Аналог азацитидину та його застосування
Номер патенту: 99308
Опубліковано: 10.08.2012
Автори: Еріксен Олє Хенрік, Мюрен Фінн, Сандвольд Маріт Ліланд, Сілверман Льюіс, Холланд Джеймс

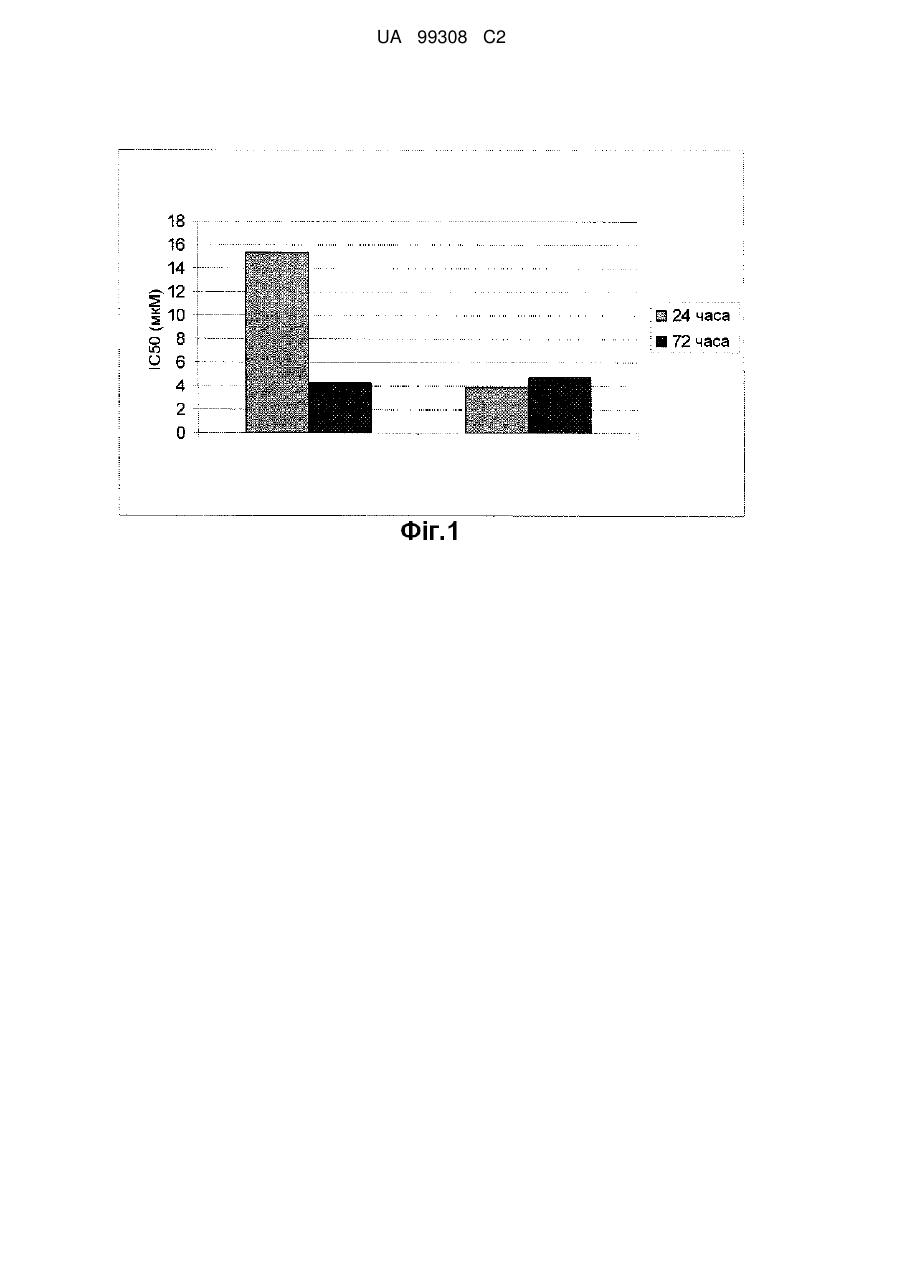







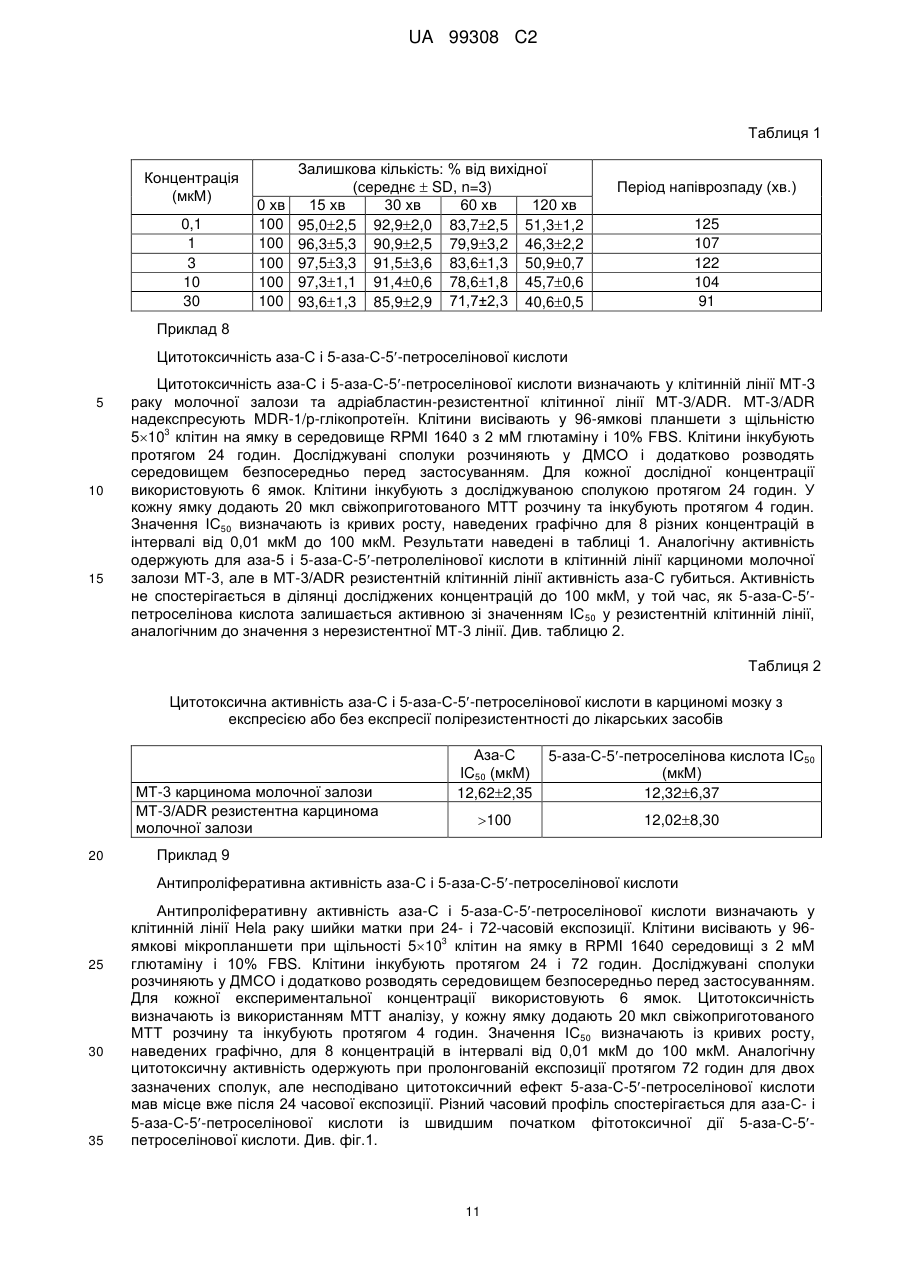




Формула / Реферат
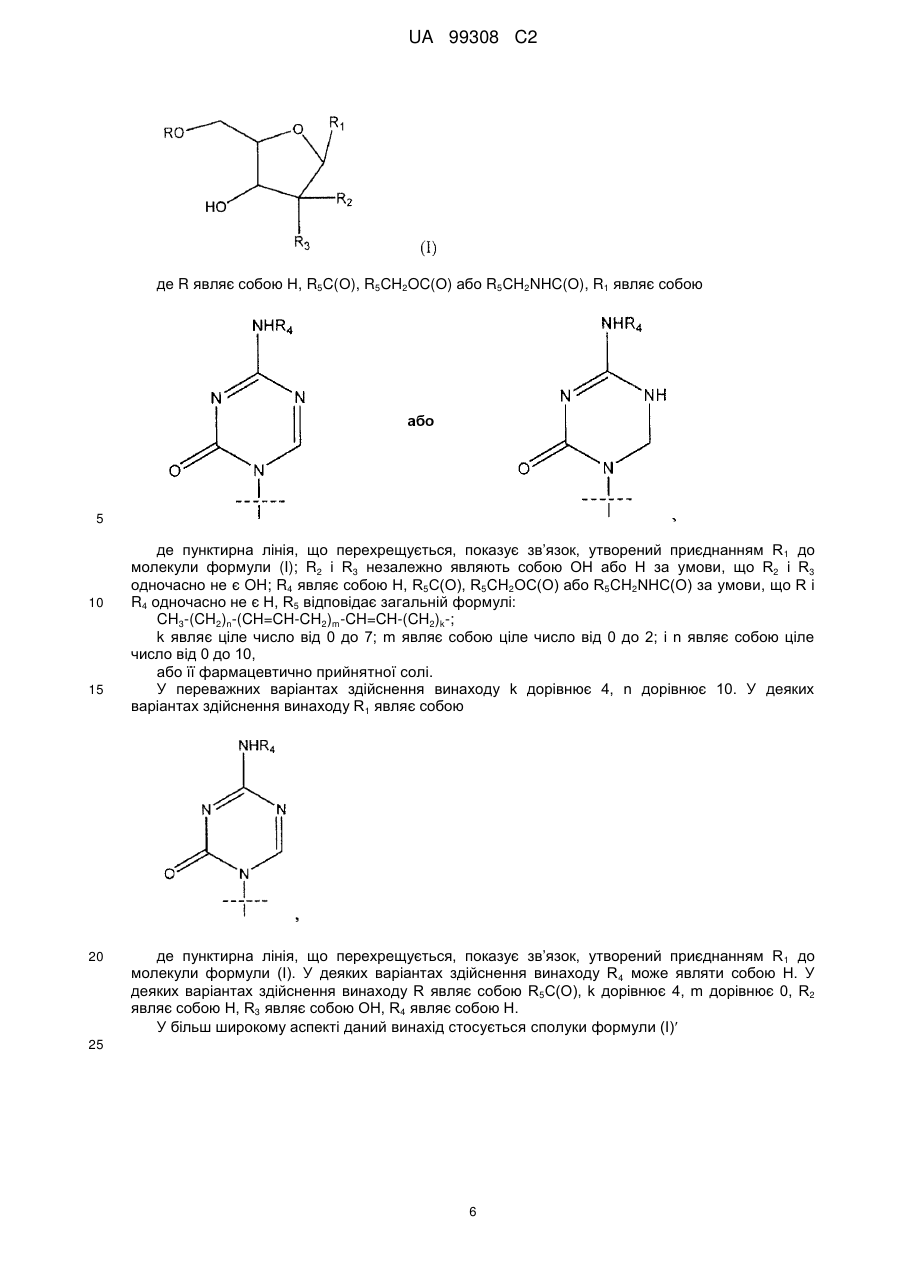
1. Сполука формули (І)
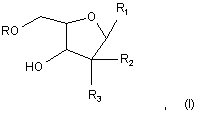
де R являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O);
R1 являє собою
 ,
,
де пунктирна лінія, що перехрещується, показує зв'язок, утворений приєднанням R1 до молекули формули (І);
R2 і R3 незалежно являють собою ОН або Η, за умови, що R2 і R3 одночасно не є ОН;
R4 являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O), за умови, що R і R4 одночасно не є Н;
R5 має загальну формулу:
CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-;
k являє собою ціле число від 0 до 7;
m являє собою ціле число від 0 до 2; і
n являє собою ціле число від 0 до 10,
або її фармацевтично прийнятна сіль.
2. Сполука за п. 1, де k дорівнює 4.
3. Сполука за п. 2, де n дорівнює 10.
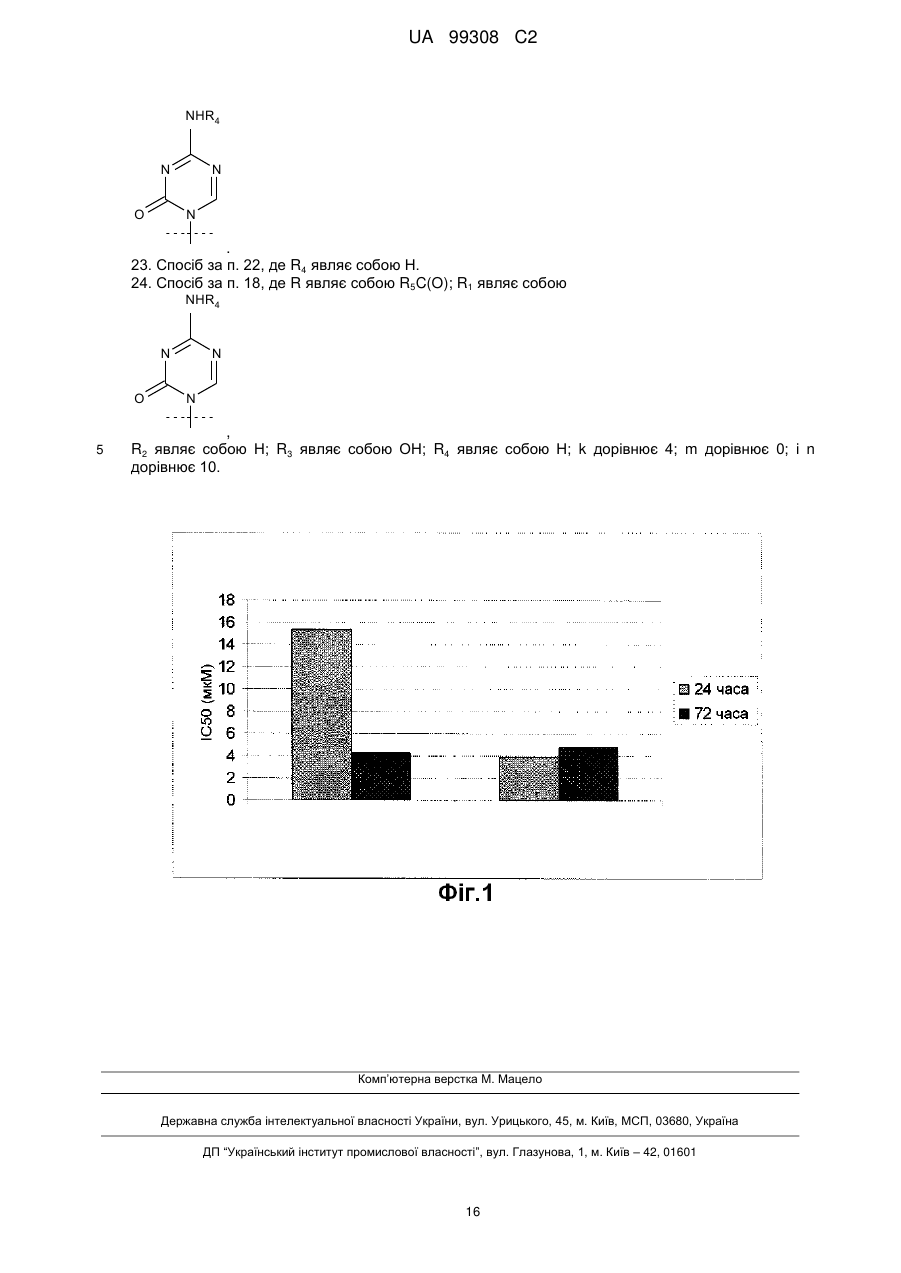
4. Сполука за п. 1, де R1 являє собою
 .
.
5. Сполука за п. 4, де R4 являє собою Н.
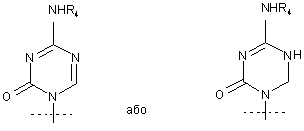
6. Сполука за п. 1, де R являє собою R5C(O); R1 являє собою
,
R2 являє собою Н; R3 являє собою ОН; R4 являє собою Н; k дорівнює 4; m дорівнює 0; і n дорівнює 10.
7. Фармацевтична композиція, що містить: сполуку за п. 1, фармацевтичний наповнювач, розріджувач і/або носій.
8. Спосіб лікування у суб'єкта новоутворення, що включає: вибір суб'єкта з новоутворенням і
введення суб'єкту сполуки формули:
де R являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O);
R1 являє собою
,
де пунктирна лінія, що перехрещується, показує зв'язок, утворений приєднанням R1 до молекули формули (І);
R2 і R3 незалежно являють собою ОН або Н, за умови, що R2 і R3 одночасно не є ОН;
R4 являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O), за умови, що R і R4 одночасно не є Н;
R5 має загальну формулу:
СН3-(СН2)n-(СН=СН-СН2)m-CH=СН-(СН2)k-;
k являє собою ціле число від 0 до 7;
m являє собою ціле число від 0 до 2; і
n являє собою ціле число від 0 до 10;
або її фармацевтично прийнятної солі в умовах, ефективних для лікування новоутворення у суб'єкта.
9. Спосіб за п. 8, де новоутворення являє собою ракове захворювання.
10. Спосіб за п. 9, де ракове захворювання являє собою солідну пухлину, гематологічний рак або злоякісну пухлину.
11. Спосіб за п. 9, де ракове захворювання являє собою лейкемію, лімфому, множинну мієлому або мієлодиспластичний синдром.
12. Спосіб за п. 10, де солідна пухлина є раком тканини, вибраної з групи, що складається з тканин молочної залози, яєчників, передміхурової залози, мозку, сечового міхура та легенів.
13. Спосіб за п. 8, де k дорівнює 4.
14. Спосіб за п. 13, де n дорівнює 10.
15. Спосіб за п. 8, де R1 являє собою
.
16. Спосіб за п. 15, де R4 являє собою Н.
17. Спосіб за п. 8, де R являє собою R5C(O); R1 являє собою
,
R2 являє собою Н; R3 являє собою ОН; R4 являє собою Н; k дорівнює 4; m дорівнює 0; і n дорівнює 10.
18. Спосіб лікування у суб'єкта запального процесу, що включає: вибір суб'єкта із запальним процесом і введення суб'єкту сполуки формули:
де R являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O); R1 являє собою
,
де пунктирна лінія, що перехрещується, показує зв'язок, утворений приєднанням R1 до молекули формули (І);
R2 і R3 незалежно являють собою ОН або Η, за умови, що R2 і R3 одночасно не є ОН;
R4 являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O), за умови, що R і R4 одночасно не є Н,
R5 являє собою С3-С26алкеніл, де R5 має загальну формулу CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-;
k являє собою ціле число від 0 до 7; m являє собою ціле число від 0 до 2 і n являє собою ціле число від 0 до 10;
або її фармацевтично прийнятної солі в умовах, ефективних для лікування запального процесу у суб'єкта.
19. Спосіб за п. 18, де запальний процес являє собою запалення легені, запалення сполучної тканини, запалення шлунково-кишкового тракту або запалення судинної сітки.
20. Спосіб за п. 18, де k дорівнює 4.
21. Спосіб за п. 20, де n дорівнює 10.
22. Спосіб за п. 18, де R1 являє собою
.
23. Спосіб за п. 22, де R4 являє собою Н.
24. Спосіб за п. 18, де R являє собою R5C(O); R1 являє собою
,
R2 являє собою Η; R3 являє собою ОН; R4 являє собою Н; k дорівнює 4; m дорівнює 0; і n дорівнює 10.
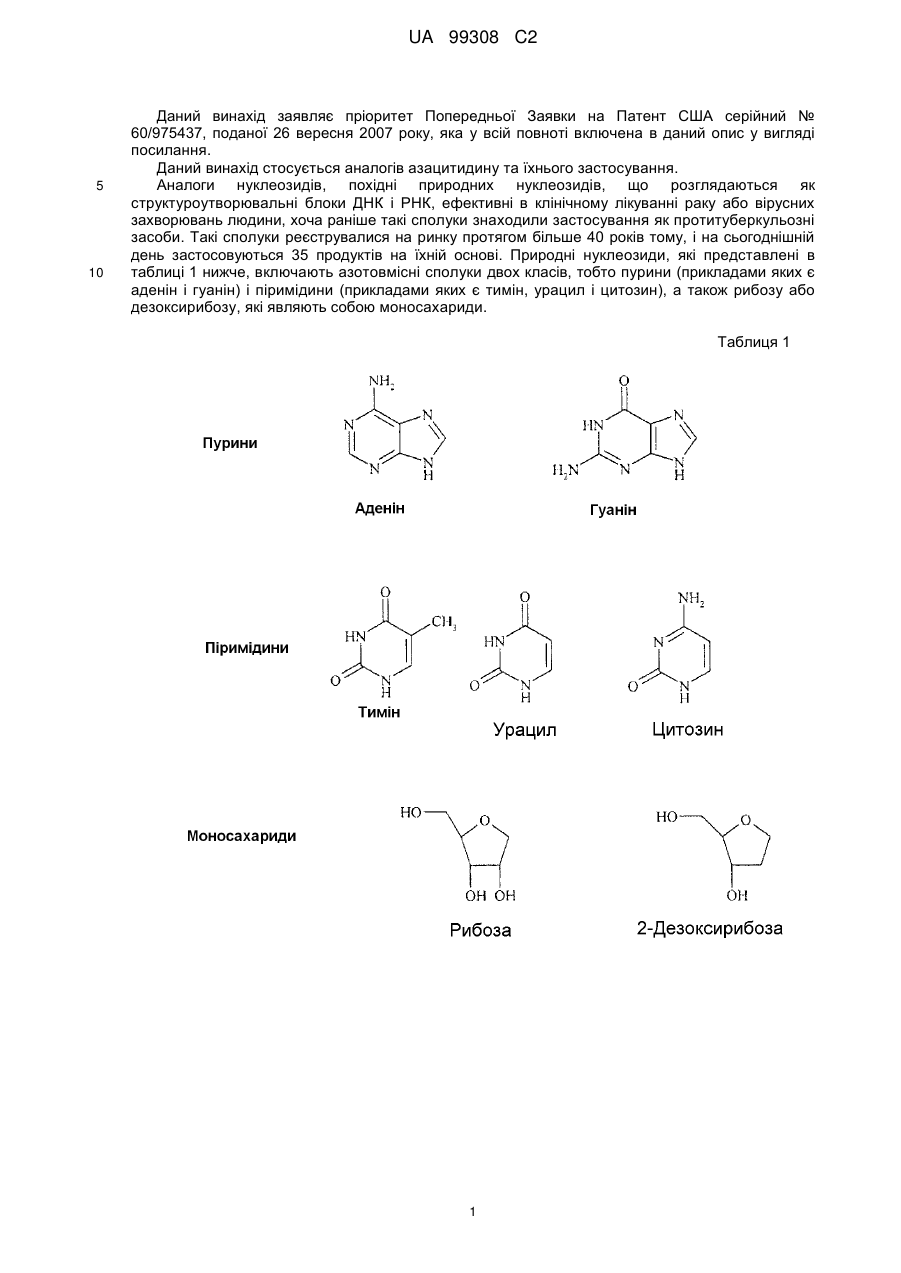
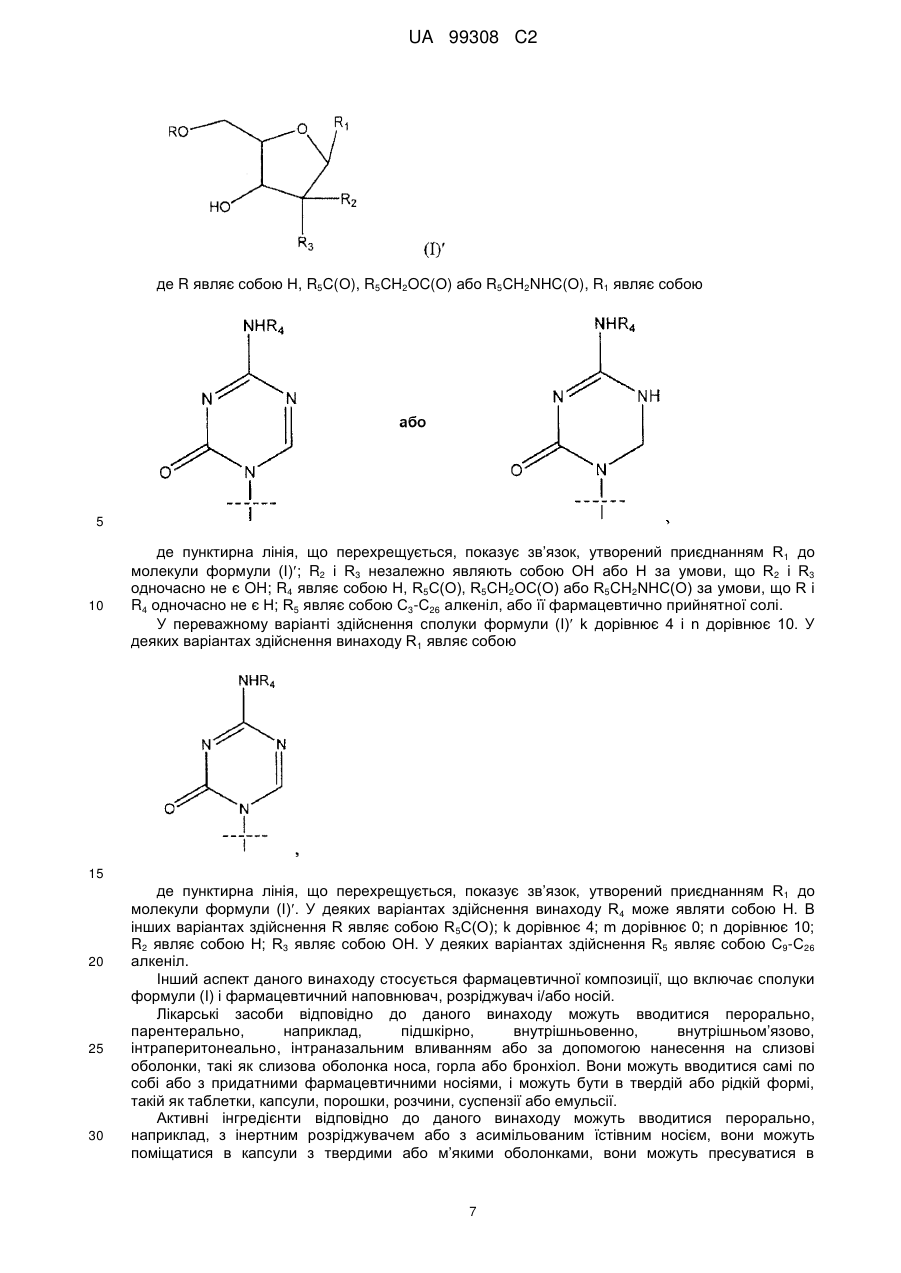
Текст
Реферат: Даний винахід стосується сполук формули (І), яка наведена нижче, де R являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O), R1 являє собою фрагмент, що відповідає формулам, наведеним нижче, де пунктирна лінія, що перехрещується, показує зв'язок, утворений приєднанням R1 до молекули формули (І); R2 і R3 незалежно являють собою ОН або Η, за умови, що R2 і R3 одночасно не є ОН; R4 являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O), за умови, що R і R4 одночасно не є Н, R5 відповідає загальній формулі: CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-; k являє собою ціле число від 0 до 7; m являє собою ціле число від 0 до 2; і n являє собою ціле число від 0 до 10; або їхніх фармацевтично прийнятних солей. Розкриті також способи одержання і застосування зазначених сполук. UA 99308 C2 (12) UA 99308 C2 UA 99308 C2 5 10 Даний винахід заявляє пріоритет Попередньої Заявки на Патент США серійний № 60/975437, поданої 26 вересня 2007 року, яка у всій повноті включена в даний опис у вигляді посилання. Даний винахід стосується аналогів азацитидину та їхнього застосування. Аналоги нуклеозидів, похідні природних нуклеозидів, що розглядаються як структуроутворювальні блоки ДНК і РНК, ефективні в клінічному лікуванні раку або вірусних захворювань людини, хоча раніше такі сполуки знаходили застосування як протитуберкульозні засоби. Такі сполуки реєструвалися на ринку протягом більше 40 років тому, і на сьогоднішній день застосовуються 35 продуктів на їхній основі. Природні нуклеозиди, які представлені в таблиці 1 нижче, включають азотовмісні сполуки двох класів, тобто пурини (прикладами яких є аденін і гуанін) і піримідини (прикладами яких є тимін, урацил і цитозин), а також рибозу або дезоксирибозу, які являють собою моносахариди. Таблиця 1 1 UA 99308 C2 Всі природні нуклеозиди існують у так званій -D-конфігурації, як показано у формулі А нижче. Азотовмісна основа і гідроксиметильний бічний ланцюг на циклі цукру знаходяться з одного боку (цис-положення) площини циклу цукру. 5 10 15 20 25 30 35 40 45 50 Формула А Для одержання нуклеозид-похідних із протираковою або противірусною активністю здійснювалися зміни хімічної структури азотовмісної основи і/або моносахариду. Наприклад, введення атомів галогенів або інших функціональних груп в азотовмісну основу, вставлення додаткових атомів азоту або стереохімічні зміни в моносахаридному циклі з перетворенням рибози в арабінозу або видалення гідроксильної групи з одержанням дезоксирибози можуть призводити до одержання продуктів із можливою корисною терапевтичною дією. У ряді сполук моносахаридний цикл був збережений, у той час як в інших він перетворений у ланцюг. Аналоги нуклеозидів являють собою малі молекули з від хорошої до чудової розчинності у воді. Широкі дослідження і зусилля, спрямовані на розробку нуклеозидних аналогів внаслідок поширення СНІДу в усьому світі, які зробили великий внесок у загальну базу знань і розуміння механізму дії, зміни профілю активності при зміні хімічної структури і т.д., також мають велике значення для галузі техніки, що стосується лікування раку. Загальним недоліком застосування багатьох лікарських засобів, включаючи нуклеозидні аналоги, є їхня низька активність і низька специфічність стосовно лікування цільового захворювання. Деякі з цих проблем можуть бути зумовлені активністю, властивою діючій речовині самого лікарського засобу, інша частина може бути пов’язана з певними механізмами резистентності (властивої пацієнту або набутої ним у процесі лікування раку, такої як, наприклад, полірезистентність до лікарських засобів (MDR)). Деякі проблеми можуть бути результатом недостатнього транспорту або клітинного поглинання, а також поганих механізмів активації. Деякі інші проблеми можуть бути пов’язані зі швидкою інактивацією і/або екскрецією лікарського засобу. Ефективність нуклеозидних аналогів значною мірою залежить від їхньої здатності імітувати природні нуклеозиди, взаємодіючи з вірусними і/або клітинними ферментами і впливаючи на визначальні процеси в метаболізмі нуклеїнових кислот або інгібуючи ці процеси. Для того, щоб привести в дію противірусну або протиракову активність нуклеозидних аналогів, вони повинні бути перетворені через їхні моно- і дифосфати у відповідні трифосфати під дією вірусних і/або клітинних кіназ. Як правило, активною сполукою є трифосфат, але у деяких похідних, таких як, наприклад, гемцитабін, навіть дифосфат може виявляти клінічно значущу дію. Для того щоб нуклеозидні аналоги досягали хворих, ракових або інфікованих вірусами клітин або тканин після ентерального або парентерального введення, нуклеозидні аналоги повинні мати сприятливі фармакокінетичні характеристики. Крім швидкої екскреції введеного лікарського засобу багато-які нуклеозидні аналоги можуть дезактивуватися і в тоці крові, і в тканинах. Наприклад, похідні цитозину, навіть на монофосфатному рівні, можуть швидко дезамінуватися внаслідок дії ферментів класу дезаміназ з одержанням неактивного аналога урацилу. Тому клітинне поглинання і, як наслідок, хороша терапевтична ефективність багатьох нуклеозидних аналогів значною мірою залежать від білків трансмембранного переміщення пов’язаних нуклеозидів (які називають концентративними і переносниками нуклеозидів різної маси). Отже, бажаними є сполуки, дія яких не засновується на таких специфічних механізмах поглинання. Ще одним фактором, що обмежує активність, особливо в галузі боротьби з раковими захворюваннями, є клітинні механізми відновлення. Коли протираковий монофосфатний нуклеозидний аналог вводиться в клітинну ДНК, він не повинен виводитися з ДНК ракової клітини внаслідок екзонуклеазної активності, пов’язаної з р53 білком. Однак видалення нуклеозидного аналога з ДНК здорової клітини є сприятливим для обмеження побічних ефектів лікарського засобу. Протягом багатьох років була розроблена велика кількість нуклеозидних аналогів, які значною мірою долають деякі або багато-яких факторів, що несприятливо впливають на їхню 2 UA 99308 C2 5 10 15 20 25 30 35 40 активність. Наприклад, ацикловір (ACV) може бути ілюстрацією сполуки з великою селективністю дії. ACV-монофостат може бути одержаний тільки під дією вірусних кіназ, а це означає, що ACV не може активуватися в неінфікованих клітинах. Незважаючи на цей факт, ACV не є дуже активним продуктом. Для того, щоб обійти стадію, що часто обмежує швидкість активації нуклеозидного аналога, стадію внутрішньоклітинного утворення монофосфату нуклеозидного аналога, було розроблено декілька фосфонатів, таких як цидофовір, і навіть монофосфатів. Для полегшення перорального поглинання або гарантовано сприятливого використання лікарського засобу в організмі, були розроблені специфічні проліки, такі як гепсера. Крім структурних змін, що вводяться в нуклеозидні аналоги для сприяння підвищеної клінічної застосовності, для підвищення їх активності були здійснені додаткові модифікації. Є декілька прикладів модифікованих нуклеозидних аналогів, одержаних внаслідок додавання ліпідних фрагментів (Патенти США №№ 6153594, 6548486, 6316425 і 6384019; Європейські Заявки на Патент №№ EP-A-56265 і EP-A-393920; WO 99/26958). Зазначений синтез може здійснюватися зв’язуванням жирних кислот, наприклад, через складний ефір, амід, карбонат або карбаматний зв’язок. Можуть бути одержані і більш складні продукти, такі як фосфоліпідпохідні нуклеозидних аналогів (див. Eur. J. Pharm. Sci. 11b Suppl 2: 15-27 (2000); EP No. 545966; Патент Канади № 2468099; Патенти США №№ 6372725 і 6670341). Такі аналоги, як описано в літературі, мають противірусну активність і особливо є придатними для терапевтичного лікування і профілактики інфекцій, викликаних ДНК-, РНК- і ретровірусами. Вони також є придатними для лікування злоякісних пухлин. Ліпід-похідні нуклеозидних аналогів можуть бути для декількох цілей. Вони можуть бути як проліки, які не є субстратом для дезаміназ, внаслідок чого нуклеозидні аналоги захищаються від дезактивації в процесі транспорту в тоці крові. Ліпідні похідні також можуть ефективніше транспортуватися через клітинну мембрану, що призводить до підвищеної внутрішньоклітинної концентрації нуклеозидного аналога. Ліпідні похідні можуть також більш підходити для застосування в дермальних препаратах, препаратах для перорального введення (див. Патент США № 6576636 і WO 01/18013) або в особливих препаратах, таких як ліпосоми (див. Патент США № 5223263), розроблених для цілеспрямованого ураження пухлин. Було показано, що противірусна або протиракова активність нуклеозидних аналогів зі збереженою -D-конфігурацією моносахаридного циклу або нуклеозидних аналогів із нециклічним бічним ланцюгом може найефективніше поліпшуватися за допомогою утворення ліпідних похідних моно-заміщених -9 С18 і С20 жирних кислот (див. Antimicrobial Agents and Chemotherapy, Vol., 53-61 (1999); Cancer Research 59: 2944-2949 (1999); Gene Therapy, 5: 419426 (1998); Antiviral Research, 45: 157-167 (2000); Biochemical Pharmacology, 67: 503-511 (2004). Переважні моно-ненасичені похідні є не тільки активнішими, ніж поліненасичені аналоги, але і більш кристалічними і хімічно стабільними стосовно окислення ліпідного ланцюга. Отже, вони є більш сприятливими сполуками з хімічної точки зору і з точки зору виробництва лікарського засобу. Було також показано, що моно-ненасичені -9 С18 і С20 жирні кислоти є придатними для підвищення терапевтичної активності у великій кількості ненуклеозидних біологічних активних сполук (див. Європейський Патент № 0977725). Відносно нову підгрупу нуклеозидних аналогів складають, так звані, аза-С-похідні. У даному класі сполук СН група в 5 положенні піримідинової основи замінена на атом азоту, як показано у формулі В нижче. 45 50 Формула В Гени, що пригнічують пухлину, які були заглушені аберрантним ДНК метилуванням, є потенційними мішенями для реактивації за допомогою цих нових хіміотерапевтичних засобів. Сильні інгібітори ДНК метилування і протилейкемічні засоби, аза-цитидин і 5-аза-2дезоксицитидинові похідні (5-аза-C, 5-аза-CdR, децитабін), можуть повторно активувати заглушені гени, що пригнічують пухлину. При високих концентраціях дані сполуки є 3 UA 99308 C2 5 цитотоксичними, але при нижчих концентраціях гіпометилування призводить до диференціації клітинних ліній. Сполукам необхідна метаболічна активація дезоксицитидинкіназою, і вони забезпечують інгібування ДНК метилтрансферази. Перешкодою застосування цих похідних як лікарських засобів є їхня швидка in vivo інактивація цитидиндеаміназою (CD). Нестабільність у водних розчинах, а також профілі побічних ефектів також обмежили їхню клінічну активність. Даний винахід спрямований на подолання цих та інших проблем попереднього рівня. Суть винаходу Один аспект даного винаходу стосується сполуки формули (I) 10 де R являє собою Н, R5C(О), R5CH2OC(О) або R5CH2NHC(О), R1 являє собою 15 20 25 30 35 40 де пунктирна лінія, що перехрещується, показує зв’язок, утворений приєднанням R 1 до молекули формули (I), R2 і R3 незалежно являють собою OH або Н за умови, що R2 і R3 одночасно не є OH; R4 являє собою Н, R5C(О), R5CH2OC(О) або R5CH2NHC(О) за умови, що R і R4 одночасно не є Н; R5 має загальну формулу: CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-; k являє собою ціле число від 0 до 7; m являє собою ціле число від 0 до 2; і n являє собою ціле число від 0 до 10, або її фармацевтично прийнятної солі. Інший аспект даного винаходу стосується фармацевтичної композиції, що включає сполуку формули (I) і фармацевтичний наповнювач, розріджувач і/або носій. Ще один аспект даного винаходу стосується способу лікування новоутворення у суб’єкта. Спосіб включає вибір суб’єкта з новоутворенням і введення зазначеному суб’єкту сполуки формули (I), яка описана вище, або її фармацевтичної солі в умовах, ефективних для лікування новоутворення у суб’єкта. Ще один аспект даного винаходу стосується способу лікування у суб’єкта запального процесу. Спосіб включає вибір суб’єкта із запальним процесом і введення суб’єкту сполуки формули (I), яка описана вище, або її фармацевтично прийнятної солі в умовах, ефективних для лікування запального стану у суб’єкта. Нестабільність аза-С у буфері і плазмі добре відома (див. публікації Israili et al., Cancer Research 36, 1453-1461 (1976); Rudek et al., J. Clin. Oncol., 23:17, 3906-3911(2005); Rustum et al., J. Chromat, 421:12, 387-91 (1987); Zhao et al., J. Chromat В, 813, 81-88 (2004), які у всій повноті введені в даний опис у вигляді посилання). Середній період напіврозпаду для аза-С в клінічних зразках плазми, як повідомлялося, становить 1,50±2,30 години (див. публікацію Rudek et al., J. Clin. Oncol., 23:17, 3906-3911(2005), яка таким чином у всій повноті введена в даний опис у вигляді посилання). В умовах in vitro при зберіганні аза-С протягом 4,5 днів навіть при -60С 4 UA 99308 C2 5 10 15 20 25 30 35 40 45 50 спостерігається втрата 20% сполуки, а при зберіганні протягом 0,5 години при кімнатній температурі втрата 10% (див. публікацію Zhao et al., J. Chromat В, 813, 81-88 (2004), яка у всій повноті введена в даний опис у вигляді посилання). Вважалося, що первинна нестабільність аза-С зумовлена швидким (перша стадія є оборотною) розкриттям 5-азапіримідинового циклу з подальшим відщепленням мурашиної кислоти (див. публікацію Chan et al., J. Pharma Sci., 68;7, 807-12 (1979), яка таким чином у всій повноті введена в даний опис у вигляді посилання). Вважається також, що іншими шляхами розкладання є дезамінування аміногрупи в положенні 4 і гідроліз глікозидного зв’язку з одержанням D-рибози і 5-азацитозину. Несподівано було виявлено, що переважні ліпідні похідні аза-С мають значною мірою кращий профіль стабільності в плазмі, ніж сам аза-С. Сполуки є стабільними (відсоток сполуки 94% від початкового вмісту) у контрольній матриці плазми людини при кімнатній температурі протягом, принаймні, 4 годин в умовах експерименту, і не спостерігається значного розкладання продуктів в постекстракційному супернатанті після осадження білків плазми. Стабільність в плазмі переважних ліпідних-похідних додатково досліджувалася при зберіганні при 37С. Показано, що розкриття циклу аза-фрагмента або інше розкладання сполуки значно знижується, коли ліпідний бічний ланцюг приєднаний до аза-С. Швидке розкладання аза-С є недоліком для його клінічного застосування. Підвищена стабільність в плазмі ліпід-похідних у порівнянні з аза-С може забезпечувати високий і тривалий рівень вмісту в плазмі пацієнта ліпід-похідного. Це може призводити до кращого розподілу в тканині/органі/пухлині, кращої клітинної експозиції і поглинання лікарського засобу в порівнянні з аза-С, з одного боку, і потім кращої експозиції пухлинної клітинної ДНК дії аза-С після внутрішньоклітинного гідролізу аза-С-5-складноефірного зв’язку. Варіантами здійснення даного винаходу є нові сполуки, молекули яких одержані через модифікацію азацитидину і деоксицитидину (наприклад, 5-аза-2-деоксицитидину) і властивості яких несподівано відрізняються від властивостей азацитидину і дезоксицитидину (наприклад, 5аза-2-дезоксицитидину). У результаті одержаний ряд сполук з активністю, яка розширює вже досягнуту протиракову активність азатидину і деоксицитидину (наприклад, 5-аза-2діоксицитидину), обмежену гематобластозами. Ці нові сполуки мають протиракову ефективність стосовно широкого спектра солідних пухлин, включаючи рак молочної залози і рак шийки матки. Сполуки також несподівано активні стосовно новоутворень, резистентних до лікування і, таким чином, можуть надати корисну терапевтичну дію в лікуванні солідних пухлин, для яких вибір терапевтичних засобів обмежений. Варіанти здійснення даного винаходу можуть застосовуватися для лікування ракових захворювань, для яких вибір і ефективність терапевтичних засобів залишаються обмеженими, і задовольняють існуючим потребам. Дані сполуки показують швидший прояв активності після обмеженої експозиції і, отже, можуть бути ефективними протягом короткого періоду в патологічному процесі. Це повинно призвести до менш тривалого лікування з введенням лікарського засобу рідше і зниження токсичності, пов’язаної із застосуванням лікарських засобів, у порівнянні з початковими лікарськими засобами. Це повинно забезпечити вищий терапевтичний індекс. Зміна структури з додаванням ліпідного (включає складний ефір, аміди, карбамати і карбонати) компонента зберігає азолцитидиновий цикл і, таким чином, забезпечує вплив молекули на епігенетичні механізми. Епігенетична модуляція надає важливий механізм для зміни експресії гена при ракові і запаленні. Ці нові сполуки мають активність при нижчих концентраціях, ніж азацитидин, і, отже, є ефективнішими. Зазначені сполуки зі зміненим спектром активності можуть модулювати епігенетичні мішені солідних пухлин і запальних процесів. Епігенетичні механізми мають велике значення для зміни протікання запальних процесів, які включають, але не виключно, запалення легень, запалення сполучних тканин, запалення шлунково-кишкового тракту і запалення судинної сітки. Дані сполуки за допомогою цільових епігенетичних механізмів можуть знижувати або сприяти зворотному розвитку запальні процеси, що відповідають за ці захворювання. Стислий опис малюнків На фіг.1 наведений графік, що показує часовий профіль цитотоксичної активності аза-С і 5аза-С-5-петроселінової кислоти. 55 Детальний опис винаходу Один аспект даного винаходу стосується сполуки формули (I) 5 UA 99308 C2 де R являє собою Н, R5C(О), R5CH2OC(О) або R5CH2NHC(О), R1 являє собою 5 10 15 20 де пунктирна лінія, що перехрещується, показує зв’язок, утворений приєднанням R 1 до молекули формули (I); R2 і R3 незалежно являють собою OH або Н за умови, що R2 і R3 одночасно не є OH; R4 являє собою Н, R5C(О), R5CH2OC(О) або R5CH2NHC(О) за умови, що R і R4 одночасно не є Н, R5 відповідає загальній формулі: CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-; k являє ціле число від 0 до 7; m являє собою ціле число від 0 до 2; і n являє собою ціле число від 0 до 10, або її фармацевтично прийнятної солі. У переважних варіантах здійснення винаходу k дорівнює 4, n дорівнює 10. У деяких варіантах здійснення винаходу R1 являє собою де пунктирна лінія, що перехрещується, показує зв’язок, утворений приєднанням R 1 до молекули формули (I). У деяких варіантах здійснення винаходу R 4 може являти собою Н. У деяких варіантах здійснення винаходу R являє собою R 5C(О), k дорівнює 4, m дорівнює 0, R2 являє собою Н, R3 являє собою ОН, R4 являє собою Н. У більш широкому аспекті даний винахід стосується сполуки формули (I) 25 6 UA 99308 C2 де R являє собою Н, R5C(О), R5CH2OC(О) або R5CH2NHC(О), R1 являє собою 5 10 де пунктирна лінія, що перехрещується, показує зв’язок, утворений приєднанням R1 до молекули формули (I); R2 і R3 незалежно являють собою OH або Н за умови, що R2 і R3 одночасно не є OH; R4 являє собою Н, R5C(О), R5CH2OC(О) або R5CH2NHC(О) за умови, що R і R4 одночасно не є Н; R5 являє собою С3-С26 алкеніл, або її фармацевтично прийнятної солі. У переважному варіанті здійснення сполуки формули (I) k дорівнює 4 і n дорівнює 10. У деяких варіантах здійснення винаходу R1 являє собою 15 20 25 30 де пунктирна лінія, що перехрещується, показує зв’язок, утворений приєднанням R1 до молекули формули (I). У деяких варіантах здійснення винаходу R4 може являти собою Н. В інших варіантах здійснення R являє собою R5C(О); k дорівнює 4; m дорівнює 0; n дорівнює 10; R2 являє собою Н; R3 являє собою ОН. У деяких варіантах здійснення R5 являє собою С9-С26 алкеніл. Інший аспект даного винаходу стосується фармацевтичної композиції, що включає сполуки формули (I) і фармацевтичний наповнювач, розріджувач і/або носій. Лікарські засоби відповідно до даного винаходу можуть вводитися перорально, парентерально, наприклад, підшкірно, внутрішньовенно, внутрішньом’язово, інтраперитонеально, інтраназальним вливанням або за допомогою нанесення на слизові оболонки, такі як слизова оболонка носа, горла або бронхіол. Вони можуть вводитися самі по собі або з придатними фармацевтичними носіями, і можуть бути в твердій або рідкій формі, такій як таблетки, капсули, порошки, розчини, суспензії або емульсії. Активні інгредієнти відповідно до даного винаходу можуть вводитися перорально, наприклад, з інертним розріджувачем або з асимільованим їстівним носієм, вони можуть поміщатися в капсули з твердими або м’якими оболонками, вони можуть пресуватися в 7 UA 99308 C2 5 10 15 20 25 30 35 40 45 50 55 таблетки або вони можуть вводитися безпосередньо з їжею раціону харчування. Для перорального введення при терапевтичному лікуванні зазначені активні інгредієнти можуть вводитися з наповнювачами і застосовуватися у формі таблеток, капсул, еліксирів, суспензій, сиропів і т.ін. Такі композиції і препарати повинні містити, принаймні, 0,1% активного інгредієнта. Відсотковий вміст активного інгредієнта у зазначених композиціях, зрозуміло, може змінюватися і традиційно може знаходитися в інтервалі від приблизно 2% до приблизно 60% маси стандартної лікарської форми. Кількість активного інгредієнта в таких терапевтично застосовних композиціях є такою, щоб була одержана придатна доза. Переважні композиції відповідно до даного винаходу одержують таким чином, що лікарська форма стандартної дози містить від приблизно 1 до 250 мг активного інгредієнта. Таблетки, капсули і т.ін. також можуть містити зв’язувальну речовину, таку як трагакантова камедь, гуміарабік, кукурудзяний крохмаль або желатин; наповнювачі, такі як гідрофосфат кальцію; дезінтегрувальний засіб, такий як кукурудзяний крохмаль, картопляний крохмаль, альгінова кислота; добавку, що підвищує ковзання, таку як стеарат магнію; і підсолоджувальну добавку, таку як сахароза, лактоза або сахарин. Коли лікарська форма стандартної дози являє собою капсулу, вона може містити крім добавок, зазначених вище, рідкий носій, такий як жирне масло. Різні інші матеріали можуть бути присутніми у вигляді покриттів або служити для модифікації фізичного стану лікарської форми стандартної дози. Наприклад, таблетки можуть бути покриті шелаком, цукром або обома цими речовинами. Сироп може крім активного інгредієнта містити сахарозу як підсолоджувальний агент, метил і пропілпарабени як консерванти, барвник і смакову добавку, таку як вишнева або апельсинова смакова добавка. Зазначені активні інгредієнти також можуть вводитися парентерально. Розчини або суспензії зазначених активних інгредієнтів можуть бути одержані у воді, придатним чином змішаною з поверхово-активною речовиною, такою як гідроксипропілцелюлоза. Дисперсії також можуть бути одержані в гліцерині, рідких поліетиленгліколях та їхніх сумішах у маслах/оліях. Типовими прикладами масел/олій є масла нафтового, тваринного, олії рослинного походження або синтетичні масла, наприклад, арахісова олія, соєва олія або мінеральне масло. Переважними рідкими носіями, зокрема в розчинах для ін’єкцій, звичайно є вода, розчин солі, водна декстроза і розчин споріднених цукрів, а також гліколі, такі як пропіленгліколь або поліетиленгліколь. У стандартних умовах зберігання і застосування зазначені препарати містять консервант для запобігання зростанню мікроорганізмів. Фармацевтичні форми, придатні для застосування у вигляді ін’єкцій, включають стерильні водні розчини або дисперсії і стерильні порошки для швидкого приготування стерильних розчинів або дисперсій для ін’єкцій. У всіх випадках форма повинна бути стерильною і повинна бути рідкою в такій мірі, щоб можна було їх застосовувати за допомогою шприца. Вона повинна бути стабільною в умовах виробництва і зберігання і повинна бути захищена від забруднюючої дії мікроорганізмів, таких як бактерії і гриби. Носій може являти собою розчинник або дисперсійне середовище, що містить, наприклад, воду, етанол, багатоатомний спирт (наприклад, гліцерин, пропіленгліколь і рідкий поліетиленгліколь), їхні придатні суміші і рослинні олії. Активні інгредієнти відповідно до даного винаходу також можуть вводитися безпосередньо в дихальні шляхи у формі аерозолю. Для застосування у вигляді аерозолів активні інгредієнти відповідно до даного винаходу в розчині або суспензії можуть розфасовуватися в аерозольний балон під тиском разом із придатними пропелентами, наприклад вуглеводневими пропелентами, такими як пропан, бутан або ізобутан, із придатними ад’ювантами. Сполуки відповідно до даного винаходу також можуть вводитися у формі без тиску, такій як аерозольний інгалятор або пульверизатор. Додатковий аспект даного винаходу стосується способу лікування у суб’єкта новоутворення. Спосіб включає вибір суб’єкта з новоутворенням і введення суб’єкту сполуки формули (I), яка описана вище, або її фармацевтично прийнятної солі в умовах, ефективних для лікування новоутворення у суб’єкта. У деяких варіантах здійснення винаходу новоутворення являє собою ракове захворювання. Ракове захворювання може являти собою солідну пухлину, гемобластоз або злоякісну пухлину кровотворної тканини. Ракове захворювання може являти собою лейкемію, лімфому, множинну мієлому або мієлодиспластичний синдром. У деяких варіантах здійснення винаходу солідна пухлина може являти собою рак тканини, такої як тканина молочної залози, тканина яєчника, тканина передміхурової залози, тканина мозку, тканина сечового міхура і тканина легень. 8 UA 99308 C2 5 10 Додатковий аспект даного винаходу стосується способу лікування у суб’єкта запального процесу. Спосіб включає вибір суб’єкта із запальним процесом і введення суб’єкту сполуки формули (I), яка описана вище, або її фармацевтично прийнятної солі в умовах, ефективних для лікування запального процесу у суб’єкта. У деяких варіантах здійснення винаходу запальний стан являє собою запалення легень, запалення сполучної тканини, запалення шлунково-кишкового тракту або запалення судинної сітки. За винятком особливо оговорених випадків, наукові і технічні терміни, що використовуються в зв’язку з даною заявкою, будуть мати значення, які звичайно вкладає в них фахівець даної галузі техніки. Крім того, за винятком випадків, коли того вимагає контекст, терміни в однині повинні включати і термін у множині, а терміни в множині повинні включати терміни в однині. Приклади Приклад 1 Реагенти, клітинні лінії і клітинна культура 15 20 25 Реагент клітинної проліферації WST-1 одержують із Roche Applied Science (Manheim, Germany), PI і Annexin V – FITC (набір для визначення відсоткової частини клітин, що зазнали апоптозу) одержують від BD Biosciences, Palo Alto, CA, 5-азатидин (5-аза-C), бромід етидію (EB), акридиновий оранжевий (АО), нітросиній тетразолій (NBT), форбол-12-міристат-13-ацетат (ТРА) одержують від Sigma Chemical Co (St. Lous, MO). Клітинні лінії промієлоцитарної лейкемії людини HL60, гістіоцитної лімфоми людини U937, хронічної мієлогенної лейкемії людини К562, Т-клітини людини лінії Jurkat, аденокарциноми молочної залози MCF-7, карциноми сечового міхура 5637, карциноми простати DU-145 одержують з Американської колекції типів культур (American Type Culture Collection). Всі клітинні лінії, за винятком клітин лінії Jurkat, витримують у RPMI 1640 середовищі (Gibco, Glasgow, UK), забезпечениму 10% термоінактивованою фетальною телячою сироваткою (FCS), 100 U/мл пеніциліну і 100 мг/мл стрептоміцину в атмосфері 5% СО2 при 37С. Клітини лінії Jurkat вирощують у RPMI 1640 середовищі, забезпеченому 1,5 г/л гідрокарбонату натрію, 4,5 г/л глюкози, 10 мМ пірувату натрію, 10% FCS, 100 U/мл пеніциліну і 100 мг/мл стрептоміцину. Приклад 2 30 35 40 45 Визначення цитотоксичності Цитотоксичність 5-азацитидинового ліпіду визначають колориметричним аналізом, який заснований на розщепленні солі тетразолію WST-1 (дисульфонат 4-[3-(4-лодофеніл)-2-(4нітрофеніл)-2Н-5-тетразоліо]-1,3-бензолу) за допомогою мітохондріальних дегідрогеназ у життєздатних клітинах. Клітини висівають у 96-ямкові плоскодонні мікропланшети з початковою 6 5 концентрацією 110 /мл (HL60 клітини) або 1,2510 /мл (U937, K562 і Jurkat) у середовищі з додаванням або без додавання 5-азацитидинового ліпіду різних концентрацій і вирощують 4 протягом від 24 до 72 годин. Клітини MCF-7, DU-145 і 5637 (110 /мл) висівають, дають їм можливість адгезувати і розростатися протягом 24 годин. 5-азацитидиновий ліпід додають у різних концентраціях і культури витримують протягом 24 або 72 годин. Культури інкубують із WST-1 реагентом протягом 1 години. Продукування формазану кількісно визначають за допомогою мікропланшет-рідера (Bio-Tek Instruments, Elx 800) при 450 нм з еталонною довжиною хвилі 650 нм. Інгібування росту визначають при порівнянні з необробленими клітинами (%). Значення IC50 обчислюють із використанням програмного забезпечення CalcuSyn (Biosoft). Приклад 3 Підрахунок апоптотичних клітин 50 55 Апоптотичні клітини ідентифікують із використанням морфологічних критеріїв і флуоресцентного сортера клітин (fluorescence-activated cell sorting – FACS) після підфарбовування Annexin V FITC. Для морфологічного аналізу 1 мкл вихідного розчину, що містить 100 мкг/мл АО і 100 мкг/мл ЕВ, додають до 25 мкг клітинної суспензії. Апоптотичні клітини і апоптичну масу аналізують за допомогою флуоресцентного мікроскопа. Відсоток 5 апоптотичних клітин розраховують після підрахунку всіх 300 клітин. Для FACS аналізують 210 6 до 510 клітин промивають PBS і потім мітять за допомогою Annexin V-FICS і йодидом пропідіуму (PI) в реагенті, що зв’язує середовище, відповідно до інструкції набору Annexin VFITC для визначення відсоткової частини клітин, що зазнали апоптозу, наданої виробником. 9 UA 99308 C2 5 Флуоресцентні сигнали FITC і PI виявляють, відповідно, при 518 нм і при 620 нм на FACSCAN (Becton Dickinson, San Jose, CA). Значення log Annexin V-FITC флуоресценції наносять на осі Х, значення log PI флуоресценції наносять на осі Y. Дані аналізують за допомогою програмного забезпечення CellQuest (Becton Dickinson). Для кожного аналізу зчитують результати для 10000 клітин. Приклад 4 Клітинний цикл 10 15 Клітини пелетують центрифугуванням і двічі промивають PBS, фіксують за допомогою охолодженого 70% (об./об.) етанолу (-20С) і зберігають при 4С протягом, принаймні, 24 годин. Клітини промивають у PBS. Клітинні пелети підфарбовують розчином барвника PI/RNase. Клітинну суспензію інкубують у темряві при кімнатній температурі протягом 30 хвилин. Вміст ДНК визначають із використанням апарату проточного цитометричного аналізу FACSCalibur (Becton Dickinson, Mount View, CA). Відсотки клітин Sub-G1, G1, S і G2/M стадій клітинного циклу визначають із використанням програми, що надає дані ДНК у вигляді гістограм (Becton Dickinson). Для кожного зразка зчитують мінімум 10000 результатів. Приклад 5 Синтез складного ефіру аза-С-5-петроселінової кислоти 20 25 30 35 40 45 50 55 Петроселінову кислоту (1,75 ммоль, 494 мг) розчиняють у толуолі (3 мл). Додають спочатку ДМФА (10 мкл), потім протягом 10 хвилин при кімнатній температурі додають оксалілхлорид (3,6 ммоль, 457 мг). Через 3 години толуол видаляють у вакуумі. Аза-С (1,57 ммоль, 427 мг) суспендують у ДМА (6 мл), додають HCl (1М в Et2O, 2,0 ммоль, 2,0 мл), витримують протягом 5 хвилин при кімнатній температурі, після чого Et2O видаляють у вакуумі. Одержаний каламутний розчин охолоджують на водно-льодяній бані і протягом 40 хвилин додають хлорангідрид, розчинений у ДМА (2 мл). Реакційну суміш перемішують протягом ночі, у процесі чого температура суміші повільно досягає кімнатної температури. Через 24 години розчинники видаляють при тиску 0,1 мбар. Залишок розподіляють між насиченим водним розчином NaHCO3 і EtOAc (25 мл кожного). Водну фазу екстрагують EtOAc (325 мл). Органічні фази об’єднують, промивають розчином солі і сушать (MgSO 4). Після видалення розчинників у вакуумі сирий продукт (600 мг) очищають флеш-хроматографією (SiO2, CH2Cl2 із 2,5, 5 і 10% MeOH). І нарешті, продукт сушать при (0,25 мбар протягом ночі. Вихід: 210 мг (24%). Приклад 6 Синтез складного ефіру аза-С-5-петроселаїдинової кислоти Петроселаїдинову кислоту (1,77 ммоль, 500 мг) розчиняють у толуолі (3 мл), додають спочатку ДМФА (10 мкл), потім протягом 10 хвилин при кімнатній температурі оксалілхлорид (3,6 ммоль, 457 мг). Через 3 години толуол видаляють у вакуумі. Аза-С (1,75 ммоль, 427 мг) суспендують у ДМА (6 мл), додають HCl (1М в Et2O, 2,0 ммоль, 2,0 мл), суміш витримують протягом 5 хвилин при кімнатній температурі, після чого Et2O видаляють у вакуумі. Одержаний каламутний розчин охолоджують на водно-льодяній бані і протягом 2 годин додають хлорангідрид, розчинений у ДМА (2 мл). Реакційну суміш перемішують протягом ночі, у процесі чого температура суміші повільно досягає кімнатної температури, після цього суміш протягом 2 годин витримують при 30С. Після охолодження до кімнатної температури залишок розподіляють між насиченим водним розчином NaHCO 3 і EtOAc (по 25 мл кожного). Водну фазу екстрагують EtOAc (325 мл). Органічні фази об’єднують, промивають розчином солі і сушать (MgSO4). Після видалення розчинників у вакуумі одержують складний ефір у вигляді білого порошку. Вихід: 500 мг. Приклад 7 Метаболічна стабільність складного ефіру 5-аза-5-петроселінової кислоти в пулі плазми людини Складний ефір 5-аза-С-5-петроселінової кислоти обережно додають у пул плазми людини в п’яти концентраціях (0,1, 1, 3, 10 і 30 мкМ, відповідно). Суміш інкубують при 37С на водяній бані, що гойдається. Відбирають по три (n=3) аліквоти інкубованих розчинів після закінчення встановленого періоду інкубації (0, 15, 30, 60 і 120 хвилин) і відразу осаджують білок плазми з використанням ацетонітрилу, що містить 0,1% мурашиної кислоти (300 мкл). Негативний контроль готують із досліджуваною сполукою і аза-С в експериментальному буфері (PBS, рН 7,4) при одній концентрації інкубації (1 мкМ). Після центрифугування супернатант безпосередньо використовують у РХ-МС-МС аналізі. Див. таблицю 1. 10 UA 99308 C2 Таблиця 1 Концентрація (мкМ) 0,1 1 3 10 30 0 хв 100 100 100 100 100 Залишкова кількість: % від вихідної (середнє SD, n=3) 15 хв 30 хв 60 хв 120 хв 95,02,5 92,92,0 83,72,5 51,31,2 96,35,3 90,92,5 79,93,2 46,32,2 97,53,3 91,53,6 83,61,3 50,90,7 97,31,1 91,40,6 78,61,8 45,70,6 93,61,3 85,92,9 71,7±2,3 40,60,5 Період напіврозпаду (хв.) 125 107 122 104 91 Приклад 8 Цитотоксичність аза-С і 5-аза-С-5-петроселінової кислоти 5 10 15 Цитотоксичність аза-С і 5-аза-С-5-петроселінової кислоти визначають у клітинній лінії МТ-3 раку молочної залози та адріабластин-резистентної клітинної лінії МТ-3/ADR. MT-3/ADR надекспресують МDR-1/p-глікопротеїн. Клітини висівають у 96-ямкові планшети з щільністю 3 510 клітин на ямку в середовище RPMI 1640 з 2 мМ глютаміну і 10% FBS. Клітини інкубують протягом 24 годин. Досліджувані сполуки розчиняють у ДМСО і додатково розводять середовищем безпосередньо перед застосуванням. Для кожної дослідної концентрації використовують 6 ямок. Клітини інкубують з досліджуваною сполукою протягом 24 годин. У кожну ямку додають 20 мкл свіжоприготованого МТТ розчину та інкубують протягом 4 годин. Значення IC50 визначають із кривих росту, наведених графічно для 8 різних концентрацій в інтервалі від 0,01 мкМ до 100 мкМ. Результати наведені в таблиці 1. Аналогічну активність одержують для аза-5 і 5-аза-С-5-петролелінової кислоти в клітинній лінії карциноми молочної залози МТ-3, але в МТ-3/ADR резистентній клітинній лінії активність аза-С губиться. Активність не спостерігається в ділянці досліджених концентрацій до 100 мкМ, у той час, як 5-аза-С-5петроселінова кислота залишається активною зі значенням IC 50 у резистентній клітинній лінії, аналогічним до значення з нерезистентної МТ-3 лінії. Див. таблицю 2. Таблиця 2 Цитотоксична активність аза-С і 5-аза-С-5-петроселінової кислоти в карциномі мозку з експресією або без експресії полірезистентності до лікарських засобів МТ-3 карцинома молочної залози МТ-3/ADR резистентна карцинома молочної залози 20 Аза-С IC50 (мкМ) 12,622,35 5-аза-С-5-петроселінова кислота IC50 (мкМ) 12,326,37 100 12,028,30 Приклад 9 Антипроліферативна активність аза-С і 5-аза-С-5-петроселінової кислоти 25 30 35 Антипроліферативну активність аза-С і 5-аза-С-5-петроселінової кислоти визначають у клітинній лінії Hela раку шийки матки при 24- і 72-часовій експозиції. Клітини висівають у 963 ямкові мікропланшети при щільності 510 клітин на ямку в RPMI 1640 середовищі з 2 мМ глютаміну і 10% FBS. Клітини інкубують протягом 24 і 72 годин. Досліджувані сполуки розчиняють у ДМСО і додатково розводять середовищем безпосередньо перед застосуванням. Для кожної експериментальної концентрації використовують 6 ямок. Цитотоксичність визначають із використанням МТТ аналізу, у кожну ямку додають 20 мкл свіжоприготованого МТТ розчину та інкубують протягом 4 годин. Значення IC50 визначають із кривих росту, наведених графічно, для 8 концентрацій в інтервалі від 0,01 мкМ до 100 мкМ. Аналогічну цитотоксичну активність одержують при пролонгованій експозиції протягом 72 годин для двох зазначених сполук, але несподівано цитотоксичний ефект 5-аза-С-5-петроселінової кислоти мав місце вже після 24 часової експозиції. Різний часовий профіль спостерігається для аза-С- і 5-аза-С-5-петроселінової кислоти із швидшим початком фітотоксичної дії 5-аза-С-5петроселінової кислоти. Див. фіг.1. 11 UA 99308 C2 Пример 10 Вплив інгібування нуклеозидного транспортера на цитотоксичну активність у ракових клітинах для аза-С і 5-аза-С-5-петроселінової кислоти 5 10 15 20 Вплив інгібування нуклеозидного транспортера на цитотоксичну активність оцінюють у мутантних клітинах Hela раку шийки матки для аза-С і 5-аза-С-5-петроселінової кислоти. У ролі інгібітора рівноважних нуклеозидних транспортерів використовують hENT1 і hENT2. Клітини 3 висівають у 96-ямкові планшети при щільності 510 клітин на ямку в RPMI 1640 середовище з 2 мМ глютаміну і 10% FBS. Клітини попередньо інкубують протягом 24 годин. У клітини за 30 хвилин до додавання досліджуваних сполук додають дипіридамол (10 мкМ). Досліджувані сполуки розчиняють у ДМСО і додатково розводять середовищем безпосередньо перед застосуванням. Для кожної експериментальної концентрації використовують 6 ямок. Клітини інкубують із досліджуваною сполукою протягом 72 годин. У кожну ямку додають 20 мкл свіжоприготованого МТТ розчину та інкубують протягом 4 годин. Значення IC50 визначають із кривих росту, наведених графічно, для 8 концентрацій в інтервалі від 0,01 мкМ до 100 мкМ. Результати наведені в таблиці 3. Активність аза-С знижується в 3 рази внаслідок додавання інгібітора нуклеозидного транспорту дипіридамолу, показуючи, що приплив і відтік аза-С у Hela клітинах у деякій мірі залежить від нуклеозидних транспортерів hENT1 і hENT2. Цитотоксична активність 5-аза-С-5-петроселінової кислоти не тільки зберігається, але і підвищується в 10 разів при блокуванні hENT1 і hENT2 нуклеозидних транспортерів за допомогою застосування дипіридамолу. Це може бути особливо важливо для пацієнтів, в яких активність аза-С відсутня внаслідок втрати експресії нуклеозидних транспортерів. Див. таблицю 3. Таблиця 3 Цитотоксична активність аза-С і 5-аза-С-5-петроселінової кислоти в клітинах Hela раку шийки матки з інгібітором нуклеозидного транспорту дипіридамолом і без нього Азацитидин 5-Аза-С-5-петроселінова кислота IC50 (мкМ) Hela 4,32 4,74 Hela з дипіридамолом 12,77 0,42 Приклад 11 25 30 35 40 45 Експресія гена естрогенового рецептора (ER) у лінії клітин раку молочної залози після обробки азацитидином або 5-аза-С-5-петроселіновою кислотою Експресію гена (визначену на рівні РНК) естрогенового рецептора бета визначають методом ПЛР у реальному часі (TagMan). Клітини раку молочної залози MCF-7 вирощують у середовищі з дефіцитом естрогену (Phenol-Red-free RPMI з 2% глютаміну і 10% фетальної телячої сироватки, обробленої сумішшю деревне вугілля-декстран). Клітини висівають у чашки площею 2 25 см та інкубують їх протягом 24 годин перед обробкою 1 мкМ азацитидину або 5-аза-С-5петроселіновою кислотою. У ролі контролю використовують один необроблений контроль. Клітини збирають після 5 денної експозиції сполуками, їх збирають трипсинізацією, промиванням і швидким заморожуванням у рідкому азоті. 6 Всю РНК екстрагують із приблизно 10 швидкозаморожених MCF-7 клітин, визначають концентрацію і чистоту РНК, РНК транскрибують у кДНК із використанням TagMan Reverse Transcription реагенту (N808-0234). Кількісне визначення в реальному часі здійснюють із використанням стандартних методик і попередньо змішаних ПЛР-реагентів. Вихідні пробні суміші одержують від Applied Biosystems, ER (ID Hs00230957_m1) і гідроцилметилбілансинтази гену “домашнього господарства” HMBS (ID Hs00609297_m1). Експресію гена вираховують із використанням порівняльного дельта-дельта Сt способу. Індукція експресії ER у 4,14 рази підвищується після експозиції 5-аза-С-5-петроселіновою кислотою, у той час як після експозиції в азацитидині вона збільшується тільки у 2,51 рази (див. таблицю 4). Це може мати велике значення для лікування гормонально активних пухлин, коли може бути відновлена гормональна чутливість (див. таблицю 4). 12 UA 99308 C2 Таблиця 4 Дельта дельта Х-кратна індукція ER гена 7,06 -2,40 5,26 8,13 -1,32 2,51 Ct ER 5 дельта 35,36 25,91 9,45 32,97 25,92 34,31 Контроль 1 мкМ 5-аза-С-5петроселінової кислоти 1 мкМ аза-С Сt HMBD 26,18 Хоча переважні варіанти здійснення були наведені та описані тут детально, фахівцю даної галузі техніки буде зрозумілим, що різні модифікації, доповнення, заміщення і т.ін. можуть бути зроблені без виділення їх із обсягу даного винаходу і, отже, повинні розглядатися як частина обсягу винаходу, яка визначена у формулі винаходу, наведеній далі. ФОРМУЛА ВИНАХОДУ 1. Сполука формули (І) O R1 RO R2 HO R3 10 , (I) де R являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O); R1 являє собою NHR4 NHR4 N 15 20 25 O N O NH N N N або , де пунктирна лінія, що перехрещується, показує зв'язок, утворений приєднанням R1 до молекули формули (І); R2 і R3 незалежно являють собою ОН або Η, за умови, що R2 і R3 одночасно не є ОН; R4 являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O), за умови, що R і R4 одночасно не є Н; R5 має загальну формулу: CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-; k являє собою ціле число від 0 до 7; m являє собою ціле число від 0 до 2; і n являє собою ціле число від 0 до 10, або її фармацевтично прийнятна сіль. 2. Сполука за п. 1, де k дорівнює 4. 3. Сполука за п. 2, де n дорівнює 10. 4. Сполука за п. 1, де R1 являє собою NHR4 N O N N . 5. Сполука за п. 4, де R4 являє собою Н. 13 UA 99308 C2 6. Сполука за п. 1, де R являє собою R5C(O); R1 являє собою NHR4 N N O 5 N , R2 являє собою Н; R3 являє собою ОН; R4 являє собою Н; k дорівнює 4; m дорівнює 0; і n дорівнює 10. 7. Фармацевтична композиція, що містить: сполуку за п. 1, фармацевтичний наповнювач, розріджувач і/або носій. 8. Спосіб лікування у суб'єкта новоутворення, що включає: вибір суб'єкта з новоутворенням і введення суб'єкту сполуки формули: O R1 RO R2 HO R3 10 , (I) де R являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O); R1 являє собою NHR4 NHR4 N O 15 20 25 30 NH N N O N N або , де пунктирна лінія, що перехрещується, показує зв'язок, утворений приєднанням R1 до молекули формули (І); R2 і R3 незалежно являють собою ОН або Н, за умови, що R2 і R3 одночасно не є ОН; R4 являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O), за умови, що R і R4 одночасно не є Н; R5 має загальну формулу: СН3-(СН2)n-(СН=СН-СН2)m-CH=СН-(СН2)k-; k являє собою ціле число від 0 до 7; m являє собою ціле число від 0 до 2; і n являє собою ціле число від 0 до 10; або її фармацевтично прийнятної солі в умовах, ефективних для лікування новоутворення у суб'єкта. 9. Спосіб за п. 8, де новоутворення являє собою ракове захворювання. 10. Спосіб за п. 9, де ракове захворювання являє собою солідну пухлину, гематологічний рак або злоякісну пухлину. 11. Спосіб за п. 9, де ракове захворювання являє собою лейкемію, лімфому, множинну мієлому або мієлодиспластичний синдром. 12. Спосіб за п. 10, де солідна пухлина є раком тканини, вибраної з групи, що складається з тканин молочної залози, яєчників, передміхурової залози, мозку, сечового міхура та легенів. 13. Спосіб за п. 8, де k дорівнює 4. 14. Спосіб за п. 13, де n дорівнює 10. 15. Спосіб за п. 8, де R1 являє собою 14 UA 99308 C2 NHR4 N N N O . 16. Спосіб за п. 15, де R4 являє собою Н. 17. Спосіб за п. 8, де R являє собою R5C(O); R1 являє собою NHR4 N N O 5 N , R2 являє собою Н; R3 являє собою ОН; R4 являє собою Н; k дорівнює 4; m дорівнює 0; і n дорівнює 10. 18. Спосіб лікування у суб'єкта запального процесу, що включає: вибір суб'єкта із запальним процесом і введення суб'єкту сполуки формули: O R1 RO R2 HO R3 10 , (I) де R являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O); R1 являє собою NHR4 NHR4 N O 15 20 25 NH N N O N N або , де пунктирна лінія, що перехрещується, показує зв'язок, утворений приєднанням R1 до молекули формули (І); R2 і R3 незалежно являють собою ОН або Η, за умови, що R2 і R3 одночасно не є ОН; R4 являє собою Н, R5C(O), R5CH2OC(O) або R5CH2NHC(O), за умови, що R і R4 одночасно не є Н, R5 являє собою С3-С26алкеніл, де R5 має загальну формулу CH3-(CH2)n-(CH=CH-CH2)m-CH=CH(CH2)k-; k являє собою ціле число від 0 до 7; m являє собою ціле число від 0 до 2 і n являє собою ціле число від 0 до 10; або її фармацевтично прийнятної солі в умовах, ефективних для лікування запального процесу у суб'єкта. 19. Спосіб за п. 18, де запальний процес являє собою запалення легені, запалення сполучної тканини, запалення шлунково-кишкового тракту або запалення судинної сітки. 20. Спосіб за п. 18, де k дорівнює 4. 21. Спосіб за п. 20, де n дорівнює 10. 22. Спосіб за п. 18, де R1 являє собою 15 UA 99308 C2 NHR4 N N N O . 23. Спосіб за п. 22, де R4 являє собою Н. 24. Спосіб за п. 18, де R являє собою R5C(O); R1 являє собою NHR4 N O 5 N N , R2 являє собою Η; R3 являє собою ОН; R4 являє собою Н; k дорівнює 4; m дорівнює 0; і n дорівнює 10. Комп’ютерна верстка M. Мацело Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 16
ДивитисяДодаткова інформація
Назва патенту англійськоюAzacytidine analogue and use thereof
Автори англійськоюSilverman, Lewis, Holland, James, Sandvold, Marit, Liland, Myhren, Finn, Eriksen, Ole, Henrik
Назва патенту російськоюАналог азацитидина и его применение
Автори російськоюСилверман Льюис, Холланд Джеймс, Сандвольд Марит Лиланд, Мюрен Финн, Эриксен Оле Хенрик
МПК / Мітки
МПК: A01N 43/04
Мітки: застосування, аналог, азацитидину
Код посилання
<a href="https://ua.patents.su/18-99308-analog-azacitidinu-ta-jjogo-zastosuvannya.html" target="_blank" rel="follow" title="База патентів України">Аналог азацитидину та його застосування</a>
Аналог локсапіну та його застосування для регулювання сну

Номер патенту: 86265
Опубліковано: 10.04.2009
Автори: Едгар Дейл М., Уайт Джеймз Ф., Сіосакі Кадзумі, Хангоер Дейвід Дж., Соломон Майкл
МПК: A61K 31/553, A61P 25/20
Мітки: локсапіну, аналог, сну, регулювання, застосування
Формула / Реферат:
1. Сполука, що має формулу:, (I)або сіль, сольват, гідрат або проліки такої сполуки.2. Сполука за п. 1, яка є фармацевтично прийнятною сіллю.3. Сполука за п. 2, причому згаданою сіллю є сіль із кислотою.4. Сполука за п. 3, причому згаданою сіллю є гідрохлорид.5. Сполука за п. 4, яка має формулу:
Аналог 17-десметилрапаміцину та спосіб його одержання (варіанти)
Номер патенту: 95895
Опубліковано: 26.09.2011
Автори: Мартін Крістін Джанет, Грегорі Метью Алан
МПК: A61P 37/06, C07D 498/18, A61P 35/00, A61K 31/436, A61P 31/10
Мітки: одержання, спосіб, варіанти, аналог, 17-десметилрапаміцину
Формула / Реферат:
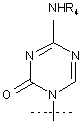
1. Аналог 17-десметилрапаміцину, що має наступну формулу:,де х є прямим зв'язком, -СН2-, -S-СН2-, -СН2-S- або -S(=O)-CH2-;або -CHR5-x-CHR6- є;R1 є =О або (Н,Н);R2 є ОН або ОМе;R3 є Н, ОН або ОМе;R4 є структурним фрагментом, вибраним із груп А, В, С,...
Аналог інсуліну, аналог інсуліну, що має здатність знижувати рівень цукру в крові

Номер патенту: 27299
Опубліковано: 15.09.2000
Автори: Чанс Рональд Юджин, Дімарчі Річард Денніс, Френк Брюс Хілл, Шілдз Джеймс Едвін
МПК: C07K 14/575, C07K 14/62, A61K 9/00, C12P 21/02, A61K 38/00, A61P 3/08, C12R 1/19, C12N 15/09, A61K 38/28, C07K 1/02
Мітки: крові, цукру, знижувати, має, здатність, рівень, аналог, інсуліну
Формула / Реферат:
(57) 1. Аналог инсулина формулы (І) или его фармацевтически приемлемая соль, отличающийся тем, что А21 является аспарагином, В1 является фенилаланином, В2 является валином, ВЗ является аспарагином, В9 является сери-ном, В10 является гистидином или асларагиновой кислотой, В27 является треонином, В28 является любой природной L-аминокислотой, В29 является L-пролином,...
Захищений від підробки документ та його застосування, захисний елемент, перебивний матеріал, його застосування та спосіб його виготовлення, спосіб виготовлення цінного документа

Номер патенту: 76591
Опубліковано: 15.08.2006
Автор: Хайм Манфред
МПК: B42D 15/00, G07D 13/00
Мітки: захисний, документа, захищений, спосіб, застосування, елемент, матеріал, цінного, документ, підробки, перебивний, виготовлення
Формула / Реферат:
1. Захищений від підробки документ, насамперед цінний папір, такий як банкнота, або напівфабрикат для виготовлення захищеного від підробки документа, що має першу і другу розташовані одна напроти іншої по різні сторони документа поверхні і оснащений захисним елементом (2, 4), який відрізняється тим, що захисний елемент з'єднаний з документом, відповідно з напівфабрикатом, таким чином, що він візуально помітний зі сторони обох його поверхонь і...
Фармацевтична композиція, що містить нуклеотидний аналог і спосіб її приготування

Номер патенту: 65576
Опубліковано: 15.04.2004
Автор: Бродхед Джоанн
МПК: A61K 31/7064, A61K 47/36, A61K 31/70, A61K 9/19
Мітки: композиція, спосіб, нуклеотидний, фармацевтична, містить, приготування, аналог
Формула / Реферат:
1. Фармацевтична композиція в ліофілізованій, висушеній розпилювальним сушінням або висушеній в вакуумі формі, що містить нуклеотидний аналог і одну або кілька склотвірних домішок, де нуклеотид є сполукою формули (І):, (І)де R1 і R2 незалежно означають водень або галоген,R3 і R4 незалежно означають феніл або С1-6-алкіл, необов'язково заміщений одним...
Попередній патент: Установка для приготування та електромагнітного розливання алюмінієвих сплавів у ливарні форми
Наступний патент: Заміщені піперидинодигідротієнопіримідини
Випадковий патент: Прилад для визначення газоутворювальної здатності