Похідні n-ацил-2,3-бензодіазепіну, що діють на центральну нервову систему, фармакологічна композиція
Номер патенту: 27227
Опубліковано: 15.08.2000
Автори: Тарнава Іштван, Моравчік Імре, Голдшмідт Каталін, Берженьі Пал, Фаркаш Шандор, Ботка Петер, Хаморі Тамаш, Андраші Ференц, Кьорьоші Йеньо








Формула / Реферат
(57) 1.Производные N-ацил-2,3-бензодиазепина общей формулы (1)
где R – С1-С6-алифатическая ацильная группа, незамещенная или многократно замещенная галоидом, С1-С5-алкилкарбамоил, бензоилрадикал или R отсутствует, когда между N(3) и С(4) атомами существует двойная связь,
R1 - водород или R1 отсутствует, когда между N(3) и С(4) атомами существует двойная связь,
R2 – С1 - Сз-алкил,
R3-водород и С1-С4-алифатическая ацильная группа,
R4 - водород, С1-С4 -алифатическая ацильная группа, незамещенная или замещенная амино, ди- С1-С4-алкиламино, пирролидино или многократно замещенная галоидом, пунктирные линии представляют двойную связь, возможно, присутствую- щую в молекуле, при условии, что когда оба ради кала R3 и R4 представляют атом водорода, то дво йная связь между N(3) и С{4) отсутствует, или их стереоизомеры, или их соли с кислотами, обладающие действием на центральную нервную систему.
2. Соединение по п. 1, выбранное из группы, состоящей из 1-(4-аминофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиа-зепина,
1-(4-аминофенил)-3-пропионил-4-метил-7,8-мети-лендиокси-3,4-дигидро-5Н-2,3-бензодиазепина,
1-(4-ацетиламинофенил)-3-ацетил-4-метил-7,8-ме-тилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина,
1-(4-пропиониламинофенил)-3-пропионил-4-ме-тил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензо-диазепина,
1-(4-пропиониламинофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиа-зепина,
1-(4-ацетиламинофенил)-3-пропионил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиа-зепина,
1-(4~трифторацетиламинофенил)-3-ацетил~4-ме-тил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензо-диазепина,
1-(4-глициламинофенил)-3-ацетил-4-метил-7,8-ме-тилендиокси-3,4-дигидро 5Н-2,3-бензодиазепинди-гидрохлорида,
N -[4-/3-ацетил-4-метил-7,8-метилендиокси-3,4-ди-гидро-5Н-2,3-бензодиазепин-1-ил/фенил ]-N3-MeTH-лмочевины,
1-[4-(N,N-диметилглициламино)фенил]3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бен-зодиазепина,
1-[4-( N,N-диэтилглициламино)фенил]3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бен-зодиазепина,
1-[4-(1-пирролдиноацетиламино)фенил]3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина и гидрофумаратов этих соединенийи
1-(4-глициламинофенил)-3-мететилкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бен-зодиазепина.
3 Фармакологическая композиция, обладающая нейропротекторным, наркозпотенцирующим, противоковультивным и миорелаксирующим действием, включающая активный ингредиент, носители и/или целевые добавки, отличающаяся тем, что в качестве активного ингредиента содержит производное М-ацил-2,3-бензодиаэепина общей формулы(1)
где R – С1 - С6 - бензоилрадикал или R отсутствует, когда между N(3) и С(4) атомами существует двойная связь;
R1 - водород или R1 отсутствует, когда между N(3) и С(4) атомами существует двойная связь, R2 – С1-Сз-алкил,
R3 - водород и С1 – С4-алифатическая ацильная группа;
R4 - водород, С1-С4-алифатическая ацильная группа, незамещенная или замещенная амино, ди- С1-С4-алкмламино, пирролидино или многократно замещенная галоидом, пунктирные линии представляют двойную связь, возможно, присутствующую в молекуле, при условии, что когда оба радикала R3 и R4 представляют атом водорода, то двойная связь между N(3) и С(4) отсутствует, или его фармацевтически приемлемую соль в эффективном количестве.
Текст
27227 Изобретение относится к новым производным N-ацил-2,3-бензодиазепина общей фо рмулы, (I) R2 O H2C 6 7 O C5H 2 1 4' R1 R 4 2 3N R (I) N C 1' C 2' 3' NR3R 4 в которой R это – С1-6 алифатическая ацильная группа, возможно замещен ная метокси-, циано, карбоксильной, амино-, С1-4-алкиламино, ди (С1-4 алкил) амино-, пирролидино-, фта лимидо- или фе нильной группой, или одним, или более галогеном(ами), или R – это бензоил, циклопропанкарбонил, С1-5-карбамоил или фенилкарбамоильная группа, или R отсутствует, когда между N(3) и С(4) атомами суще ствует двойная связь, R1 представляет атом водорода или R1 отсутствует, когда между N(3) и С(4) атомами существует двойная связь, R2 представляет С1-3 алкил, или R1 и R2 вместе представляют метиленовую гр уппу, а между N(3) и С(4) атомами нет двойной связи, R3 означает атом водорода или С 1-4-алифа тическую ацильную группу, R4 представляет атом водорода, C1-6 – алифа тическую ацильную гр уппу, во зможно замещенную метокси-, циано-, карбоксильной, амино-, С1-4-алкил-амино-, ди (С1-4 алкил) амино-, пирролидино-, фта лимидо или фенильной группой, или одним или более галогеном(ами), а также бензоил, пальмимитоил, циклопропанкарбонил, С1-5 алкил-карбамоил или фенилкарбамоильную гр уппу, а пун ктирные линии представляют возможно присутствующие ва лентные связи, при условии, что, когда оба заместителя R3 и R4 представляют ато мы водорода, между N(3) и С(4) атомами нет двойной связи, а также стереоизомеры этих со единений наряду с кислыми солями присоединения (в тех случаях, когда это возможно) и фармацевтические композиции, содержащие эти со единения. Соединения формулы (I), соответствующие настоящему изобретению, имеют асимметрическую молекулярную стр уктуру. Общая фо рмула (I) относится ко всем возможным индивидуальным изомерам и их смесям. Также в настоящем изобрете нии предлагается способ получения новых соединений общей фо рмулы (I) и кислых солей присоединения на их основе. Цель настоящего изобретения состоит в том, чтобы разработать новые соединения общей фо рмулы (I), кото рые обладают значительной акти вностью, связанной с воздействием на центральную нервную систему (ЦНС), в частности, обладают способностью вызывать релаксацию мышц и/или – противо конвульсивным действием. Единственное соединение, проявляющее такое действие, известно только среди 2,3-бензодиазепинов, а именно, это – 1-(4-аминофе нил)-4-метил-7,8-мети лендиокси -5Н-2,3-бензодиазепин (патент США № 4164740), также получено авторами настояще го изобретения. Однако в результате детального фа рмакологического скрининга было уста новлено, что выше названное соединение положительное в Ames-тесте, т.е. оно обладает мутагенным действием. Итак, конкретная цель настояще го изобретения состоит в том, чтобы новое производное бензодиазепина, которое бы сохраняло свои ценные свойства, связанные со способностью снимать мышечное напряжение и обладало противоконвульсивной активностью, но не обладало мута генным действием. Новые соединения общей формулы (I), в кото рой R, R1, R2, R3, R4 и пунктирные линии имеют значения, указанные выше, и и х фа рмацевтически приемлемые кислые соли присоединения полностью удо влетворяют это му тре бованию. Согласно насто яще му изобретению соединения общей формулы (I) получают путем: 27227 а) ацилирования соединения формулы (II) CH 3 O CH2 C O C N H2C N (II) NH 2 С1-6 алифати ческой карбоновой кислотой, возможно замещен ной метокси-, циано, карбоксильной, или фе нильной группой или одним или более галоге ном(ами), или бензойной кислотой, циклопропанокарбоновой кислотой или пальмити новой кислотой или производными этих кислот, и, если в этом есть необхо димость, реакции нового, полученного та ким образом соединения общей фо рмулы (I), в которой R 4 означает С1-6 алифа тическую ацильную группу, замещенную галогеном, с С1-4-алкиламином, ди-(С1-4 алкил) амином или пирролидином с образованием соединений общей фо рмулы (I), в которой R2, R3 и п ун ктирные линии имеют смысл, определенный выше, R4 предста вляет С 1-6-алифа тическую ацильную гр уппу, во зможно замещенную метокси-, циано-, карбоксильной, фенильной, С1-4-алкиламино-, ди (С1-4 алкил) амино- или пирролидиновой группой, или одним или более галогеном(ами), или представляет бензоил, циклопропанокарбонил или пальмитоил, R и R1 отсутствуют, а между N(3) и С(4) атомами наличествует двойная связь, b) ацилирования соединения общей фо рмулы (III) CH3 O CH2 C O C N H2C NH (III) NH R4 в которой R4 определен выше, С1-6-али фатической карбоновой кислотой, во зможно замещенной мето кси-, циано-, карбоксильной или фенильной группой или одним или более атомами галогенов, или бензойной или циклопропанкарбоновой кислотой или реакционноспособными производными на их основе, и, если в этом есть необходимость, реакция нового со единения общей формулы (I), полученной таким образом, в которой R4 предста вляет С1-6 али фатическую ацильную гр уппу, за мещен ную галогеном, с С1-4 алкиламином, ди (С1-4 алкил) амином или пирролидином, приво дящая к получению соединений общей формулы (I), в которой R1, R2, R3, R4 и пунктирные линии имеют смысл, определенный выше, R – С1-6 – алифати ческая ацильная группа, возможно замещенная метокси-, циано-, карбоксильной, фенильной, С1-4 – алкиламино-, ди(С1-4 алкил) амино- или пирролидиновой группой или одним или более атомами галогенов, или бензоил или циклопропанкарбонил, и между N(3) и С(4) ато мами отсутствует двойная связь, или с) ацилирования соединения формулы (II) N-фта лоиламинокислотой общей фо рмулой (VI) C C N (CH)n O COOH (VI) R5 в которой R5 представляет атом водорода или С 1-4- алкильную гр уппу и n равен 1 в случае a -аминокислот, тогда же, когда R5 означает атом водорода, то n варьируется в пределах от 2 до 5, что имеет место в случае аминокислот, и если в этом есть необхо димость, удаляют фта лоильную гр уппировку и получают соединения общей формулы (I), в которой R2 и пун ктирные линии имеют значения, указанные выше, R3 – означает атом водорода, R4 – это С1-6 алифа тическая ацильная группа, замещенная амино- или фталимидной группой, оба R и R1 отсутствуют, а дво йная связь существует между N(3) и С(4) атомами, d) или ацилирования соединения общей фо рмулы (III), в которой R4, определен вы ше, N-фта лоидаминокислотой общей фо рмулой (VI) в кото рой R5 предста вляет атом во дорода или С 1-4-алкильную гр уппу и n равен 1 в случае a -аминокислот, то гда же, когда R5 представляет атом водорода, то n ва рьируется в пределах от 2 до 5, что име ет место в случае b-e-аминокислот, и если в этом есть необхо димость, уда ляют фта лоильную гр уппировку и получают со единения общей формулы (I), в которой R1, R 2 и п унктирные линии имеют значе ния, определенные выше, R3 означает а том водорода, R4 имеет значения, 27227 определенные выше, за исключением водорода, R предста вляет С 1-6 али фати ческую ацильную гр уппу, замещенную амино-, или фта лимидной группой, а двойная связь между N(3) и С(4) атомами отсутствует, или е) реакции соединения формулы (II) с С1- 5 алкилизоцианатом или фе нилизоцианатом, приводящей к получению соединений общей фо рмулы (I), в кото рой R2 и пунктирные линии имеют значения, обозначенные выше, R3 означает атом водорода, R4 предста вляет С 1-5 – алкилкарбамоил или фенилкарбамоил, R1 и R2 отсутствуют, а между N(3) и С(4) ато мами суще ствует дво йная связь, или f) при взаимодействии соединения общей фо рмулы (III), в кото рой R4 определен выше, с С1-5 – алкил изоцианатом или фенил-изоцианатом, получа ют соединения общей фо рмулы (I), в кото рой R1, R2 и пунктирные линии имеют значения, определенные выше, R3 означает водород, R4 имеет значения, определенные выше, за исключением водорода, R представляет С1-5 – алкилкарбамоил или фе нилкарбамоильную гр уппу, дво йная связь между N(3) и C(4) а томами отсутствует, или g) селективным восстановлением нитросоединения формулы (IV) CH3 O CH2 C O C N H2C N (IV) NO 2 в новое соединение общей фо рмулы (V) CH3 O CH2 C O C N H2C N R (V) NO 2 в которой R представляет водород, с последующим либо ацилированием полученного таким образом соединения формулы (V), с использованием описанных выше способов b), d) или f) и восстановлением нитрогруппы полученного таким образом нового соединения общей фо рмулы (V), в которой R определен выше, в аминогруппу, либо восста новлением сначала нитрогруппы и затем ацилирования, соединения общей фо рмулы (III), полученного таким образом, в котором R представляет атом водорода, с использованием одного из вышеперечисленных процессов b), d), f) или h) с получением соединений общей формулы (I), в которой R1, R3 и R4 представляют атом водорода, R2, R и п ункти рные линии имеют смысл, определенный выше и между N(3) и С(4) ато мами двойная связь отсутствует, или i) ацилирование нового соединения общей фо рмулы (I), в которой R, R1, R2 и пунктирные линии имеют смысл, определенный выше, R3 и R4 представляют атомы водорода и нет двойной связи между N(3) и С(4) атомами, С1-6 алифати ческой карбоновой кислотой, во зможно замещенной метокси-, циано- или карбоксильной группой или одним или более атомами галогенов, или бензойной кислотой, или реакционноспособным производным на ее основе, с образованием соединений общей формулы (I), в которой R1, R2, R3 и пунктирные линии имеют смысл, определенный выше, R и R4 представляют С 1-6 али фати ческую ацильную гр уппу, во зможно замещенную метокси-, циано- или карбоксильной группой, или одним или более атомами галогенов или бензоильную группу, а двойная связь между N(3) и С(4) атомами отсутствует, или взаимодействие нового соединения формулы (I), в которой R, R1, R2 и пунктирные линии имеют смысл, определенный выше, R3 и R4 представляют атом водорода, а двойная связь между N(3) и С(4) атомами отсутствует, с С1-5 алкилизоцианатом или фенилизоцианатом с образованием соединений общей фо рмулы (I), в которой R1, R2 и пунктирные линии имеют смысл, определенный выше, R представляет С1-6 алифатическую ацильную группу, возможно замещенную метокси-, циано- или карбоксильной группой, или одним или более атомами галогенов, или бензоил, R3 представляет атом водорода, R4 представляет С1-5 алкилкарбамоил или фенилкарбамоил, и дво йная связь между N(3) и С(4) отсутствует, или j) ацилирование нового соединения общей фо рмулы (I) в которой R1, R2 и пунктирные линии имеют смысл, обозначенный выше, R3 и R4 представляют атомы водорода, и двойная связь между N(3) и С(4) атомами отсутствует с N -фталоиламинокислотой общей фо рмулы (VI), в которой R 5 предста вляет атом водорода или С1-4 алкильную гр уппу и n равно 1, в том случае, когда это a – аминокислоты, в те х случаях, когда используются b -e аминокислоты, то R5 означает атом водорода и множитель n варьируется от 2 до 5, 27227 и, если в этом есть необхо димость, удаляют фта лоильную гр уппу и получают соединения общей формулы (I), в которой R1, R2 и пунктирные линии имеют смысл, определенный выше, R представляет С1-6 алифа тическую ацильную группу, во зможно замещенную метокси-, циано- или карбоксильной группой или одним или более атомами галогенов, или бензоил, R3 представляет атом водорода, R4 представляет С1-6 алифатическую ацильную группу, замещенную амино – или фталимидной группой, и двойная связь между N(3) и С(4) атомами отсутствует, и, если в этом есть необхо димость, переводят основание общей фо рмулы (I), полученное по любому из выше перечисленных процессов с а) по j), в кислую соль присоединения. В соответствии с предпочтите льным вариантом осуще ствления способа настояще го изобретения ацилирование соединений общей фо рмулы (I), (II), (III) и (V) может быть проведено предпочтительно с использованием подходящей карбоновой кислоты в присутствии дициклогексилкарбодиимида, в температурном инте рвале от 10 до 30 °С в те чение 1 до 25 ч. В соответствии с другим предпочтительным вариантом настояще го изобретения соединения общей фо рмулы (I), (II), (III) и (V) могут быть ацилированы в температурном интервале от 0 до 150 °С с использованием подхо дяще го реакционноспособного ацильного производного, например, ангидрида карбоновой кислоты, смешанного ангидрида или хлорангидрида, в отсутствии или в растворителе, обычно используемом для таких способов ацилирования, например, в хло рофо рме или дихлорэтане, в отсутствии или в присутствии средства, свя зывающе го кислоту, та кого, как триэти ламин. Если дополнительное ацилирование осуществляется с использованием изоцианатов, то реакцию преимущественно проводят в димети лформамиде, бензоле или дихлорметане в температурном интервале от 15 до 100 °С в промежуток времени от 0,5 до 100 ч. Селективнее восста новление соединения общей формулы (IV) в соединение общей формулы (V), в которой R представляет атом водорода может быть осуще ствлено с использованием неорганического или неорганического -органического комплексного соединения гидрида металла, предпочтительно с использованием борогидрида натрия, в растворите ле или смеси растворите лей, который имеет только низкую реакционную способность по отноше нию к комплексному соединению металлогидрида, используемому в реакции, или таковая способность отсутствует. В этих реакциях растворителем выбора является С 1-4 спирт или пиридин. (Подобное селекти вное восста новление описано в пате нтах США № 4423044 или 4835152). Для восстановления нитрогруппы новых соединений общей формулы (V) в аминогруппу используют гидразин или гидразингидрат, восстановление проводят в присутствии катализато ра, такого как палладий, платина или никель Ренея в С1-4 -спирте, диоксане, тетрагидрофуране, бензоле, диметилфо рмамиде, диметилацета миде или смесях этих растворителей. Согласно предпочти тельному ва рианту осуще ствления настоящего изобретения восстановление может быть проведено в метаноле гидразином или гидразингидратом в присутствии катализатора-никеля Ренея в температурном интервале от 10 до 65 °С (патент США № 4614740), но, если в этом есть необхо димость, восстановление и удаление защи тной фталоильной группы, описанное в способе d) может быть проведено в одном и том же реакционном сосуде. N-фта лоидаминокислоты общей формулы (IV), содержащие хи ральный атом углерода, где R 5 означает С1-4 алкильную гр уппу и множитель n равен 1, могут быть получены из DL-L- и/или D-альфа -аминокислот. Соединения формулы (I) насто ящего изобретения, которые содержат основную аминогруппу, в ко торой R3 и R4 предста вляют а том во дорода или R и/или R4 представляет аминоацильную гр уппу, мо гут быть превращены в ки слые соли присоединения в со ответствии с из вестными методами. Получение соединений обшей фо рмулы (II), используемых в качестве исхо дных материалов в процессе настоящего изобретения, описано в пате нте США № 4614740, соединений общей формулы (III), в которой R4, предста вляет атом водорода - в патенте США № 4835152, а соединений общей формулы (IV) - опубликовано во французской патентной заявке № 8509793. Соединения общей фо рмулы (III), в которой R4, представляет различные ацильные группы, являются новыми соединениями. Способ их получения описан здесь далее до табл. 10, или они могут быть си нте зированы описанными здесь мето дами. Получение новых исхо дных мате риалов общей фо рмулы (V) описано в примерах. Производные (a -e)-аминокислот общей фо рмулы (VI) получены в соответствии со способами, известными из литературных данных [J. Аm. Сhem. Sос. 35, 1133 (1913), 41, 845 (1919), Berichtedev, Deutschen Chemischen Gesellshaft, 40, 498, 2649 (1907), 46,1103, 3159 (1913), 47, 3166 (1914)] или согласно известным методам, использующим реакцию фталимида калия с нужной галоидкарбоновой кислотой. Соединения общей фо рмулы (I), полученные по способу настоящего изобретения, обладают активностью в отноше нии центральной нервной системы (ЦНС), включая антиконвульсивное действие, способствуют мышечной релаксации и оказывают нейропротекто рное действие, что может быть подтверждено фармакологическими испытаниями. В сравнительном исследовании 1-(4-аминофе нил)-4-метил-7,8-метилендиокси-5Н- 2,3-бензодиазепин (патент США № 4614740 далее именуемое "соединение прото типа"), имеющий подобную стр уктуру и эффе ктивность действия, как и соединения настоящего изобретения, использова лся как соединение прото типа. Как уже упоминалось во введении, это соединение обладает ценными фармакологическими свойствами, но как доказано, является Аmes-положительным. В отличие от него соединения настоящего изобретения являются Аmes-отрицательными. Фармакологическое действие этих соединений общей фо рмулы (I) представлено в табл. 1-8. Наркоз-потенциирующее действие на мышах Наркоз-потенциирующее действие было исследовано с использова нием трех пероральных доз на 10 мышах. Значение ЕД50 соответствует дозе, пролонгирующей длительность общего наркоза, вызванного 27227 введением внутривенно гексобарбитала в количестве 50 мг/кг, в два раза у 50% животных в сравнении с контрольной группой животных, которым давали только разбавитель. Значения ЕД50 рассчитывались по методу Litchfield -Wilcoxon [J. Pharmacol Exp. Ther. 96, 99 (1949)]. Результаты представлены в табл. 1. Из данных, приведенных в табл.1 сле дует, что эффе ктивность некоторых со единений совпадает или значите льно прево схо дит эффе кти вность соединения прототи па. Соединения примеров 15(16), 45, 60, 73, 98 относятся к соединениям, отлича ющи мся особенно высокой эффективностью. Противоконвульсивное действие на мыша х Противоконвульсивное действие соединений измеряли, используя испыта ние электрошо ком (Swinyard: J. Рharmacol. Ехр. Тhеr. 106, 319 (1952)], кроме того, были использованы различные химические средства, такие как пентатетразол [Goodman: J. Рharmacol. Ехр. Тhеr, 108, 168 (1953)], стрихнин [Roskovski: J. Рharmacol. Ехр. Тher. 129, 75 (1960)] бемегрид, никотин и 4 аминопиридин. Тестируемые соединения перорально тремя дозами вводили самцам мышей линии CFLP, каждая доза давалась 10 мышам. Результа ты представлены в табл. 2. Вышеприведенные данные свидетельствуют о том, что противоконвульсивное действие некоторых соединений (примеры 15, 42, 45, 46, 73, 98, 107, 108, 109 и 115) превосходит аналогичное действие соединения прототипа. Миорелаксантная активность на мышах Определение миорелаксантной активности было проведено в двух испытаниях. В скрининговом испытании по Randall’s [J. Pharmacol. Ехр. Тher. 129, 163, (1960)] 10 мышам линии CFLP на дозу, вн утрибрюшинно вводили 3 дозы соединения. Результаты приведены в табл. 3. Для определения мышечного то нуса и координации движения был использован rоtаrоd тест. [Dunham аnd Мija J. Аm. Рharm. Assoc. 46, 208, (1957)]. Результаты, полученные с тремя выбранными соединениями высшей активности и с со единением прототипа, представлены в табл. 4. Данные, приведенные в табл. 3 и 4 свидетельствуют о том, что некоторые соединения обладают сильной миорелаксантной активностью (соединения примеров 15, 18, 42, 45, 48, 49, 62, 73, 98 и 115). Влияние на спинальную ф ункцию Было определено влияние самых активных соединений (соединение примера 15 или 16) и соединения прототипа на спинальную функцию. В табл. 5 приведены результаты, сви детельствующие о вли янии на полисинаптические сгибательные рефлексы у кошек [Farkas and Karpati. Pharm. Res. Comm. 20, S1, 141 (1988)]. Определение влияния вышеуказанных соединений на напряжения в спинальном корне у кошек было проведено на спинально иммобилизованных животных. [Farkas et al. Neuropharmacology 21. 161, (1989)] Результаты представлены в табл. 6. Значения ЕД50, ин гибирующие моносинапти ческий рефлекс Соединение прототи па : 2,20 (1,02 – 4,75) мг/кг, вн утривенно. Соединение N 15(16) : 2,30 (1,06 – 5,01) мг/кг, вн утривенно. Значения ЕД50, ин гибирующие полисинаптический рефлекс Соединение прототи па: 0,60 ( 0,32 – 1,13) мг/кг, внутривенно Соединение N 15(16) : 0,73(0,39 – 1,37) мг/кг, внутривенно. Электрофи зиологические испыта ния Данные по ингибирующему действию на участки напряжений, вызванные электростимуляцией in vitro срезов неокортекса у выживших крыс, суммирова ны в табл. 7 [Fletcher и др., Br. J. Pharmacology 95, 585 (1988)]. Не-NМDА (quisqualate) антагонистическое действие определили на срезах неокортекса крыс, используя метод Harrison и Simnionds [Вr. J. Рharmacol. 84, 381 (1981)]. Изменения напряжения постоянного тока на срезах неокорте кса крыс, вызванные перфузией quisqualate ингибировались соединением прототипа в концентрационном интервале от 10 до 50 мкМ. При определенной концентрации соединение примера 15 (16) в два ра за активнее соединения прототипа при инги бировании ответа на двухминутную перфузию 10mМ quisqualate. Однако обе молекулы (оба соединения) оказываются не в состоянии воздейство вать на реакцию, индуцируемую NМDА. Следовательно, соединение примера 15 (16) можно рассматривать как не NМDА, но quisqualate типа селективный возбуждающий аминокислотный антагонист. Острая токсичнос ть на крысах Данные по острой токсичности на крысах суммированы в табл. 8. *При токсических уровнях дозы соединения вызыва ли дозозависимое снижение мыше чного то нуса, атаксию, адинамию и поте рю установочного рефлекса. Причиной смерти служила респираторная недостаточность, развивающа яся в течение 1-2 ч после в.б. введения и в течение 10-20 ч после перорального приема. Основываясь на вышеприведенных результа тах фа рмакологических исследований, можно сделать вывод о том, что соединения, соответствующие настояще му изобретению формулы (I), обладают значите льным противоконвульсивным действием, миорелаксантной акти вностью и являются возбуждающими аминокислотными анта гонистами (нейропротекто рами). Таким образом, они полезны в качестве терапевтических средств при лечении эпилепсии, а также различных заболеваний, связанных со спазмами скелетной мускулатуры и церебральной ишемии ("удар"). Это изобретение также относится к фармацевтическим композициям, содержащим соединения общей фо рмулы (I) или фармацевти чески приемлемым кислым солям присоединения на их основе в качестве активных ингредиентов, а также к способу получения этих композиций. Для терапевти ческого использования, акти вные соединения согласно настояще му изобретению подхо 27227 дящим образом вводят в фа рмацевтические составы путем смешения с традиционно используемыми нетоксичными, инертными, твердыми или жидкими фармацевтическими носителями и/или дополнительными материалами, полезными для энтерального или паренте рального введения. В качестве носите лей можно использовать, например, воду, желатин, лактозу, крахмал, пектин, сте арат магния, стеариновую кислоту, тальк или растительные масла. В качестве добавок, например, могут быть использованы: консерванты и увла жните ли, такие как эмульгаторы, диспергаторы и ароматизирующие средства, а также буфе рные растворы. При использовании выше упомянутых но сителей и добавок активные ингредиенты изобрете ния могут быть внесены в обычные фармацевтические композиции, например, в тве рдые композиции (выпускаемые в виде таблеток, капсул, пилюль или суппозиториев) или жидкие композиции, такие как водные или масляные растворы, суспензии, эмульсии или сиропы), а также в виде растворов для инъекций, суспензий или эмульсий. Для терапевтических целей ежедневная доза соединений насто яще го изобретения обычно составляет от 0,2 до 1,5 мг/кг массы те ла, эта доза, возможно разбивается на несколько приемов. Основываясь на выше приведенных фа ктах, настоящее изобретение также обеспечивает: способ блокирования одного или более возбуждающи х аминокислотных рецепторов у млекопитающи х. Этот способ включает вве дение млекопита ющим,нуждающи мся в такого рода лечении, фармацевтически эффе ктивного количества соединений общей фо рмулы (I); способ лечения эпилепсии у животных. Этот способ включает введение млекопитающим, нуждающи мся в такого рода лечении, противо эпилептического количества соединения насто яще го и соединения общей фо рмулы (I); способ снятия спазм (судорог) скелетной мускулатуры у млекопитающи х. Этот способ включает введение млекопита ющим, нуждающи мся в та кого рода лечении, миорелаксантного количества соединения общей фо рмулы (I); способ лечения церебральной ишемии ("удара") у млекопитающих. Этот способ включает, нуждающимся в такого рода лечении, введение фармацевтически эффективного количества соединения общей формулы (I). Соединения, полученные по способу данного изобретения, иденти фицированы методом элементного анализа, чисто та этих соединений и их стр уктура контролировалась и были подтверждены методами тонкослойной хроматографии, ИКС, 1Н-Я МР, 13С-Я МР и масс -спектрометрии. Это изобрете ние детально проиллюстрировано следующи ми далее примерами, которые не носят ограничивающий ха рактер. Пример 1 1-(4-диацетиламино(-3-ацетил-4-метилен-7,8-метилендиокси-4,5-дигидро-3Н-2,3-бензодиазепин. 2,93 г (0,01 моля) 1-(4-аминопропенил)-4-метил-7,8-мети лендиокси-5Н-2,3-бензодиазепина кипятили с обратным холодильником с 20 мл уксусного анги дрида в те чение 6 ч. Этот ра створ упаривали при пониженном давлении, остаток со брали двумя порциями по 20 мл безводного этанола, полученный раствор вновь упа рили, и полученный остаток массой 4,55 г пропусти ли через хроматографи ческую колонку (адсорбент-кизельгур 60, элюент - смесь этилацета та и бензола = 4:1). Сырой продукт ра стерли с 20 мл изопропанола, в результа те получили 1,44 г (34,4%) нужного продукта, т.пл. 240-245 °С (слабое разложение), С23Н21N3O 5=419,445. Пример 2 1-(4-фо рмиламинофе нил)-4-метил-7 ,8-ме тилендиокси-5Н-2,3-бензодиазепин 3,0 г (10, 2 ммоля) 1-(4-аминофе нил-4-метил-7 ,8-ме тиле ндиокси-5Н-2,3 -бе нзоди азепина раство рили в 160 мл дихлорметана и сначала к раство ру до бави ли 2,75 г (13,3 ммоля) дицикло гексилкарбодиммида, за тем -0,51 мл (13 ,3 ммоля) 100% муравьиной кислоты, и по лученную ре акционную смесь перемеши ва ли в те чение 2 ч при комнатной температуре. Вы павший оса док N,N’-дициклогексилмочеви ны отфи льтрова ли, фи льтрат проэкстраги ровали двумя порциями по 30 мл 10% во дного ра ство ра карбоната натрия , затем двумя порциями по 30 мл дистилли рованной во ды, орга нический слой вы суши ли и упа рили при пониженном давлении. Оста ток раство рили в эти лацета те, о тфи льтрова ли и упа рили при пониженном давлении. Получен ный сырой продук т перекристаллизовали при пониженном давлении из 20 мл 50% этанола, в результа те получили 2,93 г (83 ,3%) н ужного продукта, т.пл. 152 -154°С (незначите льное разложение), С 8Н 15N 3 О3 =321,342. Приме ры с 3 по 7 Соединения примеров с 3 по 7 были получены по способу, описанному в примере 2. Пример 3 1-(4-цианоацетиламинофе нил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. С20Н15N4O3=360,380, т.пл. 241-243°С (разл.). Пример 4 1-(4-мето кси ацетила минофе нил)-4-метил-7,8-ме ти лендиокси-5Н-2 ,3-ди азепин. С20Н19N 3O 4-365,396, т.пл. 203-205°С. Пример 5 1-(4-валериламинофе нил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. С22Н23N3O3=377,450, т.пл. 217-219°С (разл.). Пример 6 1-(4-фенилацети ламинофенил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. С25Н21N3 О3=411,467, т.пл.=245-247°С (разл.). 27227 Пример 7 1-(4-циклопропанкарбониламинофенил)-4-метил-7,8-мети лендиокси-5Н-2,3-бензодиазепин. С21Н19N3 О3=361,407, т.пл. 260-262°С (разл.). Пример 8 1-(ацетиламинофе нил)-4-метил-7,8-мети лендиокси-5Н-2,3-бензодиазепин. 10 г (34 ммоля) 1-(4-аминофе нил)-4-метил-7,8-мети лендиокси-5Н-2,3-бензодиазепина перемеши вали в течение 3 ч со 100 мл уксусного ангидрида. Образовавши еся кристаллы отфильтровывали, промыли 5 х 10 мл безводного этилового спирта и высушили, в результате получили 9,2 г сырого продукта, т.пл. 252-254°С (разл.). Этот продукт обработали 45 мл горячего 99,5% этанола. После охлаждения кристаллы отфи льтровывали, промыли 3 х 10 мл этанола и высушили, в результате получили 8,68 г, 76,1% нужного продукта, т.пл. 256-258°С (разл.), С19Н17О 3=335,269. Пример 9 1-(4-пропиониламинофе нил)-4-метил-7 ,8-мети лендиокси-5Н-2,3-бензодиазепин. Это соединение было получено согласно процедуре, описанной в примере 8. С20Н19N3О3=349,396, т.пл. 228-230°С (разл.). Пример 10 1-(4-пивалоиламинофе нил)-4-метил)-7,8-метилендиокси-5Н-2,3-бензодиазепин. 1,56 мл (11,2 ммоля) триэтиламина и 1,38 мл (11,2 ммоля) пивалоилхлорида добавили к раствору 3 г (10,2 ммоля) 1-(4-аминофенил) -4-метил-7,8-метилендиокс-5Н-2,3-бензодиазепина в 160 мл дихлормета на и реакционную смесь перемешивали при 25°С в течение 1 ч. Образовавшийся осадок отфи льтровывали, промыли 3 х 5 мл дихлормета на, затем 3 х 20 мл дистиллированной воды и высушили, в результа те получили 1,59 г чи стого продукта, т.пл. 225-227°С (разл.). Другую порцию продукта выделили из органической фа зы. Фильтрат проэкстраги ровали 3 х 20 мл дистиллированной воды, затем 3 х 15 мл 4%-ным водным раствором гидроксида натрия, и в заключение 2 х 30 мл ди стиллированной воды. За тем органический слой высушили и упарили при пониженном давлении. Кристаллический остаток объединили с полученными ранее 169 г продукта и суспендировали в 20 мл горячего этанола. Продукт отфи льтровывали после охлаждения, промыли 3 х 3 мл этанола и высуши ли, в результа те получили 3,38 г (87,8%) чисто го продукта т.пл. 225-227°С (разл.), С22Н23N3О 3 = 377,450. Пример 11 1-(4-бензоиламинофе нил)-4-метил-7,8 метилендиокси-5Н-2,3-бензодиазепин. 1,0 мл (15 ммоля) бензоилхлорида и 2,1 мл (15 ммоля) триэтиламина добавили к раствору 4 г (13,6 ммоля) 1-(4-аминофенил)-4-метил-7,8-метилендиокси-2,3-диазепина в дихлорметане и реакционную смесь перемеши вали при 25°С в течение 24 ч. Раствор проэкстрагировали 3 х 30 мл дистиллированной воды, 3 х 20 мл 4% водного раствора гидроксида натрия и, в заключение, 2 х 30 мл дистиллированной воды. Органический слой высуши ли, упарили при пониженном давлении, затем кристаллический осадок обработа ли 20 мл горячего этанола, на следующий день его отфи льтровали при 0-5°С, промыли 3 х 3 мл этанола и высушили при 100°С, в результате получили 3,85 г (71,3%) чистого нужного продукта, т.пл. 246-247°С (разл.), С24Н19N3 О3 =397,40. Пример 12 1-(4-пальмитоиламинофенил)-4-метил 7,8-метилендиокси-5Н-2,3-бензодиазепин Следуя процедуре, описанной в примере 11, с перекристаллизацией сырого продукта из 50% этанола, получили чистый нужный продукт, т.пл. 138-140°С, С33Н45N3О 3= 531,747. Пример 13 1-(4-фенилкарбамоиламинофе нил)-4-метил-7,8-мети лендиокси-5Н-2,3 -бензодиазепин. 0,22 мл (2,04 ммоля) фенилизоцианата добавили к раствору 0,50 г (1,7 ммоля) 1-(4-аминофенил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепина в 4 мл диметилформамида и реакционную перемешивали при 25°С в течение часа. Затем ее разбавили 20 мл диэтилового эфи ра и отфильтровали при 5° С. Кристаллы промыли 2 х 25 мл диэтилового эфира и высуши ли при 60-100 °С. 0,70 г сы рого продукта с т.пл. 239240°С (спекается при 180 °С) кипятили с обратным холодильником в 15 мл этанола, потом отфи льтровывали после охлаждения, промыли 3 х 1 мл этанола и высушили при 100°C, в результате получили 0,55 г нужного продукта (78,6%) т.пл. 240-241 °С (разл.), С24Н20N4O 3 Х=412,456. Пример 14 1-[4-(4-карбоксибути риламино)фенил]-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. Раствор 0,50 г (1,7 ммоля) 1-(4-аминофе нил)-4-метил- 7,8-метилендиокси-5Н-2,3-бензодиазепина в 30 мл безводного дихлорметана перемеши вали с 0,18 г (1, 87 ммоля) ангидрида глутаровой кислоты при 2025°С в течение 6 ч. На следующий день образовавшиеся кристаллы отфи льтровывали при 0-5°С, промыли 3 х 2 мл ди хлорметана и высушили при 60-80°С, в результате получили 0,60 г (87%) чистого нужного продукта, т.пл. 225-227 °С (разл.), С22Н21N3 O5S=407,434. Пример 15 1 (4-аминофенил)-3-ацетил-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. К раствору 3,58 г (12,1 ммоля) 1-(4-аминофенил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепина в 100 мл хлорофо рма в начале добавили к 1,68 мл (12,1 ммоля) триэтиламина, затем при постоянном охлаждении льдом и перемешивании добави ли 1,15 мл (12,1 ммоля) уксусного ангидрида. Перемеши вание продолжали в течение 2 ч. Затем раствор проэкстрагировали 3 х 100 мл дистиллированной воды, органический слой высушили и упаривали при пониженном давлении. Кристаллический остаток перекристаллизовали из 40 мл изопропанола, в результате получили 3,50 г (85,7%) нужного продукта, т.пл. 220-222 °С. После повто рной перекристаллизации, т.пл. достигла 223-225 °С. С19Н19N3О 3=373,385, гидрохлорид : 27227 (С19Н20N3 О3)С=373,850, т.пл. 248-252 °С (разл). Пример 16 1-(4-аминофе нил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. К суспензии 1,91 г (5,37 ммоля) 1-(4-нитрофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н2,3 -бензодиазепина (продукт примера 27) в 40 мл метанола добавили около 0,2 г катализатора - никель Ренея и 1,4 мл (28 ммоля) 100% гидразингидрата, после чего реакционную смесь перемешива ли в течение 1 ч при 20-25°С. Исходное нитропроизводное растворили в течение 10-20 мин. После фильтрова ния фильтрат упарили при пониженном давлении, белый кристаллический оста ток промыли 30 мл дистиллированной воды на фильтре, затем промыли 3 х 10 мл дистиллированной воды и высуши ли на 100°С, в результате получили 1,50 г неочищенного продукта с т.пл. 218-220°С. Этот неочищенный продукт растворили в 12 мл горячего изопропанола. После охлаждения смесь отфильтровали при 5°С, промыли 3 х 1 мл изопропанола и высуши ли при 100°С, в результате получили 1,40 г (77, 35%) белого кристаллического порошка, т.пл. 221-223°С. На основе проведенных анализов и данных спектров было уста новлено, что полученный продукт идентичен продукту примера 15, полученному другим способом. Пример 17-25 Согласно способу, описанному в примере 16, были получены другие 1-(4-аминофе нил)-3-R-4-метил7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепины общей фо рмулы (I). Сведения об этих продуктах представлены в табл. 9. Новые нитросоединения общей фо рмулы (V), в которой R=Н или ацильная группа, используемые при получении продуктов примеров с 16 по 25 могут быть получены согласно способам, описанным в примерах с 26 по 36. Пример 26 1-(4-нитрофенил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. К суспензии 5,0 г (15,5 ммоля) известного 1-(4-нитрофенил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепина (патент Франции № 8509793) в 380 мл этанола сначала добавили 22,5 мл (0,278 моль) концентрированной хлористоводородной кислоты при постоянном перемешивании при этом за несколько минут образовался раствор, затем в течение 30 мин частями добавляли 11,5 г (0,3 моль) борогидрида натрия. Перемешивание продолжили еще 15 мин, затем образовавшийся осадок оранжевого цвета отфи льтровали и на фильтре проэкстрагировали 4 х 30 мл хло роформа. Объединенный фильтрат упарили при пониженном давлении, кристаллический остаток перенесли на фильтр с 200 мл дистиллированной воды, затем промыли 3 х 20 мл дистиллированной воды и высушили при 80-100°С, в результате получили 4,90 г (97,2%) нужного продукта, т.пл.: 162-164 °С, С17Н15N3O 4 = 325,331. Пример 27 1-(4-нитрофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. 2,0 г продукта примера 26 перемешали с 10 мл уксусного ангидрида при 25 °С, перемеши вание длилось 3 ч, затем добавили 50 мл дистиллированной воды и перемешивание продолжили в течение 1 ч. Образовавшийся желтый осадок отфи льтровали, промыли 3 х 10 мл дистиллированной воды и высуши ли при 80-100 °С, в результа те получили 2,6 г неочищен ного (сырого) продукта. После перекристаллизации из 10 мл этанола получили 1,94 г (85,8%) нужного продукта, т.пл. 140-142°С, С19Н17N3O 5=367, 369. Пример 28 1-(4-нитрофенил(-3-трифторацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. К раствору 1,5 г (4,61 ммоля( – часть продукта примера 26, в 30 мл безво дного дихлорметана добавили 0,75 мл (5,3 ммоля) трифторуксусной кислоты и 0,75 ммоля (триэтиламина, и реакционную смесь перемешивали при 25 °С в те чение 3 ч. Затем смесь проэкстрагировали 3 х 20 мл дистиллированной воды, и органический слой высушили и упарили при пониженном давлении. Кристаллический остаток обработали 15 мл горячего этанола, охладили, отфильтровали, промыли 3 х 1 мл этанола и высушили при 80-100°С, в результате получили 1,84 г (94,85%) нужного соединения в виде светло-желтого кристаллического продукта, т.пл. 165-167°С (разл.), С19Н14N3F 3O 5 = 421,339. Пример 29 1-(4-нитрофенил)-3-пропионил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Часть продукта примера 26 -1,54 г (4,7 ммоля) перемешивали при 25°С в течение 3 ч с 8 мл ангидрида пропионовой кислоты, затем добавили 30 мл диэти лового эфи ра, и раствор оставили на ночь при 0-5°С. Образовавшийся осадок отфи льтровали, промыли 3 х 8 мл диэтилового эфира и высуши ли, в результа те получили 1,32 г (73,7%) нужного соединения в виде светло-желтого продукта, т.пл. 189-190°С, С20Н19N3 O5=381,396. Пример 30 1-(4-нитрофенил)-3-валерил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазелин. К раствору 2,5 г (7,68 ммоля) продукта примера 26 в 40 мл безводного дихлорметана добавили 4,75 г (23 ммоля) дициклогексилкарбодиимида и 2,88 г (23 ммоля (н-валериановой кислоты и реакционную смесь поддерживали при 25°С при переменном перемешивании в течение 24 ч. Затем N,N'-дициклогексилмочевину, образовавшуюся в качестве побочного продукта отфи льтровали, фи льтрат упарили при пониженном давлении, остаток смешали с 2 х 40 мл дистиллированной воды, декантировали и влажный продукт оставили отверждаться при 50 мл 50% этанола. Тве рдое соединение отфильтровыва ли, промыли 2 х 10 мл 50% этанола и высуши ли при 80°С. Сырой продукт был перекристаллизован из 24 мл ж этанола, и кристаллы высуши ли при 100°С, в результате получили 2,20 г (70% выход) нужного продукта в ви де порошка желтого цвета, т.пл. 145-147°С, С22Н23N3O 5= 409,450. Пример 31 27227 1-(4-нитрофенил)-З-пивалоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин Согласно процедуре, описанной в примере 28, но и с использованием пивалоилхлорида вместо ангидрида трифто руксусной кислоты получили 1,68 г (89,4%) нужного продукта, т.пл. 164-166°С, С22Н23N3 O5=409,450. Пример 32 1-(4-нитрофенил)-3-бензоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-бензодиазепин. Согласно процедуре, описанной в примере 31, но с использованием в качестве ацилхлорида-бензоилхлорида, получили 1,72 г (86,9%) продукта цвета желтой охры, т.пл. 222-224°С (разл), С245Н19N3O5=429,404. Пример 33 1-(4-нитрофенил)-З-фенилацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Согласно процессу, описанному в примере 30, но с использованием 50% расчетного молярного количества дициклогексилкарбодиимида и фе нилуксусной кислоты, получили све тло -желтый продукт, т.пл. 193195°С, С25Н21N3O5=443,567. Приме ры с 34 по 36 Следуя процедуре, описанной в примере 33, были получены продукты примеров с 34 по 36, только использовались соответствующие компоненты кислот. Пример 34 1-(4-нитрофе нил)-3-циклопропанкарбонил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин, т.пл. 225-228°С, С21Н19N3 O5=393,407. Пример 35 1-(4-нитрофенил)-3-цианоацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Т.пл. 185-188°С, С20Н16N4 O5=392,380 Пример 36 1-(4-нитрофенил(-3-методсиацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Т.пл. 187-189°С, С20Н19N3 O6=397,396. Пример 37 1-(4-нитрофенил)-3-(4 -карбоксибутирил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин. Используя продукт примера 26 в качестве исхо дного продукта и, проводя ацилирование согласно примеру 14, используя в качестве ацилирующего средства ангидрид глутаровой кислоты, после окончате льной перекристаллизации неочищенного продукта из этанола, получили нужный продукт в чи стом виде. Т.пл. 148-150°С, С22Н21N3O7=439,434. Пример 38 1-(4-аминофенил)-3-фенилкарбамоил-4-метил-7,8-мети лендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. К раствору 0,70 г (2,3 ммоля) 1-(4-аминофенил)-4-метил-7,8-мети лендиокси-3,4-дигидро-5Н-2,3-бензодиазепина в 10 мл безводного бензола добавили 0,24 мл (2,3 ммоля) фенилизоцианата и реакционную смесь нагревали с обратным холодильником в течение 1 ч. После этого раствор упарили при пониженном давлении, и аморфный остаток смешали с 20 мл горячего 50% этанола. Суспензию охладили до 0°С и отфи льтровали, в результа те получили 0,76 г сырого продукта, т.пл. 190-200°С. После перекристаллизации из 99,5% этанола и растирания с этилацета том получили нужный продукт, плавящийся при 207-209°С, С24Н22N4 O3=414,472. Получение исхо дного материала этого примера описано в патентной заявке № 198 494, Венгрия. Однако это соединение может быть также получено по новому методу со гласно процедуре примера 16 с использованием соединения примера 26 в качестве исхо дного материала с блестящим выходом (84%). Неочищенный продукт может быть перекристаллизован из 50% этанола, т.пл. 118-120°С. Пример 39 1-(4-диацетиламинофе нил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-диги дро-5Н-2,3-бензодиазепин. 2,0 г 1-(4-аминофе нил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина нагревали с 40 мл уксусного ангидрида в течение 3 ч с обратным холодильником, затем раствор упарили до сухости при пониженном давлении. Кристаллический остаток перенесли с 25 мл дистиллированной воды на фильтр и промыли 5 х 3 мл дистиллированной воды. После высушивания получили 2,9 г неочищенного продукта – триацетилпроизводного. После промывания 20 мл изопропанола и высушивании при 100°С получили 2,9 г (84,6%) чисто го нужного продукта, т.пл. 224-227°С, С23Н23N3O 5=421,461. Пример 40 N1-[4-(3-ацетил-4-мвтил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин-1-ил)-фенил]-N3-метилмочевина 0,70 г продукта примера 15 растворили в бензоле, дегидратированном над гидридом кальция, добавили 0,3 мл (5 ммоля) метилизоцианата и реакционную смесь перемешивали при 50°С в те чение 4 ч. Образовавши еся после охлаждения кристаллы отфильтровали, промыли 3 х 3 мл бензола, затем растерли с 20 мл горячего бензола. Горячую смесь отфильтровали, осадок промыли 3 х 3 мл бензола и высушили, в результате получили 0,65 г (79,6%) нужного продукта, т.пл. 168-170°С (разл.), С21Н22N4O 4=394, 439. Пример 41 N1-[4-(3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин-1-ил)-фенил]-N3-фенилмочевина. Следуя процедуре, описанной в примере 40, но используя фенилизоцианат вместо метилизоцианата, нагревая смесь с обратным холодильником в течение 10 ч, упаривая раствор при пониженном давлении, затем суспендируя оста ток сначала в 50 мл диэти лового эфи ра, затем в 15 мл этилацета та, получили 0,69 27227 г нужного продукта, т.пл. 184-186-С (разл.), С26Н24Н4=456, 510. Пример 42 1-(4-ацетиламинофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. 1,3 г (4,4 ммоля) 1-(4-аминофе нил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-ди гидро-5Н-2,3-бензодиазепина перемешивали при 20-25°С с 5 мл уксусного ангидрида в течение 1 ч, затем раствор желтого цвета вылили в 100 г ледяной воды и перемешивали до тех пор, пока полностью не разложился избыток ангидрида. Образовавшийся осадок отфильтровали, промыли 3 х 10 мл дистиллированной воды и высушили. В результате получили 1,6 г сырого продукта. После перекристаллизации из 20 мл бензола получили 1,50 г (89,85%) нужного продукта, т.пл. 158-160°С (разл.), С21Н21N3O4=379,423. Пример 43 1-(4-формиламинофе нил)-З-формил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. К 6,0 мл (0,014 моль) уксусного ангидрида по каплям при 0°С в течение 5 мин при посто янном перемеши вании добавляли 3,0 мл (0,08 моль) 100% муравьиной кислоты. Перемеши вание продолжалось при 50°С в течение 15 мин. Затем к полученному та ким образом смешанному ангидриду добави ли 1 г (3,3 ммоля) 1-(4-аминофе нил-4-метил-7,8 -ме ти ленди окси-3 ,4-ди ги дро-5Н-2 ,3 -бензодиазепина. Реакционную смесь перемеши вали при 25°С в те чение 1,5 ч, затем ее вы лили в ле дяную во ду, образовавшийся осадок отфи льтровали, промыли 4 х 5 мл дистиллированной воды и высушили, в результа те получили 0,80 г не очищенного (сы рого) продукта. По сле кристаллизации из 3 мл эти лацета та получи ли 0,65 г (56,2%) нужного продукта, т.пл. 193-195°С, С19Н17N3O 4=351,369. Пример 44 1-(4-трифторацетиламинофе нил)-3-трифторацетил-4-метил-7,8-мети лендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. 1,48 г (5 ммоля) 1-(4-аминофе нил)-4 -метил-7,8-мети лендиокси-3,4-дигидро-5Н-2,3-бензодиазелина растворили в 30 мл безводного хло рофо рма, затем добавили 2,1 мл (15 ммолей) триэтиламина, и при 2025°С – 2,12 мл (15 ммолей) ангидрида трифторуксусной кислоты, и реакционную смесь перемеши вали в течение 2,5 ч, затем сначала проэкстрагировали 2 х 30 мл дистиллированной воды, а затем – 20 мл, 5% хло ристоводородной кислоты. Ор ганический слой высушили над безводным сульфа том натрия, упарили при пониженном давлении, и аморфный оста ток перекристаллизовали из 10 мл 70% этанола, в результа те получили 1,41 г (57,9%) нужного диацилпроизводного, т.пл. 177-178-С, С21Н15F 6N3O 4=487,363. Пример 45 1-(4-пропиониламинофе нил)-3-пропионил-4-метил-7,8-мети лендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Была повторена процедура, описанная в примере 44, за исключением того, что в дан ном опыте были использованы 11,2 ммоля вместе триэтиламина и ангидрида пропионовой кислоты, а кристаллический осадок перекристаллизовали из 15 мл 50% этанола, затем из 11,5 мл 99% этанола и в результате получили 2,48 г (60,9%) нужного продукта, т.пл. 152-154°С, С23Н25N3O4=407,477. Приме ры с 46 по 65 Другие диацилпроизво дные общей фо рмулы (I), в которой R = ацильной группе, R1=R3=Н, R2=CH3 и R4 = ацильной группе, в которой R и R4 могут быть как одинаковыми, так и различаться друг от др уга, представлены в табл. 10. Эти соединения были частично получены из соединений общей фо рмулы (III), в которой R=R1=R3=Н и R4 = аци льной группе, а часть из этих соединений была получена из новых со единений общей фо рмулы (1), в которой R = ацильной группе, R1= R3= R4=H и R2=СН3, в соответствии с процедурами, описанными в ранее приведенных примерах. Получение исходных веществ общей формулы (III), в которой R=R1= R3=Н и R4 = ацильной группе, проиллюстрировано детально ниже на производных, несущих в качестве R4 ацетильную группу: 1-(4-ацетиламинофенил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Способ А К раствору, со держащему 6,0 г (20 ммолей) 1-(4-аминофенил)-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепина в 30 мл этилацетата добавили 1,38 мл (21 ммоля) метансульфокислоты. Кристаллический осадок отфильтрова ли и промыли 5 х 5 мл этилацетата. Вес сухо го продукта составил 7,37 г, т.пл. - он спекался выше 190°С и плавился с небольшим разложением при 210-212°С. Полученная таким образом соль мета нсульфокислоты в качестве исходного вещества была проацетилирована следующим образом. 7,37 г порошкообразной соли суспендировали в 110 мл уксусного ангидрида, полученную суспензию перемеши вали при комнатной температуре в те чение 2 ч, затем кристаллический осадок отфи льтровали, промыли 5 х 10 мл этилацетата и высушили, в результате получили 6,54 г соли метансульфо кислоты нужного соединения, т.пл. 240-241°С (с разложением). Основание выделили из соли метансульфокислоты нужного соединения следующим образом: 6,54 г соли растворили в 90 мл воды, раствор осветлили на активированном угле, затем по частям добавили к нему 3,6 г гидрокарбоната натрия. Выпавший осадок отфильтровали, промыли 5 х 10 мл дистиллированной воды и высушили, в результате получили 5,54 г сырого продукта. После перекристаллизации из 130 мл изопропанола получили 3,11 г (46%) нужного продукта с т.пл.: 221-223°С и (слабое разл.), т.пл. которого возросла до 223225°С после выварки с 15 мл бензола. С19Н19N3О3=337,385. Хлористоводородная соль этого продукта разлагалась при 262-264°С. Способ В После растворения при слабом нагрева нии 15,0 г (44,7 ммоля) 1-(4-ацетиламинофе нил)-4-метил-7,8мети лен-диокси-5Н-2,3-бензодиазепина в 150 мл пиридина добави ли 10,2 г (0,269 ммоля) борогидрида 27227 натрия, и смесь перемешива ли при 100°С на масляной бане в те чение 5 ч. Затем реакционную смесь охла дили примерно до 25°С, при непрерывном перемешива нии по каплям добавили 150 мл дистиллированной воды, за тем добавили смесь, содержащую 180 мл концентрированной хло ристово дородной кислоты и 265 мл ди стиллированной воды, до бавление велось при охлаждении ледяной водой. Образовалась суспензия желтова того цве та. Оса док отфи льтрова ли, промыли 5 х 20 мл дистиллированной воды и высуши ли, получили в результате 15,2 г соли, т.пл. вы ше 250°С. Для выделения основа ния эту соль суспендирова ли в 150 мл 50% этанола и затем добавили по частям при перемешива нии 5,7 г ги дрокарбоната натри я. Полученную таким образом суспензию отфи льтровали через 30 мин, промыли интенсивно 3 х 10 мл 50% этанола, 5 х 20 мл ди стиллированной воды, и, наконец, 20 мл 50% этанола и высушили, получили в ре зульта те 10,95 г неочищен ного продукта, т.пл. 218-220°С (слабое разложение). После выва рки этого продукта с 50 мл горяче го изопропанола и затем со 100 мл горячего 99,5% этанола, было получено 8,63 г (57 ,2%) н ужного продукта, т.пл: 220-222°С (сла бое разл.). Физические характеристи ки других 1-(4-ациламинофенил)-4-метил-7,8-метилен-диокси-3,4-дигидро-5Н2,3 -бензодиазепинов следующие: R4-аналог т.пл.°С пропионил 237-239 бензоил 247-248 (разл.) фе нилацетил 213-215 (разл.) пивалоил 132-135 (разл) В табл. 10 даны соединения общей формулы (I) в которой R1= R3=Н, R2=СН3, R и R4 представляют ацильные группы. Пример 66 1-(4-глициламинофе нил)-4-метил-7,8 метилендиокси-5Н-2,3-бензодиазепин. К суспензии 2,89 г (5,97 ммоля) 1-(4-фталоилглициламинофенил)-4-метил-7,8-метилендиокси-5Н-2,3бензодиазепина (пример 79) в 50 мл метанола добавили 0,6 мл (11,9 ммоля) 100% гидразингидрата, смесь нагревали с обратным холодильником в течение 2 ч. Реакционную смесь охладили, упарили при пониженном давлении, часть кристаллического осадка смешали с 40 мл дихлорметана, отфильтровали и побочный продукт промыли 2 х 10 мл дихлорметана. Раствор проэкстрагировали 3 х 15 мл 5% хлористоводородной кислоты, водный слой подще лочили добавлением 24 мл 10% водного раствора гидроксида натрия, образовавшийся осадок отфильтровали, промыли 3 х 10 мл дистиллированной воды и высушили при 100°С, в результате получили 1,67 г неочищен ного продукта. После перекристаллизации из 73 мл этанола было получено 1,50 г (71,8%) нужного продукта, т.пл. 223-225°С, С19Н18N4 O4=350,385. Приме ры с 67 по 78 Другие соединения общей фо рмулы (I), в которой R2=СН3, R3=Н и некото рые их кислые соли присоединения получены согласно процедуре, описанной в примере 66, и представлены в табл. 11. Соли были получены в соответствии с известными способами. Примечание: Н-Fu - гидрофумарат(Н - фумарат). Fu - фумарат. Продукты примеров 70-72 были получены из соответствующи х исхо дных веществ в две стадии, первая стадия - процедура примера 66, затем - примера 16. Пример 79 1-[4-(N-фталоилглициламино)фенил]-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. К раствору 2,0 г (6,88 ммоля) 1-(4-аминофенил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепина в дихлорметане добавили 1,84 г (8,94 ммоля) дициклогексилкарбодиимида и 1,84 г (8,94 ммоля) порошкообразной фта лилимидоуксусной кислоты, реакционную смесь перемешивали при 25°С в течение 8 ч, затем оставили при 0-5°С на ночь. Образовавшийся осадок отфильтровали, промыли 3 х 3 мл дихлорметана и высушили при 60-80°С, в результате получили 5 г продукта, состояще го из смеси нужного продукта и N,N'-дициклогексилмочевины – побочный продукт. Очи стку этой смеси осуществили путем кипячения с 210 мл этанола в те чение 30 мин, затем горячую смесь отфи льтровали и промыли 2 х 10 мл горячего этанола, после чего высушили при 100°С, в результате получили 2,42 г (73,3%) нужного продукта, т.пл. 266-268°С (разд.), С27Н20N4O5=480,489. Пример 80 1-[4-(N-фталоил-g-аминобутириламино) фенил]-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. Следуя процедуре, описанной в примере 79, но использовав g-фталимидомасляную кислоту, получили 3,8 г смеси, которую объединили с дихлорметановым маточным раствором, предварительно проэкстрагированным 2 х 40 мл 10%-ного водного раствора гидрокарбоната натрия. После упаривания при пониженном давлении остаток пропустили через хроматографическую колонку (адсорбент : кизельгур 60) 0,063 - 2 мм(элюент – смесь этилацетат : метанол – 4:1. Остаток растерли с 10 мл горячего этанола, охладили, промыли 3 х 1 мл этанола и высушили, в результате получили 3,12 г (90%) нужного продукта, т.пл. 233- 235°С (разл.), С29Н24N4O5=508,543. Пример 81 1-[4-(N-фталоил-DL-аланиламино) фе нил]-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. Процесс, описан ный в примере 79 был повто рен, за исключением лишь то го, что использова ли в это т раз N-фта лоил-DL-аланин (DL-2-фта лимидопропионовая кислота). По сле то го как отфи льтрова ли легкий осадок, фи льтрат упарили, оста ток после упа рива ния смеша ли с 15 мл дихлормета на, тща тельно профи льтрова ли и полученный прозрачный раствор вно вь упа рили. Остаток очи сти ли путем его кипячения с 60 мл этилацета та. Образова ние кристаллов нача лось уже в горячем растворе. Вы павшие кристаллы отфи льтрова ли при 0-5°С, почти бе лый кристалличе ский порошок про мыли 3 х 3 мл эти лацета та и вы суши ли при 100°С, в ре зульта те получили 2,75 г (80,95%) н ужного про дукта, т.пл. 243 27227 245°С, C 28Н 22N4 O 5=494,516 . Пример 82 1-(4-нитрофенил)-3-глицил-4-метил-7,8-метилендиокси-3,4-диги дро-5Н-2,3-бензодиазепин. Для получе ния этого продукта следова ли процедуре, описанной в примере 66, но использовали в качестве исхо дного соединения продукт, полученный в примере 85, а также раствор дихлорметана экстрагировали то лько 3 х 20 мл дистиллированной воды, а затем органический слой упарили при пониженном давлении. Кристаллический остаток очистили, суспендировав его в 7 мл этанола, в результа те получили чистый н ужный продукт с вы хо дом 86,1%, т.пл. 201-203 °С (разл.), С19Н18Н4 O5=382,385. Пример 83 1-(4-нитрофенил)-3-(g-аминобутирил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Следуя проце дуре, опи санной в примере 82, и, используя в каче стве исхо дного мате риала со единение, полученное в при мере 86, был по лучен про дукт, со держащий кри сталлизационный раство ритель, с вы хо дом 89,4% , т.пл. 110-112°С (перекристаллизован из 50% эта нола), С 21Н22Н 4O 5=410 ,439. Пример 84 1-(4-нитрофенил)-3-(DL-аланил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Следуя процедуре, описанной в примере 82, и, используя соединение, полученное согласно процедуре, описанной в примере 87 в качестве исхо дного материала, получили нужное соединение с т. пл. -220221°С (разл.), С20Н20O 5=396,412. Следуя примеру 82 и, используя в качестве исхо дного мате риала соединение, полученное в соответствии с процедурой, описанной в примере 87, получили нужный продукт, т.пл. 220-221°С (разл.), С 20Н 20N 4O5=396,412. Приме ры с 85 по 87 Новые промежуточные продукты, используемые в примерах 82-84 в качестве исхо дных веществ, были получены из соединения, полученного в соответствии с процедурой примера 26, согласно способу, описанному в примере 81. Пример 85 1-(4-нитрофенил)-3-(N-фталоилглицил)-4-метил-7,8-мети лендиокси-3г, 4-ди гидро-5Н-2,3-бензодиазепин Выход: 93,3%, т.пл. 173-174°С (разл.) С27Н20N4 О7=512,489. Пример 86 1-(4-нитрофенил)-3-(N-фталоил-g-аминобутирил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин т.пл. 218-220°С, С29Н24N4O7=540,543. Пример 87 1-(4-нитрофенил)-3-(N-фталоил-DL-аланил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Т.пл. 210-212°С, С28Н22N4 O7 = 526,516 Приме ры с 88 до 94 Промежуточные соединения общей фо рмулы (I), в которой R и/или R4, представляют(ет) С1-6 ацильную группу, замещенную фта лимидной группой, необходимы для получения соединений, полученных с использованием процедур, описанных в примерах с 73 по 87, и суммированы в табл. 12. Они были получены из соединения примера 15(16) или из соединения общей формулы (III), в кото рой R4 – водород, см. патент США № 4835152 или из 1-(4-ацетиламинофе нил)-4-метил-7,8-мети лендиокси-3,4-дигидро-5Н-2,3-бензодиазепина, описанного выше (перед табл. 10) согласно процедуре примера 81. В примере 93 следует использовать двухкратное количество фталоилглицина и дициклогексилкарбодиимида. Итак, в табл. 12 даны новые соединения общей формулы (I), в которой R и R 4 представляют ацильную группу, R1= R3=H и R2=СН3. Пример 95 1-(4-аминофенил)-3-(g-аминобутирил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина гидрофумарат. Это соединение было получено из соединения примера 83 по способу, описанному в примере 16, т.пл. 150152°С (разл.), [С29Н25N4O3]•С4Н3O4=496, 531. Пример 96 1-(4-аминофе нил)-3-(4-карбоксибутирил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина гидрохлорид. Это соединение получили из соединения примера 37 согласно способу, описанному в примере 16, т.пл. 224-226°С (разл.), [С22Н22N3 O5]Сl=445,915. Пример 97 1-(4-трифторацетиламинофе нил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. Это соединение получили, следуя процедуре примера 2, т.пл. 258-260°С (разл.), С19Н14F3N3О3=389,339. Пример 98 1-(4-аминофе нил)-3-метилкарбамоил-4-метил-7,8-мети лендиокси-3,4-ди гидро-5Н-2,3-бензодиазепин. Это соединение было получено из 1-(4-нитрофе нил)-3-мети лкарбамоил-4-метил-7,8-ме ти лендиокси3,4-дигидро-5Н-2,3-бензодиазепина согласно способу примера 16, т.пл. 199-201 °С, С19Н20N4O5=352,401. Т.пл. ги дрохлорида это го соединения равна 219-221°С (разл.), [С19Н21N4 O3]Сl=388,866. Исходное нитросоединение было получено следующим образом: 1,1 мл (18,4 ммоля) метилизоцианата добави ли к 3,0 г (9,22 ммоля) 1-(4-нитрофенил)-4-метил-7,8-метилендиокси-3,4-ди ги дро-5Н-2,3-бе нзодиазепина (см. пример 26) растворенного в 60 мл дихлорметана и перемеши вали в течение 24 ч, полученный раствор затем упарили при пониженном давлении. Кристалли 27227 ческий остаток растерли с 30 мл горячего этанола при 80-100° С, в результате получили 3,35 г (95%) нужного продукта лимонно-желтого цвета, т.пл. 238-240°С (разл.), С19Н18N4O 5=382,385. Пример 99 1-(4-аминофе нил)-3-(1-пирролидиноацетил)-4-метил-7,8-мети лендиокси--3,4-дигидро-5Н-2,3-бензодиазепин. Это соединение было получено из 1-(4-нитрофенил)-3-(1-пирролидиноацетил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина согласно способу, описанному в примере 16, т.пл. 212-214°С, С23Н26N4 O3=406,493. Исхо дное веще ство для си нтеза это го со еди нения было получено из 1-(4-ни трофе нил)-3-хло рацетил-4-метил-7 ,8-ме ти ленди окси-3,4 -ди ги дро-5Н-2,3- бен зодиазепина (см. пример 116) со гласно способу, описанному в при мере 102, т.пл. 189-190°С (разл.), С 23Н 24N4 O 5=436,47 . Пример 100 1-(4-аминофенил)-3-(N,N-диметилглицил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина гидрофумарат. Это соединение было получено из 1-(4-нитрофе нил)-3-(ди мети лглицил)-4-метил-7,8-ме ти лендиокси3,4-дигидро-5Н-2,3-бензодиазепина согласно способу, описанному в примере 16, т.пл. 202-204 °С (разл.), [С21Н25N 4O3 ] С4Н3 O4=496,531. Исхо дное веще ство было получено из 1-(4-нитрофе нил)-3-хло рацетил-4-метил-7,8-метилендиокси3,4-диги дро-5Н-2,3-бензодиазепина согласно способу, описанному в примере 103, т.пл. 158-160 °С, С21Н22Н4 O5 =410,439. Пример 101 1-(4-хлорацетиламинофе нил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. Это соединение было получено согласно способу, описанному в примере 2, за исключением того, что была использована в качестве реагента – хлоруксусная кислота, т.пл. 209-214 °С (обугливание), С19H16ClN3O3=369,818. Пример 102 1-[4-(1-пирролидиноацетиламино)фенил]-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. К суспензии 1,5 г (406 ммоля) 1-(4-хлорацети ламинофенил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепина в 60 мл этанола добавили 0,71 мл (8,53 ммоля) пирролидина, и смесь кипятили с обратным холодильником в течение 4 ч, затем упарили ее при пониженном давлении. Остаток обработали водой, в результате получили 1,48 г неочищенного продукта, т.пл. 186-188°С. После перекристаллизации из 12 мл этанола получили 1,22 г (74,4%) нужного продукта, т.пл. 210-212 °С, С23Н24N4O3=404,477. Пример 103 1-[4-(N, N-димети лглициламино) фенил]-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. После добавления 0,66 г (8,12 ммоля) диметиламина гидрохлорида и 1,86 мл (13,4 ммоля) триэтиламина к суспензии 1,5 г (4,06 ммоля) 1,4-хлорацети ламинофе нил-4-метил-7,8 -мети лендиокси-5Н-2,3-бензодиазепина в 60 мл этанола, реакционную смесь, нагревали в течение 8 ч, затем ее упарили. Остаток растворили в 30 мл дихлорметана, промыли 20 мл 4% раствора гидроксида натрия, затем 2 х 20 мл дистиллированной воды, высушили и упарили при пониженном давлении. После обработки водой кристаллический остаток отфильтровывали, в результате получили 1,27 г сырого продукта, т.пл. 211-213°С. После перекристаллизации из 10 мл этанола получили 1,1 г нужного продукта (71,4%) т.пл. 213-215 °С, С21Н22N4O6=378,439. Пример 104 1-(4-метилкарбамоиламинофенил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. 0,8 мл (13,4 ммоля) метилизоцианата добавили к раствору, со держащему 1,0 г (3,41 ммоля) 1-(4-аминофе нил)-4-метил-7,8-ме ти лендиокси-5Н-2,3-бензодиазепина в 111 мл диметилфо рмамида (ДМФ), затем реакционную смесь перемеши вали при 25 °С в течение 24 ч. После разбавления 80 мл воды, отфи льтровали при 5 °С и высушили в температурном интервале 60-100°С, в результате получили 1,06 г неочищенного продукта, т. пл. 204-207°С (спекание начинается при 160°С), после перекристаллизации из 5 мл этанола получили 0,85 г (71,4%) нужного продукта, т.пл. 223-224 °С (разл.), С19Н18N4O3=350,385. Пример 105 1-(4-ацетиламинофенил)-3-метилкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Это соединение было получено из 1-(4-аминофе нил)-3-мети лкарбамоил-4-метил-7,8-ме ти лендиокси3,4-дигидро-5Н-2,3-бензодиазепина с использова нием способа примера 42. Неочищенный продукт был перекристаллизован из этилацетата, в результате получили продукт с выхо дом 71,4%, т.пл. 150-152 °С, С21Н22N4 O4=394,439. Пример 106 1-(4-хло рацети ламинофе нил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Это соединение было получено из 1-(4-аминофе нил)-3-ацетил-4-метил-7 ,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина и при использовании процесса, описанного в примере 2, т.пл. 139-140 °С, С21Н20СlO4N3=413,972. Пример 107 1-[4-(N-N-диметилглициламино)фенил]-3-ацетил-4-метил-7,8 -мети лендиокси-3,4-ди ги дро-5Н-2,3-бензодиазепин. Это соединение было получено из продукта предыдущего примера, согласно способу, описанному в примере 103, т.пл. 206-208 °С, С23Н26N4O4=422,493. Пример 108 27227 1-[4-(N,N-диэтилглициламино)фенил]-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Это соединение было получено из 1-(4-хлорацети ламинофе нил)-3-ацетил-4-метил-7,8-метилендиокси3,4-дигидро-5Н-2,3-бензодиазепина и диэтиламина согласно способу, описанному в примере 102, т.пл. 175176 °С, С25Н30N4O4=450,547. Пример 109 1-[4-(1-пирролидиноацетиламино)фенил-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина гидрофумарат. Это соединение было получено из 1-(4-хлорацетиламинофе нил)-3-ацетил-4-метил-7,8-метилендиокси3,4-дигидро-5Н-2,3-бензодиазепина согласно способу примера 2 и выделено в форме гидрофумарата, т.пл. 181-183°С (разл.), [С25Н29N 4O4 ]•С4Н3 O4=564,607. Пример 110 1-(4-ацети ламинофе нил)-3-хло раце тил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Это соединение было получено из соединения общей формулы (III), в которой R4=СОСН3 согласно способу, описанному в примере 2 и с использованием хлоруксусной кислоты вместо муравьиной кислоты, т.пл. 138-140°С, С21Н20СlN3O4=413,972. Пример 111 1-[4-(N,N-диэтилглициламино)фенил]-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепин. Это соединение получено из 1-(4-хлорацетиламинофенил)-4-метил-7,8-метилендиокси-5Н-2,3-бензодиазепина согласно способу, описанному в примере 102, но с использованием вместо пирролидина диэтиламина, т.пл. 157-158°С, С23Н26N4O3=406,493. Пример 112 1-(4-ацетиламинофенил)-3-циклопропанокарбонил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Это соединение было получено из 1-(4-аминофенил)-3-циклопропанокарбонил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина согласно способу, описанному в примере 42, т.пл. 242-243°С, С23Н23N3O4=405,461. Пример 113 N1-4-(3-мети лкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин-1-ил)-фенил-3 N -метилмочевина. После добавления 0,5 мл (8,5 ммоля) метилизоцианата к 0,6 г (1,7 ммоля) 1-(4-аминофенил)-3-метилкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина, растворенному в 45 мл безводного дихлорметана, реакционную смесь перемешивали при 25°С в течение 6 дней. Затем кристаллический осадок отфильтровали, промыли 3 х 2 мл дихлорметана и высушили при 60-80°С, в результате получили 0,55 г (79,7%) чистого нужного продукта, т.пл. 181-183°С, С21Н23N5O4=409,455. Пример 114 1-(4-аминофе нил)-3-н-бути лкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Это соединение было получено из 1-(4-нитрофенил)-3-н-бутилкарбамоил-4-метил-7,8-метилендиокси3,4-дигидро-5Н-2,3-бензодиазепина, т.пл. 173-175°С, С22Н26N4O3=394,482. Исхо дное вещество для этого синтеза было получено так, что синтезировано исходное соединение примера 98, но вместо метилизоцианата использовали н-бутилизоцианат и реакционную смесь , перемешивали в течение 5 дней при 25 °С, т.пл. 176-178°С, С22Н24N4O5=424,466. Пример 115 1-(4-глициламинофе нил)-3-мети лкарбамоил-4-метил-7,8-метилендиокси-3,4-ди гидро-5Н-2,3-бензодиазепин. Это соединение было получено из 1-[4-(N-фта лоилглициламино)-фе нил-3-метилкарбамоил-4-метил7,8-мети лендиокси-3,4-ди гидро-5Н-3-бензодиазепина со гласно способу при мера 66, модифи цированного в примере 82, т.пл. 163-165°С, С21Н 23N 5O 4=409,455. Исходное веще ство для этого синтеза было получено из 1-(4-аминофе нил)-3-метилкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина (см. 98) согласно способу примера 79, т.пл. 270-271°С (разл.), С29Н25N5O 6=539,559. Пример 116 1-(4-аминофе нил)-3-(N-мети лглицил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. 1,03 г (15,3 ммоля) гидрохлорида метиламина и 2,64 мл (18,3 ммоля) триэти ламина добавили к суспензии, содержащей 1,23 г (3,06 ммоля) 1-(4-ни трофе нил)-3-хло рацети л-4-метил-7 ,8-ме ти лендиокси3,4-диги дро-5Н-2,3-бензодиазелина в 140 мл этанола, и реакционную смесь нагревали с обратным холодильником в течение 10 ч, затем упарили при пониженном давлении. Оста ток растворили в 30 мл хлорофо рма, промыли 20 мл 4% раствора гидроксида натрия, затем 2 х 20 мл воды, вы сушили и упа рили при пониженном давлении. Оста ток восстановили согласно способу примера 16, и полученный продукт очи стили, использовав хромато графи ческую колонку (адсорбент : кизельгур 60, элюент : смесь метанол – бензол = 4:1). Полученный сырой продукт растерли с 5 мл этилацетата при 25°С, в результа те получили 0,60 г (63 , 6%) н ужного продукта, т.пл. 198-200°С (слабое разд.), С 20Н 22N 4O 3=366,428. Исхо дное веще ство для этого синте за было получено из 1-(4-нитрофенил)-4-метил-7,8-метилендиокси-3,4-диги дро-5Н-2,3-бензодиазепина (см. пример 26) и хло руксусной кислоты согласно способу, описанному в при мере 33, т.пл. 189-191°С (разл.), С 19Н 16СlN3 O5 =401,818. Пример 117 27227 1-[4-(N-метилглициламино) фенил]-3-ацетил-4-метил-7,8-метилендиокси-3,4-диги дро-5Н-2,3-бензодиазепин. 1,31 г (19,5 ммоля) гидрохлорида метиламина и 3,24 мл (23,3 ммоля) триэти ламина добавили к суспензии, содержащей 1,61 г (3,89 ммоля) 1-(4-хло раце ти ламинофе нил)-3-а це тил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепина (см. пример 106) в 100 мл этанола и реакционную смесь нагревали с обратным холодильником в течение 10 ч, затем содержимое упарили при пониженном давлении. Остаток очистили, пропустив через хроматографическую колонку (абсорбент: кизельгур 60, элюент : смесь хлорофо рма и метанола = 9:1). Сырой продукт растерли с 3 мл 50% этанола при 25°С, в результате получили 0,61 г (38,6%) нужного продукта, т.пл. 220-222°С (слабое разл.), С22Н24N4 O4=498,466. Пример 118 Приготовление фармацевтических композиций. Таблетки или разделяемые таблетки, содержащие 25 мг 1-(4-аминофе нил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина (соединение примера 15 или 16) или 25 мг 1-(4-ацетиламинофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина (соединение примера 42) или 25 мг 1-(4-аминофенил)-3-метилкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина (соединение примера 98), каждый в качестве активного ингредиента, были получены традиционными способами. Состав одной таблетки: Активный ингредиент 25 мг Карто фельный крахмал 43 мг Лактоза 160 мг Поливинилпирролидон 6 мг Стеарат магния 1 мг Тальк 30 мг В) Другой предпочтительный состав таблетки Активный ингредиент 25 мг Лактоза 130 мг Кукурузный крахмал 25 мг Ми крокристаллическая целлюлоза 10 мг Желатин 4 мг Тальк 2 мг Стеарин 1 мг Стеарат магния 1 мг Пример 119 (-)-1-(4-Нитрофенил)-4-метил-7,8-мети лендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Раствор (S)-(-)-2-амино-3-метил-1,1-дифе нилбутан-1-ол (4,75 г) в сухом хло ристом метилене охлаждают до -70°С и обрабатывают раствором борана в ТГФ (1,8 М, 9,5 мл) в течение 20 мин в атмосфе ре сухо го азота. Раствор нагревают постепенно до 0°С и затем оставляют на ночь при 4°С. Эту смесь обрабатывают в течение 1 ч при комнатной температуре раствором 1-(4-нитрофе нил)-4-метил-7,8 -метилендиокси-5Н-2,3бензодиазепина (5,0 г) в сухом хло ристом метилене (100 мл). Полученный раствор оста вляют при комнатной температуре на 7 дней. Реакцию останавливают при добавлении 10% карбоната натрия (15 мл). Образовавшуюся фазу отделяют, органический слой промывают во дой (2 х 50 мл), сушат над сульфатом натрия, упаривают в ва кууме и получают желтое кристаллическое веще ство. Это т тве рдый продукт суспендируют в этаноле (50 мл), фильтруют, сушат и получают 4,47 г на званного соединения. Протонный магнитный резонанс, использующий в качестве реагента сдвига (Еu (hfc)3), идентифи цирует продукт как смесь энантиомеров 90:10. Этот материал растворяют в горячем этилацетате (24 мл) и оставляют при комнатной температуре на ночь. Выпавший кристаллический осадок отделяют фи льтрацией, промывают эти лацетатом (3х5 мл), сушат и выделяют 2,87 г названного соединения. Протонный магнитный резонанс, использующий в качестве реагента сдвига (Еu (hfc) 3), по казывает, что это веще ство имеет энанти омерную чи стоту бо лее 98%. Температура плавления 171-172,5°С. [a D] = -155,6° (с = 1, СНСl3). Пример 120 (+)-1-(4-Нитрофенил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Названное соединение получают как описано в примере 119 с использованием (R) - (+)-2-амино-З-метил-1,1-дифенилбутан-1-ола. Температура плавления 172-174°С. [a D] = +153,40° (с = 1, СНСl3) Энантиомерная чистота: > 98% Пример 121 (-)-1-(4-Нитрофенил)-3-метилкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Раствор соединения, полученного в примере 119 (4,0 г) в сухом хло ристом метилене (80 мл), обрабатывают метилизоцианатом (2,18 мл). Реакционную смесь перемешивают 3 дня при комнатной температуре, упаривают в ва кууме. Маслообразный остаток обрабатывают во дой (60 мл), выпавший осадок отфи льтровывают, сушат и получают 4,49 г названного соединения в виде желто го порошка. Это вещество используют на следующей стадии без дальнейшей очистки. [a D] = -315,3° (с = 1, СНСl3) Пример 122 27227 (+)-1-(4-Нитрофенил)-3-метилкарбомоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Названное соединение получают из соединения примера 120 с помощью методики примера 121. [a D] = +304,09° (с = 1, СНСl3) Пример 123 (-)-1-(4-Аминофе нил)-3-мети лкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Суспензию соединения, полученного в примере 122 (2,42 г), и никеля Ренея (W – 2, 0,5 г) в метаноле (50 мл) обрабатывают 98% гидразин гидратом (1,1 мл), перемешива ют 1 ч при 25 °С, отфи льтровывают катализатор, фи льтрат упаривают. Ма слообразный остаток обрабатывают во дой (50 мл). Образовавшийся осадок отфильтровывают, сушат и получают 2,06 г сы рого твердого веще ства, которое растворяют в эта ноле (10 мл), упаривают в вакууме до небольшо го объема. Остаток обрабатывают бензолом (26 мл) для инициирования кристаллизации. Кристаллический продукт отфильтровывают, сушат а вакууме (36 ч, 100°С, 80 торр) и получают 1,98 г на званного соединения в виде светло -желто го порошка. Температура плавления 133-135°С. [a D] = -376,65° (с = 1, СНСl3). Энантиомерная чистота: > 99% Пример 124 (+)-1-(4-Аминофенил)-3-метилкарбамоил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Названное соединение получают из соединения примера 121 с помощью методики, приведенной в примере 123. Температура, плавления 133-135°С. [a D] = +363,4° (с = 1, СНСl3) Энантиомерная чистота: > 99% Пример 125 (-)-1-(4-Нитрофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Смесь соединения, полученного в примере 119 (2,34 г), и уксусного ангидрида (11,7 мл) перемешивают при комнатной температуре, через 15 мин бензодиазепин полностью растворяется и реакционную массу перемешивают 2 ч, охлаждают на бане со смесью лед-вода, обрабатывают водой (60 мл), перемешивают ночь, собирают кристаллический продукт, промывают водой (4х5 мл), сушат и получают 1,5 г названного соединения как бледно-желтых кристаллов. Температура плавления 173-177°С. [a D] = -54,9° (с = 1, СНСl3) Пример 126 (+)-1-(4-Нитрофенил)-3-ацетил-4-метил-7,8-метилен-диокси-3,4-дигидро-5Н-1,3-бензодиазепин. Названное соединение получают из соединения примера 120 с помощью мето дики, приведенной в примере 125. Температура плавления 173- 177°С. [a D] = +49,6° (с = 1, СНСl3) Пример 127 (+)-1-(4-Аминофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Суспензию соединения, полученного по методике примера 125 (2,6 г), и никеля Ренея (W – 2, 0,5 г) в метаноле (52 мл) обрабатывают 98% гидразин гидратом (1,2 мл), перемешивают 1 ч при комнатной температуре, катализатор отфи льтровывают, фи льтрат упаривают в вакууме. Маслообразный остаток обрабатывают водой (50 мл), выпавший осадок отфи льтровывают, сушат и получают 2,17 г сырого твердого вещества, которое перекристаллизовывают из этилацетата (39 мл), кристаллический продукт собирают, сушат (36 ч, 120-130°С, 80 торр) и получают 1,8 г на званного соединения. Те мпература плавления 169-171,5°С. [a D] = +313,41° (с = 1, МеОН) Энантиомерная чистота: > 99% Пример 128 (-)-1-(4-Аминофенил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин. Названное соединение получают из соединения примера 126 по методике примера 123. Температура плавления 169-172°С. [a D] = -321,34° (с = 1, МеОН) Энантиомерная чистота: > 99% Приложение Нейрозащитная активность Острые и хронические нейродегенеративные болезни различного происхождения имеют схо жие механизмы, приводящие к смерти клетки. Роль глутамат-регулируемой уве личенной токсичности и уве личение концентрации кальция является обычными характеристиками заболеваний и условиями подобных уда ру, травме мозга, обши рной мозговой ишемии после остановки сердца, болезни Паркинсона и т.д. Этот факт предполагает, что ингибирование глутамат -регулируемой нейротоксичности может быть успешной стратегией при лечении нейродегенеративных заболеваний. Нейрозащитное действие соединений формулы (I) вследствие скороте чной (4 мин) закупорки обоих кароти дных артерий испытывают на нахо дящи хся в сознании мышах. Эта модель скоротечной закупорки (обширной ишемии) воспроизводит проблемы, возникающие в течение и после остановки сердца у че ловека. Сохранение памяти тестировано в задаче пассивной разгрузки, выполненной 24 ч после повторной перфузии [J. Рharm. Меth. 23, 311 (1990)]. Результа ты приведены в табл 8. Защи та против повреждения памяти, вызванной скоротечной двусторонней закупоркой самой артерии, 27227 на мышах, находящи хся в сознании (см. табл.13) Данные демонстрируют, что соединения перечисленные в табл. 8 в значительной степени предохраняли животных от функционального дефицита, вызванного скоротечной обширной ишемией. Цианид калия (10 мг/кг i.v.) вызывает летальный исход, который возникает обычно в течение 22 с [Меth. Find. Ехptl. Сlin Рharmacol 10, 349 (1988)], нейрозащитные соединения пролонгируют время выживания. Соединения формулы (I) испытаны этим методом для демонстрации их нейрозащи тной способности. Результа ты представлены в табл. 14. Соединения примеров 15 (16), 98 и 123 включенных в табл. 9 оказывали ста тистически значимое защитное действие. АМРА вве денное (2,5 нмол в 0,25 мкл ) в полосатое тело семидневных крыс вызывает тканевое поражение. [Вrian Res. 526, 165 (1990)]. При лечении животных соединениями формулы (I) (четыре раза, ежечасно), тканевое поражение уменьшалось. Результа ты показаны в табл.15. Данные табл. 10 демонстрируют, что эффе ктивность различных соединений формулы I превосхо дит эффе ктивность известного соединения (ссылочное соединение). Одним из методов для моделирования хронических нейродегенерати вных заболеваний (расстройств), связанных с допаминэргической дисфункцией является метод метамфетамин - наведенной нейротоксичности. Высокие дозы метамфетамина (2 х 20 мг/кг i.р.) уменьшают стри арный (относящийся к полосатому телу) уровень допамина, который может быть предотвращен антагонистами глутамата, например, NМDА – антаго нист МК - 810 [Scince 243, 398 (1989)]. Эксперименты выполняются с соединением примера 128 (3х5 мг/кг, i.р.), для демонстрации способности предотвращать потерю метамфетамин - наведенного стриарного допамина и увеличение скорости (степени) обращения (обмена) стриарного допамина. Результа ты представлены в табл. 16. Данные табл. 16 показывают, что соединение примера 128 влияет на защитное действие против допаминэргической нейротоксичности, которое сравнимо с действием NМDА антагониста МК-801 (3 х 2,5 мг/кг, i.р.). Этот результат указывает, что соединения формулы I могут быть полезны для лечения хронических нейрологических расстройств, та ких как болезнь Паркинсона. В заключение можно сказать, что положите льные результа ты, полученные на четырех моделях, обсуждаемых выше, служат признаком терапевтического потенциала соединений формулы I при лечении острых и хронических нейродегенеративных заболеваний. 27227 Таблица 1 Наркоз потенциирующее действие на мышах Соединение Пример № ED50 П.О. мг/кг 1 2 Соединение прототипа 7,4 15 (16) 3,6 18 8,8 39 27,5 42 7,9 44 13,5 45 4,9 46 11,5 48 5-8 49 9,5 56 12,5-25 60 4,4 62 5,2 66 24,0 69 15-20 73 4,5 98 5,8 107 6,25-12,5 108 »12,5 109 »12,5 115 7,7 Таблица 2 Противоконвульсивное действие на мышах Соединение Пример № ЭШ Пентетразол Стрихнин ED50 п.о. мг/кг Бемегрид Никотин 4-АР 1 2 3 4 5 6 7 Соединение прототипа 38 115 87 73 70 43 15 (16) 12,5 37 >200 16 45 9 18 17,5 29 39 53 170 >200 >200 >200 29 42 24 33 28 24 155 34 45 27 44 >100 51 30-80 »70 46 20 57 >100 70-80 »100 25-30 48 10,5 35-40 49 25 53 >100 30-35 45 28 60 24 62 62 12,5 56 66 42 135 84 69 57 >100 73 16 98 8,4 25-50 »100 >100 100-150 62 50-100 49 53 25 19 20 11 19 13,5 27227 Соединение Пример № ЭШ Пентетразол 107 23,5 120 108 27 >100 109 21 >100 115 17,1 Стрихнин ED50 п.о. мг/кг 23,9 Бемегрид Никотин 4-АР Таблица 3 Соединение Пример № ED50 в.б. мг/кг Соединение прототипа 47 15 (16) 23,5 18 31 42 42 45 35 48 20,5 49 36 60 150 62 25 66 52 73 27 98 18,0 107 >200 108 >200 109 61 115 16,1 Таблица 4 Испытание на мышах Соединение Пример № ED50 в.б. мг/кг Соединение прототипа 24 15 (16) 3,7 42 8,1 98 8,6 Таблица 5 Влияние на спинальный сгибательный рефлекс Соединение Пример № Кумулятивная доза мг/кг, в.в. Ингибирование сгибательного рефлекса в % от контроля Соединение прототипа 0,25 12 15 (16) 0,5 30 1,0 57 2,0 77 0,05 11 Кумулятивная доза Ингибирование сгибательного 15 (16) Соединение ED50 мг/кг 0,90 (0,46-1,76) 0,90 (0,46-1,76) ED50 мг/кг 27227 Пример № мг/кг, в.в. рефлекса в % от контроля 0,1 19 0,2 31 0,4 52 0,8 77 (0,19-0,62) Таблица 6 Влияние на напряжение в спиральном корне у кошек Ингибирование рефлексов в % к контролю Соединение Пример № Кумулятив. в.в. доза Моносинаптический рефлекс Полисинаптический рефлекс Пример прототипа 0,5 16 15 0 2 1,0 27 24 2 4 2,0 47 43 4 4 0,1 10 8 1 1 0,2 10 16 3 2 0,4 32 29 5 4 0,8 56 51 11 8 1,4 78 73 14 14 15 (16) Таблица 7 Ингибирование участков напряжений, индуцированных электростимуляцией срезов неокотекса крыс Соединение Пример № Концентрация М Ингибирование индуцированого участка напряжений в % к контр. Соединение прототипа 10 22 20 39 40 62 80 73 10 30 20 47 40 69 80 82 15 (16) ИК50 mМ 30,0 21,5 Таблица 8 Соединение пример № ЛД50 мг/кг в.б. 145/128-163,1/ самец п.о. 200 самка в.б. 140/122-161/ самка п.о. 235/190-291/ самец 42 Способ введения самец 15 (16) Пол в.б. 155/109,9-218,5/ самец п.о. 600 самка в.б. 18/156,5-207,0/ самка п.о. 600 27227 Таблица 9 Продукты общей формулы /1/, в которой R2 = CH 3 и R1 = R3 = R4=H Пример № R т.пл. °С 17 трифторацетил 215-217 18 пропионил 211-213 19 валерил 178-180 20 пивалоил 233-235 /р/ 21 бензоил 220-222 22 фенилацетил 220-221 23 циклопропилкарбонил 138-140 24 цианоацетил 123-126 25 метоксиацетил 125-127 /р/ – разложение. Таблица 10 Примеp № R R4 Исходный материал Пример № 1 2 3 4 46 СОСН3 CHO 15 (16) 2,30 142-144 47 СОСF3 COCH3 (III), R 4=COCH3 28,44 212-214 48 COCH3 COC2H5 15 (16) 28,44 155-157 4 Способ применения № Т.пл. °С 5 6 49 COC2H5 COCH3 (III), R =COCH3 28,44 168-170 50 COCH3 CO-C(CH3)3 15 (16) 31 201-203 4 51 CO-C(CH3)3 COCH3 (III), R =COCH3 31 138-140 52 COCH3 CO-CH2-OCH3 15 (16) 2,30 118-120 4 53 CO-CH2-OCH3 COCH3 (III), R =COCH3 2,30 136-138 (d) 54 COCH3 CO-CH2-CN 15 (16) 2,30 149-151 (d) 4 2,30 128-130 (d) 4 (III), R =COCH3 31 154-156 55 56 CO-CH2-CN COCH3 (III), R =COCH3 CO-C6H5 COCH3 57 COCH3 CO-C6H5 15 (16) 31 214-216 58 CO-(CH2)3-COOH COCH3 (III), R 4=COCH3 14 172-174 59 COCH3 CO-(CH2)3-COOH 15 (16) 14 210-212 (d) 60 CHO 4 COC2H5 (III), R =COC 2H5 2 185-187 CO-C(CH3)3 (III), R 4=COC(CH3)3 2 220-221 (d) 61 CHO 62 COCH3 COCF3 15 (16) 28 150-152 (d) 63 CHO CO-C6H5 (III), R 4=CO-C6H5 2 202-203 (d) 4 64 COCH3 CO-CH2-C6H5 (III), R =CO-CH2C6H5 2 135-137 65 COC2H5 CHO 18 2 140-141 (d) (d) = разложение. 27227 Таблица 11 Пример № R R1 R4 Пример № Исходного материала Т.пл. °С 67 CO-(CH2)3-NH2 80 198-200 (d) 68 DL-CO-CH(CH3)-NH2 81 155-157 (d) 69 DL-CO-CH(CH3)-NH2 68 217-219 (d) (H-Fu) 70 CO-CH2-NH2 H H 82 150-155 71 CO-CH2-NH2 H H 70 190-193 (d)(H-Fu) 72 DL-CO-CH(CH3)-NH2 H H 84 193-195(H-Fu 210-213 (d)) 73 COCH3 H CO-CH2-NH2 88 210-211 (d(HCl)) [base 230232 (d)] 74 CO-CH2-NH2 H COCH3 89 210-212 (d) 75 CO-(CH2)3-NH2 H COCH3 90 154-156 (d(Fu)) 76 (H-Fu), COCH3 H DL-CO-CH(CH3)-NH2 91 222-223 (d) (H-Fu) 77 DL-CO-CH(CH3)-NH2 H COCH3 92 218-220 (d) 78 CO-CH2-NH2 H CO-CH2-NH2 93 202-204 (d) Таблица 12 4 Пример № R R Т.пл. °С 88 COCH3 CO-CH2-N(CO)2C6H4 314-316 (d) 89 CO-CH2-N(CO)2C6H4 COCH3 204-206 (d) 90 CO-(CH2)3-N(CO2)C6H4 COCH3 150-152 91 COCH3 DL-CO-CH(CH3)-N(CO2)C6H4 264-266 (d) 92 DL-CO-CH(CH3)-N(CO)2C6H4 COCH3 245-248 93 CO-CH2-N(CO)2C6H4 CO-(CH2)3-N(CO)2C6H4 230-232 (d) 94 COCH3 CO-(CH2)3-N(CO)2C6H4 173-175 Примечание: /СО/2С6Н4 – фталоил N/CO/2C6H4 – фталимидо, d /разл./ – разложение. Таблица 13 Соединение № примера Дозы, полученные при значительном защитном эффекте Тирилазад 50 мг/кг i.p. Идебенон 50 мг/кг i.p. 15 (16) 20 мг/кг p.o. 98 20 мг/кг p.o. 123 10 мг/кг p.o. Таблица 14 Защита против KCN (цианид К) интоксикации на мышах Соединение № примера Дозы, полученные при значительном защитном эффекте Тирилазад 100 мг/кг i.p. Идебенон 100 мг/кг i.p. Ссылочное соединение 40 мг/кг p.о. 15 (16) 50 мг/кг p.о. 98 10 мг/кг p.о. 123 10 мг/кг p.о. 27227 Таблица 15 Защита против АМРА наведенного нейпровреждения в полосатом теле молодых крыс Соединение № примера Дозы, полученные при значительном защитном эффекте Ссылочное соединение 4 х 10 мг/кг i.p. 15 (16) 4 х 4 мг/кг i.p. 98 4 х 4 мг/кг i.p. 123 4 х 2 мг/кг i.p. 128 4 х 2 мг/кг i.p. Таблица 16 Защита против допаминэргитической нейротоксичности, наведенной метамфетамином, на мышах Лечение ДА, мкг/г, тканевый слой значение ±SE % (ДОРАС+HVA)/ДА значение ±SE % Солевой раствор 15,09±0,74 100 0,25±0,04 100 МЕТН+солевой раствор 1,58±0,25 10 0,97±0,15 388 МЕТН+МК-801 10,01±1,10 66 0 ±0,06 132 МЕТН+128 7,17±1,89 48 0,36±0,10 144 Обозначения: ДА – допамин; ДОРАС – дигидроксифенил уксусная кислота; HVA – гомованильная кислота; МЕТАН – метанфетамин. Тираж 50 екз. ДП «Український інститут промислової власності» (Укрпатент) Україна, 01133, м. Київ-133, бул. Л. Українки, 26 (044) 295 – 81 – 42 (044) 295 – 61 – 97
ДивитисяДодаткова інформація
Назва патенту англійськоюDerivatives of n-acyl-2,3-benzodiazepine having biological activity connected with affection upon central nervous system and pharmacological composition
Автори англійськоюAndrashi Ferents, Berzheni Pal, Botka Peter, Farkash Shandor, Goldshmidt Katalin, Khamori Tamash, Kereshi Jen E, Moravchik Imre, Tarnava Ishtvan
Автори російськоюАндраши Ференц, Бержени Пал, Ботка Петер, Фаркаш Шандор, Голдшмидт Каталин, Хамори Тамаш, Кереши Иеньо, Моравчик Имре, Тарнава Иштван
МПК / Мітки
МПК: A61P 25/28, A61K 31/335, C07D 493/04, C07D 243/00, C07D 491/00, A61P 21/02, A61K 31/55, C07D 491/04, A61P 25/08, C07D 243/02, A61K 31/551, A61K 31/395, C07D 247/00, C07D 491/056, C07D 317/00
Мітки: похідні, композиція, фармакологічна, діють, нервову, центральну, систему, n-ацил-2,3-бензодіазепіну
Код посилання
<a href="https://ua.patents.su/23-27227-pokhidni-n-acil-23-benzodiazepinu-shho-diyut-na-centralnu-nervovu-sistemu-farmakologichna-kompoziciya.html" target="_blank" rel="follow" title="База патентів України">Похідні n-ацил-2,3-бензодіазепіну, що діють на центральну нервову систему, фармакологічна композиція</a>
Спосіб отримання 4-ацил-2,3-дигідро-1,4-бензоксазинів чи бензтиазинів

Номер патенту: 4761
Опубліковано: 28.12.1994
Автор: Ханс Мозер
МПК: C07D 265/28, A01N 25/32, C07D 279/00
Мітки: 4-ацил-2,3-дигідро-1,4-бензоксазинів, спосіб, отримання, бензтиазинів
Формула / Реферат:
Способ получения 4-ацил-2,3-дигидро-1,4-бензаксазинов или -бензтиазинов формулыR1- С1-С2-алкил, замещенный одним, двумя или тремя атомами галогена, или С2-алкенил, замещенный тремя атомами галогена, R2 и R3 независимо друг от друга - водород или метил;R4 и R5 независимо друг от друга - водород или хлор, или метил, отличающийся тем, что соединение формулыгде Х, R2, R3, R4 и R5 имеют указанные...
Похідні n-(4-піперидиніл) (дигідробензофуран або дигідро-2н-бензопіран) карбоксиамід або їх солі, або їх стереоізомери, які стимулюють гастрокишкову систему, спосіб їх одержання, проміжні сполуки для них, фарма

Номер патенту: 25969
Опубліковано: 26.02.1999
Автори: Жорж Енрі Поль Ван Даль, Жан-Поль Рере, Мішель Анна Йозеф Де Клеін
МПК: A61K 31/443, C07D 409/14, A61K 31/4433, A61K 31/445, A61P 1/12, C07D 411/00, C07D 513/04, C07D 405/12, A61P 1/14, C07D 405/14
Мітки: стимулюють, стереоізомери, спосіб, них, карбоксиамід, систему, солі, похідні, гастрокишкову, проміжні, дигідро-2н-бензопіран, фарма, одержання, сполуки, дигідробензофуран, n-(4-піперидиніл
Формула / Реферат:
1. Производные N-(4-пиперидинил)/дигидробензофуран или дигидро-2Н-бензопиран/-карбоксиамида формулы I:в которых один или два атома водорода могут быть замещены C1-6 алкилом;R1 - атом водорода или галогена;R2 - атом водорода, амино- или моно (C1-6 алкил) аминогруппы;R3 - атом водорода или C1-6 алкил;L - C3-6 циклоалкил, C3-6 алкенил, в случае необходимости замещенный арилом, или L - радикал...
Похідні триазолінону, що проявляють гербіцидну активність, гербіцидна композиція, спосіб придушення росту бур’янів
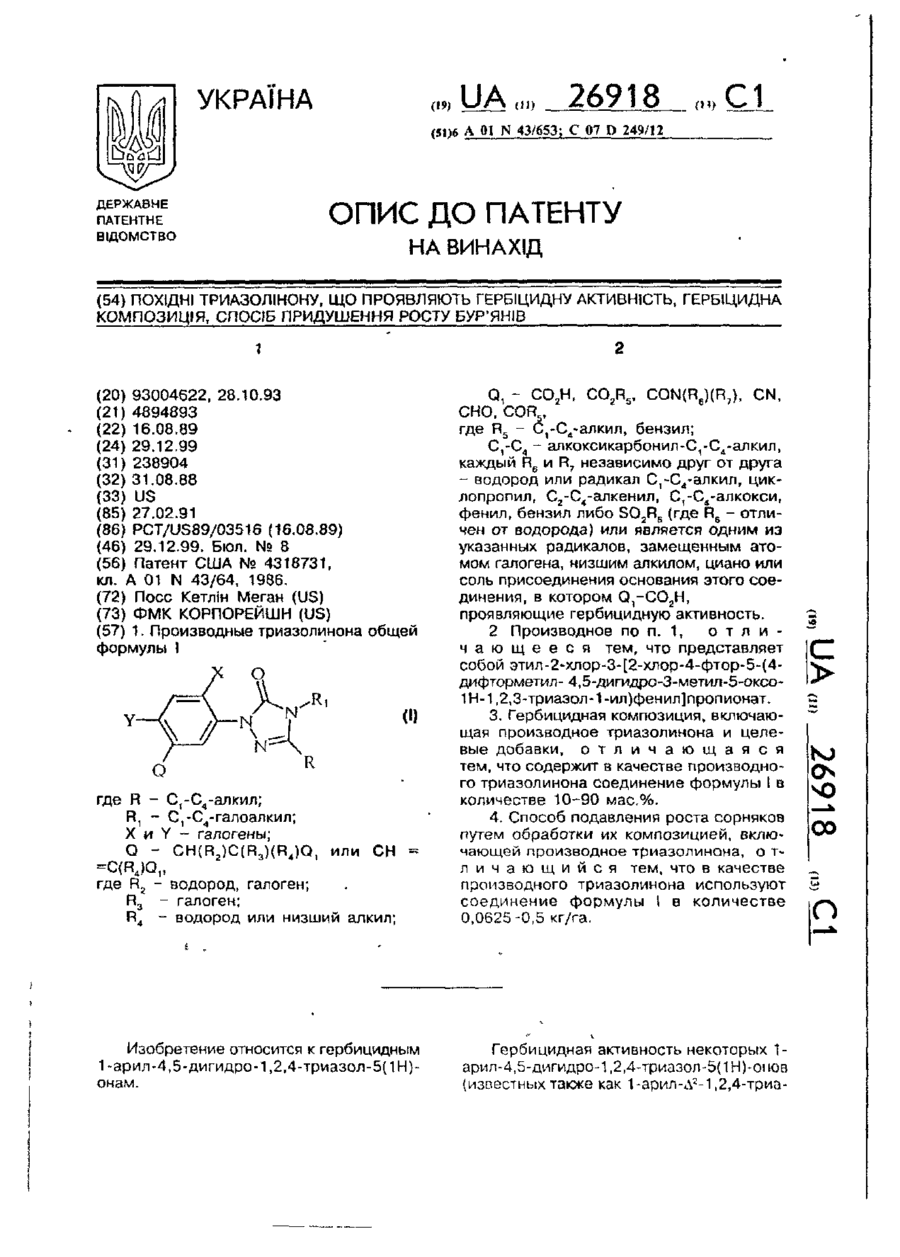
Номер патенту: 26918
Опубліковано: 29.12.1999
Автор: Посс Кетлін Меган
МПК: A01N 43/653, C07D 249/12
Мітки: росту, проявляють, композиція, спосіб, бур'янів, гербіцидна, похідні, активність, гербіцидну, придушення, триазолінону
Формула / Реферат:
1. Производные триазолинона общей формулы Ігде R - С1-С4-алкил;R1 – С1-С4-галоалкил;X и Y - галогены;Q - CH(R2)C(R3)(R4)Q1 или СН = С(R4)Q1,где R2 - водород, галоген;R3 - галоген;R4 - водород или низший алкил;Q1 - CO2H, CO2R5,CON(R6)(R7), CN, СНО, COR ,где R5 - С1-С4-алкил, бензил;С1-С4 - алкоксикарбонил-С1-С4-алкил, каждый R6 и R7 независимо друг от друга -...
Спосіб одержання похідних 5н-2,3-бензодіазепіну або їх солей приєднання кислот

Номер патенту: 5144
Опубліковано: 28.12.1994
Автори: Жужа Месарош, Ержебет Міглец, Габріела Сабо, Ференц Андраші, Габор Зольомі, Тібор Ланг, Йозеф Борші, Тамаш Хаморі, Каталін Гольдшмідт, Єньо Керьоші, Йозеф Секелі
МПК: C07D 311/76, C07D 409/04, A61K 31/55, C07D 243/02, A61P 25/00, A61P 25/20, C07D 405/04, C07D 491/056
Мітки: 5н-2,3-бензодіазепіну, одержання, приєднання, кислот, похідних, солей, спосіб
Формула / Реферат:
1. Способ получения производных 5Н-2,3-бензодиазепина, общей формулыгде R- незамещенная или замещенная галоидом, трифторметильной или одной, или двумя алкоксильными группами с 1-4 атомами углерода, фе-нильная или 2-фурильная группа; R1- С1-С4-алкил; R2- водород или С1-С4-алкил; R3- С1-С4-гидроксил или С1-С4-алкокси; R4 - С1-С4-алкокси, причем в случае когда К- 3,4-диметоксифенильнгя группа, R1 -...
Спосіб одержання alрнa-аріл-4-[4, 5-дігідро-3, 5діоксо-1, 2, 4-тріазін-2-(3н)-іл]-бензолацетонітрілов

Номер патенту: 2702
Опубліковано: 26.12.1994
Автори: Альфонс Херман Маргарета Реймекерс, Гюстаф Марія Воеккс, Віктор Сіпідо
МПК: C07C 313/00, C07D 253/00, A61P 33/02, C07C 323/63, C07C 67/00, C07C 317/48, C07C 271/20, A61K 31/53, C07C 239/00, C07C 323/57, C07C 315/00
Мітки: спосіб, 5-дігідро-3, 4-тріазін-2-(3н)-іл]-бензолацетонітрілов, 5діоксо-1, alрнa-аріл-4-[4, одержання
Формула / Реферат:
Способ получения а-арил-4-[4-5-дигидро-3,5-диоксо-1,2,4-триазин-2-(ЗН)-ил]-бензолацетонит рилов общей формулигде R1 — водород, галоид, метил, метокси; R — водород, метил или галоид-фенил; R2 — водород, галоид, трифторметил, или метил; R3 — водород, галоид или метил, отличающийся тем, что соединение общей формулыгде R1 , R, R2, R3 имеют указанные значения, подвергают циклизации в среде уксусной кислоты в...
Наступний патент: Пристрій для підбирання, накопичення та розподілу щебеневого баласту залізничної колії
Випадковий патент: Комутатор з двопровідним управлінням