Лікарська форма паліперидону для імплантування
Номер патенту: 115987
Опубліковано: 25.01.2018


















Формула / Реферат
1. Внутрішньом'язова ін'єктована депо-композиція уповільненого вивільнення, придатна для формування в організмі in situ твердого імплантата із вмістом лікарського засобу паліперидону і/або будь-якої його фармацевтично прийнятної солі в будь-якій комбінації, біосумісного співполімеру на основі молочної і гліколевої кислоти зі співвідношенням мономерів молочної кислоти до гліколевої приблизно 50:50 і розчинника диметилсульфоксиду (ДМСО), яка відрізняється тим, що біосумісний співполімер має молекулярну масу в діапазоні 31-40 кДа і характеристичну в'язкість в межах від 0,27-0,31 дл/г ±10 %.
2. Композиція за п. 1, в якій біосумісний співполімер піддається опроміненню гамма- або бета-частинками дозою в діапазоні 10-30 кГр, що вимірюється при температурі в діапазоні від -40 °С до +35 °С для коректування його молекулярної маси в межах 27-47 кДа і характеристичної в'язкості в діапазоні 0,27-0,31 дл/г ±10 %.
3. Композиція за п. 1 або 2, з наступним розподілом частинок препарату за розміром:
менше 10 % частинок розміром менше 10 мікронів;
менше 10 % частинок розміром більше 225 мікронів, і
значення d0,5 - в діапазоні 40-90 мікронів.
4. Композиція за будь-яким з пп. 1-3, в якій масове співвідношення лікарський препарат/(полімер+препарат) становить близько 33 %.
5. Композиція за будь-яким з пп. 1-4, в якій вміст препарату становить близько 13 % у ваговому співвідношенні від загального складу, а в'язкість розчину із вмістом полімеру і ДМСО знаходиться в діапазоні 1,5-2,5 Па·с.
6. Композиція за будь-яким з попередніх пунктів, що використовується для лікування шизофренії або біполярних розладів у людини.
7. Фармацевтичний набір, придатний для утворення в організмі in situ твердого імплантата, що містить заявлену в пп. 1-6 композицію, де лікарський препарат і біосумісний полімер містяться в першому контейнері, а розчинник - у другому, окремому, контейнері.
8. Фармацевтичний набір за п. 7, в якому принаймні один з контейнерів (або перший, або другий) являє собою шприц, флакон, спеціальний пристрій або картридж (одноразові або ні).
9. Спосіб виготовлення композиції відповідно до будь-якого з вищезгаданих пунктів 1-6, що включає етап створення біосумісного співполімеру з більш високою молекулярною масою, ніж необхідна для ін'єктованої внутрішньом'язово дено-композиції, з подальшою корекцією його молекулярної маси до діапазону 31-40 кДа і його характеристичної в'язкості до діапазону 0,27-0,31 дл/г гамма- або бета-випромінюванням дозами в діапазоні 10-30 кГр, що вимірюється при температурі від -40 °С до +35 °С.
10. Спосіб за п. 9, при якому доза опромінення полімеру, що вимірюється при температурі 8 °С, знаходиться в діапазоні 16-25 кГр.
11. Спосіб за п. 9 або 10, в якому біосумісний полімер з початковою молекулярною масою близько 50 кДа піддається опроміненню дозою близько 16 кГр з метою зменшення його молекулярної маси до значень в діапазоні 27-47 кДа.
12. Спосіб за п. 9 або 10, в якому біосумісний полімер з початковою молекулярною масою близько 54 кДа піддається опроміненню дозою близько 25 кГр з метою зменшення його молекулярної маси до значень в діапазоні між 31 і 40 кДа.
13. Спосіб за п. 9 або 10, в якому біосумісний полімер з початковою молекулярною масою близько 63 кДа піддається опроміненню дозою близько 30 кГр з метою зменшення його молекулярної маси до значень в діапазоні 31-40 кДа.
14. Спосіб початкового режиму дозування для введення внутрішньом'язової ін'єктованої депо-композиції уповільненого вивільнення за пп. 1-6 пацієнту, який потребує психіатричного лікування, що включає:
a) внутрішньом'язове введення пацієнту першої дози від 75 мг до 250 мг;
b) внутрішньом'язове введення пацієнту другої дози ін'єктованої депо-композиції з тривалим вивільненням між 24 і 35 днями лікування, рахуючи від дня попереднього введення;
c) введення подальшої дози внутрішньом'язової ін'єктованої депо-композиції з тривалим вивільненням, що становить від 75 мг до 250 мг, приблизно між 56 і 65 днями після введення вищезазначеної першої дози;
d) повторення кроку b) за необхідності отримати необхідну концентрацію препарату в плазмі крові перед початком введення препарату кожні 8 тижнів.
15. Спосіб за п. 14, де перша доза становить приблизно від 100 мг до 200 мг.
Текст
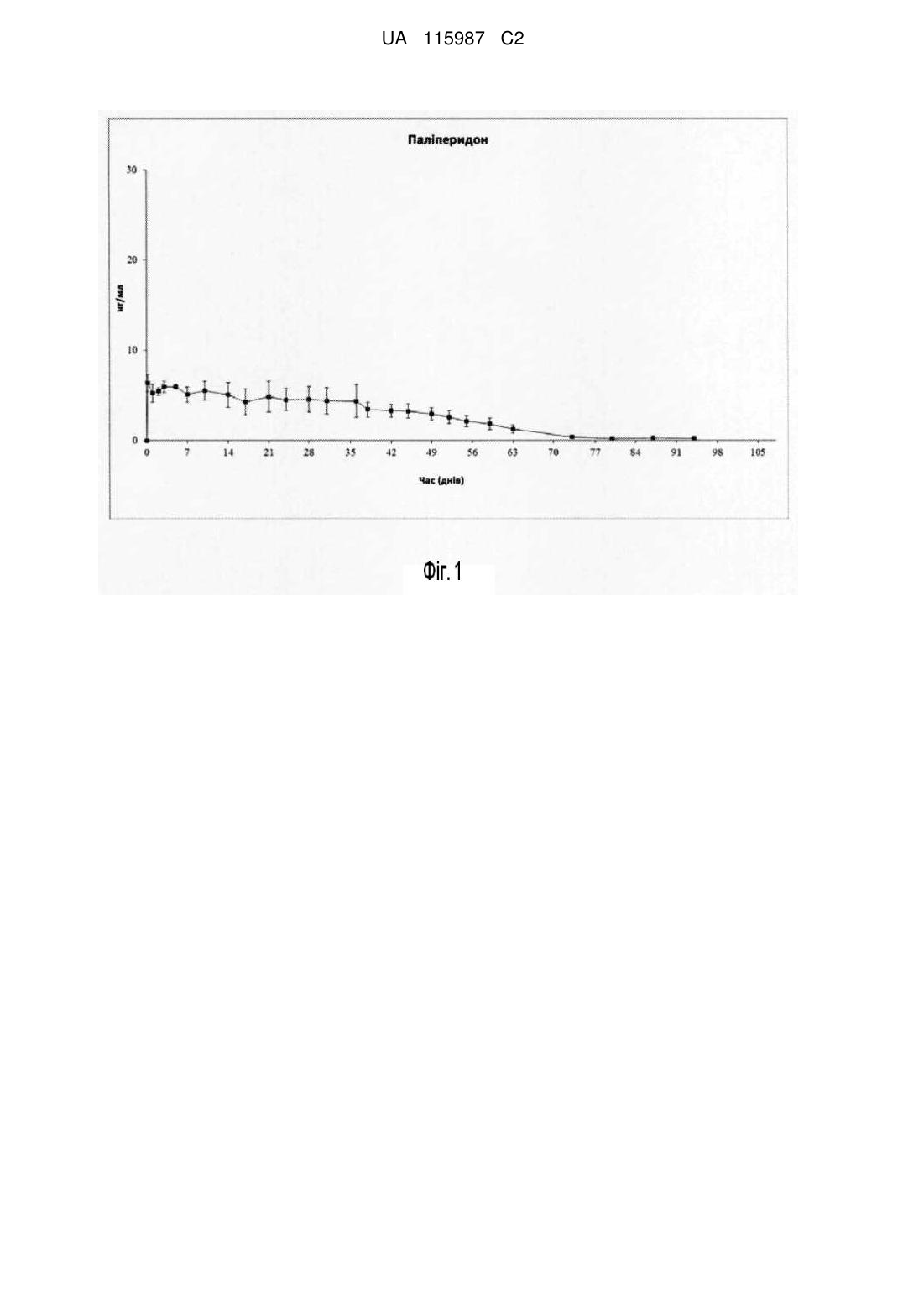
Реферат: Винахід стосується внутрішньом'язової депо-композиції для ін'єкцій, яка здатна формувати в організмі in situ твердий імплантат та складається з препарату, яким є паліперидон, та/або його фармацевтично прийнятні солі у будь-якій комбінації, біосумісного співполімеру на основі молочної і гліколевої кислот із співвідношенням мономерних форм молочної та гліколевої кислот в діапазоні від 50:50, а також розчинника DMSO (диметилсульфоксид), яка починає діяти негайно та характеризується постійним вивільненням препарату протягом щонайменше 8 тижнів і має фармакокінетичний профіль in vivo, що дозволяє вводити її один раз на 8 тижнів чи навіть довший період. UA 115987 C2 (12) UA 115987 C2 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 ОПИС ГАЛУЗЬ ТЕХНІЧНОГО ВИКОРИСТАННЯ Даний винахід стосується фармацевтичної композиції для внутрішньом’язових ін’єкцій, що містить лікарський препарат паліперидон та/або його солі, яка починає діяти негайно та здійснює вивільнення препарату безперервно протягом щонайменше 8 тижнів; фармакокінетичний профіль такої композиції in vivo дозволяє вводити її один раз на 8 тижнів, або навіть із довшими проміжками. Зокрема цей винахід стосується сумішей для утворення in situ ін’єкційних біорозкладаних імплантатів, що містять паліперидон. ПЕРЕДУМОВИ СТВОРЕННЯ ВИНАХОДУ Паліперидон є атиповим антипсихотичним засобом, що містить бендизоксазольну і піперидинову функціональні групи та діє як потужний антагоніст дофамінергічних рецепторів і селективний антагоніст серотонінових рецепторів. Однією з проблем, притаманних паліперидоновій терапії, є порушення деякими пацієнтами, хворими на шизофренію, терапевтичного режиму, особливо якщо він полягає в щоденному прийманні препарату, що призводить до нерегулярного або непостійного лікування та сприяє розвитку психотичної кризи. Крім того, внаслідок такого способу лікування спостерігаються високі перепади вмісту препарату в плазмі крові пацієнтів (вимірюються як різниця між Cmax і Cmin), які часто спричиняють зміни їхнього настрою. Таким чином, паліперидон є перспективним препаратом з точки зору включення його до складу систем пролонгованого вивільнення, завдяки яким забезпечення (лікування) пацієнтів препаратом протягом тривалого часу відбувається шляхом одноразового введення. Такі системи звільняють осіб, що опікуються пацієнтом, від необхідності дбати про щоденне лікування та дозволяють зменшити небажані коливання вмісту препарату в плазмі крові пацієнта. Іншими показаннями до застосування є маніакальний стан під час біполярного розладу та шизоафективний розлад; також можливим є позитивний ефект для пацієнтів із аутизмом, синдромом Аспергера та синдромом Турета. У багатьох хворих на ці психічні хвороби можливо досягти стабілізації симптомів за допомогою існуючих пероральних антипсихотичних засобів, проте, за оцінками дослідників, до 75 % таких пацієнтів зазнають труднощів у дотриманні щоденного режиму лікування, тобто мають низьку комплаєнтність. Недотримання режиму зазвичай призводить до погіршення симптомів, заниження відповіді на лікування, частих рецидивів і повторної госпіталізації, а також відсутності результату від реабілітаційної та психосоціальної терапії. Нещодавно було надано дозвіл на продаж паліперидону як першого антипсихотичного засобу для перорального застосування з тривалим вивільненням, яке досягається завдяки пероральній системі осмотично контрольованого вивільнення. Паліперидон з тривалим вивільненням (заявка на патент WO2006/17537) реалізується під торговельною назвою Invega Sustenna®, а його ненасичені похідні описані в заявці WO2008/128436. Триває розробка та інших лікарських форм паліперидону з тривалим вивільненням для перорального використання. Завдяки наявності вторинної гідроксильної групи паліперидон може використовуватися у вигляді попередника. У WO2009/15828 детально описані низькомолекулярні форми препаратівпопередників паліперидону, які підлягають гідролізу в шлунку. Таким чином, з огляду на наявні результати досліджень, розробка ін’єкційних депо паліперидону дуже тривалої дії є доцільною. Існує нагальна потреба у поліпшенні чинника комплаєнтності, зокрема стосовно лікування шизофренії. Розробка композицій цих препаратів у формі ін’єкційних депо для введення один раз на тиждень, чи навіть більш довготривалої дії, становитиме значний прогрес у забезпеченні постійного та рівномірного введення пацієнту ефективного лікарського засобу. EP2234617 описує сполуки-попередники паліперидону з естерним зв’язком, які забезпечують сталу концентрацію паліперидону в плазмі за умов введення один раз на місяць. Такий режим дозування дозволяє значно покращити комплаєнтність. Речовина паліперидону пальмітат була схвалена як атиповий антипсихотичний засіб для внутрішньом’язового введення один раз на місяць для лікування шизофренії та запобігання рецидивам її симптомів. Паліперидону пальмітат входить до складу лікарського засобу в субмікрокристалічній формі. Через обмеження абсорбції паліперидону пальмітату швидкістю його розчинення ця речовина демонструє кінетику вивільнення з постійною швидкістю («фліп-флоп»): уявний період напівелімінації обумовлений константою швидкості абсорбції. Додатково на уявну константу швидкості впливає об’єм введеного препарату. Було також виявлено, що найшвидше зростання початкової концентрації препарату в плазмі, що сприяє скорішому досягненню потенційно терапевтичних концентрацій, спостерігається після ін’єкції в дельтовидний м’яз. Таким чином, для сприяння швидкому досягненню терапевтичної концентрації паліперидону в крові пацієнта 1 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 рекомендовано вводити початкову дозу насичення паліперидону пальмітату в дельтовидний м’яз. Доза насичення має становити приблизно 100-150 мг-екв паліперидону у формі паліперидону пальмітату. Після введення першої, або краще другої дози насичення концентрація паліперидону в плазмі крові пацієнтів доводиться до рівноважної, для чого виконуються наступні ін’єкції препарату в дельтовидний або сідничний м’яз. Більш бажаними на цьому етапі є ін’єкції в сідничний м’яз. У US2009/163519 розглянуто відповідний режим дозування і введення для пальмітатних ефірів паліперидону довготривалої дії. Інша композиція-депо описана в міжнародній заявці WO2011/42453. Вказана специфікація описує фармацевтичну композицію для підшкірної ін’єкції, яка містить сполуку паліперидону. Ця композиція, зокрема, характеризується вивільненням паліперидону з негайним початком дії та тривалим часом вивільнення. Крім того, вказана специфікація стосується фармацевтичних композицій для підшкірних ін’єкцій, які містять сполуку паліперидону в певній концентрації. Нарешті, ще одна антипсихотична композиція-депо для ін’єкцій описана в міжнародній заявці WO2011/151355. Ця заявка присвячена композиції, яка може бути використана для введення в організм попередника антипсихотичного препарату рисперидону, подібно до паліперидону, у вигляді ін’єкційного біорозкладаного імплантату з тривалим вивільненням, який формується in situ і забезпечує терапевтичні рівні препарату в плазмі з першого дня. Композиція являє собою суспензію препарату у розчині біорозкладаного і біосумісного кополімеру або кополімерів, з використання розчинників на основі води, і вводиться у формі рідини. Коли композиція контактує з рідинами організму, полімерна матриця, яка утримує препарат, ущільнюється, формуючи щільний або напівщільний імплантат, який поступово вивільнює препарат. Виготовлення паліперидону в лікарській формі імплантату описане в заявці WO2011/151355. Проте після аналізу даних, одержаних в ході клінічних випробувань цього лікарського засобу, було виявлено, що абсорбція паліперидону після таких ін’єкцій відбувається у значно складніший спосіб, ніж очікувалося спочатку для препарату, який починає діяти негайно та безперервно вивільнюється протягом щонайменше 8 тижнів. Також було виявлено, що досягнення потенційно терапевтичного рівня паліперидону в плазмі крові пацієнтів залежить від молекулярної маси біосумісної полімерної речовини, утвореної з молочної та гліколевої кислот, а саме від співвідношення мономерів-залишків лактату та гліколату. У зв’язку зі складністю завдання щодо забезпечення оптимального профілю «концентрація в плазмі-час» під час лікування пацієнтів паліперидоном існує потреба в розробці придатного для досягнення цієї мети режиму дозування препарату для пацієнтів, яким необхідно лікування. СТИСЛИЙ ОГЛЯД ВИНАХОДУ Таким чином, композиції, описані в огляді стану проблеми, не відповідають наявним потребам у композиціях паліперидону, їхнім наборам та режимам лікування психіатричних розладів, і все ще існує потреба у композиціях та засобах, які б забезпечували контрольоване і постійне вивільнення препарату впродовж пролонгованого періоду часу, упродовж щонайменше 8 тижнів без потреби в супутньому прийомі препаратів або початкових дозах рисперидону та/або паліперидону. Вирішення цієї проблеми забезпечується композицією-депо для внутрішньом’язових ін’єкцій, яка здатна формувати in situ твердий імплантат в організмі та складається з препарату, яким є паліперидон, біосумісного кополімеру на основі молочної і гліколевої кислот із співвідношенням мономерних форм молочної та гліколевої кислот в діапазоні від 45:55 до 55:45 (найкраще близько 50:50), а також розчинника DMSO (диметилсульфоксид), і яка починає діяти негайно та характеризується постійним вивільненням препарату протягом щонайменше 8 тижнів і має фармакокінетичний профіль in vivo без значного початкового сплеску вивільнення препарату, а також відрізняється тим, що біосумісний кополімер має молекулярну масу від 27 до 47 кДа (найкраще 31-43 кДа, і навіть краще 31-40 кДа) і має характеристичну в’язкість 0,27-0,31 дл/г ± 10 %. Одним з аспектів даного винаходу є пропонований режим дозування для введення паліперидону пацієнту, який має психічний розлад і потребує лікування. Цей режим полягає у внутрішньом’язовому введенні першої дози паліперидону в кількості приблизно 75-250 мг у формі композиції з пролонгованим вивільненням в перший день лікування; наступні дози паліперидону в кількості приблизно 75-250 мг у формі композиції з пролонгованим вивільненням вводяться внутрішньом’язово приблизно в 56-65 день лікування. ДЕТАЛЬНИЙ ОПИС ВИНАХОДУ Склад даного винаходу охоплює, принаймні, полімер або полімерну матрицю, розчинник і лікарський препарат. Важливим аспектом цього винаходу є композиція-депо для внутрішньом’язових ін’єкцій, яка 2 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 здатна формувати in situ твердий імплантат в організмі та складається з препарату, яким є паліперидон або будь-яка його фармацевтично прийнятна сіль у будь-якій комбінації, біосумісного кополімеру на основі молочної і гліколевої кислот із співвідношенням мономерних форм молочної та гліколевої кислот в діапазоні від 45:55 до 55:45 (найкраще близько 50:50), а також розчинника DMSO (диметилсульфоксид), яка починає діяти негайно та характеризується постійним вивільненням препарату протягом щонайменше 8 тижнів і має фармакокінетичний профіль in vivo, що дозволяє вводити її приблизно через 56-65 днів після попередньої ін’єкції, а також відрізняється тим, що біосумісний кополімер має молекулярну масу від 27 до 47 кДа (найкраще 31-43 кДа, і навіть краще 31-40 кДа) і має характеристичну в’язкість 0,27-0,31 дл/г ± 10 %. В рамках реалізації цього винаходу рекомендовано провести опромінення біосумісного кополімеру гамма- або бета-випромінюванням дозою 10-30 кГр ±10 %, виміряною за температури від -40 ºC до +35 ºC, для доведення його молекулярної маси до значення у діапазоні 27-47 кДа (найкраще 31-43 кДа, і навіть краще 31-40 кДа), а характеристичної в’язкості до 0,27-0,31 дл/г ± 10 %. Найбільш рекомендованою дозою опромінення полімеру є 15-25 кГр ±10 %, виміряна за температури 8 ºC. Рекомендований розподіл частинок препарату за розміром у даній композиції є таким: - менше 10 % частинок, менших за 10 мікронів; - менше 10 % частинок, більших за 225 мікронів, краще - більших за 200 мікронів; - значення d0,5 в діапазоні 40-90 мікронів. Якщо не вказано інше, розподіл частинок за розміром був визначений за методом розсіювання світла з використанням лазерного дифрактора у вологому режимі. Відомо, що результати визначення розподілу частинок за розміром можуть змінюватися залежно від обробки матеріалу, наприклад, з використанням поверхнево активних речовин у високій концентрації та/або енергії сильних взаємодій (вихровий рух, ультразвук і т. д.). Якщо не згадується інше, препарат не підлягає обробці, і зразки утворюються шляхом прямого додавання в резервуар за помірного перемішування (2000-3500 об./хв.). Методика, використана в даному винаході для визначення розподілу частинок препарату за розміром, найбільш достовірно відтворює поведінку порошку препарату в ін’єкційній лікарській формі, що описується в цьому документі, порівняно з іншими методами, які передбачають дію на зразок енергії сильних взаємодій та/або використання поверхнево активних речовин у високій концентрації для приготування зразків з метою досягнення високого ступеня дезагрегації порошку, неможливого за умов ресуспендування лікарського засобу вручну. Відповідно до іншої реалізації винаходу, масове співвідношення «препарат/(полімер+препарат)» складає близько 33 %, масова частка препарату в загальній масі лікарської форми становить близько 13 %, а в’язкість розчину полімеру та DMSO знаходиться в діапазоні 1,2-2,5 Па·с (найкраще 1,5-2,1 Па·с, і навіть краще 1,7-1,8 Па·с) В іншій реалізації під час формування композиції препарат частково суспендують з коефіцієнтом розчинення препарату в DMSO нижче 10 мг/мл. Ще одна реалізація передбачає, що композиція є стерильною. Відповідно до іншого аспекту винаходу, композиція є стерильною та придатною для лікування шизофренії та біполярних розладів в організмі людини. Відповідно до іншого аспекту, винахід надається у вигляді фармацевтичного набору, придатного для формування в організмі in situ біорозкладаного імплантату, що містить заявлену композицію, до складу якого входить перший контейнер з препаратом і біорозкладаним полімером і другий, окремий контейнер з розчинником. Бажано, щоб щонайменше один із цих двох контейнерів був шприцом, флаконом, пристроєм чи картриджем, одноразового чи багаторазового використання; найбільш бажано, щоб обидва контейнери були одноразовими шприцами. Відповідно до іншого аспекту, у винаході пропонується метод виготовлення вказаної композиції, який складається з етапу одержання біосумісного кополімеру з вищою початковою полімерною молекулярною масою, ніж необхідно для утворення внутрішньом’язової композиціїдепо, а потім доведення його молекулярної маси до значення у діапазоні 27-47 кДа (найкраще 31-43 кДа, і навіть краще 31-40 кДа), а характеристичної в’язкості до 0,27-0,31 дл/г шляхом опромінення його гамма- або бета-випромінюванням дозою 10-30 кГр ±10 %, виміряною за температури від -40 ºC до +35 ºC ± 10 %. Якщо біосумісний полімер має початкову молекулярну масу близько 50 кДа, рекомендовано опромінювати його дозою приблизно 16 кГр, виміряною за температури 8 ºC, з метою зменшення його молекулярної маси до 27-47 кДа 25 (найкраще 31-43 кДа, і навіть краще 31-40 3 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 кДа). Якщо біосумісний полімер має початкову молекулярну масу близько 54 кДа, рекомендовано опромінювати його дозою приблизно 25 кГр, виміряною за температури 8 ºC, з метою зменшення його молекулярної маси до 31-43 кДа. Якщо біосумісний полімер має початкову молекулярну масу близько 63 кДа, рекомендовано опромінювати його дозою приблизно 30 кГр, виміряною за температури 8 ºC, з метою зменшення його молекулярної маси до 30-43 кДа (найкраще 31-36 кДа). Відповідно до іншого аспекту, у даному винаході пропонується режим дозування для введення винайденої ін’єкційної внутрішньом’язової композиції-депо пацієнту, який потребує лікування від психічного розладу. Цей режим полягає в наступному: a) внутрішньом’язове введення пацієнту першої дози ін’єкційної композиції-депо в кількості приблизно 75-250 мг у період між 24 і 35 днем від дня попереднього введення; b) введення наступної дози ін’єкційної композиції-депо в кількості приблизно 75-250 мг приблизно в 56-65 день після введення вищевказаної першої дози; c) за потреби повторення етапу b). Рекомендовано, щоб вищевказана перша доза складала приблизно 100-200 мг та була еквівалентною іншим дозам. Етапи a) і b) рекомендовано повторювати необхідну кількість разів. Бажано, щоб полімер або полімерна матриця були біосумісною і біорозкладаною полімерною матрицею. З метою запобігання будь-яких значних пошкоджень для організму після введення, полімери повинні бути біосумісними, нетоксичними для організму людини, не канцерогенними, і не спричиняти значного запалення тканин. Полімери повинні бути біорозкладаними для забезпечення природного розкладання у метаболічних процесах організму, так, щоб вони легко виводилися і не акумулювалися в організмі. Полімерні матриці, які мають перевагу для практичного застосування даного винаходу, обираються з кополімерів полімолочної і полігліколевої кислот з термінальною карбоксильною групою, які змішані у пропорціях від 45:55 до 55:45, найкраще близько 50:50, з середньої молекулярною масою в діапазоні 27-47 кДа, найкраще 31-43 кДа, і навіть краще 31-40 кДа, і 5 характеристичною в’язкістю в діапазоні 0,27-0,31 дл/г ± 10 %. Очевидно, що можливе використання комерційного полімеру з необхідною молекулярною масою. Однак нами було встановлено, що основний діапазон його молекулярних мас становить 31-43, а найкраще 31-40 кДа. Крім того, нами було встановлено власними засобами за допомогою спеціальних експериментів, що молекулярна маса полімеру може бути змінена шляхом опромінення дозою радіації від 10 до 30 кГр за температури від -40 ºC до +35 ºC, що для спеціаліста не випливає явним чином з урахуванням поточного рівня техніки (див. фіг. 7). Наприклад, молекулярна маса комерційно доступного полімеру на даний момент становить у середньому 50 кДа. Ми визначили метод для змінювання цієї молекулярної маси шляхом опромінення полімеру визначеною дозою радіації, яка може бути розрахована попередньо. В контрольованихумовах можливо отримати математичну модель, що визначає можливість зниження молекулярної маси полімеру із зростанням дози опромінення. Отже, для прикладу: - якщо необхідно використати полімер PLGA з молекулярною масою між 31 та 43 кДа і значенням характеристичної в’язкості в діапазоні 0,27-0,31 дл/г ± 10 %, а наявний полімер має початкову молекулярну масу в середньому 54 кДа, нами було визначено, що для зниження його молекулярної маси до значення у вищезгаданому діапазоні 31-43 кДа (найкраще 37-43 кДа) необхідна доза радіації у 25 кГр; - якщо необхідно використати полімер PLGA з молекулярною масою між 31 та 43 кДа (найкраще 31-40 кДа) і значенням характеристичної в’язкості в діапазоні 0,27-0,31 дл/г ± 10 %, а наявний полімер має початкову молекулярну масу в середньому 50 кДа, нами було визначено, що для зниження його молекулярної маси до значення у вищезгаданому діапазоні 31-43 кДа (найкраще 37-43 кДа ± 10 %) необхідна доза радіації у 16 кГр; - якщо необхідно використати полімер PLGA з молекулярною масою між 31 та 43 кДа і значенням характеристичної в’язкості в діапазоні 0,27-0,31 дл/г ± 10 %, а наявний полімер має початкову молекулярну масу в середньому 50 кДа, нами було визначено, що для зниження його молекулярної маси до значення у вищезгаданому діапазоні 31-43 кДа (найкраще 31-37 кДа) необхідна доза радіації у 25 кГр; - якщо необхідно використати полімер PLGA з молекулярною масою між 31 та 43 кДа (найкраще 31-40 кДа) і значенням характеристичної в’язкості у діапазоні 0,27-0,31 дл/г ± 10 %, а наявний полімер має початкову молекулярну масу в середньому 38 кДа, нами було визначено, що немає необхідності використовувати будь-яку дозу радіації; 4 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 - якщо необхідно використати полімер PLGA з молекулярною масою між 31 та 43 кДа (найкраще 31-40 кДа) і значенням характеристичної в’язкості в діапазоні 0,27-0,31 дл/г ± 10 %, а наявний полімер має початкову молекулярну масу в середньому 63 кДа, нами було визначено, що для зниження його молекулярної маси до значення у вищезгаданому діапазоні 31-43 кДа (найкраще 31-40 кДа, і навіть краще 31-36 кДа) необхідна доза радіації у 30 кГр. У цих експериментальних випробуваннях температура для полімеру під час опромінення становила приблизно 8 ºC. Однак можливе використання інших температур, зокрема, нижче 35 ºC, або нижче 25 ºC, хоча при цьому взаємозв’язок між дозою радіації і отриманою молекулярною масою може змінюватися. Вказана процедура особливо підходить для виробництва композицій, описаних у даному винаході. Крім того, наповнення шприца щільним полімером становить серйозну проблему при виробництві ін’єкційних композицій. Полімер, який виготовляється як нестерильний продукт, потребує стерилізації для отримання композиції, яку можна вводити людині. Вірогідно, найкращим вирішенням цієї технічної проблеми може бути стерилізація полімеру за допомогою гамма- або бетаопромінення. Опромінення становить складну проблему при роботі з біорозкладаними полімерами, оскільки опромінення може розщеплювати ланцюги до більш коротких фрагментів. Керування молекулярною масою полімеру є критичним параметром для контролю кінцевих характеристик продукту після процесу стерилізації. Як вказано вище, зниження розміру ланцюгу за допомогою опромінення може бути математично модельовано і контрольовано з метою передбачення кінцевої молекулярної маси полімеру, що використовується як сировина і має молекулярну масу вище, ніж необхідно. Таким чином, якщо визначено пакувальну вагу полімеру для наповнення контейнеру (наприклад, пакувальна вага полімеру в шприці) та біологічного навантаження, присутнього у полімері як сировинному матеріалі, обирається необхідна для стерилізації полімеру доза опромінення для заданої пакувальної ваги (як визначено нормами ISO 11137). Після цього можна використати математичну модель, яка описує втрату молекулярної маси для певного полімеру залежно від дози опромінення, для визначення початкової молекулярної маси полімеру, що використовується як сировина, для отримання, після процесу опромінення, полімеру з бажаною кінцевою молекулярною масою для лікарської форми. Оскільки доступність полімеру з конкретною молекулярною масою може бути дещо обмеженою, можливо, за альтернативу, обрати доступний полімер з молекулярною масою, вищою за ту, яка потрібна відповідно до визначеної дози опромінення, і скоригувати дозу опромінення у бік більших значень для отримання стерильного полімеру з необхідною молекулярною масою. Концентрація полімерного компоненту у композиціях, відповідно до винаходу, знаходиться у діапазоні24-50 % (вираженому як відсоток ваги полімеру до загальної ваги композиції), і, більш бажано, 25-27 %. У даному винаході, при визначенні специфікації, термін «внутрішня» або «характеристична» в’язкість (ηinh) полімеру визначається як відношення натурального логарифму відносної в’язкості ηr до масової концентрації полімеру c, тобто: ηinh= (lnηr)/c а відносна в’язкість (ηr) є відношенням в’язкості розчину η до в’язкості розчинника ηs, тобто: ηr= η/ ηs Якщо не встановлено інше, всюди у даній специфікації слід розуміти значення характеристичної в’язкості та молекулярної маси у тому значенні, як вони вимірюються у прикладі 1. Значення характеристичної в’язкості у даній специфікації, відповідно до загальноприйнятого у галузі, є непрямим показником молекулярної маси полімеру. В цьому розумінні, зменшення характеристичної в’язкості полімеру, визначене для певної концентрації і заданого розчинника, з однаковим мономерним складом і термінальними кінцевими групами, є показником зменшення молекулярної маси полімеру (IUPAC. Basic definitions of terms relating to polymers 1974. Pure Appl. Chem. 40, 477-491 (1974). Мають переваги такі розчинники, які є нетоксичними, біосумісними і підходять для парентерального введення. Розчинники, для яких припускається можлива токсичність, не повинні використовуватися для ін’єкцій будь-яких матеріалів у живі організми. Більші переваги мають певні розчинники, які є біосумісними і не спричиняють жодних важких подразнень тканин або некрозів у місці ін’єкції. Таким чином, розчинники повинні мати, переважно, клас ІІ або ІІІ, або, що більш прийнятно, клас ІІІ відповідно до керівних настанов ICH. Для формування імплантату in-situ, розчинник повинен, переважно, швидко дифундувати з полімерного розчину у навколишні тканини, при попаданні у фізіологічні рідини. Таким чином, як розчинник переваги має DMSO. Лікарський препарат - це, переважно, паліперидон та/або його фармацевтично прийнятні 5 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 солі або їхні комбінації. Бажано, щоб цей препарат частково суспендувався в розчиннику. «Часткове суспендування» означає, що бажана розчинність препарату в розчиннику DMSO має бути нижче 10 мг/мл під час утворення композиції або імплантату, і нижче 10 мг/мл в загальному об’ємі DMSO (визначено за температури 25 ºC). Перевагою такої низької розчинності є те, що початковий сплеск концентрації препарату, під час дифузії розчинника до зовнішнього водного середовища, значно зменшується. Крім того, у кінцевих композиціях, відповідно до винаходу, препарат знаходиться переважно у концентраціях між 4 та 16 мас. %, виражених як відсоток препарату по відношенню до загальної маси композиції. Більш бажано, щоб вміст препарату знаходився в діапазоні між 7 і 15 %, і найбільш бажано - близько 13 % по відношенню до загальної маси композиції. Одним з факторів, що має вплив на початкове вивільнення композиції у даному винаході, є в’язкість полімерного розчину. «Полімерний розчин», який визначається як комбінація полімерної матриці та розчинника, у якому вона розчинена, має рекомендовану в’язкість у діапазоні 1,5-2,5 Па·с, більш бажано 1,5-2,3 Па·с, і ще більш бажано 1,5-2,1 Па·с ± 10 %. Другим фактором, що має вплив на контроль початкового вивільнення композицій винаходу, є молекулярна маса біосумісного кополімеру, яка повинна знаходитися між 27 і 47, більш бажано між 31 і 43 кДа, і ще більш бажано між 31 і 40 кДа. Належний баланс у цій композиції між розчинністю препарату в розчиннику та молекулярною масою полімеру в імплантаті (яка обумовлює процес осадження полімеру та кінцеві структурні властивості імплантату) дозволяє композиції після внутрішньом’язової ін’єкції вивільняти паліперидон шляхом дифузії в фазу розчинника в обмеженій кількості. Одразу після введення композиції у внутрішньом’язову тканину, DMSO швидко розчиняється в навколишньому водному середовищі. Відносне збільшення концентрації полімеру в DMSO вище значення розчинності полімеру в цьому розчиннику призводить до утворення преципітату полімеру, який утримує в собі не розчинений у розчиннику паліперидон. Молекулярна маса полімеру має величезний вплив на цьому критичному етапі, оскільки надто низькомолекулярні ланцюги характеризуються довшим часом осадження порівняно з ланцюгами, молекулярна маса яких лежить у відповідних межах. Така затримка осадження призводить до посилення взаємодії препарату з навколишніми рідинами, у які він вивільнюється. Таким чином, низька молекулярна маса ланцюгів може спричинити надмірне вивільнення паліперидону після ін’єкції та потенційне досягнення токсичноїконцентрації препарату в плазмі в перші дні після введення й/або утворення композиції, яка не здатна забезпечити вивільнення препарату протягом мінімального 8-тижневого інтервалу між ін’єкціями. Молекулярна маса полімеру також може впливати на вивільнення препарату із введеного внутрішньом’язово імплантату після дифузії розчинника та осадження полімеру. Молекулярна маса в указаному діапазоні не здатна підтримувати відповідну швидкість вивільнення паліперидону шляхом дифузії. Крім того, більш високомолекулярні ланцюги потребують довшого часу для гідролізу у внутрішньом’язовій тканині, внаслідок якого утворюються розчинні фракції та вивільнюється препарат, затриманий у полімерній матриці. Надмірний залишковий вміст препарату, який може бути вивільнений, може спричинити одержання небажано високої концентрації активної речовини в плазмі або такої концентрації в плазмі через 60 днів після введення, яка може певним чином взаємодіяти з наступною дозою, оскільки дана лікарська форма призначена для багаторазового 20 введення людині з інтервалами у 8 тижнів чи більше. Вираз «близько 50:50», використаний у цьому описі, стосується співвідношення мономерівзалишків лактату та гліколату в біосумісному кополімері, утвореного з молочної та гліколевої кислот. В контексті винаходу він застосовується до показника співвідношення мономерів, виміряного зі стандартною технічною похибкою ± 10 %. Важливим аспектом цього винаходу є композиція-депо для внутрішньом’язових ін’єкцій, яка здатна формувати in situ твердий імплантат в організмі та складається з препарату, яким є паліперидон або будь-яка його фармацевтично прийнятна сіль у будь-якій комбінації, біосумісного кополімеру на основі молочної і гліколевої кислот із співвідношенням мономерних форм молочної та гліколевої кислот в діапазоні від 45:55 до 55:45 (найкраще близько 50:50), а також розчинника DMSO (диметилсульфоксид), яка починає діяти негайно та характеризується постійним вивільненням препарату протягом щонайменше 8 тижнів і має фармакокінетичний профіль in vivo, що дозволяє вводити її приблизно через 56-65 днів після попередньої ін’єкції, а також відрізняється тим, що біосумісний кополімер має молекулярну масу від 31 до 43 кДа (найкраще 31-40 кДа) і має характеристичну в’язкість 0,27-0,31 дл/г. У названих раніше композиціях опромінення полімеру відбувається за температури, нижчої за 35 ºC, найкраще нижче за 25 ºC, і навіть краще нижче за 8 ºC. У даній композиції рекомендований розподіл частинок препарату за розміром є 10 таким: 6 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 - менше 10 % частинок, менших за 10 мікронів; - менше 10 % частинок, більших за 200 мікронів, краще більших за 225 мікронів; - значення d0,5 в діапазоні 40-90 мікронів. Відповідно до реалізації винаходу, масове співвідношення «препарат/(полімер+препарат)» складає близько 33 %, масова частка препарату в загальній масі лікарської форми становить близько 13 %, а в’язкість розчину полімеру та DMSO знаходиться в діапазоні 1,5-2,5 Па·с (найкраще 1,5-2,1 Па·с, і навіть краще 1,7-1,8 Па·с). Відповідно до іншої реалізації, під час формування композиції препарат частково суспендують з коефіцієнтом розчинення препарату в DMSO нижче 10 мг/мл. Відповідно до ще однієї реалізації, композиція є стерильною та придатною для лікування шизофренії та біполярних розладів в організмі людини. Відповідно до іншого аспекту, винахід надається у вигляді фармацевтичного набору, придатного для формування в організмі in situ біорозкладаного імплантату, що містить заявлену композицію, до складу якого входить перший контейнер з препаратом і біорозкладаним полімером і другий, окремий контейнер з розчинником. Бажано, щоб щонайменше один із цих двох контейнерів був шприцом, флаконом, пристроєм чи картриджем, одноразового чи багаторазового використання; найбільш бажано, щоб обидва контейнери були одноразовими шприцами. Цей аспект винаходу передбачає створення набору, що складається з першого контейнера, яким бажано є шприц, флакон, пристрій або картридж, одноразового чи багаторазового використання, що містить полімер у твердій формі, наприклад, PLGA (кополімер молочної та гліколевої кислот), і препарат у відповідній кількості, і другого контейнера, бажано також шприца, флакона, пристрою чи картриджа, одноразового чи багаторазового використання, що містить розчинник, який змішується з водою. За необхідності вміст обох контейнерів об’єднують, наприклад, через з’єднувач, або за допомогою шприців з ніпельномуфтовими фланцями, і змішують один з одним для утворення композиції відповідно до винаходу, наприклад, прямим і зворотним рухом поршнів у шприцах. Ілюстрація до переважного способу реалізації наведена на фіг. 5 (шприці з’єднані через перехідний пристрій) та на фіг. 6 (шприці об’єднані через пряме різьбове з’єднання). Відповідно до іншого аспекту, у даному винаході пропонується метод виготовлення заявленої композиції, який складається з етапу одержання біосумісного кополімеру з вищою полімерною молекулярною масою, ніж необхідно для утворення внутрішньом’язової композиціїдепо, а потім доведення його молекулярної маси до діапазону 31-43 кДа (найкраще 31-40 кДа), а характеристичної в’язкості до 0,27-0,31 дл/г шляхом опромінення його гамма- або бетавипромінюванням дозою 10-30 кГр, виміряною за температури від -40 ºC до +35 ºC. Якщо біосумісний полімер має початкову молекулярну масу близько 54 кДа, рекомендовано опромінювати його дозою приблизно 25 кГр з метою зменшення його молекулярної маси до 3143 кДа (найкраще 37-43 кДа). Якщо біосумісний полімер має початкову молекулярну масу близько 50 кДа, рекомендовано опромінювати його дозою приблизно 16 кГр з метою зменшення його молекулярної маси до 3543 кДа (найкраще 37-43 кДа). Якщо біосумісний полімер має початкову молекулярну масу близько 50 кДа, рекомендовано опромінювати його дозою приблизно 25 кГр з метою зменшення його молекулярної маси до 3137 кДа. Якщо біосумісний полімер має початкову молекулярну масу близько 63 кДа, рекомендовано опромінювати його дозою приблизно 30 кГр з метою зменшення його молекулярної маси до 3143 кДа (найкраще 30-36 кДа). Відповідно до іншого аспекту, даний винахід пропонує режим дозування для введення винайденої ін’єкційної внутрішньом’язової композиції-депо пацієнту, який потребує лікування від психічного розладу. Цей режим полягає в наступному: a) внутрішньом’язове введення пацієнту першої дози ін’єкційної композиції-депо в кількості приблизно 75-250 мг у період між 24 і 35 днем від дня попереднього введення; b) введення наступної дози ін’єкційної композиції-депо в кількості приблизно 75-250 мг приблизно в 56-65 день після введення вищевказаної першої дози; c) за потреби повторення етапу b). Рекомендовано, щоб вищевказана перша доза складала приблизно 100-200 мг та 20 була еквівалентною іншим дозам. Рекомендований спосіб реалізації передбачає, що композиція-депо в стані готового лікарського засобу є стерильною. В рамках іншої рекомендованої реалізації біосумісний полімер стерилізують опроміненням перед процесом асептичного наповнення, бажана доза становить 15-30 кГр. 7 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 Стосовно даного винаходу, без обмежень і в зв’язку з наведеними прикладами, всі технічні параметри передбачають стандартну технічну похибку ± 10 %. У контексті даного винаходу, без обмежень і в зв’язку з наведеними прикладами, важливо навести пояснення стосовно можливої необхідності використання стартового режиму з метою прискореного досягнення бажаних рівнів препарату в плазмі до початку 8-тижневого режиму дозування. Цей стартовий режим може бути, окрім іншого, таким, як описано в пункті 15 або наведено нижче. Перше внутрішньом’язове введення композиції у день 0 у дозі від 75 до 250 мг, після якого друге введення між 5-м і 10-м днями у дозі в діапазоні від 75 до 250 мг, після якого третє введення між 28-м і 35-м днями після першого введення, з дозою у діапазоні 75-250 мг, після якого послідовні дози композиції один раз на 8 тижнів. - Перше внутрішньом’язове введення композиції у день 0 у дозі від 75 до 250 мг, після якого друге введення між 28-м і 35-м днями у дозі в діапазоні від 75 до 250 мг, після якого третє введення між 56-м і 65-м днями після першого введення, з дозою у діапазоні 75-250 мг, після якого послідовні дози композиції один раз на 8 тижнів. - Будь-які інші комбінації дозувань та інтервалів, які потрібні для отримання необхідних рівнів препарату в плазмі перед початком введень 1 раз на 8 тижнів. КОРОТКИЙ ОПИС РИСУНКІВ Фіг. 1. Профіль концентрацій паліперидону в організмі собаки після введення лікарської форми паліперидону, описаної в прикладі 1, собакам породи бігль (n = 3). Доза дорівнює 2,5 мг/кг. Результати виражені у вигляді залежності концентрації паліперидону в плазмі (нг/мл) від часу. У таблиці вказана площа під кривою (ППК) залежності концентрації паліперидону в плазмі від часу. Включені загальна ППК і ППК в залежності від трьох різних часових проміжків. Одиниця вимірювання значень - год*нг/мл. Фіг. 2. Профіль концентрацій паліперидону в організмі собаки після введення лікарської форми паліперидону, описаної в прикладі 2, двом когортам собак породи бігль (n = 6у кожній когорті). Дози дорівнювали 2,5 і 5 мг/кг. Результати виражені у вигляді залежності концентрації паліперидону в плазмі (нг/мл) від часу. У таблиці вказана площа під кривою (ППК) залежності концентрації паліперидону в плазмі від часу. Включені загальна ППК і ППК в залежності від трьох різних часових проміжків. Одиниця вимірювання значень - год*нг/мл. Фіг. 3. Профіль концентрацій паліперидону в організмі собаки після введення лікарської форми паліперидону, описаної в прикладі 3, собакам породи бігль (n = 3). Доза дорівнює 7,5 мг/кг. Результати виражені у вигляді залежності концентрації паліперидону в плазмі (нг/мл) від часу. У таблиці вказана площа під кривою (ППК) залежності концентрації паліперидону в плазмі від часу. Включені загальна ППК і ППК в залежності від трьох різних часових проміжків. Одиниця вимірювання значень - год*нг/мл. Фіг. 4. Профіль концентрацій паліперидону в організмі собаки після введення лікарської форми паліперидону, описаної в прикладі 3, собакам породи бігль (n = 3). Доза дорівнює 2,5 мг/кг. Результати виражені у вигляді залежності концентрації паліперидону в плазмі (нг/мл) від часу. У таблиці вказана площа під кривою (ППК) залежності концентрації паліперидону в плазмі від часу. Включені загальна ППК і ППК в залежності від трьох різних часових проміжків. Одиниця вимірювання значень - год*нг/мл. Фіг. 5. Креслення набору, придатного для приготування композицій паліперидону, який складається з двох шприців з ніпелями, поєднаних перехідним пристроєм. Полімер+паліперидон містяться в одному шприці, а DMSO - в другому. Фіг. 6. Креслення набору, придатного для приготування композицій паліперидону, який складається з шприца з муфтою, з’єднаного зі шприцом з ніпелем. Полімер+паліперидон можуть міститися в одному шприці, а DMSO - в другому. Бажано, щоб шприц з муфтою містив полімер+паліперидон в твердому стані, а шприц з ніпелем був заповнений DMSO. Фіг. 7. Відсоток втрати молекулярної маси в експериментальній установці. Молекулярна маса полімеру може бути змінена шляхом його опромінення визначеною дозою радіації. У таблиці наведені відсотки втрати молекулярної маси полімеру в залежності від дози радіації. ПРИКЛАДИ Наступні приклади надані задля ілюстрації даного винаходу і не повинні розглядатися як такі, що його обмежують. У контексті даного винаходу, без обмеження і у зв’язку з наведеними прикладами in vivo, термін «початковий сплеск» або початкове вивільнення означає зростання концентрацій паліперидону в плазмі крові з моменту ін’єкції до третього дня після введення. Аналогічно, «адекватним профілем концентрації препарату в плазмі» вважають такий, який не перевищує 8 UA 115987 C2 5 10 15 45 % від ППК для паліперидону, яка спостерігається між моментом ін’єкції та 21 добою, між 35 % і 45 % від ППК для паліперидону, яка спостерігається від 21 доби до 49 доби, і не перевищує 35 % від ППК для паліперидону, яка спостерігається після 49 доби. Вказані відсотки відображають адекватний баланс між різними періодами, під час яких паліперидон вивільнюється з імплантату, необхідний для одержання композиції для введення кожні 8 тижнів або кожні 60 днів з можливістю забезпечення терапевтичних концентрацій паліперидону в плазмі крові людини з моменту першого дня ін’єкції, з можливістю отримання бажаних середніх концентрацій паліперидону в плазмі впродовж періоду між ін’єкціями, а також зі зменшеним відношенням максимальних і мінімальних концентрацій паліперидону в плазмі, які можуть спричинити токсичність або недостатню ефективність. Також у контексті даного винаходу, без обмеження і у зв’язку з наведеними прикладами, прийнятними концентраціями паліперидону в плазмі під час початкового сплескового вивільнення є такі, що не перевищують 75 нг/мл у собак породи бігль за введеної дози паліперидону 2,5 мг/кг. Приклад 1. Композиція-депо з Resomer® 503 В даному прикладі було створено наступну композицію. Шприц з муфтою 2,25 мл Шприц з ніпелем 2,25 мл 20 25 30 35 40 45 50 Інгредієнт Кополімер молочної та гліколевої кислот (з термінальною N-групою) з 50 % вмістом кожного органічного мономеру та молекулярною масою 32,5 кДа. Паліперидон Інгредієнт Диметилсульфоксид Кількість (мг) 50 25 Кількість (мг) 117 Розмір частинок паліперидону був визначений за методом розсіювання світла та продемонстрував такий розподіл: d(0,1) = 17,41 мкм, d(0,5) = 51,61 мкм, d(0,9) = 175,32 мкм. Полімер був охарактеризований за його молекулярною масою відповідно до наступної методики. Обладнання Гель-проникаючий хроматограф з потрійним детектором (лазерна дифракція, віскозиметрія, індекс рефракції)- Viscotek® GPCmax VE 2001 GPC З МОДУЛЕМ РОЗЧИННИК/ЗРАЗОК - Viscotek® TDA 305 З ПОТРІЙНОЮ МАТРИЦЕЮ ДЕТЕКТОРА Реактиви - Тетрагідрофуран (THF) класу GPC (гель-проникаюча хроматографія), стабілізований бутилгідрокситолуолом (BHT) в кількості 250 ppm. - Полістирольний стандарт з вузьким молекулярно-масовим розподілом (бажано з молекулярною масою 90 або 99 кДа) Приготування зразків - 1-2 мг/мл - стандартний зразок - 10 мг/мл - досліджуваний зразок: по 3 зразки для кожного досліджуваного полімеру Попередня обробка Підготовлюють і стабілізують колонку та детектори за допомогою мобільної фази (THF) до досягнення робочої швидкості потоку 1 мл/хв., та прочищають віскозиметр і детектори рефракції, перевіряючи в кінці, щоб усі сигнали були стабільними і адекватними. Умови проведення хроматографії - Колонка: 2 послідовні колонки i-MBMMW-3078 (CLM1012, Viscotek) - Колонка затримки: середня затримка (CLM9002, Viscotek) - Температура колонки: 30 ºC - Швидкість потоку 1 мл/хв. - Об’єм проби, що вводиться: 100 мкл - Час роботи: 35 хвилин - Елюент: стабілізований THF, попередньо нагрітий до 30 ºC при перемішуванні зі швидкістю 100 об./хв. Верифікація системи - Вводять 100 мкл елюенту і перевіряють відсутність відгуку сигналів, пов’язаних з визначенням молекулярної маси. - Вводять 100 мкл полістирольного стандарту з вузьким молекулярно-масовим розподілом і перевіряють адекватність вимірів. Повторюють не менше двох разів. 9 UA 115987 C2 5 10 15 20 25 30 35 40 45 50 55 60 Критерії прийнятності: ±5 % від номінальної молекулярної маси і ±3 % від характеристичної в’язкості, заявлених у сертифікаті стандарту виробника. Калібрування Не потрібне, якщо система успішно пройшла верифікацію і попередні хроматографічні умови не змінювалися. У випадку, якщо калібрування необхідне: - Вводять 100 мкл полістирольного стандарту з вузьким молекулярно-масовим розподілом не менше, ніж два рази. - Використовують дані першого зразку для потрійного калібрування і створення нових мультидетекторів - гомополімерний метод. - В цей метод слід включити всі дані, потрібні для внутрішнього калібрування, такі як стандартні значення молекулярної маси, характеристичної в’язкості, dn/dc, dA/dc та індексу рефракції розчинника. Після калібрування системи відповідно до потреб обладнання зберігають новий метод. За допомогою нового методу перевіряють адекватність вимірювання для другого введення стандарту. Процедура Ввести 100 мкл досліджуваного зразка в трьох повторностях. Молекулярна маса полімеру, визначена відповідно до вказаного методу, становила 32,5 кДа. Визначена за схожим методом характеристична в’язкість полімеру становила 0,27 дл/г. Важливо зазначити, що значення характеристичної в’язкості відповідають одержаним за описаною методикою, зокрема у відношенні температурних умов і використовуваного елюента. Будь-яка зміна умов вимірювання означає одержання інших значень, які прямо залежать від них. Лікарська форма паліперидону для ін’єкцій була утворена шляхом з’єднання шприців із ніпелем и муфтою, після чого прямим і зворотним рухом поршнів полімер був повністю розчинений з утворенням гомогенної суспензії паліперидону в розчині полімеру. Концентрація препарату в плазмі крові собаки породи бігль in vivo після внутрішньом’язової ін’єкції: Композиція паліперидону, вказана в цьому прикладі, була введена внутрішньом’язово собакам породи бігль середньою масою 10 кг. Введена кількість відповідала дозі 25 мг паліперидону, композиція якого вводилася внутрішньом’язово в ліву задню лапу тварини за допомогою шприца з голкою калібру 20G. Загальна кількість собак дорівнювала 3. Після ін’єкції вимірювалася концентрація препарату в плазмі у 0, 4 год., 1 д., 2 д., 3 д., 5 д., 7 д., 10 д., 14 д., 17 д., 21 д., 24 д., 28 д., 31 д., 36 д., 38 д., 42 д., 45 д., 49 д., 52 д., 55 д., 59 д., 63 д., 73 д., 80 д., 87 д., 94 д., 108 д. і 118 д. Кінетика концентрацій у плазмі, що відповідає паліперидону, оцінювалася за методом тандемної мас-спектрометрії-ВЕРХ. Профіль концентрацій паліперидону в плазмі зображений на фіг. 1. Результати виражені у вигляді залежності концентрації паліперидону в плазмі (нг/мл) від часу. На цьому малюнку можна побачити, що ін’єкція композиції в кількості, еквівалентній 25 мг паліперидону, собаці породи бігль призводила до дуже надійно регульованого початкового сплеску вивільнення, після якого слідувало повільне стале зниження зі збереженням рівня препарату в плазмі з 1 дня й надалі. Профіль концентрацій у плазмі для паліперидону, як пояснювалося раніше, можна вважати адекватним, оскільки він пов’язаний із дуже низьким ризиком досягнення токсичного рівня препарату в плазмі одразу після ін’єкції. Ця властивість є критично важливою для лікарської форми, призначеної для введення один раз на щонайменше 8 тижнів, оскільки доза, яку потрібно вводити, є високою, порівняно з існуючими режимами щомісячного введення, а тому ризик досягнення токсичних рівнів препарату в плазмі через невеликі коливання фракції препарату, що вивільняється на цьому етапі, є потенційно дуже високим. В цій лікарській формі вивільнення препарату в ході осадження полімеру регулюється розчинністю препарату в розчиннику, кількістю розчинника (відношенням кількості препарату до кількості розчинника) і концентрацією та молекулярною масою полімеру. Результати також вказують на те, що дана лікарська форма здатна пролонговано вивільняти паліперидон протягом понад двох місяців зі зниженими коливаннями різниці між значеннями піку та западини концентрації препарату в плазмі. Для досягнення цієї мети найважливішим є адекватне регулювання співвідношення кількості препарату та полімеру, а також молекулярної маси полімеру. Одразу після введення композиції у внутрішньом’язову тканину, DMSO швидко розчиняється в навколишньому водному середовищі. Відносне збільшення концентрації полімеру в DMSO вище значення розчинності полімеру в цьому розчиннику призводить до утворення преципітату 10 UA 115987 C2 5 10 15 20 25 30 35 полімеру, який утримує в собі не розчинений у розчиннику паліперидон. Молекулярна маса полімеру має величезний вплив на цьому критичному етапі, оскільки надто низькомолекулярні ланцюги характеризуються довшим часом осадження порівняно з ланцюгами, молекулярна маса яких лежить у відповідних межах. Така затримка осадження призводить до посилення взаємодії препарату з навколишніми рідинами, у які він вивільнюється. Таким чином, низька молекулярна маса ланцюгів може спричинити надмірне вивільнення паліперидону після ін’єкції та потенційне досягнення токсичної концентрації препарату в плазмі в перші дні після введення. Молекулярна маса полімеру також може впливати на вивільнення препарату із введеного внутрішньом’язово імплантату після дифузії розчинника та осадження полімеру. Молекулярна маса в указаному діапазоні не здатна підтримувати відповідну швидкість вивільнення паліперидону шляхом дифузії. Ця фаза є особливо тривалою для лікарської форми, призначеної для введення один раз на два місяці, порівняно з лікарською формою, призначеною для введення один раз на 4 тижні, тому контроль молекулярної маси полімеру набуває надзвичайно великого значення. Крім того, надмірна молекулярна маса ланцюгів може призвести до підвищення в’язкості композиції понад рівень, припустимий для її введення в необхідній концентрації (вище за 3,0 Па·с), оскільки після введення у внутрішньом’язову тканину вона потребуватиме довшого часу для гідролізу з утворенням розчинних фракцій, які можуть вивільняти препарат, затриманий у полімерній матриці. Надмірний залишковий вміст препарату, який може бути вивільнений, може спричинити одержання небажано низької концентрації паліперидону в плазмі в період з 24 по 49 день, що може виявитися нижчою за терапевтичну концентрацію препарату в плазмі. Приклад 2. Композиція-депо з Resomer® 504, опроміненим дозою 16 кГр 2 дози. Приготування стерильних лікарських форм для використання in vivo може потребувати використання певних процедур, таких як опромінення, які можуть пошкодити структуру молекул полімеру. У цьому зразку показано, що втрату молекулярної маси полімеру внаслідок його опромінення можна виміряти та контролювати з метою одержання стерильного полімеру з необхідними характеристиками, здатного забезпечити адекватний профіль концентрації паліперидону в плазмі. У зразку кополімер молочної та гліколевої кислот з 50 % вмістом кожної з двох органічних кислот-мономерів і молекулярною масою 50 кДа був стерилізований бетавипромінюванням дозою 16 кГр за контрольованої температури та вологості. Молекулярна маса одержаного в результаті полімеру була визначена за методом, описаним у прикладі 1. Молекулярна маса після опромінення становила 40 кДа. Інгредієнт Кополімер молочної та гліколевої кислот (з блокованою N-кінцевою групою) з 50 % вмістом кожного з двох органічних кислот-мономерів та молекулярною Шприц з муфтою масою 50 кДа був опромінений у 2,25 мл нефасованому стані бетавипромінюванням дозою 16 кГр, внаслідок чого його молекулярна маса була доведена до 40 кДа. Паліперидон Шприц з ніпелем Інгредієнт 2,25 мл Диметилсульфоксид 40 Кількість (мг) 100 50 Кількість (мг) 234 Розмір частинок паліперидону був визначений за методом розсіювання світла та продемонстрував такий розподіл: d(0,1) = 17,41 мкм, d(0,5) = 51,61 мкм, d(0,9) = 175,32 мкм. 5 Характеристична в’язкість опроміненого полімеру, обчислена за методом, викладеним у прикладі 1, становила 0,31 дл/г. Лікарська форма паліперидону для ін’єкцій була утворена шляхом з’єднання шприців із 11 UA 115987 C2 5 10 15 20 25 30 35 40 ніпелем і муфтою, після чого прямим і зворотним рухом поршнів полімер був повністю розчинений з утворенням гомогенної суспензії паліперидону в розчині полімеру. Концентрація препарату в плазмі крові собаки породи бігль in vivo після внутрішньом’язової ін’єкції: Композиція паліперидону, вказана в цьому прикладі, була введена внутрішньом’язово двом когортам собак породи бігль середньою масою 10 кг. Введена кількість відповідала дозі 25 мг паліперидону в одній когорті та дозі 50 мг паліперидону в другій когорті. Композиція вводилася внутрішньом’язово в ліву задню лапу тварини за допомогою шприца з голкою калібру 20G. Загальна кількість собак у кожній когорті дорівнювала 3. Після ін’єкції вимірювалася концентрація препарату в плазмі у 0, 4 год., 1 д., 2 д., 3 д., 5 д., 7 д., 10 д., 14 д., 17 д., 21 д., 24 д., 28 д., 31 д., 36 д., 38 д., 42 д., 45 д., 49 д., 52 д., 55 д., 59 д., 63 д., 73 д., 80 д., 87 д., 94 д., 108 д. і 118 д. Кінетика концентрацій у плазмі, що відповідає паліперидону, оцінювалася за методом тандемної мас-спектрометрії-ВЕРХ. Профіль концентрацій паліперидону в плазмі зображений на фіг. 2. Результати виражені у вигляді залежності концентрації паліперидону в плазмі (нг/мл) від часу. Як можна побачити на цьому рисунку, внаслідок введення собакам породи бігль композиції в кількості, еквівалентній 25 мг паліперидону, був одержаний профіль концентрації препарату в плазмі, аналогічний наведеному в прикладі 1, де використовувався неопромінений полімер. Цей приклад демонструє можливість регулювання молекулярної маси полімеру за допомогою процесу стерилізації з одержанням стерильного полімеру, який надає лікарській формі необхідні характеристики стосовно вивільнення препарату. Можливі зміни кінцевих груп полімеру після опромінення не спричиняють суттєвої зміни характеристик вивільнення препарату in vivo у вказаному діапазоні, що свідчить про вирішальне значення молекулярної маси для регулювання процесу деградації в цій композиції. Приклад 3. Композиція-депо з Lakeshore Biomaterials® 5050DLG 5E, опроміненим дозою 25 кГр. Приготування стерильних лікарських форм для використання in vivo може потребувати використання певних процедур, таких як опромінення, які можуть пошкодити структуру молекул полімеру. У цьому зразку показано, що втрату молекулярної маси полімеру внаслідок його опромінення можна виміряти та контролювати з метою одержання стерильного полімеру з необхідними характеристиками, здатного забезпечити адекватний профіль концентрації паліперидону в плазмі. У зразку кополімер молочної та гліколевої кислот з 50 % вмістом кожної з двох органічних кислот-мономерів і молекулярною масою 54 кДа був стерилізований бетавипромінюванням дозою 25 кГр за контрольованої температури та вологості. Молекулярна маса одержаного в результаті полімеру була визначена за методом, описаним у прикладі 1. Молекулярна маса після опромінення становила 42 кДа. Інгредієнт Кополімер молочної та гліколевої кислот (з блокованою N-кінцевою групою) з 50 % вмістом кожного з двох органічних кислот-мономерів та молекулярною Шприц з муфтою 2,25 масою 54 кДа був опромінений у нефасованому мл стані бетавипромінюванням дозою 25 кГр, внаслідок чого його молекулярна маса була доведена до 42 кДа. Паліперидон Шприц з ніпелем 2,25 Інгредієнт мл Диметилсульфоксид 12 Кількість (мг) 100 50 Кількість (мг) 234 UA 115987 C2 5 10 15 20 25 Розмір частинок паліперидону був визначений за методом розсіювання світла та продемонстрував такий розподіл: d(0,1) = 17,41 мкм, d(0,5) = 51,61 мкм, d(0,9) = 175,32 мкм. Характеристична в’язкість опроміненого полімеру, обчислена за методом, викладеним у прикладі 1, становила 0,31 дл/г. Лікарська форма паліперидону для ін’єкцій була утворена шляхом з’єднання шприців із ніпелем и муфтою, після чого прямим і зворотним рухом поршнів полімер був повністю розчинений з утворенням гомогенної суспензії паліперидону в розчині полімеру. Концентрація препарату в плазмі крові собаки породи бігль in vivo після внутрішньом'язової ін'єкції: Композиція паліперидону, вказана в цьому прикладі, була введена внутрішньом'язово собакам породи бігль середньою масою 10 кг. Введена кількість відповідала дозі 75 мг паліперидону. Композиція вводилася внутрішньом'язово в ліву задню лапу тварини за допомогою шприца з голкою калібру 20G. Загальна кількість собак дорівнювала 3. Після ін'єкції вимірювалася концентрація препарату в плазмі у 0, 4 год., 1 д., 3 д., 5 д., 7 д., 10 д., 14 д., 17 д., 21 д., 24 д., 28 д., 31 д., 34 д., 41 д., 49 д. і 56 д. Кінетика концентрацій у плазмі, що відповідає паліперидону, оцінювалася за методом тандемної мас-спектрометрії-ВЕРХ. Профіль концентрацій паліперидону в плазмі зображений на фіг. 3. Результати виражені у вигляді залежності концентрації паліперидону в плазмі (нг/мл) від часу. Як можна побачити на цьому рисунку, внаслідок введення собакам породи бігль композиції в кількості, еквівалентній 75 мг паліперидону, був одержаний профіль концентрації препарату в плазмі, аналогічний наведеному в прикладі 1, де використовувався неопромінений полімер, і в прикладі 2, де використовувався полімер, опромінений дозою 16 кГр. Цей приклад демонструє можливість регулювання молекулярної маси полімеру за допомогою процесу стерилізації з одержанням стерильного полімеру, який надає лікарській формі необхідні характеристики стосовно вивільнення препарату. Можливі зміни кінцевих груп полімеру після опромінення не спричиняють суттєвої зміни характеристик вивільнення препарату in vivo у вказаному діапазоні, що свідчить про вирішальне значення молекулярної маси для регулювання процесу деградації в цій композиції. 30 35 40 Приклад 4. Композиція-депо з Resomer® 504, опроміненим дозою 25 кГр В цьому прикладі продемонстровано, що надмірне зменшення молекулярної маси полімеру може призвести до скорочення профілю концентрації паліперидону в плазмі, внаслідок чого така лікарська форма стає непридатною для введення раз на 8 чи більше тижнів. Кополімер молочної та гліколевої кислот з 50 % вмістом кожної з двох органічних кислотмономерів і молекулярною масою 38 кДа був стерилізований бетавипромінюванням дозою 25 кГр за контрольованої температури та вологості. Молекулярна маса одержаного в результаті полімеру була визначена за методом, описаним у прикладі 1. Молекулярна маса після опромінення становила 29 кДа. Шприц з муфтою 2,25 мл Шприц з ніпелем 2,25 мл Інгредієнт Кополімер молочної та гліколевої кислот (з блокованою N-кінцевою групою) з 50 % вмістом кожної з двох органічних кислотмономерів та молекулярною масою 38 кДа був опромінений у нефасованому стані бета-випромінюванням дозою 25 кГр, внаслідок чого його молекулярна маса була доведена до 29 кДа. Паліперидон Інгредієнт Диметилсульфоксид Кількість (мг) 50 25 Кількість (мг) 117 Розмір частинок паліперидону був визначений за методом розсіювання світла та продемонстрував такий розподіл: d(0,1) = 17,41 мкм, d(0,5) = 51,61 мкм, d(0,9) = 15 175,32 мкм. 13 UA 115987 C2 5 10 15 20 Характеристична в'язкість опроміненого полімеру, обчислена за методом, викладеним у прикладі 1, становила 0,27 дл/г. Лікарська форма паліперидону для ін'єкцій була утворена шляхом з'єднання шприців із ніпелем и муфтою, після чого прямим і зворотним рухом поршнів полімер був повністю розчинений з утворенням гомогенної суспензії паліперидону в розчині полімеру. Концентрація препарату в плазмі крові собаки породи бігль in vivo після внутрішньом'язової ін'єкції: Композиція паліперидону, вказана в цьому прикладі, була введена внутрішньом'язово собакам породи бігль середньою масою 10 кг. Композиція в кількості, еквівалентній дозі 2,5 мг/кг паліперидону, вводилася внутрішньом'язово в ліву задню лапу тварини за допомогою шприца з голкою калібру 20G. Загальна кількість собак у кожній когорті дорівнювала 3. Після ін'єкції вимірювалася концентрація препарату в плазмі у 0, 4 год., 1 д., 2 д., 3 д., 5 д., 7 д., 10 д., 14 д., 17 д., 21 д., 24 д., 28 д., 31 д., 35 д., 38 д., 42 д., 45 д., 49 д., 52 д., 56 д., 59 д., 63 д., 70 д., 77 д. Кінетика концентрацій у плазмі, що відповідає паліперидону, була оцінена та зображена на фіг. 4. Результати виражені у вигляді залежності концентрації паліперидону в плазмі (нг/мл) від часу. Як можна побачити на цьому малюнку, введення собакам породи бігль композиції в кількості, еквівалентній 2,5 мг/кг, призвело до скорочення профілю концентрації препарату в плазмі. Зменшення молекулярної маси на 4 кДа (з 33 до 29 кДа) значно підсилює дифузію препарату крізь полімерну матрицю, що негативно впливає на здатність препарату вивільнюватися через 30 днів після введення. ФОРМУЛА ВИНАХОДУ 25 30 35 40 45 50 55 1. Внутрішньом'язова ін'єктована депо-композиція уповільненого вивільнення, придатна для формування в організмі in situ твердого імплантата із вмістом лікарського засобу паліперидону і/або будь-якої його фармацевтично прийнятної солі в будь-якій комбінації, біосумісного співполімеру на основі молочної і гліколевої кислоти зі співвідношенням мономерів молочної кислоти до гліколевої приблизно 50:50 і розчинника диметилсульфоксиду (ДМСО), яка відрізняється тим, що біосумісний співполімер має молекулярну масу в діапазоні 31-40 кДа і характеристичну в'язкість в межах від 0,27-0,31 дл/г ±10 %. 2. Композиція за п. 1, в якій біосумісний співполімер піддається опроміненню гамма- або бетачастинками дозою в діапазоні 10-30 кГр, що вимірюється при температурі в діапазоні від -40 °С до +35 °С для коректування його молекулярної маси в межах 27-47 кДа і характеристичної в'язкості в діапазоні 0,27-0,31 дл/г ±10 %. 3. Композиція за п. 1 або 2, з наступним розподілом частинок препарату за розміром: менше 10 % частинок розміром менше 10 мікронів; менше 10 % частинок розміром більше 225 мікронів, і значення d0,5 - в діапазоні 40-90 мікронів. 4. Композиція за будь-яким з пп. 1-3, в якій масове співвідношення лікарський препарат/(полімер+препарат) становить близько 33 %. 5. Композиція за будь-яким з пп. 1-4, в якій вміст препарату становить близько 13 % у ваговому співвідношенні від загального складу, а в'язкість розчину із вмістом полімеру і ДМСО знаходиться в діапазоні 1,5-2,5 Па·с. 6. Композиція за будь-яким з попередніх пунктів, що використовується для лікування шизофренії або біполярних розладів у людини. 7. Фармацевтичний набір, придатний для утворення в організмі in situ твердого імплантата, що містить заявлену в пп. 1-6 композицію, де лікарський препарат і біосумісний полімер містяться в першому контейнері, а розчинник - у другому, окремому, контейнері. 8. Фармацевтичний набір за п. 7, в якому принаймні один з контейнерів (або перший, або другий) являє собою шприц, флакон, спеціальний пристрій або картридж (одноразові або ні). 9. Спосіб виготовлення композиції відповідно до будь-якого з вищезгаданих пунктів 1-6, що включає етап створення біосумісного співполімеру з більш високою молекулярною масою, ніж необхідна для ін'єктованої внутрішньом'язово дено-композиції, з подальшою корекцією його молекулярної маси до діапазону 31-40 кДа і його характеристичної в'язкості до діапазону 0,27 14 UA 115987 C2 5 10 15 20 25 0,31 дл/г гамма- або бета-випромінюванням дозами в діапазоні 10-30 кГр, що вимірюється при температурі від -40 °С до +35 °С. 10. Спосіб за п. 9, при якому доза опромінення полімеру, що вимірюється при температурі 8 °С, знаходиться в діапазоні 16-25 кГр. 11. Спосіб за п. 9 або 10, в якому біосумісний полімер з початковою молекулярною масою близько 50 кДа піддається опроміненню дозою близько 16 кГр з метою зменшення його молекулярної маси до значень в діапазоні 27-47 кДа. 12. Спосіб за п. 9 або 10, в якому біосумісний полімер з початковою молекулярною масою близько 54 кДа піддається опроміненню дозою близько 25 кГр з метою зменшення його молекулярної маси до значень в діапазоні між 31 і 40 кДа. 13. Спосіб за п. 9 або 10, в якому біосумісний полімер з початковою молекулярною масою близько 63 кДа піддається опроміненню дозою близько 30 кГр з метою зменшення його молекулярної маси до значень в діапазоні 31-40 кДа. 14. Спосіб початкового режиму дозування для введення внутрішньом'язової ін'єктованої депокомпозиції уповільненого вивільнення за пп. 1-6 пацієнту, який потребує психіатричного лікування, що включає: a) внутрішньом'язове введення пацієнту першої дози від 75 мг до 250 мг; b) внутрішньом'язове введення пацієнту другої дози ін'єктованої депо-композиції з тривалим вивільненням між 24 і 35 днями лікування, рахуючи від дня попереднього введення; c) введення подальшої дози внутрішньом'язової ін'єктованої депо-композиції з тривалим вивільненням, що становить від 75 мг до 250 мг, приблизно між 56 і 65 днями після введення вищезазначеної першої дози; d) повторення кроку b) за необхідності отримати необхідну концентрацію препарату в плазмі крові перед початком введення препарату кожні 8 тижнів. 15. Спосіб за п. 14, де перша доза становить приблизно від 100 мг до 200 мг. 15 UA 115987 C2 16 UA 115987 C2 Комп’ютерна верстка Л. Бурлак Міністерство економічного розвитку і торгівлі України, вул. М. Грушевського, 12/2, м. Київ, 01008, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 17
ДивитисяДодаткова інформація
Назва патенту англійськоюPaliperidone implant formulation
Автори англійськоюGutierro Aduriz, Ibon, Franco Rodriguez, Guillermo
Автори російськоюГутьерро Адурис Ибон, Франко Родригес Гильермо
МПК / Мітки
МПК: A61K 9/00, A61K 31/519, A61K 47/34
Мітки: лікарська, форма, паліперидону, імплантування
Код посилання
<a href="https://ua.patents.su/19-115987-likarska-forma-paliperidonu-dlya-implantuvannya.html" target="_blank" rel="follow" title="База патентів України">Лікарська форма паліперидону для імплантування</a>
Спосіб дозування ін’єкційної форми паліперидону пальмітату тривалої дії

Номер патенту: 99163
Опубліковано: 25.07.2012
Автори: Вермелен Ан Маргріт Корнелія, Ваутерс Альфонс Жанна
МПК: A61K 31/519, A61P 25/18
Мітки: тривалої, ін'єкційної, дозування, форми, дії, пальмітату, паліперидону, спосіб
Формула / Реферат:
1. Спосіб дозування для введення паліперидону пальмітату психіатричному пацієнту, потребуючому лікування, що включає:(1) внутрішньом'язове введення в ділянку дельтоподібного м'яза пацієнта першої насичуючої дози, що складає від приблизно 100 мг-екв. до приблизно 150 мг-екв. паліперидону в формі паліперидону пальмітату у вигляді складу з тривалим вивільненням, в перший день лікування;(2) внутрішньом'язове введення в ділянку...
Пероральна лікарська форма, яка містить розиглітазон та метформін

Номер патенту: 82537
Опубліковано: 25.04.2008
Автори: Коулз Пітер Джон, Мадд Пол Норман, мол., Маккензі Дональд Колін
МПК: A61K 9/28, A61P 3/10, A61K 31/155, A61K 31/4439
Мітки: лікарська, містить, розиглітазон, метформін, пероральна, яка, форма
Формула / Реферат:
1. Пероральна лікарська форма, яка має ядро, що піддається ерозії, яке містить 5-[4-[2-(N-метил-N-(2-піридил)аміно)етокси]бензил]тіазолідин-2,4-діон або його фармацевтично прийнятну сіль чи сольват та метформін або його фармацевтично прийнятну сіль чи сольват, причому ядро має непроникне покриття з одним або декількома отворами, що ведуть до ядра, яка відрізняється тим, що покриття є таким, що піддається ерозії при рН від 4,5 до 8.2....
Стабільна лікарська форма похідного тіадіазолу

Номер патенту: 97672
Опубліковано: 12.03.2012
Автори: Тасіро Йосікадзу, Мішра Дінеш Шямдео, Чжуан Хун, Ватанабе Йосуке, Кусано Хіроко
МПК: A61K 35/00, A61K 9/19, A61K 31/433
Мітки: стабільна, форма, похідного, тіадіазолу, лікарська
Формула / Реферат:
1. Фармацевтична композиція, яка містить N-{4-(2,2-диметилпропіоніл)-(5R)-5-[(2-етиламіноетансульфоніламіно)метил]-5-феніл-4,5-дигідро-[1,3,4]тіадіазол-2-іл}-2,2-диметилпропіонамід, буфер та/або сіль, вибрані з групи, до якої входять тартрат, фосфат, цитрат, мезилат, фосфат натрію та сульфат натрію, та фармацевтично прийнятний носій, розріджувач або наповнювач, у водному розчині, причому рН згаданої композиції є нижчим ніж 5,4 та вищим ніж...
Лікарська форма

Номер патенту: 111834
Опубліковано: 24.06.2016
Автор: Мохаммад Хассан
МПК: A61P 25/04, A61K 9/26, A61K 9/16
Формула / Реферат:
1. Лікарська форма, що містить:не розтягнуті, екструдовані з розплаву частинки, що містять лікарський засіб, що являє собою агоніст опіатів, і матрицю;де вказані екструдовані з розплаву частинки присутні у вигляді дисперсної фази у вказаній матриці, де вказана матриця містить диспергуючу фазу, що включає гелеутворювальну речовину, і де вказані частинки додатково містять співполімер з алкільних естерів акрилової кислоти і...
Тверда лікарська форма для птиці і спосіб її виготовлення

Номер патенту: 86572
Опубліковано: 12.05.2009
Автори: Устянич Анатолій Євгенович, Устянич Євген Петрович, Устянич Марта Анатолієвна
МПК: A61D 99/00, A61K 9/16
Мітки: тверда, птиці, виготовлення, лікарська, спосіб, форма
Формула / Реферат:
1. Тверда лікарська форма для птиці, що складається з капілярно-пористих гранул, насичених рідинним лікарським препаратом чи розчином, покритих захисним покриттям, обпудрених і висушених, яка відрізняється тим, що як капілярно-пористі гранули для насичення лікарським препаратом чи розчином використовують індиферентні в організмі птиці поризовані каменеподібні тіла - силікагель, алюмосилікат, кульки поризованого скла, кераміки, як захисне чи...
Попередній патент: Ультратонкодисперсна композиція вапнякового молока
Наступний патент: Електронний пристрій для одержання пари
Випадковий патент: Композиція інгредієнтів кондитерського виробу "alcoids" і спосіб його виготовлення