Малеатний співкристал 2-{етил[3-({4-[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}-1н-піразол-3-іл)аміно]хіназолін-7-іл}окси)пропіл]аміно}етилдигідрофосфату та його застосування для лікування злоякісних новоутворень








Формула / Реферат
1. Малеатний співкристал AZD1152, де AZD1152 являє собою
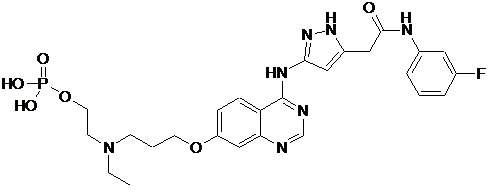 .
.
2. Кристалічна форма малеатного співкристала AZD1152, як визначено в пункті 1.
3. Кристалічна форма малеатного співкристала AZD1152 за пунктом 2, де вказаний співкристал має порошкову рентгенограму з характерними піками при 2-тета = 12,9° і 15,2° та/або 10,2°.
4. Спосіб одержання малеатного співкристала AZD1152, як визначено в пункті 1, що включає стадію змішування розчину вільної форми AZD1152 з малеїновою кислотою в придатному розчиннику, такому як метанол, диметилсульфоксид (ДМСО) або їх суміші.
5. Фармацевтична композиція, яка містить малеатний співкристал AZD1152, як визначено в пункті 1, у сполученні з фармацевтично прийнятним розріджувачем або носієм.
6. Малеатний співкристал AZD1152, за пунктом 1, для застосування в терапії.
7. Застосування малеатного співкристала AZD1152, як визначено в пункті 1, для приготування лікарського засобу для лікування захворювання, при якому є корисним інгібування однієї або декількох Аурора-кіназ.
8. Застосування малеатного співкристала AZD1152, як визначено в пункті 1, для приготування лікарського засобу для лікування гіперпроліферативних захворювань, таких як злоякісне новоутворення.
Текст
1. Малеатний співкристал AZD1152, де AZD1152 являє собою H N N HN F N . 2. Кристалічна форма малеатного співкристала AZD1152, як визначено в пункті 1. 3. Кристалічна форма малеатного співкристала AZD1152 за пунктом 2, де вказаний співкристал має порошкову рентгенограму з характерними піками при 2-тета = 12,9° і 15,2° та/або 10,2°. 4. Спосіб одержання малеатного співкристала AZD1152, як визначено в пункті 1, що включає стадію змішування розчину вільної форми AZD1152 з малеїновою кислотою в придатному розчиннику, такому як метанол, диметилсульфоксид (ДМСО) або їх суміші. 5. Фармацевтична композиція, яка містить малеатний співкристал AZD1152, як визначено в пункті 1, у сполученні з фармацевтично прийнятним розріджувачем або носієм. 6. Малеатний співкристал AZD1152, за пунктом 1, для застосування в терапії. 7. Застосування малеатного співкристала AZD1152, як визначено в пункті 1, для приготування лікарського засобу для лікування захворювання, при якому є корисним інгібування однієї або декількох Аурора-кіназ. 8. Застосування малеатного співкристала AZD1152, як визначено в пункті 1, для приготування лікарського засобу для лікування гіперпроліферативних захворювань, таких як злоякісне новоутворення. Даний винахід стосується нового співкристала й більш переважно нової співкристалічної форми 2-{етил[3-({4-[(5-{2-[(3-фторфеніл)аміно]-2оксоетил}-1H-піразол-3-іл)аміно]хіназолін-7 іл}окси)пропіл]аміно}етил дигідрофосфату (у даному винаході позначається як AZD1152), який є інгібітором аурора-кінази, придатним для лікування гіперпроліферативних захворювань, таких як (13) O 95108 N C2 N (11) HO P O HO H N UA O O (19) (21) a200813934 (22) 14.05.2007 (24) 11.07.2011 (86) PCT/GB2007/001771, 14.05.2007 (31) 0609621.8 (32) 16.05.2006 (33) GB 3 злоякісне новоутворення. Особливо винахід стосується малеатного співкристала AZD1152, способу одержання малеатного співкристала AZD1152, фармацевтичних композицій, які містять малеатний співкристал AZD1152, застосування малеатного співкристала AZD1152 для приготування лікарського засобу для лікування гіперпроліферативних захворювань, таких як злоякісне новоутворення, і способів лікування гіперпроліферативних захворювань, таких як злоякісне новоутворення, у людини або тварини шляхом введення терапевтично ефективної кількості малеатного співкристала AZD1152. Даний винахід також стосується специфічної форми малеатного співкристала AZD1152. Злоякісні пухлини (і інші гіперпроліферативні захворювання) характеризуються неконтрольованою проліферацією клітин, що відбувається при втраті нормальної регуляції проліферації клітин. Ця втрата часто є наслідком генетичного ушкодження шляхів передачі сигналів у клітинах, які контролюють проходження клітин через клітинний цикл. Вважають, що в еукаріот здійснення клітинного циклу контролюється упорядкованим каскадом фосфорилування білків. Зараз ідентифіковані деякі сімейства протеїнкіназ, які відіграють вирішальну роль у цьому каскаді. Активність багатьох цих кіназ підвищена в тканинах пухлин людини в порівнянні з нормальними тканинами. Це може відбуватися або шляхом підвищення рівнів експресії білка (наприклад, у результаті ампліфікації гена), або шляхом зміни експресії коактиваторів або інгібувальних білків. Із цих регуляторів клітинного циклу спочатку були ідентифіковані циклін-залежні кінази (або CDK) і вони найбільш широко вивчені. Недавно були ідентифіковані протеїнкінази, які за своєю структурою відрізняються від сімейства CDK і які відіграють надзвичайно важливу роль у регуляції клітинного циклу. Ці кінази, як встановлено, є важливими при канцерогенезі й вони включають гомологи білків людини Ip11 Drosophila aurora і S. cerevisiae. Три гомологи людини цих генів аурораА, аурора-В і аурора-С (також відомі як аурора2, аурора1 і аурора3 відповідно) кодують серинтреонін протеїн-кінази, які регулюють клітинний цикл (сумарний огляд Adams і ін., 2001, в Trends in Cell Biology. 11(2): 49-54). Їх максимальна експресія й активність кіназ виявляється в G2-фазі й мітозі. У деяких спостереженнях було показано, що аурора-білки людини залучені в злоякісні новоутворення. Ген аурора-А картований на хромосомі 20q13, у ділянці, яка багаторазово ампліфікована у пухлинах людини, включаючи як пухлини молочної залози, так і пухлини ободової кишки. Аурора-А може бути головним геном-мішенню такого амплікону, тому що ДНК аурора-А ампліфікована й мРНК понадекспресується більше ніж на 50% у первинних пухлинах прямої і ободової кишки людини. Виявлено, що в цих пухлинах рівні білка аурора-А значно підвищені в порівнянні із суміжними нормальними тканинами. Крім того, трансфекція фібробластів щурів ауророю-А людини приводить до трансформації, що підтверджується здатністю рости в м'якому агарі й утворювати пухлини в 95108 4 безшерстих мишей (Bischoff і ін., 1998, The EMBO Journal. 17(11): 3052-3065). В інших роботах (Zhou і ін., 1998, Nature Genetics. 20(2): 189-93) було показано, що штучна понадекспресія аурори-А приводить до підвищення кількості центросом і до зростання анеуплоїдії, що є відомою подією в розвитку злоякісного новоутворення. Також було показано, що існує підвищена експресія аурора-В (Adams i ін., 2001, Chromsoma. 110(2):65-74) і аурора-С (Kimura i ін., 1999, Journal of Biological Chemistry, 274(11): 7334-40) у пухлинних клітинах у порівнянні з нормальними клітками. Аурора-В понадекспресується в ракових клітинах і показано, що підвищені рівні аурора-В корелюють із пізніми стадіями раку ободової і прямої кишки (Katayama і ін. (1999) J. Natl. Cancer Inst. 91:1160). Крім того, в одному дослідженні доведено, що понадекспресія аурора-В індікує анеуплоїдію шляхом підвищеного фосфорилування гістону H3 у серпні в положенні 10 і що клітини, які понадекспресують аурора-В, утворюють більш агресивні пухлини, які поширюють метастази (Ota, Т. і ін., 2002, Cancer Res. 62: 5168-5177). Аурора-В є «пасажирським» білком хромосоми, що утворює стабільний комплекс принаймні із трьома іншими «пасажирськими» білками, сурвівіном, INCENP і бореаліном (Carmena Μ. і ін. 2003, Nat. Rev. Мої. Cell Biol. 4: 842-854). Сурвівін також підвищено регулюється при злоякісному новоутворенні й містить домен BIR (повтор інгібітору апоптозних білків бакуловірусів (ІАР)) і, отже, може захищати пухлинні клітини від апоптозу та/або мітотичної катастрофи. Відносно аурора-С, вважають, що його понадекспресія обмежена яєчками, але також було виявлено, що він понадекспресується в різних ракових лініях. (Katayama H і ін., 2003, Cancer і Metastasis Reviews 22: 451-464). Також було показано, що скасування експресії й дії аурора-А при лікуванні антисмисловими олігонуклеотидами клітинних ліній пухлин людини (WO 97/22702 і WO 99/37788) приводить до затримки клітинного циклу й виявляє антипроліферативну дію в цих клітинних лініях пухлин. Крім того, було показано, що невеликі молекули інгібіторів аурора-А і аурора-В мають антипроліферативну дію в пухлинних клітинах людини (Keen i ін., 2001, постер № 2455, щорічні збори Американської асоціації дослідження раку), тому що мають селективне скасування експресії аурора-В тільки шляхом обробки міРНК (Ditchfield і ін., 2003, journal of Cell Biology, 161(2): 267-280). Це свідчить про те, що інгібування дії аурора-А та/або аурора-В буде мати антипроліферативну дію, яка може бути корисною для лікування пухлин людини й інших гіперпроліферативних захворювань. Інгібування аурора-кіназ як терапевтичний підхід при цих захворюваннях може мати значні переваги в порівнянні з націлюванням на шляхи передачі сигналів, які передують клітинному циклу (наприклад, ті, які активуються тирозинкіназами рецептора фактора росту, такого як фактор росту епідермісу (EGFR) або інших рецепторів). Оскільки клітинний цикл в остаточному підсумку регулюється всіма такими різними шляхами передачі сигналів, то передбачається, що лікування, спрямоване на клітинний цикл, таке як 5 інгібування аурора-кіназ, буде діяти на всі проліферуючі пухлинні клітини, тоді як підходи, спрямовані на специфічні сигнальні молекули (наприклад, EGFR), як думають, будуть активні тільки в тих пухлинних клітинах, які експресують такі рецептори. Також припускають наявність істотного "перехресного взаємозв'язку" між таким шляхами передачі сигналів, коли інгібування одного компонента може компенсуватися іншим. Інгібітори аурора-кінази описані в міжнародних патентних заявках WO 03/55491 і WO 2004/058781, і, зокрема в WO 2004/058781 описана сполука, яка має наступну структурну формулу, у даному винаході позначається як AZD1152: AZD1152 являє собою проліки, які швидко й повністю перетворюються (у плазмі людини) в активний компонент і в даному винаході позначається як AZD1152 HQPA: AZD1152 HQPA являє собою АТФконкурентний і оборотний інгібітор аурора-кіназ із ефективною активністю по відношенню до аурора A, B-INCENP і С-INCENP (Kі 1369±419,2 нМ, 0,359±0,386 нМ і 17,03±12,2 нМ відповідно). Було виявлено, що AZD1152 інгібує ріст пухлини на панелі раку прямої і ободової кишки людини (SW620, НСТ116, Соlо205) і ксенотрансплантатованих пухлинах легенів (А549, Calu-6) зі статистичною значимістю. AZD1152 описаний в WO 2004/058781 як дигідрохлоридна сіль, а також у вигляді вільної основи в гідрованих формах. Особливо, вільна форма описана в тригідратної - тетрагідратної формі. Для приготування гідратні форми є проблематичними, оскільки їх необхідно контролювати в процесі приготування, висушування, зберігання й обробки. Додатково, складним є одержання й збереження зразка сполуки з постійним стехіометричним складом відносно процентного вмісту води. Для вищеописаних форм AZD1152, і особливо для вільної форми, молекули води дуже слабко зв'язані з кожною молекулою AZD1152, тому ступінь 95108 6 асоціації й дисоціації молекул води з лікарським засобом AZD1152 істотно змінюється залежно від температури й відносної вологості. Отже, для даної ваги AZD1152, фактична кількість AZD1152 на основі кількості молекул AZD1152 буде залежати від температури й відносної вологості, тому що вміст води буде змінюватися. Отже, ефективність будь-якої дози, визначеної відповідно до ваги, також буде залежати від діючої температури й відносної вологості. Для оцінки коливань рівнів води, зв'язаної в AZD1152, залежно від вологості, використовують сорбцію динамічного випаровування. У заявці WO 2004/058781 у загальному описані фармацевтично прийнятні солі заявлених сполук. AZD1152 описаний тільки у вигляді дигідрохлоридної солі, а також у вільній формі. Інші форми AZD1152 не розкриті. Зокрема, в WO 2004/058781 не описаний будь-який інший співкристал AZD1152 і, звичайно, не обговорюється, що будь-які конкретні співкристали конкретних сполук несподівано будуть мати переваги й, зокрема, не будуть мати переваги, які можуть зняти проблеми, обговорювані в даній заявці. Непередбачено й несподівано нами було виявлено, що малеатний співкристал AZD1152 існує в безводній формі, яка по суті є негігроскопічною. Крім того, незважаючи на те, що стехіометричне відношення лікарського засобу в малеаті може змінюватися в діапазоні, наприклад, від 0,8:1 до 1,2:1, або від 0,9:1 до 1,1:1, нами було виявлено, що малеатний співкристал AZD1152. розкритий у даному винаході, має відтворене стехіометричне відношення лікарського засобу до малеату по суті 1:1. Отже, на ефективність вагової дози AZD1152 малеатного співкристала значно менший вплив робить температура й відносна вологість у порівнянні з вільною формою, дигідрохлоридною формою й іншими співкристалічними формами AZD1152, що може бути оцінено. Додатково, для малеатного співкристала AZD1152 значно спрощується процес приготування, оскільки зменшується необхідність контролювати рівні вологості при здійсненні процесу приготування. Безводна природа малеатного співкристала додатково означає, що представляється можливість його приготування у вигляді лікарського засобу за допомогою обмежених водних умов та/або високої температури зовнішнього середовища, оскільки знижено ризик гідратації/дегідратації в умовах обробки, у яких використовуються водні умови середовища, тобто волога грануляція. Додатково, нами було відкрито, що малеатний співкристал AZD1152 несподівано містить меншу кількість домішок, ніж вільна форма. Зокрема, доведено, що певні стійкі домішки, які присутні у вільній формі, несподівано присутні в значно меншій кількості після перетворення вільної форми в малеатну форму. Таким чином, даний винахід забезпечує малеатний співкристал AZD1152. Щоб уникнути різночитань, терміни "малеатний співкристал AZD1152", "AZD1152 малеатний співкристал" або "AZD1152 малеат" (або будь-який інший ідентичний термін, який використовується у даній заявці) стосується всіх форм асоціації між 7 AZD1152 і малеїновою кислотою, включаючи сольові форми. Зокрема, ці терміни охоплюють: (і) неіонну асоціацію між AZD1152 і малеїновою кислотою (тобто в яких не відбувається переносу протона між лікарським засобом і малеїновою кислотою); або (іі) іонну взаємодію, де відбувається перенос протона між AZD1152 і малеїновою кислотою з утворенням малеатної солі AZD1152, або (ііі) суміші вищеописаних (і) і (іі). У переважному варіанті здійснення винаходу, малеатний співкристал містить неіонну асоціацію між лікарським засобом AZD1152 і малеїновою кислотою (тобто де не відбувається переносу протона між лікарським засобом і малеїновою кислотою). В альтернативному варіанті здійснення винаходу, малеатний співкристал являє собою малеатну сіль AZD1152. У переважному варіанті здійснення, малеатний співкристал AZD1152 утворений шляхом змішування вільної форми AZD1152 з малеїновою кислотою в підходящому розчиннику, такому як метанол, диметилсульфоксид (ДМСО) або суміші ДМСО з метанолом, ацетонітрилом, і іншими подібними розчинниками. Малеатний співкристал може бути виділений шляхом дозволу здійснення кристалізації й наступного виділення отриманого кристалічного матеріалу. Ідентичність малеатного співкристала AZ1152 згідно із даним винаходом може бути підтверджена за допомогою аналізу ядерного магнітного резонансу (ЯМР). Слід також взяти до уваги, що сполука або співкристал за винаходом можуть проявляти таутомерію й що наведені в даному описі формули можуть представляти тільки одну з можливих таутомерних форм. Мається на увазі, що винахід охоплює будь-яку таутомерну форму, яка має інгібувальну дію по відношенню до Аурора-кінази, і, особливо, інгібувальну дію по відношенню до Аурора-А та/або В кінази й не обмежується тільки будь-якою з таутомерних форм, яка використовується в наведених формулах. Даний винахід також стосується переважної кристалічної форми малеатного співкристала AZD1152. Цю кристалічну форму одержують шляхом кристалізації малеатного кристала AZD1152 з органічного розчинника, такого як суміш метанолу й диметилсульфоксиду (ДМСО). Подальші експериментальні відомості розкриті в прикладах. Таким чином, даний винахід забезпечує кристалічну форму малеатного співкристала AZD1152. Кристалічна форма малеатного співкристала AZD1152 характеризується тим, що вона забезпечує порошкову рентгенограму, по суті таку ж, що й наведена на Фігурі 1. Найбільш видні піки порошкової дифракції рентгенівських променів для кристалічної форми малеатного співкристала AZD1152 представлені в таблиці 1: 95108 8 Таблиця 1 Кут 2- Відносна інтентета° сивність % 5,118 100 6,446 18,8 8,158 28,1 10,171 13,1 11,917 5,9 12,861 18 13,849 48,3 14,909 25,2 15,234 36 15,738 27,6 16,506 18,8 16,884 23 17,232 42,5 18,134 13,8 19,327 62,7 19,82 38,8 20,082 61 20,582 61,4 21,008 32 21,663 87,3 Кут 2тета° 22,425 23,228 23,583 23,994 24,271 24,671 25,32 25,574 25,813 26,21 27,122 27,946 28,418 28,847 29,725 30,521 31,74 33,424 36,181 38,106 Відносна інтенсивність % 18,8 26,8 37,7 25,7 24,1 23,3 19,1 32,9 43,8 35,3 16 45,4 31,6 21,7 26,1 18,8 15,4 14,7 15,3 10,8 Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала AZD1152, де вказаний співкристал має порошкову рентгенограму принаймні з одним характерним піком при 2-тета приблизно = 15,2°. Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала AZD1152, де вказаний співкристал має порошкову рентгенограму с характерними піками при 2-тета приблизно = 12,9°. Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала AZD1152, де вказаний співкристал має порошкову рентгенограму с характерними піками при 2-тета приблизно = 15,2 або 10,2°. Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала AZD1152, де вказаний співкристал має порошкову рентгенограму с характерними піками при 2-тета приблизно = 18,1°. Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала AZD1152, де вказаний співкристал має порошкову рентгенограму принаймні з одним характерним піком при 2-тета приблизно = 10,2°, 12,9°, 15,2° або 18,1°. Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала AZD1152, де вказаний співкристал має порошкову рентгенограму с характерними піками при 2-тета приблизно = 12,9° і 15,2° та/або 10,2°. Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала AZD1152, де вказаний співкристал має порошкову рентгенограму с характерними піками при 2-тета приблизно = 10,2°, 12,9°, 15,2° і 18,1°. Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала 9 AZD1152, де вказаний співкристал має порошкову рентгенограму с характерними піками приблизно при будь-якому одному або комбінаціях значень 2тета°, наведених у таблиці 1. Відповідно до даного винаходу, забезпечується кристалічна форма малеатного співкристала AZD1152, де вказаний співкристал має порошкову рентгенограму по суті таку ж, що й порошкова рентгенограма, наведена на Фігурі 1. Як вказано в даній заявці, даний винахід стосується кристалічної форми малеатного співкристала AZD1152, і ступінь кристалічності, як визначено згідно із даними дифракції рентгенівських променів, підходяще більше, ніж приблизно 60%, більш підходяще, більше, ніж приблизно 80%, переважно більше, ніж приблизно 90%. У попередніх абзацах при визначенні піків порошкової рентгенограми для кристалічних форм малеатного співкристала AZD1152 термін "при значеннях приблизно", який використовується у виразі "... при 2-тета приблизно = ..."вказує на те, що точне положення піків (тобто наведені значення кутів 2-тета) не повинні тлумачитися як абсолютні значення, оскільки, що є очевидним для фахівця в даній галузі техніки, точне положення піків може незначно вирізнятися при вимірюванні на різних приладах, або різних зразків, або внаслідок незначних коливань використовуваних умов вимірювання. Це також вказувалося в попередніх параграфах, що кристалічні форми малеатного співкристала AZD1152 характеризуються порошковою рентгенограмою 'по суті' такою ж, що й порошкова рентгенограма, наведена на Фігурі 1, і мають головним чином найбільш виражені піки (значення кута 2-тета), наведені в таблиці 1 і особливо при 2-тета приблизно = 10,2°, 12,9°, 15,2° або 18,1°. Слід взяти до уваги, що використання терміна 'по суті' у цьому контексті також призначено для вказівки того, що значення кутів 2-тета порошкових рентгенограм можуть незначно вирізнятися при вимірюванні на різних приладах, або різних зразків, або внаслідок незначних коливань використовуваних умов вимірювання, таким чином, піки, показані на Фігурі 1 або наведені в таблиці 1, також не повинні тлумачитися як абсолютні значення. В цьому відношенні, у даній галузі техніки відомо, що може бути отримана порошкова рентгенограма, що містить одну або декілька похибок вимірювань, які залежать від умов вимірювання (таких як використовуване обладнання, приготування зразків або прилад). Зокрема, добре відомо, що напруга при порошковій рентгенограмі може відрізнятися залежно від умов вимірювання й приготування зразків. Наприклад, фахівці в галузі порошкової рентгенограми можуть підтвердити, що на відносну інтенсивність піків можуть впливати, наприклад, частинки розміром близько 30 мікронів і неодиничні формати зображення, які можуть впливати на аналіз зразків. Фахівець у даній галузі також буде враховувати той факт, що на положення відбиттів може впливати точна висота, на яку встановлюється зразок у дифрактометрі й калібрування нуля дифрактометра. Площинність поверхні зразка також може мати незначний вплив. Отже, фахівець у даній галузі техніки візьме до 95108 10 уваги, що дані порошкової рентгенограми, наведені в даному винаході, не повинні тлумачитися як абсолютні (для додаткової інформації див. Jenkins, R & Snyder, R.L. 'Introduction to X-Ray Powder Diffractometry' John Wiley & Sons, 1996). Таким чином, слід взяти до уваги, що кристалічна форма малеатного співкристала AZD1152, описана в даному винаході, не обмежується кристалами, які охарактеризовані порошковою рентгенограмою, ідентичній до порошкової рентгенограми, наведеної на Фігурі 1, і будь-які кристали, які характеризуються порошковою рентгенограмою, по суті такою ж, що й наведено на Фігурі 1, підпадають під обсяг даного винаходу. Фахівець в галузі порошкової рентгенограми здатний оцінити практичну ідентичність порошкових рентгенограм. У цілому, погрішність виміру кута дифракції на порошковій рентгенограмі для кута 2-тета становить близько 0,5° або менше (або, більш підходяще, 2-тета приблизно = 0,2° або менше), і такий порядок похибки вимірювань слід враховувати при аналізі порошкових рентгенограм на Фігурі 1 і при тлумаченні положень піків з посиланням на вищеописаний текст і на таблицю 1. Отже, якщо вказано, наприклад, що співкристал має порошкову рентгенограму принаймні з одним характерним піком при 2-тета приблизно = 15,2° (або будь-яким з інших кутів, вказаних вище), то це може бути витлумачене як, 2-тета = 15,2° плюс або мінус 0,5°, або 2-тета = 15,2° плюс або мінус 0,2°. Відповідно до іншого аспекту винаходу, забезпечується спосіб одержання малеатного співкристала AZD1152, як визначено в даній заявці, де вказаний спосіб включає стадію змішування розчину вільної форми AZD1152 з малеїновою кислотою в підходящому розчиннику, такому, як метанол, N-метил-2-піролідинон, диметил сульфоксид (DMSO) або суміш DMSO із метанолом, ацетонітрилом, та інші подібні розчинники. Спосіб надалі може включати стадії кристалізації й, необов'язково, виділення утвореного при цьому кристалічного малеатного співкристала AZD1152. Спосіб додатково може включати подальші стадії промивання малеатного співкристала AZD1152 підходящим розчинником; і висушування малеатного співкристала AZD1152. Підходяще, вільна форма AZD1152 розчинена в підходящому розчиннику (такому як диметилсульфоксид, метанол, їх суміші або N-метил-2піролідинон) і в більшості випадків змішаний з розчином малеїнової кислоти (яка розчинена в такому ж або сумісному розчиннику). Альтернативно, тверда малеїнова кислота може бути додана до розчину вільної форми AZD1152 (або навпаки, тобто розчин вільної форми AZD1152 може бути доданий до твердої малеїнової кислоти). Підходяще, розчин перемішують для полегшення змішування вільної форми AZD1152 і доданої малеїнової кислоти. Матеріали (ідеально, але не тільки винятково в співвідношенні 1:1) можуть бути змішані при температурі навколишнього середовища, хоча методика також може бути здійснена при підвищених температурах. Можна використовувати будь-який підходящий спосіб, відомий у даній галузі техніки, для виділен 11 ня кристалічної малеатної форми AZD1152. Підходяще, малеатний співкристал AZD1152 збирають шляхом фільтрації. Переважно, промитий малеатний співкристал AZD1152 висушують у вакуумі. У цілому, бажаним є співвідношення вільна форма AZD1152:малеїнова кислота 1:1. Це бажане співвідношення 1:1 може бути отримане шляхом змішування вільної форми AZD1152 і малеїнової кислоти в композиціях у будь-яких співвідношеннях у межах інтервалу 0,6-1,4 вільна форма AZD1152:1,0 малеїнова кислота. Підходяще, співвідношення вільна форма AZD1152: малеїнова кислота в суміші знаходиться в інтервалі 0,91,1 і переважно 1,0-1,1 вільна форма AZD1152:1,01,1 малеїнова кислота. Як правило, можна використовувати надлишок малеїнової кислоти й, зокрема, співвідношення вільна форма AZD1152: малеїнова кислота в суміші знаходиться в інтервалі 0,61,0 і переважно 0,9-1,0 вільна форма AZD1152:1,0 малеїнова кислота. AZD1152 малеатний співкристал звичайно самокристалізується, але фахівцеві в даній галузі техніки слід взяти до уваги, що можна використовувати затравлювання, якщо це є необхідним або бажаним, для промотування утворення співкристалів. Відповідно до подальшого варіанта здійснення винаходу, забезпечується фармацевтична композиція, що містить малеатний співкристал AZD1152, як визначено в даній заявці, у сполученні з фармацевтично прийнятним розріджувачем або носієм. Композиції за винаходом можуть знаходитися у формах, що підходять для перорального застосування (наприклад, у вигляді таблеток, коржів, твердих або м'яких капсул, водних або масляних суспензій, емульсій, диспергованих порошків або гранул, сиропів або еліксирів), для місцевого введення (наприклад, у вигляді паст, мазей, гелів, водних або масляних розчинів або суспензій), для введення шляхом інгаляції (наприклад, у вигляді тонкоподрібненого порошку або рідкого аерозолю), для введення шляхом вдування (наприклад, у вигляді тонкоподрібненого порошку), або для парентерального введення (наприклад, у вигляді стерильного водного або масляного розчину для внутрішньовенного, підшкірного, внутрім'язового введення, або у вигляді супозиторію для ректального введення). Композиції за винаходом можуть бути отримані звичайними способами при використанні звичайних фармацевтичних наповнювачів, добре відомих у даній галузі. Таким чином, композиції, призначені для перорального введення, можуть містити, наприклад, один або кілька барвників, підсолоджувачів, ароматизаторів та/або консервантів. Підходящі фармацевтично прийнятні наповнювачі для таблетки включають, наприклад, інертні розріджувачі, такі як лактоза, карбонат натрію, фосфат кальцію або карбонат кальцію, гранулюючі і дезінтегруючі засоби, такі як кукурудзяний крохмаль або альгінова кислота; зв'язувальні речовини, такі як крохмаль; замаслювачі, такі як стеарат 95108 12 магнію, стеаринова кислота або тальк; консерванти, такі як етил або пропіл п-гідроксибензоат, і антиоксиданти, такі як аскорбінова кислота. Лікарські препарати у вигляді таблеток можуть бути без покриття або можуть мати покриття або для модифікації їх розпаду й наступного всмоктування активної речовини в шлунково-кишковому тракті, або для поліпшення їх стабільності та/або зовнішнього вигляду, для цього використовуються звичайні засоби для покриття й методики, які добре відомі в даній галузі. Композиції для перорального застосування можуть знаходитися у вигляді твердих желатинових капсул, у яких активний компонент змішаний з інертним твердим розріджувачем, наприклад, карбонатом кальцію, фосфатом кальцію або каоліном, або у вигляді м'яких желатинових капсул, у яких активний компонент змішаний з водою або маслом, таким як арахісова олія, рідкий парафін, олія соєвих бобів, кокосова олія або, переважно, маслинова олія, або іншим підходящим наповнювачем. Водні суспензії, як правило, містять активний компонент у формі тонкоподрібненого порошку разом з одним або більше суспендуючими засобами, такими як карбоксиметилцелюлоза натрію, метилцелюлоза, гідроксипропілметилцелюлоза, альгінат натрію, полівініл-піролідон, трагакантова камедь і аравійська камедь; диспергуючими або змочувальними засобами, такими як лецитин або продукти конденсації алкіленоксиду з жирними кислотами (наприклад, поліксиетиленстеарат), або продукти конденсації етиленоксиду із довголанцюговими аліфатичними спиртами, наприклад, гептадекаетиленоксицетанол, або продукти конденсації етиленоксиду з похідними неповних складних ефірів жирних кислот і гекситу, такі як поліоксіетиленсорбітмоноолеат, або продукти конденсації етиленоксиду із довголанцюговими аліфатичними спиртами, наприклад, гептадекаетиленоксицетанол, або продукти конденсації етиленоксиду з похідними неповних складних ефірів жирних кислот і гекситу, такі як поліоксіетиленсорбітмоноолеат, або продукти конденсації етиленоксиду з похідними неповних складних ефірів жирних кислот з ангідридами гекситу, наприклад, поліетиленсорбітмоноолеат. Водні суспензії також можуть містити один або більше консервантів (таких як етил або пропіл п-гідроксибензоат, антиоксидантів (таких як аскорбінова кислота), барвників, ароматизаторів та/або підсолоджувачів (таких як сахароза, сахарин або аспартам). Масляні суспензії можуть бути приготовлені шляхом суспендування активного компонента в рослинній олії (такій як арахісова олія, маслинова олія, кунжутна олія або кокосова олія) або в мінеральному маслі (такому як рідкий парафін). Масляні суспензії також можуть містити загусники, такі як бджолиний віск, твердий парафін або цетиловий спирт. Також можуть додаватися підсолоджувачі, такі як перераховані вище, і ароматизатори для одержання смачного препарату для перорального введення. Ці композиції також можуть бути захищені від розкладання шляхом додавання антиоксиданту, такого як аскорбінова кислота. 13 Дисперговані або ліофілізовані порошки й гранули, що підходять для одержання водної суспензії або розчину шляхом додавання води, як правило, містять активний компонент разом з диспергуючим або змочувальним засобом, суспендуючим засобом і одним або більше консервантами. Прикладами підходящих диспергуючих або змочувальних засобів і суспендуючих засобів є засоби, згадані вище. Також можуть включатися додаткові наповнювачі, такі як підсолоджувачі, ароматизатори й барвники. Фармацевтичні композиції за винаходом також можуть знаходитися у вигляді емульсій масло-уводі. Масляною фазою може бути рослинна олія, така як маслинова олія або арахісова олія, або мінеральне масло, таке як, наприклад, рідкий парафін або суміш будь-яких перерахованих засобів. Підходящими емульгуючими засобами можуть бути, наприклад, природні смоли, такі як аравійська камедь або трагакантова камедь, природні фосфатиди, такі як соя, лецитин, складні ефіри або похідні неповних складних ефірів жирних кислот з ангідридами гекситу (наприклад, сорбітмоноолеат) і продукти конденсації вказаних неповних складних ефірів з етиленоксидом, такі як поліоксіетиленсорбітмоноолеат. Емульсії також можуть містити підсолоджувані, ароматизатори й консерванти. Сиропи й еліксири також можуть бути приготовлені з підсолоджувачами, такими як гліцерин, пропіленгліколь, сорбіт, аспартам або сахароза, а також можуть містити засіб, що зменшує подразнення, консервант, ароматизатор та/або барвник. Фармацевтичні композиції також можуть знаходитися у формі стерильного розчину для ін'єкцій або масляної суспензії, розчинів, емульсій або особливих систем, які можуть бути приготовлені відповідно до відомих методик, використовуючи один або більше підходящих диспергуючих або змочувальних засобів і суспендуючих засобів, описаних вище. Стерильний препарат для ін'єкцій також може являти собою стерильний розчин для ін'єкції або суспензію в нетоксичному розріджувачі або розчиннику, що є прийнятним для парентерального введення, наприклад, розчин у поліетиленгліколі. Лікарські препарати у вигляді супозиторіїв можуть бути приготовлені шляхом змішування активного компонента з підходящим неподразливим наповнювачем, який є твердим при звичайних температурах, але рідким при ректальній температурі, і тому він розплавляється в прямій кишці, вивільняючи лікарський засіб. Підходящими наповнювачами є, наприклад, олія какао й поліетиленгліколі. Лікарські препарати для місцевого застосування, такі як креми, мазі, гелі, водні або масляні розчини або суспензії, як правило, можуть бути отримані шляхом приготування активного компонента зі звичайним наповнювачем або розріджувачем, що є підходящим для місцевого введення, використовуючи звичайну методику, відому в даній галузі техніки. Композиція для введення шляхом вдування може знаходитися у вигляді тонкоподрібненого порошку, що містить частинки із середнім діамет 95108 14 ром, наприклад, 30 мкм або менше, переважно 5 мкм або менше, і більш переважно в інтервалі 5 мкм - 1 мкм і порошок містить або сам активний компонент або розведений одним або декількома фізіологічно прийнятними носіями, такими як лактоза. Потім порошок для вдування звичайно поміщають у капсулу, що містить, наприклад, 1-50 мг активного компонента для застосування в пристрої для турбоінгаляції, такому як застосовується для вдмухування відомої речовини кромоглікату натрію. Композиції для введення шляхом інгаляції можуть знаходитися у вигляді звичайного аерозолю під підвищеним тиском, пристосованого для розподілу активного компонента або у вигляді аерозолю, що містить тонкоподрібнену тверду речовину або рідкі краплі. Можуть застосовуватися звичайні пропеленти для аерозолю, такі як летучі фторовані вуглеводні або вуглеводні, і аерозольний пристрій являє собою звичайний пристрій для розподілу дозованої кількості активного компонента. Додаткова інформація щодо приготування лікарських препаратів знаходиться в главі 25.2, том 5 «Загальної медичної хімії» (Comprehensive Medicinal Chemistry) (Corwin Hansch; голова редакційної колегії), Pergamon Press 1990. Отже, в подальшому аспекті даного винаходу забезпечується малеатний співкристал AZD1152 для застосування для лікування. Додатково забезпечується малеатний співкристал AZD1152 для застосування як лікарський засіб. Інший аспект даного винаходу забезпечує малеатний співкристал AZD1152 для застосування як лікарський засіб для лікування гіперпроліферативних захворювань, таких як злоякісне новоутворення і зокрема, раку прямої і ободової кишки, молочної залози, легені, передміхурової залози, сечового міхура, нирок або підшлункової залози або лейкозу або лімфоми. Лейкози й лімфоми, згадані в даному винаході, можуть бути пухлинами мієлоїдного походження, такими як гострий мієлолейкоз, або лімфоїдного походження. Додатково, малеатний співкристал AZD1152 забезпечується для застосування в способі терапевтичного лікування теплокровної тварини, такої як людина. Інший аспект винаходу забезпечує малеатний співкристал AZD1152 для застосування в способі лікування гіперпроліферативних захворювань, таких як злоякісне новоутворення і, зокрема, рак прямої і ободової кишки, молочної залози, легені, передміхурової залози, сечового міхура, нирок або підшлункової залози або лейкозу або лімфоми. В іншому аспекті винаходу, забезпечується застосування малеатного співкристала AZD1152 для приготування лікарського засобу для лікування захворювання, при якому є корисним інгібування однієї або декількох аурора-кіназ. Зокрема, вважають, що може бути корисним інгібування аурора А кінази та/або аурора В кінази. Переважно корисним є інгібування аурора В кінази. В іншому аспекті винаходу, забезпечується застосування малеатного співкристала AZD1152 для приготування лікарського засобу для лікування гіперпроліфера 15 тивних захворювань, таких як злоякісне новоутворення і зокрема, раку прямої і ободової кишки, молочної залози, легені, передміхурової залози, сечового міхура, нирок або підшлункової залози або лейкозу або лімфоми. Ще в іншому аспекті, забезпечується малеатний співкристал AZD1152 для застосування в способі лікування людини, що страждає від захворювання, при якому є корисним інгібування однієї або декількох аурора-кіназ, що включає стадії введення особини, яка потребує цього, терапевтично ефективної кількості малеатного співкристала AZD1152. Зокрема, вважають, що може бути корисним інгібування аурора А кінази та/або аурора В кінази. Переважно корисним є інгібування аурора В кінази. Надалі забезпечується малеатний співкристал AZD1152 для застосування в способі лікування людини, що страждає від гіперпроліферативного захворювання, такого як злоякісне новоутворення, і зокрема, раку прямої і ободової кишки, молочної залози, легені, передміхурової залози, сечового міхура, нирок або підшлункової залози або лейкозу або лімфоми, що включає стадії введення особини, яка потребує цього, терапевтично ефективної кількості малеатного співкристала AZD1152. Застосування малеатного співкристала AZD1152 у будь-якому зі способів лікування людини, описаних вище, також становить аспекти даного винаходу. Для вищеописаних застосувань для лікування доза, яку вводять, змінюється залежно від застосовуваної сполуки, способу введення, необхідного лікування, вказаного розладу, віку й статі тварини або хворого. Таким чином, доза визначається відповідно до добре відомих підходів у медицині. Малеатний співкристал AZD1152, які застосовується для лікування або профілактики, звичайно вводиться в добовій дозі в інтервалі, наприклад, від 0,05 мг/кг до 50 мг/кг ваги тіла пацієнта, що, при необхідності, може бути розділена на кілька прийомів. У цілому, при парентеральному введенні застосовуються більш низькі дози. Так, наприклад, для внутрішньовенного введення звичайно застосовують дозу в інтервалі, наприклад, від 0,05 мг/кг до 25 мг/кг ваги тіла. Подібним чином, для введення шляхом інгаляції застосовується доза в інтервалі, наприклад, від 0,05 мг/кг до 25 мг/кг ваги тіла. Лікування, описане у винаході, може застосовуватися у вигляді монотерапії, або, додатково до сполуки за винаходом, можна також застосовувати звичайні хірургічні методи або радіотерапію або хіміотерапію. Така хіміотерапія може включати один або кілька наступних класів протипухлинних засобів: (і) антипроліферативні/протипухлинні лікарські засоби і їх комбінації, які застосовуються в онкології, такі як алкілувальні засоби (наприклад, цисплатин, карбоплатин, циклофосфамід, азотний іприт, мельфалан, хлорамбуцил, бусульфан і нітрозосечовини); антиметаболіти (наприклад, антифолати, такі як фторпіримідини, такі як 5-фторурацил і тегафур, ралтитрексед, метотрексат, арабінозид цитозину й гідроксисечовина; протипухлинні антибіотики (наприклад, антрацикліни, такі як адриамі 95108 16 цин, блеоміцин, доксорубіцин, дауноміцин, епірубіцин, ідарубіцин, мітоміцин-С, дактиноміцин і мітраміцин); антимітотичні засоби (наприклад, алкалоїди барвінку, такі як вінкристин, вінбластин, віндезин і вінорелбін і таксоїди, такі як таксол і таксотер); і інгібітори топоізомерази (наприклад, епіподофілотоксини, такі етопозид і теніпозид, амсакрин, топотекан і камптотецин); (іі) цитостатичні засоби, такі як антиестрогени (наприклад, тамоксифен, тореміфен, ралоксифен, дролоксифен і йодоксифен), інгібітори рецептора естрогену (наприклад, фульвестратрант); антиандрогени (наприклад, бікалутамід, флутамід, нілутамід і ципротерон ацетат), антагоністи LHRH або агоністи LHRH (наприклад, гозерелін, лейпрорелін і бузерелін), прогестогени (наприклад, мегестрол ацетат), інгібітори ароматази (наприклад, анастрозол, летрозол, воразол і ексеместан) і інгібітори 5-редуктази, такі як фінастерид; (ііі) засоби, які інгібують інвазію злоякісних клітин (наприклад, інгібітори металопротеїнази, такі як маримастат, і інгібітори функції рецептора урокіназного активатора плазминогена); (iv) інгібітори дії фактора росту, наприклад, такі інгібітори включають антитіла до фактора росту, антитіла до рецептора фактора росту (наприклад, анти-erbb2 антитіло трастузумаб [Herceptin] і анти-erbb1 антитіло цетуксимаб [С225]), інгібітори фарнезилтрансферази, інгібітори тирозинкінази й інгібітори серин-треонін кінази, наприклад, інгібітори сімейства фактора росту епідермісу (наприклад, інгібітори EGFR сімейства тирозинкіназ, такі як N-(3-хлор-4-фторфеніл)-7-метокси-6-(3морфолінопропокси)хіназолін-4-амін (гефитиниб, AZD1839), N-(3-етинилфеніл)-6,7-біс(2метоксіетокси)хіназолін-4-амін (ерлотиніб, OSI774) і 6-акриламідо-N-(3-хлор-4-фторфеніл)-7-(3морфолінопропокси)хіназолін-4-амін (СІ 1033)), наприклад, інгібітори сімейства фактора росту похідних тромбоцитів і, наприклад, інгібітори сімейства фактора росту гепатоцитів; (ν) антиангіогенні речовини, такі як ті, які інгібують дію фактора росту ендотелію судин, (наприклад, антитіло до фактора росту клітин ендотелію судин бевацизумаб [Avastin], сполуки, які описані в міжнародних заявках на патент WO 97/22596, WO 97/30035, WO 97/32856 і WO 98/13354) і сполуки, які діють за іншим механізмом (наприклад, ліномід, інгібітори дії інтегрину ν3 і ангіостатин); (vi) речовини, які пошкоджують судини, такі як комбретастатин А4 і сполуки, описані в міжнародних заявках на патент WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 і WO 02/08213; (vii) антисмислова терапія, наприклад, така, що спрямована на перераховані вище мішені, такі як ISIS 2503, антисмислова терапія на основі гена ras; (viii) способи генної терапії, включаючи, наприклад, способи заміни аберантних генів, такі як способи аберації р53 або аберації BRCA1 або BRCA2, GDEPT (пролікарська терапія, спрямована на ген ферменту) способи з використанням деамінази цитозину, тимідинкінази або бактеріальної нітроредуктази й способи підвищення стійкості пацієнта 17 до хіміотерапії або радіотерапії, такі як генна терапія резистентності до багатьох лікарських засобів; і (іх) способи імунотерапії, включаючи, наприклад, способи підвищення імуногенності пухлинних клітин пацієнта в умовах ex vivo і in vivo, такі як трансфекція цитокінами, такими як інтерлейкін 2, інтерлейкін 4 або фактор стимуляції колоній гранулоцитів-макрофагів, способи зниження активності Т-клітин, способи з використанням трансфектованих імунних клітин, таких як цитокінтрансфектовані дендритні клітини, способи з використанням цитокін-трансфектованих ліній пухлинних клітин і способи з використанням антиідіотипових антитіл. Додатково, малеатний співкристал AZD1152 можна використовувати в комбінації з одним або декількома інгібіторами клітинного циклу. Зокрема, з інгібіторами клітинного циклу, які інгібують bub1, bubR1 або CDK. Таке комбіноване лікування можна здійснювати шляхом одночасного, послідовного або окремого введення окремих компонентів для лікування. Такі комбіновані продукти містять сполуки відповідно до даного винаходу в межах застосовуваних доз, описаних у даному винаході, і іншу фармацевтично активну речовину в межах застосовуваних доз. Відповідно до варіанта здійснення винаходу, забезпечується комбінація, придатна для лікування порушень проліферації клітин (таких так злоякісне новоутворення), що містить малеатний співкристал AZD1152, як визначено в даній заявці вище, і додатковий протипухлинний засіб, як визначено в даній заявці вище. Відповідно до цього аспекту винаходу, забезпечується фармацевтичний продукт, що містить малеатний співкристал AZD1152, як визначено в даній заявці вище, і додатковий протипухлинний засіб, як визначено в даній заявці вище, для спільного лікування порушень проліферації клітин (таких так злоякісне новоутворення). Додатково до їх застосування в терапевтичній медицині, малеатний співкристал AZD1152 також корисний як фармацевтичні засоби для розвитку й стандартизації тестованих систем в умовах in vitro і in vivo для оцінки дій інгібіторів активності клітинного циклу в лабораторних тварин, таких як коти, собаки, кролики, мавпи, щури й миші, як компонент пошуку нових терапевтичних засобів. В інших вищевказаних варіантах фармацевтичної композиції, способу, методу, застосування й приготування лікарського засобу також застосовуються альтернативні й переважні варіанти здійснення сполук, описані в даному винаході. Малеатний співкристал (и) за винаходом інгібують серин-треонін кіназну активність ауроракіназ, зокрема, аурора А кінази та/або аурора В кінази й у такий спосіб інгібують клітинний цикл і проліферацію клітин. Особливий інтерес представляють сполуки, які інгібують аурора В кіназу. Ці властивості можуть бути оцінені, наприклад, за допомогою однієї або декількох методик, наведених надалі. 95108 18 (а) Дослідження інгібування аурора А кінази в умовах in vitro У цьому дослідженні визначали здатність тестованої сполуки інгібувати серин-треонін кіназну активність. ДНК, що кодує аурора А, може бути отримана шляхом синтезу цілого гена або шляхом клонування. Потім цю ДНК можна експресувати у підходящій експресійній системі для одержання поліпептиду, що має серин-треонін-кіназну активність. Для аурори А із кДНК виділяли кодувальну послідовність за допомогою полімеразної ланцюгової реакції (ПЛР) і клонували в бакуловірусному експресійному векторі pFastBac HTc з ділянками для рестрикційної ендонуклеази Ваm1 і Not1 (GibcoBRL/Life technologies). 5'-ПЛР-праймер містив послідовність розпізнавання для рестрикційної ендонуклеази Ваm1 5' до кодувальної послідовності аурора А. Цього досягали вставкою гена аурора-А в рамку із шістьома залишками гістидину, спейсерною ділянкою й сайтом розщеплення протеазою rTEV, що кодується вектором pFastBac HTc. 3'-ПЛР-праймер заміняли стопкодоном аурора А с додатковою кодувальною послідовністю перед стоп-кодоном і послідовністю розпізнавання для рестикційної ендонуклеази Not1. Ця додаткова кодувальна послідовність (5' ТАС CCA TAC GAT GTT CCA GAT TAC GCT ТСТ ТАА 3') кодує поліпептид, що має послідовність YPYDVPDYAS. Ця послідовність, що має походження від білка гемаглютиніну вірусу грипу, часто використовується як епітопна послідовність-мітка, що може визначатися за допомогою специфічних моноклональних антитіл. Таким чином, рекомбінантний вектор pFastBac кодує білок аурора А, мічений на N-кінцевій ділянці 6 залишками гістидину, а на С-кінцевій ділянці мічений епітопом гемаглютиніну вірусу група. Подробиці способів зборки рекомбінантних молекул ДНК можна знайти в стандартних посібниках, наприклад, Sambrook і ін., 1989, Molecular Cloning - A Laboratory Manual, 2-ий випуск, Cold Spring Harbor Laboratory press і Ausubel і ін., 1999, Current Protocols in Molecular Biology, John Wiley і Sons Inc. Одержання рекомбінантного вірусу можна здійснювати відповідно до протоколу виробника від GibcoBRL. Коротко, вектором pFastBac-1, що містить ген Аурора А, трансформували клітини Е. coli DHlOBac, що містять бакуловірусний геном (бакмідна ДНК) і за допомогою транспозиції в клітинах, ділянка вектора pFastBac, що містить ген стійкості до гентаміцину й ген Аурора А, що включає бакуловірусний поліедриновий промотор, переносили безпосередньо на бакмідну ДНК. Шляхом відбору з гентаміцином, канаміцином, тетрацикліном і X-gal одержували білі колонії, які повинні містити рекомбінантну бакмідну ДНК, що кодує аурора А. Бакмідну ДНК екстрагували в невеликій кількості з культури декількох білих колоній ВН10Вас і трасфектували клітини Spodoptera frugiperda Sf21, які ростуть на живильному середовищі ТС 100 (GibcoBRL), що містить 10%-ну сироватку, за допомогою реагенту CellFECTIN (GibcoBRL) відповідно до інструкцій виробника. Вірусні частки збирали шляхом збору клітинного культурального середовища через 72 години після трансфекції. 0,5 мкл цього середовища застосову 19 вали для інфікування 100 мл суспензійної культури 7 Sf21, що містить 1x10 клітин/мл. Клітинне культуральне середовище збирали через 48 годин після інфікування й визначали титр вірусу за допомогою стандартної методики дослідження бляшок. Вірусні штами використовували для інфікування клітин Sf9 і "High 5" при множинності зараження (МЗ) 3 для встановлення експресії рекомбінантного білка аурора А. Для експресії активності кінази аурора А у великій кількості клітини комах Sf21 вирощували при 28°С у живильному середовищі ТС 100, доповненому 10%-ною фетальною телячою сироваткою (Viralex) і 0,2% F68 Pluronic (Sigma) на обертовому пристосуванні Wheaton при 3 об./хв. Коли щіль6 ність клітин досягала 1,2x10 клітин/мл, їх заражали рекомбінантним вірусом аурора А, очищеним від бляшок, при множинності зараження 1 і збирали через 48 годин. Всі наступні стадії очищення здійснювали при 4°С. Заморожений осад клітин комах після центрифугування, що містить у цілому 8 2,0x10 клітин, розморожували й розводили лізуючим буфером (25 мМ HEPES (N-[2гідроксіетил]піперазин-N'-[2-етансульфонова кислота]) рН 7,4 при 4°С, 100 мМ KCl, 25 мМ NaF, 1 мМ Na3VO4, 1 мМ PMSF (фенілметилсульфонілфторид), 2 мМ 2-меркаптоетанол, 2 мМ імідазол, 1 мкг/мл апротиніну, 1 мкг/мл пепстатину, 1 мкг/мл лейпептину) при використанні 1,0 мл на 3x10 клітин. Лізис здійснювали при використанні ультразвукового гомогенізатора, після чого лізат центрифугували при 41 тис. g протягом 35 хвилин. Отриманий супернатант наносили на хроматографічну колонку з діаметром 5 мм, що містить 500 мкл Ni NTA (нітрило-триоцтова кислота) агарози (Qiagen, продукт № 30250), що врівноважували в буфері для лізису. Базового рівня УФ поглинання для елюенту досягали після промивання колонки за допомогою 12 мл буфера для лізису й наступного промивання за допомогою 7 мл буфера для промивання (25 мМ HEPES рН 7,4 при 4°С, 100 мМ KCl, 20 мМ імідазол, 2 мМ 2-меркаптоетанол). Зв'язаний білок аурора А елюювали із колонки при використанні буфера для елюювання (25 мМ HEPES рН 7,4 при 4°С, 100 мМ KCl, 400 мМ імідазол, 2 мМ 2-меркаптоетанол). Збирали фракцію елюювання (2,5 мл), що відповідає піку УФ поглинання. Елюйовану фракцію, що містить активну аурора А кіназу, піддавали повному діалізу проти діалізного буфера (25 мМ HEPES рН 7,4 при 4°С, 45% гліцерин (об./об.), 100 мм KCl, 0,25% Nonidet P40 (об./об.), 1 мм дитіотреїт). Кожну нову партію ферменту аурора А титрували в аналізі шляхом розведення за допомогою розріджувача для ферменту (25 мМ Трис-НСІ рН 7,5, 12,5 мМ KCl, 0,6 мМ DTT). Для типової партії (яка може бути отримана від Upstate), маточний фермент розводили 1 мкл на мл за допомогою розріджувача для ферменту й 20 мкл розведеного ферменту використовували для кожної аналітичної лунки. Досліджувані сполуки (при концентрації 10 мМ у диметилсульфоксиді (ДМСО)) розводили водою й 10 мкл розведеної сполуки переносили в лунки аналітичних планшетів. "Загальні" і "холості" лунки контролів містили 2,5% ДМСО замість спо 95108 20 луки. До всіх лунок додавали двадцять мікролітрів свіжорозведеного ферменту, за винятком "холостих" лунок. До "холостих" лунок додавали 20 мікролітрів розріджувача для ферменту. Після цього до всіх досліджуваних лунок додавали двадцять мікролітрів реакційної суміші (25 мМ Трис-НСІ, 12,7 мМ KCl, 2,5 мМ NaF, 0,6 мМ дитіотреїт, 6,25 мМ МnСl2, 7,5 мМ АТФ, 6,25 мкМ пептидного субстрату [біотинLRRWSLGLRRWSLGLRRWSLGLRRWSLG]), що 33 містить 0,2 мкКі [ Ρ]ΑΤΦ (Amersham Pharmacia, питома активність 2500 Кі/ммоль) для початку реакції. Планшети інкубували при кімнатній температурі протягом 60 хвилин. Для зупинки реакції до всіх лунок додавали 100 мкл 20% об./об. ортофосфорної кислоти. Пептидний субстрат уловлювали на позитивно заряджену нітроцелюлозну фільтрувальну основу Р30 (Whatman) при використанні пристрою для збору вмісту планшета на 96 лунок (TomTek), а потім аналізували на вбудовування Ρ за допомогою бета-лічильника для планшета. Значення "холостого" (без ферменту) і "загального" (без сполуки) контролів використовували для визначення інтервалу розведення досліджуваної сполуки, що забезпечує 50%-не інгібування активності ферменту (значення ІС50). (б) Аналіз інгібування аурора В кінази в умовах in vitro У цьому аналізі визначали здатність досліджуваної сполуки інгібувати серин-треонін кіназну активність. ДНК, що кодує аурора В, може бути отримана шляхом синтезу цілого гена або шляхом клонування. Потім цю ДНК можна експресувати у прийнятній експресійній системі для одержання поліпептиду з активністю серин-треонін кінази. Для аурори В із кДНК виділяли кодувальну послідовність шляхом полімеразної ланцюгової реакції (ПЛР) і клонували в pFastBac системі способом, подібним описаному вище для аурора-А (тобто, за допомогою безпосередньої експресії білка аурора В, міченого 6-гістидином). Для експресії активності аурора В кінази в великих кількостях клітини комах Sf21 вирощували при 28°С у середовищі ТС 100 з додаванням 10%ної фетальної телячої сироватки (Viralex) і 0,2% F68 Pluronic (Sigma) на обертовому пристосуванні Wheaton при 3 об./хв. Коли щільність клітин дося6 гала 1,2x10 клітин/мл, їх заражали рекомбінантним вірусом аурора В, очищеним від бляшок, при множинності зараження 1 і збирали через 48 годин. Всі наступні стадії очищення здійснювали при 4°С. Заморожений осад клітин комах після центрифугування, що містить у цілому 2,0x10 клітин, розморожували й розводили лізуючим буфером (50 мМ HEPES (N-[2-гідроксіетил]піперазин-N'[2-етансульфонова кислота]) рН 7,5 при 4°С, 1 мМ Na3VO4, 1 мМ PMSF (фенілметилсульфонілфторид), 1 мМ дитіотреїт, 1 мкг/мл апротиніну, 1 мкг/мл пепстатину, 1 мкг/мл лейпептину) при вико7 ристанні 1,0 мл на 2x10 клітин. Лізис здійснювали при використанні ультразвукового гомогенізатора, після чого лізат центрифугували при 41 тис. g протягом 35 хвилин. Отриманий супернатант наносили на хроматографічну колонку з діаметром 5 мм, що містить 1,0 мл СМ сефарози Fast Flow 21 (Amersham Pharmacia Biotech), що врівноважували в буфері для лізису. Базового рівня УФ поглинання для елюенту досягали після промивання колонки за допомогою 12 мл буфера для лізису й наступного промивання за допомогою 7 мл буфера для промивання (50 мМ HEPES рН 7,4 при 4°С, 1 мМ дитіотреїт). Зв'язаний білок аурора В елюювали із колонки при використанні градієнта буфера для елюювання (50 мМ HEPES рН 7,4 при 4°С, 0,6 Μ NaCl, 1 мМ дитіотреїт, починаючи від 0% буфера для елюювання до 100% буфера для елюювання протягом 15 хвилин при швидкості потоку 0,5 мл/хв). Збирали фракції елюювання (1,0 мл), що відповідають піку УФ поглинання. Фракції елюювання піддавали повному діалізу проти діалітичного буфера (25 мМ HEPES рН 7,4 при 4°С, 45% гліцерин (об./об.), 100 мМ KCl, 0,05% (об./об.) IGEPAL CA630 (Sigma Aldrich), 1 мм дитіотреїт). У діалізованих фракціях визначали активність аурора В кінази. Фермент аурора B-INCENP (як поставляється Upstate) готували шляхом активування аурора В (5 мкМ) в 50 мМ МТрис-НСІ рН 7,5, 0,1 мМ EGTA, 0,1% 2-мераптоетанола, 0,1 мМ ванадату натрію, 10 мМ ацетату магнію, 0,1 мМ АТФ з 0,1 мг/мл GST-INCENP [826-919] при 30°С протягом 30 хвилин. Кожну нову партію ферменту аурора BINCENP титрували в дослідженні шляхом розведення за допомогою розріджувача ферменту (25 мМ Трис-НСІ рН 7,5, 12,5 мМ KCl, 0,6 мМ DTT). Для типової партії, маточний фермент розводили 15 мкл на мл розріджувачем ферменту й 20 мкл розріджувача ферменту використовували для кожного досліджуваного лунку. Тестовані сполуки (у концентрації 10 мМ у диметилсульфоксиді (ДМСО) розводили водою й 10 мкл розведеної сполуки переносили в лунки досліджуваним планшетів. "Загальні"' і "холості" лунки контролів містили 2,5% ДМСО замість сполуки. В усі лунки додавали двадцять мікролітрів свіжорозведеного ферменту, за виключення "холостих" лунок. Двадцять мікролітрів розведеного ферменту додавали в "холості" лунки. Після цього до всіх досліджуваних лунок додавали двадцять мікролітрів реакційної суміші (25 мМ Трис-НСI, 12,7 мМ KCl, 2,5 мМ NaF, 0,6 мМ дитіотреїт, 6,25 мМ МnСl2, 15 мМ АТФ, 6,25 мкМ пептидного субстрату [біотинLRRWSLGLRRWSLGLRRWSLGLRRWSLG]), що 33 містить 0,2 мкКі [ Ρ]ΑΤΦ (Amersham Pharmacia, питома активність 2500 Кі/ммоль) для початку реакції. Планшети інкубували при кімнатній температурі протягом 60 хвилин. Для зупинки реакції до всіх лунок додавали 100 мкл 20% об./об. ортофосфорної кислоти. Пептидний субстрат уловлювали на позитивно заряджену нітроцелюлозну фільтрувальну основу Р30 (Whatman), використовуючи пристрій для збору вмісту планшета на 96 лунок 33 (TomTek), і потім аналізували на вбудовування Р за допомогою бета-лічильника для планшета. Значення "холостого" (без ферменту) і "загального" (без сполуки) контролів використовували для визначення інтервалу розведення тестованої сполуки, що забезпечує 50%-не інгібування активності ферменту (значення ІС50). 95108 22 (в) Клітинний фенотип в умовах in vitro і дослідження субстратного фосфорилування Це дослідження використовували для визначення клітинних дій сполук на колонії пухлинних клітин людини SW620 в умовах in vitro. Сполуки звичайно викликають інгібування рівнів фосфогістону H3 і підвищення площі ядра в клітинах. 4 10 SW620 клітин на лунку поміщали в 100 мкл DMEM середовища (що містить 10% FCS і 1% глутаміну) (DMEM являє собою модифіковане Дульбекко середовище Ігла (Sigma D6546)) в costar планшетах на 96 лунок і залишали для прикріплення при 37°С і 5% СО2 протягом ночі. Після цього клітини дозували зі сполукою, розведеною в середовищі (у кожну лунку додавали 50 мкл для одержання 0,00015 мкМ - 1 мкМ концентрацій сполуки) і через 24 години після обробки сполукою клітини фіксували. Спочатку клітини досліджували за допомогою світлового мікроскопа й відзначали будь-які зміни морфології клітин. Після цього в кожну лунку додавали 100 мкл 3,7% формальдегіду, і планшет залишали принаймні на 30 хвилин при кімнатній температурі. Для видалення фіксажу планшет декантували й вибивали на паперову серветку й після цього планшет один раз промивали в PBS (фосфатно-буферний сольовий розчин Дульбекко (Sigma D8537)), використовуючи автоматичний промивач планшет. Додавали 100 мкл PBS і 0,5% тритон Х-100 і планшети поміщали в шейкер протягом 5 хвилин. Планшети промивали в 100 мкл PBS і розчин виливали. Додавали 50 мкл первинного антитіла, 1:500 кролячого анти-фосфогістону H3 в PBS 1% BSA (бичачий сироватковий альбумін) і 0,5% твін. Анти-фосфоістон H3 кролячої поліклональний 06-750 одержували від Upstate Biotechnology. Планшети залишали на 1 годину при кімнатній температурі на шейкері. Наступного дня, антитіло відкидали й планшети два рази промивали PBS. У темному приміщенні додавали 50 мкл вторинного антитіла, 1:10,000 Hoechst і 1:200 Alexa Fluor 488 цапиного антикролячого IgGA (№ кат. 11008 молекулярні зонди) в PBS 1% BSA, 0,5% твін. Планшети обертали олов'яною фольгою й струшували протягом 1 години при кімнатній температурі. Антитіло відкидали й планшети два рази промивали PBS. У кожну лунку додавали 200 мкл PBS, і планшети струшували протягом 10 хвилин, PBS видаляли. У кожну лунку додавали 100 мкл PBS і планшети запечатували для підготовки до аналізу. Аналіз здійснювали відповідно до алгоритму Arrayscan Target Activation для вимірювання клітинних рівнів фосфогістону H3 і змін площі ядра. Результати виражали у вигляді ефективної концентрації, необхідної для забезпечення 50%-ного інгібування рівнів фосфогістону H3 і, подібним чином, для 50%-ного підвищення площі ядра в клітинах (значення ЕС50). Далі винахід ілюструється необмежувальними прикладами, даними й фігурами, у яких, якщо спеціально не вказано інакше: (і) виходи представлені тільки з метою ілюстрації й необов'язково, що можуть бути досягнуті максимальні значення; 23 (іі) якщо продукт використовували для затравлювання, то він може бути отриманий за допомогою раніше відомого процесу, такого як описані в WO 2004/058781; (ііі) ідентичність AZD1152 малеатного співкристала, приготовленого, як описано в даній заявці, 1 підтверджували за допомогою Η ЯМР при 400 МГц у гексадейтерованому диметилсульфоксиді з додаванням тетраметилсилану (ТМС) для порівняння (ТМС = 0,00 част, на млн.). Як описано в даній заявці, AZD1152 і AZD1152 HQPA розкриті в WO 2004/058781. Подробиці процесу описані в WO2004/058781 стосовно AZD1152, AZD1152 HQPA і всі проміжні сполуки, необхідні для вказаних сполук, повністю включені в даний винахід як посилання. Спосіб одержання 1 Стадія 1 Одержання 7-(3гідроксипропокси)хіназолін-4(3H)-он 2-Аміно-4-фторбензойну кислоту й 1,3пропандіол перемішували разом і нагрівали до 120°С. Додавали ацетат формамідину й суміш перемішували протягом 3,5 годин, одержуючи 7фторхіназолін-4-он. Після цього до суміші додавали розчин гідроксиду калію в 1,3-пропандіолі протягом 2 годин і 50 хвилин, і потім охолоджували до 15°С. Після цього, суміш нагрівали до 125°С протягом 5 годин, потім охолоджували до 75°С. До реакційної суміші поступово додавали розведену соляну кислоту (приблизно 6 мас.%) до досягнення рН 4,5. Суміш охолоджували до 0°С протягом 6 годин і витримували при цій температурі додатково протягом години, після цього неочищений продукт виділяли шляхом центрифугування. Неочищену речовину промивали водою й висушували у вакуумі, потім розчиняли в метанолі при обережному нагріванні в колбі зі зворотним холодильником і частковим концентруванням при зниженому тиску при температурі 42°С. Потім цей розчин охолоджували до 0°С протягом 3 годин і отриманий продукт виділяли шляхом фільтрації, після цього висушували у вакуумі. 7-(3Гідроксипропокси)хіназолін-4(3H)-он відновлювати у виходом 73%. 1 Η ЯМР (ДМСО d6): 11,90 (br s, 1Н), 8,04 (s, 1H), 8,00 (d, 1H), 7,10 (m, 2H), 4,17 (t, 2H), 3,58 (t,2H), 1,92 (m, 2H): + MC (позитивн. ЕРІ): 221 (M+H) Стадія 2 Одержання 4-хлор-7-(3хлорпропокси)хіназоліну 7-(3-Гідроксипропокси)хіназолін-4(3H)-он, толуол і N,N-діізопропіл-формамід (DIPF) змішували разом і нагрівали до 76°С, потім додавали тіонілхлорид протягом 1 години при 76°С. Потім додатково додавали тіонілхлорид протягом 1 години, після цього температуру підтримували рівною 76°С протягом 1 години. Суміш нагрівали в колбі зі зворотним холодильником протягом 11 годин для одержання прозорого розчину, що охолоджували до 38°С і піддавали вакуумній перегонці для видалення толуолу й тіонілхлориду. Потім додавали толуол і розчин витримували при 35°С, у цей час його освітляли за допомогою фільтра (целіт або харболіт і активоване вугілля). Отриманий розчин частково концентрували, потім додавали гептан і 95108 24 суміш охолоджували до 0°С і перемішували протягом 23 годин. Утворену ясно-коричневу суспензію виділяли шляхом фільтрації, промивали холодним гептаном, після цього висушували у вакуумі при 30°С, одержуючи 4-хлор-7-(3хлорпропокси)хіназолін (63,6%) 1 Η ЯМР (ДМСО d6): 13,25 (br s, 1Н), 8,34 (s, 1H), 8,06 (d, 1H), 7,17 (m, 2H), 4,21 (t, 2H), 3,83 (t, 2H), 2,23 (m, 2H): + MC (позитивн. ЕРІ): 257, 259 (M+H) . Стадія 3 Одержання (3-{[7-(3хлорпропокси)хіназолін-4-іл]аміно}-1Н-піразол-5іл)оцтової кислоти 4-хлор-7-(3-хлорпропокси)хіназолін додавали до 1 молярного еквівалента розчину (3-аміно-1Hпіразол-5-іл)оцтової кислоти в Nметилпіролідиноні (NMP) і потім залишали протягом 12 годин. Відбувалася кристалізація продукту без затравлювання й із затравлюванням і без додавання й з додаванням ацетонітрилу як антирозчинника. Отриману тверду речовину виділяли шляхом фільтрації, промивали Nметилпіролідиноном і ацетонітрилом і після цього висушували у вакуумі, одержуючи гідрохлорид (3{[7-(3-хлорпропокси)хіназолін-4-іл]аміно}-1Hпіразол-5-іл)оцтової кислоти, у вигляді не зовсім білої твердої речовини: 1 Η ЯМР (ДМСО d6; містить NMP як сольват): 8,92 (s, 1Н), 8,8 (d, 1Н), 7,46 (d, 1Н), 7,38 (d, 1Н), 6,7 (s, 1Н), 4,32 (t, 2Н), 3,85 (t, 2Н), 3,73 (s, 2Н), 3,3 (t, 2Н), 2,7 (s, 3H), 2,51 (m, 6Н), 2,27 (m, 2Н), 2,18 (t, 2Н), 1,93 (m, 2Н). + МС (позитивн. ЕРІ): 362,1015 (М+Н) . Стадія 4 Одержання 2-(3-{[7-(3хлорпропокси)хіназолін-4-іл]аміно}-1H-піразол-5іл)-N-(3-фторфеніл)ацетамід 4-Диметиламінопіридин (DMAP), Nметилморфолін і 3-фторанілін (у великому надлишку) додавали до суспензії гідрохлориду (3-{[7-(3хлорпропокси)хіназолін-4-іл]аміно}-1H-піразол-5іл)оцтової кислоти, в N,N-диметилацетаміді (DMA) і отриману суспензію перемішували при або нижче кімнатної температури. Після цього розчин гідрохлориду 1-етил-3-(3диметиламінопропіл)карбодііміду (EDCI.HCl), попередньо розчинений у воді, контрольовано додавали протягом 8 годин таким чином, щоб підтримувати реакцію при температурі навколишнього середовища. Суміш затравлювали невеликою кількістю продукту й залишали перемішуватися протягом декількох годин. Також додавали антирозчинник ацетонітрил, після цього воду, для осадження додаткової кількості продукту. Матеріал виділяли шляхом фільтрації й осад на фільтрі промивали сумішшю Ν,Ν-диметилацетамід: вода: ацетонітрил, теплий ацетонітрил і потім висушували (у вакуумі або під потоком азоту), одержуючи 2-(3-{[7-(3-хлорпропокси)хіназолін-4-іл]аміно}-1Hпіразол-5-іл)-N-(3-фторфеніл)ацетамід. 1 Η ЯМР (ДМСО d6; містить DMA, що залишився): 10,4 (s, 1Н), 8,9 (s, 1Н), 8,8 (d, 1Н), 7,59 (m, 1Н), 7,46 (d, 1Н), 7,33 (m, 2Н), 7,29 (d, 1Н), 6,85 (m, 1Н), 6,75 (s, 1H), 4,35 (t, 2Н), 3,85 (t, 4Н), 2,95 (s), 2,83 (s), 2,56 (s), 2,25 (m, 2Н), 1,95 (s): + МС (позитивн. ЕРІ): 455 (М+Н) . 25 Стадія 5 - Одержання 2-{3-[(7-{3-[етил(2гідрокеіетил)аміно]пропокси}-хіназолін-4-іл)аміно]1H-піразол-5-іл}-N-(3-фторфеніл)ацетаміду (AZD1152 НQРА) 2-(3-{[7-(3-Хлорпропокси)хіназолін-4-іл]аміно}1Н-піразол-5-іл)-N-(3-фторфеніл)ацетамід і 2(етиламіно)етанол (12 молярних еквівалентів) додавали до N,N-диметилацетаміду в інертній атмосфері (яка забезпечувалася азотом) і суміш нагрівали до 90°С при перемішуванні. Через 12 годин, додавали воду контрольовано і партію затравлювали продуктом доти, поки вона була гарячою. Суміш охолоджували до 20°С обережним контрольовано для кристалізації продукту в необхідній формі. Після цього продукт фільтрували й промивали сумішшю вода/N,N-диметилацетамід і ацетонітрил. Після цього, осад на фільтрі суспендували протягом періоду часу теплим ацетонітрилом (40°С), фільтрували, промивали додатковою кількістю ацетонітрилу й потім висушували (у вакуумі або під потоком азоту), одержуючи безводний 2-{3[(7-{3-[етил(2-гідроксіетил)аміно]пропокси}хіназолін-4-іл)аміно]-1Н-піразол-5-іл}-N-(3фторфеніл)ацетамід у вигляді не зовсім білої твердої речовини у виходом ~90%. 1 Η ЯМР (ДМСО d6): 10,55 (s, 1H), 9,45 (br s, 1H), 8,98 (s, 1H), 8,8 (d, 1H), 7,63 (m, 1H), 7,47 (d, 1H), 7,37 (m, 2H), 7,32 (d, 1H), 6,9 (m, 1H), 6,77 (s, 1H), 4,32 (t, 2H), 3,83 (br s, 2H), 3,76 (t, 2H), 3,35 (m, 2H), 3,25 (m, 4H), 2,25 (m, 2H), 1,25 (t, 3H): + MC (позитивн. ЕРІ): 508,4 (M+H) . Стадія 6 - Одержання моно(трет-бутил) 2-[[3({4-[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}-1Hпіразол-3-іл)аміно]-хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфату [AZD1152 t-Bu P(5) складний ефір] 2-{3-[(7-{3-[Етил(2гідроксіетил)аміно]пропокси}-хіназолін-4-іл)аміно]1H-піразол-5-іл}-N-(3-фторфеніл)ацетамід і піридин, гідрохлорид змішували в Ν,Νдиметилацетаміді й розчин охолоджували до 15°С. Після цього додавали ди-трет-бутил діетилфосфорамідит (1,5-2,1 молярних еквівалентів), при цьому температуру підтримували. Реакційну суміш обробляли in situ з 30 мас.% перекисом водню (близько 4,2 мольних еквівалентів), при цьому температуру підтримували нижче температури навколишнього середовища. Перекис водню, що залишився, руйнували шляхом додавання метабісульфіту натрію (у вигляді 10% мас./об., водного розчину), при цьому температуру підтримували нижче 40°С. Отриманий розчин ди-трет-бутил 2[[3-({4-[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}-1Hпіразол-3-іл)аміно]хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфату потім нагрівали до 40°С і додавали розчин гідроксиду натрію (2М) для доведення рН 5-6,5. Температуру й рН підтримували приблизно протягом 90 хвилин із затравлюванням. Потім додавали воду й додатково доводили до значення рН в інтервалі рН 8-9 для оптимізації відновлення. Теплу реакційну суміш відразу фільтрували, одержуючи моно-третбутил 2-[[3-({4-[(5-{2-[(3-фторфеніл)аміно]-2оксоетил}-1Н-піразол-3-іл)аміно]хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфат, який про 95108 26 мивали сумішшю N,N-диметилацетамід/вода й водою й на завершення висушували (у вакуумі й у потоці підходящого інертного газу), одержуючи моно(трет-бутил) 2-[[3-({4-[(5-{2-[(3фторфеніл)аміно]-2-оксоетил}-1Н-піразол-3іл)аміно]-хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфат у вигляді не зовсім білої твердої речовини з виходом в інтервалі 86-93%. 1 Η ЯМР (ДМСО d6): 10,48 (s, 1Н), 9,75 (br s, 1H), 8,98 (s, 1H), 8,85 (d, 1H), 7,67 (m, 1H), 7,48(d, 1H), 7,37 (m, 2H), 7,3 (d, 1H), 6,87 (m, 1H), 6,83 (s, 1H), 4,34 (t, 2H), 4,28 (m, 2H), 3,88 (s, 2H), 3,53 (m, 2H), 3,43 (m, 2H), 3,33 (m, 2H), 2,3 (m, 2H), 1,47 (s, 9H), 1,32 (t,3H): + MC (позитивн. ЕРІ): (М+Н) 644,2761 фрагмент (менше бутилу) 588,2147. Стадія 7 - Одержання 2-{етил[3-({4-[(5-{2-[(3фторфеніл)аміно]-2-оксоетил}-1H-піразол-3іл)аміно]хіназолін-7-іл}окси)пропіл]аміно}етил дигідрофосфату (AZD1152) Моно(трет-бутил) 2-[[3-({4-[(5-{2-[(3фторфеніл)аміно]-2-оксоетил}-1H-піразол-3іл)аміно]-хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфат суспендували в суміші 1:1 вода/тетрагідрофуран (ТГФ) і обробляли при підвищених температурах (переважно 50-60°С) з надлишком у діапазоні від 1,5 до 3,0 молярних еквівалентів соляної кислоти протягом періоду близько 1 години. Після цього гарячий розчин підлуговували, використовуючи 2,0М гідроксид натрію до діапазону рН 4,5-5,5, охолоджували до 60°С і затравлювали. До суспензії контрольовано додавали воду при контрольованому охолодженні кристалізованої суміші до кімнатної температури й продукт виділяли шляхом фільтрації. Осад на фільтрі промивали водою й висушували у вакуумі. Після висушування, твердий 2-{етил[3-({4[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}-1Hпіразол-3-іл)аміно]хіназолін-7іл}окси)пропіл]аміно}етил дигідрофосфат урівноважували в умовах навколишнього середовища до постійної ваги, одержуючи гідровану форму у вигляді ясно-жовтої голчастої речовини. 1 Η ЯМР (ДМСО d6): + МС (позитивн. ЕРІ): 587,8 (М+Н) 1 Η ЯМР (ДМСО d6): 10,53 (s, 1Н), 8,57 (s, 1Н), 8,54 (d, 1Н), 7,62 (d, 1H), 7,37 (m, 2H), 7,27 (s, 1H), 7,21 (d, 1H), 6,88 (m, 1H), 6,65 (s, 1H), 4,27 (t, 2H), 4,05 (m, 2H), 3,75 (s, 2H), 3,24 (m, 2H), 3,21 (t, 2H), 3,13 (q, 2H), 2,18 (m, 2H), 1,24 (t, 3H): + MC (позитивн. ЕРІ): 588 (M+H) . C26H31FN7O6P+ 3,0H2O необхідно С, 48,7%; Η, 5,8%; Ν, 15,3%; Виявлено С, 48,8%; Η, 5,35%; Ν, 15,15%. Стадія 8 - Одержання 2-{етил[3-({4-[(5-{2-[(3фторфеніл)аміно]-2-оксоетил}-1H-піразол-3іл)аміно]хіназолін-7-іл}окси)пропіл]аміно}етил дигідрофосфат. малеату [AZD1152 малеат] 2-бутендіоєву кислоту (Z) (1,57 молярних еквівалентів; 449,80 мкмоль; 52,21 мг) розчиняли в метанолі (123,54 мкмоль; 5,00 мл; 3,96 г) і до цього розчину додавали попередньо підготовлений метанольний розчин AZD1152 (у вигляді вільної форми тригідрату - 1,00 молярний еквівалент, 286,14 27 мкмоль; 40,00 мл; 31,87 г), після цього додатково додавали метанол (123,54 мкмоль; 5,00 мл; 3,96 г). Суміш залишали перемішуватися протягом ночі при кімнатній температурі. Одержували білу суспензію й тверду речовину відновлювали шляхом фільтрації, після цього висушували у вакуумі. При аналізі за допомогою ЯМР підтверджували, що співкристал являв собою малеат. Альтернативна стадія 8: Неочищений AZD1152 (оцінюється при 7,44 г @ 100%, 11,61 мілімоль) додавали до диметилсульфоксиду (36 мл) і залишали при температурі навколишнього середовища, одержуючи яснокоричневий розчин. До цього розчину додавали розчин малеїнової кислоти (1,76 г, 15,16 мілімоль, 1,31 мольних еквівалентів) у метанолі (36 мл) і суміш залишали відстоюватися протягом ночі при температурі навколишнього середовища. Наступного дня аліквоту прозорого розчину переносили у флакон, розмічали й залишали запечатаним протягом декількох годин. Утворювалося відкладення білої твердої речовини і її переносили в колбу й залишали перемішуватися. Поступово розчин ставав каламутним і осаджувалася тверда речовина. Завись залишали відстоюватися протягом декількох днів і на завершення фільтрували. Осад на фільтрі промивали сумішшю 1:1 диметилсульфоксид/метанол (усього 15 мл), суспендували in situ з метанолом (3x25 мл) і, після цього висушували у вакуумі. ЯМР підтверджував, що тверду речовину являла собою малеатний співкристал AZD1152 (з виходом приблизно 78,7%). Спосіб одержання 2 Стадія 1 Одержання 2-(3-{[7-(3хлорпропокси)хіназолін-4-іл]аміно}-1Н-піразол-5іл)-N-(3-фторфеніл)ацетаміду До суспензії гідрохлориду (3-{[7-(3хлорпропокси)хіназолін-4-іл]аміно}-1H-піразол-5іл)оцтової кислоти, (отриманого, як описано в способі одержання 1 вище) в Ν,Ν-диметилацетаміді (DMA) додавали 4-диметиламінопіридин (DMAP), при цьому температуру підтримували рівною 1525°С (ідеально 15°С), потім додавали Nметилморфолін, при цьому також підтримували температуру. 3-Фторанілін (у великому надлишку, що ідеально становить 10-15 мольних еквівалентів) додавали при такій швидкості, щоб температура підтримувалася нижче 25°С. Тим часом гідрохлорид 1-етил-3-(3диметиламінопропіл)карбодііміду (EDCI.HCl) розчиняли у воді, одержуючи розчин приблизно 42% мас./об. (кількість присутньої води є важливою для підсумкової кристалізації пізніше в процесі). Цей розчин додавали контрольовано до суспензії протягом 8 годин таким чином, щоб підтримувати реакцію в інтервалі 20-25°С; після цього суміш затравлювали кристалами переважної форми продукту (ідеально в кількості приблизно 1% очікуваного виходу). Суміш перемішували приблизно протягом 16 годин, при цьому підтримували температуру (ідеально 20-25°С), потім контрольовано додавали антирозчинники ацетонітрил, після цього воду й для підтримки температури в інтервалі 20-25°С, після цього продовжували перемішувати приблизно протягом 21 годин; це оптимізує відновлення й 95108 28 утворення продукту. Матеріал виділяли шляхом фільтрації й осад на фільтрі промивали сумішшю N,N-диметилацетамід: вода: ацетонітрил (об'ємні співвідношення 5:3:2), ацетонітрил і потім висушували (у вакуумі або під потоком азоту), одержуючи 2-(3-{[7-(3-хлорпропокси)хіназолін-4-іл]аміно}-1Hпіразол-5-іл)-N-(3-фторфеніл)ацетамід, що містить невелику кількість DMA із приблизним виходом 7678%. 1 Η ЯМР (ДМСО d6; містить DMA, що залишився): 10,4 (s, 1H), 8,9 (s, 1Н), 8,8 (d, 1H), 7,59 (m, 1Н), 7,46 (d, 1Н), 7,33 (m, 2Н), 7,29 (d, 1Н), 6,85 (m, 1Н), 6,75 (s, 1Н), 4,35 (t, 2Н), 3,85 (t, 4Н), 2,95 (s), 2,83 (s), 2,56 (s), 2,25 (m, 2Н), 1,95 (s): + МС (позитивн. ЕРІ): 455 (М+Н) . Стадія 2 - (Одержання 2-{3-[(7-{3-[етил(2гідроксіетил)аміно]пропокси}-хіназолін-4-іл)аміно]1H-піразол-5-іл}-N-(3-фторфеніл)ацетаміду (AZD1152 НQРА) 2-(3-{[7-(3-Хлорпропокси)хіназолін-4-іл]аміно}1H-піразол-5-іл)-N-(3-фторфеніл)ацетамід і 2(етиламіно)етанол (ідеально 12 молярних еквівалентів) додавали до N,N-диметилацетаміду в інертній атмосфері (яка забезпечується азотом) і суміш нагрівали до 90°С при перемішуванні. Через 12-16 годин (ідеально 12 годин) реакцію знову охолоджували приблизно до 85°С і контрольовано додавали воду для підтримки температури в інтервалі 80-85°С. Партію доводили до 80°С і затравлювали кристалами переважної форми продукту (ідеально в кількості приблизно 1% очікуваного виходу). Суміш охолоджували до 20°С обережним контрольовано приблизно протягом 20 годин таким чином, щоб продукт кристалізувався в необхідній формі й достатньому розмірі, забезпечуючи гарну швидкість фільтрування. Після цього продукт фільтрували й промивали сумішшю вода/N,Nдиметилацетамід і ацетонітрил і підходяще обробляли рідиною, одержуючи гідровану форму продукту. Після цього, осад на фільтрі суспендували in situ протягом періоду (ідеально 2 години) з теплим ацетонітрилом (ідеально при температурі 40°С), потім фільтрували, промивали додатковою кількістю ацетонітрилу й потім висушували (у вакуумі або під потоком азоту), одержуючи майже безводний 2-{3-[(7-{3-[етил(2гідроксіетил)аміно]пропокси}-хіназолін-4-іл)аміно]1H-піразол-5-іл}-N-(3-фторфеніл)ацетамід у вигляді не зовсім білої твердої речовини з виходом 8590%. 1 Η ЯМР (ДМСО d6): 10,55 (s, 1Н), 9,45 (br s, 1H), 8,98 (s, 1H), 8,8 (d, 1H), 7,63 (m, 1H), 7,47 (d, 1H), 7,37 (m, 2H), 7,32 (d, 1H), 6,9 (m, 1H), 6,77 (s, 1H), 4,32 (t, 2H), 3,83 (br s, 2H), 3,76 (t, 2H), 3,35 (m, 2H), 3,25 (m, 4H), 2,25 (m, 2H), 1,25 (t, 3H): + MC (позитивн. ЕРІ): 508,4 (M+H) . Стадія 3 - Одержання моно(трет-бутил) 2-[[3({4-[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}-1Hпіразол-3-іл)аміно]-хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфату [AZD1152 t-Bu P(5) складний ефір] До суспензії гідрохлориду піридину, в N,Nдиметилацетаміді завантажували розчин 2-{3-[(7{3-[етил(2-гідроксіетил)аміно]пропокси}-хіназолін4-іл)аміно]-1H-піразол-5-іл}-N-(3 29 фторфеніл)ацетаміду й ди-трет-бутял діетилфосфорамідиту (ідеально 1 молярний еквівалент) в N,N-диметилацетаміді протягом подовженого періоду (ідеально 3 години) і температуру підтримували в інтервалі від -20 до -10°С (ідеально -15°С). Після цього додатково додавали ди-трет-бутил діетилфосфорамідит (ідеально 0,5 молярних еквіваленти) протягом 1 години, температуру також підтримували в інтервалі від -20 до -10°С (ідеально -15°С). Реакційну суміш обробляли in situ 30 мас.% перекисом водню (приблизно 4,2 мольних еквівалентів), при цьому температуру підтримували нижче -10°С (ідеально від -12 до -8°С) і витримували протягом періоду часу при цій температурі (ідеально 16 годин). Перекис водню, що залишився, руйнували шляхом додавання метабісульфіту натрію (у вигляді 10% мас./об. водного розчину), при цьому температуру підтримували нижче 40°С. Після цього отриманий розчин ди-трет-бутил 2-[[3-({4-[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}1H-піразол-3-іл)аміно]хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфату нагрівали до 40°С і додавали гідроксид натрію (ідеально 2М) для доведення рН 5,5-6,5 (ідеально рН 6) із затравлюванням з підходящою кристалічною речовиною. Температуру підтримували й інтервал рН 5-6 підтримували шляхом додавання додаткової кількості розчину гідроксиду натрію принаймні протягом 2 годин. Потім завантажували воду й після цього значення рН доводили до діапазону рН 8-9 (ідеально рН 8,8), при цьому підтримували температуру (ідеально 40°С, але в інтервалі 35-45°С) протягом 16 годин для того, щоб оптимізувати відновлення. Теплу реакційну суміш відразу фільтрували, одержуючи моно(трет-бутил 2-[[3-({4-[(5-{2[(3-фторфеніл)аміно]-2-оксоетил}-1H-тразол-3іл)аміно]хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфат, що декілька разів промивали водою й на завершення висушували (або у вакуумі й у потоці підходящого інертного газу), одержуючи моно(трет-бутил) 2-[[3({4-[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}-1Нпіразол-3-іл)аміно]-хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфат у вигляді не зовсім білої твердої речовини з виходом в інтервалі 86-93%. 1 Η ЯМР (ДМСО d6): 10,48 (s, 1H), 9,75 (br s, 1H), 8,98 (s, 1H), 8,85 (d, 1H), 7,67 (m, 1H), 7,48(d, 1H), 7,37 (m, 2H), 7,3 (d, 1H), 6,87 (m, 1H), 6,83 (s, 1H), 4,34 (t, 2H), 4,28 (m, 2H), 3,88 (s, 2H), 3,53 (m, 2H), 3,43 (m, 2H), 3,33 (m, 2H), 2,3 (m, 2H), 1,47 (s, 9H), 1,32 (t,3H): + MC (позитивн. ЕРІ): (M+H) 644,2761 фрагмент (менше бутилу) 588,2147. Стадія 4 - Одержання 2-{етил[3-({4-[(5-{2-[(3фторфеніл)аміно]-2-оксоетил}-1H-піразол-3іл)аміно]хіназолін-7-іл}окси)пропіл]аміно}етил дигідрофоефату (AZD1152) Моно(трет-бутил)2-[[3-({4-[(5-{2-[(3фторфеніл)аміно]-2-оксоетил}-1H-піразол-3іл)аміно]-хіназолін-7іл}окси)пропіл](етил)аміно]етил фосфат суспендували в суміші вода/тетрагідрофуран (ТГФ) і обробляли надлишком в інтервалі від 1,5 до 3,0 моляр 95108 30 них еквівалентів соляної кислоти (ідеально в концентрації 2М і що містить 1,5 мольних еквівалентів). Суміш нагрівали до 55-65°С (ідеально 60°С) і витримували при 60°С приблизно протягом 1 години. Після цього гарячий розчин підлуговували за допомогою гідроксиду натрію (переважна концентрація 2М і вміст 1,7 мольних еквівалентів), одержуючи значення рН в інтервалі рН 5,0-5,5 і потім затравлювали при 55-65°С (ідеально 60°С) із кристалами переважної форми продукту (ідеально в кількості приблизно 0,05%мас. очікуваного виходу). Суміш перемішували при цій температурі принаймні протягом однієї години, потім додавали воду й завись перемішували й охолоджували контрольовано приблизно протягом 12 годин, потім перемішували при температурі навколишнього середовища принаймні протягом 4 годин і після цього продукт виділяли шляхом фільтрації. Осад на фільтрі промивали послідовно водою, потім ТГФ і висушували або у вакуумі або використовуючи методику зволоження, за допомогою чого інертний газ, зволожений парами води, пропускали через тверду речовину до одержання постійної ваги. Після висушування у вакуумі тверду речовину 2-{етил[3-({4-[(5-{2-[(3-фторфеніл)аміно]-2оксоетил}-1Н-піразол-3-іл)аміно]хіназолін-7іл}окси)пропіл]аміно}етил дигідрофосфат урівноважували в умовах навколишнього середовища до постійної ваги, одержуючи гідровану форму у вигляді ясно-жовтої голчастої речовини з виходом приблизно 81%. 1 Η ЯМР (ДМСО d6): + МС (позитивн. ЕРІ): 587,8 (М+Н) 1 Η ЯМР (ДМСО d6): 10,53 (s, 1Н), 8,57 (s, 1Н), 8,54 (d, 1Н), 7,62 (d, 1H), 7,37 (m, 2H), 7,27 (s, 1H), 7,21 (d, 1H), 6,88 (m, 1H), 6,65 (s, 1H), 4,27 (t, 2H), 4,05 (m, 2H), 3,75 (s, 2H), 3,24 (m, 2H), 3,21 (t, 2H), 3,13 (q, 2H), 2,18 (m, 2H), 1,24 (t, 3H): + MC (позитивн. ЕРІ): 588 (M+H) . C26H31FN7O6P+3,0H2O необхідно С, 48,7%; Η, 5,8%; Ν, 15,3%; Виявлено С, 48,8%; Η, 5,35%; Ν, 15,15%. Стадія 5 - Одержання 2-{етил[3-({4-[(5-{2-[(3фторфеніл)аміно]-2-оксоетил}-1H-піразол-3іл)аміно]хіназолін-7-іл}окси)пропіл]аміно}етил дигідрофоефат. малеату [AZD1152 малеат] 2-бутендіоєву кислоту (Ζ) (1,57 молярних еквіваленти; 449,80 мкмоль; 52,21 мг) розчиняли в метанолі (123,54 мкмоль; 5,00 мл; 3,96 г) і до цього розчину додавали раніше приготовлений метанольний розчин AZD1152 (у вигляді вільної форми тригідрату - 1,00 молярний еквівалент, 286,14 мкмоль; 40,00 мл; 31,87 г), потім додатково додавали метанол (123,54 мкмоль; 5,00 мл; 3,96 г). Суміш залишали перемішуватися протягом ночі при кімнатній температурі. Одержували білу суспензію й тверду речовину відновлювали шляхом фільтрації, після цього висушували у вакуумі. При аналізі за допомогою ЯМР підтверджували, що співкристал являв собою малеат AZD1152. Альтернативна стадія до вищеописаної стадії 5: Неочищений AZD1152 (оцінюється при 7,44 г @ 100%, 11,61 мілімоль) додавали до диметилсульфоксиду (36 мл) і залишали при температурі 31 навколишнього середовища, одержуючи яснокоричневий розчин. До цього розчину додавали розчин малеїнової кислоти (1,76 г, 15,16 мілімоль, 1,31 мольних еквівалентів) у метанолі (36 мл) і суміш залишали відстоюватися протягом ночі при температурі навколишнього середовища. Наступного дня аліквоту прозорого розчину переносили у флакон, розмічали й залишали запечатаним протягом декількох годин. Утворювалося відкладення білої твердої речовини і її переносили в колбу й залишали перемішуватися. Поступово розчин ставав каламутним і осаджувалася тверда речовина. Завись залишали відстоюватися протягом декількох днів і на завершення фільтрували. Осад на фільтрі промивали сумішшю 1:1 диметилсульфоксид/метанол (усього 15 мл), суспендували in situ з метанолом (3x25 мл) і, після цього висушували у вакуумі. При аналізі за допомогою ЯМР підтверджували, що співкристал являв собою малеат AZD1152 (з виходом приблизно 78,7%). Подальша альтернатива до вищеописаної стадії 5: AZD1152 (у вигляді вільної форми тригідрату 1,00 молярний еквівалент, 8,51 мкмоль; 5,74 г) і 2бутендіоєву кислоту (Ζ) (1,20 молярних еквівалентів; 10,2 мкмоль; 1,19 г) розчиняли в диметилсульфоксиді (35 мл) і нагрівали до 60°С. До гарячої суміші додавали анти-розчинник ацетонітрил (20 мл), потім суміш затравлювали кристалами переважної форми продукту AZD1152 Малеат (0,005 молярних еквівалентів; 42,6 мікромолів; 30,7 мг ідеально в кількості приблизно 0,5 мас.% очікуваного виходу). Реакційну суміш витримували при 60°С протягом 4 годин, потім додатково завантажений ацетонітрил (40 мл) додавали протягом 3 годин. Суміш залишали струшуватися при 60°С протягом 12-20 годин (ідеально 16 годин). Реакцію охолоджували до 20°С контрольовано і продукт виділяли шляхом фільтрації. Осад на фільтрі промивали сумішшю диметилсульфоксид/ацетонітрил 95108 32 (об'ємне співвідношення 1:2), потім ацетонітрилом. Тверду речовину висушували у вакуумі або в потоці теплого інертного газу (ідеально 40°С), одержуючи AZD1152 Малеат у вигляді білої твердої речовини з виходом приблизно 88%. Короткий опис фігур Фігура 1: Порошкова рентгенограма для малеатного співкристала AZD1152 (отриманого відповідно до вищеописаного способу 1) - значення 2 вказані на горизонтальній осі й відносній інтенсивності лінії (імпульс) вказані на вертикальній осі. Фігура 2: Термограма диференціальної сканувальної калориметрії для AZD1152 малеатного співкристала, отриманого відповідно до вищеописаного способу 1. Фігура 3: Термограма диференціальної сканувальної калориметрії для AZD1152 малеатного співкристала, отриманого відповідно до вищеописаного способу 2. Фігура 4: Графік ізотерми сорбції динамічного випаровування для партії AZD1152 малеатного співкристала, отриманого відповідно до вищеописаного способу 1. Фігура 5: Графік ізотерми сорбції динамічного випаровування для партії AZD1152 малеатний співкристала, отриманого відповідно до вищеописаного способу 2. Фігура 6: Графік ізотерми сорбції динамічного випаровування для вільної форми AZD1152. Фігура 7: Порошкова рентгенограма для малеатного співкристала AZD1152 (отриманого відповідно до вищеописаного способу 2) - значення 2 вказані на горизонтальній осі й відносній інтенсивності лінії (імпульс) вказані на вертикальній осі. Характерні дані Спектроскопія ядерного магнітного резонансу Структура й наближене співвідношення компонентів у співкристалі може бути підтверджене за допомогою протонної ЯМР спектроскопії. Типові дані наведені нижче. 33 Диференціальна сканирующая калориметрія Диференціальну сканувальну калориметрію (ДСК) здійснювали на AZD1152 малеатному співкристалі, отриманому відповідно до способів приготування 1 і 2, використовуючи Mettler DSC820e. Зразки, які звичайно були менші, ніж 5 мг речовини, що міститься в 40 мкл алюмінієвого лотка, обладнаного проколюваною кришкою, нагрівали при температурі в інтервалі від 25° С до 325° С при постійній швидкості нагрівання 10°С на хвилину. 95108 34 Як продувний газ використовували азот - швидкість потоку 100 мл на хвилину. Отримані результати для партії AZD1152 малеатного співкристала, приготовленого відповідно до вищеописаного способу 1 (див.Фігуру 2), вказують на те, що малеатний співкристал проявляє більшу, круту ендотерму з початковою температурою 180°С внаслідок плавлення. Після розплавлювання спостерігається велика ендотермічна подія внаслідок розкладання малеїнової кислоти 35 після розплавлювання. Слід взяти до уваги, що значення початкової температури та/або температури піку, визначені за допомогою ДСК, можуть незначно відрізнятися при аналізі на різних приладах, для різних методів або різних зразків, і, отже, наведені значення не повинні тлумачитися як абсолютні. Отримані результати для AZD1152 малеатного співкристала, приготовленого відповідно до вищеописаного способу 2 (див. Фігуру 3), вказують нате, що малеатний співкристал проявляє більшу, круту ендотерму з початковою температурою 183°С внаслідок плавлення. Після розплавлювання спостерігається велика ендотермічна подія внаслідок розкладання малеїнової кислоти після розплавлювання. Слід взяти до уваги, що значення початкової температури та/або температури піка, визначені за допомогою ДСК, можуть незначно відрізнятися при аналізі на різних приладах, для різних методів або різних зразків, і, отже, наведені значення не повинні тлумачитися як абсолютні. Сорбція динамічного випаровування Аналітичний прилад: аналізатор Surface Measurements Systems Dynamic Vapour Sorption. Близько 5 мг речовини, що міститься у кварцовій посудині, при заданій температурі піддавали дії зволоженого азоту при швидкості потоку 200 мл/хвилину при 25°С при наступних значеннях відносної вологості (ОВ): 0, 20, 40, 60, 80, 95, 80, 60, 40, 20, 0% ОВ у двох повторах. За вагою матеріалу в умовах конкретної відносної вологості спостерігали доти, поки він був стабільним відповідно до вагового критерію 0,002% середньої зміни ваги протягом 10 хвилин. Якщо вага усе ще продовжувала змінюватися, то речовину залишали в умовах конкретної відносної вологості доти, поки вага не ставала стабільною (максимально аж до 12 годин). Результати для партії AZD1152 малеатного співкристала, приготовленого відповідно до способу 1, показані на Фігурі 4. Результати для партії AZD1152 малеатного співкристала, приготовленого відповідно до способу 2, показані на Фігурі 5. Результати сорбції динамічного випаровування, наведені на Фігурах 4 і 5, вказують на те, що зразок є негігроскопічним і нагромадження ваги обумовлено поверхневою абсорбцією. Втрата ваги, яка спостерігається на Фігурі 5 протягом циклу 1, обумовлена невеликою кількістю розчинника, що присутній у зразку виробника. Це підтверджується зменшеною вагою при 0%ВВ. Після упарювання розчинника, матеріал підтримує свою вагу при 0%ВВ. Це спостереження повністю розуміється середнім фахівцем у даній галузі техніки. Сорбція динамічного випаровування вільної форми AZD1152 наведена на Фігурі 6. Сорбція динамічного випаровування вільної форми AZD1152 вказує на те, що рівень води до деякої міри залежить від відносної вологості при зберіганні. Ця зміна рівня води обумовлено різними гідрованими станами, які можуть змінюватися від дегідрованого стану до тетрагідрованого стану й вище. Рентгенівська порошкова дифрактомeтрія 95108 36 Як вказано вище, порошкова рентгенограма для малеатного співкристала AZD1152 наведена на Фігурі 1. У вищеописаному способі 2 представлений додатковий метод приготування AZD1152 малеату. Порошкова рентгенограма для малеатного співкристала AZD1152, отриманого відповідно до способу 2, наведена на Фігурі 7. Основні піки представлені в таблиці 2 нижче. Таблиця 2 Кут (2- Відносна волотета) гість (%) 5,13 80,3 6,45 57,9 8,14 40,5 10,18 9,1 11,96 7,8 12,85 58,4 13,85 14,3 14,96 12,5 15,27 25,7 15,75 13 16,53 19,7 17,29 23,6 19,31 100 19,78 55,1 20,10 99,5 20,60 40,5 21,01 20,5 21,68 82,3 Кут (2- Відносна волотета) гість (%) 22,45 13,5 23,22 21,8 23,58 33,8 23,98 20,8 24,30 14,8 24,65 16,1 25,56 27,8 25,83 52,5 26,37 35,6 27,98 38,4 28,44 34,3 28,91 16,9 29,72 29,6 30,34 13 32,72 14,5 36,21 22,3 38,18 11,9 Як вказано вище, у даній галузі техніки відомо, що може бути отримана порошкова рентгенограма, що містить одну або декілька похибок вимірювання, які залежать від умов вимірювання (таких як використовуване обладнання, приготування зразка або прилад). Зокрема, добре відомо, що напруга при порошковій рентгенограмі може відрізнятися залежно від умов вимірювання й приготування зразків. Наприклад, фахівці в галузі порошкової рентгенограми можуть підтвердити, що на відносну інтенсивність піків можуть впливати, наприклад, частинки розміром близько 30 мікронів і неодиничні формати зображення, які можуть впливати на аналіз зразків. Фахівець у даній галузі також буде враховувати той факт, що на положення відбиттів може впливати точна висота, на яку встановлюється зразок у дифрактометрі й калібрування нуля дифрактометра. Площинність поверхні зразка також може мати незначний вплив. Отже, фахівець у даній галузі техніки візьме до уваги, що дані порошкової рентгенограми, наведені в даному винаході, не повинні тлумачитися як абсолютні (для додаткової інформації див. Jenkins, R & Snyder, R.L. 'Introduction to X-Ray Powder Diffractometry' John Wiley & Sons, 1996). Також, як вказано вище, у цілому, похибка вимірювання кута дифракції на порошковій рентгенограмі для кута 2-тета становить близько 0,5° або менше (або, більш підходяще, для кута 2-тета близько 0,2° або менше) і такий порядок похибки вимірювання слід враховувати при аналізі порош 37 кових рентгенограм на Фігурі 1 і при тлумаченні положень піків з посиланням на вищеописаний текст і на таблицю 1. Враховуючи викладене вище, фахівець у даній галузі техніки візьме до уваги, що дані, представлені на Фігурі 7 і таблиці 2, вказують на те, що ма 95108 38 леатний співкристал AZD1152, отриманий відповідно до способу 2, являє собою таку ж кристалічну форму, що й малеатний співкристал AZD1152, отриманий відповідно до способу 1 (і наведений на Фігурі 1 і таблиці 1). 39 95108 40 41 Комп’ютерна верстка О. Гапоненко 95108 Підписне 42 Тираж 24 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMaleate co-crystal of azd1152 and use thereof for the treatment of cancer
Автори англійськоюSependa George Joseph, Storey Richard
Автори російськоюСепенда Джордж Джозеф, Стори Ричард
МПК / Мітки
МПК: A61K 31/661, A61P 35/00, C07F 9/09, C07D 403/12
Мітки: лікування, малеатний, застосування, співкристал, злоякісних, новоутворень, 2-{етил[3-({4-[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}-1н-піразол-3-іл)аміно]хіназолін-7-іл}окси)пропіл]аміно}етилдигідрофосфату
Код посилання
<a href="https://ua.patents.su/21-95108-maleatnijj-spivkristal-2-etil3-4-5-2-3-ftorfenilamino-2-oksoetil-1n-pirazol-3-ilaminokhinazolin-7-iloksipropilaminoetildigidrofosfatu-ta-jjogo-zastosuvannya-dlya-likuvannya-zloyaki.html" target="_blank" rel="follow" title="База патентів України">Малеатний співкристал 2-{етил[3-({4-[(5-{2-[(3-фторфеніл)аміно]-2-оксоетил}-1н-піразол-3-іл)аміно]хіназолін-7-іл}окси)пропіл]аміно}етилдигідрофосфату та його застосування для лікування злоякісних новоутворень</a>
Дималеат 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4-(n,n-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((s)-тетрагідрофуран-3-ілокси)хіназоліну, спосіб його одержання та застосування
Номер патенту: 91401
Опубліковано: 26.07.2010
Автори: Шнаубельт Юрген, Кулінна Крістіан, Ралль Вернер, Зігер Петер, Зойка Райнер
МПК: A61K 31/505, C07D 405/12, A61P 35/00
Мітки: застосування, дималеат, спосіб, одержання, 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4-(n,n-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((s)-тетрагідрофуран-3-ілокси)хіназоліну
Формула / Реферат:
1. Спосіб одержання дималеату 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3-ілокси)хіназоліну, який включає наступні стадії:а) сполуку загальної формули (V) (V)піддають у відповідних розчинниках після відповідної активації взаємодії з ді-(С1-С4алкіл)фосфонооцтовою кислотою таб)...
Застосування r-(+)-a-(2,3-диметоксифеніл)-1-[2-(4-фторфеніл)етил]-4-піперидинметанолу для лікування розладів сну

Номер патенту: 74773
Опубліковано: 15.02.2006
Автори: Хічкок Джаніс М., Соренсен Стефен М., Мондадорі Чезаре
МПК: A61K 31/445, A61P 25/18, A61P 25/20
Мітки: застосування, лікування, сну, r-(+)-a-(2,3-диметоксифеніл)-1-[2-(4-фторфеніл)етил]-4-піперидинметанолу, розладів
Формула / Реферат:
1. Спосіб лікування хворого з розладом сну шляхом введення хворому терапевтично ефективної кількості R-(+)--(2,3-диметоксифеніл)-1-[2-(4-фторфеніл)етил]-4-піперидинметанолу або його фармацевтично прийнятної солі.2. Спосіб за п. 1, в якому розладом сну є безсоння.3. Спосіб за п. 1, в якому розладом сну є обструктивне апное уві сні.4. Спосіб за п. 1, в...
Спосіб одержання [(1s)-[1a,2b,3b,4a(s)]]-4-[7-[[1-(3-хлор-2-тієніл)метил] пропіл]аміно]-3н-імідазо[4,5-b]піридин-3-іл]-n-етил-2,3-дигідроксициклопентанкарбоксаміду, способи одержання проміжних сполук для його одержання та проміжні сполуки

Номер патенту: 54479
Опубліковано: 17.03.2003
Автори: Ванасс Бенуа Дж., Цуей Чінг Т., Паунер Торі Х., О'Брайєн Майкл К., Шах Харшавадан К., Вальтер Френсіс Л., Леон Патрік, Гарсіа Ерве, Томпсон Майкл Д., Рейллі Лоренс
МПК: C07D 409/12, C07D 471/04, C07D 409/14, A61P 9/12, C07D 213/69, A61P 9/00, A61P 3/06
Мітки: способи, проміжні, спосіб, сполуки, проміжних, одержання, пропіл]аміно]-3н-імідазо[4,5-b]піридин-3-іл]-n-етил-2,3-дигідроксициклопентанкарбоксаміду, сполук, 1s)-[1a,2b,3b,4a(s)]]-4-[7-[[1-(3-хлор-2-тієніл)метил
Формула / Реферат:
1. Спосіб одержання [(1S)-[1a,2b,3b,4a(S*)]]-4-[7-[[1-(3-хлор-2-тієніл)метил]пропіл]аміно]-3H-імідазо[4,5-b]піридин-3-іл]-N-етил-2,3-дигідроксициклопентанкарбоксаміду, який включає взаємодію [(1S)-[1a,2b,3b,4a(S*)]]-4-[[3-аміно-4-[[1-(3-хлор-2-тієніл)метил]пропіл]аміно]-2-піридиніл]аміно]-N-етил-2,3-дигідроксициклопентанкарбоксаміду зі складним ефіром ортоформіату, ацетатом формамідину або диметилацеталем диметилформаміду.2. Спосіб за...
Спосіб хірургічного лікування злоякісних периампулярних новоутворень

Номер патенту: 59729
Опубліковано: 25.05.2011
Автори: Хомяк Ігор Васильович, Зелінський Артем Ігорович, Копчак Володимир Михайлович, Перерва Людмила Олександрівна, Дувалко Олександр Васильович, Копчак Костянтин Володимирович
МПК: A61B 17/00
Мітки: хірургічного, злоякісних, периампулярних, новоутворень, спосіб, лікування
Формула / Реферат:
Спосіб хірургічного лікування злоякісних периампулярних новоутворень, який включає виконання панкреатодуоденальної резекції з мобілізацією дванадцятипалої кишки та видаленням панкреатодуоденального комплексу, який відрізняється тим, що спочатку пересікають всі венозні судини панкреатодуоденального комплексу, а потім мобілізують дванадцятипалу кишку в напрямку від брижових судин направо, як останній етап видалення панкреатодуоденального...
Спосіб комплексного лікування злоякісних ііі-іv а стадій новоутворень вульви

Номер патенту: 34931
Опубліковано: 26.08.2008
Автор: Ткачук Тетяна Євгенівна
МПК: A61P 35/00
Мітки: злоякісних, лікування, стадій, спосіб, новоутворень, вульви, iii-iv, комплексного
Формула / Реферат:
1. Спосіб комплексного лікування злоякісних III-IV А стадій новоутворень вульви, що включає неоад'ювантну поліхіміотерапію, хірургічне втручання та променеву терапію, який відрізняється тим, що хірургічне втручання виконують у звуженому обсязі розширеної вульвектомії, а етап неоад'ювантної поліхіміотерапії здійснюють при лімфотропному дозованому введенні метатрексату, циклофосфану, 5-фторурацилу та блеоцину, при цьому 5-фторурацил вводять...
Попередній патент: Нейтроночутливий p-i-n діод з керованою чутливістю
Наступний патент: Спосіб одержання аналогу рибної ікри
Випадковий патент: Захисна композиція з 6-(тризаміщених феніл)-4-аміно-2-піридинкарбоксилатних гербіцидів і клоквінтоцету для злакових культур