Промисловий спосіб синтезу 17-ацетокси-11b-[4-(диметиламіно)-феніл]-21-метокси-19-норпрегн-4,9-дієн-3,20-діону і проміжні сполуки даного способу
Номер патенту: 100241
Опубліковано: 10.12.2012
Автори: Хорват Золтан, Чоргеї Янош, Шанта Чаба, Балог Габор, Боді Йожеф, Селеш Янош, Махо Шандор, Араньї Анталь, Терді Ласло, Туба Золтан, Мольнар Чаба, Вішкі Дьйордь

















Формула / Реферат
1. Промисловий спосіб синтезу 17-ацетокси-11β-[4-(диметиламіно)-феніл]-21-метокси-19-норпрегн-4,9-дієн-3,20-діону формули (І):
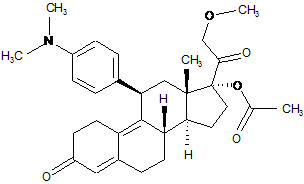 , (І)
, (І)
з 3,3-[1,2-етандіїл-біс(окси)]-оестр-5(10),9(11)-дієн-17-ону формули (II):
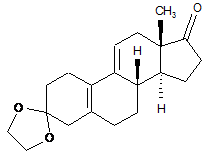 , (ІІ)
, (ІІ)
який відрізняється тим, що здійснюють стадії:
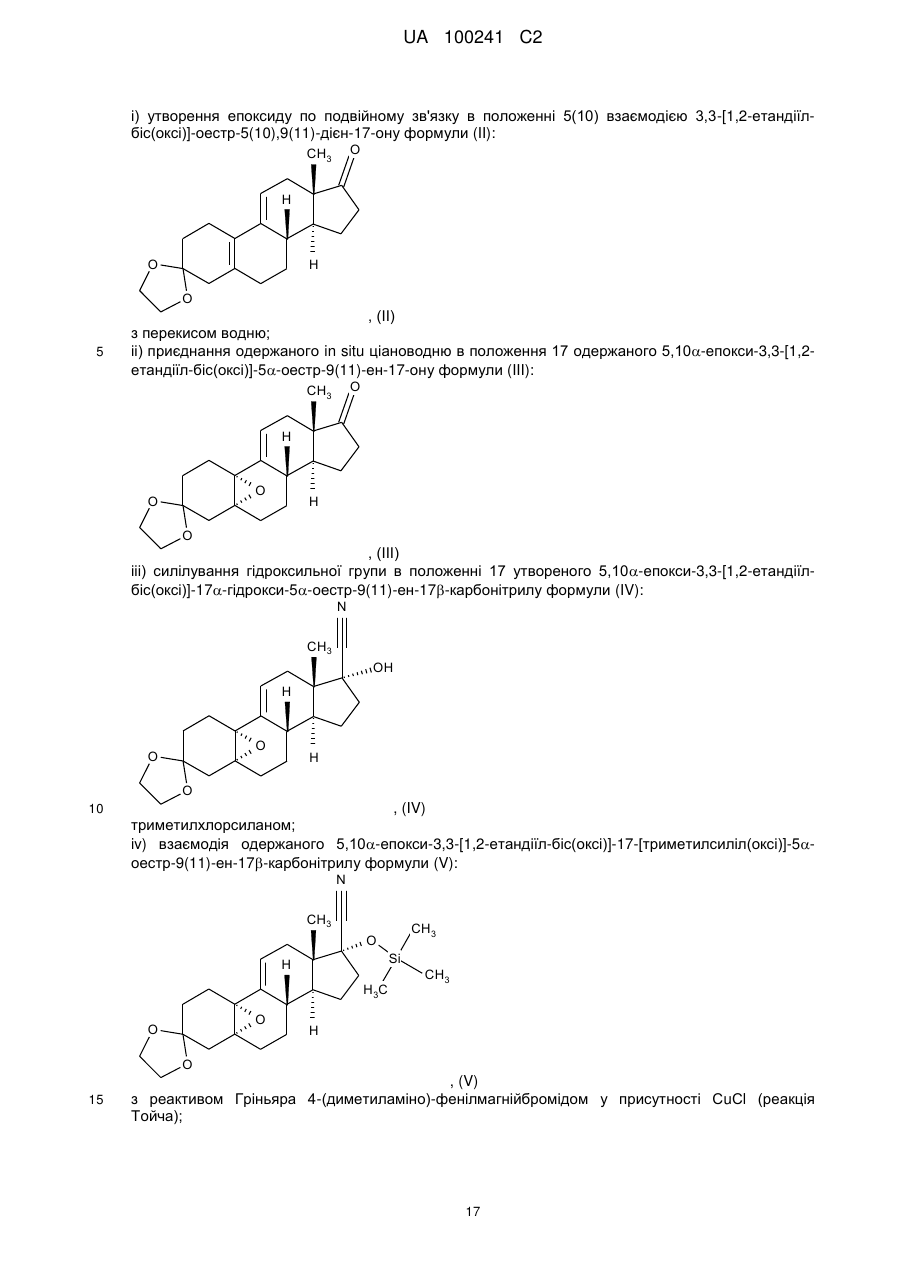
і) утворення епоксиду по подвійному зв'язку в положенні 5(10) взаємодією 3,3-[1,2-етандіїл-біс(окси)]-оестр-5(10),9(11)-дієн-17-ону формули (II):
, (ІІ)
з перекисом водню;
іі) приєднання одержаного in situ ціановодню в положення 17 одержаного 5,10a-епокси-3,3-[1,2-етандіїл-біс(окси)]-5a-оестр-9(11)-ен-17-ону формули (III):
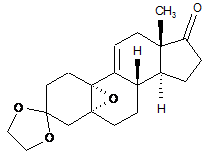 , (ІІІ)
, (ІІІ)
ііі) силілування гідроксильної групи в положенні 17 утвореного 5,10a-епокси-3,3-[1,2-етандіїл-біс(окси)]-17a-гідрокси-5a-оестр-9(11)-ен-17b-карбонітрилу формули (IV):
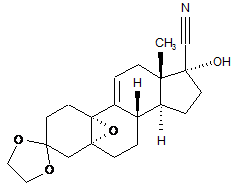 , (IV)
, (IV)
триметилхлорсиланом;
iv) взаємодія одержаного 5,10a-епокси-3,3-[1,2-етандіїл-біс(окси)]-17-[триметилсиліл(окси)]-5a-оестр-9(11)-ен-17b-карбонітрилу формули (V):
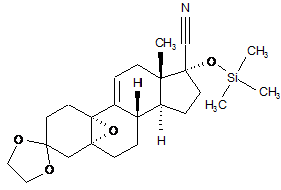 , (V)
, (V)
з реактивом Гріньяра 4-(диметиламіно)-фенілмагнійбромідом у присутності СuСl (реакція Тойча);
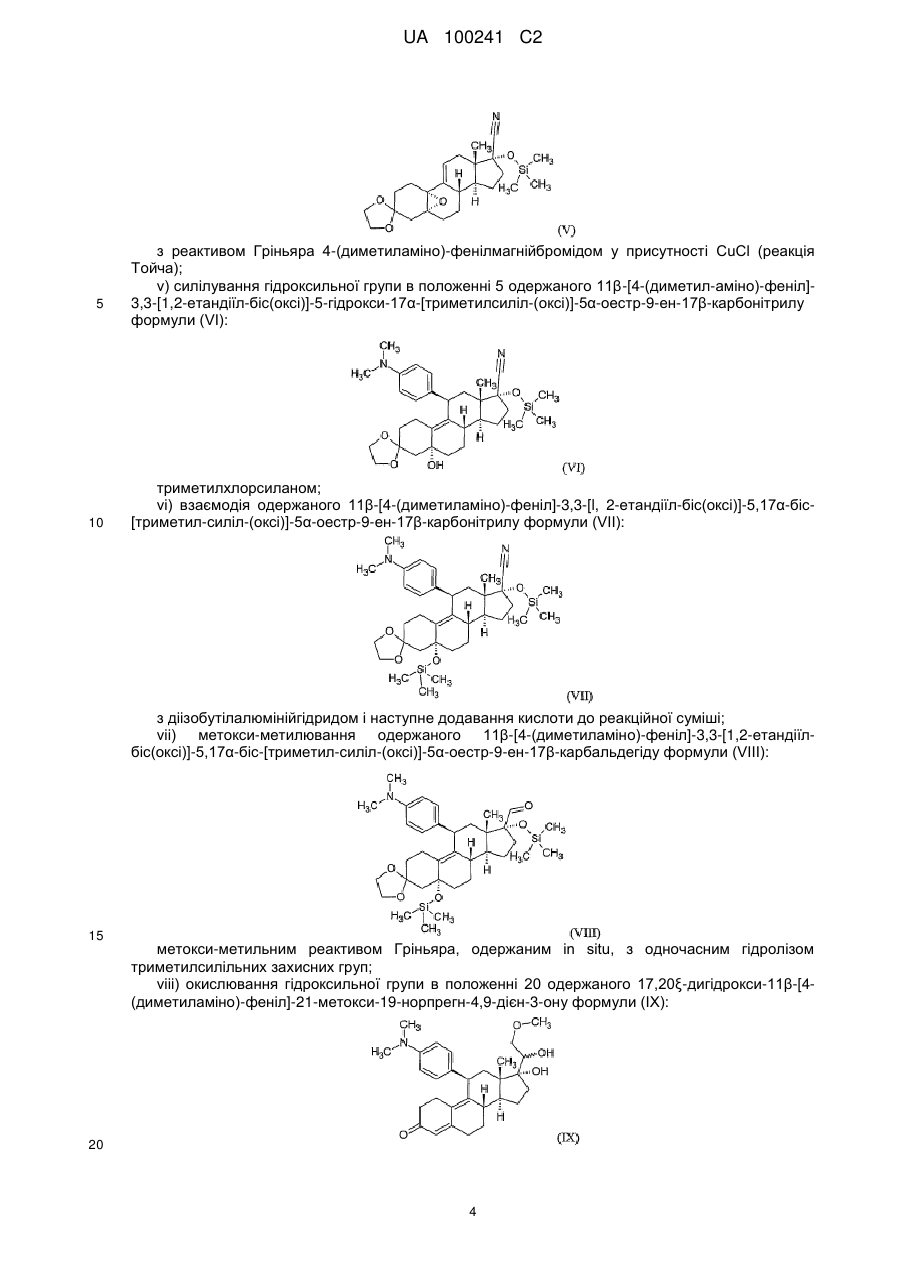
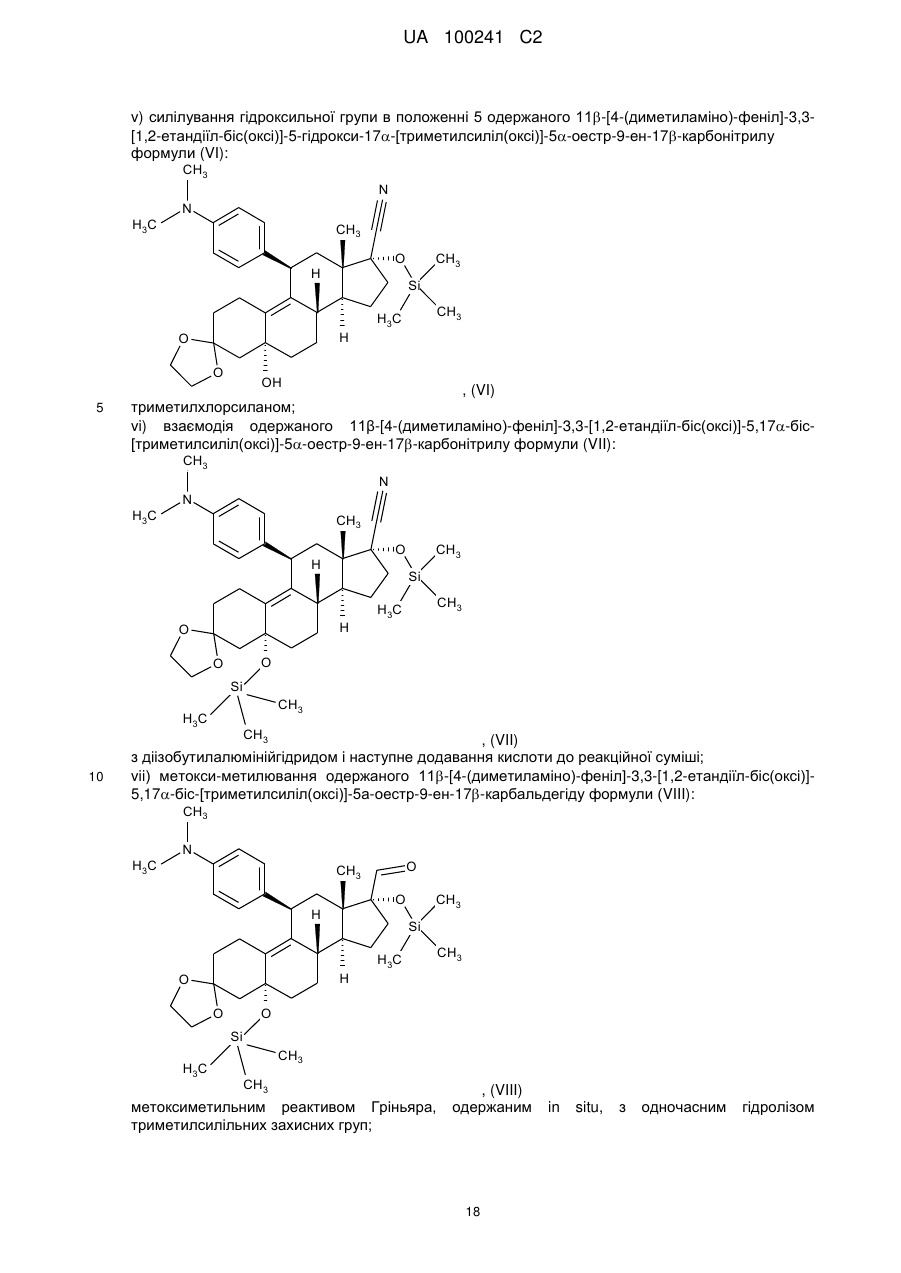
v) силілування гідроксильної групи в положенні 5 одержаного 11b-[4-(диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(окси)]-5-гідрокси-17a-[триметилсиліл(окси)]-5a-оестр-9-ен-17b-карбонітрилу формули (VI):
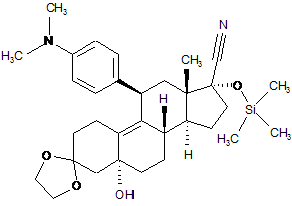 , (VI)
, (VI)
триметилхлорсиланом;
vi) взаємодія одержаного 11β-[4-(диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(окси)]-5,17a-біс-[триметилсиліл(окси)]-5a-оестр-9-ен-17b-карбонітрилу формули (VII):
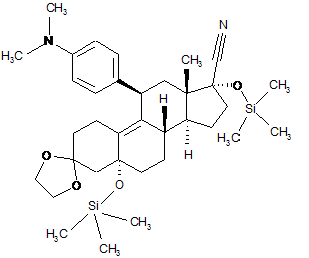 , (VII)
, (VII)
з діізобутилалюмінійгідридом і наступне додавання кислоти до реакційної суміші;
vii) метокси-метилювання одержаного 11b-[4-(диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(окси)]-5,17a-біс-[триметилсиліл(окси)]-5а-оестр-9-ен-17b-карбальдегіду формули (VIII):
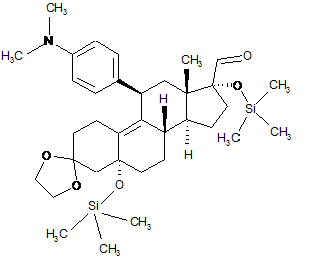 , (VIII)
, (VIII)
метоксиметильним реактивом Гріньяра, одержаним in situ, з одночасним гідролізом триметилсилільних захисних груп;
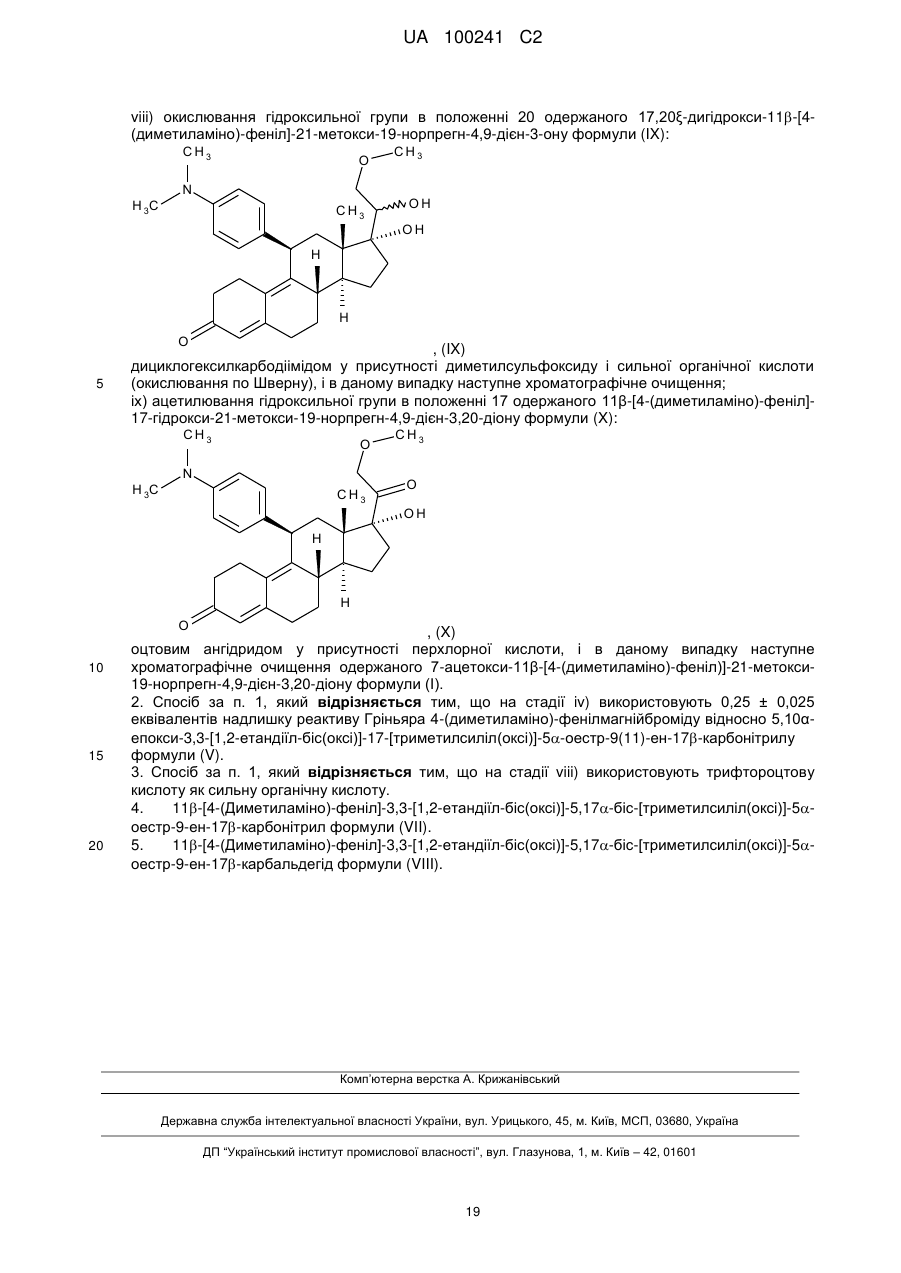
viii) окислювання гідроксильної групи в положенні 20 одержаного 17,20ξ-дигідрокси-11b-[4-(диметиламіно)-феніл]-21-метокси-19-норпрегн-4,9-дієн-3-ону формули (IX):
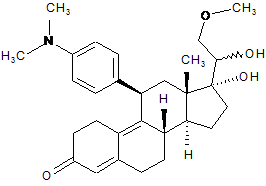 , (IX)
, (IX)
дициклогексилкарбодіімідом у присутності диметилсульфоксиду і сильної органічної кислоти (окислювання по Шверну), і в даному випадку наступне хроматографічне очищення;
іх) ацетилювання гідроксильної групи в положенні 17 одержаного 11β-[4-(диметиламіно)-феніл]-17-гідрокси-21-метокси-19-норпрегн-4,9-дієн-3,20-діону формули (X):
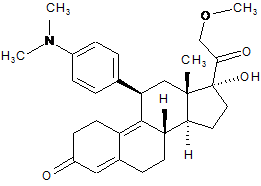 , (X)
, (X)
оцтовим ангідридом у присутності перхлорної кислоти, і в даному випадку наступне хроматографічне очищення одержаного 7-ацетокси-11β-[4-(диметиламіно)-феніл)]-21-метокси-19-норпрегн-4,9-дієн-3,20-діону формули (І).
2. Спосіб за п. 1, який відрізняється тим, що на стадії iv) використовують 0,25 ± 0,025 еквівалентів надлишку реактиву Гріньяра 4-(диметиламіно)-фенілмагнійброміду відносно 5,10α-епокси-3,3-[1,2-етандіїл-біс(окси)]-17-[триметилсиліл(окси)]-5a-оестр-9(11)-ен-17b-карбонітрилу формули (V).
3. Спосіб за п. 1, який відрізняється тим, що на стадії viii) використовують трифтороцтову кислоту як сильну органічну кислоту.
4. 11b-[4-(Диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(окси)]-5,17a-біс-[триметилсиліл(окси)]-5a-оестр-9-ен-17b-карбонітрил формули (VII).
5. 11b-[4-(Диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(окси)]-5,17a-біс-[триметилсиліл(окси)]-5a-оестр-9-ен-17b-карбальдегід формули (VIII).
Текст
Реферат: Даний винахід належить до способу синтезу відомого 17-ацетокси-11-[4-(диметиламіно)феніл]-21-метокси-19-норпрегн-4,9-дієн-3,20-діону (надалі позначається CDB-4124) формули (І) з 3,3-[1,2-етандіїл-біс(оксі)]-оестр-5(10),9(11)-дієн-17-ону формули (II). Сполука CDB-4124 належить до групи антигормонів. Спосіб за дійсним винаходом полягає в наступному: і) утворення епоксиду по подвійному зв'язку в положенні 5(10) взаємодією 3,3-[1,2-етандіїлбіс(оксі)]-оестр-5(10),9(11)-дієн-17-ону формули (II) з перекисом водню; іі) приєднання одержаного in situ ціановодню в положення 17 одержаного 5,10-епокси-3,3-[1,2-етандіїлбіс(оксі)]-5а-оестр-9(11 )-ен-17-ону формули (III); ііі) силілування гідроксильної групи в положенні 17 утвореного 5,10α-епокси-3,3-[1,2-етандіїл-біс(оксі)]-17-гідрокси-5-оестр-9(11)ен-17-карбонітрилу формули (IV) триметилхлорсиланом; iv) взаємодія одержаного 5,10епокси-3,3-[1,2-етандіїл-біс(оксі)]-17-[триметилсиліл(оксі)]-5-оестр-9(11)-ен-17-карбонітрилу формули (V) з реактивом Гріньяра 4-(диметиламіно)-фенілмагнійбромідом у присутності СuСl (реакція Тойча); v) силілування гідроксильної групи в положенні 5 одержаного 11-[4(диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(оксі)]-5-гідрокси-17-[триметил-силіл-(оксі)]-5а UA 100241 C2 (12) UA 100241 C2 оестр-9-ен-17β-карбонітрилу формули (VI) триметилхлорсиланом; vi) взаємодія одержаного 11β-[4-(диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(оксі)]-5,17-біс-[триметилсиліл(оксі)]-5оестр-9-ен-17-карбонітрилу формули (VII) з діізобутилалюмінійгідридом і наступне додавання кислоти до реакційної суміші; vii) метоксиметилювання одержаного 11β-[4-(диметиламіно)феніл]-3,3-[1,2-етандіїл-біс(оксі)]-5,17-біс-[триметилсиліл(оксі)]-5-оестр-9-ен-17карбальдегіду формули (VIII) метокси-метильним реактивом Гріньяра, одержаним in situ, з одночасним гідролізом триметилсилільних захисних груп; viii) окислювання гідроксильної групи в положенні 20 одержаного 17,20ξ-дигідрокси-11β-[4-(диметиламіно)-феніл]-21-метокси-19норпрегн-4,9-дієн-3-ону формули (IX) дициклогексилкарбодіімідом у присутності диметилсульфоксиду і сильної органічної кислоти (окислювання по Шверну), і в даному випадку наступне хроматографічне очищення; іх) ацетилювання гідроксильної групи в положенні 17 одержаного 11β-[4-(диметиламіно)-феніл]-17-гідрокси-21-метокси-19-норпрегн-4,9-дієн-3,20діону формули (X) оцтовим ангідридом у присутності перхлорної кислоти, і в даному випадку одержаний 7-ацетокси-11β-[4-(диметиламіно)-феніл)]-21-метокси-19-нопрегна-4,9-дієн-3,20-діон формули (І) очищають хроматографічно. Даний винахід також належить до нових проміжних сполук формули (VII) і (VIII). UA 100241 C2 Галузь техніки, до якої належить винахід Даний винахід відноситься до способу синтезу відомого 17-ацетокси-11β-[4-(диметиламіно)феніл]-21-метокси-19-норпрегн-4,9-дієн-3,20-діону (надалі іменований CDB-4124) формули (I): 5 10 15 20 25 30 35 40 45 з 3,3-[1,2-етандіїл-біс(оксі)]-оестр-5(10),9(11)-дієн-17-ону (надалі іменований кето-кеталь) формули (II): Попередній рівень техніки Сполука CDB-4124 належить до групи антигормонів. Антигормони можуть дезактивувати дію гормонів в організмі за рахунок інгібування зв'язування гормонів, наприклад, чоловічих і жіночих статевих гормонів або гормонів, продукованих наднирником, з місцем прикладання в органімішені; унаслідок цього, індуковані гормонами функції можуть бути блоковані введенням антигормонів. Сполуки, які інгібують синтез прогестерону або його зв'язування з рецептором, можна потенційно використовувати в контрацепції й у випадках патологій, де прогестерон відіграє помітну роль. Ідеальна анти-прогестогенна сполука: - специфічна (приєднується тільки до того рецептора, який необхідно блокувати), - має високу спорідненість до рецептора і повільно дисоціює, - не чинить іншого біологічного або фармакологічного ефекту. Перший анти-прогестин, використаний у клінічній практиці, був описаний у 1981 [EP 57115] і називався міфепристон. Після цього були синтезовані деякі аналоги і перевірений зв'язок структура-активність для одержаних сполук, особливо селективність, головним чином співвідношення анти-прогестинної і анти-глюкокортикоїдної активності. В даний час жодна з відомих анти-прогестогенних сполук не задовольняє цілком вимогам до селективності. Сполука CDB-4124, яку можна синтезувати способом за дійсним винаходом, згідно проведених до дійсного моменту клінічних іспитів являє собою таку перспективну сполуку, промисловий синтез якої є економічно вигідним. У літературі описані кілька способів лабораторного синтезу CDB-4124 формули (I), які розрізняються за вихідними матеріалами або порядком стадій реакції. Синтез різних функціональних груп здійснюють схожими способами.Згадані способи синтезу характеризуються тим, що в них звичайно не враховують аспекти безпеки при масштабуванні, особливо займистість розчинників (реакційних середовищ), а також те, що дані розчинники можуть бути шкідливі для здоров'я і що деякі реактиви можуть бути дорогими. Метою перших синтезів було одержання такої кількості сполуки/сполук, яка була б достатньою для проведення фармакологічних тестів. Для задовільнення усіх вимог чистоти для терапевтичного використання сполуки необхідна подальша розробка. Промислове здійснення економічного синтезу звичайно являє собою модифікацію оригінального способу або способів, або може являти собою поліпшений синтез. Перший синтез сполуки CDB-4124 був описаний у патенті WO 97/41145, об'єктом якого був синтез 11β- і 21-заміщених 19-норпрогестеронових похідних і їх аналогів. Дані сполуки володіють значною антипрогестогенною активністю. На Схемі 3 проілюстрований синтез сполуки CDB-4124. Вихідна речовина для даного синтезу являла собою 17α-[(бромметил)-диметил-силіл-окси]3,3-[1,2-етандіїл-біс-(оксі)]-5(10),9(11)-дієн-17β-карбонітрил, який можна одержувати з 1 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 комерційно доступного 3,3-[1,2-етандіїл-біс-(оксі)]-17α-гідрокси-оестр-5(10),9(11)-дієн-17βкарбонітрилу (Davos Chemical Inc. у Нью-Джерсі) з виходом 69,5 % силілуванням гідроксильної групи в положенні 17 (бромметил)-диметилсилілхлоридом. Очищення здійснювали флешхроматографією. Вихідну речовину вводили в реакцію з діізопропіламідом літію в тетрагідрофурановому розчині при -78 °C, а виділення продукту здійснювали екстрагуванням етилацетатом і очищенням ефіром. 21-Бром сполуку, одержану з виходом 60,4 %, вводили в реакцію з ацетатом калію (99 %), потім гідролізували гідрокарбонатом калію, одержуючи 21-гідрокси похідне з виходом 57,6 %. Для захисту кетогруп у положенні 3 і 20 застосовували утворення біс-кеталя (з виходом 62,5 %). Ключову стадію синтезу – монометилювання по положенню 21 17α,21-дигідрокси похідного, захищеного по положеннях 3 і 20 – здійснювали в присутності 1:1 суміші тетрафторборатної солі триметилоксонію і "протонної губки" [1,8-біс(диметиламіно)нафталіну]. Одержаний продукт – 3,3;20:20-біс[1,2-етандіїл-біс(оксі)]-17-гідрокси-21-метокси-19-норпрегн-5(10),9(11)-дієн – виділяли з дихлорметану з виходом 79 % і використовували в наступній стадії. Утворення епоксиду по подвійному зв'язку в положенні 5(10) одержаного сирого 21-метокси похідного здійснювали взаємодією з перекисом водню в присутності тригідрату гексафторацетону. Згідно даних ЯМР-спектроскопії, одержаний продукт містив чотири типи епоксидів. (Основний продукт являв собою 5α,10α-епоксид з виходом 66 %.) Одержану сиру суміш епоксидів використовували в реакції Гріньяра, каталізованої іоном міді(I). Після виділення з ефірного розчину продукт очищали колонковою флешхроматографією. Гідроліз дикетальної захисної групи в 11β-[4-(диметиламіно)-феніл] похідному здійснювали сумішшю трифтороцтова кислота/вода (3:1) у тетрагідрофурані. Продукт одержували з виходом 96,3 % після екстракції дихлорметаном, розпарювання й обробки маслоподібного залишку водою. Остання стадія синтезу являла собою ацетилювання гідроксильної групи в положенні 17, яке здійснювали сумішшю трифтороцтового ангідриду й оцтової кислоти в дихлорметані в присутності п-толуолсульфокислоти (каталізатор) при 0 °C. По закінченні реакції суміш розбавляли водою, нейтралізували розчином гідроксиду амонію, екстрагували дихлорметаном і промивали розсолом. Об'єднані органічні шари упарювали й очищали залишок колонковою флеш-хроматографією, одержуючи сполуку CDB-4124 з виходом 75,8 %. Вихідна речовина для синтезу, описаного у вищевказаному патенті, являла собою 17α[(бромметил)-диметил-силіл-окси]-3,3-[1,2-етандіїл-біс-(оксі)]-5(10),9(11)-дієн-17β-карбонітрил, який синтезували з кето-кеталя додаванням ціанідного іона з наступним силілуванням гідроксильної групи. 17-Силіл-окси-бром сполуку перетворювали в 21-бром похідне діізопропіламідом літію при -78 °C. Введення метокси-групи в положення 21 здійснювали не напряму в кілька стадій – 21-бром сполука, 21-ацетокси похідне – через 21-гідрокси сполука – при спільному використанні 6 еквівалентів (щодо вихідної речовини) тетрафторборатної солі триметилоксонію і "протонної губки" (SNAP реакція). Зазначений спосіб довгий і дорогий, видалення надлишку "протонної губки" важке, у багатьох випадках необхідне багаторазове очищення продукту. Утворення епоксиду по подвійному зв'язку в положенні 5(10) дало – згідно даних ЯМР-спектроскопії – чотири типи епоксидів, з яких тільки 66 % являли собою бажаний 5α,10α-епоксид. Незважаючи на той факт, що сирий продукт містив близько 34 % небажаного продукту (β-епоксид), його використовували в реакції Гріньяра. У реакції Гріньяра використовували п'ятикратний надлишок 4-бром-диметиланіліну, що сприяло утворенню монометильного похідного і димерізації цього реактиву, тому виділення й очищення продукту були ускладнені, вихід виділеного продукту знижувався. Зі стратегічної точки зору, епоксидування – що приводить до одержання сирої суміші – на сьомій стадії синтетичного ланцюжка не є економічно вигідним. Флеш-хроматографію використовували на 4 стадіях 11стадійного синтезу. У деяких випадках при виділенні й очищенні проміжних сполук використовувався ефір, що є небезпечним в умовах промислового здійснення синтезу. Вихід кінцевого продукту знижувався внаслідок стадій очищення, тому загальний вихід синтезу складав тільки 3,22 %. На схемах 1 і 2 патенту WO 01/47945 показані дві інші послідовності реакцій синтезу сполуки CDB-4124. Вихідна речовина в обох випадках синтезу являла собою 3,3-[1,2-етандіїл-біс(оксі)]-17αгідрокси-оестр-5(10),9(11)-дієн-17β-карбонітрил, який силілували описаним вище способом, але 21-галогенові похідні – хлор і бром – також утворювалися в тій самій реакційній суміші. Наступні стадії, заміщення атома брому ацетокси-групою і гідроліз, були ідентичні до відомих способів. 2 UA 100241 C2 5 10 15 20 25 30 35 Наступні стадії синтезу, показаного на Схемі 1, ідентичні до стадій, описаних у попередньому патенті. Відповідно до Схеми 2, синтезували 3-монокетальне похідне 21-гідрокси похідного, після чого проводили SNAP реакцію для введення 21-метокси функції і кето-групи в положення 20 (тимчасовий захист) відновленням літійалюмінійгідридом. Утворення епоксиду здійснювали в положенні 5(10) одержаного 20-гідрокси похідного. Розкриття епоксидного кільця і видалення кетальної групи було ідентично до описаних раніше способів. Йод-оксибензойну кислоту використовували для повторного окислювання гідроксильної групи в положенні 20. Кінцеву стадію, синтез бажаного 17-ацетокси продукту, здійснювали відповідно до способу, представленого на Схемі 1. Даний синтез проводили в 12 стадій, із загальним виходом 3,89 %. Хоча об'єднання перших двох стадій синтезу було гарним рішенням, але, наприклад, утворення епоксидного похідного – через стадію очищення, що приводила до зниженого виходу – на більш пізній фазі синтетичного ланцюжка було не сучасним, дорогим. Інше невигідне рішення полягало в тому, що для очищення проміжних сполук і кінцевого продукту використовували флеш-хроматографію. У декількох випадках виділення продуктів здійснювали обробкою ефіром, який не можна використовувати у великомасштабному синтезі. Введення метокси-групи здійснювали в кілька стадій – як описано раніше. Видалення "протонної губки", використовуваної в SNAP реакції, досягалося тільки багаторазовим очищенням. Відновлення літійалюмінійгідридом – яке застосовували для тимчасового захисту оксо-функціональної групи в положенні 20 – є особливо небезпечним у промисловому масштабі. Наступна регенерація оксо-групи в положенні 20 шляхом окислювання йод-оксибензойною кислотою є дорогою і тому непридатною для промислового одержання. Суть винаходу Відповідно до перерахованих вище фактів, не існує такого відомого способу, що є придатним для здійснення синтезу CDB-4124 у промисловому масштабі в простих реакційних умовах. Наша мета полягала в розробці способу, що легко масштабується, промислове здійснення якого безпечне, економічне і при якому чистота діючої речовини задовольняє вимогам фармакопеї. Несподівано було виявлено, що наступний спосіб задовольняє вищевказані вимоги: і) утворення епоксиду по подвійному зв'язку в положенні 5(10) взаємодією 3,3-[1,2-етандіїлбіс(оксі)]-оестр-5(10),9(11)-дієн-17-ону формули (II): з перекисом водню; ii) приєднання одержаного in situ ціановодню в положення 17 одержаного 5,10α-епокси-3,3[1,2-етандіїл-біс(оксі)]-5α-оестр-9(11)-ен-17-ону формули (III): iii) силілування гидроксильної групи в положенні 17 утвореного 5,10α-епокси-3,3-[1,2етандіїл-біс(оксі)]-17α гідрокси-5α-оестр-9(11)-ен-17β-карбонітрилу формули (IV): 40 триметилхлорсиланом; iv) взаємодія одержаного 5,10α-епокси-3,3-[1,2-етандіїл-біс(оксі)]-17-[триметил-силіл-окси]5α-оестр-9(11)-ен-17β-карбонітрилу формули (V): 3 UA 100241 C2 5 10 з реактивом Гріньяра 4-(диметиламіно)-фенілмагнійбромідом у присутності CuCl (реакція Тойча); v) силілування гідроксильної групи в положенні 5 одержаного 11β-[4-(диметил-аміно)-феніл]3,3-[1,2-етандіїл-біс(оксі)]-5-гідрокси-17α-[триметилсиліл-(оксі)]-5α-оестр-9-ен-17β-карбонітрилу формули (VI): триметилхлорсиланом; vi) взаємодія одержаного 11β-[4-(диметиламіно)-феніл]-3,3-[l, 2-етандіїл-біс(оксі)]-5,17α-біс[триметил-силіл-(оксі)]-5α-оестр-9-ен-17β-карбонітрилу формули (VII): з діізобутілалюмінійгідридом і наступне додавання кислоти до реакційної суміші; vii) метокси-метилювання одержаного 11β-[4-(диметиламіно)-феніл]-3,3-[1,2-етандіїлбіс(оксі)]-5,17α-біс-[триметил-силіл-(оксі)]-5α-оестр-9-ен-17β-карбальдегіду формули (VIII): 15 метокси-метильним реактивом Гріньяра, одержаним in situ, з одночасним гідролізом триметилсилільних захисних груп; viii) окислювання гідроксильної групи в положенні 20 одержаного 17,20ξ-дигідрокси-11β-[4(диметиламіно)-феніл]-21-метокси-19-норпрегн-4,9-дієн-3-ону формули (IX): 20 4 UA 100241 C2 дициклогексилкарбодіімідом у присутності диметилсульфоксиду і сильної органічної кислоти (окислювання по Шверну), і в даному випадку наступне хроматографічне очищення; ix) ацетилювання гідроксильної групи в положенні 17 одержаного 11β-[4-(диметиламіно)феніл]-17-гідрокси-21-метокси-19-норпрегн-4,9-дієн-3,20-діону формули (X): 5 10 15 20 25 30 35 40 45 50 оцтовим ангідридом у присутності перхлорної кислоти, і в даному випадку одержаний 7ацетокси-11β-[4-(диметиламіно)-феніл)]-21-метокси-19-норпрегн-4,9-дієн-3,20-діон формули (I) очищають хроматографічно. Кето-кеталь формули (II) переважно вводять у реакцію з 50 %-ним розчином перекису водню в сухому дихлорметані в присутності піридину і гексахлорацетону при 0-1 °C протягом 20-24 год. Після завершення реакції суміш розбавляють дихлорметаном, надлишок перекису водню розкладають, органічний шар відокремлюють, водну фазу двічі екстрагують дихлорметаном, об'єднані органічні шари промивають водою, висушують і випарюють. Маслоподібний залишок обробляють сумішшю 1:3 етилацетат/діізопропіловий ефір. Одержаний кето-кеталь-епоксид формули (III) – чистота якого складає 98,8 % і який містить 95,3 % 5α,10α-епоксиду згідно даних ВЕРХ – використовують у стадії ii) без додаткового очищення. Стадію ii) переважно здійснюють суспендуванням кето-кеталь-епоксиду формули (III) в метанолі, при 20-25 °C додають порошкоподібний ціанід калію, потім після обережного додавання оцтової кислоти реакційну суміш нагрівають до 50-55 °C. Реакційну суміш охолоджують до 20-25 °C протягом 1 год., потім перемішують при цій самій температурі протягом 5 год. Після завершення реакції суміш розбавляють водою, перемішують 1 год. і відфільтровують кристалічний епокси-карбонітрил формули (IV), що випав в осад. Одержаний продукт можна використовувати в стадії iii) без додаткового очищення. На стадії iii) епокси-карбонітрил формули (IV) переважно розчиняють у дихлорметані при інтенсивному перемішуванні, потім після висушування перевіряють вміст води. До сухого розчину додають імідазол, потім протягом 1 год. при 20-25 °C додають триметилхлорсилан. Після завершення реакції розчин розбавляють дихлорметаном і розкладають надлишок триметилхлорсилану додаванням води. Органічний шар відокремлюють, промивають водою, висушують і випарюють. Залишок кристалізують з метанолу, фільтрують і сушать. Одержаний у такий спосіб TMSO-карбонітрил формули (V) (73,3 %) можна використовувати в наступній стадії без додаткового очищення. TMSO-карбонітрил формули (V), одержаний на стадії iv), переважно вводять в реакцію в такий спосіб. Спочатку до сухого тетрагідрофурану додають магній і 1,2-диброметан. Температура реакційної суміші починає підвищуватися, вказуючи на успішне здійснення активації. Потім 4-бром-диметиланілін і невелику кількість розчину 1,2-диброметану в сухому тетрагідрофурані і толуол додають при перемішуванні в реакційну суміш, що містить магній (реактив Гріньяра). Закипання реакційної суміші вказує на успішне здійснення активації. Потім до розчину реактиву Гріньяра додають CuCl, і після 5 хв перемішування суміш охолоджують до 8-13 °C. Додають розчин TMSO-карбонітрилу в дихлорметані, підтримуючи температуру між 1015 °C. Після завершення реакції суміш додають до перемішуваного та охолодженого 10 %-ного розчину хлориду амонію, що містить піросульфіт лужного металу. Теплий розчин охолоджують до кімнатної температури, розбавляють дихлорметаном, відокремлюють органічний шар і екстрагують водну фазу дихлорметаном. Об'єднані органічні шари промивають водою, обробляють силікагелем, сушать над безводним сульфатом натрію, фільтрують і упарюють у вакуумі. Залишок кристалізують з метанолу. Кристалічний продукт відокремлюють і сушать. Одержаний A-TMSO-карбонітрил формули (VI) (80,79 %) можна використовувати в наступній стадії без додаткового очищення. Стадію v) переважно здійснюють розчиненням одержаного A-TMSO-карбонітрилу формули (VI) у дихлорметані при 20-25 °C, потім після додавання імідазолу гідроксильну групу в положенні 5 силілують триметилхлорсиланом. Час реакції близько 2 год., потім суміш розбавляють дихлорметаном і водою, органічний шар відокремлюють, промивають водою, сушать над безводним сульфатом натрію, фільтрують і упарюють. Залишок обробляють 5 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 метанолом, одержаний кристалічний A-біс-TMSO-карбонітрил формули (VII) (89,09 %) відфільтровують і висушують. Його можна використовувати в наступній стадії без додаткового очищення. На стадії vi) одержаний A-біс-TMSO-карбонітрил формули (VII) розчиняють у суміші метилтрет-бутилового ефіру і тетрагідрофурану, охолоджують до (-15) - (-20)°C і 1M розчин DIBAL-H (діізобутилалюмінійгідрид) у циклогексані додають протягом близько 30 хв, підтримуючи температуру, потім реакційну суміш перемішують ще 1 год. при тій самій температурі. Після завершення реакції суміш води й оцтової кислоти (2:1) додають при (-5)-(-10)°C, потім суміш перемішують протягом 20 хв. Органічний шар відокремлюють, промивають водою, 0,3 M розчином гідрокарбонату натрію і водою. Органічний шар упарюють без висушування при 4045 °C, залишок розчиняють у метанолі й упарюють до заданого об'єму (дивися приклади). Кристалічну суспензію охолоджують до 5-10 °C, фільтрують після 1 год. стояння, промивають метанолом з температурою 0-(-5)°C і сушать. Одержаний A-біс-TMSO-карбальдегід формули (VIII) (83,6 %) можна використовувати в наступній стадії без додаткового очищення. Одержаний A-біс-TMSO-карбальдегід формули (VIII) на стадії vii) перетворюють у 21метокси похідне з подовженням ланцюга. Це перетворення здійснюють активацією магнієвих стружок, як описано вище, у сухому тетрагідрофурані 1,2-диброметаном, потім додають хлорид ртуті(II) з утворенням амальгами. Одержану суміш розбавляють толуолом, потім перевіряють активність амальгами, як описано в Прикладах 7, 8 і 19. Після перевірки активності реактиву додають розчин метоксиметилхлориду в толуолі. Одночасно з цим A-біс-TMSO-карбальдегід розчиняють у толуолі і додають цей розчин до розчину амальгами протягом 30 хв при 0-5 °C. Після завершення реакції суміш додають до 1M водного розчину гідросульфату калію, підтримуючи температуру нижче 30 °C. Після перемішування протягом 2 год. шари розділяють, водну фазу додають до суміші 1M розчину гідрокарбонату натрію і дихлорметану і перемішують 10-15 хв. Органічний шар відокремлюють, водну фазу екстрагують дихлорметаном, об'єднані органічні шари сушать і обробляють вугіллям, фільтрують і упарюють. Залишок являє собою твердий діол формули (IX) (84,1 %), який можна використовувати в наступній стадії без додаткового очищення. Одержаний діол формули (IX) далі вводять у реакцію відповідно до стадії viii) дійсного винаходу. Переважно його розчиняють у сухому толуолі і додають в атмосфері азоту диметилсульфоксид, піридин і трифтороцтову кислоту при 20-25 °C. Потім до суміші додають розчин дициклогексилкарбодііміду в толуолі (окислювання по Шверну). Реакційну суміш перемішують при 40 °C протягом 2 год., потім охолоджують до 20-25 °C і додають 1M водний розчин гідросульфату калію. Після перемішування протягом 30 хв кристалічну сполуку, що випала в осад, відфільтровують і промивають 1M водним розчином гідросульфату калію. Дві фази фільтрату розділяють, водну фазу додають до 1M розчину гідроксиду натрію, сирий продукт, що випав в осад, відфільтровують, промивають водою і сушать. Одержаний кетон формули (X) (79,5 %) використовують у наступній стадії після очищення. Синтез чистого CDB-4124 формули (I), що задовольняє вимогам чистоти для терапевтичного застосування, включає дві стадії очищення методом ВЕРХ. Перша являє собою очищення кетону формули (X). Ацетилювання очищеного кетону формули (X) у результаті дає сирий CDB-4124, очищення якого методом ВЕРХ дає діючу речовину з чистотою 99 %. Хроматографію кетону формули (X) і сирого CDB-4124 переважно здійснюють з використанням силікагелю як нерухомої фази і 53:35:12 суміші циклогексан/метил-третбутиловий ефір/ацетон як елюента як у лабораторному, так і в промисловому способі. Н-гексан і н-гептан також можна використовувати замість циклогексану. Співвідношення розчинників у елюенті може варіюватися у визначених межах (циклогексан, н-гексан, н-гептан: 40-60 %; метил-трет-бутиловий ефір: 25-45 %; ацетон: 10-20 %). Наприкінці стадії viii) кетон формули (X) переважно очищають у такий спосіб: силікагелевим адсорбентом (ZEOPREP C-GEL C-490L, виробництво ZEOCHEM; розмір частинок 15-35 мкм; висота шару близько 60 см) заповнюють ВЕРХ колонку методом суспезійного набивання і колонку врівноважують елюентом (53:35:12 суміш циклогексан/метил-трет-бутиловий ефір/ацетон). Сирий кетон формули (X) розчиняють у суміші ацетону і метил-трет-бутилового ефіру, і до розчину додають циклогексан. Одержаний у такий спосіб розчин фільтрують і наносять на колонку шляхом вколювання. Використовують УФ-детектування. Відокремлюють першу фракцію, і фракції, що містять чисту сполуку, збирають і упарюють. Відповідно до іншого способу, після упарювання фракцій відганяють від залишку дихлорметан і розчиняють продукт у дихлорметані. Вміст домішок в обох випадках: менш 4 %. Одержаний дихлорметановий розчин можна використовувати в наступній стадії. CDB-4124 формули (I) синтезують з очищеного кетону формули (X) відповідно до стадії ix) 6 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 дійсного винаходу з використанням оцтового ангідриду в присутності перхлорної кислоти: 70 %ну перхлорну кислоту додають до перемішуваного та охолодженого до ((-20)-(-25)°C) оцтового ангідриду з такою швидкістю, щоб температура не піднімалася вище (-15)°C. Потім додають розчин очищеного 11β-[4-(диметиламіно)-феніл)]-17-гідрокси-21-метокси-19-норпрегн-4,9-дієн3,20-діону формули (X) у дихлорметані. Після завершення реакції суміш розбавляють дихлорметаном, охолоджують до (-10)°C і додають воду для розкладання оцтового ангідриду. Рівень pН суміші доводять до 7-8 додаванням розчину гідроксиду амонію. Потім водну фазу відокремлюють, екстрагують дихлорметаном, об'єднані органічні шари промивають водою, сушать і упарюють. Одержаний сирий кінцевий продукт, CDB-4124 формули (I) очищають методом ВЕРХ відповідно до описаного вище способу. Переваги способу за дійсним винаходом в порівнянні з відомими способами можна підсумовувати наступним чином: a) Вихідну речовину для синтезу [кето-кеталь формули (II)] можна легко синтезувати з оестр-4-ен-3,17-діону відомими способами. b) Відповідно до заявленого авторами способу, утворення епоксиду по подвійному зв'язку в положенні 5(10) являє собою першу стадію синтезу. З одержуваної ізомерної суміші епоксидів тільки 5α,10α-епоксид приводить до утворення необхідної сполуки, відповідно, інші ізомери є побічними продуктами. Відповідно до описаного авторами способу, дану стадію, що приводить до великих втрат речовини, проводять на початку синтетичного ланцюжка; тому відбувається втрата вихідної речовини, а не втрата проміжної сполуки з більш пізньої стадії, що є більш цінним. Спосіб за дійсним винаходом є більш економічним. Інший недолік епоксидування, проведеного на більш пізній стадії синтетичного ланцюжка у відомих методиках, полягає в більш складному очищенні одержаної сполуки. c) Відповідно до описаного авторами способу, введення стратегічно важливої 21-метоксигрупи здійснюють у дві стадії через нові проміжні сполуки формули (VII) і (VIII). Застосування нової проміжної сполуки формули (VII) уможливило утворення альдегідної групи в положенні 21, і одержана нова сполука формули (VIII) забезпечувала просте і промислово застосовне введення метокси-групи в положення 21. Силілований ціангідрин формули (VII) взаємодіє з розчином DIBAL-H у циклогексані з одержанням A-біс-TMSO-карбальдегіду формули (VIII), який реагує з метокси-метилхлоридом або бромідом по реакції Гріньяра з одержанням діолу формули (IX). Окислення 20-гідрокси похідного, одержаного в реакції Гріньяра, до 20-кето похідного здійснюють окисленням по Шверну – замість йод-оксибензойної кислоти, яку використовують у літературних прикладах – дициклогексилкарбодіімідом у присутності диметилсульфоксиду і сильної органічної кислоти. Перевага даного способу за дійсним винаходом полягає в тому, що використовуваний реактив стійкий і його застосування економічно вигідне. d) Вихідною речовиною в методиках, описаних в літературі, є силілований ціангідрин, з якого синтезували 21-метокси похідне в чотири стадії – послідовно окружним шляхом. Ціангідрин перетворювали в 21-хлор або 21-бром похідне літійдіізопропіламідом при -78 °C, потім галогенові замісники заміняли на ацетокси-групу і гідролізували останню з одержанням 21гідрокси похідного, яке вводили в реакцію з тетрафторборатом триметилоксонію (використовуючи "протонну губку") в SNAP реакції, одержуючи 21-метокси похідне. e) У способі за дійсним винаходом відсутні небезпечні реактиви, такі як алюмогідрид літію, і вкрай вогненебезпечні розчинники, такі як ефір. f) Інша перевага способу за дійсним винаходом полягає в тому, що одержувані проміжні сполуки в більшості випадків досить чисті для використання в наступних стадіях без очищення. g) Хоча хроматографічне очищення використовують тільки в двох останніх стадіях способу за дійсним винаходом, чистотаодержаної діючої речовини CDB-4124 складає 99 %, що задовольняє вимоги для терапевтичного застосування. h) Інша перевага способу за дійсним винаходом полягає у високих виходах в індивідуальних синтетичних стадіях (48,65 %; 73,36 %; 80,79 %; 89,09 %; 83,6 %; 84,1 %; 99,56 %; 83,78 %). Загальний вихід 11-стадійного синтезу складає 8,23 %, на відміну від загальних виходів за відомими методиками (3,22 % і 3,89 %). і) Умови реакцій, використовувані в деяких стадіях способу за дійсним винаходом, відрізняються від умов, використовуваних у відомих методиках, тому виходи і чистота одержаних продуктів вище. Наприклад, у реакції Гріньяра – заміщення в положенні 11 – співвідношення стероїдної вихідної речовини і 4-бромдиметиланіліну складає 1:1.25, на відміну від співвідношення 1:5, що застосовується у відомих методиках. Таке проведення реакції менш витратне, а виділення продукту легше, оскільки утворюється менше домішок. Спосіб за дійсним винаходом проілюстрований наступними не обмежуючими прикладами. 7 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 Відомості, що підтверджують можливість здійснення винаходу Приклад 1 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-5α-оестр-9(11)-ен-17-он [сполука формули (III)] В атмосфері азоту розчиняли 3,3-[1,2-етандіїл-біс(оксі)]-оестр-5(10),9(11)-дієн-17-он (46,7 г, 149 ммоль) при інтенсивному перемішуванні в суміші піридину (2,46 мл, 0,2 моль-еквівалент) і дихлорметану (234 мл) і охолоджували одержаний розчин до (-6)-(-8)°C. Після додавання гексахлорацетону (5,46 мл, 35,95 ммоль) до перемішуваного розчину додавали 50 %-ний перекис водню, що має температуру 0-(-2)°C (60 мл, 1058 ммоль) з такою швидкістю, щоб температура не піднімалася вище 0 °C. Реакційну суміш перемішували при 1-(-1)°C протягом 20-24 год., потім розбавляли дихлорметаном, що має температуру 0-5 °C (390 мл), надлишок перекису водню розкладали додаванням розчину пентагідрату тіосульфату натрію (327 г, 1318 ммоль, 8,87 моль-еквівалент) у крижаній воді (1500 мл). Реакційну суміш перемішували протягом 1,5 год., потім відокремлювали органічну фазу. Водну фазу екстрагували дихлорметаном, об'єднані органічні шари промивали водою, сушили над безводним сульфатом натрію, фільтрували й випарювали. Маслоподібний залишок кристалізували із суміші 1:3 етилацетат/діізопропіловий ефір (435 мл), що містить 0,1 % піридину. Одержаний у такий спосіб продукт сушили, одержуючи 23,87 г (48,66 %) цільової сполуки. Чистота цільової сполуки складала 98,5-98,8 % (визначено методом ВЕРХ); вміст α-епоксиду складав 95,3 %. Температура плавлення: 153-155 °C. 25 [α] D=127,5° (c=1 %, хлороформ) 1 ЯМР: H ЯМР (500 MГц, CDCl3 (ТМС), δ (м.д.)): 0,88 (3H, д, 18-CH3); 1,91 (1H, дд, Hx-4); 2,17 (1H, д, Hy-4); 3,86-3,98 (4H, м, O-CH2-CH2-O); 6,05 (1H, м, H-11) 13 C ЯМР (125 MГц, CDCl3 (ТМС) δ (м.д.)): 14,8 (C-18); 40,3 (C-4); 60,1 (C-10); 61,6 (C-5); 64,1 і 64,3 (O-CH2-CH2-O); 107,0 (C-3); 125,7 (C-11); 136,7 (C-9); 221,1 (C-17) Приклад 2 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-17α-гідрокси-5α-оестр-9(11)-ен-17β-карбонітрил [сполука формули (IV)] 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-5α-оестр-9(11)-ен-17-он (33 г, 0,1 моль), одержаний у Прикладі 1, суспендували в метанолі (132 мл), потім додавали порошкоподібний ціанід калію (19,5 г, 0,3 моль) при 20-25 °C. Після обережного додавання оцтової кислоти (11,5 мл, 0,2 моль) гетерогенну реакційну суміш нагрівали до 55 °C протягом 15 хв, потім охолоджували до 25 °C протягом 1год і перемішували при цій температурі ще 5 год. Після завершення реакції додавали воду (132 мл) протягом 30 хв, одержаний кристалічний продукт відфільтровували, промивали водою і використовували в наступній стадії без висушування. Температура плавлення висушеного зразка: 143-144 °C. 25 [α] D = +13,5° (c=1 %, хлороформ) 1 ЯМР: H ЯМР (500 MГц, CDCl3 (TMS), δ (м.д.): 0,93 (3H, с, 18-CH3); 3,09 (1H, с, OH); 3,86 і 3,98 (4H, м, O-CH2-CH2-O); 6,07 (1H, м, H-11) 13 C ЯМР (125 MГц, CDCl3 (ТМС), δ (м.д.): 16,8 (C-18); 60,2 (C-10); 61,9 (C-5); 64,0 і 64,2 (OCH2-CH2-O); 77,3 (C-17); 106,9 (C-3); 120,7 (C-20); 125,9 (C-11); 135,7 (C-9) Приклад 3 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-17α-[(триметил-силіл)-окси]-5α-оестр-9(11)-ен-17βкарбонітрил [сполука формули (V)] 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-17α-гідрокси-5α-оестр-9(11)-ен-17βкарбонітрил, одержаний у Прикладі 2, розчиняли при інтенсивному перемішуванні в дихлорметані (300 мл), одержаний розчин сушили над безводним сульфатом натрію, потім відганяли з розчину 200 мл дихлорметану. До одержаного в такий спосіб розчину додавали імідазол (10,1 г, 0,148 моль), потім додавали по краплях триметилхлорсилан (15,5 мл, 0,121 моль) при 20-25 °C протягом 20 хв. Після перемішування протягом 1 год., розчин розбавляли дихлорметаном (66 мл) і водою (66 мл). Органічний шар відокремлювали, промивали водою, сушили над безводним сульфатом натрію, фільтрували й упарювали. Залишок обробляли метанолом (60 мл), охолоджували до 0 °C, кристалічний продукт, що випав в осад, відфільтровували, промивали метанолом, що мав температуру 0 °C, і сушили при 40 °C у вакуумі, одержуючи 31,5 г (73,36 %) цільової сполуки. Одержаний продукт використовували в наступній стадії. Температура плавлення: 167-170 °C. 25 [α] D = +12,5° (c=1 %, хлороформ) 1 ЯМР: H ЯМР (300 MГц, CDCl3 (ТМС), δ (м.д.)): 0,15 (9H, с, 17-O-Si(CH3)3); 0,83 (3H, д, 18CH3); 1,83 (1H, дд, Hx-4); 2,08 (1H, д, Hy-4); 3,76-3.94 (4H, м, O-CH2-CH2-O); 6,01 (1H, м, H-11) 13 C ЯМР (75 MГц, CDCl3 (ТМС). δ (м.д.)): 0,9 (17-O-Si(CH3)3); 16,3 (C-18); 40,1 (C-4); 59,9 (C10); 61,5 (C-5); 63,9 і 64,1 (O-CH2-CH2-O); 78,2 (C-17); 106,8 (C-3); 120,5 (C-20); 126,3 (C-11); 135,4 (C-9) 8 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 Приклад 4 11β-[4-(Диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс(оксі)]-5-гідрокси-17α-[(триметил-силіл)окси]-5α-оестр-9-ен-17β-карбонітрил [сполука формули (VI)] В атмосфері азоту магнієві стружки (3,3 г, 0,136 моль), сухий тетрагідрофуран (24 мл) і 1,2диброметан (0,12 мл, 0,00131 моль) поміщали в колбу, оснащену мішалкою, термометром, краплинною лійкою, підведенням і відведенням газу, при 20-25 °C. Після перемішування протягом 5-10 хв, температура починала підвищуватися, указуючи на успішне здійснення активації. Одночасно при 25 °C в атмосфері азоту готували наступний розчин: сухий тетрагідрофуран (15 мл), сухий толуол (84 мл), 4-бром-N, N-диметиланілін (25 г, 0,125 моль) і 1,2-диброметан (0,16 мл, 0,00186 моль). 2 мл даного розчину додавали до розчину, що містить магнієві стружки, і одержану в такий спосіб перемішувану реакційну суміш нагрівали до 60 °C. Якщо інтенсивне кипіння реакційної суміші вказувало на успішне здійснення активації, тоді додавали по краплях залишок розчину 4-бром-N, N-диметиланіліну після охолодження і підтримували охолодженням температуру 14-16 °C ще протягом 2 год. Хлорид міді(I) (0,4 г, 4,04 ммоль) додавали до одержаного розчину реактиву Гріньяра, потім реакційну суміш перемішували при 20-25 °C протягом 5 хв. Після охолодження до 8-13 °C 5,10αепокси-3,3-[1,2-етандіїл-біс(оксі)]-17α-[(триметилсиліл)-окси]-5α-оестр-9(11)-ен-17β-карбонітрил (42,96 г, 0,1 моль) у дихлорметані (180 мл) прикапували до перемішуваного та охолоджуваного розчину з такою швидкістю, щоб температура не перевищувала 10-15 °C. Потім охолодження припиняли і реакційну суміш перемішували ще 4 год. Після завершення реакції, реакційну суміш додавали до інтенсивно перемішуваного розчину хлориду амонію (100 мл, 10 %-ний водний розчин), який містив піросульфіт натрію (0,4 г, 2,1 ммоль), розбавляли дихлорметаном (100 мл), перемішували і залишали для розділення. Після відокремлення органічного шару, водну фазу екстрагували дихлорметаном, об'єднані органічні шари промивали водою, сушили над безводним сульфатом натрію, фільтрували й упарювали. Залишок перекристалізували з метанолу, одержуючи 44,5 г (80,79 %) цільової сполуки. Температура плавлення: 243-256 °C. 25 [α] D= -12,4° (c=1 %, хлороформ) 1 ЯМР: H ЯМР (300 MГц, CDCl3 (ТМС), δ (м.д.)): 0,24 (9H, с, 17-O-Si(CH3)3); 0,55 (3H, с, 18CH3); 1,67 (1H, д, Hx-4); 2,02 (1H, дд, Hy-4); 2,91 (6H, с, N-CH3); 3,87-4,07 (4H, м, O-CH2-CH2-O); 4,29 (1H, д, H-11); 4,42 (1H, д, OH); 6,64 (2H, м, H-3' і H-5'); 7,05 (2H, м, H-2' і H-6') 13 C ЯМР (75 MГц, CDCl3 (ТМС), δ (м.д.)): 1,1 (17-O-Si(CH3)3); 16,9 (C-18); 38,8 (C-11); 40,7 (NCH3); 47,5 (C-4); 64,1 і 64,5 (O-CH2-CH2-O); 70,1 (C-5); 78,9 (C-17); 108,8 (C-3); 112,6 (C-3' і C-5'); 121,0 (C-20); 127,6 (C-2' і C-6'); 133,9, 134,0, 134,1 (C-9, C-10, C-1'); 148,4 (C-4') Одержаний у такий спосіб продукт використовували в наступній стадії без додаткового очищення. Приклад 5 11β-[4-(Диметиламіно)-феніл)]-3.3-[1,2-етандіїл-біс(оксі)]-5,17α-біс-[(триметил-силіл)-окси]5α-оестр-9-ен-17β-карбонітрил [сполука формули (VII)] 11β-[4-(Диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс(оксі)]-5-гідрокси-17α- [(триметил-силіл)окси]-5α-оестр-9-ен-17β-карбонітрил (55 г, 0,1 моль) та імідазол (10,2 г, 0,15 моль) розчиняли при перемішуванні в дихлорметані (225 мл) при 20-25 °C. До розчину прикапували триметилхлорсилан (15,75 мл, 0,123 моль) протягом 20 хв. Під час додавання реагенту починав осаджуватися гідрохлорид імідазолу, вказуючи на протікання реакції. Після перемішування протягом 2 год. реакційну суміш розбавляли дихлорметаном (100 мл) і водою (100 мл), перемішували протягом декількох хвилин, залишали для розділення шарів, потім органічний шар відокремлювали, промивали водою, сушили над безводним сульфатом натрію, фільтрували й упарювали. Залишок кристалізували з метанолу, відфільтрований продукт сушили у вакуумі, одержуючи 55,5 г (89,09 %) цільової сполуки. Температура плавлення: 164166 °C. 25 [α] D = +14,7° (c=1 %, хлороформ) 1 ЯМР: H ЯМР (300 MГц, ДМСО-d6_(ТМС) δ (м.д.)): 0,11 (9H, с, 17-O-Si(CH3)3); 0,22 (9H, с, 5O-Si(CH3)3); 0,45 (3H, с, 18-CH3); 1,63 (1H, д, Hx-4); 2,07 (1H, дд, Hy-4); 2,84 (6H, с, N-CH3); 3,653,90 (4H, м, O-CH2-CH2-O); 4,21 (1H, д, H-11); 6,64 (2H, м, H-3' і H-5'); 7,03 (2H, м, H-2' і H-6') 13 C ЯМР (75 MГц, ДМСО-d6 (ТМС), δ (м.д.)): 0,9 (17-O-Si(CH3)3); 2,5 (5-O-Si(CH3)3); 16,7 (C18); 37,8 (C-11); 40,1 (N-CH3); 48,6 (C-4); 62,7 і 64,0 (O-CH2-CH2-O); 73,0 (C-5); 78,5 (C-17); 107,6 (C-3); 112,3 (C-3' і C-5'); 120,7 (C-20); 127,4 (C-2' і C-6'); 132,3, 133,2, 134,9 (C-9, C-10, C-1'); 148,1 (C-4') 60 9 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 Приклад 6 11β-[4-Диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс(оксі)]-5,17α-біс-[(триметил-силіл)окси]5α-оестр-9-ен-17β-карбальдегід [сполука формули (VIII)] В атмосфері азоту 11β-[4-(диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс-(оксі)]-5,17α-біс[(триметил-силіл)-окси]-5α-оестр-9-ен-17β-карбонітрил (40 г, 62,4 ммоль) розчиняли в суміші метил-трет-бутилового ефіру (220 мл) і тетрагідрофурану (17 мл). Одержаний розчин охолоджували до (-15)-(-20)°C, потім додавали 1M розчин DIBAL-H (діізобутилгідрид алюмінію) у циклогексані (160 мл) протягом 30 хв. при (-15)-(-20)°C. Реакційну суміш перемішували 1 год., потім в атмосфері азоту додавали при інтенсивному перемішуванні суміш води (160 мл) і оцтової кислоти (80 мл), що має температуру (-5)-(-10)°C протягом 15-20 хв. Одержану в такий спосіб реакційну суміш перемішували при 20-25 °C протягом 30 хв, потім органічний шар відокремлювали, промивали водою (200 мл), 0,3M розчином гідрокарбонату натрію (2 × 200 мл) і водою (200 мл). Органічний шар без висушування упарювали у вакуумі при 40-45 °C. Залишок розчиняли в метанолі (140 мл) і упарювали у вакуумі до об'єму 30 мл. Одержану кристалічну суспензію охолоджували до 5-10 °C, фільтрували після 1 год. стояння, промивали і сушили при температурі нижче 60 °C у вакуумі, одержуючи 33,6 г (83,6 %) цільової сполуки, яку використовували в наступній стадії. Температура плавлення: 154-158 °C. 25 [α] D = +7,7° (c=1 %, хлороформ). 1 ЯМР: H ЯМР (500 MГц, CDCl3 (ТМС), δ (м.д.)): 0,12 (9H, с, 17-O-Si(CH3)3); 0,19 (9H, с, 5-OSi(CH3)3); 0,33 (3H, с, 18-CH3); 2,88 (6H, с, N-CH3); 3,86 і 3,98 (4H, м, O-CH2-CH2-O); 4,21 (1H, м, H-11); 6,61 (2H, м, H-3' і H-5'); 7,00 (2H, м, H-2' і H-6'); 9,56 (1H, с, H-20) 13 C ЯМР (125 MГц, CDCl3 (ТМС), δ (м.д.)): 1,9 (17-O-Si(CH3)3); 2,6 (5-O-Si(CH3)3); 15,7 (C-18); 38,8 (C-11); 40,7 (N-CH3); 63,4 і 64,4 (O-CH2-CH2-O); 73,7 (C-5); 91,1 (C-17); 108,5 (C-3); 112,8 (C3' і C-5'); 127,6 (C-2' і C-6'); 133,6 (C-10); 134,3 (C-1'); 135,9 (C-9); 148,3 (C-4'); 203,3 (C-20) Приклад 7 11β-[4-(Диметиламіно)-феніл)]-17,20ξ-дигідрокси-21-метокси-19-норпрегн-4,9-дієн-3-он [сполука формули (IX)] В атмосфері азоту магнієві стружки (4,2 г, 173 ммоль), сухий тетрагідрофуран (60 мл) і 1,2диброметан (2,4 мл, 28 ммоль) поміщали в 4-горлу колбу об'ємом 500 мл, оснащену мішалкою, термометром, краплинною лійкою, зворотним холодильником, підведенням і відведенням газу, при 20-25 °C. Після декількох хвилин перемішування суміш досягала температури кипіння. Потім реакційну суміш охолоджували до 35-40 °C і додавали хлорид ртуті(II) (0,23 г, 0,85 ммоль), після перемішування протягом 15 хв суміш охолоджували до 20-25 °C і додавали сухий толуол (20 мл). Метоксиметилхлорид (12,8 мл, 168 ммоль) розчиняли в сухому толуолі (50 мл) і 6 мл одержаного в такий спосіб розчину додавали до суміші в 4-горлу колбу. Через кілька хвилин температура реакційної суміші піднімалася до 35 °C. Реакційну суміш охолоджували до 0-(-5)°C і додавали залишок розчину метоксиметилхлориду в толуолі протягом 2-2,5 год., підтримуючи температуру 0-(-5)°C. По закінченні додавання, протягом 1 год. додавали розчин 11β-[(4-диметиламіно)-феніл]-3,3-[(1,2-етандіїл)-біс(оксі)]-5,17α-біс-[(триметилсиліл-(оксі)]-5α-оестр-9-ен-17β-карбальдегіду (20,0 г, 32 ммоль) у сухому толуолі (80 мл), підтримуючи температуру 0-(-5)°C. Після завершення реакції, реакційну суміш додавали до 1M водного розчину гідросульфату калію (200 мл) з такою швидкістю, щоб температура не піднімалася вище 30 °C. Суміш перемішували при 20-25 °C протягом 2 год., потім органічний шар відокремлювали і промивали 1M розчином гідросульфату калію (1×10 мл). Об'єднані водні фази додавали до перемішуваної суміші 1M розчину гідрокарбонату натрію (225 мл) і дихлорметану (75 мл). Після перемішування протягом 10-15 хв відокремлювали органічний шар. Водну фазу екстрагували дихлорметаном (5 × 50 мл), об'єднані органічні шари сушили над безводним сульфатом натрію (2 г), фільтрували, промивали дихлорметаном (2×20 мл), і фільтрат перемішували з вугіллям (2,5 г) протягом 10 хв. Вугілля відфільтровували, промивали дихлорметаном (2 × 20 мл), і фільтрат упарювали, одержуючи 12,51 г (84,1 %) цільової сполуки. Температура плавлення: 105 °C (розм'якшення). 25 [α] D = +157,7° (c=1 %, дихлорметан) 1 ЯМР: H ЯМР ((основний діастереомер), 500 MГц, CDCl3 (ТМС), δ (м.д.)): 0,49 (3H, с, 18CH3); 2,91 (6H, с, N-CH3); 3,37 (3H, с, O-CH3); 3,49 (1H, м, Hx-21); 3,57 (1H, м, Hy-21); 3,81 (1H, м, H-20); 4,30 (1H, м, H-11); 5,73 (1H, с, H-4); 6,67 (2H, м, H-3' і H-5'); 7,04 (2H, м, H-2' і H-6') 13 C ЯМР ((основний діастереомер), 125 MГц, CDCl3 (ТМС), δ (м.д.)): 16,5 (C-18); 39,5 (C-11); 40,7 (N-CH3); 59,2 (O-CH3); 71,9 (C-20); 74,9 (C-21); 84,4 (C-17); 112,8 (C-3' і C-5'); 122,5 (C-4); 127,6 (C-2' і C-6'); 128,6 (C-10); 132,0 (C-1'); 147,2 (C-9); 148,5 (C-4'); 157,1 (C-5); 199,7 (C-3) Приклад 8 11β-[4-(Диметиламіно)-феніл)]-17,20ξ-дигідрокси-21-метокси-19-норпрегн-4,9-дієн-3-он 10 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 [сполука формули (IX)] В атмосфері азоту магнієві стружки (4,2 г, 173 ммоль), сухий тетрагідрофуран (60 мл) і 1,2диброметан (2,4 мл, 28 ммоль) поміщали в 4-горлу колбу об'ємом 500 мл, оснащену мішалкою, термометром, краплинною лійкою, зворотним холодильником, підведенням і відведенням газу, при 20-25 °C. Після декількох хвилин перемішування суміш досягала температури кипіння. Потім реакційну суміш охолоджували до 35-40 °C і додавали хлорид ртуті(II) (0,23 г, 0,85 ммоль), після перемішування протягом 15 хв суміш охолоджували до 20-25 °C і додавали сухий толуол (20 мл). Метоксиметилбромід (13,7 мл, 168 ммоль) розчиняли в сухому толуолі (50 мл), і 6 мл одержаного в такий спосіб розчину додавали до суміші в 4-горлу колбу. Через кілька хвилин температура реакційної суміші піднімалася до 30-35 °C. Реакційну суміш охолоджували до 10-15 °C і додавали залишок розчину метоксиметилброміду в толуолі протягом 2-2,5 год., підтримуючи температуру 10-15 °C. По закінченні додавання, протягом 1 год. додавали розчин 11β-[(4-диметиламіно)-феніл]-3,3-[(1,2-етандіїл)-біс(оксі)]-5,17α-біс[(триметил-силіл-(оксі)]-5α-оестр-9-ен-17β-карбальдегіду (20,0 г, 32 ммоль) у сухому толуолі (80 мл), підтримуючи температуру 10-15 °C. Після завершення реакції реакційну суміш додавали до 1M водного розчину гідросульфату калію (200 мл) з такою швидкістю, щоб температура не піднімалася вище 30 °C. Суміш перемішували при 20-25 °C протягом 2 год., потім органічний шар відокремлювали і промивали 1M розчином гідросульфату калію (1×10 мл). Об'єднані водні фази додавали до перемішуваної суміші 1M розчину гідрокарбонату натрію (225 мл) і дихлорметану (75 мл). Після 10-15 хв перемішування відокремлювали органічний шар. Водну фазу екстрагували дихлорметаном (5 × 50 мл), об'єднані органічні шари сушили над безводним сульфатом натрію (2 г), фільтрували, промивали дихлорметаном (2 × 20 мл), і фільтрат перемішували з вугіллям (2,5 г) протягом 10 хв. Вугілля відфільтровували, промивали дихлорметаном (2 × 20 мл), і фільтрат упарювали, одержуючи 10,76 г (72,3 %) цільової сполуки. Невеликий зразок продукту перемішували з н-пентаном, температура плавлення: 105 °C (склоподібна структура, розм'якшення). 25 [α] D = +157,7° (c=1 %, дихлорметан) Приклад 9 11β-[4-(Диметиламіно)-феніл)]-17-гідрокси-21-метокси-19-норпрегн-4,9-дієн-3,20-діон [сполука формули (X)] В атмосфері азоту 17,20ξ-дигідрокси-11β-[(4-диметиламіно)-феніл]-21-метокси-19-норпрегн4,9-дієн-3-он (11,94 г, 25,7 ммоль) і сухий толуол (68 мл) поміщали в 4-горлу колбу об'ємом 500 мл, оснащену мішалкою, термометром, краплинною лійкою, зворотним холодильником, підведенням і відведенням газу. До одержаного в такий спосіб розчину додавали сухий диметилсульфоксид (9,9 мл, 139,5 ммоль), піридин (2,9 мл, 36,0 ммоль) і трифтороцтову кислоту (0,99 мл, 12,85 ммоль) при 20-25 °C. Потім до реакційної суміші додавали розчин дициклогексилкарбодііміду (10,6 г, 51,4 ммоль) у толуолі (54 мл), і одержану в такий спосіб суміш перемішували при 40 °C. Час реакції складав 3 год. По закінченні реакції, реакційну суміш охолоджували до 20-25 °C і додавали 1M розчин гідросульфату калію (78 мл). Після 30 хв перемішування кристали, що випали в осад, відфільтровували і промивали 1M розчином гідросульфату калію (4 × 19 мл). Дві фази фільтрату розділяли, водну фазу додавали до 1M розчину гідроксиду натрію (254 мл) при 10-20 °C. Після 30 хв перемішування сирий продукт, що випав в осад, відфільтровували, промивали водою і сушили, одержуючи 9,45 г (79,5 %) цільової сполуки. Температура плавлення: 105-110 °C. Сирий продукт очищали методом ВЕРХ відповідно до способу, описаного в наступному прикладі. 1 ЯМР: H ЯМР (500 MГц, ДМСО-d6 (ТМС), δ (м.д.)): 0,20 (3H, с, 18-CH3); 2,82 (6H, с, N-CH3); 3,26 (3H, с, O-CH3); 4,20 (1H, д, Hx-21); 4,35 (1H, м, H-11); 4,49 (1H, д, Hy-21); 5,37 (1H, с, OH); 5,67 (1H, с, H-4); 6,62 (2H, м, H-3' і H-5'); 7,00 (2H, м, H-2' і H-6') 13 C ЯМР (125 MГц, ДМСО-d6 (ТМС), δ (м.д.)): 15,8 (C-18); 38,7 (C-11); 40,1 (N-CH3); 58,3 (OCH3); 75,4 (C-21); 88,6 (C-17); 112,5 (C-3' і C-5'); 122,0 (C-4); 127,2 (C-2' і C-6'); 128,1 (C-10); 132,0 (C-1'); 146,6 (C-9); 148,2 (C-4'); 156,5 (C-5); 197,9 (C-3); 208,9 (C-20) Приклад 10 Очищення сирого 11β-[4-(диметиламіно)-феніл)]-17-гідрокси-21-метокси-19- норпрегн-4,9дієн-3,20-діону [сполука формули (X)] методом ВЕРХ (лабораторний масштаб) Силікагелем (510 г, ZEOPREP C-GEL C-490L, розмір частинок 15-35 мкм; висота шару близько 60 см) заповнювали ВЕРХ колонку з аксіальним стисненням діаметром 5 см методом суспензійного набивання, і колонку врівноважували елюентом – сумішшю 45:40:15 циклогексан/метил-трет-бутиловий ефір/ацетон. 10,0 г сирої цільової сполуки (вміст діючої речовини: 80 %) розчиняли в суміші ацетону (30 мл) і метил-трет-бутилового ефіру (80 мл), і до 11 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 перемішуваного розчину додавали циклогексан (90 мл). Одержаний у такий спосіб розчин фільтрували і наносили на колонку шляхом вколювання. Продукт елюювали зі швидкістю потоку 85 мл/хв, використовуючи УФ-детектування. Перша фракція складала близько 50 мл, основна фракція, що містить чисту цільову сполуку, складала близько 600 мл. Тверду цільову сполуку одержували упарюванням елюйованої основної фракції. Вихід: 7,4 г (74 %) чистої цільової сполуки. Вміст домішок: менш 4 %. Температура плавлення: 108-110 °C. 25 [α] D = +199,2° (c=1 %, хлороформ) 1 ЯМР: H ЯМР (500 MГц, ДМСО-d6 (ТМС), δ (м.д.)): 0,20 (3H, с, 18-CH3); 2,82 (6H, с, N-CH3); 3,26 (3H, с, O-CH3); 4,20 (1H, д, Hx-21); 4,35 (1H, м, H-11); 4,49 (1H, д, Hy21); 5,37 (1H, с, OH); 5,67 (1H, с, H-4); 6,62 (2H, м, H-3' і H-5'); 7,00 (2H, м, H-2' і H-6') 13 C ЯМР (125 MГц, ДМСО-d6 (ТМС), δ (м.д.)): 15,8 (C-18); 38,7 (C-1l); 40,1 (N-CH3); 58,3 (OCH3); 75,4 (C-21); 88,6 (C-17); 112,5 (C-3' і C-5'); 122,0 (C-4); 127,2 (C-2' і C-6'); 128,1 (C-10); 132,0 (C-1'); 146,6 (C-9); 148,2 (C-4'); 156,5 (C-5); 197,9 (C-3); 208,9 (C-20) Приклад 11 17-Ацетокси-11β-[4-(диметиламіно)-феніл)]-21-метокси-19-норпрегн-4,9-дієн-3,20-діон [сполука формули (I)] 70 %-ну перхлорну кислоту (6 мл) додавали до перемішуваного та охолоджуваного ((-20)-(25)°C) оцтового ангідриду (45 мл) з такою швидкістю, щоб температура не піднімалася вище (15)°C. Потім додавали розчин 11β-[4-(диметиламіно)-феніл)]-17-гідрокси-21-метокси-19норпрегн-4,9-дієн-3,20-діону (15,5 г) у дихлорметані (60 мл) при (-20)-(-25)°C. По закінченні реакції – яку контролювали методом тонкошарової хроматографії – реакційну суміш розбавляли дихлорметаном (50 мл), охолоджували до (-10)°C і додавали пропущену через іонообмінник воду (52 мл) для розкладання оцтового ангідриду. Після 10 хв перемішування додавали 25 %ний розчин гідроксиду амонію (77 мл) з такою швидкістю, щоб температура не піднімалася вище 25 °C (pН=7-8). Потім карбамідний побічний продукт, що випав в осад, відфільтровували, водну фазу відокремлювали, екстрагували дихлорметаном (2 × 30 мл), і об'єднані органічні шари упарювали, одержуючи 16,2 г (95,8 %) цільової сполуки, яку очищали методом ВЕРХ відповідно до способу, описаного в наступному Прикладі. 1 ЯМР: H ЯМР (500 MГц, CDCl3 (ТМС), δ (м.д.)): 0,40 (3H, с, 18-CH3); 2,10 (3H, с, O-CO-CH3); 2,90 (6H, с, N-CH3); 3,41 (3H, с, O-CH3); 4,09 (1H, д, Hx-21); 4,38 (1H, м, H-11); 4,29 (1H, д, Hy-21); 5,77 (1H, ушир., H-4); 6,62 (2H, м, H-3' і H-5'); 6,96 (2H, м, H-2' і H-6') 13 C ЯМР (125 MГц, CDCl3 (ТМС), δ (м.д.)): 15,6 (C-18); 21,1 (O-CO-CH3); (39,3 (C-11); 40,6 (NCH3); 59,4 (O-CH3); 76,0 (C-21); 93,9 (C-17); 112,8 (C-3' і C-5'); 123,0 (C-4); 127,3 (C-2' і C-6'); 129,4 (C-10); 131,3 (C-1'); 145,5 (C-9); 148,7 (C-4'); 156,4 (C-5); 170,7 (O-CO-CH3); 199,4 (C-3); 202,7 (C20) Приклад 12 Очищення сирого CDB-4124 методом ВЕРХ (елюент: циклогексан:метил/трет-бутиловий ефір:ацетон = 60:30:10) (лабораторний масштаб) [сполука формули (I)] Силікагелем (510 г, ZEOPREP C-GEL C-490L, розмір частинок 15-35 мкм; висота шару близько 60 см) заповнювали ВЕРХ колонку з аксіальним стисненням діаметром 5 см методом суспезійного набивання, і колонку врівноважували елюентом – сумішшю 60:30:10 циклогексан/метил-трет-бутиловий ефір/ацетон. 5,1 г сирої сполуки формули (I) (CDB-4124), одержаної в попередньому прикладі (вміст домішок: менше 4 %), розчиняли в елюенті (100 мл), фільтрували і наносили на колонку шляхом вколювання. Продукт елюювали зі швидкістю потоку 85 мл/хв, застосовуючи УФ-детектування. Перша фракція складала близько 40 мл, основна фракція, що містить чистий CDB-4124, складала близько 560 мл. Тверду цільову сполуку одержували упарюванням елюйованої основної фракції. Вихід: 4,25 г (83,33 %), вміст домішок: менш 0,5 %. Температура плавлення: 118 °C. 25 [α] D = +127,2° (c=1 %, хлороформ) 1 ЯМР: H ЯМР (500 MГц, CDCl3 (ТМС), δ (м.д.)): 0,40 (3H, с, 18-CH3); 2,10 (3H, с, O-CO-CH3); 2,90 (6H, с, N-CH3); 3,41 (3H, с, O-CH3); 4,09 (1H, д, Hx-21); 4,38 (1H, м, H-11); 4,29 (1H, д, Hy-21); 5,77 (1H, ушир., H-4); 6,62 (2H, м, H-3' і H-5'); 6,96 (2H, м, H-2' і H-6') 13 C ЯМР (125 MГц, CDCl3 (ТМС), δ (м.д.)): 15,6 (C-18); 21,1 (O-CO-CH3); (39,3 (C-11); 40,6 (NCH3); 59,4 (O-CH3); 76,0 (C-21); 93,9 (C-17); 112,8 (C-3' і C-5'); 123,0 (C-4); 127,3 (C-2' і C-6'); 129,4 (C-10); 131,3 (C-1'); 145,5 (C-9); 148,7 (C-4'); 156,4 (C-5); 170,7 (O-CO-CH3); 199,4 (C-3); 202,7 (C20) Приклад 13 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-5α-оестр-9(11)-ен-17-он [сполука формули (III)] Обладнання: кислототривкий сталевий реактор ємністю 500 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. 12 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 В атмосфері азоту розчиняли 3,3-[1,2-етандіїл-біс(оксі)]-оестр-5(10),9(11)-дієн-17-он (21,0 кг) при інтенсивному перемішуванні в суміші піридину (1,106 л, 0,2 моль-еквівалент) і сухого дихлорметану (105,2 л), і одержаний розчин охолоджували до (-6)-(-8)°C. Після додавання гексахлорацетону (2,455 л) до перемішуваного розчину додавали 50 %-ний перекис водню, що має температуру 0-(-2)°C (26,97 л) з такою швидкістю, щоб температура не піднімалася вище 0 °C. Реакційну суміш перемішували при 1-(-1)°C протягом 20-24 год., потім розбавляли дихлорметаном, що має температуру 0-5 °C (175 л), розкладали надлишок перекису водню додаванням розчину пентагідрату тіосульфату натрію (8,87 моль-еквівалент) у крижаній воді (150 л). Реакційну суміш перемішували протягом 1,5 год., потім відокремлювали органічну фазу. Водну фазу екстрагували дихлорметаном, об'єднані органічні шари промивали водою, сушили над безводним сульфатом натрію, фільтрували й упарювали. Маслоподібний залишок кристалізували із суміші 1:3 етилацетат/діізопропіловий ефір (195,6 л), що містила 0,1 % піридину. Одержаний у такий спосіб продукт сушили, одержуючи 11,500 кг (52,13 %) цільової сполуки. Чистота цільової сполуки складала 98,5-98,9 % (визначено методом ВЕРХ); вміст αепоксиду 96,2 %. Температура плавлення: 151-154 °C. 25 [α] D=125,0° (c=1 %, хлороформ) Приклад 14 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-17α-гідрокси-5α-оестр-9(11)-ен-17β-карбонітрил [сполука формули (IV)] Обладнання: емальований реактор ємністю 250 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-5α-оестр9(11)-ен-17-он (9,9 кг), одержаний у Прикладі 13, суспендували в метанолі (39,6 л), потім додавали порошкоподібний ціанід калію (5,85 кг, 0,3 моль) при 20-25 °C. Після обережного додавання оцтової кислоти (3,48 л) гетерогенну реакційну суміш нагрівали до 55 °C протягом 15 хв, потім охолоджували до 25 °C протягом 1 год., і перемішували при цій температурі ще 5 год. По закінченні реакції додавали воду (39,6 л) протягом 30 хв, одержаний кристалічний продукт відфільтровували, промивали водою і використовували в наступній стадії без висушування. Температура кипіння висушеного зразка: 140-143 °C. 25 [α] D = +13,0° (c=1 %, хлороформ) Приклад 15 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-17α-[(триметил-силіл)-окси]-5α-оестр-9(11)-ен-17βкарбонітрил [сполука формули (V)] Обладнання: емальований реактор ємністю 250 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. 5,10α-Епокси-3,3-[1,2-етандіїл-біс(оксі)]-17α-гідрокси-5α-оестр-9(11)-ен-17βкарбонітрил, одержаний у Прикладі 14, розчиняли при інтенсивному перемішуванні в дихлорметані (90 л), одержаний розчин сушили над безводним сульфатом натрію, потім 60 л дихлорметану відганяли з розчину. Імідазол (0,303 кг) додавали до одержаного в такий спосіб розчину, потім прикапували триметилхлорсилан (7,2 л) при 20-25 °C протягом 20 хв. Після перемішування протягом 1 год., розчин розбавляли дихлорметаном (19,8 л) і водою (19,8 л). Органічний шар відокремлювали, промивали водою, сушили над безводним сульфатом натрію, фільтрували й упарювали. Залишок обробляли метанолом (18 л), охолоджували до 0 °C, кристалічний продукт, що випав в осад, відфільтровували, промивали метанолом, що мав температуру 0 °C, і сушили при 40 °C у вакуумі, одержуючи 10,1 кг (78,4 %) цільової сполуки. Одержаний продукт використовували в наступній стадії без додаткового очищення. Температура плавлення: 167170 °C. 25 [α] D = +12,5° (c=1 %, хлороформ) Приклад 16 11β-[4-(Диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс(оксі)]-5-гідрокси-17α-[(триметил-силіл)окси]-5α-оестр-9-ен-17β-карбонітрил [сполука формули (VI)] Обладнання: емальований реактор ємністю 250 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. В атмосфері азоту магнієві стружки (0,768 кг), сухий тетрагідрофуран (5,59 л) і 1,2диброметан (27,94 мл) поміщали в реактор при 20-25 °C. Після перемішування протягом 510 хв, температура починала підвищуватися, вказуючи на успішне здійснення активації. Одночасно при 25 °C в атмосфері азоту готували наступний розчин: сухий тетрагідрофуран (3,5 л), сухий толуол (19,6 л), 4-бром-N, N-диметиланілін (5,8 кг) і 1,2-диброметан (34,25 мл). 400 мл цього розчину додавали до розчину, що містить магнієві стружки, і одержану в такий спосіб перемішувану реакційну суміш нагрівали до 60 °C. Інтенсивне кипіння реакційної суміші 13 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 вказувало на успішне здійснення активації, залишок розчину 4-бром-N, N-диметиланіліну додавали по краплях після охолодження, і охолодженням підтримували температуру рівною 1416 °C ще протягом 2 год. Хлорид міді(I) (93,11 г) додавали до одержаного розчину реактиву Гріньяра, потім реакційну суміш перемішували при 20-25 °C протягом 5 хв. Після охолодження до 8-13 °C прикапували до перемішуваного і охолоджуваного розчину 5,10α-епокси-3,3-[1,2-етандіїл-біс(оксі)]-17α[(триметилсиліл)-окси]-5α-оестр-9(11)-ен-17β-карбонітрил (10,0 кг) у дихлорметані (42 л) з такою швидкістю, щоб підтримувати температуру 10-15 °C. Потім припиняли охолодження і перемішували реакційну суміш ще 4 год. По закінченні реакції, суміш додавали до інтенсивно перемішуваного розчину хлориду амонію (23,3 л, 10 %-ний водний розчин), що містить піросульфіт натрію (93,1 г), розбавляли дихлорметаном (23,3 л), перемішували і залишали розшаровуватися. Після відділення органічного шару, водну фазу екстрагували дихлорметаном, об'єднані органічні шари промивали водою, сушили над безводним сульфатом натрію, фільтрували й упарювали. Залишок перекристалізували з метанолу, одержуючи 10,5 кг (85,71 %) цільової сполуки. Температура плавлення: 243-256 °C. 25 [α] D = -12,4° (c=1 %, хлороформ) Одержаний у такий спосіб продукт використовували в наступній стадії без додаткового очищення. Приклад 17 11β-[4-(Диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс(оксі)]-5,17α-біс-[(триметил-силіл)-окси]5α-оестр-9-ен-17β-карбонітрил [сполука формули (VII)] Обладнання: емальований реактор ємністю 250 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. 11β-[4-(Диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс(оксі)]-5-гідрокси-17α-[(триметил-силіл)окси]-5α-оестр-9-ен-17β-карбонітрил (10,45 кг) та імідазол (1,93 кг) розчиняли при перемішуванні в дихлорметані (42,75 л) при 20-25 °C. До розчину прикапували триметилхлорсилан (3,0 л) протягом 20 хв. Під час додавання реактиву починав випадати в осад гідрохлорид імідазолу, вказуючи на протікання реакції. Після перемішування протягом 2 год., реакційну суміш розбавляли дихлорметаном (19 л) і водою (19 л), перемішували кілька хвилин, залишали розшаровуватися, потім органічний шар відокремлювали, промивали водою, сушили над безводним сульфатом натрію, фільтрували й упарювали. Залишок кристалізували з метанолу, відфільтрований продукт сушили у вакуумі, одержуючи 10,25 кг (87,0 %) цільової сполуки. Температура плавлення: 164-166 °C. 25 [α] D = +14,7° (c=1 %, хлороформ) Приклад 18 11β-[4-(Диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс(оксі)]-5,17α-біс-[(триметил-силіл)-окси]5α-оестр-9-ен-17β-карбальдегід [сполука формули (VIII)] Обладнання: емальований реактор ємністю 250 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. В атмосфері азоту 11β-[4-(диметиламіно)-феніл)]-3,3-[1,2-етандіїл-біс-(оксі)]-5,17α-біс[(триметил-силіл)-окси]-5α-оестр-9-ен-17β-карбонітрил (8 кг) розчиняли в суміші метил-третбутилового ефіру (44 л) і тетрагідрофурану (3,4 л). Одержаний розчин охолоджували до (-15)(-20)°C, потім додавали 1M розчин DIBAL-H у циклогексані (32 л) протягом 30 хв при (-15)(-20)°C. Реакційну суміш перемішували протягом 1 год., потім при інтенсивному перемішуванні в атмосфері азоту додавали суміш води (32 л) і оцтової кислоти (16 л), що має температуру (-5)(-10)°C, протягом 15-20 хв. Одержану в такий спосіб реакційну суміш перемішували при 20-25 °C протягом 30 хв, потім органічний шар відокремлювали, промивали водою (40 л), 0,3M розчином гідрокарбонату натрію (2 × 40 л) і водою (40 л). Органічний шар без висушування упарювали у вакуумі при 40-45 °C. Залишок розчиняли в метанолі (28 л) і упарювали у вакуумі до об'єму 6 л. Одержану кристалічну суспензію охолоджували до 5-10 °C, фільтрували після 1 год. стояння, промивали і сушили при температурі нижче 60 °C у вакуумі, одержуючи 6,95 кг (86,46 %) цільової сполуки, яку використовували в наступній стадії без додаткового очищення. Температура плавлення: 154-158 °C. 25 [α] D = +7,7° (c=1 %, хлороформ) Приклад 19 11β-[4-(Диметиламіно)-феніл)]-17,20ξ-дигідрокси-21-метокси-19-норпрегн-4,9-дієн-3-он [сполука формули (IX)] Обладнання: емальований реактор ємністю 250 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. 14 UA 100241 C2 5 10 15 20 25 30 35 40 45 50 55 60 В атмосфері азоту магнієві стружки (1,05 кг), сухий тетрагідрофуран (15 л) і 1,2-диброметан (600 мл) додавали в реакційний апарат при 20-25 °C. Після декількох хвилин перемішування суміш досягала температури кипіння. Потім реакційну суміш охолоджували до 35-40 °C і додавали хлорид ртуті(II) (57,5 г), після перемішування протягом 15 хв суміш охолоджували до 20-25 °C і додавали сухий толуол (5 л). Метоксиметилхлорид (3,2 л) розчиняли в сухому толуолі (12,5 л), і 1,5 л одержаного в такий спосіб розчину додавали до реакційної суміші. Через кілька хвилин температура реакційної суміші піднімалася до 35 °C. Реакційну суміш охолоджували до 0-(-5)°C і залишок розчину метоксиметилхлориду в толуолі додавали протягом 2-2,5 год., підтримуючи температуру 0-(-5)°C. По закінченні додавання, протягом 1 год. додавали розчин 11β-[(4-диметиламіно)-феніл]-3,3-[(1,2-етандіїл)-біс(оксі)]-5,17α-біс-[(триметил-силіл-(оксі)]-5αоестр-9-ен-17β-карбальдегіду (5,0 кг) у сухому толуолі (20 л), підтримуючи температуру 0-(-5)°C. По закінченні реакції, реакційну суміш додавали до 1M водного розчину гідросульфату калію (50 л) з такою швидкістю, щоб температура не піднімалася вище 30 °C. Суміш перемішували при 20-25 °C протягом 2 год. потім органічний шар відокремлювали і промивали 1M розчином гідросульфату калію (1 × 2,5 л). Об'єднані водні фази додавали до перемішуваної суміші 1M розчину гідрокарбонату натрію (56 л) і дихлорметану (19 л). Після 10-15 хв перемішування відокремлювали органічний шар. Водну фазу екстрагували дихлорметаном (5 × 12,5 л), об'єднані органічні шари сушили над безводним сульфатом натрію (500 г), фільтрували, промивали дихлорметаном (2 × 2 л), і фільтрат перемішували з вугіллям (625 г) протягом 10 хв. Вугілля відфільтровували, промивали дихлорметаном (2 × 5 л), і фільтрат упарювали, одержуючи 3,3 кг (88,73 %) цільової сполуки. Температура плавлення: 105 °C (розм'якшення). 25 [α] D = +156,2° (c=1 %, дихлорметан). Приклад 20 11β-[4-(Диметиламіно)-феніл)]-17-гідрокси-21-метокси-19-норпрегн-4,9-дієн-3,20-діон [сполука формули (X)] Обладнання: емальований реактор ємністю 250 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. В атмосфері азоту 17,20ξ-дигідрокси-11β-[(4-диметиламіно)-феніл]-21-метокси-19-норпрегн4,9-дієн-3-он (3,2 кг) і сухий толуол (18 л) поміщали в реактор. До одержаного в такий спосіб розчину додавали сухий диметилсульфоксид (2,7 л), піридин (0,78 л) і трифтороцтову кислоту (0,265 л) при 20-25 °C. Потім до реакційної суміші додавали розчин дициклогексилкарбодііміду (2,84 кг) у толуолі (14,5 л), і одержану в такий спосіб суміш перемішували при 40 °C. Час реакції складав 3 год. По закінченні реакції реакційну суміш охолоджували до 20-25 °C і додавали 1M розчин гідросульфату калію (21 л). Після 30 хв перемішування кристали, що випали в осад, відфільтровували і промивали 1M розчином гідросульфату калію (4 × 5 л). Дві фази фільтрату розділяли, водну фазу додавали до 1M розчину гідроксиду натрію (68 л) при 10-20 °C. Після 30 хв. перемішування сирий продукт, що випав в осад, відфільтровували, промивали водою і сушили, одержуючи 2,556 кг (80,0 %) цільової сполуки. Сирий продукт очищали методом ВЕРХ відповідно до способу, описаного в наступному прикладі. Приклад 21 ' Очищення сирого 11β-[4-(диметиламіно )-феніл)]-17-гідрокси-21-метокси-19- норпрегн-4,9дієн-3,20-діону [сполука формули (X)] методом ВЕРХ (промисловий масштаб) Силікагелем (8 кг, ZEOPREP C-GEL C-490L, розмір частинок 15-35 мкм; висота шару близько 60 см) заповнювали ВЕРХ колонку з аксіальним стисненням діаметром 20 см методом суспезійного набивання, і колонку врівноважували елюентом – сумішшю 53:35:12 циклогексан/метил-трет-бутиловий ефір/ацетон. 160 г сирої цільової сполуки (вміст діючої речовини: 80 %) розчиняли в суміші ацетону (0,48 л) і метил-трет-бутилового ефіру (1,28 л), і до перемішуваного розчину додавали циклогексан (1,44 л). Одержаний у такий спосіб розчин фільтрували і наносили на колонку шляхом вколювання. Продукт елюювали при швидкості потоку 80 л/г, застосовуючи УФ-детектування. Перша фракція складала близько 1 л, основна фракція, що містить чисту цільову сполуку, складала близько 14 л. Тверду цільову сполуку одержували упарюванням елюйованої основної фракції, але переважно після упарювання основної фракції дихлорметан відганяли з залишку, і продукт розчиняли в дихлорметані. Одержаний дихлорметановий розчин використовували в наступній стадії. Вихід: 120 г (75 %) чистої, твердої цільової сполуки або дихлорметанового розчину, що містить діючу речовину. Вміст домішок: менш 4 %. Температура плавлення: 105-110 °C. 25 [α] D = +199,2° (c=1 %, хлороформ) Приклад 22 Сирий 17-ацетокси-11β-[4-(диметиламіно)-феніл)]-21-метокси-19-норпрегн-4,9-дієн-3,20-діон 15 UA 100241 C2 5 10 15 20 25 30 [сполука формули (I)] Обладнання: емальований реактор ємністю 250 літрів, оснащений пропелерною мішалкою з різними швидкостями обертання, зворотним холодильником і термометром. 70 %-ну перхлорну кислоту (1,8 л) додавали до перемішуваного і охолоджуваного ((-20)(-25)°C) оцтового ангідриду (13,5 л) з такою швидкістю, щоб температура не піднімалася вище (-15)°C. Потім додавали розчин 11β-[4-(диметиламіно)-феніл)]-17-гідрокси-21-метокси-19норпрегн-4,9-дієн-3,20-діону (4,65 кг) у дихлорметані (18 л) при (-20)-(-25)°C. По закінченні реакції – яку контролювали методом тонкошарової хроматографії – реакційну суміш розбавляли дихлорметаном (15 л), охолоджували до (-10)°C і додавали пропущену через іонообмінник воду (15,5 л) для розкладання оцтового ангідриду. Після 10 хв перемішування додавали 25 %-ний розчин гідроксиду амонію (23 л) з такою швидкістю, щоб температура не піднімалася вище 25 °C (pН=7-8). Потім побічний карбамідний продукт, що випав в осад, відфільтровували, відокремлювали водну фазу, екстрагували дихлорметаном (2 × 9 л), і об'єднані органічні шари упарювали, одержуючи 4,73 кг (93,79 %) цільової сполуки (CDB-4124), яку очищали методом ВЕРХ відповідно до способу, описаного в наступному Прикладі. Приклад 23 Очищення сирого CDB-4124 методом ВЕРХ (промисловий масштаб) [сполука формули (I)] Силікагелем (8 кг, ZEOPREP C-GEL C-490L, розмір частинок 15-35 мкм; висота шару близько 60 см) заповнювали ВЕРХ колонку з аксіальним стисненням діаметром 20 см методом суспезійного набивання, і врівноважували колонку елюентом – сумішшю 53:35:12 циклогексан/метил-трет-бутиловий ефір/ацетон. 80 г сирої сполуки формули (I) (CDB-4124), одержані в попередньому прикладі (вміст домішок: менш 4 %), розчиняли в елюенті (1,6 л), фільтрували і наносили на колонку шляхом вколювання. Продукт елюювали при швидкості потоку 80 л/год., застосовуючи УФ-детектування. Перша фракція складала близько 0,7 л, основна фракція, що містить чистий CDB-4124, складала близько 10 л. Тверду цільову сполуку одержували упарюванням елюйованої основної фракції, або її можна одержувати у вигляді метанольного розчину після упарювання основної фракції і розчинення продукту в метанолі. Вихід: 70 г твердої цільової сполуки або метанольного розчину, що містить діючу речовину. Вміст домішок: менш 0,5 %. Температура плавлення: 118 °C. 25 [α] D = +127,2° (c=1 %, хлороформ) ФОРМУЛА ВИНАХОДУ 35 1. Промисловий спосіб синтезу 17-ацетокси-11β-[4-(диметиламіно)-феніл]-21-метокси-19норпрегн-4,9-дієн-3,20-діону формули (І): CH3 O CH3 N H3C O CH3 O H CH3 O H O , (І) з 3,3-[1,2-етандіїл-біс(оксі)]-оестр-5(10),9(11)-дієн-17-ону формули (II): CH3 O H O H O , (ІІ) який відрізняється тим, що здійснюють стадії: 16 UA 100241 C2 і) утворення епоксиду по подвійному зв'язку в положенні 5(10) взаємодією 3,3-[1,2-етандіїлбіс(оксі)]-оестр-5(10),9(11)-дієн-17-ону формули (II): O CH3 H O H O 5 , (ІІ) з перекисом водню; іі) приєднання одержаного in situ ціановодню в положення 17 одержаного 5,10-епокси-3,3-[1,2етандіїл-біс(оксі)]-5-оестр-9(11)-ен-17-ону формули (III): O CH3 H O O H O , (ІІІ) ііі) силілування гідроксильної групи в положенні 17 утвореного 5,10-епокси-3,3-[1,2-етандіїлбіс(оксі)]-17-гідрокси-5-оестр-9(11)-ен-17-карбонітрилу формули (IV): N CH3 OH H O O H O 10 , (IV) триметилхлорсиланом; iv) взаємодія одержаного 5,10-епокси-3,3-[1,2-етандіїл-біс(оксі)]-17-[триметилсиліл(оксі)]-5оестр-9(11)-ен-17-карбонітрилу формули (V): N CH3 CH3 O Si H H3C O O CH3 H O 15 , (V) з реактивом Гріньяра 4-(диметиламіно)-фенілмагнійбромідом у присутності СuСl (реакція Тойча); 17 UA 100241 C2 v) силілування гідроксильної групи в положенні 5 одержаного 11-[4-(диметиламіно)-феніл]-3,3[1,2-етандіїл-біс(оксі)]-5-гідрокси-17-[триметилсиліл(оксі)]-5-оестр-9-ен-17-карбонітрилу формули (VI): CH3 N N H3C CH3 CH3 O H Si CH3 H3C H O O 5 OH , (VI) триметилхлорсиланом; vi) взаємодія одержаного 11β-[4-(диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(оксі)]-5,17-біс[триметилсиліл(оксі)]-5-оестр-9-ен-17-карбонітрилу формули (VII): CH3 N N H3C CH3 CH3 O H Si CH3 H3C H O O O Si CH3 H3C 10 CH3 , (VII) з діізобутилалюмінійгідридом і наступне додавання кислоти до реакційної суміші; vii) метокси-метилювання одержаного 11-[4-(диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(оксі)]5,17-біс-[триметилсиліл(оксі)]-5а-оестр-9-ен-17-карбальдегіду формули (VIII): CH3 N H3C O CH3 CH3 O H Si H3C CH3 H O O O Si H3C CH3 CH3 метоксиметильним реактивом Гріньяра, триметилсилільних захисних груп; , (VIII) одержаним 18 in situ, з одночасним гідролізом UA 100241 C2 viii) окислювання гідроксильної групи в положенні 20 одержаного 17,20ξ-дигідрокси-11-[4(диметиламіно)-феніл]-21-метокси-19-норпрегн-4,9-дієн-3-ону формули (IX): CH3 O N H 3C CH3 CH3 OH OH H H O 5 , (IX) дициклогексилкарбодіімідом у присутності диметилсульфоксиду і сильної органічної кислоти (окислювання по Шверну), і в даному випадку наступне хроматографічне очищення; іх) ацетилювання гідроксильної групи в положенні 17 одержаного 11β-[4-(диметиламіно)-феніл]17-гідрокси-21-метокси-19-норпрегн-4,9-дієн-3,20-діону формули (X): CH3 O N H 3C CH3 CH3 O OH H H O 10 15 20 , (X) оцтовим ангідридом у присутності перхлорної кислоти, і в даному випадку наступне хроматографічне очищення одержаного 7-ацетокси-11β-[4-(диметиламіно)-феніл)]-21-метокси19-норпрегн-4,9-дієн-3,20-діону формули (І). 2. Спосіб за п. 1, який відрізняється тим, що на стадії iv) використовують 0,25 ± 0,025 еквівалентів надлишку реактиву Гріньяра 4-(диметиламіно)-фенілмагнійброміду відносно 5,10αепокси-3,3-[1,2-етандіїл-біс(оксі)]-17-[триметилсиліл(оксі)]-5-оестр-9(11)-ен-17-карбонітрилу формули (V). 3. Спосіб за п. 1, який відрізняється тим, що на стадії viii) використовують трифтороцтову кислоту як сильну органічну кислоту. 4. 11-[4-(Диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(оксі)]-5,17-біс-[триметилсиліл(оксі)]-5оестр-9-ен-17-карбонітрил формули (VII). 5. 11-[4-(Диметиламіно)-феніл]-3,3-[1,2-етандіїл-біс(оксі)]-5,17-біс-[триметилсиліл(оксі)]-5оестр-9-ен-17-карбальдегід формули (VIII). Комп’ютерна верстка А. Крижанівський Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 19
ДивитисяДодаткова інформація
Назва патенту англійськоюIndustrial method for the synthesis of 17-acetoxy-11b-[4-(dimethylamino)-phenyl]-21-methoxy-19-norpregna-4,9-dien-3,20-dione and the key intermediates of the process
Автори англійськоюBodi, Jozsef, Visky Gyoergy, Szeles, Janos, MAKHO SHANDOR, Santa, Csaba, Csoergei Janos, Tuba Zoltan, Terdy, Laszlo, Molnar Csaba, Aranyi, Antal, Horvath, Zoltan, Balogh Gabor
Назва патенту російськоюПромышленный способ синтеза 17-ацетокси-11b-[4-(диметиламино)-фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона и промежуточные соединения данного способа
Автори російськоюБоди Йожеф, Вишки Дьйордь, Селеш Янош, Махо Шандор, Шанта Чаба, Чоргеи Янош, Туба Зольтан, Терди Ласло, Мольнар Чаба, Араньи Анталь, Хорват Зольтан, Балог Габор
МПК / Мітки
МПК: C07J 51/00, C07J 21/00, C07J 41/00
Мітки: синтезу, сполуки, проміжні, промисловий, 17-ацетокси-11b-[4-(диметиламіно)-феніл]-21-метокси-19-норпрегн-4,9-дієн-3,20-діону, способу, спосіб, даного
Код посилання
<a href="https://ua.patents.su/21-100241-promislovijj-sposib-sintezu-17-acetoksi-11b-4-dimetilamino-fenil-21-metoksi-19-norpregn-49-diehn-320-dionu-i-promizhni-spoluki-danogo-sposobu.html" target="_blank" rel="follow" title="База патентів України">Промисловий спосіб синтезу 17-ацетокси-11b-[4-(диметиламіно)-феніл]-21-метокси-19-норпрегн-4,9-дієн-3,20-діону і проміжні сполуки даного способу</a>
Промисловий спосіб синтезу несольватованого 17a-ацетокси-11b-[4-(n,n-диметиламіно)-феніл]-19-норпрегна-4,9-дієн-3,20-діону й проміжні сполуки за даним способом

Номер патенту: 94269
Опубліковано: 26.04.2011
Автори: Мад'ярі Ендрене, Чоргеї Янош, Мольнар Чаба, Данчі Лайошне, Туба Золтан, Вішкі Дьйордь
МПК: C07J 71/00, C07J 31/00, C07J 41/00, C07J 21/00
Мітки: спосіб, 17a-ацетокси-11b-[4-(n,n-диметиламіно)-феніл]-19-норпрегна-4,9-дієн-3,20-діону, несольватованого, проміжні, синтезу, промисловий, способом, сполуки, даним
Формула / Реферат:
1. Промисловий спосіб синтезу несольватованого 17α-ацетокси-11β-[4-(N,N-диметиламіно)-феніл]-19-норпрегна-4,9-дієн-3,20-діону формули (І), що включає утворення кеталю з 3-(етилендіокси)-17α-гідрокси-19-норпрегна-5(10),9(11)-дієн-20-ону формули (VI), утворення епоксиду в положенні 5,10 одержаного 3,3,20,20-біс(етилендіокси)-17α-гідрокси-19-норпрегна-5(10),9(11)-дієну формули (V), реакцію одержаного...
Спосіб одержання похідних 3-[4-[2-(феноксазин-10-іл)метокси]феніл]пропіонової кислоти (варіанти) та проміжні сполуки для їх отримання

Номер патенту: 60386
Опубліковано: 15.10.2003
Автори: Маміллапаллі Рамабхадра Сарма, Батчу Чандра Секхар, Потлапаллі Раджендер Кумар, Сіріпрагада Махендер Рао, Чебійям Прабхакар, Гаддам Ом Редді
МПК: C07C 67/31, C07D 265/38, C07C 51/347
Мітки: проміжні, 3-[4-[2-(феноксазин-10-іл)метокси]феніл]пропіонової, кислоти, отримання, одержання, спосіб, варіанти, сполуки, похідних
Формула / Реферат:
1. Спосіб одержання сполуки формули (1)(1),де R1 є гідрогеном чи нижчою алкільною групою, і Х є гідрогеном чи атомом галогену, в якому здійснюють:(і) бензилювання п-гідроксибензальдегіду формули (7)(7)з одержанням сполуки формули (8)(8),(іі) реакцію сполуки формули (8) з алкілгалогенацетатом у присутності основи при температурі у діапазоні від -10°С до 60°С з одержанням...
Оптично чисті солі (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-піридиніл)метил]сульфініл]-1н-бензімідазолу, спосіб їх одержання, проміжні сполуки та фармацевтична композиція

Номер патенту: 60289
Опубліковано: 15.10.2003
Автори: Вон Унге Свєркєр, Ліндберг Пер Леннарт
МПК: A61P 29/00, C07D 401/12, A61K 31/44, A61P 1/00, A61P 31/04, A61K 31/4439, A61P 1/04, A61P 19/06, A61K 31/4427, A61P 17/06
Мітки: фармацевтична, одержання, композиція, солі, оптично, проміжні, сполуки, 5-метокси-2-[[(4-метокси-3,5-диметил-2-піридиніл)метил]сульфініл]-1н-бензімідазолу, чисті, спосіб
Формула / Реферат:
1. Оптически чистые Na+, Мg2+, Li+, Κ+ или Са2+ соли (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил]сульфинил]-1Н-бензимидазола.2. Соединение по пункту 1, представляющее собой Na+, Мg2+ или Са2+ соль (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил]сульфинил]-1Н-бензимидазола.3. Соединение по пункту 1, представляющее собой Мg2+ соль...
Спосіб одержання 5-[4-[[3-метил-4-оксо-3,4-дигідрохіназолін-2-іл]метокси]бензил]тіазолідин-2,4-діону та проміжної сполуки

Номер патенту: 70962
Опубліковано: 15.11.2004
Автори: Чебійям Прабхакар, Потлапаллі Раджендер Кумар, Маміллапаллі Рамабхадра Сарма, Сатіш Баларам Маханті, Гаддам Ом Редді, Гаде Чінна Баккі
МПК: C07D 277/24, C07B 61/00, C07D 277/20, C07D 277/34, C07D 417/12
Мітки: проміжної, спосіб, сполуки, одержання, 5-[4-[[3-метил-4-оксо-3,4-дигідрохіназолін-2-іл]метокси]бензил]тіазолідин-2,4-діону
Формула / Реферат:
1. Спосіб одержання 5-[4-[[З-метил-4-оксо-З,4-дигідрохіназолін-2-іл]метокси]бензил]тіазолідин-2,4-діону формули (1) , (1)що включаєа) відновлення сполуки формули (2') , (2')де R означає (С1-С4)алкільну групу, та, за бажанням, повторну етерифікацію з використанням...
Спосіб синтезу інгібіторів сох-2 (циклооксигенази-2) та проміжні сполуки для їх одержання
Номер патенту: 57143
Опубліковано: 16.06.2003
Автори: Девіс Ян В., Корлі Едвард Г., Пай Філіп Дж., Россен Кай, Ларсен Роберт Д.
МПК: A61P 43/00, A61K 31/444, A61P 35/00, C07C 251/12, A61P 31/12, A61K 31/44, C07D 213/61, A61P 29/00, C07C 251/30, A61P 31/16, A61P 35/04, A61K 31/4418, C07D 213/34
Мітки: циклооксигенази-2, одержання, інгібіторів, синтезу, сох-2, сполуки, проміжні, спосіб
Формула / Реферат:
1. Спосіб синтезу сполуки формули (І), (І)деприсутні 0-2 групи R;кожний з R, R' і R", незалежно, являє собою С1-10-алкіл, С-6-10-арил, аралкіл, галоген, -S(O)mH, -S(O)mС1-6-алкіл, -S(О)m-арил, нітро, аміно, С1-6-алкіламіно, ді-С1-6-алкіламіно, -S(O)mNH2, -S(О)mNH-С1-6-алкіл, -S(O)mNНС(O)СF3 і ціано, причому алкільна і арильна групи і алкільна і арильна частини аралкілу, -S(O)m-С1-6-алкілу, -S(O)m-арилу,...
Попередній патент: Спосіб одержання луб’яних волокон
Наступний патент: Похідна оксопіразину, проміжні сполуки, гербіцид на основі похідної оксопіразину та спосіб боротьби з небажаною рослинністю
Випадковий патент: Гральні карти