 години, або доки реакція не була завершеною, та температуру повільно підвищували до 10°C. 1М НСІ водн. додавали для гасіння реакції. Насичений NaHCO3 водн. додавали до рН приблизно 8. Продукт екстрагували ДХМ. Комбіновані органічні екстракти сушили Na2SO4 та концентрували у вакуумі, отримуючи (VIII). Продукт необов'язково очищали колонковою хроматографією. Загальний спосіб С1 a) 1-(5-Флуорпіридин-2-іл)етанол Т рифенілфосфін (1,3 еквів.) у ТГФ додавали при 0°C до NCS (1,3 еквів.) у ТГФ в атмосфері аргону. Утворену суміш перемішували при температурі довкілля протягом 30 хвил. (VII) або (VIII) (1 еквів.) додавали при 0°C та реакційну суміш перемішували при температурі довкілля, доки реакція не була завершеною (за РХ-МС, ВЕРХ або ТШХ). Розчинник випарювали з наступним додаванням гексану та видаленням осаду фільтруванням. Фільтрат концентрували у вакуумі, отримуючи (IX) або (X). Сирий продукт, якщо необхідно, очищали препаративною ВЕРХ або колонковою флешхроматографією. Загальний спосіб С2 Хлорангідрид ціанурової кислоти (0,6 еквів.) розчиняли в етилацетаті. ДМФ (1,5 еквів.) додавали та суміш перемішували при кімнатній температурі протягом 10 хвил. Реакційну суміш охолоджували до 0°C. (VII) або (VIII) (1 еквів.) розчиняли в етилацетаті та додавали краплями протягом 10 хвил. Утворену суміш перемішували при кімнатній температурі протягом ночі. Ізопропанол (приблизно 0,25 мл/ммоль (VII) або (VIII)) додавали. Осад відфільтровували та промивали ЕtOАс. Фільтрат концентрували, отримуючи заголовну сполуку (IX) або (X). Приклад 1 (2R)-2-[(2-Аміно-5-{[1-(5-флуорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол При -78°C до розчину 5-флуор-2формілпіридину (0,66 г, 5,3 ммоль) у ТГФ (20 мл) додавали краплями метиллітій (1,6 Μ у діетилетері, 4,0 мл). Після перемішування протягом 1,5 годин при -78°C додавали насичений розчин амоній хлориду (25 мл), а потім воду (25 мл). Суміш екстрагували хлороформом та органічну фазу сушили магній сульфатом та концентрували у вакуумі. Залишок очищали колонковою флешхроматографією (елюент: хлороформ:метанол 98:2), отримуючи заголовну сполуку (0,25 г, 33% виходу). 1 -1 Н ЯМР (400 МГц, CDCI3): млн 8,38 (d, 1H), 7,42 (dt, 1H), 7,33 (dd, 1H), 4,90 (q, 1H), 3,90 (s, 1H), + 1,50 (d, 3H); МС (ІЕР) m/z 142 [М+1] . b) 2-(1-Хлоретил)-5-флуорпіридин Суміш трифенілфосфіну (0,92 г, 3,5 ммоль) та ССl4 (4 мл) перемішували протягом 10 хвил. та тоді додавали розчин 1-(5-флуорпіридин-2іл)етанолу (0,25 г, 1,7 ммоль) у ДХМ (2 мл). Через 18 годин додавали пентан (20 мл), твердий матеріал видаляли фільтруванням та фільтрат концентрували у вакуумі. Залишок очищали колонковою флеш-хроматографією (елюент гептан:етилацетат 3:1), отримуючи заголовну сполуку (8 мг, 3% виходу). 1 -1 Н ЯМР (400 МГц, CDCI3): млн 8,35 (d, 1H), 7,44 (dd, 1H), 7,36 (dt, 1H), 5,09 (q, 1H), 1,81 (d, 3H). с) (2R)-2-[(2-Аміно-5-{[1-(5-флуорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол 21 95966 22 новані органічні екстракти сушили натрій сульфатом та концентрували у вакуумі. Продукт очищали колонковою флеш-хроматографією (елюент гептан:ЕtоАс градієнт), отримуючи 7,9 г (64% виходу) заголовної сполуки. 1 -1 Н ЯМР (300 МГц, CDCI3) млн 8,62 (m, 1H); 8,00 (m, 1H); 7,80 (m, 1H); 2,70 (s, 3H). b) (1S)-1-(5-Хлорпіридин-2-іл)етанол Заголовну сполуку отримували загальним способом А, починаючи з (2R)-2-[(2-аміно-5меркапто[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-олу (21 мг, 0,072 ммоль) та 2-(1хлоретил)-5-флуорпіридину (8 мг, 0,048 ммоль). Очистка препаративною ВЕРХ дала 4 мг (21% виходу) заголовної сполуки як суміш діастереомерів. 1 -1 Н ЯМР (400 МГц, ДМСО-d6): млн 8,44 (t, 1H), 7,91 (s, 2H), 7,55-7,64 (m, 1H), 7,47-7,55 (m, 1H), 6,83 (d, 1H), 4,99-5,11 (m, 1H), 4,59 (q, 1H), 3,41-3,28 (m, 2H), 1,60 (dd, 3H), 1,48-1,57 (m, 1H), 1,25-1,42 (m, 2H), 0,72-0,86 (m, 6H); MC (IEP) m/z + 423 [M+1] . Приклад 2 (2R)-2-[(2-Аміно-5-{[(1S)-1-(5-хлорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол Заголовну сполуку отримували загальним способом В2, починаючи з 1-(5-хлорпіридин-2іл)етанон (780 мг, 5 ммоль). Очистка колонковою флеш-хроматографією дала 695 мг (88% виходу) заголовної сполуки із 92% ен. 1 Н ЯМР (300 МГц, CDCI3): 8,47 (s, 1H); 7,65 (d, 1H); 7,26 (d, 1H); 4,87 (q, 1H); 3,87 (br s, 1H); 1,47 + (d, 3H); МС (ІЕР) m/z 140 та 142[М+1] . c) 5-Хлор-2-[(1R)-1-хлоретил]піридин Заголовну сполуку отримували загальним способом С2, починаючи з (1S)-1-(5-хлорпіридин-2іл)етанолу (695 мг, 4,41 ммоль). Сирий продукт застосовували на наступному етапі без очистки. 1 -1 Н ЯМР (400 МГц, CDCI3): млн 8,46 (d, 1H), 7,64 (dd, 1H), 7,41 (d, 1H), 5,08 (q, 1H), 1,80 (d, 3H); + МС (ІЕР) m/z 176 та 178 [М+1] . d) (2R)-2-[(2-Аміно-5-{[(1S)-1-(5-хлорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол а) 1-(5-Хлорпіридин-2-іл)етанон 5-Хлорпіридин-2-карбонітрил (10,7 г, 77 ммоль) розчиняли у діетил-етері (65 мл) та ТГФ (35 мл) в атмосфері азоту. Суміш охолоджували до температури -63°C. Метилмагній бромід (ЗМ у ТГФ, 35 мл, 105 ммоль) додавали протягом 30 хвил. Реакційну суміш тоді перемішували при 60°C протягом 45 хвил та тоді нагрівали до кімнатної температури. ТГФ (50 мл) додавали для розчинення будь-якого осадженого матеріалу та реакційну суміш перемішували протягом 1 години. 2М НСІ водн. (100 мл) додавали та реакційну суміш перемішували протягом 4 годин. рН тоді доводили до 7 натрій гідрогенкарбонатом. Фази розділяли та продукт екстрагували з водної фази ДХМ. Комбі Заголовну сполуку отримували загальним способом А, починаючи з (2R)-2-[(2-аміно-5меркапто[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-олу (572 мг, 1,91 ммоль) та 5хлор-2-[(1R)-1-хлоретил]піридину (376 мг, 2,1 ммоль). Очистка препаративною ВЕРХ дала 99 мг (12% виходу) заголовної сполуки. 1 -1 Н ЯМР (400 МГц, CD3OD): млн 8,49 (d, 1H), 7,79 (dd, 1H), 7,66 (d, 1H), 5,22 (q, 1Н), 4,46 (br s, 1H), 3,57-3,40 (m, 2H), 1,78-1,66 (m, 4H), 1,61-1,40 23 95966 24 (m, 2H), 1,03-0,93 (m, 6H); МС (ІЕР) m/z 439 та 441 + [M+1] . Приклад 3 (2R)-2-[(2-Аміно-5-{[(1S)-1-(5-флуорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]пipимiдин-7-ιл)aмiнo]4-мeтилпeнтaн-1-oл Заголовну сполуку із 87% ен отримували загальним способом С2, починаючи з (1S)-1-(5флуорпіридин-2-іл)етанолу (407 мг, 2,9 ммоль). Сирий продукт застосовували на наступному етапі без очистки. 1 Н ЯМР (300 МГц, CDCI3): 8,44-8,40 (m, 1H); 7,6-7,4 (m, 2H); 5,16 (q, 1H), 1,86 (d, 3H). d) (2R)-2-[(2-Аміно-5-{[(1S)-1-(5-флyopпіридин2-іл)етил]тіо}[1,3]тіоaзоло[4,5-d]піримідин-7іл)аміно]-4-метилпентан-1-oл а) 1-(5-Флуорпіридин-2-іл)етанон 5-Флуор-піридин-2-карбонітрил (29 г, 240 ммоль) розчиняли у ТГФ (150 мл) в атмосфері азоту. Реакційну суміш охолоджували до температури -64°C. Метил магній бромід (ЗМ у ТГФ, 105 мл, 315 ммоль) додавали протягом 40 хвил. Реакційну суміш перемішували при -65°C протягом 1,5 годин, тоді нагрівали до кімнатної температури. ΤΓΦ (50 мл) додавали та суміш перемішували ще 3 години. 2М хлоридну кислоту (водн., 100 мл) додавали до слабкої кислотності та реакційну суміш перемішували при кімнатній температурі протягом ночі. Натрій гідрогенкарбонат тоді додавали для нейтралізації реакційної суміші. Фази розділяли та водну фазу екстрагували ДХМ. Комбіновані органічні екстракти промивали розсолом, сушили натрій сульфатом та концентрували у вакуумі. Сирий продукт очищали колонковою флешхроматографією, отримуючи 18 г (55% виходу) заголовної сполуки. 1 Н ЯМР (300 МГц, CDCI3): 8,50 (m, 1Н); 8,10 (m, 1Н); 7,52 (m, 1Н); 2,70 (s, 3H). b) (1S)-1-(5-Флуорпіридин-2-іл)етанол Заголовну сполуку отримували загальним способом В2, починаючи з 1-(5-флуорпіридин-2іл)етанону (2,70 г, 19,4 ммоль). Очистка колонковою флеш-хроматографією дала 0,85 г (31% виходу) заголовної сполуки із 94% ен. 1 Н ЯМР (300 МГц, CDCI3): 8,38 (m, 1H); 7,5-7,2 (m, 2H); 4,89 (q, 1H); 3,9 (br s, 1H); 1,49 (d, 3H). c) 2-[(1R)-1-Хлоретил]-5-флуорпіридин Заголовну сполуку отримували загальним способом А, починаючи з (2R)-2-[(2-аміно-5меркапто[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-олу (550 мг, 1,8 ммоль) та 2[(1R)-1-хлоретил]-5-флуортридину (0,46 г, 2,9 ммоль). Продукт очищали колонковою флешхроматографією, а потім препаративною ВЕРХ (Колонка: Reprosil, елюент ізопропанолтептан 20:80, швидкість потоку: 16 мл/хвил), збираючи перший ельований ізомер, отримуючи 135 мг (18% виходу) заголовної сполуки. 1 -1 Н ЯМР (400 МГц, ДМСО-d6) млн 8,51 (d, 1H), 7,98 (s, 2H), 7,65 (dt, 1H); 7,58 (dd, 1Н), 6,88 (d, 1Н); 5,12 (q, 1Н); 4,66 (t, 1Н); 4,27 (br s, 1H); 3,413,27 (m, 2H), 1,66 (d, 3H), 1,65-1,55 (m, 1H); 1,481,35 (m, 2H), 0,88 (d, 3H), 0,85 (d, 3H); MC (IEP) m/z + 423 [M+1] . Приклад 4 (2R)-2-[(2-аміно-5-{[1-(3-хлорпіридин-4іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол a) (1S)-1-(3-Хлорпіридин-4-іл)етанол 25 Заголовну сполуку із 90% ен отримували загальним способом В1, починаючи з 1-(3хлорпіридин-4-іл)етанону (0,90 г, 5,78 ммоль). 1 Н ЯМР (300 МГц, CDCI3) 8,47 (s, 1H); 7,65 (d, 1H); 7,26 (d, 1H); 4,87 (q, 1H); 3,87 (br s, 1H); 1,47 + (d, 3H); MC (IEP) m/z 158 та 160[М+1] . b) 3-Хлор-4-[(1R)-1-хлоретил]піридин Заголовну сполуку отримували загальним способом С2, починаючи з (1S)-1-(3-хлорпіридин-4іл)етанолу (570 мг, 3,62 ммоль). Сирий продукт застосовували на наступному етапі без очистки. + MC (IEP) m/z 176 та 178 [М+1] . c) (2R)-2-[(2-Аміно-5-{[1-(3-хлорпіридин-4іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол (ізомер 1 та ізомер 2) Заголовну сполуку отримували загальним способом А, починаючи з (2R)-2-[(2-аміно-5меркапто[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-олу (860 мг, 2,87 ммоль) та 3хлор-4-[(1R)-1-хлоретил]піридину з попереднього етапу. Очистка флеш-хроматографією дала заголовну сполуку (105 мг, 8% виходу) як діастереомерну суміш. + МС (ІЕР) m/z 439 та 441 [М+1] . Приклад 5 (2r)-2-[(2-Аміно-5-{[(1S)-1-(3-флуорпіридин-2іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол 95966 26 а) 1-(6-Бром-З-флуор-піридин-2-іл)етанон 2-Бром-5-флуор-піридин (11 г, 62,5 ммоль) розчиняли у діетил-етері при кімнатній температурі в атмосфері азоту. Реакційну суміш охолоджували до температури -66°C. Бутиллітій (2,5 Μ у гексані, 26 мл, 65 ммоль) додавали краплями протягом 0,5 годин. Утворену реакційну суміш залишали при 65°C протягом 1 години. Ν,Ν-Диметилацетамід (6,5 мл, 70 ммоль) додавали протягом 10 хвил. та реакційну суміш перемішували при -65°C протягом 2 годин. 1М водну хлоридну кислоту (50 мл) додавали та суміш нагрівали до кімнатної температури. рН доводили до 7 додатковою хлоридною кислотою. Водну фазу екстрагували діетил-етером три рази. Комбіновані органічні фази промивали розсолом, сушили натрій сульфатом та концентрували у вакуумі. Очистка колонковою флешхроматографією (елюент гептан:діетил-етер градієнт) дала 4,6 г (34% виходу) заголовної сполуки. 1 Н ЯМР (300 МГц, ДМСО-d6): 8,0-7,8 (m, 2H); + 2,57 (s, 3H); МС (ІЕР) m/z 218 та 220 [М+1] . b) (1S)-1-(6-Бром-3-флуор-піридин-2-іл)етанол Заголовну сполуку отримували загальним способом В2, починаючи з 1-(6-бром-3-флуорпіридин-2-іл)етанону (1,76 г, 8,19 ммоль). Продукт очищали колонковою флеш-хроматографією (елюент: гептан: етилацетат градієнт), отримуючи 1,31 г (73% виходу) заголовної сполуки із 80% ен. 1 Н ЯМР (300 МГц, CDCI3) 7,38 (m, 1H); 7,26 (m, 1H); 5,06 (q, 1H); 3,38 (br s, 1H); 1,47 (d, 3H); МС + + (ІЕР) m/z 220 та 222 [M+1] , m/z 202 [М-Н2О] . c) (1S)-1-(3-Флуор-піридин-2-іл)етанол 27 (1S)-1-(6-Бром-3-флуор-піридин-2-іл)етанолу (1,3 г, 5,9 ммоль), триетиламін (1,6 мл, 11,5 ммоль) та паладій на вугіллі (0,64 г, 0,34 ммоль) змішували у ДХМ (25 мл). Колбу промивали воднем у 4 цикли та тоді залишали при 2,5 атм тиску водню при кімнатній температурі протягом 24 годин. Суміш фільтрували та твердий матеріал промивали ДХМ. Фільтрат промивали водою та розсолом та сушили натрій сульфатом та концентрували у вакуумі. Сирий продукт очищали колонковою флеш-хроматографією (елюент ДХМ:метанол градієнт), отримуючи 0,54 г (65% виходу) заголовної сполуки. 1 Н ЯМР (300 МГц, CDCI3): 8,38 (m, 1H); 7,39 (m, 1H); 7,26 (m, 1H); 5,11 (q, 1H); 4,16 (br s, 1Н); 1,49 (d, 3H). d) 2-((R)-1-Хлоретил)-3-флуop-піридин Заголовну сполуку (0,24 г) отримували загальним способом С2, починаючи з (1S)-1-(3-флуорпіридин-2-іл)етанолу (254 мг, 1,8 ммоль). 1 Н ЯМР (300 МГц, CDCI3): 8,46 (m, 1H); 7,47 (m, 1H); 7,34 (m, 1H); 5,48 (q, 1H), 1,94 (d, 3H); MC + (IEP) m/z 160 та 162 [M+1] . е) (2R)-2-[(2-Аміно-5-{[(1S)-1-(3-флуорпіридин2-іл)етил]тіо}[1,3]тіоазоло[4,5-d]nipимідин-7іл)аміно]-4-метилпентан-1-ол 95966 28 (2R)-2-[(2-Аміно-5-{[(1S)-1-(6-хлорпіридин-3іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7іл)(метил)аміно]пентан-1-ол а) (1S)-1-(6-Хлорпіридин-3-іл)етанол Заголовну сполуку отримували загальним способом В1, застосовуючи (-)DІРСІ та 1-(6хлорпіридин-3-іл)етанон (0,80 г, 5,14 ммоль), отримуючи 0,71 г (88% виходу) заголовної сполуки. 1 -1 Н ЯМР (CDCI3) млн 8,40-8,28 (m, 1H), 7,757,63 (m, 1H), 7,35-7,24 (m, 1H), 5,04-4,79 (m, 1H), + 1,63-1,45 (m, 3H); MC (IEP) m/z 158 та 160[М+1] . b) 2-Хлор-5-[(1R)-1-хлоретил]піридин Заголовну сполуку отримували загальним способом С 1, застосовуючи(1S)-1-(6-хлорпіридин-3іл)етанол (0,20 г, 1,27 ммоль), отримуючи 0,16 г (72% виходу) заголовної сполуки. 1 -1 Н ЯМР (CDCI3) млн 8,45-8,35 (m, 1H), 7,797,70 (m, 1H), 7,39-7,29 (m, 1H), 5,07 (q, 1H), 1,85+ 1,78 (m, 3H); MC (IEP) m/z 176 та 178[М+1] . c) N-(Етоксикарбоніл)-D-норвалін Заголовну сполуку отр имували загальним способом А, починаючи з (2R)-2-[(2-аміно-5меркапто[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-олу (348 мг, 1,16 ммоль) та 2((R)-1-хлоретил)-3-флуор-піридину (240 мг, 1,5 ммоль). Очистка колонковою флешхроматографією (елюент ДХМ: метанол градієнт) дала 190 мг (47% виходу) заголовної сполуки з дн 60%. Цього матеріалу 55 мг далі очищали препаративноюВЕРХ. Ельований останнім ізомер збирали, отримуючи 21 мг заголовної сполуки. 1 -1 Н ЯМР (400 МГц, ДМСО-d6) млн 8,40 (dt, 1H), 7,98 (s, 2H), 7,70 (m, 1H), 7,40 (m, 1Н); 6,92 (d, 1Н); 5,45 (q, 1Н); 4,65 (t, 1Н); 4,27 (br s, 1H); 3,453,30 (m, 2H), 1,69 (d, 3H), 1,66-1,58 (m, 1H), 1,501,35 (m, 2H), 0,88 (d, 3H), 0,85 (d, 3H); MC (IEP) m/z + 423 [M+1] . Приклад 6 D-Норвалін (10,0 r, 85,3 ммоль) розчиняли у водному натрій гідроксиді (4М, 25 мл). Етил хлорформіат (10,6 мл, 111 ммоль) та водний натрій гідроксид (4М, 25 мл) додавали протягом 15 хвил. при 0°C. Реакційну суміш нагрівали до кімнатної температури та перемішували при цій температурі протягом 4 годин. Реакційну суміш промивали діетил-етером три рази та тоді підкислювали водною хлоридною кислотою (2М). Продукт екстрагували 29 діетил-етером три рази. Комбіновані органічні фази сушили магній сульфатом та концентрували у вакуумі, отримуючи заголовну сполуку з кількісним виходом. 1 -1 Н ЯМР (CDCI3) млн 6,43 (br s, 1H), 5,22 (d, 1H), 4,37 (q, 1H), 4,13 (q, 2H), 1,84 (m, 1H), 1,68 (секстет, 1Н), 1,42 (секстет, 1Н), 1,25 (t, 3H), 0,95 + (t, 3H); MC (XI) 144 (100%), 190[M+1] . d) (2R)-2-(Метиламіно)пентан-1-ол 95966 30 f) (2Н)-2-[(2-Аміно-5-меркапто[1,3]тіоазоло[4,5d]піримідин-7-іл)(метил)аміно]-пентан-1-ол Алюмогідрид літію (6,5 г, 171 ммоль) суспендували у ТГФ при 0°C в атмосфері азоту. N(Етоксикарбоніл)-D-норвалін розчиняли у ТГФ та додавали краплями при 0°C. Реакційну суміш гріли при кипінні під зворотним холодильником протягом ночі. Після охолодження до кімнатної температури, додавали насичений водний натрій сульфат для утворення кашки. Утворену суміш фільтрували через целіт. Твердий матеріал промивали ДХМ, доки усі продукт не екстрагувалися. Комбінований фільтрат сушили натрій сульфатом та концентрували у вакуумі. Перегонка при 0,1 мбар, збираючи фракцію між 75-85°C, дала 7,1 г (71% виходу) заголовної сполуки. 1 Н ЯМР (CDCI3) 3,63 (dd, 1H); 3,30 (dd, 1H); 2,51 (m, 1H); 2,41 (s, 3H); 2,09 (br s, 2H); 1,50-1,28 + (m, 4H); 0,93 (t, 3H); МС (XI) 86 (100%), 118 [M+1] . e) (2Н)-2-{[2-Аміно-5(бензилтіо)[1,3]тіоазоло[4,5-d]піримідин-7іл](метил)аміно}-пентан-1-ол Круглодонна колба була з холодильником сухий лід-етанол та зануреною у баню охолодження сухий лід-етанол. Аміак (200 мл) конденсували у колбу з наступним додаванням (2R)-2-{[2-аміно-5(бензилтіо)[1,3]тіоазоло[4,5-d]піримідин-7іл](метил)аміно}пентан-1-олу (5,43 г, 13,9 ммоль). Утвореній суміші давали нагрітися до -33°C та металевий натрій додавали малими шматками до появи синього кольору та його утримування протягом 30 с. Реакцію тоді гасили ложкою твердого амоній хлориду. Аміак випарювали та воду (250 мл) додавали до залишку. Утворену суміш нейтралізували 1М хлоридною кислотою (водн.). Осаджений продукт збирали фільтруванням, промивали водою та ацетонітрилом та сушили у вакуумі, отримуючи 3,38 г (81% виходу) заголовної сполуки. 1 Н ЯМР (ДМСО-d6) 12,81 (br s, 1Н); 8,45 (br s, 2Н), 4,84 (br s, 1H), 3,55-3,40 (m, 2H), 3,02 (s,3H), 1,48 (m, 2H), 1,21 (m, 2H), 0,87 (t, 3H); MC (ІЕР) m/z + 300 [М+1] . g) (2R)-2-[(2-Аміно-5-{[(1S)-1-(6-хлорпіридин-3іл)етил]тіо}[1,3]тіоaзоло[4,5-d]піримідин-7іл)(метил)аміно]пентан-1-ол 5-(Бензилтіо)-7-хлор[1,3]тіоазоло[4,5d]піримідин-2-амін (6,0 г, 19,4 ммоль) розчиняли у NMP (25 мл). DIPEA (6,8 мл, 38,8 ммоль) та додавали (2R)-2-(метиламіно)пентан-і-олу (3,4 г, 29,1 ммоль) та суміш гріли до 120°C протягом 3 діб. Ще додавали (2R)-2-(метиламіно)пентан-1-ол (350 мг, 2,99 ммоль) та DIPEA (1 мл, 5,74 ммоль) та реакційну суміш гріли протягом 6 годин при 120°C. Після охолодження до кімнатної температури, суміш виливали на лід. Осаджений продукт збирали фільтруванням та очищали колонковою флешхроматографією (елюент ДХМ: етилацетат градієнт), отримуючи заголовну сполуку (5,74 г, 76% виходу). 1 Н ЯМР (ДМСО-d6) 7,98 (br s, 2H), 7,41 (m, 2H), 7,29 (m, 2H), 7,22 (m, 1H), 4,73 (t, 1Н), 4,54 (br s, 1Н), 4,33 (m, 2Н), 3,55-3,40 (m, 2Н), 3,01 (s, 3H), 1,52-1,44 (m, 2H), 1,25-1,10 (m, 2Н), 0,84 (t, 3H); МС + (ІЕР) m/z 390 [М+1] . Заголовну сполуку отр имували загальним способом А, починаючи з (2R)-2-[(2-аміно-5меркапто[1,3]тіоазоло[4,5-d]піримідин-7іл)(метил)аміно]пентан-1-олу (96 мг, 0,32 ммоль) та 2-хлор-5-[(1R)-1-хлоретил]піридину (85 мг, 0,48 ммоль). Очистка колонковою флешхроматографією (елюент ДХМ: етилацетат) дала 22 мг (16% виходу) заголовної сполуки. 1 -1 Н ЯМР (400 МГц, ДМСО-d6) млн 8,51 (d, 1Н), 7,99 (br s, 2H), 7,96 (dd, 1H), 7,45 (d, 1H); 4,98 (q, 1H); 4,73 (t, 1H); 4,46 (br s, 1H); 3,53-3,38 (m, 2H), 2,97 (s, 3H), 1,66 (d, 3H), 1,48 (m, 2H), 1,17 (m, + 2H), 0,84 (t, 3H); MC (IEP) m/z 439 та 441 [M+1] . Приклад 7 (2R)-2-[(2-Аміно-5-{[(1S)-1-(6-хлорпіридин-3іл)етил]тіо}[1,3]тіоазоло[4,5-d]пipимiдин-7iл)aмiнo]пeнтaн-1-oл 31 а) (2R)-2-{[2-Аміно-5(бензилтіо)[1,3]тіоазоло[4,5-d]піримідин-7іл]аміно}пентан-1-ол 5-(Бензилтіо)-7-хлор[1,3]тіоазоло[4,5гі]піримідин-2-амін (6,0 г, 19,4 ммоль) розчиняли у NMP (30 мл). DIPEA (8,4 мл, 48,5 ммоль) та додавали 2-аміно-(2R)-1-пентанол (3,5 г, 33,9 ммоль) та суміш гріли до 110°C протягом 4 діб. Після охолодження до кімнатної температури, суміш виливали у воду (200 мл). Осаджений продукт збирали фільтруванням, промивали водою та застосовували на наступному етапі без очистки (7,0 г, 97% виходу). + MC (IEP) m/z 376 [M+1] . b) (2R)-2-[(2-Аміно-5-меркапто[1,3]тіоазоло[4,5d]піримідин-7-іл)аміно]пентан-1-ол 95966 32 Заголовну сполуку отримували загальним способом А, починаючи з (2r)-2-[(2-аміно-5меркапто[1,3]тіоазоло[4,5-d]піримідин-7іл)аміно]пентан-1-олу (70 мг, 0,245 ммоль) та 2хлор-5-[(1R)-1-хлоретил]піридину (Приклад 6b, 52 мг, 0,29 ммоль). Очистка препаративною ВЕРХ дала 23 мг (22% виходу) заголовної сполуки. 1 Н ЯМР (400 МГц, ДМСО-d6) 8,50 (d, 1H); 7,99 (br s, 2H); 7,95 (dd, 1H); 7,44 (d, 1H); 6,91 (d, 1Н); 4,95 (q, 1Н); 4,11 (m, 1Н); 3,41 (dd, 1Н); 3,31 (dd, 1Н); 1,64 (d, 3H); 1,56 (m, 1Н); 1,41 (m, 1Н); 1,4-1,2 (m, 2Н); 0,85 (t, 3H); МС (ІЕР) m/z 425 та 427 + [М+1] . Приклад 8 2-{(1S)-1-[(2-Аміно-7-{[(1R)-1-(гідроксиметил)-3метилбутил]аміно}[1,3]тіоазоло[4,5-d]піримідин-5іл)тіо]етил}ізонікотинонітрил a) 2-((S)-1-Гідрокси-етил)-ізонікотинонітрил Круглодонна колба була з холодильником сухий лід-етанол та зануреною у баню охолодження сухий лід-етанол. Аміак (250 мл) конденсували у колбу з наступним додаванням (2R)-2-{[2-аміно-5(бензилтіо)[1,3]тіоазоло[4,5-d]піримідин-7іл]аміно}пентан-1-олу (6,8 г, 18,1 ммоль). Утвореній суміші давали нагрітися до -33°C та металевий натрій додавали малими шматками до появи синього кольору та його утримування протягом 30 с. Реакцію тоді гасили ложкою твердого амоній хлориду. Аміак випарювали та воду (250 мл) додавали до залишку. Утворену суміш нейтралізували 1М хлоридною кислотою (водн.). Осаджений продукт збирали фільтруванням, промивали водою та сушили у вакуумі, отримуючи 4,15 г (80% виходу) заголовної сполуки. + MC (IEP) m/z 286 [M+1] . c) (2R)-2-[(2-Аміно-5-{[(1S)-1-(6-хлорпіридин-3іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7іл)аміно]пентан-1-ол Заголовну сполуку отримували загальним способом В, починаючи з 2-ацетил-ізонікотинонітрилу (2,00 г, 13,7 ммоль) та (-)-DIPCI (6,58 г, 20,53 ммоль), отримуючи заголовну сполуку (625 мг, 4,22 ммоль) із 90% ен. 1 Н ЯМР (CDCI3) 8,72 (d, 1Н), 7,62 (s, 1H), 7,44 (dd, 1Η), 4,96 (q, 1Н), 1,54 (d, 3H). b) 2-((R)-1-Хлор-етил)-ізонікотинонітрил Заголовну сполуку отримували з 2-((S)-1гідрокси-етил)-ізонікотинонітрилу (620 мг, 4,18 ммоль) загальним способом С, за винятком того, що застосовували 1,0 еквів. NCS та 1,0 еквів. PPh3. Через 24 годин реакції ще 0,2 еквів. кожного з NCS та PPh3 додавали та температуру збільшували до 30°C протягом 12 годин. Обробка та очистка колонковою флеш-хроматографією дали заголовну сполуку (46 мг, 7% виходу) із 82% ен. 1 Н ЯМР (CDCI3) 8,74 (d, 1Н), 7,76 (s, 1Н), 7,46 (dd, 1Н), 5,16 (q, 1H), 1,88 (d, 3H). 33 с) 2-{(1S)-1-[(2-Аміно-7-{[(1R)-1(гідроксиметил)-3-метилбутил]аміно}[1,3]тіоазоло[4,5-d]піримідин-5іл)тіо]етил}ізонікотинонітрил Заголовну сполуку отримували загальним способом А, починаючи з 2-((R)-1-хлор-етил)ізонікотинонітрилу (46 мг, 0,28 ммоль) та (2R)-2[(2-аміно-5-меркапто[1,3]тіоазоло[4,5-d]піримідин7-іл)аміно]-4-метилпентан-1-олу (69 мг, 0,23 ммоль). Очистка хіральною ВЕРХ (Колонка: Chiralpak AD 50x150 мм, елюент: гептан:ізопропанол 85:15), швидкість потоку 60 мл/хвил) дала заголовну сполуку (20 мг, 20% виходу). 1 -1 Н ЯМР (400 МГц, ДМСО-d6) млн 8,78 (d, 1H) 8,00 (s, 1H) 7,95 (s, 1H) 7,72 (m, 1H) 6,90 (m, 1H) 5,14 (q, 1H) 4,64 (t, 1H) 4,23 (br s, 1H) 3,23-3,41 (m, 2H) 1,67 (d, 3H) 1,64-1,52 (m, 1H) 1,32-1,48 (m, + 2H) 0,87 (dd, 6H); MC (IEP) m/z 430 [M+1] . Приклад 9 Приклад 9а (2R)-2-[(2-аміно-5-{[(1R)-1-(3-хлорпіридин-4іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол та Приклад 9b (2R)-2-[(2-аміно-5-{[(1S)-1-(3-хлорпіридин-4іл)етил]тіо}[1,3]тіоазоло[4,5-d]піримідин-7-іл)аміно]4-метилпентан-1-ол Діастереомерну суміш (2R)-2-[(2-аміно-5-{[1-(3хлорпіридин-4-іл)етил]тіо}[1,3]тіоазоло[4,5d]піримідин-7-іл)аміно]-4-метилпентан-1-олу (105 мг) з прикладу 4 розділяли препаративною ВЕРХ, отримуючи 14 мг ельованого першим ізомеру (Приклад 9а): 1 -1 Н ЯМР (400 МГц, ДМСО-d6) млн 8,59 (s, 1H), 8,48 (d, 1H), 8,00 (s, 2H), 7,68 (d, 1H); 6,93 (d, 1H); 5,27 (q, 1H); 4,63 (t, 1H); 4,16 (br s, 1H); 3,423,30 (m, 2H), 1,64 (d, 3H), 1,58-1,30 (m, 3H), 0,84 (d, + 3H), 0,74 (d, 3H); MC (IEP) m/z439 та 441 [M+1] . та 15 мг ельованого останнім ізомеру (Приклад 9b): 95966 34 Н ЯМР (400 МГц, ДМСО-d6) млн 8,60 (s, 1H), 8,48 (d, 1H), 8,00 (s, 2H), 7,67 (d, 1Н); 6,92 (d, 1Н); 5,28 (q, 1Н); 4,56 (t, 1Н); 4,22 (br s, 1H); 3,333,12 (m, 2H), 1,62 (d, 3H), 1,60-1,30 (m, 3H), 0,88 (d, + 3H), 0,85 (d, 3H); MC (IEP) m/z 439 та 441 [M+1] . Фармакологічний скринінг Матеріали Рекомбінантний фракталкін людини (hCX3CL1) та рекомбінантний інтерлейкін-8 людини (IL-8 або hCXCL8) отримували від PeproTech Inc., UK Реко125 125 мбінантний [ І]-фракталкін (людини) та [ І] hlL-8 зі специфічною активністю 2200 Кі/ммоль, отримували від NEN Life Science Products, Inc., UK. Fluo4-AM отримували від Molecular Probes, US. Усі інші хімікати були аналітичного гатунку. Клітини Повну кДНК людини CX3CR1 (G enBank № U20350) отримували з мРНК мозку людини (Superscript, Life Technologies) та лігували у вектор pCR-Blunt II ТОРО (InVitrogen). Вставку, відповідну hCX3CR1 виділяли та субклонували у pcDNA3,1zeo. Плазмідну ДНК отримували, застосовуючи комплект Plasmid Midi Kit (Qiagen). Застосовуючи реагент трансфекції Superfect (Qiagen) згідно з протоколом виробника експресійну плазміду для hCX3CR1 тоді уводили у суспензію лінії клітин нирок ембріона людини (HEKS) 293, що містять вектор для стабільної експресії химерного G-білку Gqi5. Стабільний клон створювали, застосовуючи селекцію зеоцином (500 мкг/мл) та гігроміцином (100 мкг/мл). Для наступних застосувань клітини тримали у суміші модифіковане Дульбекко середовище Ігла/живильне середовище Гама F12 (DMEM/F12), що містить піридоксин та доповнене 10 об.% сироватки зародка теляти, 2 мМ Lглютаміну, 100 Од/мл пеніциліну та 100 мг/мл стрептоміцину, 250 мкг/мл зеоцину та 100 мкг/мл гігроміцину. Клітини, що експресують CXCR2 людини, отримані від AstraZeneca Charnwood, культивують у ЕМЕМ, що містить Glutamax та доповнене 10% сироватки зародка теляти (від РАА, Austria), 1% замінних амінокислот (NEAA), 100 Од/мл пеніциліну та 100 мкг/мл стрептоміцину (PEST) та 500 мкг/мл генетицинуА3418. Отримання мембран Клітини вирощують при 37°С та 5% СО2 та збирають при конфлюєнтності 60-80% у буфер, що містить 10 мМ Трис-НСІ рН 7,4, 5 мМ EDTA, 0,1 мг/мл бацитрацину. Клітини центрифугують при 300хg протягом 10 хвил та пелету ресуспендують у буфері для збирання (10 мМ Трис-НСІ, рН 7,4, 5 мМ етилендіамінтетра-оцтова кислота (ЕДТА) та 0,1 мг/мл бацитрацину), поєднують та гомогенізують, застосовуючи гомогенізатор Даунса. Гомогенат центрифугують при 48000хg протягом 10 хвил та ресуспендують у буфері для збирання, застосовуючи Ultra-Turrax T8. Аліквоти мембран тримають при -80°С. Концентрацію білку визначають у мікротирувальних планшетах, як описано Harrington (1990, Anal. Biochem. 186, 285-287). Дослідження зв'язування рецептору in vitro Дослідження конкурентного зв'язування 125 [ І]фракталкіну проводили у глибоких 96лункових планшетах на 2 мл (Beckman, Germany), 1 -1 35 у загальному об'ємі 1000 мкл/лунку. Кожна лунка 125 містила 10 пМ [ І]-фракталкіну та еквівалент мембран для концентрації рецептору 1 пМ у буфері для дослідження (50 мМ Гепес-КОН, рН 7,4, 10 мМ МgСІ2, 1 мМ ЕДТА, 0,1 мас/об.% желатину). Десять концентрацій (2 точки/лог. од.) тест-сполук спочатку розчиняли у ДМСО та давали досягти кінцевої концентрації 1 об.% ДМСО. Дослідження починали додаванням мембран та інкубували при 25°C протягом 24 годин. Реакції зупиняли швидким фільтруванням через скляні фільтри Whatman GF/B, попередньо оброблені 0,3% поліетиліміном, та наступним промиванням охолодженим льодом буфером (10 мМ Гепес-КОН рН 7,4, 500 мМ NaCI), застосовуючи збирач для зв'язування рецепторів Brandel. Додавали сцинтиляційну суміш та радіоактивність визначали у лічильнику рідинної сцинтиляції Packard 2500TR. (Perkin Elmer, USA) 125 Дослідження конкурентного зв'язування [ l]hlL-8 проводять у білих 96-лункових планшетах з прозорим дном з кінцевим об'ємом 200 мкл та ко125 жна лунка містить 150 пМ [ l]-hlL-8 (специфічна активність 2200 Кі/ммоль), препарату мембранSPA, еквівалентного 20 пМ рецепторів, та 1,5 мг кульок SPA у буфері для дослідження [50 мМ ГЕПЕС-КОН рН 7,4, 10 мМ МgСІ2, 1 мМ ЕДТА, 0,5мас./об.% желатину]. Тест-сполуки обробляли як вищезазначено. Неспецифічне зв'язування визначають при наявності 500 нМ неміченого hlL-8. Агоніст hlL-8 (крива концентрація-реакція від 3 пМ до 30 нМ), застосовують як сполуку для порівняння при кожному тесті. Пептидна крива не містить ДМСО. Реакцію сполучення починають додаванням 140 мкл препарату мембран-SPA, та зразки інкубують у темряві при кімнатній температурі протягом 4 годин. Планшети для дослідження підраховують у лічильнику рідинної сцинтиляції (Wallac Micro Beta TriLux 1450 від PerkinElmer, USA). 35 Зв'язування [ S]GTPS 35 Дослідження зв'язування [ S]GTPyS проводили у мікротирувальних планшетах з прозорим дном з 10 концентраціями інгібітору (2 конц./лог. од.), розбавленого ДМСО (кінцева концентрація 1%), при кімнатній температурі. Мембрани, що експресують рецептор hCX3CR1 (кінцева концентрація 20 мкг білку/лунку) додавали разом з кульками SPA (кінцева концентрація 1 мг/лунку), усі суспендовані у буфері зв'язування GTPS (50 мМ Трис-НСІ, 100 мМ NaCI, 0,1% желатину, 15 мкг Комп’ютерна верстка О. Гапоненко 95966 36 сапоніну/мл та 3 мкМ GDP, рН 7,4 при кімнатній температурі). Мембрани, кульки SPA та ліки попередньо інкубували 30 хвил перед додаванням 310 пМ фракталкіну для максимальної стимуляції. Базову активність визначали як активність без стимуляції фракталкіном (буфер зв'язування GTPS). Через ще 30 хвил реакцію починали з додаванням 35 [ S]GTPS до кінцевої концентрації 0,1 нМ та кінцевого об'єму дослідження 0,2 мл. Експеримент завершували через 30 хвилин центрифугуванням при 2000 об/хвил протягом 2x5 хвилин (різні напрями) та радіоактивність визначали у лічильнику рідинної сцинтиляції (Wallac MicroBeta TriLux 1450). Результати Звичайні значення Кі CX3CR1 для сполук заявленого винаходу є у межах приблизно 0,1-1000 нМ. Інші значення Кі CX3CR1 є у межах приблизно 0,1 нМ - 500 нМ. Наступні значення Кі CX3CR1 є у межах приблизно 0,1 нМ - 25 нМ. Результати від дослідження зв'язування in vitro hCX3CR1 для кінцевої сполуки показані у таблиці. Таблиця Приклад № 1 2 3 4 5 6 7 8 9a 9b Кi (нМ) 21 6,2 6,5 Не тестовано* 23 203 149 81 He тестовано 35*) Приклад 4 представляє суміш діастереомерів, що є розділеними у прикладі 9. 1 Сполуки заявленого винаходу, де R - Me (містить розгалужений тіоалкілпіридил у 5-позиції) є більш потужними антагоністами стосовно рецептору CX3CR1 та/або менш потужними антагоністами стосовно рецептору CX3CR2, ніж відповідні 1 сполуки для порівняння, де R - Н. Така посилена селективність стосовно антагонізму рецептору CX3CR1, як очікують, призводитиме до значної терапевтичної корисності. Підписне Тираж 23 прим. Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійською5,7-disubstituted[1,3]thiazolo[4,5]pyrimidin-2(3h)-amine derivatives and their use in therapy
Автори англійськоюJohansson Rolf, Karlstrom, Sofia, Nordvall, Gunnar, KERS ANNIKA, Kers, Annika, Rein Tobias, Slivo Can
Автори російськоюЙоганссон Рольф, Карлстрьом София, Нордволл Гуннар, Керс Анника, Рейн Тобиас, Сливо Кен
МПК / Мітки
МПК: A61P 11/06, A61P 25/00, A61K 31/519, A61P 9/10, C07D 513/04
Мітки: похідні, 5,7-дизаміщених[1,3]тіоазоло[4,5-d]піримідин-2(3н)аміну, застосування, терапії
Код посилання
<a href="https://ua.patents.su/18-95966-pokhidni-57-dizamishhenikh13tioazolo45-dpirimidin-23naminu-ta-kh-zastosuvannya-u-terapi.html" target="_blank" rel="follow" title="База патентів України">Похідні 5,7-дизаміщених[1,3]тіоазоло[4,5-d]піримідин-2(3н)аміну та їх застосування у терапії</a>
5,7-дизаміщені похідні [1,3]тіазоло[4,5-d]піримідин-2(3н)-ону та їх застосування у терапії

Номер патенту: 95811
Опубліковано: 12.09.2011
Автори: Рейн Тобіас, Йоганссон Рольф, Нордволл Ґуннар, Карлстрьом Софія, Сліво Кен, Керс Анніка
МПК: C07D 513/04, A61P 25/00, A61P 9/10, A61K 31/519, A61P 11/06
Мітки: 1,3]тіазоло[4,5-d]піримідин-2(3н)-ону, застосування, похідні, терапії, 5,7-дизаміщені
Формула / Реферат:
1. Сполука формули (І), (I) де: R1 - CH3 або CF3; R2 - галоген, CN або С1-6алкіл; R3 - H або CH3; R4 - H або CH3; n = 0, 1 або 2;як вільна основа або її фармацевтично прийнятна сіль, сольват або сольват солі. 2. Сполука за...
Похідні піримідин-2-аміну і їх застосування як антагоністів аденозинового рецептора a2b

Номер патенту: 82563
Опубліковано: 25.04.2008
Автори: Естеве Тріас Крістіна, Відаль Хуан Бернат
МПК: C07D 471/04, A61K 31/506, C07D 405/14, C07D 401/14, A61P 11/06, C07D 409/14
Мітки: аденозинового, застосування, антагоністів, рецептора, піримідин-2-аміну, похідні
Формула / Реферат:
1. Сполука формули (І), (І)у якійR1 означає моноциклічну або поліциклічну арильну або гетероарильну групу, яка необов'язково містить 1, 2 або 3 замісники, вибрані із групи, яка включає атоми галогенів, лінійний або розгалужений, необов'язково заміщений нижчий алкіл, гідроксигрупу, лінійну або розгалужену, необов'язково заміщену нижчу алкоксигрупу, -SH,...
Похідні піроло[3,2-d]-піримідин-4-ону та їх застосування у терапії

Номер патенту: 89968
Опубліковано: 25.03.2010
Автори: Півонка Доналд, Ло-Альфредссон Івонн, Тіден Анна-Карін, Беґевіґ Андерс
МПК: C07D 487/04, A61K 31/519, A61P 9/10
Мітки: терапії, піроло[3,2-d]-піримідин-4-ону, похідні, застосування
Формула / Реферат:
1. Сполука формули (І), (I)де:принаймні один з Х та Y - S, а інший - О або S;L - безпосередній зв'язок або С1-7алкілен, вказаний алкілен, як варіант, містить гетероатом, вибраний з О, S(O)n та NR6, вказаний алкілен, як варіант, містить один або два подвійні зв'язки карбон-карбон, і вказаний алкілен, як варіант, заміщено одним або більше...
Похідні піридо[2,3-d]піримідину, їх одержання, їх застосування у терапії

Номер патенту: 92021
Опубліковано: 27.09.2010
Автори: Казелла П'єр, Перро П'єр, Мюно Клод, Буррі Бернар, Жегам Самір
МПК: A61P 35/00, C07D 471/04, A61K 31/519
Мітки: застосування, похідні, одержання, терапії, піридо[2,3-d]піримідину
Формула / Реферат:
1. Сполука формули (І), в якій Аr2 і R1 є відповідно: 2,6-дихлорофеніл та трет-бутил або 2-бромо-6-хлорфеніл та трет-бутил; або 2,6-дихлорофеніл та етил; або 2,6-дибромофеніл та трет-бутил; або 2,6-дибромофеніл та етил; або...
Похідні тетрагідроізохінолілсульфонамідів, їх одержання й застосування в терапії

Номер патенту: 84771
Опубліковано: 25.11.2008
Автори: Діас Мартін Хуан Антоніо, Хімінес Баргуено Марія Долорес
МПК: A61K 31/4725, A61P 3/04, C07D 405/06, C07D 217/04, C07D 217/02, A61K 31/5377, A61K 31/472, A61K 31/496, A61P 3/10, C07D 409/06, C07D 217/06, A61P 25/00, C07D 401/12
Мітки: терапії, похідні, тетрагідроізохінолілсульфонамідів, застосування, одержання
Формула / Реферат:
1. Сполука формули І:, Іде:n може приймати значення від 1 до 6;-(С)n- являє собою С1-6-алкіліден, за необхідності заміщений замісниками в числі від 1 до 4, вибраними з атома галогену, гідрокси, нітро, ціано, аміно, С1-3-моноалкіламіно, С2-6-діалкіламіно або С1-3-алкокси;R1 являє собою:атом водню;С1-6-алкіл;R2 являс...
Попередній патент: Конденсовані гетероциклічні похідні та їх застосування
Наступний патент: Препаративна форма для локального місцевого введення, що містить індоксакарб
Випадковий патент: Підкопуюче обладнання для зрізання та евакуації ґрунту з-під трубопроводу