Покращена фармацевтична композиція, що містить антагоніст кальцієвих каналів – лерканідипін, спосіб її одержання
Номер патенту: 99136
Опубліковано: 25.07.2012
Автори: Коутріс Макіс, Іліопоулоу Атіна, Матсінгоу Крістіна, Самара Вікі, Каравас Євангелос








Формула / Реферат
1. Фармацевтична композиція для перорального введення, що містить лерканідипін або його фармацевтично прийнятну сіль та поліморф, як активний інгредієнт, та колоїдний двоокис кремнію, а саме Аеросил™, від 5 до 25 % за масою, для підвищення біологічної засвоюваності та/або покращення розчинності.
2. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить від 7 до 20 % за масою колоїдного двоокису кремнію.
3. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить від 3 до 25 % за масою лерканідипіну або його фармацевтично прийнятної солі та поліморфу, переважно від 5 до 10 % за масою, більш переважно приблизно 8 % за масою.
4. Фармацевтична композиція за п. 1, яка відрізняється тим, що масове співвідношення лерканідипіну або його фармацевтично прийнятної солі та поліморфу та колоїдного двоокису кремнію становить від 10/1 до 1/60.
5. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить зв'язуючу речовину, переважно полівінілпіролідон (ПВП), де вказана зв'язуюча речовина наявна у кількості від 5 до 20 % за масою, переважно у кількості до приблизно 15 % за масою.
6. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить агент розпаду, переважно крохмаль гліколяту натрію, де вказаний агент розпаду присутній у кількості від 5 до 15 % за масою, переважно приблизно 10 % за масою.
7. Фармацевтична композиція за п. 1, яка відрізняється тим, що вона містить зволожувач, переважно співполімер поліоксіетилену та поліоксипропілену (полоксамер), де вказаний зволожувач присутній у кількостях аж до 5 % за масою, переважно приблизно 2,5 % за масою.
8. Фармацевтична композиція за п. 1, яка відрізняється тим, що вона містить розріджувач, переважно мікрокристалічну целюлозу та/або лактозу, більш переважно у моногідратній формі, де вказаний розріджувач присутній у кількості від 40 до 65 % за масою, переважно від 45 до 60 % за масою.
9. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить внутрішню фазу та зовнішню фазу.
10. Фармацевтична композиція за п. 13, яка відрізняється тим, що вказана зовнішня фаза містить та переважно складається зі стеарату магнію.
11. Фармацевтична композиція за п. 1, яка відрізняється тим, що лерканідипін знаходиться у гідрохлоридній формі, переважно кристалічній.
12. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить лерканідипіну гідрохлорид, моногідрат лактози, мікрокристалічну целюлозу, крохмаль гліколяту натрію, полівінілпіролідон, колоїдний двоокис кремнію, співполімер поліоксіетилен-поліоксипропілену, стеарат магнію.
13. Фармацевтична композиція за п. 1, яка відрізняється тим, що вона знаходиться у твердій лікарській формі, такій як таблетка, капсула чи саше.
14. Фармацевтична композиція за п. 13, яка відрізняється тим, що кожна тверда лікарська форма містить від 7 до 9 мг чи від 14 до 18 мг лерканідипіну гідрохлориду, переважно приблизно 8 мг чи приблизно 16 мг лерканідипіну гідрохлориду.
15. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить приблизно 16 мг лерканідипіну гідрохлориду, приблизно 80 мг моногідрату лактози, приблизно 16 мг мікрокристалічної целюлози, приблизно 20 мг крохмалю гліколяту натрію, приблизно 30 мг полівінілпіролідону, приблизно 31 мг колоїдного двоокису кремнію, приблизно 5 мг співполімеру поліоксіетилен-поліоксипропілену, приблизно 2 мг стеарату магнію.
16. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить приблизно 8 мг лерканідипіну гідрохлориду, приблизно 45 мг моногідрату лактози, приблизно 8 мг мікрокристалічної целюлози, приблизно 10 мг крохмалю гліколяту натрію, приблизно 10 мг полівінілпіролідону, приблизно 15,50 мг колоїдного двоокису кремнію, приблизно 2,50 мг співполімеру поліоксіетилен-поліоксипропілену, приблизно 1 мг стеарату магнію.
17. Застосування композиції за будь-яким з попередніх пунктів для збільшення, переважно на 15-25 %, біологічної засвоюваності лерканідипіну, переважно лерканідипіну гідрохлориду.
18. Спосіб одержання твердої лікарської форми для перорального введення, вибраної з таблетки, капсули чи саше, що містить лерканідипін або його фармацевтично прийнятну сіль як активний інгредієнт та ефективну кількість колоїдного двоокису кремнію за п. 1 для підвищення біологічної засвоюваності, який включає:
- розчинення загальної кількості вказаного активного інгредієнта, частини загальної кількості вказаного колоїдного двоокису кремнію та зв'язуючої речовини у розчиннику вода/EtOH;
- додавання до утвореного розчину решти частини колоїдного двоокису кремнію та, необов'язкового, ексципієнта, такого як розріджувач, зв'язуюча речовина, агент розпаду, глідант, змазка, та проведення вологого гранулювання;
- розчинення зволожуючого агента, такого як полоксамер, у невеликій кількості розчинника вода/EtOH та змішування з першим розчином до одержання однорідної маси;
- висушування зволоженої маси;
- просіювання висушеної маси та додавання до просіяної суміші загальної кількості щонайменше одного необов'язкового ексципієнта, такого як зв'язуюча речовина, розріджувач, агент розпаду, змазка та/або глідант, та змішування до досягнення однорідності; та
- формування одержаної суміші у тверду лікарську форму шляхом її пресування у бажану форму таблетки або шляхом наповнення капсул чи саше.
19. Тверда лікарська форма, яку одержують способом за п. 18 та яка містить від 5 до 25 % за масою колоїдного двоокису кремнію.
Текст
Реферат: Даний винахід належить до композицій твердих форм дозування, що містять терапевтично ефективну кількість антагоніста кальцієвих каналів - похідного дигідропіридину, такого як лерканідипін чи його сіль, у комбінації з колоїдним двооксидом кремнію, таким як Аеросил™, для підвищення біологічної засвоюваності та покращення стійкості, та до способу їх одержання шляхом вологої грануляції. UA 99136 C2 (12) UA 99136 C2 UA 99136 C2 5 10 15 20 25 30 35 40 45 50 55 Галузь винаходу Даний винахід відноситься до покращених форм дозування, таких, як таблетки та капсули, зокрема, до композиції для перорального введення, що має підвищену біологічну засвоюваність, яка містить терапевтично ефективну кількість антагоніста кальцієвих каналів похідного дигідропіридину, та більш конкретно, лерканідипіну чи його солі, похідного та поліморфу, та спосіб її одержання. Передумови створення винаходу Антагоніст кальцієвих каналів - сполуки дигідропіридину, таких, як амлодипін, ніфедипін, лацидипін та лерканідипін, є сполуками, про які відомо, що вони надзвичайно корисні, серед іншого, для лікування гіпертонії та коронарної хвороби серця. Лерканідипін (метил 1,1-N-триметил-N-(3, 3-дифенілпропіл)-аміноетил-1, 4-дигідро-6диметил(3-нітрофеніл)пирідин-3,5-дикарбоксилат) є сильно ліпофільним антагоністом кальцієвих каналів - похідним дигідропіридину довготривалої дії, що має високу судинну селективність. Його зазвичай застосовують у дозі 10 мг - 20 мг один раз на день (що продають в Італії під торгівельною маркою Занедіп®), максимальна доза становить приблизно 30 мг на день. Лерканідипін швидко всмоктується після перорального введення та пікові рівні у плазмі наявні через 1,5-3 години після приймання дози, проте він має швидкий пресистемний метаболізм. Антагоністи кальцієвих каналів - похідні дигідропіридину – мають низку розчинність у воді, що призводить до низької біологічної засвоюваності активного інгредієнта. Лікарські препарати з низькою розчинністю у воді (під якою мають на увазі розчинність, що становить менш ніж 0,1 мас. відсотків у воді при 20 °C) викликають додаткові проблеми при складанні композицій через їх низьку швидкість та низьку розчинність у водних середовищах, включаючи шлунково-кишкові рідини, що призводить до низького поглинання у великому колі кровообігу після перорального всмоктування. Для одержання композиції, що містить такий лікарський препарат, що дозволить максимальне поглинання з шлунково-кишкового тракту, необхідно включити до композиції ознаку, що підвищить розчинність лікарського препарату таким чином, щоб він розчинявся у шлунково-кишкових рідинах. Вже відомі різні способи промислового одержання лікарських форм для перорального застосування, що містять антагоністи кальцієвих каналів - похідні дигідропіридину, та зокрема лерканідипін чи його фармацевтично прийнятні солі як активний інгредієнт, оскільки він має корисні терапевтичні властивості. Проте, з рівня техніки відомо про істотні труднощі при одержанні твердих композицій для перорального застосування, що мали б бажану біологічну засвоюваність, через низьку розчинність вказаного активного інгредієнта. Відомо, що активні сполуки в аморфній формі часто мають більш високу біологічну засвоюваність, ніж відповідні кристалічні активні сполуки. У DE-A-3 024 858 описано форму дозування, що містить нікрадипін, низькорозчинний дигідропирідин, що застосовують в аморфній формі для підвищення розчиненності та поглинання. Аморфні активні інгредієнти зазвичай мають бути обережно включені до композиції, оскільки вони мають тенденцію до рекристалізації, що призводить до неможливості відтворення біологічної засвоюваніості, чи її значного зменшення через певні періоди зберігання, що викликано розпадом продуктів. У EP 0 385 582 описано композицію ніфедипіну, що має розмір частинок менш ніж 100 мікрон. Хоча контроль за розчиненням ніфедипіну здійснюють шляхом обробки речовини для одержання великої площі питомої поверхні, невеликі кристали активного інгредієнта мають тенденцію до агломерації та перетворення до більших розмірів частинок. У EP 0 557 244 описано композицію, що місить ніфедипін, що був мікронізований до невеликих кристалів для підвищення розчинності, разом із гідрофільним гель-утворюючим полімером для уповільнення та контролю швидкості розчинення та поглинання. Проте, найменший розмір, до якого може бути мікронізований ніфедипін за допомогою традиційного обладнання, становить приблизно 1 мікрон, та цей розмір частинок все ще не настільки малий, щоб дозволити повне розчинення та поглинання ніфедипіну. Додатково, при відсутності ретельного контролю однакового розміру кристалів у кожній партії таблеток, характеристики вивільнення можуть біти різними у кожній партії. У GB 1 456 618 описано покращення розчинення та поглинання ніфедипіну шляхом одержання твердого розчину ніфедипіну у полієтиленгліколі у присутності поверхнево-активної речовини. У EP 0 448 091 описано дигідропирідин із поверхнево-активною речовиною, проте великі кількості поверхнево-активних речовин зазвичай викликають подразнення шлунку пацієнтів. 1 UA 99136 C2 5 10 15 20 25 30 35 40 45 50 55 Додатково, застосування поверхнево-активних речовин, солюбілізаторів та деяких ексцепієнтів, що мають певну поверхню, часто призводить до одержання форм введення, в яких продукти є небажано великими. Для полегшення всмоктування, такі таблетки чи капсули часто перетворюють на конкретні форми, такі, як, наприклад, еліпсоїдні чи продольні форми, проте це також вже не призводить до позитивних результатів для продуктів з масою більш ніж 400 мг. Більш часте приймання продуктів меншого розміру також не є задовільним рішенням. Хоча кожен з вищенаведених патентних документів являє собою спробу вирішення проблем нестійкості, пов'язаних із фармацевтичними композиціями, що містять антагоністи кальцієвих каналів - похідні дигідропіридину, все ще існує потреба у покращенні біологічної засвоюваності таких активних інгредієнтів та, зокрема, у наданні їх біологічній засвоюваності незалежності від умов годування/голоду. Стислий опис винаходу Тому метою даного винаходу є забезпечення покращеної твердої форми дозування для перорального введення, що містить антагоніст кальцієвих каналів - похідне дигідропіридину, зокрема лерканідипін чи його сіль, як активний інгредієнт, та в якій відсутні недоліки, відомі з рівня техніки, що підвищує біологічну засвоюваність активного інгредієнта та надає йому незалежність від умов годування/голодування. Іншим аспектом даного винаходу є забезпечення твердої форми дозування для перорального введення, що містить антагоніст кальцієвих каналів - похідне дигідропіридину, зокрема лерканідипін чи його сіль, як активний інгредієнт, що є біологічно засвоюваним та ефективним та має достатній термін існування, добрі фармакотехнічні властивості, підвищену сумісність із пацієнтом та зменшену кількість можливих бічних ефектів. Додатково, іншим аспектом даного винаходу є забезпечення твердої форми дозування для перорального введення, що містить антагоніст кальцієвих каналів - похідне дигідропіридину, зокрема лерканідипін чи його сіль, як активний інгредієнт, яка може бути одержана у формах дозування різної концентрації шляхом пропорційного регулювання кількості ексципієнтів та активного інгредієнта, таким чином забезпечуючи фармакотехнічну лінійність, без впливу на профіль розчинення та біологічну засвоюваність активного інгредієнта. Додатковим аспектом даного винаходу є забезпечення способу забезпечення стійкої твердої лікарської форми для перорального введення, що містить антагоніст кальцієвих каналів похідне дигідропіридину, зокрема лерканідипін чи його сіль, як активний інгредієнт, таким чином, підвищуючи біологічну засвоюваність активного інгредієнта, залишаючись стійкою протягом тривалого періоду та покращюучи фармакотехнічні характеристики композиції. Відповідно до вищезазначених цілей даного винаходу, забезпечено фармацевтичну композицію для перорального введення, що містить антагоніст кальцієвих каналів - похідне дигідропіридину, зокрема лерканідипін чи його фармацевтично прийнятну сіль, похідне та поліморф як активний інгредієнт, та ефективну кількість колоїдного двооксиду кремнію, такого, як аеросил, як агента, що підвищує біологічну засвоюваність. Зокрема, переважний об'єкт даного винаходу представлений фармацевтичною композицією для перорального введення, що містить лерканідипін чи його фармацевтично прийнятну сіль, похідне та поліморф, та від 5 % до 25 %, переважно від 7 % до 20 % за масою, колоїдного двооксиду кремнію. Як буде очевидним з опису та прикладів, композиції за даним винаходом забезпечують біологічну засвоюваність активного начала, що на приблизно 15-25 % вища за біологічну засвоюваність, яку досягають за допомогою композицій, що на даний час доступні на ринку; таким чином, у конкретному випадку лерканідипіну, можливо одержати біологічну засвоюваність, еквівалентну біологічній засвоюваності Занедіпу® 10 мг та Занедіпу® 20 мг кількістю лерканідипін HCl, що становить від приблизно 8 мг до 16 мг, відповідно. Додатково, композиція за даним винаходом має знижену залежність біологічної засвоюваності від умов годування/голодування чи така залежність в неї відсутня. Іншими втіленнями даного винаходу є, таким чином, застосування композицій за даним винаходом для збільшення, переважно на приблизно 15-25 % біологічної засвоюваності лерканідипіну HCl та/або для зменшення та/або усунення їх залежності від того, чи була композиція введена пацієнту за умов голодування чи за умов годування. Відповідно до іншого втілення даного винаходу, забезпечено спосіб одержання твердих форм дозування для перорального введення, таких, як таблетки, капсули та саше, що містять антагоніст кальцієвих каналів - похідне дигідропіридину, зокрема лерканідипін чи його фармацевтично прийнятну сіль, похідне та поліморф, як активний інгредієнт, який включає: 2 UA 99136 C2 5 10 15 20 25 30 35 40 45 50 55 - розчинення усієї кількості вказаного активного інгредієнта, частини від усієї кількості колоїдного двооксиду кремнію для підвищення біологічної засвоюваності та необов'язково зв'язуючої речовини у розчиннику вода/EtOH; - додавання до утвореного розчину частини колоїдного двооксиду кремнію, що залишилася, та необов'язкового ексципієнта, такого, як розріджувач, зв'язуюча речовина, агент розпаду, глідант, зволожувач, та проведення вологого гранулювання; - розчинення зволожуючого агента у невеликій кількості розчинника вода/EtOH та перемішування з першим розчином; - висушування зволоженої маси; - просіювання висушеної маси та додавання до просіяної суміші загальної кількості щонайменше одного необов'язкового ексципієнта, такого, як зв'язуюча речовина, зволожувач, розріджувач, агент розпаду, зволожувач та/або глідант та змішування до досягнення однорідності, та - складання одержаної в результаті суміші у тверду форму дозування або шляхом її пресування у бажану форму таблетки, або шляхом наповнення капсул чи саше. Додаткові переважні втілення за даним винаходом визначені у залежних пунктах формули 2-16. Інші об'єкти та переваги даного винаходу будуть очевидними фахівцям у цій галузі, приймаючи до уваги наступний детальний опис. Стислий опис креслень На Фіг. 1 та 2 показано середні значення у плазмі для композиції Прикладу 1 відповідно до даного винаходу. На Фіг. 3 та 4 показано середні значення у плазмі для композиції Прикладу 3 відповідно до даного винаходу. На Фіг. 5, 6, та 7 показано спектри порошкової рентгенівської кристалографії аморфного лерканідипіну HCl, плацебо композиції Прикладу 3, композиції Прикладу 3 відповідно до даного винаходу. На Фіг. 8 показано спектри скануючої електронної мікроскопії аморфного лерканідипіну HCl. На Фіг. 9 показано спектри скануючої електронної мікроскопії тонкодисперсного аморфного лерканідипіну HCl та колоїдного двооксиду кремнію відповідно до даного винаходу. На Фіг. 10 показано профіль розчинення композиції Прикладу 4 (що містить 16 мг лерканідипіну HCl) порівняно із Занедіпом® (що містить 20 мг лерканідипіну HCl). На Фіг. 11 показано профіль розчинення композиції Прикладу 5 (що містить 8 мг лерканідипіну HCl) порівняно із Занедіпом® (що містить 10 мг лерканідипіну HCl). На Фіг. 12 та 13 показано спектри порошкової рентгенівської кристалографії композицій Прикладу 4 та 5, відповідно. На Фіг. 14 та 15 показано профілі розчинення за симульованих умов годування/голодування композиції Прикладу 4 та Занедіпу® 20 мг, відповідно. Детальний опис винаходу Для цілей даного винаходу, фармацевтичну композицію, що містить низькорозчинний активний інгредієнт (антагоніст кальцієвих каналів - похідне дигідропіридину наприклад, лерканідипін чи його фармацевтично прийнятну сіль), вважають "стійкою", якщо вказаний інгредієнт розпадається більш чи менш повільно, ніж взятий окремо та/або у відомих фармацевтичних композиціях. Додатково, її вважають такою, що надає біологічну засвоюваність активному інгредієнту незалежно від умов годування/голодування, коли вона забезпечує істотно аналогічну біологічну засвоюваність активного інгредієнта незалежно від того, чи вводять її пацієнту натщесерце чи при годуванні. Активний інгредієнт (антагоніст кальцієвих каналів - похідне дигідропіридину, зокрема лерканідипін чи його фармацевтично прийнятна сіль), який містить форма дозування, має "підвищену біологічну засвоюваність", навіть при введенні у формі дозування, та вивільняється з форми дозування, поглинається та після цього досягає більш високих рівнів концентрації у плазмі, ніж продукти, наявні на ринку, що містять аналогічну кількість аналогічного активного інгредієнта та призначені для аналогічного застосування. Хоча фармацевтична композиція може існувати у різних формах, переважними твердими формами є таблетки, капсули та таблетки у вигляді капсул. Несподівано було знайдено, що мету даного винаходу досягають шляхом застосування колоїдного двооксиду кремнію, такого, як Аеросил™ для підвищення біологічної засвоюваності активного інгредієнта. 3 UA 99136 C2 5 10 15 20 25 30 35 40 45 50 55 Як вже було зазначено, антагоністи кальцієвих каналів - похідні дигідропіридину, такі, як лерканідипін HCl, мають низьку розчинність, що впливає на погану біологічну засвоюваність активної речовини. Колоїдний двоокис кремнію є субмікроскопічним колоїдним двоокисом кремнію з розміром часток, що становить приблизно 15 нм. Це легкий, незв'язаний, блакитно-білий, незернистий аморфний порошок, що не має запаху та смаку. Колоїдний двооксид кремнію широко застосовують у фармацевтиці. Його невеликі за розміром частинки та велика питома поверхня надають бажані характеристики текучості, які застосовують для покращення властивостей текучості сухих порошків. Коли колоїдний двооксид кремнію включають у фармацевтичну композицію відповідно до даного винаходу, утворюється тонка дисперсія аморфних часток активного інгредієнта на поверхні колоїдного двооксиду кремнію, що призводить до утворення однофазної системи. Вказана однофазна система покращує розчинність активного інгредієнта. Активний інгредієнт (антагоніст кальцієвих каналів - похідне дигідропіридину, таке, як лерканідипін чи його сіль, похідне та поліморф) та придатну кількість колоїдного двооксиду кремнію, такого, як Аеросил™, розчиняють у розчиннику для утворення тонкої дисперсії, а потім змішують із зв'язуючою речовиною. Частину колоїдного двооксиду кремнію, що залишилася, та необов'язковий ексципієнт додають до розчину та проводять вологе гранулювання. Після висушування зволоженої маси та просіювання висушеної маси, додають будь-який необов'язковий додатковий ексципієнт. Після цього композицію перемішують до досягнення однорідності. Утворена в результаті композиція може бути потім спресована. Додатково, до вказаної вище композиції необов'язково може бути доданий будь-який ексципієнт, за умови, що він сумісний з активним інгредієнтом композиції, для усунення проблем, пов'язаних із низькою текучістю та несприятливими фармакотехнічними характеристиками цих речовин, та для підвищення розчинності лікарської речовини та терміну напіввивідення лікарського продукту, та забезпечення продукту, що проявляє відмінну біологічну засвоюваність. Даний винахід може бути застосований у композиції таблеток, капсул, таблеток у вигляді капсул, саше чи інших твердих форм дозування для перорального чи сублінгвального введення активного інгредієнта, що має проблеми із розчинністю та біологічною засвоюваністю. Додатково, можливо одержати форми дозування різної концентрації із застосуванням відповідної кількості тієї ж самої композиції, таким чином, обмежуючи витрати на виробництво та мінімізуючи кількість, та відповідно витрати на, проведення клінічних стадій, необхідних для схвалення продукту органами влади. Спосіб одержання препарату відповідно до даного винаходу є більш простим та дешевим, ніж будь-який інший традиційний спосіб. Тому, у першому втіленні, даний винахід забезпечує фармацевтичну композицію, що містить від приблизно 0,5 % до 30 % за масою лерканідипіну чи його солі та від приблизно 3 % до 30 % за масою колоїдного двооксиду кремнію. Масове співвідношення лерканідипіну чи його солі та колоїдного двооксиду кремнію становить переважно 10:1-1:60. Більш переважні фармацевтичні композиції відповідно до даного винаходу містять приблизно 3 % - 30 %, більш переважно 5 % - 25 % та найбільш переважно 7 % - 20 % за масою колоїдного двооксиду кремнію, такого, як Аеросил™. Такі переважні композиції містять від 3 % до 25 % за масою лерканідипіну чи його фармацевтично прийнятної солі, похідного та поліморфу, переважно від 5 % до 10 % за масою, більш переважно приблизно 8 % за масою. Зв'язуюча речовина, якщо вона наявна, може бути загалом присутня у кількості від 5 % до 20 % за масою, переважно у кількостях аж до приблизно 15 % за масою; зволожувач, якщо він присутній, може бути загалом присутній у кількості аж до приблизно 5 % за масою, переважно приблизно 2,5 % за масою. Фармацевтичні композиції за даним винаходом стандартно містять розріджувач, що може бути присутній у кількостях від 40 % до 65 % за масою, переважно від 45 % до 60 % за масою. Такі композиції можуть також містити агент розпаду, що переважно присутній у кількостях від 5 % до 15 % за масою, більш переважно приблизно 10 % за масою. Відповідно до іншого втілення, фармацевтична композиція за даним винаходом містить внутрішню фазу та зовнішню фазу; відповідно до переважного втілення, вказана зовнішня фаза містить, та переважно складається зі стеарату магнію. Переважні фармацевтичні композиції наявні у формі твердих форм дозування для перорального чи сублінгвального введення, таких, як таблетки, капсули, таблетки у вигляді 4 UA 99136 C2 5 10 15 20 25 30 35 40 45 50 55 капсул, троше, пастилок, пігулок, лепешок та інш., усіх розмірів та форм, покритих чи непокритих. Усі відсоткові значення, наведені у цій заявці, є масовими відсотками, виходячи із загальної маси композиції, якщо не вказано інше. Іншим втіленням даного винаходу є застосування способу вологої грануляції для одержання твердих форм дозування для перорального введення, таких, як таблетки, капсули та саше, що містять лерканідипін чи його сіль, похідне та поліморф. Вказаний спосіб вологої грануляції включає: - розчинення усієї кількості антагоністу кальцієвих каналів - похідного дигідропіридину, зокрема лерканідипіну чи його фармацевтично прийнятної солі як активного інгредієнта, частини від усієї кількості колоїдного двооксиду кремнію (переважно від 40 до 60 % від загальної маси) для підвищення біологічної засвоюваності та необов'язково зв'язуючої речовини у розчиннику вода/EtOH; - додавання до утвореного розчину частини колоїдного двооксиду кремнію, що залишилася, та необов'язкового ексципієнта, такого, як розріджувач, зв'язуючої речовини, агента розпаду, гліданта, зволожувача та проведення вологого гранулювання; - розчинення зволожуючого агента у невеликій кількості розчинника вода/EtOH та перемішування з першим розчином; - висушування зволоженої маси; - просіювання висушеної маси та додавання до просіяної суміші загальної кількості щонайменше одного необов'язкового ексципієнта, такого, як зв'язуюча речовина, зволожувач, розріджувач, агент розпаду, зволожувач та/або глідант та змішування до досягнення однорідності, та - складання одержаної в результаті суміші у тверду форму дозування або шляхом її пресування у бажану форму таблетки, або шляхом наповнення капсул чи саше. Фармацевтичні композиції за даним винаходом можуть також містити один чи більше додаткових інгредієнтів композиції, вибраних з широкого різноманіття ексципієнтів. Відповідно до бажаних властивостей композиції, може бути вибрана будь-яка кількість інгредієнтів, окремо чи у комбінації, виходячи з їх відомого застосування при одержанні композицій у твердих формах дозування. Такі інгредієнти включають, не обмежуючись наведеним, розріджувачі, зв'язуючі речовини, речовини, що полегшують пресування, агенти розпаду, поверхнево-активні речовини, зволожувачі, антиоксиданти, гліданти, змащувачі, ароматизатори, поглиначі води, барвники, підсолоджувачі, покривні агенти та консерванти. Необов'язкові ексципієнти мають бути сумісними із антагоністом кальцієвих каналів похідним дигідропіридину чи його сіллю, таким чином, щоб не перешкоджати його дії у композиції. Розріджувачами можуть бути, наприклад, карбонат кальцію, двохосновний фосфат кальцію, трьохосновний фосфат кальцію, сульфат кальцію, мікрокристалічна целюлоза, мікрокристалічна силікатована целюлоза, порошок целюлози, декстрати, декстроза, фруктоза, лактітол, безводна лактоза, моногідрат лактози, дигідрат лактози, тригідрат лактози, манітол, сорбітол, крохмаль, пептизований крохмаль, сахароза, тальк, ксиліт, мальтоза, мальтодекстрин, мальтітол. Зв'язуючими речовинами можуть бути, наприклад, гуміарабік, альгінова кислота, карбомер, кальцій карбоксиметилцелюлоза, натрій карбоксиметилцелюлоза, мікрокристалічна целюлоза, порошок целюлози, етилцелюлоза, желатин, рідка глюкоза, гуарова камедь, гідроксиетилцелюлоза, гідроксипропіл целюлоза, гідроксипропілметил целюлоза, мальтодекстрин, метилцелюлоза, полідекстроза, поліетиленоксид, повідон, альгінат натрію, крохмаль, пептизований крохмаль, сахароза. Агентами розпаду можуть бути, наприклад, альгінова кислота, двооксид вуглецю, кальцій карбоксиметилцелюлоза, натрій карбоксиметилцелюлоза, мікрокристалічна целюлоза, порошок целюлози, натрій кроскармелоза, кросповідон, натрій докузат, гуарова камедь, гідроксипропілцелюлоза, метилцелюлоза, калій полакрилін, полоксамер, повідон, альгінат натрію, натрій гліцин карбонат, натрій лаурил сульфат, натрій крохмаль гліколат, крохмаль, пептизований крохмаль. Зволожувачами можуть бути сополімери поліоксиетилен-поліоксипропілену та блоксополімери, комерційно доступні як Плюронік™ чи Полоксамер™, етоксильовані холестерини, комерційно доступні як Солюлан™ вітамінні похідні, наприклад, похідні вітаміну E, такі, як токоферол поліетиленгліколь сукцинат (TPGS), натрій додецилсульфат чи натрій 5 UA 99136 C2 5 10 15 20 лаурилсульфат; жовчна кислота чи її сіль, наприклад холева кислота, гліколева кислота чи її сіль. Глідантами можуть бути наприклад, силікат кальцію, порошок целюлози, крохмаль, тальк, колоїдний двооксид кремнію. Змащувачами можуть бути, наприклад, поліетиленгліколь 4000, поліетиленгліколь 6000, натрій лаурил сульфат, крохмаль, тальк, стеарат магнію, гліцерил бегенат, гідрогенізована касторова олія, стеаринова кислота, гліцерил пальмітостеарат, гліцерил моностеарат, натрій стеарил фумарат. Відповідно до іншого втілення, фармацевтична композиція за даним винаходом знаходиться у твердій формі дозування та кожна з форм містить від 7 до 9 мг чи від 14 до 18 мг лерканідипін гідрохлориду, переважно приблизно 8 мг чи приблизно 16 мг лерканідипін гідрохлориду. Зокрема, відповідно до особливо переважних втілень, кожна з форм може містити: приблизно 16 мг лерканідипін гідрохлориду, приблизно 80 мг моногідрату лактози, приблизно 16 мг мікрокристалічної целюлози, приблизно 20 мг натрій крохмаль гліколату, приблизно 30 мг полівінілпіролідону, приблизно 31 мг колоїдного двооксиду кремнію, приблизно 5 мг сополімеру поліоксиетилен-поліоксипропілену, приблизно 2 мг стеарату магнію; чи приблизно 8 мг лерканідипін гідрохлориду, приблизно 45 мг моногідрату лактози, приблизно 8 мг мікрокристалічної целюлози, приблизно 10 мг натрій крохмаль гліколату, приблизно 10 мг полівінілпіролідону, приблизно 15,50 мг колоїдного двооксиду кремнію, приблизно 2,50 мг сополімеру поліоксиетилен-поліоксипропілен, приблизно 1 мг стеарату магнію. Наступні приклади ілюструють переважні втілення відповідно до даного винаходу, не обмежуючи обсяг чи суть даного винаходу: Приклади Приклад 1: Таблетка 20 мг лерканідипіну (Композиція 1) 25 Інгредієнти Внутрішня фаза Лерканідипін НСl Мікроцеллак Мікрокристалічна целюлоза Крохмаль 1500 Прімойєл НРС Очищена вода ЕtOН Зовнішня фаза Стеарат магнію 30 35 % 20 мг таблетка 10,00 40,00 25,00 17,00 2,00 5,00 20,00 80,00 50,00 34,00 4,00 10,00 30,00 24,00 1,00 Загальна маса 2,00 200,00 Таблетки вищевказаного складу були одержані відповідно до наступного способу одержання: HPC розчиняли у розчиннику вода/EtOH. Лерканідипін HCl змішували із мікроцеллаком, мікрокристалічною целюлозою, крохмалем 1500 та прімойєлом з утворенням гомогенної суміші. Вищезазначене суміш змішували із розчином HPC. Гранульовану масу висушували. Остаточно до висушеної гранули додавали стеарат магнію та змішували до повної гомогенності. Утворену у результаті гранулу пресували у таблетки. Одержані таблетки аналізували на твердість, крихкість, розпад та вміст води. Усі аналізи проводили відповідно до European Pharmacopoeia 5.1 та вони відповідали специфікаціям. Приклад 2: Таблетка 20 мг лерканідипіну (Композиція 2) Інгредієнти Внутрішня фаза Лерканідипін HCl Моногідрат лактози Мікрокристалічна целюлоза Крохмаль1500 Прімойєл Твін 20 Очищена вода EtOH % 10,00 35,00 22,50 15,00 8,00 1,00 6 20 мг таблетка 20,00 70,00 45,00 30,00 16,00 2,00 45,00 19,80 UA 99136 C2 Зовнішня фаза Мікрокристалічна целюлоза Стеарат магнію 5 10 20 % 20 мг таблетка 10,00 48,00 8,00 10,00 5,00 15,50 2,50 20,00 96,00 16,00 20,00 10,00 31,00 5,00 43,30 34,70 1,00 Загальна маса 2,00 200,00 Таблетки вищевказаного складу були одержані відповідно до наступного способу одержання: лерканідипін HCl та половину кількості Аеросилу™ розчиняли/диспергували у розчиннику вода/EtOH та після цього додавали ПВП. Моногідрат лактози та частину Аеросилу™, що залишилася, перемішували, додавали до попереднього розчину та перемішували. Потім до вищезазначеного розчину додавали прімойєл. Полоксамер розчиняли у невеликій кількості розчинника вода/EtOH та перемішували із попереднім розчином. Гранульовану масу висушували та просіювали. Остаточно, до гранули додавали стеарат магнію та перемішували до досягнення повної гомогенності. Утворену у результаті гранулу пресували у таблетки. Приклад 4: Таблетка 16 мг лерканідипіну (Композиція 4) Інгредієнти Внутрішня фаза Лерканідипін HCl Моногідрат лактози Мікрокристалічна целюлоза Натрій крохмаль гліколат (Прімойєл) Полівінілпіролідон (повідон/коллідон K30) Колоїдний двооксид кремнію (Аеросил) Сополімер поліоксиетилену та поліоксипропілену(Полоксамер 407) Очищена вода EtOH Зовнішня фаза Стеарат магнію Усього 25 15,00 2,00 200,00 Таблетки вищевказаного складу були одержані відповідно до наступного способу одержання: Твін 20 розчиняли у 20мг води (розчин 1). Моногідрат лактози, лерканідипін HCl та половину кількості прімойєлу розчиняли у кількості води та EtOH, що залишилася, та перемішували (розчин 2). Розчини 1 та 2 поєднували та потім додавали суміш мікрокристалічної целюлози, крохмаль 1500 та кількість прімойєлу, що залишилася, та перемішували. Гранульовану масу висушували. До висушеної гранули додавали мікрокристалічну целюлозу та перемішували. Остаточно, до гранули додавали стеарат магнію та перемішували до досягнення повної гомогенності. Утворену у результаті гранулу пресували у таблетки. Приклад 3: Таблетка 20 мг лерканідипіну (Композиція 3) Інгредієнти Внутрішня фаза Лерканідипін HCl Моногідрат лактози Мікрокристалічна целюлоза Прімойєл ПВП Аеросил Полоксамер Очищена вода EtOH Зовнішня фаза Стеарат магнію 15 7,50 1,00 Загальна маса Приклад 5: Таблетка 8 мг лерканідипіну (Композиція 5) 7 % мг таблетка 8,00 40,00 8,00 10,00 15,00 15,50 2,50 16,00 80,00 16,00 20,00 30,00 31,00 5,00 94,50 40,00 1,00 100,00 2,00 200,00 UA 99136 C2 Інгредієнти Внутрішня фаза Лерканідипін HCl Моногідрат лактози Мікрокристалічна целюлоза Натрій крохмаль гліколат (Прімойєл) Полівінілпіролідон (повідон/коллідон K30) Колоїдний двооксид кремнію (Аеросил) Сополімер поліоксиетилену та поліоксипропілену(Полоксамер 407) Очищена вода EtOH Зовнішня фаза Стеарат магнію Усього 5 10 15 % мг таблетка 8,00 45,00 8,00 10,00 10,00 15,50 2,50 8,00 45,00 8,00 10,00 10,00 15,50 2,50 47,25 20,00 1,00 100,00 1,00 100,00 Таблетки композицій прикладів 4 та 5 одержували шляхом застосування способу, аналогічного способу, що наведений у прикладі 3. Вони істотно відрізняються, оскільки лерканідипін HCl знаходиться у кристалічній формі (а не в аморфній) та присутній у меншій кількості (тобто, 8 % за масою, а не 10 %); додатково, вони також мають менший вміст моногідрату лактози та більший вміст ПВП. Порівняльні дослідження Одним з найбільш критичних аналізів є аналіз розчинності, оскільки він значною мірою корелює із біологічною засвоюваністю продукту. Для способу розчинення застосовували прилад із лопатями II при 75об./хв., 37 °C ± 0,5 °C, протягом 30 хвилин, а як середовище розчинення застосовували буфер із pH=1,2. Результати швидкості розчинення для кожної проаналізованої композиції представлено у Таблиці 1. результати вказують на те, що усі три композиції розчинилися не повністю протягом приблизно 30 хвилин. Таблиця 1 Профіль розчинення композицій Прикладів 1, 2 та 3 Час (хв) 5 10 15 20 25 30 20 25 30 Композиція 1 12,25 25,99 35,94 42,08 47,91 55,29 Композиція 2 49,31 60,18 65,80 70,11 73,29 74,83 Композиція 3 19,96 31,33 69,07 80,69 88,01 90,73 Загальновідомою проблемою для фармацевтичних композицій, що містять низькорозчинні активні інгредієнти є те, що навіть якщо аналіз розчинності забезпечує задовільні результати, багато разів результати, отримані in vivo відрізняються від очікуваних. Для цього виду ліків, що мають погане поглинання (менш, ніж 10 %) через низьку розчинність та високий пресистемний метаболізм, аналіз розчинності не є настільки різним, тільки тому результати фармакокінетичних досліджень є репрезентативними у тому, що стосується композицій. Іншими цілями даного винаходу було одержання фармацевтичної композиція, що є стійкою, вказаний активний інгредієнт не розпадається та залишається в аморфній формі протягом тривалого періоду зберігання. Тому перед початком клінічних випробувань три композиції упаковували у ПВХ/PE/ПВДХ алюмінієві блістерні упаковки та піддавали стандартним (25 °C±2 °C/60 %±5 % RH), та прискореним (40 °C±2 °C/75 %±5 % RH) дослідженням стійкості відповідно до поточних інструкцій ICH. Результати досліджень стійкості через шість місяців відображено у таблиці, наведеній нижче. Результати вказують на те, що лерканідипін є більш стійким, коли у композицію включено колоїдний двооксид кремнію. Конкретні аналізи та результати описані у таблиці стійкості (ТАБЛИЦЯ 2). 8 UA 99136 C2 ТАБЛИЦЯ 2: Результати досліджень стійкості для композицій 1, 2 та 3 безпосередньо після одержання та через 6 місяців після зберігання за нормальних та прискорених умов. 0 МІСЯЦІВ ДОМІШКИ Домішки A NMT 0,15 % Домішки B NMT 0,15 % Домішки C NMT 0,15 % Домішки D NMT 0,50 % Невідомі NMT 0,20 % Усього NMT 1,2 % Комп. 1 НД НД 0,04 % 0,04 % НД 0,08 % Комп. 2 НД НД 0,05 % 0,04 % НД 0,09 % Домішки A NMT 0,15 % Домішки B NMT 0,15 % Домішки C NMT 0,15 % Домішки D NMT 0,50 % Невідомі NMT 0,20 % Усього NMT 1,2 % 6 МІСЯЦІВ Комп. 1 Комп. 2 25 °C±2 °C/60 %±5 % RH НД НД НД НД 0,04 % 0,06 % 0,04 % 0,04 % НД НД 0,08 % 0,10 % Домішки A NMT 0,15 % Домішки B NMT 0,15 % Домішки C NMT 0,15 % Домішки D NMT 0,50 % Невідомі NMT 0,20 % Усього NMT 1,2 % 40 °C±2 °C/75 %±5 % RH НД НД НД 0,06 % 0,18 % НД НД 0,24 % ДОМІШКИ 510 15 20 25 30 0,01 % 0,06 % 0,17 % Комп. 3 НД НД 0,03 % 0,04 % НД 0,07 % Комп. 3 НД НД 0,03 % 0,04 % НД 0,07 % НД НД 0,05 % 0,16 % НД 0,24 % 0,21 % Відповідно до іншого аспекту даного винаходу, активна речовина має залишатися в аморфному стані після пресування та не повинна перетворюватися на кристалічну форму. Як показано на Фіг. 5 за допомогою порошкової рентгенівської кристалографії лерканідипін є повністю аморфним, оскільки зареєстрований лише широкий пік, максимум якого розташовано при приблизно 2θ = 20 град. Кристалічні властивості залишаються також незмінними через шість місяців за аналогічних умов, коли суміш включають у фармацевтичну композицію з іншими ексципієнтами. Після одержання чи через 6 місяців зберігання піків, які б відповідали кристалічній формі лерканідипіну, не спостерігали, що вказує на стабілізацію суміші. Таблетки композиції Прикладу 3 мають основні піки, одержані при приблизно 2θ=12.7, 16.6, 19.2, 19.7, 20.2, 21.4, 23.0, 36.4, 37.8 градусів, які також були знайдені у таблетках плацебо (Фіг. 6 та 7). Рентгенограми порошкової рентгенівської кристалографії композицій 4 та 5 наведено на фігурах 12 та 13, відповідно. Біологічну засвоюваність та фармакокінетичний профіль усіх п'яти композицій за даним винаходом визначали у дослідженнях однократних доз "in vivo". Дослідження однократних доз проводили на 12 здорових добровольцях за допомогою композиції, одержаної з аморфного лерканідипіну HCL відповідно до Прикладів 1, 2 та 3. Реперною сполукою була таблетка 20мг лерканідипіну HCL (Carmen 20 мг), що складалася з активного інгредієнта, лактози, мікрокристалічної целюлози, натрій крохмаль гліколату, повідону, стеарату магнію та рожевого опадрай (Композиція B); тобто, таблетка має таку ж саму композицію, що й Занедіп® 20 мг. Кожен пацієнт одержував однократну пероральну дозу 20мг композиції 1 Прикладу 1 та таблетку Композиції B, що дорівнює 20 мг активного інгредієнта, у різні періоди часу. Проби крові були узяті у різні періоди часу та визначали концентрації лерканідипіну у плазмі. У фармакокінетичному аналізі композиції 1 відповідно до Прикладу 1, R-лерканідипін та Sлерканідипін вимірювали окремо (хіральний спосіб). У Таблиці 3 показано основні фармакокінетичні параметри, одержані в результаті аналізу. 9 UA 99136 C2 ТАБЛИЦЯ 3 Фармакокінетичний аналіз композиції 1 порівняно з реперним продуктом (B) lnAUC0-t (нг год./мл) R-Лерканідипін Співвідношення мінімальних середньоквадратичних помилок (спол.1/B)% 90 % C.I. (спол. 1/B)% Коефіцієнт варіації один відносно одного% S-лерканідипін Співвідношення мінімальних середньоквадратичних помилок спол. /B)% 90 % C.I. (спол. 1/B)% Коефіцієнт варіації один відносно одного% 5 10 15 20 50,1 Cмакс (нг/мл lnAUC0-inf (нг год./мл) 57,1 40,2-62,4 30,4 51,5 41,5-63,9 29,8 41,7-78,2 44,4 48,1 48,9 56,5 37,4-61,8 35,0 38,1-62,7 34,6 39,3-81,1 52,1 де: C макс = (пікова концентрація) є найвищою концентрацією, досягнутою ліками у плазмі після дозування; AUC0-t =(площа під кривою) є загальною площею під кривою час – концентрація у плазмі, від часу 0 до останньої вимірюваної концентрації, згідно до розрахунків за допомогою лінійного трапецоїдного способу; вона являє собою міру біологічної засвоюваності ліків. AUC0-inf =(площа під кривою) є загальною площею під кривою час – концентрація у плазмі від часу 0 до нескінченості. AUCinf розраховують як суму AUC 0-t плюс співвідношення останньої вимірюваної концентрації у плазмі та константи швидкості елімінування. Ці дані вказують на те, що властивості двох композицій можна порівнювати у тому, що стосується основних фармакокінетичних параметрів. Відповідно, було знайдено, що композиція 1 (20 мг таблетка) має відносну біологічну засвоюваність, що становить приблизно 50 % порівняно із таблеткою лерканідипіну HCl 20 мг, що наявна на ринку (Фіг. 1 та 2). Додатково, дослідження однократних доз було проведено на 12 здорових добровольцях у рандомізованому двофакторному перехресному дослідженні, де порівнювали форму дозування композиції Прикладу 2 із формою дозування композиції B. Проби плазми виділяли та аналізували на наявність лерканідипіну через проміжки часу. У фармакокінетичному аналізі композиції 2 відповідно до Прикладу 2, вимірювали рацемічну суміш лерканідипіну. У Таблиці 4 показано основні фармакокінетичні параметри, одержані з аналізу. ТАБЛИЦЯ 4 Фармакокінетичний аналіз композиції 2 порівняно з реперним продуктом (B) lnAUC0-t (нг год./мл) lnAUC0-inf (нг год./мл) Співвідношення мінімальних середньоквадратичних 84,0 помилок (спол. 2/B)% 90 % C.I. (спол. 2/B)% 69,0-102,0 Коефіцієнт варіації один відносно одного% 27,3 87,0 71,0-106,0 27,8 Cмакс (нг/мл) 125,0 105,0-149,0 24,1 25 30 Відповідно, було знайдено, що композиція 2 (20 мг таблетка) має відносну біологічну засвоюваність, що становить 84 %, порівняно із таблеткою лерканідипіну HCl 20 мг, що наявна на ринку. Додатково, дослідження однократних доз проводили на 72 здорових добровольцях у рандомізованому двофакторному перехресному дослідженні, де порівнювали форму дозування композиції Прикладу 3 з формою дозування композиції B. Проби плазми видаляли та аналізували на наявність лерканідипіну через проміжки часу. 10 UA 99136 C2 У фармакокінетичному аналізі композиції 3 відповідно до Прикладу 3, окремо вимірювали Rлерканідипін та S-лерканідипін (хиральний спосіб). У Таблиці 5 показано основні фармакокінетичні параметри, одержані з аналізу. ТАБЛИЦЯ 5 Фармакокінетичний аналіз композиції 3 порівняно з реперним продуктом (B) lnAUC0-t lnAUC0-inf (нг (нг год./мл) год./мл) R-Лерканідипін Співвідношення мінімальних середньоквадратичних помилок (спол. 3/B)% Cмакс (нг/мл) Коефіцієнт варіації один відносно одного% S-Лерканідипін Співвідношення мінімальних середньоквадратичних помилок (спол. 3/B)% 90 % C.I. (спол. 3/B)% Коефіцієнт варіації один відносно одного% 140,6 187,1 129,6157,7 34,5 128,0154,6 32,8 168,2208,1 37,5 144,4 142,5 181,1 131,8158,2 31,9 90 % C.I. (спол. 3/B)% 143,0 130,4155,7 30,7 164,0200,0 34,8 5 10 15 20 25 30 35 40 Відповідно, було знайдено, що композиція 3 (20 мг таблетка) має відносну біологічну засвоюваність, що становить приблизно 144 % порівняно із таблеткою лерканідипіну HCl 20 мг, що наявна на ринку (Фіг. 3 та 4). Результати, одержані in vivo, вказують на те, що рівень концентрації активної речовини у плазмі для композиції 1 становить приблизно 50 % від рівня реперного продукту. Композиція 2 показала 68 % підвищення концентрації у плазмі порівняно із композицією 1. Несподівано, композиція 3, що становить приблизно 144 % від рівня реперного продукту, має збільшення на 71 % та 188 % порівняно із композиціями 2 та 1, відповідно. Композицію 3 за даним винаходом можна вважати, таким чином, з точки зору фармакологічної дії, найбільш біологічно засвоюваною твердою речовиною серед композицій, наявних на даний час. Підвищену біологічну засвоюваність композиції 3 можна віднести до утворення дисперсії між колоїдним двооксидом кремнію та лерканідипін HCl. Оптимальну дисперсію перевіряли шляхом скануючої електронної мікроскопії (SEM), та як можна побачити на Фіг. 8 та 9, частинок чи агломератів лікарської речовини не спостерігали. Дуже велика поверхня Аеросилу™, на якій адсорбовано активний інгредієнт, не призвела до збільшення питомої поверхні та до надлишку біологічної засвоюваності. Наявність зв'язуючої речовини сприяє гомогенному розподілу активного інгредієнта на поверхні частинок Аеросилу™. Додаткове порівняльне дослідження проводили для порівняння впливу їжі на композиції прикладів 4 та 5 по відношенню до Занедіпу® 20 мг та 10 мг, відповідно. Більш детально, композицію прикладу 4 вводили 12 здоровим людям з 240 мл води, за 15 хвилин до введення стандартного сніданку чи натщесерце; аналогічно вводили композицію прикладу 5. Занедіп® 20 мг вводили тієї ж самій кількості/типу людей з 240 мл води, за 15 хвилин до введення стандартного сніданку; аналогічно вводили Занедіп® 10 мг. Вплив їжі на Занедіп® за аналогічних умов годування потім розраховували непрямим способом. В усіх випадках сніданок складався з: 1 англійського кексу з маслом, 1 яєчні, 1 шматка американського сиру, 1 шматка канадського бекону, 1 тарілки мілко нарізаної смаженої картоплі, 240 мл цільного молока, 180 апельсинового соку. Схема відбору проб була такою: 0.333, 0.75, 1, 1.333, 1.667, 2, 2.333, 2.667, 3, 3.5, 4.5, 6, 8, 10, 14, 18, 24, 30 та 36 годин. Результати наведено нижче. i). Вплив їжі на 16 мг композиції прикладу 4: AUCпри годуванні/AUCнатщесерце=103,6 % Cмакс при годуванні/Cмакс натщесерце=106,9 % ii). Вплив їжі на 8 мг композиції прикладу 5: AUCпри годуванні/AUCнатщесерце=125,8 % 11 UA 99136 C2 5 10 15 20 25 Cмакс при годуванні/Cмакс натщесерце=118,0 % iii). Вплив їжі на Занедіп® 20 мг: AUCпри годуванні/AUCнатщесерце=125,2 % Cмакс при годуванні/Cмакс натщесерце=160,3 % iv). Вплив їжі на Занедіп® 10 мг: AUCпри годуванні/AUCнатщесерце=163,4 % Cмакс при годуванні/Cмакс натщесерце=178,5 % Виходячи з даних результатів, очевидно, за цих експериментальних умов, для 16 мг композиції прикладу 4 вплив їжі відсутній, тоді як вплив їжі на 8 мг композиції прикладу 5 був значно знижений. Такі результати також підтримані даними in vitro за симульованих умов годування та голодування, що вказують на приблизно подвійну солюбілізацію (191 %) ліків за умов годування при введенні як Занедіпу® 20 мг (див. фігуру 15); з іншого боку, 16 мг композиції відповідно до даного винаходу, проаналізованих за аналогічних умов симуляції, показують приблизно таку ж саму кількість ліків, розчинених за умов годування та голодування (див. фігуру 14). Профілі розчинення, одержані як для 16мг, так і для 8 мг композицій, забезпечують додаткове підтвердження покращених властивостей композиції за даним винаходом порівняно із Занедіпом® (див. фігури 10 та 11, відповідно). Незалежно від того, чи була добровольцям введена така ж сама (20 мг) чи інша доза (16/8 мг), композиції відповідно до даного винаходу показали підвищену біологічну засвоюваність порівняно із реперним продуктом, наявним на ринку. Цей факт надає можливість одержувати фармацевтичну композицію, що містить меншу кількість активного інгредієнта, ніж реперний продукт, але аналогічним чином призводить до кращої сумісності із пацієнтом та має меншу кількість бічних ефектів. Оскільки даний винахід був описаний щодо конкретних втілень, фахівцю у цій галузі буде очевидно, що різні зміни та модифікації можуть бути здійснені у даному винаході без виходу за обсяг та без відхилення від суті даного винаходу, як визначено у формулі винаходу, що додається. ФОРМУЛА ВИНАХОДУ 30 35 40 45 50 55 1. Фармацевтична композиція для перорального введення, що містить лерканідипін або його фармацевтично прийнятну сіль та поліморф, як активний інгредієнт, та колоїдний двоокис кремнію, а саме Аеросил™, від 5 до 25 % за масою, для підвищення біологічної засвоюваності та/або покращення розчинності. 2. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить від 7 до 20 % за масою колоїдного двоокису кремнію. 3. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить від 3 до 25 % за масою лерканідипіну або його фармацевтично прийнятної солі та поліморфу, переважно від 5 до 10 % за масою, більш переважно приблизно 8 % за масою. 4. Фармацевтична композиція за п. 1, яка відрізняється тим, що масове співвідношення лерканідипіну або його фармацевтично прийнятної солі та поліморфу та колоїдного двоокису кремнію становить від 10/1 до 1/60. 5. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить зв'язуючу речовину, переважно полівінілпіролідон (ПВП), де вказана зв'язуюча речовина наявна у кількості від 5 до 20 % за масою, переважно у кількості до приблизно 15 % за масою. 6. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить агент розпаду, переважно крохмаль гліколяту натрію, де вказаний агент розпаду присутній у кількості від 5 до 15 % за масою, переважно приблизно 10 % за масою. 7. Фармацевтична композиція за п. 1, яка відрізняється тим, що вона містить зволожувач, переважно співполімер поліоксіетилену та поліоксипропілену (полоксамер), де вказаний зволожувач присутній у кількостях аж до 5 % за масою, переважно приблизно 2,5 % за масою. 8. Фармацевтична композиція за п. 1, яка відрізняється тим, що вона містить розріджувач, переважно мікрокристалічну целюлозу та/або лактозу, більш переважно у моногідратній формі, де вказаний розріджувач присутній у кількості від 40 до 65 % за масою, переважно від 45 до 60 % за масою. 9. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить внутрішню фазу та зовнішню фазу. 10. Фармацевтична композиція за п. 13, яка відрізняється тим, що вказана зовнішня фаза містить та переважно складається зі стеарату магнію. 12 UA 99136 C2 5 10 15 20 25 30 35 40 11. Фармацевтична композиція за п. 1, яка відрізняється тим, що лерканідипін знаходиться у гідрохлоридній формі, переважно кристалічній. 12. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить лерканідипіну гідрохлорид, моногідрат лактози, мікрокристалічну целюлозу, крохмаль гліколяту натрію, полівінілпіролідон, колоїдний двоокис кремнію, співполімер поліоксіетилен-поліоксипропілену, стеарат магнію. 13. Фармацевтична композиція за п. 1, яка відрізняється тим, що вона знаходиться у твердій лікарській формі, такій як таблетка, капсула чи саше. 14. Фармацевтична композиція за п. 13, яка відрізняється тим, що кожна тверда лікарська форма містить від 7 до 9 мг чи від 14 до 18 мг лерканідипіну гідрохлориду, переважно приблизно 8 мг чи приблизно 16 мг лерканідипіну гідрохлориду. 15. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить приблизно 16 мг лерканідипіну гідрохлориду, приблизно 80 мг моногідрату лактози, приблизно 16 мг мікрокристалічної целюлози, приблизно 20 мг крохмалю гліколяту натрію, приблизно 30 мг полівінілпіролідону, приблизно 31 мг колоїдного двоокису кремнію, приблизно 5 мг співполімеру поліоксіетилен-поліоксипропілену, приблизно 2 мг стеарату магнію. 16. Фармацевтична композиція за п. 1, яка відрізняється тим, що містить приблизно 8 мг лерканідипіну гідрохлориду, приблизно 45 мг моногідрату лактози, приблизно 8 мг мікрокристалічної целюлози, приблизно 10 мг крохмалю гліколяту натрію, приблизно 10 мг полівінілпіролідону, приблизно 15,50 мг колоїдного двоокису кремнію, приблизно 2,50 мг співполімеру поліоксіетилен-поліоксипропілену, приблизно 1 мг стеарату магнію. 17. Застосування композиції за будь-яким з попередніх пунктів для збільшення, переважно на 15-25 %, біологічної засвоюваності лерканідипіну, переважно лерканідипіну гідрохлориду. 18. Спосіб одержання твердої лікарської форми для перорального введення, вибраної з таблетки, капсули чи саше, що містить лерканідипін або його фармацевтично прийнятну сіль як активний інгредієнт та ефективну кількість колоїдного двоокису кремнію за п. 1 для підвищення біологічної засвоюваності, який включає: - розчинення загальної кількості вказаного активного інгредієнта, частини загальної кількості вказаного колоїдного двоокису кремнію та зв'язуючої речовини у розчиннику вода/EtOH; - додавання до утвореного розчину решти частини колоїдного двоокису кремнію та, необов'язкового, ексципієнта, такого як розріджувач, зв'язуюча речовина, агент розпаду, глідант, змазка, та проведення вологого гранулювання; - розчинення зволожуючого агента, такого як полоксамер, у невеликій кількості розчинника вода/EtOH та змішування з першим розчином до одержання однорідної маси; - висушування зволоженої маси; - просіювання висушеної маси та додавання до просіяної суміші загальної кількості щонайменше одного необов'язкового ексципієнта, такого як зв'язуюча речовина, розріджувач, агент розпаду, змазка та/або глідант, та змішування до досягнення однорідності; та - формування одержаної суміші у тверду лікарську форму шляхом її пресування у бажану форму таблетки або шляхом наповнення капсул чи саше. 19. Тверда лікарська форма, яку одержують способом за п. 18 та яка містить від 5 до 25 % за масою колоїдного двоокису кремнію. 13 UA 99136 C2 14 UA 99136 C2 15 UA 99136 C2 16 UA 99136 C2 17 UA 99136 C2 18 UA 99136 C2 19 UA 99136 C2 Комп’ютерна верстка В. Мацело Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 20
ДивитисяДодаткова інформація
Назва патенту англійськоюImproved pharmaceutical composition comprising a calcium channel antagonist such as lercanidipine, process for the preparation thereof
Автори англійськоюKaravas, Evangelos, Koutris, Makis, Samara, Vicky, Matsingou, Christina, Iliopoulou, Athina
Назва патенту російськоюУлучшенная фармацевтическая композиция, содержащая антагонист кальциевых каналов - лерканидипин, способ ее получения
Автори російськоюКаравас Евангелос, Коутрис Макис, Самара Вики, Матсингоу Кристина, Илиопоулоу Атина
МПК / Мітки
МПК: A61P 9/00, A61K 9/20, A61K 31/4422, A61K 47/04
Мітки: спосіб, каналів, лерканідипін, композиція, містить, одержання, фармацевтична, кальцієвих, покращена, антагоніст
Код посилання
<a href="https://ua.patents.su/22-99136-pokrashhena-farmacevtichna-kompoziciya-shho-mistit-antagonist-kalciehvikh-kanaliv-lerkanidipin-sposib-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Покращена фармацевтична композиція, що містить антагоніст кальцієвих каналів – лерканідипін, спосіб її одержання</a>
Фармацевтична композиція, що містить дигідропіридиновий антагоніст кальцієвого каналу лерканідипін і колоїдний діоксид кремнію, спосіб її приготування

Номер патенту: 97161
Опубліковано: 10.01.2012
Автори: Коутрі Іоанна, Коутріс Ефсіміос, Каравас Євангєлос, Стасакі Єлєні
МПК: A61K 31/4422, A61K 47/04, A61P 9/00, A61K 9/20
Мітки: колоїдний, приготування, спосіб, кальцієвого, діоксид, композиція, каналу, дигідропіридиновий, антагоніст, містить, фармацевтична, лерканідипін, кремнію
Формула / Реферат:
1. Фармацевтична композиція для перорального прийому, що включає дигідропіридиновий антагоніст кальцієвого каналу лерканідипін або фармацевтично прийнятну його сіль, похідне і поліморф, як діючий компонент, і ефективну кількість колоїдного кремнію діоксиду Aerosil™ від 5 до 25 % для збільшення біодоступності та/або поліпшення розчинності.2. Фармацевтична композиція за п. 1, де вагове відношення зазначеного дигідропіридинового...
Ефіри 2,6-диметил-1,4-дигідропіридин-3-карбонової кислоти, що є блокаторами кальцієвих каналів, спосіб їх одержання та фармацевтична композиція
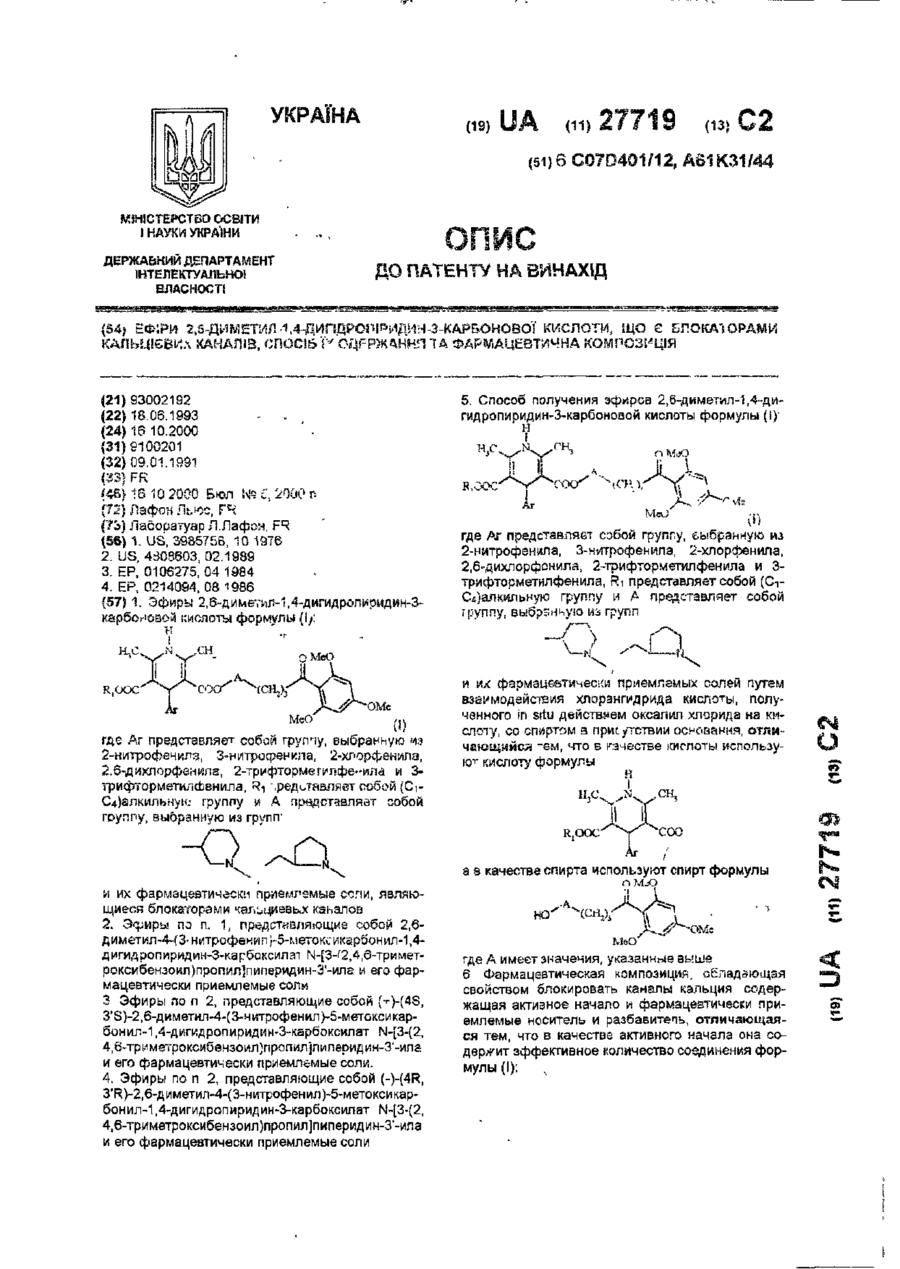
Номер патенту: 27719
Опубліковано: 16.10.2000
Автор: Лафон Льюс
МПК: C07D 401/12, A61K 31/435, A61K 31/451, C07D 211/90, A61K 31/4427, A61P 9/12, A61K 31/4433, C07D 409/14, A61K 31/445, A61P 9/08, A61K 31/455
Мітки: кальцієвих, спосіб, фармацевтична, блокаторами, композиція, ефіри, 2,6-диметил-1,4-дигідропіридин-3-карбонової, одержання, кислоти, каналів
Текст:
..."COOH в которой Аг и Ri имеют указанное выше значение, а в качестве спирта используют спир г формулы ОМеО Mel) где А имеет значение, указанное выше Аддитивные соли получают по классическим методикам реакцией соединения формулы (I) с фармацевтически приемлемой кислотой в подходящем растворителе И, напротив, основания могут быть получаны и; зддитизных солей обработкой их сильным основанием Кисго^ы формулы (ПІ моп.'т быть получше! классическим...
Фармацевтична композиція з контрольованим вивільненням, що містить лерканідипін

Номер патенту: 89177
Опубліковано: 11.01.2010
Автори: Хольм Пер, Норлінг Томас
МПК: A61P 9/12, A61K 9/22, A61K 31/4422
Мітки: містить, фармацевтична, контрольованим, вивільненням, композиція, лерканідипін
Формула / Реферат:
1. Фармацевтична композиція з контрольованим вивільненням, яка містить лерканідипін або його фармацевтично прийнятну сіль як активну речовину, і фармацевтично прийнятний наповнювач, при цьому активний інгредієнт: повністю розчинений у наповнювачі, формуючи твердий розчин при температурі навколишнього середовища; або частково розчинений у наповнювачі, формуючи суміш твердої дисперсії і твердого розчину при температурі...
Композиція, яка містить інгібітор іf-каналів синусного вузла та інгібітор кальцієвих каналів, та її застосування для лікування стенокардії

Номер патенту: 91507
Опубліковано: 10.08.2010
Автори: Бенатар Відаль, Леребур-Піжоньєр Гі
МПК: A61K 31/55, A61K 31/4422, A61P 9/10
Мітки: каналів, застосування, стенокардії, іf-каналів, лікування, інгібітор, кальцієвих, композиція, яка, містить, синусного, вузла
Фармацевтична композиція, яка містить серцевину та ентеросолюбільну оболонку, фармацевтична композиція у вигляді сфероїдальних гранул, спосіб одержання сфероїдальних гранул та спосіб одержання фармацевтичної ко

Номер патенту: 69413
Опубліковано: 15.09.2004
Автори: Вілі Гері Дж., Уллах Ісмат
МПК: A61K 9/62, A61P 31/18, A61K 9/16, A61K 31/7068
Мітки: фармацевтична, спосіб, одержання, фармацевтично, серцевину, гранул, яка, сфероїдальних, композиція, ентеросолюбільну, оболонку, містить, вигляді
Формула / Реферат:
1. Фармацевтична композиція, яка містить серцевину та ентеросолюбільну оболонку вказаної серцевини, причому серцевина містить нестійкий у кислому середовищі лікарський препарат у кількості приблизно від 80 до 100 мас.% від маси вказаної композиції, дезінтегрант у кількості до 10 мас.% включно від маси вказаної композиції та зв'язувальну речовину у кількості до 10 мас.% включно від маси вказаної композиції, причому вказана зв'язувальна...
Попередній патент: Скловолоконна структура для зміцнення абразивів зі зв’язкою, смоляна композиція, призначена для покриття скловолоконної структури, та виріб
Наступний патент: Термостабільна композиція для тварин, що містить суміш ферментів
Випадковий патент: Цукерка "какаду"