Гідрат 1-{(2s)-2-аміно-4-[2,4-bis(трифторметил)-5,8-дигідропіридо[3,4-d]піримідин-7(6н)-іл]-4-оксобутил}-5,5-дифторпіперидин-2-онтартрату
Номер патенту: 106445
Опубліковано: 26.08.2014
Автори: Кім Кюу Юн, Лі Цзи Хє, Парк Кі Сук, Кім Бон Чан, Юнь Цзюн Мінь







Формула / Реферат
1. 1.5 Гідрат 1-{(2S)-2-аміно-4-[2,4-біс(трифторметил)-5,8-дигідропіридо[3,4-d]піримідин-7(6Н)-іл]-4-оксобутил}-5,5-дифторпіперидин-2-он солі винної кислоти, яка має вміст води в межах від 3,5 до 5,5 %.
2. 1.5 Гідрат за пунктом 1, де характерні пікові значення (2θ) на дифрактограмі порошкової рентгенівської дифрактометрії (XRD) становлять 15, 18, 20, 21 і 23°.
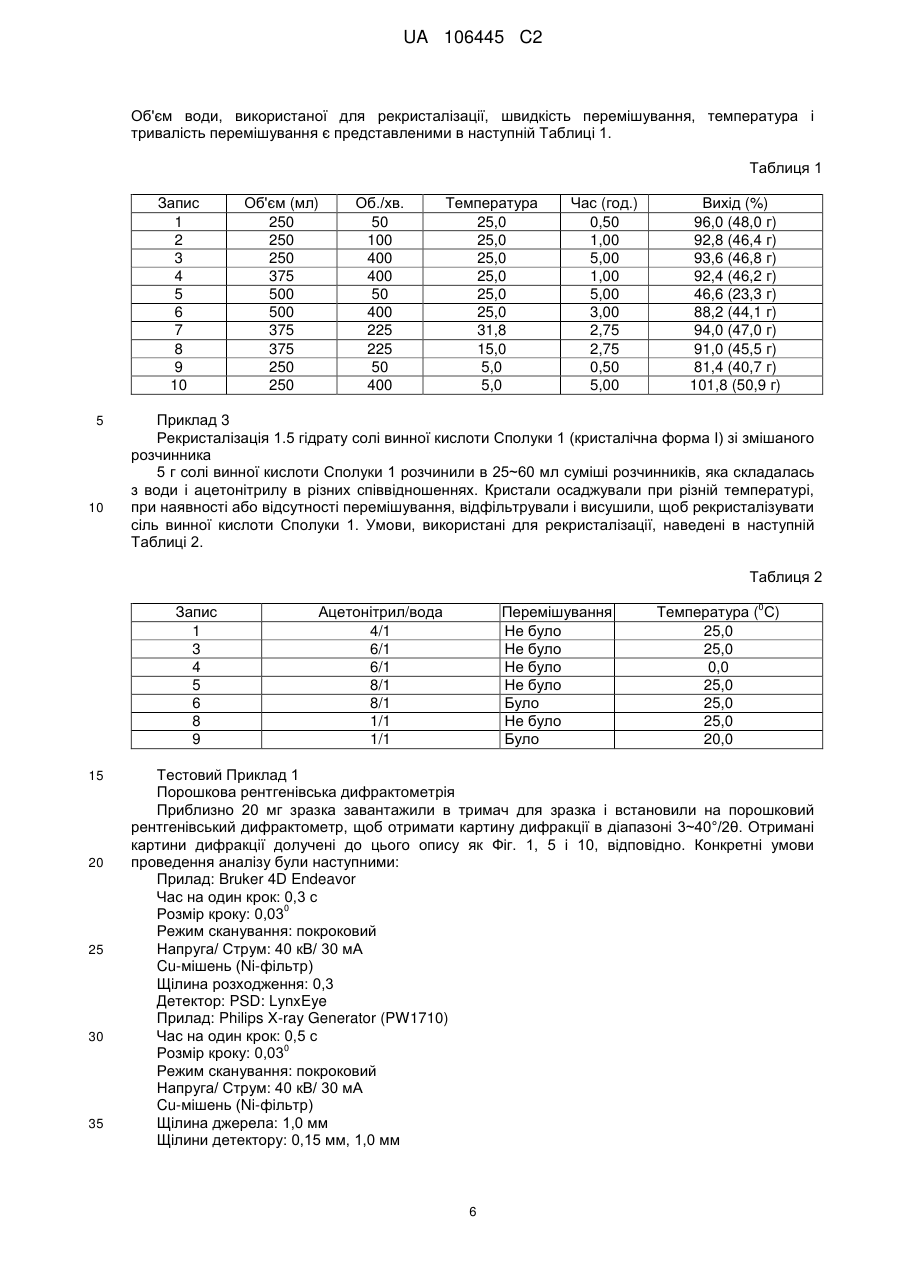
3. Процес отримання 1.5 гідрату за пунктом 1, який відрізняється тим, що 1-{(2S)-2-аміно-4-[2,4-біс(трифторметил)-5,8-дигідропіридо[3,4-d]1-піримідин-7(6Н)-іл]-4-оксобутил}-5,5-дифторпіперидин-2-он солі винної кислоти рекристалізують з розчиннику, вибраного з: вода, ацетонітрил/вода, етиловий спирт/вода, етиловий спирт/гексан або етилацетат/гексан.
4. Процес за пунктом 3, в якому використовують як розчинник для рекристалізації воду.
5. Фармацевтична композиція для пригнічення дипептидилпептидази-IV (DPP-IV), яка містить 1.5 гідрат за пунктом 1 як активний компонент разом з фармацевтично прийнятним носієм.
6. Композиція за пунктом 5, яка використовується для лікування або профілактики діабету або ожиріння.
Текст
Реферат: Даний винахід стосується 1.5 гідрату 1-{(2S)-2-аміно-4-[2,4-біс(трифторметил)-5,8дигідропіридо[3,4-d]піримідин-7(6Н)-іл]-4-оксобутил}-5,5-дифторпіперидин-2-онтартрату, процесу його отримання і фармацевтичної композиції для пригнічення дипептидилпептидази-IV (DPP-IV), яка містить вказану сполуку як активний компонент. UA 106445 C2 (12) UA 106445 C2 UA 106445 C2 Даний винахід стосується 1.5 гідрату 1-{(2S)-2-аміно-4-[2,4-bis(трифторметил)-5,8дигідропіридо[3,4-d]піримідин-7(6H)-іл]-4-оксо-бутил}-5,5-дифторпіперидин-2-он тартрата, представленого наступною формулою 1 (далі буде називатись "Сполука 1"), і процесу його приготування. F F F N N F F F F F N N O 5 10 15 20 25 30 35 40 45 NH2 O (1) Рівень техніки Сполука 1 є описаною в корейській патентній заявці № 10-2006-0029138, яку включено сюди за посиланням у всій її повноті, і демонструє виключну інгібіторну активність проти дипептидил пептидази-IV (DPP IV), завдяки чому використовується в якості агенту для лікування діабету. Типові приклади захворювань, які викликає DPP IV, можуть включати, не обмежуючись ними, діабет, ожиріння і т.п. Що стосується діабету, він є особливо корисним для лікування і профілактики діабету ІІ типу. Термін "лікування", як він тут використовується, означає, що, коли ця сполука використовується для індивідів, які демонструють симптоми хвороби, вона може перервати або відстрочити прогресування хвороби, а термін "профілактика", як він тут використовується, означає, що, коли ця сполука використовується для індивідів, які не демонструють симптоми хвороби, але мають ризик її появи, вона може перервати або відстрочити ознаки хвороби. Дослідження фізичних і хімічних властивостей нової сполуки є необхідним для ефективної і успішної розробки нового лікарського препарату. Зокрема, шляхом вивчення наявності поліморфів і псевдополіморфів цієї сполуки і відмінностей в фізичних і хімічних властивостях між відповідними поліморфами можна вибрати найкращу кристалічну форму з точки зору фармацевтичного аспекту (Remington's Pharmaceutics, Chapter 75 Preformulation); (Byrn, S.R., Solid State Chemistry of Drugs, Academic Press, New York, 1982). Коли поліморфи знаходяться в розчині, вони є хімічно ідентичними, але в твердому стані вони відповідно мають чітко відмінні рентгенівські дифракційні картини і демонструють відмінності в різних фізичних і хімічних властивостях. Зокрема, відповідні поліморфи можуть мати різну біодоступність через відмінності в швидкості розчинення і демонструвати неочікувані властивості в аспекті термодинамічної стабільності. Коли певна лікарська сполука є присутньою у формі поліморфів, можна отримати кристалічні форми різної структури в залежності від умов рекристалізації, таких як використовуваний для рекристалізації розчинник, концентрація сполуки, швидкості нагрівання і охолодження, температури, швидкості перемішування і т.п., під час процедур, застосованих для приготування лікарського препарату. Відповідно, для того, щоб отримати таку саму кристалічну форму, необхідна особлива увага до ведення технологічних процедур. Гідрати як одна з форм псевдополіморфів містять молекулу води всередині кристалу лікарської сполуки і мають форму кристалу, відмінну від ангідриду. Різницю у структурі кристалу можна побачити за допомогою рентгенівської дифракційної картини. Оскільки в гідратах змінюються тільки фізичні властивості, такі як кристалічність, гігроскопічні властивості, температура плавлення, розчинність, швидкість розчинення і т.п., без будь-якої зміни хімічних властивостей, які забезпечують фармакологічні ефекти, вони мають дуже важливе значення в фармацевтичному аспекті, подібно до поліморфів (Morris, K. R. et al., Int. J. Pharm., 108, 1994, 15-206). Знання, які можна почерпнути на сьогоднішній день з різних джерел, що мають відношення до галузі техніки, до якої належить даний винахід, показують відсутність загальної тенденції в тому, щоб віддавати перевагу гідратам перед ангідридами або навпаки, для поліпшення фармацевтичних властивостей, включаючи стабільність лікарського препарату, гігроскопічних властивостей і т.п. Насамкінець, визначення форм, що мають оптимальні фармацевтичні властивості для відповідних сполук, має здійснюватись спеціалістом в цій галузі шляхом постійного вивчення кожного окремого випадку. 1 UA 106445 C2 5 10 15 20 25 30 35 40 45 50 55 Зокрема, ніколи не можна було очікувати серед будь-яких припустимих форм певного лікарського препарату, тобто вільної сполуки, солі, ангідриду і гідрату, яка саме може демонструвати стабільність з гігроскопічними властивостями, які не змінюються в залежності від оточуючої вологості. Більше того, серед гідратів неможна передбачити найбільш стабільне число гідратації. І, навіть коли число гідратації є таким самим, все ще неможливо передбачити, яка форма кристалу буде найстабільнішою. Це явище, яке виявляється нестабільно, оскільки на нього неможна очікувати і оскільки воно належить до експериментальної царини і його можна підтвердити тільки повторними експериментами. Суть винаходу Отже, винахідники провели інтенсивне дослідження, щоб забезпечити стабільний поліморф або псевдополіморф Сполуки 1. Як результат, нами було неочікувано встановлено, що 1.5 гідрат тартратної солі Сполуки 1 демонструє відмінну стабільність при зміні відносної вологості, порівняно з ангідридом або іншими гідратами, що мають подібне число гідратації, і, отже, завершили даний винахід. До сьогоднішнього дня кристалічна форма Сполуки 1 ніколи відкрито не розкривалась. Відповідно, даний винахід пропонує 1.5 гідрат тартратної солі Сполуки 1. Додатково, даний винахід пропонує процес приготування 1.5 гідрат тартратної солі Сполуки 1. Докладний опис винаходу Оскільки 1.5 гідрат тартратної солі Сполуки 1, отриманої у вигляді кристалічної форми, яку можна розробити у відповідності до даного винаходу, є кращою, ніж подібні кристалічні форми з точки зору стабільності, зокрема стабільності при зберіганні, його можна дуже доцільно використати при приготуванні фармацевтичної композиції, що містить Сполуку 1 в якості активного компоненту. Тобто, 1.5 гідрат за цим винаходом не втрачає жодної молекули води в кристалі і не акцептує більше молекул води в широкому діапазоні відносної вологості, підтримуючи свій вміст води, і відповідно демонструє суттєву відсутність зміни ваги в залежності від зміни вологості. У випадку нестабільних кристалічних форм вміст води в них може змінюватись в залежності від оточуючого середовища або добавок під час зберігання і в процесі приготування лікарської форми. Наприклад, при визначенні стандартного матеріалу і зразка для цілей квантифікації, коли експеримент проводиться не в сушильній камері, певна експериментальна похибка може спричинити до проблем з контролем якості. Однак, оскільки в 1.5 гідраті за цим винаходом вміст води чутливо не змінюється в залежності від оточуючого середовища в широкому діапазоні відносної вологості, продукт з незмінними стандартними параметрами завжди може бути отриманий під час зберігання і в процесі приготування лікарської форми, і більше того, похибка в контролі якості є дуже незначною. Як показано вище, 1.5 гідрат за цим винаходом забезпечує велику перевагу з точки зору користування і контролю якості. Більше того, 1.5 гідрат солі винної кислоти Сполуки 1 за цим винаходом не виявляє будьякої зміни в кристалічній формі в залежності від зміни вологості. З іншого боку, винахідниками було встановлено, що 0.5 гідрат і ангідрид солі винної кислоти Сполуки 1 абсорбують багато води, коли відносна волога підвищується, перетворюючись на більш стабільний 1.5 гідрат за цим винаходом. Було також встановлено, що навіть в експерименті з прискореного тестування стабільності (40 °C/75 % ВВ) 0.5 гідрат перетворюється на 1.5 гідрат через кілька годин. До того ж, є можливість контролювати приготування 1.5 гідрату солі винної кислоти Сполуки 1 різними способами, як запропоновано в даному винаході. Короткий опис малюнків На Фіг. 1 представлено спектр рентгенівської порошкової дифракції (XRD) кристалічної форми І як 1.5 гідрату солі винної кислоти Сполуки 1. На Фіг. 2 представлено спектр інфрачервоної спектроскопії (FT-IR) кристалічної форми І як 1.5 гідрату солі винної кислоти Сполуки 1. На Фіг. 3 представлено результат диференціальної скануючої калориметрії (DSC) або термогравіметричного аналізу (TG) кристалічної форми І як 1.5 гідрату солі винної кислоти Сполуки 1. На Фіг. 4 представлено динамічну ізотерму адсорбції/десорбції пари кристалічної форми І як 1.5 гідрату солі винної кислоти Сполуки 1. На Фіг. 5 представлено спектр рентгенівської порошкової дифракції (XRD) кристалічної форми ІІ як 0.5 гідрату солі винної кислоти Сполуки 1. На Фіг. 6 представлено спектр інфрачервоної спектроскопії (FT-IR) кристалічної форми ІІ як 0.5 гідрату солі винної кислоти Сполуки 1. 2 UA 106445 C2 5 10 15 20 25 30 35 40 45 50 55 60 На Фіг. 7 представлено результат диференціальної скануючої калориметрії (DSC) або термогравіметричного аналізу (TG) кристалічної форми ІІ як 0.5 гідрату солі винної кислоти Сполуки 1. На Фіг. 8 представлено динамічну ізотерму адсорбції/десорбції пари кристалічної форми ІІ як 0.5 гідрату солі винної кислоти Сполуки 1. На Фіг. 9 представлено спектр рентгенівської порошкової дифракції (XRD) кристалічної форми ІІ як 0.5 гідрату солі винної кислоти Сполуки 1 під час дослідження адсорбції вологи. На Фіг. 10 представлено спектр рентгенівської порошкової дифракції (XRD) кристалічної форми ІІІ як ангідриду солі винної кислоти Сполуки 1. На Фіг. 11 представлено спектр інфрачервоної спектроскопії (FT-IR) кристалічної форми ІІІ як ангідриду солі винної кислоти Сполуки 1. На Фіг. 12 представлено результат диференціальної скануючої калориметрії (DSC) або термогравіметричного аналізу (TG) кристалічної форми ІІІ як ангідриду солі винної кислоти Сполуки 1. На Фіг. 13 представлено динамічну ізотерму адсорбції/десорбції пари кристалічної форми ІІІ як ангідриду солі винної кислоти Сполуки 1. На Фіг. 14 представлено криву адсорбції вологи кристалічної форми ІІІ як ангідриду солі винної кислоти Сполуки 1 з витримкою при нормальній температурі і нормальній вологості. На Фіг. 15 представлено спектр рентгенівської порошкової дифракції (XRD) кристалічної форми ІІІ як ангідриду солі винної кислоти Сполуки 1 під час дослідження адсорбції вологи. На Фіг. 16 представлено спектр рентгенівської порошкової дифракції (XRD), отриманий після проведення експерименту Тестового Прикладу 8, який показує XRD кристалічної форми ІІ, XRD зразка кристалічної форми ІІ після 8-тижневого зберігання при 60 °C/5 % ВВ, XRD зразка кристалічної форми ІІ після 8-тижневого зберігання при 40 °C/75 % ВВ, і XRD кристалічної форми I, в порядку знизу догори. Кращий варіант здійснення Коли ідентифікували характерні кристалічні форми за цим винаходом, в даному описі 1.5 гідрат солі винної кислоти Сполуки 1 позначили як "кристалічна форма 1", 0.5 гідрат позначили як "кристалічна форма ІІ", і ангідрид як "кристалічна форма ІІІ". При цьому, вміст води в 1.5 гідраті становить 3,0~5,5 %, в 0.5 гідраті 1,0~2,5 % і в ангідриді 0~1,0 %. Кристалічності кристалічних форм І, ІІ і ІІІ відрізняються одна від одної, як можна бачити з діаграм рентгенівської дифракції, показаних на Фіг. 1, 5 і 10. Як встановлено з аналізу методом диференціальної скануючої калориметрії для кристалічної форми І, ендотермічний пік, який спостерігається при вивільненні води, що міститься в кристалічній решітці, при 90~130 °C, має широку ділянку, в яку включена температура плавлення, і ендотермічний пік трапляється ще раз в межах 130~160 °C, коли сіль винної кислоти Сполуки 1 піддається хімічній дегідратації. До того ж, термогравіметричнй аналіз показує втрату ваги біля 4,0 %, яка еквівалентна воді, що містилась в решітці на першій ендотермічній ділянці диференціальної скануючої калориметрії, і втрату ваги біля 2,5 %, яка еквівалентна воді, видаленій дегідратацією зі структури солі винної кислоти Сполуки 1 на другій ендотермічній ділянці (Фіг. 3). Аналіз у відношенні кристалічних форм ІІ і ІІІ методом диференціальної скануючої калориметрії показує три ендотермічні точки. Конкретно, перша ендотермічна точка трапляється при вивільненні води, що міститься в решітці, друга ендотермічна точка трапляється при температурі плавлення і третя ендотермічна точка досягається через реакцію хімічної дегідратації. Більше того, згідно термогравіметричному аналізу перша ендотермічна ділянка показує втрату ваги, яка еквівалентна воді, що міститься в решітці (близько 1,3 % в кристалічній формі ІІ і близько 0,6 % в кристалічній формі ІІІ), а третя ендотермічна ділянка показує втрату ваги біля 2,4 %, яка еквівалентна воді, видаленій дегідратацією (Фіг. 7 і 12). Оскільки вміст води в решітці солі винної кислоти Сполуки 1, як показано вище, відповідав вмісту води, визначеному методом Карла-Фішера (Mettler Toledo DL37 KF Coulometer), було доведено, що ендотермічні піки викликаються випаровуванням молекул води. Далі, ми маємо намір пояснити, що кристалічна форма І за цим винаходом має неочікувані відмінні характеристики в будь-якому аспекті, у порівнянні з іншими кристалічними формами ІІ або ІІІ, шляхом більш докладного порівняння характеристик відповідних кристалічних форм. Кристалічна форма І Даний винахід стосується кристалічної форми 1.5 гідрату солі винної кислоти Сполуки 1. В результаті аналізу її властивостей виявилось, що кристалічна форма І демонструє наступні характеристики: (а) Вміст води в кристалічній формі І знаходиться в межах 3,0~5,5 %. 3 UA 106445 C2 5 10 15 20 25 30 35 40 45 50 55 60 (b) Характерні пікові значення (2θ) спектру XRD, виміряні при CuKα, 40кВ, 30мА, становлять 15, 18, 20, 21 і 23°. (с) Інфрачервоний (IR) спектр показує характерне поглинання приблизно при 3591, 3401, -1 3128, 1712, 1655, 1636, 1229, 1205, 1129 і 1058 см . (d) Спектр диференціальної скануючої калориметрії показує ендотермічні піки в двох широких діапазонах температур близько 90~130 °C і 130~160 °C. (е) Вміст води, визначений методом Карла-Фішера становить біля 4,0 %. (f) Коли температура піднімається з 25 °C до 250 °C, втрата ваги біля 4,0 % і біля 2,5 % спостерігається в діапазоні 70~110 °C і 140~170 °C, відповідно. (Це результат, отриманий з TG на Фіг. 3. Оскільки зміна енергії трапляється трохи пізніше, ніж зміна ваги, загалом спостерігається різниця між результатами DSC і TG.). (g) Тартрат Сполуки 1 можна кристалізувати з води, ацетонітрилу/ води, етилового спирту/води, етилового спирту/гексану або з розчинника етилацетат/гексан; переважно, вона кристалізується з води. (h) Кристалічну форму І можна отримати, піддавши кристалічну форму ІІ або ІІІ адсорбції вологи. (і) Зміна ваги є відсутньою або, якщо вона є, становить 0,8 % або менше в залежності від зміни зовнішньої вологості в межах 5~95 % ВВ, і ця кристалічна форма не змінюється зі зміною вологості. Кристалічна форма ІІ Для того, щоб знайти інші кристалічні форми, ніж кристалічна форма І, винахідники приготували 0.5 гідрат солі винної кислоти Сполуки 1 (кристалічна форма ІІ). Хоча 0.5 гідрат є такою самою сіллю винної кислоти тієї самої сполуки і має подібне число гідратації, він демонструє нестабільність при зберіганні у порівнянні з 1.5 гідратом (кристалічна форма І). (а) Вміст води в кристалічній формі ІІ знаходиться в межах 1,0~2,5 %. (b) Характерні пікові значення (2θ) спектру XRD, виміряні при CuKα, 40кВ, 30мА, становлять 14, 15, 17, 18, 19, 21 і 23°. (с) Інфрачервоний (IR) спектр показує характерне поглинання приблизно при 3455, 2891, -1 1721, 1655, 1571, 1228, 1209, 1131, 1086 і 1059 см . (d) Спектр диференціальної скануючої калориметрії показує ендотермічні піки в трьох широких діапазонах температур близько 80~115 °C, 115~135 °C і 135~173 °C, і температуру плавлення приблизно при 117 °C. (е) Вміст води, визначений методом Карла-Фішера становить біля 2,0 %. (f) Коли температура піднімається з 25 °C до 250 °C, втрата ваги біля 1,3 % і біля 2,4 % спостерігається в діапазоні 70~104 °C і 137~168 °C, відповідно. (g) Зміна маси на 4,0 % або більше трапляється при зміні зовнішньої вологості в межах 5~95 % ВВ, і волога швидко абсорбується з 45 % ВВ, приводячи до збільшення маси на 3,7 % при 75 % ВВ. Коли вміст вологи підвищується до 4,0 % або більше, кристалічна форма ІІ перетворюється на кристалічну форму І. Було показано, що кристалічна форма ІІ перетворюється на кристалічну форму І від близько 60 % ВВ (дивись Фіг. 8 і 9). Тобто, кристалічна форма змінюється зі зміною вологості, щоб досягти більш стабільної кристалічної форми І. До того ж, вона перетворюється на кристалічну форму І в межах 8 тижнів в ході прискореного тестування стабільності (40 °C/75 % ВВ). Кристалічна форма ІІІ Для того, щоб знайти інші кристалічні форми, ніж кристалічна форма І, винахідники приготували ангідрид солі винної кислоти Сполуки 1 (кристалічна форма ІІІ). Ангідрид демонструє нестабільність при зберіганні у порівнянні з 1.5 гідратом (кристалічна форма І). (а) Вміст води в кристалічній формі ІІІ знаходиться в межах 0~1,0 %. (b) Характерні пікові значення (2θ) спектру XRD, виміряні при CuKα, 40кВ, 30мА, становлять 6, 17, 21, 23, 24, 26 і 30°. (с) Інфрачервоний (IR) спектр показує характерне поглинання приблизно при 3470, 3187, -1 2940, 1640, 1570, 1229, 1206, 1130 і 1056 см . (d) Спектр диференціальної скануючої калориметрії показує ендотермічні піки в трьох широких діапазонах температур близько 65~100 °C, 100~130 °C і 132~170 °C, і температуру плавлення приблизно при 104 °C. (е) Вміст води, визначений методом Карла-Фішера становить біля 0,1 %. (f) Коли температура піднімається з 25 °C до 250 °C, втрата ваги біля 0,6 % і біля 2,4 % спостерігається в діапазоні 62~110 °C і 120~173 °C, відповідно. (g) Зміна маси на 3,5 % або більше трапляється при зміні зовнішньої вологості в межах 5~95 % ВВ, і волога швидко абсорбується від 5 % ВВ, так що кристалічна форма ІІІ 4 UA 106445 C2 5 10 15 20 25 30 35 40 45 50 перетворюється на кристалічну форму І від близько 15 % ВВ (дивись Фіг. 13, 14 і 15). Тобто, кристалічна форма змінюється зі зміною вологості, щоб досягти більш стабільної кристалічної форми І. Вищенаведені результати дозволяють припустити, що в діапазоні відносної вологості, в якому препарат звичайно використовується, кристалічні форми ІІ і ІІІ є нестабільними і автоматично перетворюються на кристалічну форму І. Така тенденція є більш вираженою у випадку кристалічної форми ІІІ. 1.5 Гідрат солі винної кислоти Сполуки 1 за цим винаходом демонструє сильну інгібіторну активність у відношенні DPP-IV у такий самий спосіб, як відповідна вільна основа, описана в корейській патентній заявці № 10-2006-0029138. До того ж, 1.5 гідрат за даним винаходом показує поліпшені фізичні і хімічні властивості у порівнянні з кристалічними формами, які мають інший стан гідратації. Відповідно, 1.5 гідратом за даним винаходом легше користуватись, з ним зручно проводити контроль якості і готувати лікарську форму, у порівнянні з кристалічними формами, які мають інший стан гідратації. 1.5 Гідрат солі винної кислоти Сполуки 1 за цим винаходом демонструє сильну інгібіторну активність у відношенні DPP-IV, як вже зазначалось, і відповідно його можна приготувати для зручного введення у фармацевтичній і ветеринарній галузі. Приготування лікарської форми може здійснюватись у відповідності до методик і методів, відомих в цій галузі у відношенні інших препаратів, що володіють інгібіторною активністю у відношенні DPP-IV, зокрема з посиланням на опис корейської патентної заявки № 10-2006-0029138, включеної сюди за посиланням у всій її повноті. Відповідно, фармацевтична композиція для пригнічення DPP-IV, яка містить 1.5 гідрат за цим винаходом в якості активного компонента, разом з фармацевтично прийнятним носієм, охоплюється об'ємом даного винаходу. Композиція за цим винаходом характеризується тим, що використовується зокрема для лікування і профілактики діабету або ожиріння. Даний винахід ілюструється більш докладно за допомогою наступних Прикладів і Тестових Прикладів. Однак ці наступні Приклади і Тестові Приклади наведені тільки для кращого розуміння даного винаходу і жодним чином не обмежують його об'єм. Приклад 1 Приготування 1.5 гідрату солі винної кислоти Сполуки 1 (кристалічна форма І) Сполука 2 Сіль винної кислоти Сполуки 1 1,87 кг Сполуки 2 розчинили в приблизно 9 л етилового спирту. Додали 0,94 кг SOCl2 при 0 0~10 С, а потім перемішували підтримуючи низьку температуру. Після концентрації під зниженим тиском концентрат розчинили в 11,2 л МТВЕ (метил t-бутиловий ефір), і отриману суміш відрегулювали до рН 7~8 за допомогою розчину 10 N NaOH. Після розділення шарів водний шар екстрагували приблизно 3,7 л МТВЕ, а потім концентрували під зниженим тиском. Отриманий коричневий каламутний розчин розчинили в 12 л етилового спирту, до цього додали 0,47 кг L-винної кислоти, розчиненої приблизно в 1,5 л води, і перемішували впродовж 1 години. Отриманий кристалічний шлам профільтрували, промили водою і етиловим спиртом (1:8), після чого висушили, щоб отримати 1,13 кг (вихід 97,5 %) титульної сполуки. 1 H ЯМР (500 МГц, CD3OD) δ 2,38 (m, 2H), 2,59 (m, 2H), 2,82 – 2,99 (m, 2H), 3,11 (bt, 1H), 3,21 (bt, 1H), 3,50 – 3,55 (m, 1H), 3,72 – 3,91 (m, 5H), 3,98 (t, J=5,2 Гц, 1H), 4,38 (s, 2H), 4,97 – 5,00 (m, 2H). Приклад 2 Рекристалізація 1.5 гідрату солі винної кислоти Сполуки 1 (кристалічна форма І) з води 59 г солі винної кислоти Сполуки 1, отриманої в Прикладі 1, додали до 250~500 мл води і розчинили у воді, відрегулювавши цей розчин до рН 6~7 за допомогою 10 N NaOH. Додали 11,7 г L-винної кислоти, розчиненої 23,5 мл води, і отримали кристали, змінюючи температуру, швидкість перемішування і тривалість перемішування, як показано в наступній Таблиці 1. Потім кристали відфільтрували і висушили, щоб отримати кристалічну форму І. Швидкість 0 перемішування змінювали в межах 50~400 об./хв., температуру змінювали в межах 5~32 С. 5 UA 106445 C2 Об'єм води, використаної для рекристалізації, швидкість перемішування, температура і тривалість перемішування є представленими в наступній Таблиці 1. Таблиця 1 Запис 1 2 3 4 5 6 7 8 9 10 5 10 Об'єм (мл) 250 250 250 375 500 500 375 375 250 250 Об./хв. 50 100 400 400 50 400 225 225 50 400 Температура 25,0 25,0 25,0 25,0 25,0 25,0 31,8 15,0 5,0 5,0 Час (год.) 0,50 1,00 5,00 1,00 5,00 3,00 2,75 2,75 0,50 5,00 Вихід (%) 96,0 (48,0 г) 92,8 (46,4 г) 93,6 (46,8 г) 92,4 (46,2 г) 46,6 (23,3 г) 88,2 (44,1 г) 94,0 (47,0 г) 91,0 (45,5 г) 81,4 (40,7 г) 101,8 (50,9 г) Приклад 3 Рекристалізація 1.5 гідрату солі винної кислоти Сполуки 1 (кристалічна форма І) зі змішаного розчинника 5 г солі винної кислоти Сполуки 1 розчинили в 25~60 мл суміші розчинників, яка складалась з води і ацетонітрилу в різних співвідношеннях. Кристали осаджували при різній температурі, при наявності або відсутності перемішування, відфільтрували і висушили, щоб рекристалізувати сіль винної кислоти Сполуки 1. Умови, використані для рекристалізації, наведені в наступній Таблиці 2. Таблиця 2 Запис 1 3 4 5 6 8 9 15 20 25 30 35 Ацетонітрил/вода 4/1 6/1 6/1 8/1 8/1 1/1 1/1 Перемішування Не було Не було Не було Не було Було Не було Було 0 Температура ( С) 25,0 25,0 0,0 25,0 25,0 25,0 20,0 Тестовий Приклад 1 Порошкова рентгенівська дифрактометрія Приблизно 20 мг зразка завантажили в тримач для зразка і встановили на порошковий рентгенівський дифрактометр, щоб отримати картину дифракції в діапазоні 3~40°/2θ. Отримані картини дифракції долучені до цього опису як Фіг. 1, 5 і 10, відповідно. Конкретні умови проведення аналізу були наступними: Прилад: Bruker 4D Endeavor Час на один крок: 0,3 с 0 Розмір кроку: 0,03 Режим сканування: покроковий Напруга/ Струм: 40 кВ/ 30 мА Cu-мішень (Ni-фільтр) Щілина розходження: 0,3 Детектор: PSD: LynxEye Прилад: Philips X-ray Generator (PW1710) Час на один крок: 0,5 с 0 Розмір кроку: 0,03 Режим сканування: покроковий Напруга/ Струм: 40 кВ/ 30 мА Cu-мішень (Ni-фільтр) Щілина джерела: 1,0 мм Щілини детектору: 0,15 мм, 1,0 мм 6 UA 106445 C2 5 10 15 20 25 30 35 40 45 50 55 Тестовий Приклад 2 Інфрачервона спектроскопія Інфрачервоні спектри для відповідних кристалічних форм у відповідності до даного винаходу були отримані з використанням ZASCO FT-IR 4200, оснащеного детектором DTGS. -1 Розділення відповідних спектрів становило 4 см , а кількість сканувань становила 16. В цьому Тестовому Прикладі 1~2 мг зразка поміщали на приставку ATR (порушене повне внутрішнє відбиття) і включали прилад, щоб отримати спектр. Фонові дані отримувались при роботі приладу без будь-якого матеріалу в ATR. Отримані у такий спосіб спектри долучені до даного опису як Фіг. 2, 6 і 11, відповідно. Тестовий Приклад 3 Диференціальна скануюча калориметрія (DSC) Диференціальна скануюча калориметрія (DSC) проводилась з використанням Mettler e Toledo's DSC821 . Приблизно 2~3 мг зразка завантажили в алюмінієву чашку і її вагу точно зареєстрували. Чашку закрили кришкою, через яку прокололи отвір. Чашку встановили на 0 0 приладі і нагрівали від 25 до 250 С зі швидкістю 10 С/хв. під продувкою азотом. Метал індій використовувався як стандарт для калібрування. Отримані у такий спосіб спектри долучені до даного опису як Фіг. 3, 7 і 12, відповідно. Тестовий Приклад 4 Термогравіметрія (TG) Термогравіметрія (TG) проводилась з використанням Mettler Toledo TGA850. Приблизно 4~5 мг зразка завантажили в алюмінієву чашку. Чашку встановили на приладі і нагрівали від 25 до 0 0 250 С зі швидкістю 10 С/хв. під продувкою азотом. Nickel and Aluminum™ використовувались як стандарт для калібрування. Отримані у такий спосіб результати долучені до даного опису як Фіг. 3, 7 і 12, відповідно, разом з результатами DSC аналізу з Тестового Прикладу 3. Тестовий Приклад 5 Аналіз динамічної адсорбції/десорбції пари Дані динамічної адсорбції/десорбції пари були отримані на аналізаторі VTI-SA Vapor Sorption 0 Analyzer. Підтримуючи 25 С, адсорбцію і десорбцію пари повторили тричі з інтервалами 5 % ВВ в діапазоні відносної вологості (ВВ) 5~95 %. Зразки перед аналізом не висушувались. Рівноважним стандартом, використаним для аналізу, був таким, щоб зміна ваги в межах 2 хвилин була меншою ніж 0,01 %. В якості результату аналізу, Фіг. 4 показує ізотерму адсорбції/десорбції пари кристалічної форми І, з якої можна бачити, що кристалічна форма І демонструє зміну ваги 0,8 % або менше при зміні зовнішньої вологості в межах 5~95 % ВВ. Тобто, кристалічна форма І за цим винаходом є дуже стабільною при зміні відносної вологості. Фіг. 8 показує результат як ізотерму адсорбції/десорбції пари кристалічної форми ІІ, з якої можна бачити, що в кристалічній формі ІІ вага зростає на 4,3 %, коли відносна вологість підвищується до 95 % в початковому тесті на адсорбцію вологи. Після десорбції вологи кристалічна форма ІІ дає такий самий графік поведінки у відношенні вологи, що й кристалічна форма І. Такий результат підтверджує, що кристалічна форма ІІ була перетворена на кристалічну форму І в ході початкової адсорбції вологи. Фіг. 13 показує результат як ізотерму адсорбції/десорбції пари кристалічної форми ІІІ, з якої можна бачити, що в кристалічній формі ІІІ вага зростає на 3,6 %, коли відносна вологість підвищується до 95 %. Більше того, можна бачити, що зменшення вологості викликає десорбцію вологи 0,6 % або менше, так що кристалічна форма ІІІ починає містити таку саму кількість вологи, що й кристалічна форма І. Тестовий Приклад 6 Тест з використанням порошкової рентгенівської дифракції кристалічної форми ІІ під час адсорбції вологи Приблизно 50 мг кристалічної форми ІІ помістили в скляний флакон, який піддавали відносній вологості 11 %, 32 %, 53 %, 64 %, 78 % і 97 %, відповідно, впродовж двох або більше днів, щоб викликати адсорбцію вологи, а потім піддавали порошковій рентгенівській дифракції в умовах, представлених в Тестовому Прикладі 1, щоб ідентифікувати будь-яку зміну кристалічної форми під час адсорбції вологи (дивись Фіг. 9). Відповідна відносна вологість забезпечувалась шляхом приготування насиченого водного розчину солі, як показано в наступній Таблиці 3, і встановлення цього розчину в ексикаторі, який після цього герметизували. 7 UA 106445 C2 Таблиця 3 Відносна вологість 11 % Відносна вологість 32 % Відносна вологість 53 % Відносна вологість 64 % Відносна вологість 78 % Відносна вологість 97 % 5 10 15 20 25 Насичений водний розчин LiCl Насичений водний розчин MgCl2 Насичений водний розчин Mg(NO3)2∙6H2O Насичений водний розчин NaNO2 Насичений водний розчин NaCl Насичений водний розчин KNO3 Тестовий Приклад 7 Тест з використанням порошкової рентгенівської дифракції кристалічної форми ІІІ під час адсорбції вологи Кристалічну форму ІІІ помістили на XRD тримач і піддавали XRD тестуванню через різний час (через 30 хвилин, 1 годину і 5 годин), дозволяючи адсорбцію вологи при кімнатній температурі і атмосферному тиску (дивись Фіг. 15). Крім того, реєстрували зміну ваги при кімнатній температурі і атмосферному тиску, щоб побудувати графік (дивись Фіг. 14). Можна було бачити, що кристалічна форма ІІІ швидко абсорбує воду, так що вона перетворюється на кристалічну форму І в межах приблизно 1 години. Тестовий Приклад 8 Термальна стабільність кристалічної форми І і кристалічної форми ІІ Приблизно по 50 мг кристалічної форми І і кристалічної форми ІІ помістили в пляшечки з 0 0 ПЕВЩ, які тримали при 40±2 С, 75±5 % ВВ або 60±2 С, 5±5 % ВВ. Через 2 тижні, 4 тижні і 8 тижнів зразки видаляли з пляшечок і аналізували з використанням XRD для ідентифікації будьякої зміни кристалічної форми ВЕРХ для визначення стабільності. Для аналізу з використанням ВЕРХ зразок розчиняли в суміші ацетонітрил/ вода/ трифтороцтова кислота = 30/70/0,1 (о/о/о), а потім піддавали аналізу. Умови ВЕРХ аналізу були наступними: Умови проведення ВЕРХ: Колонка: Atlantis dC18 (4,6 мм внутрішній діаметр х 250 мм довжина, розмір часток 5 мкм, Waters) 0 Температура колонки: 10 С Мобільна фаза: Мобільна фаза А: MeCN/TFA=100/0,1 (о/о) Мобільна фаза В: H2O/TFA=100/0,1 (о/о) Градієнтні умови: Час (хв.) Початковий момент 25 35 40 55 30 35 A (%) 38 38 80 38 38 B (%) 62 62 20 62 62 Швидкість потоку: 0,7 мл/хв. Реєстрація: 256 нм, УФ Об'єм інжекції: 10 мкл Тривалість аналізу: 55 хвилин Результати тестування стабільності для кристалічної форми І і кристалічної форми ІІ є показаними в Таблиці 4. Таблиця 4 Час (тижні) 0 2 4 8 0 40 С/75 % ВВ Кристалічна Кристалічна форма I форма II 99,4 98,4 99,3 98,4 99,2 98,3 99,2 98,2 8 0 60 С/5 % ВВ Кристалічна Кристалічна форма форма I II 99,4 98,4 98,8 98,1 98,6 97,8 98,4 97,5 UA 106445 C2 5 10 15 20 Як показано в Таблиці 4, можна підтвердити, що при витримці кристалічної форми І і 0 0 кристалічної форми ІІ при 40±2 С, 75±5 % ВВ або 60±2 С, 5±5 % ВВ вони демонструють відмінну стабільність до 8 тижнів. Однак згідно результату XRD аналізу кристалічна форма І не виявляє жодних змін до 8 тижнів, тоді як кристалічна форма ІІ перетворилась на кристалічну 0 форму І на 8 тижні при 40 С/75 % ВВ (дивись Фіг. 16). Тестовий Приклад 9 Визначення щільності кристалічної форми І і кристалічної форми ІІ Приблизно по 20~30 мл кристалічної форми І і кристалічної форми ІІ помістили в 50-мл мірний циліндр. При цьому зразки вводили повільно і так, щоб вони тісно не контактували один з одним. Після зчитування шкали на мірному циліндрі визначали його вагу, щоб обчислити насипну щільність до ущільнення. Після визначення насипної щільності мірний циліндр зі зразками встановили на денситометр ERWEKA і піддали імпульсам 250 разів, щоб визначити щільність після ущільнення (Таблиця 5). Результат визначення показує, що насипна щільність до ущільнення і після ущільнення кристалічної форми І була більшою, ніж у кристалічної форми ІІ. Коефіцієнт здатності пресуватись було обчислено за насипною щільністю і щільністю після ущільнення. Він виявився нижчим для кристалічної форми І ніж для кристалічної форми ІІ. Оскільки загалом вважається, що чим нижчим є коефіцієнт здатності пресуватись, тим кращою є текучість, то, порівнюючи коефіцієнти здатності пресуватись, можна дійти висновку, що кристалічна форма І буде більш легко піддаватись обробці, включаючи таблетування, порівняно з кристалічною формою ІІ. Таблиця 5 Визначення щільності і розміру часток кристалічної форми І і кристалічної форми ІІ Щільність Кристалічна форма І ІІ Насипна Після ущільнення 0,45 0,34 0,54 0,44 Коефіцієнт здатності пресуватись 16,22 23,47 25 ФОРМУЛА ВИНАХОДУ 30 35 40 1. 1.5 Гідрат 1-{(2S)-2-аміно-4-[2,4-біс(трифторметил)-5,8-дигідропіридо[3,4-d]піримідин-7(6Н)іл]-4-оксобутил}-5,5-дифторпіперидин-2-он солі винної кислоти, яка має вміст води в межах від 3,5 до 5,5 %. 2. 1.5 Гідрат за пунктом 1, де характерні пікові значення (2θ) на дифрактограмі порошкової рентгенівської дифрактометрії (XRD) становлять 15, 18, 20, 21 і 23°. 3. Процес отримання 1.5 гідрату за пунктом 1, який відрізняється тим, що 1-{(2S)-2-аміно-4[2,4-біс(трифторметил)-5,8-дигідропіридо[3,4-d]1-піримідин-7(6Н)-іл]-4-оксобутил}-5,5дифторпіперидин-2-он солі винної кислоти рекристалізують з розчиннику, вибраного з: вода, ацетонітрил/вода, етиловий спирт/вода, етиловий спирт/гексан або етилацетат/гексан. 4. Процес за пунктом 3, в якому використовують як розчинник для рекристалізації воду. 5. Фармацевтична композиція для пригнічення дипептидилпептидази-IV (DPP-IV), яка містить 1.5 гідрат за пунктом 1 як активний компонент разом з фармацевтично прийнятним носієм. 6. Композиція за пунктом 5, яка використовується для лікування або профілактики діабету або ожиріння. 9 UA 106445 C2 10 UA 106445 C2 11 UA 106445 C2 12 UA 106445 C2 13 UA 106445 C2 14 UA 106445 C2 15 UA 106445 C2 16 UA 106445 C2 Комп’ютерна верстка І. Скворцова Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 17
ДивитисяДодаткова інформація
Назва патенту англійськоюHydrate of 1-{(2s)-2-amino-4-[2,4-bis(trifluoromethyl)-5,8-di- hydropyrido[3,4-d]pyrimidin-7(6h)-yl]-4-oxobutyl}-5,5-difluoro-piperidin-2-one tartrate
Автори англійськоюPark, Ki Sook, Yun, Jung Min, Kim, Bong Chan, Kim, Kyu Young, Lee, Ji Hye
Автори російськоюПарк Ки Сук, Юнь Цзюн Минь, Ким Бон Чан, Ким Кюу Юн, Ли Цзи Хе
МПК / Мітки
МПК: A61P 3/04, C07D 471/04, A61K 31/519, A61P 3/10
Мітки: 1-{(2s)-2-аміно-4-[2,4-bis(трифторметил)-5,8-дигідропіридо[3,4-d]піримідин-7(6н)-іл]-4-оксобутил}-5,5-дифторпіперидин-2-онтартрату, гідрат
Код посилання
<a href="https://ua.patents.su/19-106445-gidrat-1-2s-2-amino-4-24-bistriftormetil-58-digidropirido34-dpirimidin-76n-il-4-oksobutil-55-diftorpiperidin-2-ontartratu.html" target="_blank" rel="follow" title="База патентів України">Гідрат 1-{(2s)-2-аміно-4-[2,4-bis(трифторметил)-5,8-дигідропіридо[3,4-d]піримідин-7(6н)-іл]-4-оксобутил}-5,5-дифторпіперидин-2-онтартрату</a>
Кристалічні гідрати 2-хлор-5-[3,6-дигідро-3-метил-2,6-діоксо-4-(трифторметил)-1-(2н)-піримідиніл]-4-фтор-n-[[метил-(1-метилетил)аміно]сульфоніл]бензаміду, способи одержання гідрату, гербіцид, що містить гідрат,

Номер патенту: 99114
Опубліковано: 25.07.2012
Автори: Вольф Бернд, Шмідт Томас, Міхель Альфред, Цагар Сірілл, Майєр Гуідо, Кокс Герхард, Закселль Хайді Емілія, Льор Сандра, Райнхард Роберт, Веверс Ян Хендрік, Зайтц Вернер, Кайл Міхаель, Зіверніх Бернд, Гебхардт Йоахім, Хампрехт Герхард, Ерк Петер
МПК: A01P 13/00, C07D 239/54, A01N 43/48
Мітки: гербіцид, кристалічні, містить, 2-хлор-5-[3,6-дигідро-3-метил-2,6-діоксо-4-(трифторметил)-1-(2н)-піримідиніл]-4-фтор-n-[[метил-(1-метилетил)аміно]сульфоніл]бензаміду, одержання, гідрат, гідрату, гідрати, способи
Формула / Реферат:
1. Кристалічні гідрати 2-хлор-5-[3,6-дигідро-3-метил-2,6-діоксо-4-(трифторметил)-1-(2Н)-піримідиніл]-4-фтор-N-[[метил-(1-метилетил)аміно]сульфоніл]бензаміду, що містять від 0,8 до 1,2 моль води, в перерахуванні на 1 моль 2-хлор-5-[3,6-дигідро-3-метил-2,6-діоксо-4-(трифторметил)-1-(2Н)-піримідиніл]-4-фтор-N-[[метил-(1-метилетил)аміно]сульфоніл]бензаміду.2. Гідрати за п. 1 з піком плавлення в межах від 100 до 140 °C.3....
Спосіб одержання 4-{4-[({[4-хлор-3-(трифторметил)феніл]аміно}карбоніл)аміно]фенокси}-n-метилпіридин-2-карбоксаміду

Номер патенту: 90691
Опубліковано: 25.05.2010
Автори: Морс Клаус, Хайльманн Вернер, Герінг Райнхольд, Льогерс Міхаель, Ленц Яна, Штіль Юрген, Мюллер-Гліманн Маттіас, Берве Матіас, Кун Олівер, Маттхойс Майк
МПК: C07D 213/81
Мітки: спосіб, 4-{4-[({[4-хлор-3-(трифторметил)феніл]аміно}карбоніл)аміно]фенокси}-n-метилпіридин-2-карбоксаміду, одержання
Формула / Реферат:
1. Спосіб одержання сполуки формули (І), (І)при якому на першій стадії піддають реакції сполуку формули (V) (V)з 4-хлор-3-трифторметилфенілізоціанатом у нехлорованому органічному розчиннику, інертному по відношенню до ізоціанатів, спочатку завантажують сполуку формули (V) при...
Кристалічна форма кальцію біс[(е)-7-[4-(4-флуорфеніл)-6-ізопропіл-2-[метил(метилсульфоніл)аміно]піримідин-5-іл](3r,5s)-3,5-дигідроксигепт-6-енової кислоти]

Номер патенту: 85062
Опубліковано: 25.12.2008
Автори: Кребб Джеффрі Норман, Джоунс Девід Він Калверт, Сіттерн Пітер Ентоні, Горбері Джон, Бут Ребекка Джейн
МПК: A61P 3/06, C07D 239/42, A61K 31/505
Мітки: форма, кальцію, кислоти, кристалічна, біс[(е)-7-[4-(4-флуорфеніл)-6-ізопропіл-2-[метил(метилсульфоніл)аміно]піримідин-5-іл](3r,5s)-3,5-дигідроксигепт-6-енової
Формула / Реферат:
1. Кристалічна гідратована форма сполуки: сіль кальцію біс[(Е)-7-[4-(4-флуорфеніл)-6-ізопропіл-2-[метил(метилсульфоніл)аміно]піримідин-5-іл](3R,5S)-3,5-дигідроксигепт-6-енової кислоти] формули І І,яка має рентгенодифрактограму порошку з піками при 2-тета (2q) = 8,8, 13,1 та 21,5°.2. Кристалічна гідратована форма за п. 1, яка має рентгенодифрактограму...
Гідрат солі малеїнової кислоти 5-[4-[2-(n-метил-n-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону, спосіб його одержання та фармацевтична композиція

Номер патенту: 63980
Опубліковано: 16.02.2004
Автори: Лі Девід К., Блеклер Пол Девід Джеймс, Сейсс Майкл Джон
МПК: A61K 31/4439, A61P 3/10, C07D 417/12
Мітки: 5-[4-[2-(n-метил-n-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону, одержання, кислоти, композиція, гідрат, солі, фармацевтична, спосіб, малеїнової
Формула / Реферат:
1. Гідрат солі малеїнової кислоти 5-[4-[2-(N-метил-N-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діону, який відрізняється тим, щоі) містить воду в кількості від 0,2 до 1,1% мас., ііі) має інфрачервоний спектр, який включає піки при 764 і 579 см-1;і/абоiiі) має картину порошкової дифракції рентгенівських променів (РЛПД), яка по суті відповідна приведеній на фіг. II.2. Гідрат по п.1, в якому вміст води...
8-феніл-7,8-дигідропіридо[2,3-d]піримідин-7-они та їх застосування для виготовлення фармацевтичних препаратів

Номер патенту: 95444
Опубліковано: 10.08.2011
Автори: Невінс Нейса, Купер Ентоні Вільям Джеймс, Каллахан Джеймс Френсіс, Боем Джеффрі Ч., Лівіа Стефано, Нортон Бет А., Ван Зехонг
МПК: C07D 487/02, A61K 31/519, C07D 471/02, A61K 31/4745
Мітки: застосування, виготовлення, 8-феніл-7,8-дигідропіридо[2,3-d]піримідин-7-они, фармацевтичних, препаратів
Формула / Реферат:
1. Сполука, вибрана з групи, що...
Попередній патент: Спосіб отримання ливарних сплавів на основі цирконію або титану
Наступний патент: Пристрій для вимірювання концентрації газів
Випадковий патент: Горизонтальний твердомір ґрунту