Спосіб одержання транс-1-((1r,3s)-6-хлор-3-феніліндан-1-іл)-3,3-диметилпіперазину
Номер патенту: 106191
Опубліковано: 11.08.2014
Автори: Бресен Петер, Робен Давід, Даль Аллан Карстен, Вехльк Нільсен Хрістіна, Сюте Крістіна





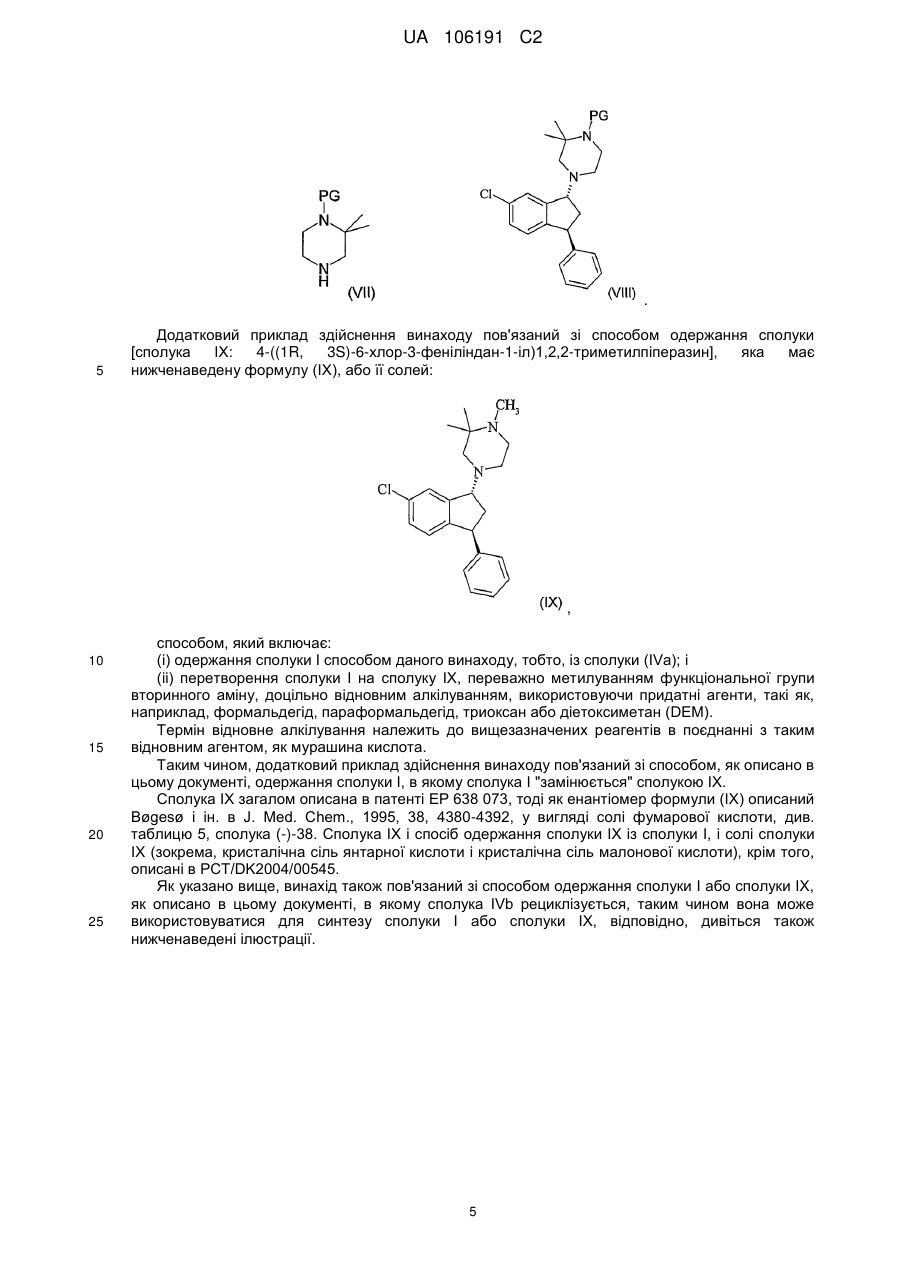
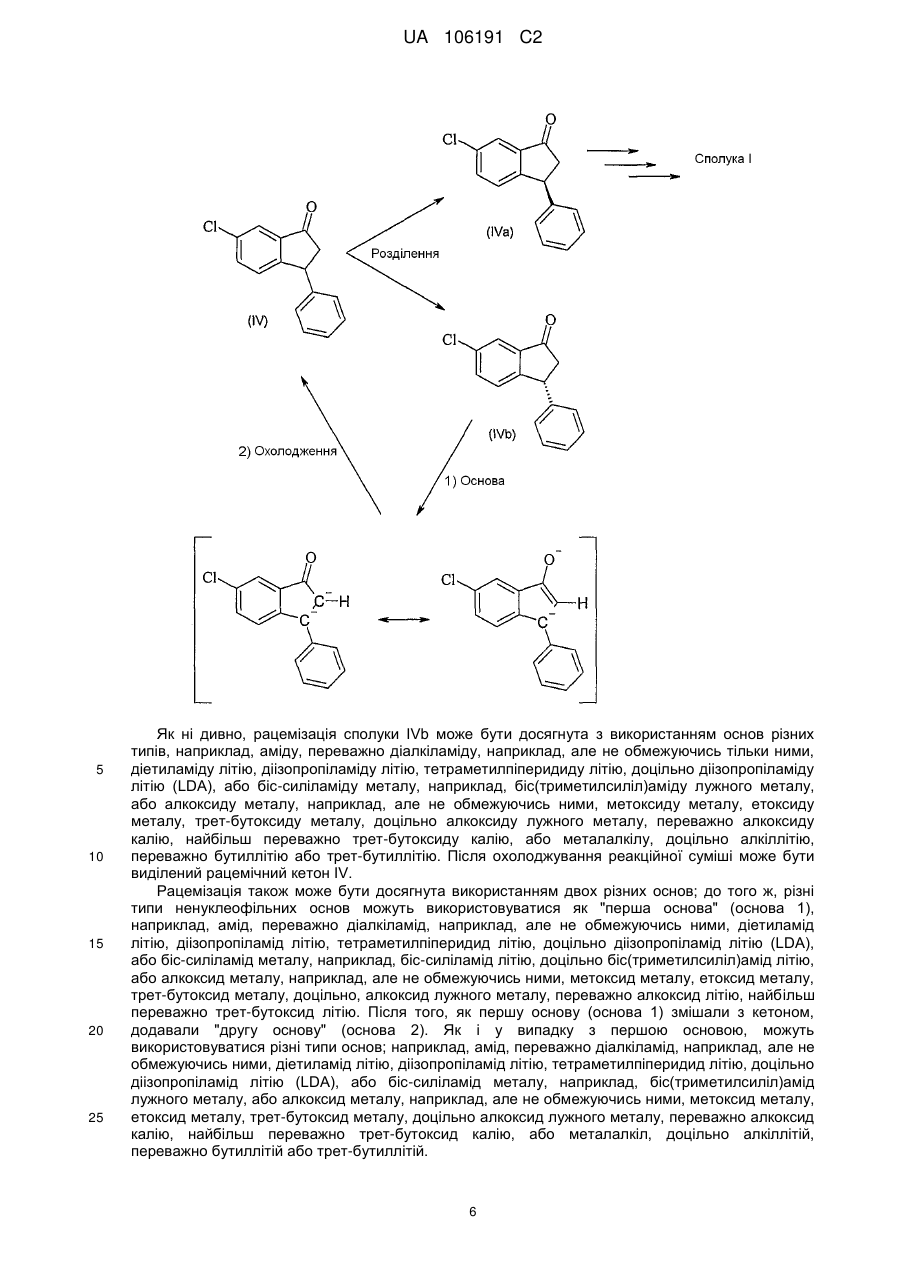
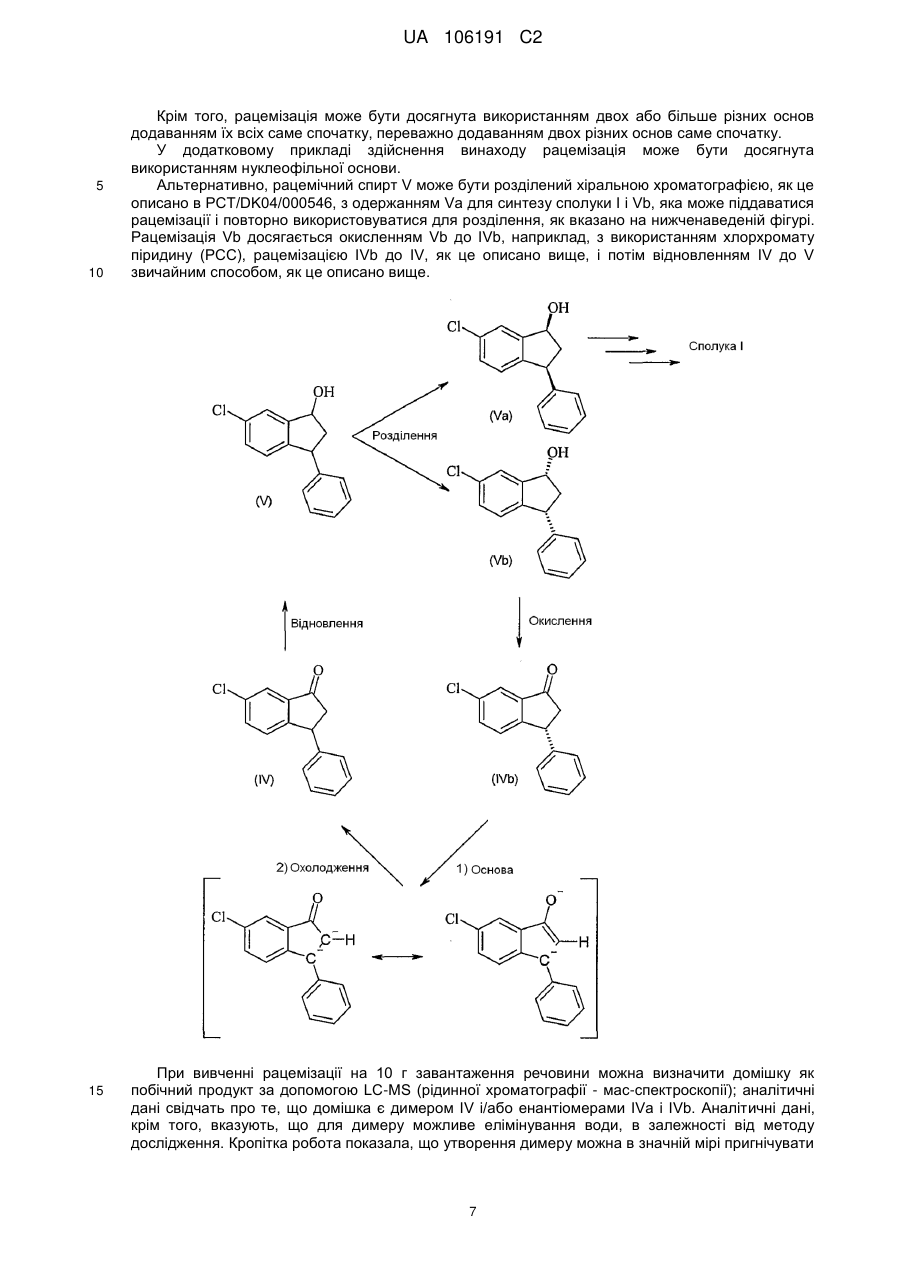









Формула / Реферат
1. Спосіб одержання сполуки формули І (сполука І) або її солі, в якому здійснюють:
одержання сполуки IVa розділенням рацемічної сполуки IV з використанням хіральної хроматографії
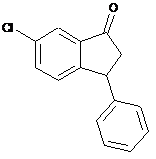 , (IV)
, (IV) 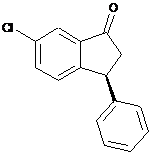 , (IVa)
, (IVa)
відновлення сполуки формули IVa в присутності боргідриду у розчиннику у відповідний спирт Va з цис-конфігурацією
 , (Va)
, (Va)
перетворення в присутності тіонілхлориду, тіонілброміду, мезил(метансульфоніл)хлориду або тозил(4-толуолсульфоніл)хлориду спиртової групи цис-спирту формули Va в групу LG, що видаляється, з одержанням сполуки формули VI
 , (VI)
, (VI)
взаємодію сполуки VI з 2,2-диметилпіперазином з одержанням сполуки формули І
 (I).
(I).
2. Спосіб за п. 1, в якому LG є галогеном, наприклад Сl або Вr, переважно Сl, або сульфонатом, наприклад тозилатом або мезилатом.
3. Спосіб за п. 1, в якому сполука LG є галогеном, наприклад Сl, і розчинник є алканом, наприклад гептаном.
4. Спосіб за п. 1, в якому сполуку І осаджують у вигляді солі.
5. Спосіб за п. 4, в якому сіль, що утворилася, є сіллю фумарової кислоти, сіллю малеїнової кислоти або сіллю соляної кислоти і сполуки І.
6. Спосіб за п. 1, в якому сполуку І виділяють у вигляді вільної основи.
7. Спосіб за п. 1, в якому згадану хіральну хроматографію здійснюють з використанням хіральної рідинної хроматографії.
8. Спосіб за п. 7, в якому згадану хіральну хроматографію здійснюють на хіральній нерухомій фазі.
9. Спосіб за п. 8, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою модифікованої амілози.
10. Спосіб за п. 9, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою трис-[(S)-a-метилбензилкарбамат]амілози.
11. Спосіб за п. 10, в якому для згаданої хіральної хроматографії застосовують розчинник, що включає суміш н-гептану і етанолу, і, необов'язково, N,N-діетиламін.
12. Спосіб за п. 11, в якому згадану хіральну хроматографію здійснюють з використанням хроматографії з суб- або суперкритичною рухомою фазою, переважно хроматографії з суперкритичною рухомою фазою.
13. Спосіб за п. 12, в якому згадану хіральну хроматографію здійснюють на хіральній нерухомій фазі.
14. Спосіб за п. 13, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою, що складається з хірального полімеру, або на колонці з силікагелем з іммобілізованим хіральним полімером, або на колонці з силікагелем з ковалентно зв'язаним хіральним мономером.
15. Спосіб за п. 14, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою трис-(3,5-диметилфенілкарбамат)амілози або трис-[(S)-a-метилбензилкарбамат]амілози, або на колонці з силікагелем, з іммобілізованою трис-(3,5-диметилфенілкарбамат)амілозою, або на колонці з силікагелем, покритим оболонкою трис-(3,5-диметилфенілкарбамат)целюлози або трис-(4-метилбензоат)целюлози, або на колонці з силікагелем з ковалентно зв'язаним аміном 3,5-динітробензоїлтетрагідрофенантрену.
16. Спосіб за будь-яким одним з пп. 14, 15, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою трис-(3,5-диметилфенілкарбамат)амілози.
17. Спосіб за будь-яким одним з пп. 14-16, в якому згадану хіральну хроматографію здійснюють з модифікатором, вибраним з групи: метанол, етанол, ізопропанол або ацетонітрил, що необов'язково містить діетиламін, необов'язково 0,1 % діетиламіну.
18. Спосіб за будь-яким одним з пп. 1-17, в якому додатково здійснюють рециклізацію сполуки IVb перетворенням енантіомерно збагаченої сполуки IVb у переважно рацемічну сполуку IV, де IVb і IV є:
 , (IVb) (IV).
, (IVb) (IV).
19. Спосіб за п. 18, в якому рацемізацію здійснюють використанням основи або суміші двох або більше основ.
20. Спосіб за п. 18 або 19, в якому рацемізацію здійснюють застосуванням одного еквівалента або більше ненуклеофільної основи ("першої основи"), за яким іде додавання каталітичної кількості або одного еквівалента або більше тієї ж самої або іншої основи ("другої основи").
Текст
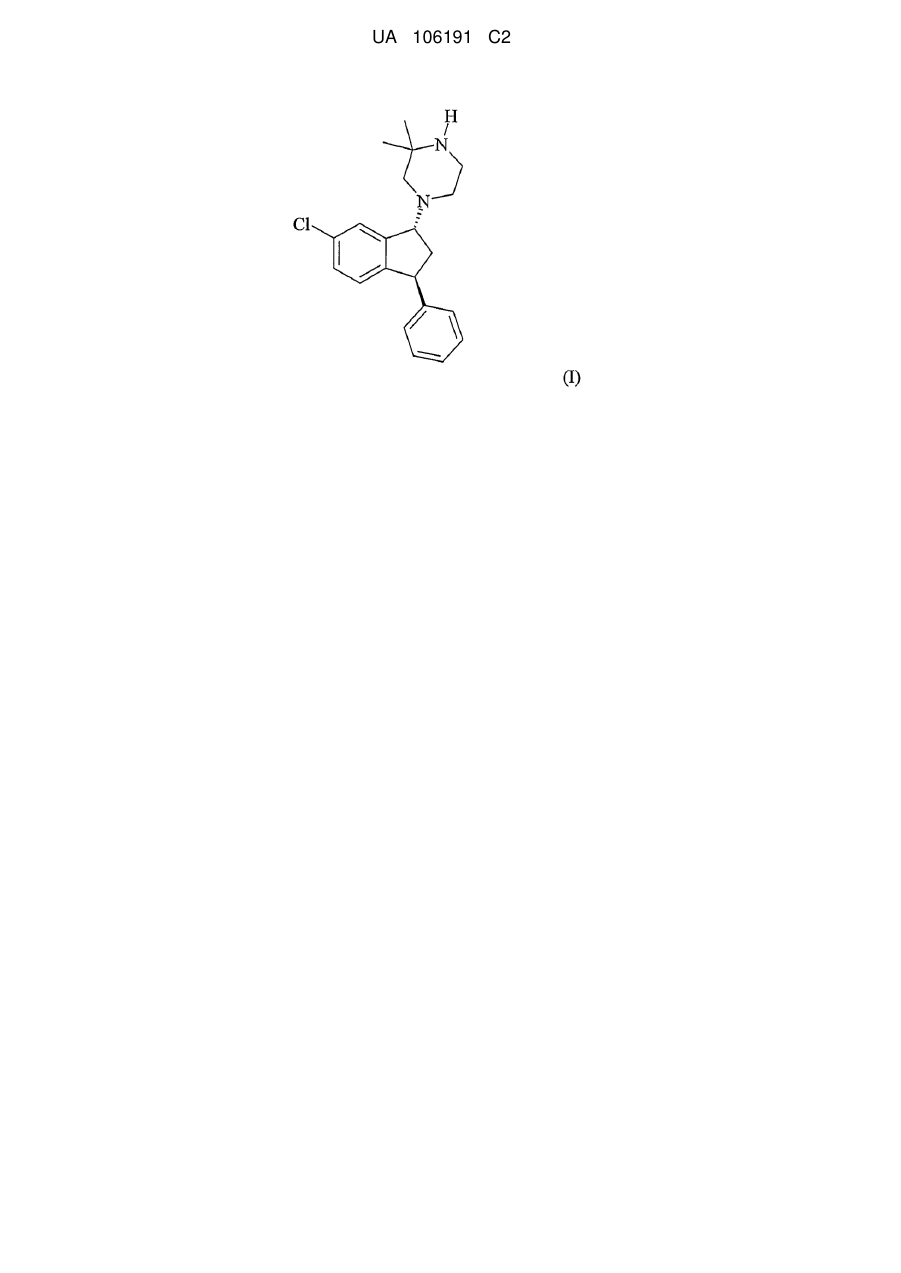
Реферат: Описаний спосіб одержання транс-1-((R,3S)-6-хлор-3-феніліндан-1-іл)-3,3-диметилпіперазину (формула І) і його солей і аналогічний спосіб одержання 4-((1R,3S)-6-хлор-3-феніліндан-1-іл)1,2,2-триметилпіперазину (формула IX) і його солей, цей спосіб включає перетворення сполуки формули IVa на сполуку формули І або сполуку формули IX, відповідно. UA 106191 C2 (12) UA 106191 C2 UA 106191 C2 5 Даний винахід належить до способу одержання транс-1-((1R, 3S)-6-хлор-3-феніліндан-1-іл)3,3-диметилпіперазину (сполука I) і його солей. Рівень техніки винаходу Сполука, яка є предметом даного винаходу (сполука I, транс-1-((1R, 3S)-6-хлор-3феніліндан-1-іл)-3,3-диметилпіперазин) має загальну формулу (I) . 10 15 20 25 30 35 40 45 Сполука I і її солі, включаючи фумарати і малеати, і її застосування в медицині, наприклад, при шизофренії або інших захворюваннях, які викликають психотичні симптоми, описані в РСТ/DK04/000546 (WO05/016901). Як описано в РСТ/DK04/000546, винахідники встановили, що сполука I виявляє високу спорідненість до рецепторів дофаміну (DA), D1 рецепторів, DA D2 рецепторів і до альфа1 адреноцепторів. Крім того, встановлено, що сполука I є антагоністом рецепторів дофаміну D1 і D2 і рецепторів серотоніну 5-НТ2A. Як далі описано в РСТ/DK04/000546, сполука I є відносно слабим інгібітором CYP2D6 (тобто знижувало можливість взаємодії ліків між собою) і має відносно слабий вплив на QT-інтервал на моделі кроликів (тобто знижувало можливість встановлення пролонгування QT-інтервалу, викликаного ліками, і виникнення летальної аритмії серця, двоспрямованої веретеноподібної шлуночкової тахікардії (TdP) у людей). Крім того, активність сполуки I як 5-НТ2-антагоніста свідчить про те, що сполука I може мати відносно невеликий ризик екстрапірамідального побічного ефекту. Вищевизначені властивості, наприклад, тести зв'язування (що включають альфа-1, DA D1 або D2, або рецептори серотоніну 5-НТ2А), інгібування CYP2D6 і QT-інтервал можуть бути визначені, як описано в РСТ/DK04/000546, проведіть порівняння саме розділу "Приклад" стор. 19-24 тексту даної заявки як галузь застосування РСТ/DK04/000546. Крім того, винахідники встановили, що сполука I не викликає дистонію при тестуванні на свинях, сенсибілізованих галоперидолом, що вказує на те, що сполука I не має EPS (екстрапірамідальні симптоми) сприйнятливості/схильності у людей. РСТ/DK04/000546 описує наступні медичні застосування сполуки I: захворювання центральної нервової системи, що включає психоз, а саме, шизофренія (наприклад, позитивний, негативний і/або депресивні симптоми), або інші захворювання, включає психотичні симптоми, такі як, наприклад, шизофренія, шизофреноформний розлад, шізоафективний розлад, галюцинаторний розлад, короткочасний психотичний розлад, загальний психотичний розлад, крім того інші психотичні розлади або захворювання, які викликають психотичні симптоми, наприклад розлад як маніакальний біполярний синдром. Також описане застосування сполуки I для лікування тривожних розладів, афективних розладів, що включають депресію, порушення сну, мігрень, нейролептично індукований паркінсонізм, або залежності від кокаїну, від нікотину, від алкоголю і інших розладів, зумовлених залежністю. Як указано в РСТ/DK04/000546, група сполук, структурно родинних сполуці I, тобто трансізомери 3-арил-1-(1-піперазиніл)інданів, заміщені у 2- і/або 3-положенні піперазинового кільця, описані в патенті ЕР 638 073; Bøgesø і ін. в J. Med. Chem., 1995, 38, 4380-4392 і Klaus Р. Вogeso в "Drug Hunting, the Medical Chemistry of 1-Piperazino-3-phenilindans and Related Compounds", 1998, ISBN 87-88085-10-41. Наприклад, енантіомерно чиста сполука, що відповідає формулі (I), але відрізняється тим, що вона має N-метильну групу замість N-водневої групи в піперазині, розкрито в Bøgesø і ін. в J. Med. Chem., 1995, 38, 4380-4392, див. таблицю 5, сполука (-)-38. Жодне з окремо взятих посилань з РСТ/DK04/000546 не розкриває специфічну енантіомерну форму вищезазначеної сполуки (сполука I) або її медичне застосування. Тільки транс-ізомер у вигляді рацемату сполуки I неявно розкритий як проміжна сполука в синтезі сполуки 38 в 1 UA 106191 C2 5 10 15 20 Bøgesø і ін. в J. Med. Chem., 1995, 38, 4380-4392, в той час, як не описане медичне застосування сполуки I або відповідного ній рацемату. Сполука I як проміжна сполука розкрита в РСТ/DK04/000545 (WO05/016900). Докладний опис винаходу Даний винахід в одному аспекті належить до нового способу одержання сполуки I, в якому хіральність вводиться на більш ранній стадії одержання, в порівнянні зі способом, описаним в РСТ/DK04/000546. Введення хіральності на одну стадію раніше є перевагою, оскільки в цьому випадку наступна стадія проходить ефективно, тобто по величині виходу і по витрачанню реагентів і розчинників, і по зниженню кількості побічних продуктів. У РСТ/DK04/000546 хіральність вводиться розділенням рацемічної нижчеприведеної проміжної сполуки V або ферментативно, або хіральною ВЕРХ. Дані винахідники розробили спосіб синтезу, в якому енантіомер формули (I) одержують послідовними перетвореннями з вихідного чистого енантіомера IV, тобто сполуки IVa ((S)-6-хлор-3-феніліндан-1-ону, див. нижче). Таким чином, в цьому способі проміжна сполука формули IV розділяється, тобто піддається хіральній хроматографії, з одержанням енантіомера формули IVa. Крім того, дані винахідники розробили спосіб рацемізації небажаного енантіомера (сполука IVb, див. нижче), який потім може бути повторно використаний при розділенні. Це впливає величезний чином на ефективність всього синтезу, оскільки ефективність стадій до розділення підвищується так само, як і подальших стадій. Таким чином, енантіомер формули (I) може бути одержаний в способі, який включає наступні стадії: . 25 30 Ціанистий бензил реагує з 2,5-дихлорбензонітрилом в присутності основи; придатним є трет-бутоксид калію (t-BuOK), у придатному розчиннику, такому як 1,2-диметоксіетан (DME), подальша реакція з метилхлорацетатом (МСА) приводить до спонтанного закриття циклу і одностадійного утворення сполуки формули (II). Сполука формули (II) потім піддається кислотному гідролізу з утворенням сполуки формули (III), відповідними для цього умовами є нагрівання в суміші оцтової кислоти, сірчаної кислоти і води, з подальшим декарбоксилуванням, тобто нагріванням сполуки формули (III) у придатному розчиннику, такому як толуол, з триетиламіном або N-метилпіролідин-2-оном (NMP), з утворенням сполуки формули (IV) . 35 Сполука формули (IV) розділяється для одержання необхідного енантіомера (формула IVа) для подальшого синтезу сполуки I, і небажаного енантіомера (формула IVb), який може бути підданий рацемізації і рециклізації: 40 2 UA 106191 C2 . 5 10 15 20 25 30 35 40 45 Розділення IV може бути здійснене, наприклад, з використанням хіральної хроматографії, переважна рідинна хроматографія, або хроматографія з суб- або суперкритичною рухомою фазою. Хіральна рідинна хроматографія може здійснюватися, наприклад, на хіральній нерухомій фазі, доцільно на колонці з силікагелем, на якому іммобілізований хіральний полімер, або переважно на колонці з силікагелем, покритим оболонкою, яка складається з хірального полімеру, наприклад, модифікованої целюлози або модифікованої амілози, такої як трис-[(S)-αметилбензилкарбамат]амілози, переважно на колонці з силікагелем, покритим оболонкою трис[(S)-α-метилбензилкарбамат]амілози. Придатним розчинником для застосування в хіральній рідинній хроматографії є такий розчинник, як, наприклад, спирт (переважно, С1-4-спирт), нітрил, простий ефір або алкан (переважно, С5-10-алкан), або їх суміші, доцільно використовувати етанол, метанол, ізопропанол, ацетонітрил, метил-трет-бутиловий ефір або н-гептан, або їх суміші. Кислотні або основні модифікатори можуть додаватися в розчинник, наприклад, мурашина кислота, оцтова кислота, трифтороцтова кислота, триетиламін або N, N-діетиламін. Хроматографія з суб- або суперкритичною рухомою фазою може здійснюватися, наприклад, на хіральній нерухомій фазі, доцільно на колонці з силікагелем, на якому іммобілізований хіральний полімер, або на колонці з силікагелем, покритим оболонкою, яка складається з хірального полімеру, наприклад, модифікованої амілози, такої як трис-[(S)-αметилбензилкарбамат]амілози або переважно трис-(3,5-диметилфенілкарбамат)амілози, найбільш переважно трис-(3,5-диметилфенілкарбамат)амілози, або модифікованої целюлози, такої як трис-(4-метилбензоат)целюлоза або переважно трис-(3,5диметилфенілкарбамат)целюлоза, найбільш переважно трис-(3,5диметилфенілкарбамат)целюлоза, оболонкою, що покриває силікагель. Можуть застосовуватися інші типи хіральних нерухомих фаз, наприклад, колонки типу Pirkle, доцільна колонка з силікагелем, який ковалентно зв'язаний з амін 3,5динітробензоїлтетрагідрофенантреном. Як елюент для хроматографії з суб- або суперкритичною рухомою фазою може застосовуватися суб- або суперкритичний рухомий діоксид вуглецю, доцільний суперкритичний діоксид вуглецю, що містить модифікатор. Модифікатор вибирається з числа нижчих спиртів, таких як метанол, етанол, пропанол і ізопропанол, або, наприклад, може застосовуватися ацетонітрил. Можуть бути додані амін, такий як діетиламін, при бажанні, 0,1 % діетиламін, триетиламін, пропіламін і диметилізопропіламін, і при бажанні, кислота, така як мурашина кислота, оцтова кислота і трифтороцтова кислота. У додатковому прикладі здійснення винаходу модифікатор вибирається з числа нижчих спиртів, таких як метанол, етанол, пропанол і ізопропанол, або, наприклад, може застосовуватися ацетонітрил, при умові, що модифікатор сумісний з колонкою. Хіральна хроматографія може бути збільшена в масштабі при використанні відповідних технологій, наприклад, технології псевдорухомого шару (SMB), або технології суб- або суперкритичної рухомої фази (порівн.G. B. Cox (ed.) Preparative Enantioselective Chromatography, Blackwell Publishing Ltd., Oxford, UK, 2005). Сполука формули (IVa) потім відновлюється, наприклад, за допомогою комплексного металогідриду, такого як борогідрид, доцільно, за допомогою борогідриду натрію (NaBH 4), або такого як літійалюмогідрид, в розчиннику, такому як спирт (наприклад, С 1-5-спирт), наприклад, етанол або ізопропанол, і переважно при температурі в інтервалі від близько -30 °C до +30 °C, наприклад, при нижче 30 °C, нижче 20 °C, нижче 10 °C або переважно нижче 5 °C, з утворенням сполуки формули (Va) з цис-конфігурацією: 50 3 UA 106191 C2 . 5 Спиртова група цис-спирту формули (Va) перетворюється у придатну групу, що видаляється, таку як, наприклад, галоген, наприклад, Сl або Br, переважно Cl, або сульфонат, наприклад, мезилат (метансульфонілат) або тозилат (4-толуолсульфонілат), по реакції з таким реагентом, як тіонілхлорид, мезил(метансульфоніл)хлорид або тозил(4толуолсульфоніл)хлорид в інертному розчиннику, наприклад, в простому ефірі, доцільно, в тетрагідрофурані. Сполука, що одержується в результаті, має формулу (VI), де LG є групою, що видаляється: 10 . У переважному прикладі здійснення винаходу LG=Cl, тобто цис-хлорид формули (VIa): . 15 20 25 Сполука VI, наприклад, з LG=Cl, далі реагує з 2,2-диметилпіперазином у придатному розчиннику, наприклад, кетон, такому як метилізобутилкетон або метилетилкетон, переважно метилізобутилкетон, в присутності основи, такої як, наприклад, карбонат калію, з одержанням сполуки I. Альтернативно, піперазинова частина молекули може бути введена реакцією сполуки VI із сполукою нижченаведеної формули (VII), де РG є захисною групою, такою як, але не обмежується тільки ними, наприклад, фенілметоксикарбоніл (що часто позначається як Cbz або Z), трет-бутилоксикарбоніл (що часто позначається як ВОС), етоксикарбоніл або бензил, таким чином одержують сполуку нижченаведеної формули (VIII). Подальше зняття захисних груп сполуки VIII дозволяє одержати сполуку I, 4 UA 106191 C2 . 5 Додатковий приклад здійснення винаходу пов'язаний зі способом одержання сполуки [сполука IX: 4-((1R, 3S)-6-хлор-3-феніліндан-1-іл)1,2,2-триметилпіперазин], яка має нижченаведену формулу (IX), або її солей: , 10 15 20 25 способом, який включає: (i) одержання сполуки I способом даного винаходу, тобто, із сполуки (IVa); і (ii) перетворення сполуки I на сполуку IX, переважно метилуванням функціональної групи вторинного аміну, доцільно відновним алкілуванням, використовуючи придатні агенти, такі як, наприклад, формальдегід, параформальдегід, триоксан або діетоксиметан (DEM). Термін відновне алкілування належить до вищезазначених реагентів в поєднанні з таким відновним агентом, як мурашина кислота. Таким чином, додатковий приклад здійснення винаходу пов'язаний зі способом, як описано в цьому документі, одержання сполуки I, в якому сполука I "замінюється" сполукою IX. Сполука IX загалом описана в патенті ЕР 638 073, тоді як енантіомер формули (IX) описаний Bøgesø і ін. в J. Med. Chem., 1995, 38, 4380-4392, у вигляді солі фумарової кислоти, див. таблицю 5, сполука (-)-38. Сполука IX і спосіб одержання сполуки IX із сполуки I, і солі сполуки IX (зокрема, кристалічна сіль янтарної кислоти і кристалічна сіль малонової кислоти), крім того, описані в PCT/DK2004/00545. Як указано вище, винахід також пов'язаний зі способом одержання сполуки I або сполуки IX, як описано в цьому документі, в якому сполука IVb рециклізується, таким чином вона може використовуватися для синтезу сполуки I або сполуки IX, відповідно, дивіться також нижченаведені ілюстрації. 5 UA 106191 C2 5 10 15 20 25 Як ні дивно, рацемізація сполуки IVb може бути досягнута з використанням основ різних типів, наприклад, аміду, переважно діалкіламіду, наприклад, але не обмежуючись тільки ними, діетиламіду літію, діізопропіламіду літію, тетраметилпіперидиду літію, доцільно діізопропіламіду літію (LDA), або біс-силіламіду металу, наприклад, біс(триметилсиліл)аміду лужного металу, або алкоксиду металу, наприклад, але не обмежуючись ними, метоксиду металу, етоксиду металу, трет-бутоксиду металу, доцільно алкоксиду лужного металу, переважно алкоксиду калію, найбільш переважно трет-бутоксиду калію, або металалкілу, доцільно алкіллітію, переважно бутиллітію або трет-бутиллітію. Після охолоджування реакційної суміші може бути виділений рацемічний кетон IV. Рацемізація також може бути досягнута використанням двох різних основ; до того ж, різні типи ненуклеофільних основ можуть використовуватися як "перша основа" (основа 1), наприклад, амід, переважно діалкіламід, наприклад, але не обмежуючись ними, діетиламід літію, діізопропіламід літію, тетраметилпіперидид літію, доцільно діізопропіламід літію (LDA), або біс-силіламід металу, наприклад, біс-силіламід літію, доцільно біс(триметилсиліл)амід літію, або алкоксид металу, наприклад, але не обмежуючись ними, метоксид металу, етоксид металу, трет-бутоксид металу, доцільно, алкоксид лужного металу, переважно алкоксид літію, найбільш переважно трет-бутоксид літію. Після того, як першу основу (основа 1) змішали з кетоном, додавали "другу основу" (основа 2). Як і у випадку з першою основою, можуть використовуватися різні типи основ; наприклад, амід, переважно діалкіламід, наприклад, але не обмежуючись ними, діетиламід літію, діізопропіламід літію, тетраметилпіперидид літію, доцільно діізопропіламід літію (LDA), або біс-силіламід металу, наприклад, біс(триметилсиліл)амід лужного металу, або алкоксид металу, наприклад, але не обмежуючись ними, метоксид металу, етоксид металу, трет-бутоксид металу, доцільно алкоксид лужного металу, переважно алкоксид калію, найбільш переважно трет-бутоксид калію, або металалкіл, доцільно алкіллітій, переважно бутиллітій або трет-бутиллітій. 6 UA 106191 C2 5 10 15 Крім того, рацемізація може бути досягнута використанням двох або більше різних основ додаванням їх всіх саме спочатку, переважно додаванням двох різних основ саме спочатку. У додатковому прикладі здійснення винаходу рацемізація може бути досягнута використанням нуклеофільної основи. Альтернативно, рацемічний спирт V може бути розділений хіральною хроматографією, як це описано в PCT/DK04/000546, з одержанням Va для синтезу сполуки I і Vb, яка може піддаватися рацемізації і повторно використовуватися для розділення, як вказано на нижченаведеній фігурі. Рацемізація Vb досягається окисленням Vb до IVb, наприклад, з використанням хлорхромату піридину (PCC), рацемізацією IVb до IV, як це описано вище, і потім відновленням IV до V звичайним способом, як це описано вище. При вивченні рацемізації на 10 г завантаження речовини можна визначити домішку як побічний продукт за допомогою LC-MS (рідинної хроматографії - мас-спектроскопії); аналітичні дані свідчать про те, що домішка є димером IV і/або енантіомерами IVa і IVb. Аналітичні дані, крім того, вказують, що для димеру можливе елімінування води, в залежності від методу дослідження. Кропітка робота показала, що утворення димеру можна в значній мірі пригнічувати 7 UA 106191 C2 5 10 15 20 25 30 35 40 45 50 55 ретельним підбором умов рацемізації, і вміст димеру в продукті може бути, крім того, знижений перекристалізацією продукту з придатного розчинника, наприклад, спирту, переважно етанолу або 2-пропанолу. У способі синтезу сполуки I може утворитися деяка кількість діастереомеру сполуки I (тобто 1-((1S, 3S)-6-хлор-3-феніліндан-1-іл)-3,3-диметилпіперазину) як домішка до кінцевого продукту. Ця домішка в основному зумовлена утворенням деякої кількості транс-форми (VI) (тобто (1S, 3R)-3,5-дихлор-1-феніліндану, коли LG=Cl) на стадії, коли утворюється сполука VI. Тому домішку можна звести до мінімуму кристалізацією необхідної цис-форми сполуки VI з суміші транс і цис (VI); у випадку, коли LG=Cl в сполуці VI, це може бути зроблено перемішуванням суміші з придатним розчинником, наприклад, алканом (С 5-10-алканом), таким як гептан, внаслідок чого необхідна цис-форма VI випадає в осад і небажана транс-форма сполуки VI переходить в розчин. Необхідна цис-форма сполуки VI (тобто, коли LG=Cl) виділяється, наприклад, фільтруванням, і переважно промивається обговорюваним розчинником і висушується. Цис-форма сполуки I також може бути відділена виділенням сполуки I у вигляді вільної основи з придатного розчинника. Винахід в додаткових аспектах також пов'язаний з проміжними сполуками, як описано в цьому документі для синтезу сполуки I, зокрема з проміжними сполуками IVa і IVb. У цьому контексті зрозуміло при встановленні стереоізомерних форм, що стереоізомер є основним компонентом. Зокрема, при встановленні енантіомерної форми, коли сполука має енантіомерний надлишок, обговорюваного енантіомера. Таким чином, один з прикладів здійснення винаходу пов'язаний із сполукою формули (IVa), що переважно має енантіомерний надлишок принаймні 60 % (60 % енантіомерний надлишок означає, що відношення сполуки IV до її енантіомера становить 80:20 в обговорюваній суміші), принаймні 70 %, принаймні 80 %, принаймні 85 %, принаймні 90 %, принаймні 96 %, переважно принаймні 98 %. Один з прикладів здійснення винаходу пов'язаний, в основному, з чистою сполукою IVа. Додатковий приклад здійснення винаходу пов'язаний із сполукою формули (IVb), що переважно має енантіомерний надлишок принаймні 60 %. Винахід в додатковому аспекті пов'язаний із сполукою I або її сіллю (тобто її сіллю з кислотами НСl, фумаровою, малеїновою), одержаними способом винаходу і з медичним застосуванням їх, зокрема, за медичними показаннями, як це розкрито в даному документі, наприклад, як антипсихотичного засобу, як наприклад при шизофренії. Також у винаході присутня фармацевтична композиція сполуки I або її солі, одержана способом винаходу. У даному контексті, зокрема для фармацевтичного застосування сполуки I, зрозуміло, що коли визначена енантіомерна форма, як відповідна формулі (I), тоді сполука є переважно, загалом і в цілому, стехіометрично чистою, переважно енантіомерний надлишок становить принаймні 60 %, принаймні 70 % і більш переважно принаймні 80 % (80 % енантіомерний надлишок означає, що відношення сполуки I до її енантіомера становить 90:10 в обговорюваній суміші), принаймні 90 %, принаймні 96 %, переважно принаймні 98 %. У переважному прикладі здійснення винаходу діастереомерний надлишок сполуки I становить принаймні 90 % (90 % діастереомерна чистота означає відношення сполуки I до цис-1-((1S, 3S)-6-хлор-3-феніліндан-1іл)-3,3-диметилпіперазину дорівнює 95:5), принаймні 95 %, принаймні 97 % або принаймні 98 %. Таким чином, спосіб за винаходом може включати стадію, в результаті якої сполука I або її сіль складають фармацевтичну композицію. Сполука, сіль або композиція сполуки I можуть вводитися будь-яким зручним способом, наприклад, перорально, трансбукально, сублінгвально, парентерально, і сполука або сіль можуть бути представлені в будь-якій зручній формі для такого введення, наприклад, у формі таблеток, капсул, порошків, сиропів або розчинів, або дисперсій для ін'єкцій. У одному з прикладів здійснення винаходу, сполука або сіль винаходу вводиться у вигляді твердої фармацевтичної форми, доцільно як таблетка або капсула. Способи приготування твердих фармацевтичних препаратів добре відомі з рівня техніки. Так, таблетки можуть бути приготовані змішуванням активного інгредієнта з традиційними допоміжними лікарськими речовинами, наповнювачами і розріджувачами і з подальшим пресуванням суміші у відповідній машині для таблетування. Приклади допоміжних лікарських засобів, наповнювачів і розріджувачів включають кукурудзяний крохмаль, лактозу, тальк, стеарат магнію, желатин, лактозу, камеді і ним подібні. Будь-які інші допоміжні лікарські речовини або добавки, такі як фарбувальні речовини, ароматизуючі речовини, консервувальні речовини і т. д., також можуть використовуватися, при умові, що вони сумісні з активними інгредієнтами. 8 UA 106191 C2 5 10 15 20 25 30 35 40 45 50 55 60 Розчини для ін'єкцій можуть бути приготовані розчиненням солі винаходу і, по можливості, добавок в зразку розчинника для ін'єкції, переважно стерильній воді, доведенням розчину до бажаного об'єму, стерилізацією розчину і заповненням ним відповідних ампул або флаконів. Будь-які придатні добавки, що стандартно застосовуються відповідно до рівня техніки, можуть бути додані, такі як тонізуючі агенти, консервувальні речовини, антиоксиданти, солюбілізуючі агенти і т. д. Щоденна доза сполуки вищенаведеної формули (I), розрахована для вільної основи, доцільна в межах від 1,0 до 160 мг/день, більш доцільна в межах від 1 до 100 мг, наприклад, в межах від 2 до 55 мг. Термін "лікування", як він використовується в цьому документі в зв'язку із захворюванням або розладами, включає також профілактику як можливий випадок. Винахід буде проілюстрований нижченаведеними прикладами, які не обмежують його об'єму. ПРИКЛАДИ АНАЛІТИЧНІ МЕТОДИ Енантіомерний надлишок сполук (IV), (IVa) і (IVb) визначали хроматографією з суперкритичною рухомою фазою, використовуючи хроматограф Gilson SF3 Supercritical Fluid Chromatography System, детекцію здійснювали використанням детектора Gilson UV/VIS-831 при ® 254 нм. Також або використовували хроматографічну колонку CHIRALPAK AD-H, 0,46 см ID х 25 см L при кімнатній температурі при наступних умовах: Елюент:Етанол з 0,1 % діетиламіном як модифікатором (30 %), швидкість потоку 3 мл/хв. і тиск 200 бар. Час утримання двох енантіомерів становить 2,36 хв. (IVa) і 2,99 хв. (IVb); або використовували хроматографічну ® колонку CHIRALCEL ОD-H, 0,46 см ID х 25 см L при кімнатній температурі при наступних умовах: Елюент:Етанол (30 %) як модифікатор, швидкість потоку 4 мл/хв. і тиск 200 бар. Енантіомерний надлишок сполуки (Va) в Прикладі 8 визначали хроматографією з суперкритичною рухомою фазою, використовуючи хроматограф Gilson SF3 Supercritical Fluid ® Chromatography System з хроматографічною колонкою CHIRALPAK AD-H, 0,46 см ID х 25 см L при кімнатній температурі. Елюент:Етанол з 0,1 % діетиламіном як модифікатором (30 %), швидкість потоку 3 мл/хв. і тиск 200 бар. Детекцію здійснювали використанням детектора Gilson UV/VIS-831 при 254 нм. Час утримання двох енантіомерів становить 2,41 хв. (Va) і 3,06 хв. (Vb). Енантіомерний надлишок сполуки (Va) в Прикладі 1а визначали хіральної ВЕРХ, ® використовуючи хроматографічну колонку CHIRALCEL ОD, 0,46 см ID х 25 см L, 10 мкм при 40 °C. Як рухому фазу застосовували н-гексан/етанол 95:5 (об./об.) при швидкості потоку 1,0 мл/хв., детекцію здійснювали з використанням УФ-детектора при 220 нм. Енантіомерний надлишок сполуки (I) визначали за допомогою капілярного електрофорезу (СЕ) на плавленому кварці, використовуючи наступні умови: Капіляр: 50 мкм ID х 48,5 см L, буфер, що проганяється: 1,25 мМ β-циклодекстрин в 25 мМ дигідрофосфаті натрію, рН 1,5, напруга: 16 кВ, температура: 22 °C, введення проби: 40 мбар за 4 сек., детекція: колонкова діодна матрична детекція при 195 нм, концентрація зразка: 500 мкг/мл. У цій системі сполука I має час утримання приблизно 10 хв., і інший енантіомер має час утримання приблизно 11 хв. Енантіомерний надлишок сполуки (IХ) визначали за допомогою капілярного електрофорезу (СЕ) на плавленому кварці, використовуючи наступні умови: Капіляр: 50 мкм ID х 64,5 см L, буфер, що проганяється: 3,0 мМ β-циклодекстрин і 10 мМ гідроксипропіл-β-циклодекстрин в 50 мМ дигідрофосфаті натрію, рН 1,5, напруга: 15 кВ, температура: 22 °C, введення проби: 40 мбар за 4 сек., детекція: колонкова діодна матрична детекція при 192 нм, концентрація зразка: 100 мкг/мл. У цій системі сполука IХ має час утримання приблизно 47 хв., і енантіомер має час утримання приблизно 46 хв. Інші два діастереомери 4-((1R, 3R)-6-хлор-3-феніліндан-1-іл)-1,2,2триметилпіперазин і 4-((1S, 3S)-6-хлор-3-феніліндан-1-іл)-1,2,2-триметилпіперазин мають часи утримання приблизно 49 хв. і 52 хв., відповідно. 1 Н- ЯМР спектри знімали при 500,13 МГц на приладі Bruker Avance AV-500, або на приладі Bruker Avance DRX500, або при 250,13 МГц на приладі Bruker Avance DPX-250, або на приладі Bruker AC-250. Хлороформ (99,8 % D) або диметилсульфоксид (99,8 % D) застосовували як розчинники, і тетраметилсилан (TMS) використовували як внутрішній стандарт. 1 Відношення цис/транс сполуки (I) і сполуки (IХ) визначали, використовуючи Н-ЯМР, як це описане в Bøgesø і ін. в J. Med. Chem., 1995, 38, 4380-4392 (стор. 4388, права колонка). 1 Відношення цис/транс сполуки (VI) визначали за допомогою Н-ЯМР в DMSO-d6, використовуючи інтеграли сигналу при 5,6 м. ч. для цис-ізомеру і сигналу при 5,75 м. ч. для 1 транс-ізомеру, або за допомогою Н-ЯМР в хлороформі, використовуючи інтеграли сигналу при 5,3 м. ч. для цис-ізомеру і сигналу при 5,5 м. ч. для транс-ізомеру. Звичайно, вміст небажаного ізомеру, що приблизно складає 1 %, може бути визначений за допомогою ЯМР. 9 UA 106191 C2 5 10 15 20 25 30 35 40 45 50 55 60 Температури плавлення вимірювали з використанням Диференціального Скануючого Калориметра (DSC). Прилад являв собою TA-Instruments DSC-Q1000 або TA-Instruments DSC2920, відкалібрований при 5°/хв., щоб показувати температуру плавлення як початкове значення температури процесу плавлення. Близько 2 мг зразка нагрівали зі швидкістю 5°/хв. в нещільно закритому резервуарі в потоці азоту. Елементний аналіз здійснювали, використовуючи Vario El аналізатор, настроєний на вимірювання вмісту елементів С, Н і N. Одержана величина являла собою середнє з двох визначень, в кожному з яких використовувалося приблизно 4 мг речовини. Оптичне обертання вимірювали, використовуючи поляриметр Perkin Elmer модель 241, встановлювалася концентрація речовини 1 % в метанолі, за винятком інших випадків. Рідинна хроматографія - мас-спектрометрія (LC-MS) проводилася з використанням колонки Waters Symmetry С-18, 0,46 см ID х 3 см L, 3,5 мкм, при 60 °C. Елюентом є градієнт (А) вода з 0,05 % трифтороцтовою кислотою і (В) ацетонітрил з 5 % водою і 0,035 % трифтороцтовою кислотою, що починається з 90 % А і 10 % В і закінчується 100 % В за 2 хв., при швидкості елюювання 3 мл/хв. Детекцію здійснювали з використанням детектора Shimadzu при 254 нм. Мас-спектр реєстрували на мас-спектрометрі Sciex API300. СИНТЕЗ Синтез ключових вихідних речовин Сполуку V синтезували зі сполукою IV відновленням борогідридом натрію (NaBH 4), застосування методу описане в Bøgesø J. Med. Chem., 1983, 26, 935, з використанням етанолу як розчинника і проведенням реакції приблизно при 0 °C. Обидві сполуки описані в Bøgesø і ін., J. Med. Chem., 1995, 38, 4380-4392. Сполуку IV синтезували зі сполукою II, застосовуючи загальну методику, описану в Sommer і ін., J. Org. Chem. 1990, 55, 4822, який також описує сполуку II і її синтез. Приклад 0а Синтез (S)-6-хлор-3-феніліндан-1-ону (IVа) і (R)-6-хлор-3-феніліндан-1-ону (IVb) із застосуванням хіральної хроматографії Рацемічний 6-хлор-3-феніліндан-1-он (IV) розділяли препаративною хроматографією, ® використовуючи колонку CHIRALPAK AS-V. Суміш н-гептану, етанолу і N, N-діетиламіну застосовували як рухому фазу, детекцію здійснювали з використанням УФ-детектора при 220 нм. Рацемічний кетон (IV) вводився у вигляді розчину в елюенті; прийнятні об'єми розчину вводили через доцільні інтервали. Всі фракції, які містять сполуку (IVa) з більше ніж 98 % енантіомерним надлишком, об'єднувати упарювали досуха, використовуючи роторний випарник. Всі фракції, які містять сполуку (IVb) або суміш сполук (IVa) і (IVb), об'єднували і упарювали досуха, використовуючи роторний випарник. Приклад 0b Синтез енантіомерно чистого (R)-6-хлор-3-феніліндан-1-ону (IVb) окисленням (1R, 3R)-6хлор-3-феніліндан-1-олу (Vb) (1R, 3R)-6-хлор-3-феніліндан-1-ол (Vb), виділений, як в прикладі 1а, (20 г) розчиняли в дихлорметані (400 мл) і додавали піридинхлорхромат (РСС) (26,5 г). Суміш перемішували 11/2 години при кімнатній температурі. Суміш фільтрували, маслоподібний залишок в реакційній посудині промивали дихлорметаном. Об'єднані органічні фракції упарювали досуха на роторному випарнику, одержували масло чорного кольору (25 г). Додавали етилацетат (200 мл) і гідроксид натрію (2М у воді, 200 мл). Фази розділяли, водну фазу екстрагували двічі етилацетатом (200 мл). Об'єднані органічні фази промивали тричі гідроксидом натрію (2М у воді, 100 мл), двічі водою (100 мл) і один раз - сольовим розчином (100 мл), і на закінчення сушили сульфатом натрію. За упарюванням досуха йшло сушіння у вакуумному термостаті при 20 40 °C, в результаті одержували 15 г кристалічної речовини. [α] D -61° (с=1,0, метанол). 90 % енантіомерний надлишок, згідно з даними хірального аналізу. Приклад 0с Рацемізація (R)-6-хлор-3-феніліндан-1-ону (IVb) Діізопропіламін (5,1 мл) розчиняли в сухому тетрагідрофурані (THF) (50 мл) і розчин перемішували під азотом при охолоджуванні в бані ацетон/сухий лід. Повільно додавали бутиллітій (1,6 М в гексані, 22,6 мл), причому після охолоджування баню замінювали на лід/вода. Після перемішування протягом 11/2 години додавали протягом 30 хвилин синтезований в прикладі 0b (R)-6-хлор-3-феніліндан-1-он (IVb) (7,05 г, 90 % енантіомерний надлишок), розчинений в сухому THF (60 мл), і перемішування на охолоджувальній бані продовжували 17 хв. Потім додавали трет-бутоксид калію (1,0 М в THF, 28,8 мл) за 17 хвилин, і потім перемішування продовжувалося протягом ще двох годин на бані лід/вода. Реакційну суміш нейтралізували соляною кислотою (4М, 50 мл) і потім THF видаляли з суміші на 10 UA 106191 C2 5 10 15 20 25 30 35 40 45 50 55 роторному випарнику. Додавали воду (200 мл) і діетиловий ефір (350 мл), і фази розділяли. Водну фазу двічі екстрагували діетиловим ефіром (200 мл, потім 100 мл). Об'єднані органічні фази промивали двічі водою (100 мл), один раз - сольовим розчином (100 мл) і сушили сульфатом натрію. За упарюванням досуха на роторному випарнику йшло висушування у 20 вакуумі при 40 °C, в результаті одержували 6,70 г твердої речовини червоного кольору. [α] D 2,34° (с=1,0, метанол). Продукт мав енантіомерний надлишок 2 %, відповідно до хірального аналізу, і містив 6 % побічного продукту (див. основний текст), згідно з даними ВЕРХ. Неочищений продукт (4,99 г) перекристалізовували з абсолютного етанолу (40 мл), в результаті 20 одержували 3,71 г твердої речовини червоного кольору. [α] D -0,84° (с=1,0, метанол). Вміст побічного продукту становив 2,6 % (див. основний текст), згідно з даними ВЕРХ. Приклад 1а Синтез (1S, 3S)-6-хлор-3-феніліндан-1-олу (Va) і (1R, 3R)-6-хлор-3-феніліндан-1-олу (Vb) з використанням хіральної хроматографії Рацемічний цис-6-хлор-3-феніліндан-1-ол (Vb) (492 г) розділяли препаративною ® хроматографією, використовуючи колонку CHIRALPAK AD, 10 см ID х 50 см L, 10 мкм при 40 °C. Метанол використовували як мобільну фазу при швидкості потоку 190 мл/хв., детекцію здійснювали з використанням УФ-детектора при 287 нм. Рацемічний спирт (V) вводився у вигляді 50000 м. ч. розчину в метанолі; 90 мл вводилися з інтервалами в 28 хв. Всі фракції, які містили сполуку (Va) з енантіомерним лишком більше ніж 98 %, об'єднували і упарювали досуха, використовуючи роторний випарник, потім йшло висушування у вакуумі при 40 °C. Вихід твердої речовини становив 220 г. Дані елементного аналізу і ЯМР-спектра відповідали структурі, енантіомерний надлишок перевищував 98 %, згідно з даними хіральної ВЕРХ, 20 [α]D +44,5° (с=1,0, метанол). Аналогічним чином, фракції, які містять сполуку (Vb), об'єднували і упарювали досуха, одержуючи 214 г сполуки (Vb). Приклад 1b Синтез (1S, 3S)-6-хлор-3-феніліндан-1-олу (Va) шляхом відновлення енантіомерно чистої сполуки (IVa) (S)-6-хлор-3-феніліндан-1-он (IVa) шляхом відновлення борогідридом натрію може бути перетворений на сполуку (Va), застосовуючи спосіб, описаний в Bøgesø J. Med. Chem., 1983, 26, 935, використовуючи етанол як розчинник і здійснюючи реакцію приблизно при 0 °C. Приклад 2 Синтез (1S, 3S)-3,5-дихлор-1-феніліндану (VI, LG=Cl) Цис-(1S, 3S)-6-хлор-3-феніліндан-1-ол (Va) (204 г), одержаний, як описано в Прикладі Iа, розчиняли в THF (1500 мл) і охолоджували до -5 °C. Тіонілхлорид (119 г) додавали по краплях у вигляді розчину в THF (500 мл) протягом 1 год. Суміш перемішували при кімнатній температурі протягом ночі. До реакційної суміші додавали лід (100 г). Коли лід розтанув, розділяли водну фазу А і органічну фазу В, і органічну фазу В двічі промивали насиченим розчином бікарбонату натрію (200 мл). Фази з бікарбонатом натрію об'єднували з водною фазою А, доводили до рН 9 гідроксидом натрію (28 %), і використовували ще раз для однократного промивання органічної фази В. Одержану водну фазу С і органічну фазу В розділяли, і водну фазу С екстрагували етилацетатом. Етилацетатну фазу об'єднували з органічною фазою В, сушили сульфатом магнію і упарювали досуха, використовуючи роторний випарник, одержували згадану в заголовку сполуку у вигляді масла. Вихід 240 г, які повністю використали в прикладі 5. Цис/транс відношення 77:23, згідно з даними ЯМР. Приклад 3 Синтез 3,3-диметилпіперазин-2-ону Карбонат калію (390 г) і етилендіамін (1001 г) перемішували з толуолом (1,50 л). Додавали розчин етил-2-бромізобутирату (500 г) в толуолі (750 мл). Суспензію нагрівали із зворотним холодильником протягом ночі і фільтрували. Осад на фільтрі промивали толуолом (500 мл). Об'єднані фільтрати (об'єм 4,0 л) нагрівали на водяній бані і переганяли при 0,3 атм, використовуючи насадку Кляйзена; перші 1200 мл дистиляту збирали при 35 °C (температура в суміші 75 °C). Додатково додавали толуол (600 мл) і інші 1200 мл дистиляту збирали при 76 °C (температура в суміші 80 °C). Знову додавали толуол (750 мл) і 1100 мл дистиляту збирали при 66 °C (температура в суміші 71 °C). Суміш перемішували на льодяній бані і вводили кристалзатравку, внаслідок чого продукт випадав в осад. Продукт відділяли фільтруванням, промивали толуолом, сушили протягом ночі у вакуумному термостаті при 50 °C. Вихід 3,3диметилпіперазин-2-ону 171 г (52 %). ЯМР-спектр відповідає структурі. 11 UA 106191 C2 5 10 15 20 25 30 35 40 45 50 55 Приклад 4 Синтез 2,2-диметилпіперазину Суміш 3,3-диметилпіперазин-2-ону (8,28 кг, 64,6 моль) і тетрагідрофурану (THF) (60 кг) нагрівали до 50-60 °C, одержували злегка каламутний розчин. THF (50 кг) перемішували під азотом і LiAlH4 (250 г в розчинній пластиковій упаковці) додавали, що приводило до слабого виділення газу. Після припинення виділення газу додавали ще LiAlH 4 (загальна використана кількість 3,0 кг, 79,1 моль), і внаслідок екзотермічної реакції температура зростала від 22 °C до 50 °C. Розчин 3,3-диметилпіперазин-2-ону повільно додавали протягом 2 годин при 41-59 °C. Суспензію перемішували ще протягом години при 59 °C (температура нагрівальної оболонки 60 °C). Суміш охолоджували і додавали воду (3 л) протягом двох годин, підтримуючи температуру нижче 25 °C (необхідно охолоджувати зовнішньою охолоджуючою оболонкою з температурою 0 °C). Потім додавали гідроксид натрію (15 %, 3,50 кг) протягом 20 хв. при 23 °C, охолоджування необхідне. Ще додатково додавали воду (9 л) протягом півгодини (охолодження необхідне), і суміш перемішували протягом ночі під азотом. Додавали фільтраційну речовину Celit (4 кг) і суміш фільтрували. Осад на фільтрі промивали THF (40 кг). Об'єднані фільтрати концентрували в реакторі, поки температура в реакторі не досягала 70 °C (температура дистиляції 66 °C) при 800 мбар. Залишок (12,8 кг) далі концентрували на роторному випарнику приблизно до 10 л. На закінчення, суміш фракціонували дистиляцією при атмосферному тиску і продукт збирали при 163-4 °C. Вихід 5,3 кг (72 %). ЯМР-спектр відповідає структурі. Приклад 5 Синтез транс-1-((1R, 3S)-6-хлор-3-феніліндан-1-іл)-3,3-диметилпіперазину (сполука I) кислої солі малеїнової кислоти Цис-(1S, 3S)-3,5-дихлор-1-феніліндан (VI, LG=Cl) (240 г) розчиняли в бутан-2-оні (1800 мл). Карбонат калію (272 г) і 2,2-диметилпіперазин (одержаний в Прикладі 4) (113 г) додавали і суміш нагрівали із зворотним холодильником до температури кипіння протягом 40 год. До реакційної суміші додавали діетиловий ефір (2 л) і соляну кислоту (1М, 6 л). Фази розділяли і рН водної фази знижували від 8 до 1 за допомогою концентрованої соляної кислоти. Водна фаза використовувалася для ще одного промивання органічної фази, щоб пересвідчитися, що весь продукт у водній фазі. Гідроксид натрію (28 %) додавали до водної фази доти, поки рН не стане дорівнювати 10, і водну фазу двічі екстрагували діетиловим ефіром (2 л). Екстракти з діетиловим ефіром об'єднували, сушили сульфатом натрію і упарювали досуха, використовуючи роторний випарник. Вихід згаданої в заголовку сполуки становив 251 г у вигляді масла. Цис/транс відношення становить 18:82, відповідно до даних ЯМР-спектра. Неочищене масло (прибл. 20 г) додатково очищали флеш-хроматографією на силікагелі (елюент: етилацетат/етанол/триетиламін 90:5:5), за якою йшло упаривание досуха на роторному випарнику. Вихід 12 г озаглавленої сполуки у вигляді масла (цис/транс відношення становить 10:90, відповідно до даних ЯМР-спектра). Масло розчиняли в етанолі (100 мл), і до розчину додавали розчин малеїнової кислоти в етанолі до досягнення рН 3. Одержану суміш перемішували при кімнатній температурі протягом 16 годин, і осад, що утворився, відділяли фільтруванням. Об'єм етанолу зменшували і іншу порцію осаду відділяли. Вихід твердої речовини (цис-ізомер не виявлявся, відповідно до даних ЯМР-спектра) згаданої в заголовку сполуки становив 3,5 г. Енантіомерний надлишок відповідно до даних СЕ становить >99 %. Температура плавлення 175-178 °C. ЯМР-спектр відповідає структурі. Приклад 6 Скринінг умов розділення 6-хлор-3-феніліндан-1-ону (IV) хроматографією з суперкритичною рухомою фазою Проводили скринінг серії колонок на можливість розділення сполуки (IV), використовуючи хроматограф Gilson SF3 Supercritical Fluid Chromatography System. Елюент: Різні розчинники з 0,1 % діетиламіном як модифікатором (30 %), швидкість потоку 4 мл/хв. і тиск 200 бар, і колонка працювала при кімнатній температурі. Детекцію здійснювали з використанням детектора Gilson UV/VIS-831 при 254 нм. Час утримання двох енантіомерів (RT 1 і RT2) і ширину кожного з двох піків на середині висоти (ω1 і ω2) розраховували з використанням програмного забезпечення Gilson Unipoint, версія 3,2. Нижченаведена таблиця містить розраховані величини розділення (Rs) для окремих колонок, як серійних, так і модифікованих; Rs розраховували за формулою: Rs=2(RT2-RT1)/(ω1 + ω2). 12 UA 106191 C2 Колонка Найменування ® Chiralpak AD-H ® Chiralpak AS-H ® Chiralcel OD-H ® Chiralcel OJ-H ® Chiralpak IA ® (R, R)-Whelk-O 1 Rs Геометричні а параметри 4,6, 250, 5 4,6, 250, 5 4,6, 250, 5 4,6, 250, 5 4,6, 250, 5 4,6, 250, 5 Метанол Етанол Ізопропанол Ацетонітрил 17,1 3,5 4,0 0,7 5,4 1,0 10,0 4,0 3,2 0,0 2,8 1,1 2,0 4,9 2,8 0,0 0,6 1,7 19,1 3,6 2,6 а Внутрішній діаметр (мм), довжина колонки (мм), розмір частинок (мкм) 5 10 15 20 25 30 35 40 45 Приклад 7 Розділення 6-хлор-3-феніліндан-1-ону (IV) хроматографією з суперкритичною рухомою фазою Розділення здійснювали, використовуючи хроматограф Berger Multigram II Prep-SFC з ® колонкою CHIRALPAK AD-H, 20 мм ID х 250 мм L, 5 мкм. Елюент:Етанол використовували як модифікатор (20 %), швидкість потоку 50 мл/хв. і тиск 100 бар, і колонка працювала при 35 °C. Детекцію здійснювали з використанням УФ-детектора при 230 нм. Застосовували Berger пристрій для збирання фракцій і для декомпресії. Робота апаратури контролювалася програмним забезпеченням SFC Pronto. Два енантіомери мали час утримання 3,9 хв. (IVa) і 4,8 хв. (IVb). Рацемічний кетон (IV) вводився у вигляді розчину в ацетонітрилі (55 г (IV) в 800 мл ацетонітрилу); 500 мл цього розчину вводилися з інтервалами 132 сек. Всі фракції, що містять сполуку (IVa), об'єднували і декомпресія дозволяла одержати розчин (IVa) в етанолі, і всі фракції, що містять сполуку (IVb), об'єднували і також піддавали декомпресії. Сполуку (IVa) виділяли упарюванням розчину на роторному випарнику і висушуванням залишку у вакуумному термостаті при 40 °C. Вихід 25,6 г (47 %) твердої речовини. Температура 20 плавлення 110,8 °C, ЯМР-спектр відповідає структурі, [α]D +72,65° (с=1,0, метанол). Вміст С, Н, розраховано для С15Н11OCl: С - 74,23, Н - 4,57; знайдено: С - 74,09, Н - 4,70. Енантіомерний надлишок >99 % згідно з даними хірального аналізу. Сполуку (IVb) виділяли таким же чином, одержали 23,9 г (43 %) твердої речовини. 20 Температура плавлення 110,6 °C. ЯМР-спектр відповідає структурі, [α]D -70,33° (с=1,0, метанол). Вміст С, Н, розраховано для С15Н11OCl: С - 74,23, Н - 4,57; знайдено: С - 73,79, Н 4,70. Енантіомерний надлишок >99 % згідно з даними хірального аналізу. Приклад 8 Синтез (1S, 3S)-6-хлор-3-феніліндан-1-олу (Va) відновленням енантіомерно чистої сполуки (IVa) (S)-6-хлор-3-феніліндан-1-он (IVa) (виділений, як в прикладі 7(23 г)) додавали маленькими порціями до суспензії борогідриду натрію (1,6 г) в етанолі (160 мл) при 3-5 °C. Після того, як додавання було завершене, суміш залишали до досягнення кімнатної температури. Реакційну суміш перемішували 2,75 годин, після чого упарювали досуха. Залишок розчиняли в суміші води (150 мл) і етилацетату (200 мл), фази розділяли і водну фазу екстрагували етилацетатом (100 мл). Органічні фази об'єднували, промивали водою (100 мл), сушили сульфатом магнію, фільтрували і упарювали досуха. Залишок перекристалізовували з гептану (250 мл), в результаті одержували 20,9 г (90 %) згаданого в заголовку продукту у вигляді твердої речовини. 20 Температура плавлення 108,9 °C, ЯМР-спектр відповідає структурі, [α]D +48,30 ° (с=1,0, метанол). Вміст С, Н, розраховано для С15Н13OCl: С - 73,62, Н - 5,35; знайдено: С - 73,55, Н 5,29. Енантіомерний надлишок >99 % згідно з даними хірального аналізу. Приклад 9 Синтез (1S, 3S)-3,5-дихлор-1-феніліндану (VI, LG=Cl) Розчин (1S, 3S)-6-хлор-3-феніліндан-1-олу (Va) (17 г) (синтезований, як в прикладі 8) в тетрагідрофурані (130 мл) охолоджували в льодяній бані. Тіонілхлорид (9,9 г) в тетрагідрофурані (50 мл) додавали по краплях при 4-5 °C, і потім суміш перемішували протягом ночі при температурі навколишнього середовища. Додавали суміш води з льодом (приблизно 25 мл) і перемішування продовжували доти, поки розтанув весь лід. Фази розділяли, органічну фазу двічі промивали бікарбонатом натрію (5 % у воді, 25 мл). Водні фази потім об'єднували, екстрагували органічною фазою і потім екстрагували етилацетатом (50 мл). Органічні фази потім об'єднували, сушили сульфатом магнію і упарювали досуха, використовуючи роторний випарник, одержували озаглавлену сполуку у вигляді масла. Вихід 18,7 г (102 %) неочищеної 13 UA 106191 C2 5 10 15 20 25 30 35 40 45 50 55 60 суміші (1S, 3S)-3,5-дихлор-1-феніліндану та (1S, 3R)-3,5-дихлор-1-феніліндану. Вміст (1S, 3S)3,5-дихлор-1-феніліндану становить 18 % за даними ЯМР-спектра (в неочищеній суміші або в перерахунку на чисту суміш діастереомерів). Приклад 10 Синтез транс-1-((1R, 3S)-6-хлор-3-феніліндан-1-іл)-3,3-диметилпіперазину (сполука I) Суміш (1S, 3S)-3,5-дихлор-1-феніліндану (VI, LG=Cl) (18 г) (синтезованого, як в прикладі 9), карбонату калію (20,8 г), 2,2-диметилпіперазину і метилетилкетону (135 мл) нагрівали до кипіння із зворотним холодильником протягом ночі. Після охолоджування до кімнатної температури додавали діетиловий ефір (150 мл) і соляну кислоту (1М, 450 мл), і суміш перемішували декілька хвилин. Фази розділяли і рН водної фази доводили від 1 до 12, використовуючи гідроксид натрію (28 %). Водну фазу екстрагували діетиловим ефіром (два рази 170 мл). Всі органічні фази об'єднували, сушили сульфатом магнію, фільтрували і упарювали, використовуючи роторний випарник. Вихід 20,7 г (89 %) озаглавленої сполуки у вигляді масла. Вміст цис-ізомеру становить 19 % відповідно до даних ЯМР-спектра. Приклад 11 Синтез транс-4-((1R, 3S)-6-хлор-3-феніліндан-1-іл)-1,2,2-триметилпіперазину (IX) кислої солі фумарової кислоти Транс-1-((1R, 3S)-6-хлор-3-феніліндан-1-іл)-3,3-диметилпіперазину (I, синтезована, як в прикладі 10) перемішували з мурашиною кислотою (15,2 мл) і формальдегідом (37 % у воді, 12,5 мл) і нагрівали на масляній бані (температура 110 °C) протягом 11/2 години. Воду додавали до реакційної суміші після охолоджування до кімнатної температури, рН доводили приблизно до 14 гідроксидом натрію (28 %). Продукт екстрагували діетиловим ефіром і потім етилацетатом, додаючи гідроксид натрію (28 %) між екстракціями, якщо рН понижчався за 12. Органічні фази об'єднували, сушили сульфатом натрію, фільтрували і упарювали досуха, використовуючи роторний випарник. Вихід 10,9 г (100 %) сполуки (IX) у вигляді масла, що містить 20 % цис-форми, відповідно до даних ЯМР-спектра. Маслоподібну речовину (10 г) нагрівали з 1-пропанолом (150 мл), одержуючи розчин. Фумарову кислоту (3,3 г) додавали і нагрівання продовжували до повного розчинення. Суміш охолоджували до кімнатної температури і вносили кристал-затравку, внаслідок чого продукт випадає в осад. Тверду речовину відділяли фільтруванням, промивали невеликою кількістю 1пропанолу і сушили у вакуумному термостаті при 40 °C. Вихід 6,85 г (52 %). Температура 20 плавлення 193,3 °C, ЯМР-спектр відповідає структурі, [α]D -15,2° (с=1,0, метанол). Містить 4 % цис-форми згідно з СЕ, два інші діастереомери не детектувалися (тобто їх вміст нижче 1 %). Вміст С, Н, N розрахований для С26Н31N2О4Cl: С - 66,30, Н - 6,63, N-5,95; знайдено: С - 65,96, Н 6,61, N-5,57. Енантіомерний надлишок >98 % відповідно до даних СЕ. Приклад 12 Синтез енантіомерно чистого (R)-6-хлор-3-феніліндан-1-ону (IVb) окисленням (1R, 3R)-6хлор-3-феніліндан-1-олу (Vb) Хлорхромат піридину (66,1 г) додавали до розчину (1R, 3R)-6-хлор-3-феніліндан-1-олу (Vb) (виділеного, як в прикладі 1а) (50,0 г) в дихлорметані (840 мл), і суміш перемішували при температурі навколишнього середовища дві години. Суміш фільтрували і залишок в реакційній судині промивали двічі дихлорметаном (200 мл), який також застосовували для промивання осаду на фільтрі. Об'єднані фільтрати упарювали досуха, використовуючи роторний випарник. Залишок перемішували з гідроксидом натрію (2М, 1 л) і етилацетатом (750 мл) протягом 1/2 години. Фази розділяли і водну фазу екстрагували етилацетатом (500 мл). Об'єднані органічні фази промивали двічі гідроксидом натрію (2М, 250 мл) і 25 % хлоридом натрію (250 мл). Потім органічну фазу перемішували з сульфатом магнію (60 г), активованим вугіллям (1,4 г) і силікагелем 60 (0,06-0,2 мм, 5 г), фільтрували і упарювали досуха, використовуючи роторний випарник. Залишок (31,5 г) перекристалізовували з 2-пропанолу (125 мл); продукт виділяли фільтруванням і промивали 2-пропанолом (40 мл). Висушування у вакуумному термостаті при 50 °C приводило до одержання 26,0 г (53 %) продукту у вигляді твердої речовини. Температура 20 плавлення 110,8 °C, ЯМР-спектр відповідає структурі, [α]D -75,6° (с=1,0, метанол). Вміст С, Н, N, розрахований для С15Н11ОCl: С - 74,23, Н - 4,57, N-0,00; знайдено: С - 73,89, Н - 4,71, N-0,05. Енантіомерний надлишок 99,2 % згідно з даними хірального аналізу. Реакцію повторювали двічі, одержуючи 48 г продукту з енантіомерним надлишком 99,6 % і 48 г продукту з енантіомерним надлишком 98,9 %, відповідно. Приклад 13 Скринінг основ для рацемізації (R)-6-хлор-3-феніліндан-1-ону (IVb) Використовували основи фірми Aldrich: бутил-літій (BuLi) номер за каталогом 18,617-1, третбутил-літій (tBuLi) номер за каталогом 18,619-8, трет-бутоксид калію (КОtBu) номер за 14 UA 106191 C2 5 10 15 20 каталогом 32,865-0, трет-бутоксид літію (LiОtBu) номер за каталогом 398195, трет-бутоксид натрію (NaОtBu) номер за каталогом 35,927-0, біс(триметилсиліл)амід літію (LiHMDS) номер за каталогом 225770, біс(триметилсиліл)амід натрію (NaHMDS) номер за каталогом 24,558-5, і біс(триметилсиліл)амід калію (КHMDS) номер за каталогом 324671. Діізопропіламід літію одержували з діізопропіламіду (Sigma-Aldrich номер за каталогом 386464) і BuLi безпосередньо перед застосуванням в кожному експерименті. Нижченаведена методика є типовою для експериментів і ілюструє застосування LDA і застосування двох різних основ: Суміш діізопропіламіду (437 мкл) і тетрагідрофурану (THF) (4 мл) перемішували в потоці азоту і охолоджували на бані з сухим льодом і ацетоном. Бутил-літій (BuLi) (1,6 М в гексані, 1,60 мл) додавали протягом 5 хвилин. Перемішування продовжували 10 хвилин і потім замінювали охолоджувальну баню на водно-льодяну баню. Після перемішування ще протягом 10 хв. додавали розчин (R)-6-хлор-3-феніліндан-1-ону (IVb) (синтезованого, як в прикладі 12) (0,50 г) в THF (4 мл) по краплях протягом 5 хвилин до розчину LDA ("перша основа", основа 1), і перемішування на водно-льодяній бані продовжували 1/2 години. Потім додавали BuLi (1,6 М в гексані) (1,61 мл) ("друга основа", основа 2) по краплях протягом 5 хвилин, а після 1 перемішування на водно-льодяній бані, що продовжувалося 2 /2 години, додавали соляну кислоту (4М, 4 мл). Після перемішування протягом 10 хв. фази розділяли і водну фазу екстрагували етилацетатом (два рази по 10 мл). Об'єднані органічні фази промивали хлоридом натрію (25 %, 10 мл), сушили сульфатом магнію, фільтрували і упарювали досуха, використовуючи роторний випарник. Вихід 0,47 г (94 %) маслоподібної речовини, хімічна чистота 83 %, згідно з даними LC-MS, і енантіомерний надлишок становить 1 % згідно з даними хірального аналізу. Нижченаведена таблиця підсумовує одержані результати: 25 Запис в журнал Еквівалент основи 1 Основа 1 1 2 3 4 5 6 7 8 9 10 11 12 6) 13 6) 14 6) 15 2,25 2,25 2,25 2.25 1,25 1,25 1,25 1,25 1,25 1,25 1,25 1,25 1,25 1,25 1,25 LDA KOtBu BuLi tBuLi 4) LDA 4) LDA 4) LDA LiHMDS LiOtBu 5) LDA 5) LDA 5) LDA 5) LDA 5) LDA 5) LDA 3) 1) Еквівалент основи 2 Основа 2 Час год. N/А N/А N/А N/А 1,25 1 1,25 1 1 1 1 1 1 1 1 N/А N/А N/А N/А BuLi KOtBu tBuLi KOtBu KOtBu BuLi BuLi BuLi BuLi BuLi BuLi 2,5 2,5 2,5 2,5 2,5 2,5 2,5 2,5 2,5 0,5 1 2,5 0,5 1 2,5 1) Чистота сировинного продукту % 91 38 39 40 83 88 88 86 93 82 93 90 85 85 86 Енантіомерний надли2) шок % 2 -10 5 -13 1 0 -4 1 0 2 2 -3 2 2 -1 Вказаний час перемішування при 0 °C після змішування всіх компонентів. Енантіомерний надлишок; негативний знак вказує, що IV в надлишку, домішки в зразку можуть заважати аналізу. 3) LDA одержували, використовуючи 2,50 еквівалентів діізопропіламіну і 2,25 еквівалентів BuLi. 4) LDA одержували, використовуючи 1,50 еквівалентів діізопропіламіну і 1,25 еквівалентів BuLi. 5) LDA одержували, використовуючи 1,25 еквівалентів діізопропіламіну і 1,50 еквівалентів BuLi. 6) У цьому експерименті весь BuLi, вказаний також як кількість основи 2, додавали на початку експерименту, до додання сполуки IVb. 2) 15 UA 106191 C2 5 10 15 20 Приклад 14 Збільшення рацемізації (R)-6-хлор-3-феніліндан-1-ону (IVb) Використані основи такі ж, як в прикладі 13. Типова методика є наступною: Діізопропіламін (6,25 г) розчиняли в тетрагідрофурані (THF) (160 мл) і суміш охолоджували на бані сухий лід/ацетон під час перемішування під азотом. Бутиллітій (1,6 М в гексані, 33 мл) повільно додавали, підтримуючи температуру нижче -60 °C. Перемішування продовжували 5 хвилин на бані сухий лід/ацетон, яку потім замінювали банею лід/вода. Суміш перемішували 10 хв. при температурі від -10 до 0 °C, після чого повільно додавали розчин (R)-6-хлор-3феніліндан-1-ону (IVb) (синтезований, як в прикладі 12) (10,0 г) в THF (80 мл), підтримуючи температуру нижче 5 °C. Після перемішування приблизно протягом 1/2 години повільно додавали бутиллітій (1,6 М в гексані, 33 мл), підтримуючи температуру нижче 5 °C. Після перемішування при 0-5 °C протягом 21/2 години повільно додавали соляну кислоту (4М, 100 мл). Фази розділяли і водну фазу екстрагували два рази етилацетатом (100 мл). Об'єднані органічні фази промивали 25 % хлоридом натрію (100 мл) і перемішували 10 хвилин з сульфатом магнію (26 г), активованим вугіллям (1 г) і силікагелем (2,6 г). Після фільтрування органічну фазу упарювали досуха, використовуючи роторний випарник. Залишок перекристалізовували з 2-пропанолу (40 мл). Продукт відділяли фільтруванням, промивали льодяним 2-пропанолом (20 мл) і сушили у вакуумному термостаті при 50 °C протягом ночі. 20 Вихід 5,85 г (60 %). Температура плавлення 95,2 °C, ЯМР-спектр відповідає структурі, [α]D 1,1 ° (с=1,0, метанол). Вміст С, Н, розраховано для С15Н11ОCl: С - 74,23, Н - 4,57; знайдено: С 74,29, Н - 4,62. Енантіомерний надлишок 1,0 % згідно з даними хірального аналізу. Чистота 97 % згідно з LC-MS. Нижченаведена таблиця підсумовує одержані результати: 25 Запис в журнал 1 2 3 4 5 6) 6 Енантіо1) Еквівалент Еквівалент Час мерний Основа 1 Основа 2 Вихід % Чистота % 2) основи 1 основи 2 год. надлишок % 3) 2,25 LDA N/А N/А 2,5 53 97 -3 4) 1,25 LDA 1,25 BuLi 2,5 60 97 -1 4) 1,25 LDA 1 KOtBu 2,5 76 87 0 1,25 LiOtBu 1 KOtBu 2,5 70 79 -2 5) 1,25 LDA 1 BuLi 2,5 70 97 0 6) 1 1,25 LDA 1,25 BuLi /2 59 96 0 1) Вказаний час перемішування при 0 °C після змішування всіх компонентів. Енантіомерний надлишок; негативний знак вказує, що IV в надлишку. 3) LDA одержували, використовуючи 2,50 еквівалентів діізопропіламіну і 2,25 еквівалентів BuLi. 4) LDA одержували, використовуючи 1,50 еквівалентів діізопропіламіну і 1,25 еквівалентів BuLi. 5) LDA одержували, використовуючи 1,25 еквівалентів діізопропіламіну і 1,50 еквівалентів BuLi. 6) У цьому експерименті весь BuLi, вказаний також як кількість основи 2, додавали на початку експерименту, до додання сполуки IVb, тобто застосовували 1,25 еквівалентів діізопропіламіну і всього 2,50 еквівалентів BuLi. 2) ФОРМУЛА ВИНАХОДУ 30 1. Спосіб одержання сполуки формули І (сполука І) або її солі, в якому здійснюють: одержання сполуки IVa розділенням рацемічної сполуки IV з використанням хіральної хроматографії 16 UA 106191 C2 O O Cl Cl , (IV) , (IVa) відновлення сполуки формули IVa в присутності боргідриду у розчиннику у відповідний спирт Va з цис-конфігурацією OH Cl 5 , (Va) перетворення в присутності тіонілхлориду, тіонілброміду, мезил(метансульфоніл)хлориду або тозил(4-толуолсульфоніл)хлориду спиртової групи цис-спирту формули Va в групу LG, що видаляється, з одержанням сполуки формули VI LG Cl , (VI) взаємодію сполуки VI з 2,2-диметилпіперазином з одержанням сполуки формули І H N N Cl 10 15 (I). 2. Спосіб за п. 1, в якому LG є галогеном, наприклад Сl або Вr, переважно Сl, або сульфонатом, наприклад тозилатом або мезилатом. 3. Спосіб за п. 1, в якому сполука LG є галогеном, наприклад Сl, і розчинник є алканом, наприклад гептаном. 4. Спосіб за п. 1, в якому сполуку І осаджують у вигляді солі. 5. Спосіб за п. 4, в якому сіль, що утворилася, є сіллю фумарової кислоти, сіллю малеїнової кислоти або сіллю соляної кислоти і сполуки І. 17 UA 106191 C2 5 10 15 20 25 30 6. Спосіб за п. 1, в якому сполуку І виділяють у вигляді вільної основи. 7. Спосіб за п. 1, в якому згадану хіральну хроматографію здійснюють з використанням хіральної рідинної хроматографії. 8. Спосіб за п. 7, в якому згадану хіральну хроматографію здійснюють на хіральній нерухомій фазі. 9. Спосіб за п. 8, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою модифікованої амілози. 10. Спосіб за п. 9, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою трис-[(S)--метилбензилкарбамат]амілози. 11. Спосіб за п. 10, в якому для згаданої хіральної хроматографії застосовують розчинник, що включає суміш н-гептану і етанолу, і, необов'язково, N,N-діетиламін. 12. Спосіб за п. 11, в якому згадану хіральну хроматографію здійснюють з використанням хроматографії з суб- або суперкритичною рухомою фазою, переважно хроматографії з суперкритичною рухомою фазою. 13. Спосіб за п. 12, в якому згадану хіральну хроматографію здійснюють на хіральній нерухомій фазі. 14. Спосіб за п. 13, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою, що складається з хірального полімеру, або на колонці з силікагелем з іммобілізованим хіральним полімером, або на колонці з силікагелем з ковалентно зв'язаним хіральним мономером. 15. Спосіб за п. 14, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою трис-(3,5-диметилфенілкарбамат)амілози або трис-[(S)-метилбензилкарбамат]амілози, або на колонці з силікагелем, з іммобілізованою трис-(3,5диметилфенілкарбамат)амілозою, або на колонці з силікагелем, покритим оболонкою трис-(3,5диметилфенілкарбамат)целюлози або трис-(4-метилбензоат)целюлози, або на колонці з силікагелем з ковалентно зв'язаним аміном 3,5-динітробензоїлтетрагідрофенантрену. 16. Спосіб за будь-яким одним з пп. 14, 15, в якому згадану хіральну хроматографію здійснюють на колонці з силікагелем, покритим оболонкою трис-(3,5-диметилфенілкарбамат)амілози. 17. Спосіб за будь-яким одним з пп. 14-16, в якому згадану хіральну хроматографію здійснюють з модифікатором, вибраним з групи: метанол, етанол, ізопропанол або ацетонітрил, що необов'язково містить діетиламін, необов'язково 0,1 % діетиламіну. 18. Спосіб за будь-яким одним з пп. 1-17, в якому додатково здійснюють рециклізацію сполуки IVb перетворенням енантіомерно збагаченої сполуки IVb у переважно рацемічну сполуку IV, де IVb і IV є: O Cl 35 40 O Cl , (IVb) (IV). 19. Спосіб за п. 18, в якому рацемізацію здійснюють використанням основи або суміші двох або більше основ. 20. Спосіб за п. 18 або 19, в якому рацемізацію здійснюють застосуванням одного еквівалента або більше ненуклеофільної основи ("першої основи"), за яким іде додавання каталітичної кількості або одного еквівалента або більше тієї ж самої або іншої основи ("другої основи"). Комп’ютерна верстка М. Ломалова Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 18
ДивитисяДодаткова інформація
Автори англійськоюSuteu Christina
Автори російськоюСюте Кристина
МПК / Мітки
МПК: C07C 49/697, C07D 295/073
Мітки: спосіб, одержання, транс-1-((1r,3s)-6-хлор-3-феніліндан-1-іл)-3,3-диметилпіперазину
Код посилання
<a href="https://ua.patents.su/20-106191-sposib-oderzhannya-trans-1-1r3s-6-khlor-3-fenilindan-1-il-33-dimetilpiperazinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання транс-1-((1r,3s)-6-хлор-3-феніліндан-1-іл)-3,3-диметилпіперазину</a>
Сукцинатна і малонатна солі транс-4-((1r,3s)-6-хлор-3-феніліндан-1-іл)-1,2,2-триметилпіперазину та їх застосування як лікарського засобу

Номер патенту: 89617
Опубліковано: 25.02.2010
Автори: Сван Хенрік, Лопес де Дієґо Хейді, Нільсен Оле, Лінґсьо Ларс Оле, Дахл Аллан Карстен, Банґ-Андерсен Бенні, Колліер Шеррі Лінн, Хауелз Марк, Рінґор Лоне Мунк
МПК: C07C 25/00, A61K 31/495, A61P 25/18, C07D 295/06, C12P 41/00, C07C 35/00, C12P 7/02
Мітки: застосування, транс-4-((1r,3s)-6-хлор-3-феніліндан-1-іл)-1,2,2-триметилпіперазину, лікарського, сукцинатна, солі, малонатна, засобу
Формула / Реферат:
1. Сукцинатна сіль або малонатна сіль сполуки формули (І) (I)[транс-4-((1R,3S)-6-хлор-3-феніліндан-1-іл)-1,2,2-триметилпіперазин].2. Сукцинатна сіль за п. 1, яка являє собою гідросукцинатну сіль сполуки формули (І).3. Кристалічна гідросукцинатна сіль сполуки формули (І), визначеної в п. 1.4. Сіль за п. 3, яка являє собою кристалічну...
Кристалічна основа транс-1-((1r,3s)-3-феніл-6-хлороіндан-1-іл)-3,3-диметилпіперазину

Номер патенту: 95901
Опубліковано: 26.09.2011
Автори: Банг-Андерсен Бенні, Лопес де Дієго Хейді
МПК: A61K 31/495, C07D 295/073, A61P 25/18
Мітки: кристалічна, основа, транс-1-((1r,3s)-3-феніл-6-хлороіндан-1-іл)-3,3-диметилпіперазину
Формула / Реферат:
1. Кристалічна основа сполуки формули (І), сполуки І, транс-l-((1R,3S)-3-феніл-6-хлороіндан-1-іл)-3,3-диметилпіперазину, (I) яка характеризується порошковою рентгенівською дифрактограмою, одержаною з використанням СuKа1 опромінення (λ=1,5406 Å), що показує сигнали при наступних 2θ-кутах: 6,1, 11,1, 12,1, 16,2, 16,8,...
Проміжні сполуки для отримання транс-5-хлор-2-метил-2,3,3а,12в-тетрагідро-1н-дибенз[2,3:6,7]оксепіно[4,5-с]піролу
Номер патенту: 93043
Опубліковано: 10.01.2011
Автори: ван дер Лінден Якобус Йоганнес Марія, Рідер Майкл Р., Кемперман Ґерардус Йоганнес
МПК: C07D 491/04, C07D 313/00
Мітки: транс-5-хлор-2-метил-2,3,3а,12в-тетрагідро-1н-дибенз[2,3:6,7]оксепіно[4,5-с]піролу, отримання, проміжні, сполуки
Формула / Реферат:
1. Транс-амінокислотне похідне формули І або формули II,або енантіомер кожного транс-амінокислотного похідного, що має протилежну абсолютну конфігурацію, або рацемічна суміш кожного транс-амінокислотного похідного, або його сіль. 2. Транс-амінокислотне похідне, вибране з...
Спосіб одержання 5-хлор-4-амінозаміщеного похідного 3(2н)-піридазинону (варіанти) та проміжні сполуки

Номер патенту: 64003
Опубліковано: 16.02.2004
Автори: ШЕРЕШ Петер, Раткаі Зольтан, Каранчі Тамаш, БАРКОЦІ Йожеф, БАЛАЖ Ласло, Грефф Зольтан, Струхар Ілона, ДОМАН Імре, Котаі-Надь Петер, ШІМІГ Дьюла
МПК: C07D 237/22, C07D 237/20
Мітки: одержання, проміжні, варіанти, 5-хлор-4-амінозаміщеного, спосіб, похідного, сполуки, 3(2н)-піридазинону
Формула / Реферат:
1. Спосіб одержання 5-хлор-4-{3-[N-[2-(3,4-диметоксифеніл)етил]-N-метиламіно]пропіламіно}-3(2Н)-піридазинону формули (І) (І)і його фармацевтично прийнятних кислотно-адитивних солей, в якому здійснюють реакцію сполуки загальної формули (II), (ІІ)де Х означає відщеплювану групу, з N-метилгомовератриламіном формули (VI) (VI)і, якщо бажано, перетворення одержаної таким чином сполуки формули (І) в її...
Спосіб одержання пропінілового естеру (r)(+)-2-[4-(5-хлор-3-фторпіридин-2-ілокси)фенокси]пропіонової кислоти

Номер патенту: 64718
Опубліковано: 15.03.2004
Автори: Зайферт Готфрід, Стінг Андреа Рольф, Урвілер Бернхард
МПК: C07D 213/61, C07D 213/643, A01P 13/00
Мітки: спосіб, r)(+)-2-[4-(5-хлор-3-фторпіридин-2-ілокси)фенокси]пропіонової, одержання, пропінілового, кислоти, естеру
Формула / Реферат:
1. Спосіб одержання пропінілового естеру (R)(+)-2-[4-(5-хлор-3-фторпіридин-2-ілокси)фенокси]пропіонової кислоти формули (І) , (I)який відрізняється тим, що сполуку формули (II) перетворюють , (II)в інертному органічному розчиннику разом з М2СО3, де М являє собою натрій чи...
Попередній патент: Пристрій для демонтажу вертикальних опор
Випадковий патент: Свердло-різець