Спосіб твердофазного синтезу пептидів та застосування активованої твердої фази в способі
Номер патенту: 86023
Опубліковано: 25.03.2009
Автори: Брахт Франц-Петер, Хаберл Удо, Франк Ханс-Георг, Рібка Андреас






Формула / Реферат
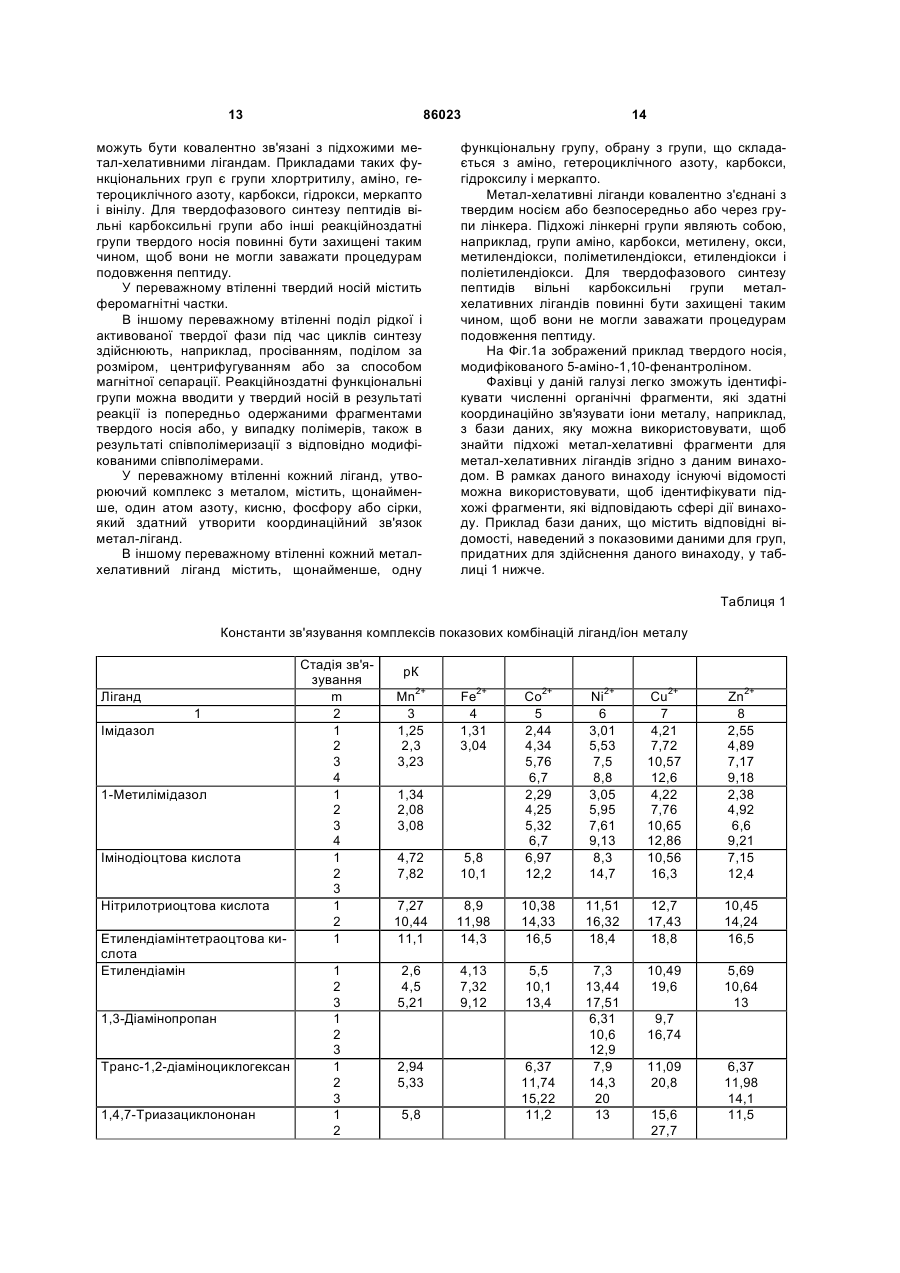
1. Застосування активованої твердої фази, що містить твердий носій, метал-хелатні ліганди, ковалентно зв'язані з твердим носієм, іони металу Мn+ з n = 1-3, координаційно зв'язані із згаданими метал-хелатними лігандами, причому вказана активована тверда фаза здатна забезпечувати координаційні ділянки для координаційного та оборотного приєднання якірної частини пептиду, для твердофазового синтезу пептидів на вказаній активованій твердій фазі, при цьому пептид являє собою «зростаючий пептид» і здатний піддаватися процедурам подовження пептиду.
2. Застосування за п. 1, яке відрізняється тим, що використано твердий носій на основі діоксиду кремнію, скла або целюлози, чи полімеру, вибраного з групи, що складається з полістирольних смол, меламінових смол і полівінілових спиртів.
3. Застосування за будь-яким з пп. 1-2, яке відрізняється тим, що кожний метал-хелатний ліганд містить щонайменше один атом азоту, кисню, фосфору або сірки, який здатний утворювати координаційний зв'язок ліганд-метал.
4. Застосування за будь-яким з пп. 1-3, яке відрізняється тим, що кожний метал-хелатний ліганд містить щонайменше одну функціональну групу, вибрану з групи, що складається з аміно, гетероциклічного азоту, карбокси, гідроксилу і меркапто.
5. Застосування за будь-яким з пп. 1-4, яке відрізняється тим, що кожний метал-хелатний ліганд, ковалентно зв'язаний з твердим носієм, містить щонайменше один фрагмент, вибраний з групи, що складається з трифенілфосфінових фрагментів, амінопуринових фрагментів, переважно 6-амінопуринових фрагментів, фталоціанінових фрагментів, 1,10-фенантролінових фрагментів, переважно 5-аміно-1,10-фенантролінових фрагментів, терпіридинових фрагментів, переважно 4'-аміно-[2,2';6',2'']терпіридинових фрагментів, триазациклононанових фрагментів, переважно [1,4,7]-триазациклононанових фрагментів, і тетраазациклододеканілових фрагментів, переважно [1,4,7,10]-тетраазациклододеканілових фрагментів.
6. Застосування за будь-яким з пп. 1-5, яке відрізняється тим, що іони металу Мn+ вибрані з групи, що складається з іонів Mn2+, Cu2+, Ni2+, Co2+, Zn2+, Mg2+, Ca2+, Fe2+, Fe3+ і лантаніду, особливо переважно Мn+ вибраний з Cu2+, Ni2+, Co2+, Zn2+, Mg2+.
7. Застосування за п. 1, яке відрізняється тим, що якірна частина пептиду може бути відокремлена від вказаної активованої твердої фази доданням конкурентного ліганду.
8. Застосування за п. 7, яке відрізняється тим, що конкурентний ліганд містить щонайменше один фрагмент, здатний утворювати комплекс з іонами металів, переважно азотовмісний фрагмент, вибраний з групи, що складається з імідазольних, N-метилімідазольних, амінопуринових, фенантролінових, біпіридинових, терпіридинових, триазациклононанових, тетраазациклододеканових фрагментів, фрагментів імінодіоцтової кислоти, фрагментів нітрилотриоцтової кислоти і фрагментів етилендіамінтетраоцтової кислоти.
9. Застосування за п. 8, яке відрізняється тим, що моно- або олігомерні амінокислоти можуть бути приєднані до С- або N-кінця зростаючого пептиду за протоколом послідовних реакцій типу протоколу Мерифілда.
10. Застосування за будь-яким з пп. 7-9, яке відрізняється тим, що якірна частина пептиду містить щонайменше один фрагмент, що утворює комплекс з іоном металу, причому кожний із вказаних фрагментів включає щонайменше одну групу, що містить азот, кисень, фосфор або сірку, яка здатна координувати іони металу активованої твердої фази.
11. Застосування за будь-яким з пп. 7-10, яке відрізняється тим, що азотовмісну групу, яка здатна координувати іони металу активованої твердої фази, вибрано з групи, що складається з аміно, гідроксильних, карбоксильних, меркапто, імідазолільних, N-метилімідазолільних, амінопуринільних фрагментів, фенантролільних фрагментів, піридильних фрагментів, біпіридильних фрагментів, терпіридинільних фрагментів, триазациклононанонільних фрагментів, тетраазациклододеканільних фрагментів, фрагментів імінодіоцтової кислоти, фрагментів нітрилотриоцтової кислоти і фрагментів етилендіамінотетраоцтової кислоти.
12. Застосування за будь-яким з пп. 7-11, яке відрізняється тим, що якірна частина пептидного ланцюга може бути розміщена на С-кінці і/або щонайменше в одному амінокислотному бічному ланцюзі пептиду.
13. Застосування за п. 12, яке відрізняється тим, що щонайменше одну амінокислоту якірної частини на С-кінці пептиду можна подовжити на одну або більше амінокислот, що робить можливим визначення за допомогою систем детектування.
14. Застосування за будь-яким з пп. 7-13, яке відрізняється тим, що якірну частину пептиду може бути подовжено на її N-кінці амінокислотною послідовністю, що забезпечує ділянку розпізнавання для специфічної протеази.
15. Застосування за будь-яким з пп. 7-14, яке відрізняється тим, що після відщеплення пептид може бути повторно приєднаний до активованої твердої фази розбавленням реакційної суміші, що містить конкурентний ліганд, згідно з протоколом послідовних реакцій типу протоколу Мерифілда.
16. Застосування за п. 1, яке відрізняється тим, що додатково включає стадію рефолдингу неправильно скручених структур і/або деагрегації міжмолекулярних агрегатів необов'язково захищеного пептиду, при якому якірна частина пептиду координаційно та оборотно приєднана до активованої твердої фази, і повторного створення правильно скрученої структури пептиду, що включає стадії:
а) дії на пептид щонайменше одного хаотропного або денатурувального реагенту, і
b) подальшої дії ряду розчинників, щоб поступово зменшити хаотропність і забезпечити відтворювальні умови рефолдингу та повторного створення вторинної і третинної структур.
17. Застосування за п. 16, яке відрізняється тим, що вторинна і третинна структури пептиду можуть підтримуватись ковалентними зв'язками між реакційноздатними бічними ланцюгами вказаного пептиду шляхом обробки пептиду підхожими реагентами, що включає утворення вказаних ковалентних зв'язків до відщеплення пептиду від активованої твердої фази.
18. Застосування за п. 17, яке відрізняється тим, що ковалентні зв'язки являють собою дисульфідні зв'язки, амідні зв'язки або стабільні ароматичні чи аліфатичні гідразони.
19. Застосування за пп. 7, 16 і 17 для синтезу пептиду з послідовністю
HHHH-XX-TIVESCNRWITFAQSIISTLT-βAla-G-G-βAla-TKKTQLQLEHLLLDLQMCLNGINN-XX, (І)
де X = d-аланін і βАlа = бета-аланін, що включає утворення дисульфідного зв'язку між залишками цистеїну, внаслідок чого утворюється
,
де X і βАlа є такими, як описано вище.
20. Спосіб твердофазного синтезу пептидів, який відрізняється тим, що його проводять за допомогою нековалентного приєднання зростаючого пептидного ланцюга до активованої твердої фази, причому активний компонент відповідної твердої фази утворений метал-хелатними комплексами з вільними координаційними ділянками для нековалентного приєднання зростаючого пептидного ланцюга до активованої твердої фази через хелатні групи, які присутні на якірній частині зростаючого пептидного ланцюга, при цьому метал-хелатні комплекси утворені комплексами між іоном металу і метал-хелатним лігандом, який безпосередньо або через лінкерну молекулу ковалентно зв'язаний з твердою фазою.
21. Спосіб за п. 20, який відрізняється тим, що повністю утворений комплекс металу для повторюваних циклів синтезу у процесі синтезу пептидів включає тверду фазу, метал-хелатний ліганд, іон металу, який розміщений між метал-хелатним лігандом і хелатними групами якірної частини пептидного ланцюга, і хелатні групи, що присутні у N- або С-кінцевих положеннях і/або у бічних ланцюгах моно- чи олігомерних амінокислот, утворюючих якірну частину пептидного ланцюга, яку нарощують східчастим чином внаслідок повторюваних циклів синтезу.
22. Спосіб за будь-яким одним з пп. 20-21, який відрізняється тим, що нековалентне та координаційне приєднання іона металу до метал-хелатного ліганду характеризується більшою силою, ніж сила приєднання до хелатних груп.
23. Спосіб за будь-яким з пп. 20-22, який відрізняється тим, що структура комплексу з металом складається з:
а) метал-хелатних лігандів, які ковалентно приєднані до твердої фази і здатні утворювати комплекс з іонами металів через атоми N, О, Р і/або S;
b) іонів металу Men+, де n = 1-3, переважно 2, які утворюють комплекс з метал-хелатними лігандами, при цьому залишаючи доступними вільні координаційні ділянки;
с) одиночних або олігомерних природних чи неприродних амінокислот, що містять природні або хімічно модифіковані бічні ланцюги чи N- або С-кінцеві модифікації, до яких приєднані хелатні групи і які таким чином здатні утворювати комплекс з вільними координаційними ділянками, наданими частково насиченими комплексами метал-хелатних лігандів з іонами Меn+ на активованій твердій фазі, описаній в а) та b) і пп. 1-4, при цьому хелатні групи здатні утворювати комплекс з іонами металів через атоми N, О, Р і/або S і при цьому щонайменше одна, переважно 1-3, хелатні групи можуть бути присутні у бічному ланцюзі для того, щоб утворювати стабільну якірну частину для приєднання зростаючого пептидного ланцюга.
24. Спосіб за будь-яким з пп. 20-23, який відрізняється тим, що тверда фаза характеризується присутністю функціональних хімічних груп, які вибирають з аміногруп, груп, що містять гетероциклічний азот, карбоксигруп, гідроксильних, тіолових груп або інших функціональних залишків, для яких самих по собі відомі реакції зв'язування, таким чином вказані функціональні хімічні групи та реакції зв'язування використовують, щоб ковалентно зв'язати тверду фазу з метал-хелатним лігандом за пп. 20-23.
25. Спосіб за будь-яким з пп. 20-24, який відрізняється тим, що метал-хелатні ліганди містять функціональну групу, яка робить можливим хімічне зв'язування з функціональними хімічними групами на поверхні твердої фази за п. 24, і при якому ті ж метал-хелатні ліганди, а також хелатні групи амінокислотних бічних ланцюгів за п. 23 містять одну або більше, переважно 1-3, функціональні групи, які здатні утворювати комплекс з іонами металів за п. 5 і які вибирають з аміногруп, груп, що містять гетероциклічний азот, азагруп, карбоксигруп, фрагментів, що містять сірку або фосфор.
26. Спосіб за будь-яким з пп. 20-25, який відрізняється тим, що іони металів утворюють комплекс з метал-хелатними лігандами на твердій фазі, причому вказані іони металів вибирають з іонів Mn2+, Ni2+, Cu2+, Co2+ або Zn2+, Ca2+, Fe2+, Fe3+ або іонів лантаніду.
27. Спосіб за будь-яким з пп. 20-26, який відрізняється тим, що послідовні реакції використовують, щоб приєднати додаткові моно- або олігомерні амінокислоти до С- або N-кінця повністю утвореного комплексу з металом за п. 21, при цьому відповідно захищені амінокислотні похідні або олігомерні фрагменти, які приєднують у кожному циклі, можуть бути вибрані чи складені з будь-яких природних або неприродних амінокислот.
28. Спосіб за будь-яким з пп. 20-27, який відрізняється тим, що конкурентний хелатний реагент додають до суміші реагентів стадії конденсації протоколу реакцій типу протоколу Мерифілда для того, щоб конкурентно відщепити зростаючий пептидний ланцюг від твердої фази під час вказаної стадії, при цьому підхожі конкурентні хелатні реагенти мають приблизно таку ж спорідненість до вільних координаційних ділянок на активованій твердій фазі, як індивідуальні хелатні групи бічних ланцюгів моно- або олігомерних амінокислот, що використовуються для приєднання зростаючого пептидного ланцюга до твердої фази, причому конкурентні хелатні реагенти є розчинними у суміші реагентів стадії конденсації та не взаємодіють або іншим чином не впливають на інгредієнти суміші реагентів стадії конденсації.
29. Спосіб за п. 28, який відрізняється тим, що реакційну суміш стадії конденсації, що містить конкурентний хелатний реагент, розбавляють перед наступними стадіями промивання для того, щоб повторно приєднати моно- або олігомерні амінокислоти, що утворюють якірну частину пептидного ланцюга, до активованої твердої фази перед подальшими стадіями, такими як полоскання чи промивання.
30. Спосіб за будь-яким з пп. 20-29, який відрізняється тим, що відщеплений і деблокований неочищений продукт синтезу пептидів, що несе моно- та олігомерні амінокислоти з бічними ланцюгами або N- чи С-кінцевими фрагментами з хелатними групами за п. 23, здатний утворювати комплекс металу з твердою фазою за будь-яким з пп. 20- 26 і може бути далі оброблений шляхом:
а) очищення дією на розчин неочищеного продукту активованою твердою фазою за пп. 20-26 в умовах, при яких відбувається повторне приєднання необхідного продукту до твердої фази, і відмиванням домішок, таких як залишки захисних груп та очищувачі або небажані побічні продукти, надлишком розчинника при вибірковому збереженні на активованій твердій фазі продукту, що утворив комплекс, і далі
b) видалення можливих небажаних неправильно скручених структур продукту і міжмолекулярних агрегатів молекул продукту при дії на зв'язаний продукт хаотропними або денатурувальними реагентами, наприклад, сечовиною, детергентами, такими як додецилсульфат натрію, високими концентраціями солей, меркаптоетанолом або іншими реагентами, в результаті чого зв'язаний продукт переходить у денатурований стан, який характеризується руйнуванням вторинної і третинної структур, тоді як зв'язок з твердою фазою зберігається.
31. Спосіб за п. 30, який відрізняється тим, що очищений, зв'язаний і денатурований продукт піддають дії ряду розчинників, причому ряд розчинників підібраний і оптимізований для відповідного продукту, щоб поступово зменшити хаотропність і створити відтворювальні умови рефолдингу молекул зв'язаного продукту, а також контрольованої повторної появи вторинної і третинної структур при збереженні молекул продукту зв'язаними з твердою фазою.
32. Спосіб за п. 30 і/або 31, який відрізняється тим, що ковалентні зв'язки між бічними ланцюгами амінокислот, переважно замикання дисульфідних зв'язків з вільних сульфгідрильних груп, утворення амідних зв'язків або утворення стабільних ароматичних чи аліфатичних гідразонів здійснюють пропусканням сумішей реагентів через повторно скручений продукт, який зв'язаний з твердою фазою.
33. Спосіб за п. 24, який відрізняється тим, що тверду фазу вибирають з діоксиду кремнію, целюлози або з полімерів, переважно із полістирольної смоли, поперечнозшитої з дивінілбензолом, хлортритильної смоли, з модифікованих, переважно карбоксильованих, частинок меламіну або модифікованого, переважно карбоксильованого, полімерного носія на основі полівінілового спирту.
34. Спосіб за п. 24 і/або 33, який відрізняється тим, що матеріал смоли містить феромагнітні частинки, що дозволяє застосовувати технології магнітної сепарації.
35. Спосіб за будь-яким з пп. 20-27, який відрізняється тим, що моно- або олігомерні амінокислоти містять імідазольні бічні ланцюги, переважно не менше шести залишків гістидину, двох або більше залишків гістидину, більш переважно 6-10 залишків гістидину.
36. Спосіб за пп. 23 і 35, який відрізняється тим, що моно- або олігомерні амінокислоти, що використовують, подовжують на N-кінці короткими амінокислотними послідовностями, що забезпечують ділянку розпізнавання для специфічної протеази.
37. Спосіб за будь-яким з пп. 23, 35 і/або 36, який відрізняється тим, що модифіковані або немодифіковані за п. 35 моно- чи олігомерні амінокислоти подовжують на С-кінці на одну або більше амінокислот, що дозволяє проводити визначення системами детектування, такими як біотиніловані амінокислоти, які приєднують, щоб мати можливість визначати за допомогою авідиноподібних взаємодій.
38. Спосіб за будь-яким з пп. 28 і/або 29 і/або 35-37, який відрізняється тим, що конкурентний хелатний реагент містить структурні фрагменти, що містять метал-хелатні фрагменти, які мають електронні пари для координаційних фрагментів, одержаних з імідазолілу, N-метилімідазолілу, імінодіоцтової кислоти, нітрилотриоцтової кислоти, етилендіамінтетраоцтової кислоти, амінопурину, фенантроліну, біпіридилу, терпіридинілу, триазациклононану або тетраазациклододекану.
39. Спосіб за будь-яким з пп. 20-26, який відрізняється тим, що хелатний ліганд містить структурні фрагменти, що мають електронні пари для координаційних зв'язків, такі як трифенілфосфін, 6-амінопурин, фталоціанін.
40. Спосіб за п. 39, який відрізняється тим, що ліганд являє собою 5-аміно-1,10-фенантролін або амінотерпіридин чи триазациклононан або тетраазациклододекан чи їхні похідні.
41. Спосіб за будь-яким з пп. 20-40, який відрізняється тим, що хелатні групи бічних ланцюгів амінокислот вибирають з імідазоло, амінофрагментів, гідроксильного, карбоксильного, тіолового фрагментів, фрагментів нітрилотриоцтової кислоти, фрагментів імінодіоцтової кислоти, фенантролінового фрагмента, піридинового, біпіридинового, терпіридинового, триазациклононанового, тетраазациклододеканового або пуринового фрагментів чи похідних перелічених фрагментів, які здатні утворювати комплекси з металом за будь-яким з пп. 20-26.
42. Спосіб за будь-яким з пп. 20-41, який відрізняється тим, що повністю автоматизований, сумісний з роботами для синтезу і при якому поділ рідини та твердої фази під час циклів синтезу здійснюють відомими способами, переважно просіванням, поділом за розміром частинок, центрифугуванням або технологією магнітної сепарації.
Текст
1. Застосування активованої твердої фази, що містить твердий носій, метал-хелатні ліганди, ковалентно зв'язані з твердим носієм, іони металу Мn+ з n = 1-3, координаційно зв'язані із згаданими метал-хелатними лігандами, причому вказана активована тверда фаза здатна забезпечувати координаційні ділянки для координаційного та оборотного приєднання якірної частини пептиду, для твердофазового синтезу пептидів на вказаній активованій твердій фазі, при цьому пептид являє собою «зростаючий пептид» і здатний піддаватися процедурам подовження пептиду. 2. Застосування за п. 1, яке відрізняється тим, що використано твердий носій на основі діоксиду кремнію, скла або целюлози, чи полімеру, вибраного з групи, що складається з полістирольних смол, меламінових смол і полівінілових спиртів. 3. Застосування за будь-яким з пп. 1-2, яке відрізняється тим, що кожний метал-хелатний ліганд містить щонайменше один атом азоту, кисню, фо UA (21) a200512300 (22) 24.05.2004 (24) 25.03.2009 (86) PCT/EP2004/005568, 24.05.2004 (31) 03017839.6 (32) 05.08.2003 (33) EP (31) 04006195.4 (32) 16.03.2004 (33) EP (31) 60/472,724 (32) 23.05.2003 (33) US (46) 25.03.2009, Бюл.№ 6, 2009 р. (72) РІБКА АНДРЕАС, ФРАНК ХАНС-ГЕОРГ, БРАХТ ФРАНЦ-ПЕТЕР, ХАБЕРЛ УДО (73) АПЛАГЕН ГМБХ (56) US 4877830 A, 31.10.1989 Kaplan B. E. et al. Solid-phase synthesis and characterization of carcinoembryonic antigen (cea) domains // Journal of Peptide Research, Munksgaard International Publishers, Copenhagen, DK. - 1998. Vol. 52, No. 4. - P. 249-260 Comely A. C. et al. Transition metal complexes as linkers for solid phase synthesis: chromium carbonyl complexes as linkers for arenes //Journal of the Chemical Society, Perkin Transactions 1. - 2001. - P. 2526-2531 Ueda E. et al. Current and prospective applications of metal ion-protein binding //Journal of Chromatography A, Elsevier Science, NL. -2003. -Vol. 988, No. 1. -P.123 Cheng K.L. et al. Ligand sorption and chromatographic separation of metals with xad-2 resin // Mikrochimica Acta. - 1978. - Vol. 1, No. 1-2. P. 55-68 Lemercier G. et al. On-column refolding of an insoluble histidine tag recombinant exopolyphosphatase from trypanosoma brucei overexpressed in escherichia coli // Journal of Chromatography B: Biomedical Sciences & Applications, Elsevier Science Publishers, NL. - 2003. - Vol. 786, No. 1-2. - P. 305-309 Birger Anspach F. et al. Immobilized-metal-chelate regenerable carriers: (ii) stability of copper chelate: 2 (19) 1 3 сфору або сірки, який здатний утворювати координаційний зв'язок ліганд-метал. 4. Застосування за будь-яким з пп. 1-3, яке відрізняється тим, що кожний метал-хелатний ліганд містить щонайменше одну функціональну групу, вибрану з групи, що складається з аміно, гетероциклічного азоту, карбокси, гідроксилу і меркапто. 5. Застосування за будь-яким з пп. 1-4, яке відрізняється тим, що кожний метал-хелатний ліганд, ковалентно зв'язаний з твердим носієм, містить щонайменше один фрагмент, вибраний з групи, що складається з трифенілфосфінових фрагментів, амінопуринових фрагментів, переважно 6амінопуринових фрагментів, фталоціанінових фрагментів, 1,10-фенантролінових фрагментів, переважно 5-аміно-1,10-фенантролінових фрагментів, терпіридинових фрагментів, переважно 4'аміно-[2,2';6',2'']терпіридинових фрагментів, триазациклононанових фрагментів, переважно [1,4,7]триазациклононанових фрагментів, і тетраазациклододеканілових фрагментів, переважно [1,4,7,10]тетраазациклододеканілових фрагментів. 6. Застосування за будь-яким з пп. 1-5, яке відрізняється тим, що іони металу Мn+ вибрані з групи, що складається з іонів Mn2+, Cu2+, Ni2+, Co2+, Zn2+, Mg2+, Ca2+, Fe2+, Fe3+ і лантаніду, особливо переважно Мn+ вибраний з Cu2+, Ni2+, Co2+, Zn2+, Mg2+. 7. Застосування за п. 1, яке відрізняється тим, що якірна частина пептиду може бути відокремлена від вказаної активованої твердої фази доданням конкурентного ліганду. 8. Застосування за п. 7, яке відрізняється тим, що конкурентний ліганд містить щонайменше один фрагмент, здатний утворювати комплекс з іонами металів, переважно азотовмісний фрагмент, вибраний з групи, що складається з імідазольних, Nметилімідазольних, амінопуринових, фенантролінових, біпіридинових, терпіридинових, триазациклононанових, тетраазациклододеканових фрагментів, фрагментів імінодіоцтової кислоти, фрагментів нітрилотриоцтової кислоти і фрагментів етилендіамінтетраоцтової кислоти. 9. Застосування за п. 8, яке відрізняється тим, що моно- або олігомерні амінокислоти можуть бути приєднані до С- або N-кінця зростаючого пептиду за протоколом послідовних реакцій типу протоколу Мерифілда. 10. Застосування за будь-яким з пп. 7-9, яке відрізняється тим, що якірна частина пептиду містить щонайменше один фрагмент, що утворює комплекс з іоном металу, причому кожний із вказаних фрагментів включає щонайменше одну групу, що містить азот, кисень, фосфор або сірку, яка здатна координувати іони металу активованої твердої фази. 11. Застосування за будь-яким з пп. 7-10, яке відрізняється тим, що азотовмісну групу, яка здатна координувати іони металу активованої твердої фази, вибрано з групи, що складається з аміно, гідроксильних, карбоксильних, меркапто, імідазолільних, N-метилімідазолільних, амінопуринільних 86023 4 фрагментів, фенантролільних фрагментів, піридильних фрагментів, біпіридильних фрагментів, терпіридинільних фрагментів, триазациклононанонільних фрагментів, тетраазациклододеканільних фрагментів, фрагментів імінодіоцтової кислоти, фрагментів нітрилотриоцтової кислоти і фрагментів етилендіамінотетраоцтової кислоти. 12. Застосування за будь-яким з пп. 7-11, яке відрізняється тим, що якірна частина пептидного ланцюга може бути розміщена на С-кінці і/або щонайменше в одному амінокислотному бічному ланцюзі пептиду. 13. Застосування за п. 12, яке відрізняється тим, що щонайменше одну амінокислоту якірної частини на С-кінці пептиду можна подовжити на одну або більше амінокислот, що робить можливим визначення за допомогою систем детектування. 14. Застосування за будь-яким з пп. 7-13, яке відрізняється тим, що якірну частину пептиду може бути подовжено на її N-кінці амінокислотною послідовністю, що забезпечує ділянку розпізнавання для специфічної протеази. 15. Застосування за будь-яким з пп. 7-14, яке відрізняється тим, що після відщеплення пептид може бути повторно приєднаний до активованої твердої фази розбавленням реакційної суміші, що містить конкурентний ліганд, згідно з протоколом послідовних реакцій типу протоколу Мерифілда. 16. Застосування за п. 1, яке відрізняється тим, що додатково включає стадію рефолдингу неправильно скручених структур і/або деагрегації міжмолекулярних агрегатів необов'язково захищеного пептиду, при якому якірна частина пептиду координаційно та оборотно приєднана до активованої твердої фази, і повторного створення правильно скрученої структури пептиду, що включає стадії: а) дії на пептид щонайменше одного хаотропного або денатурувального реагенту, і b) подальшої дії ряду розчинників, щоб поступово зменшити хаотропність і забезпечити відтворювальні умови рефолдингу та повторного створення вторинної і третинної структур. 17. Застосування за п. 16, яке відрізняється тим, що вторинна і третинна структури пептиду можуть підтримуватись ковалентними зв'язками між реакційноздатними бічними ланцюгами вказаного пептиду шляхом обробки пептиду підхожими реагентами, що включає утворення вказаних ковалентних зв'язків до відщеплення пептиду від активованої твердої фази. 18. Застосування за п. 17, яке відрізняється тим, що ковалентні зв'язки являють собою дисульфідні зв'язки, амідні зв'язки або стабільні ароматичні чи аліфатичні гідразони. 19. Застосування за пп. 7, 16 і 17 для синтезу пептиду з послідовністю HHHH-XX-TIVESCNRWITFAQSIISTLT-βAla-G-G-βAlaTKKTQLQLEHLLLDLQMCLNGINN-XX, (І) де X = d-аланін і βАlа = бета-аланін, що включає утворення дисульфідного зв'язку між залишками цистеїну, внаслідок чого утворюється , 5 де X і βАlа є такими, як описано вище. 20. Спосіб твердофазного синтезу пептидів, який відрізняється тим, що його проводять за допомогою нековалентного приєднання зростаючого пептидного ланцюга до активованої твердої фази, причому активний компонент відповідної твердої фази утворений метал-хелатними комплексами з вільними координаційними ділянками для нековалентного приєднання зростаючого пептидного ланцюга до активованої твердої фази через хелатні групи, які присутні на якірній частині зростаючого пептидного ланцюга, при цьому метал-хелатні комплекси утворені комплексами між іоном металу і метал-хелатним лігандом, який безпосередньо або через лінкерну молекулу ковалентно зв'язаний з твердою фазою. 21. Спосіб за п. 20, який відрізняється тим, що повністю утворений комплекс металу для повторюваних циклів синтезу у процесі синтезу пептидів включає тверду фазу, метал-хелатний ліганд, іон металу, який розміщений між метал-хелатним лігандом і хелатними групами якірної частини пептидного ланцюга, і хелатні групи, що присутні у Nабо С-кінцевих положеннях і/або у бічних ланцюгах моно- чи олігомерних амінокислот, утворюючих якірну частину пептидного ланцюга, яку нарощують східчастим чином внаслідок повторюваних циклів синтезу. 22. Спосіб за будь-яким одним з пп. 20-21, який відрізняється тим, що нековалентне та координаційне приєднання іона металу до металхелатного ліганду характеризується більшою силою, ніж сила приєднання до хелатних груп. 23. Спосіб за будь-яким з пп. 20-22, який відрізняється тим, що структура комплексу з металом складається з: а) метал-хелатних лігандів, які ковалентно приєднані до твердої фази і здатні утворювати комплекс з іонами металів через атоми N, О, Р і/або S; b) іонів металу Men+, де n = 1-3, переважно 2, які утворюють комплекс з метал-хелатними лігандами, при цьому залишаючи доступними вільнікоординаційні ділянки; с) одиночних або олігомерних природних чи неприродних амінокислот, що містять природні або хімічно модифіковані бічні ланцюги чи N- або Скінцеві модифікації, до яких приєднані хелатні групи і які таким чином здатні утворювати комплекс з вільними координаційними ділянками, наданими частково насиченими комплексами металхелатних лігандів з іонами Меn+ на активованій твердій фазі, описаній в а) та b) і пп. 1-4, при цьому хелатні групи здатні утворювати комплекс з іонами металів через атоми N, О, Р і/або S і при цьому щонайменше одна, переважно 1-3, хелатні групи можуть бути присутні у бічному ланцюзі для того, щоб утворювати стабільну якірну частину для приєднання зростаючого пептидного ланцюга. 24. Спосіб за будь-яким з пп. 20-23, який відрізняється тим, що тверда фаза характеризується присутністю функціональних хімічних груп, які вибирають з аміногруп, груп, що містять гетероциклічний азот, карбоксигруп, гідроксильних, тіолових груп або інших функціональних за 86023 6 лишків, для яких самих по собі відомі реакції зв'язування, таким чином вказані функціональні хімічні групи та реакції зв'язування використовують, щоб ковалентно зв'язати тверду фазу з металхелатним лігандом за пп. 20-23. 25. Спосіб за будь-яким з пп. 20-24, який відрізняється тим, що метал-хелатні ліганди містять функціональну групу, яка робить можливим хімічне зв'язування з функціональними хімічними групами на поверхні твердої фази за п. 24, і при якому ті ж метал-хелатні ліганди, а також хелатні групи амінокислотних бічних ланцюгів за п. 23 містять одну або більше, переважно 1-3, функціональні групи, які здатні утворювати комплекс з іонами металів за п. 5 і які вибирають з аміногруп, груп, що містять гетероциклічний азот, азагруп, карбоксигруп, фрагментів, що містять сірку або фосфор. 26. Спосіб за будь-яким з пп. 20-25, який відрізняється тим, що іони металів утворюють комплекс з метал-хелатними лігандами на твердій фазі, причому вказані іони металів вибирають з іонів Mn2+, Ni2+, Cu2+, Co2+ або Zn2+, Ca2+, Fe2+, Fe3+ або іонів лантаніду. 27. Спосіб за будь-яким з пп. 20-26, який відрізняється тим, що послідовні реакції використовують, щоб приєднати додаткові моно- або олігомерні амінокислоти до С- або N-кінця повністю утвореного комплексу з металом за п. 21, при цьому відповідно захищені амінокислотні похідні або олігомерні фрагменти, які приєднують у кожному циклі, можуть бути вибрані чи складені з будь-яких природних або неприродних амінокислот. 28. Спосіб за будь-яким з пп. 20-27, який відрізняється тим, що конкурентний хелатний реагент додають до суміші реагентів стадії конденсації протоколу реакцій типу протоколу Мерифілда для того, щоб конкурентно відщепити зростаючий пептидний ланцюг від твердої фази під час вказаної стадії, при цьому підхожі конкурентні хелатні реагенти мають приблизно таку ж спорідненість до вільних координаційних ділянок на активованій твердій фазі, як індивідуальні хелатні групи бічних ланцюгів моно- або олігомерних амінокислот, що використовуються для приєднання зростаючого пептидного ланцюга до твердої фази, причому конкурентні хелатні реагенти є розчинними у суміші реагентів стадії конденсації та не взаємодіють або іншим чином не впливають на інгредієнти суміші реагентів стадії конденсації. 29. Спосіб за п. 28, який відрізняється тим, що реакційну суміш стадії конденсації, що містить конкурентний хелатний реагент, розбавляють перед наступними стадіями промивання для того, щоб повторно приєднати моно- або олігомерні амінокислоти, що утворюють якірну частину пептидного ланцюга, до активованої твердої фази перед подальшими стадіями, такими як полоскання чи промивання. 30. Спосіб за будь-яким з пп. 20-29, який відрізняється тим, що відщеплений і деблокований неочищений продукт синтезу пептидів, що несе моно- та олігомерні амінокислоти з бічними ланцюгами або N- чи С-кінцевими фрагментами з хелатними групами за п. 23, здатний утворювати 7 86023 8 комплекс металу з твердою фазою за будь-яким з пп. 20- 26 і може бути далі оброблений шляхом: а) очищення дією на розчин неочищеного продукту активованою твердою фазою за пп. 20-26 в умовах, при яких відбувається повторне приєднання необхідного продукту до твердої фази, і відмиванням домішок, таких як залишки захисних груп та очищувачі або небажані побічні продукти, надлишком розчинника при вибірковому збереженні на активованій твердій фазі продукту, що утворив комплекс, і далі b) видалення можливих небажаних неправильно скручених структур продукту і міжмолекулярних агрегатів молекул продукту при дії на зв'язаний продукт хаотропними або денатурувальними реагентами, наприклад, сечовиною, детергентами, такими як додецилсульфат натрію, високими концентраціями солей, меркаптоетанолом або іншими реагентами, в результаті чого зв'язаний продукт переходить у денатурований стан, який характеризується руйнуванням вторинної і третинної структур, тоді як зв'язок з твердою фазою зберігається. 31. Спосіб за п. 30, який відрізняється тим, що очищений, зв'язаний і денатурований продукт піддають дії ряду розчинників, причому ряд розчинників підібраний і оптимізований для відповідного продукту, щоб поступово зменшити хаотропність і створити відтворювальні умови рефолдингу молекул зв'язаного продукту, а також контрольованої повторної появи вторинної і третинної структур при збереженні молекул продукту зв'язаними з твердою фазою. 32. Спосіб за п. 30 і/або 31, який відрізняється тим, що ковалентні зв'язки між бічними ланцюгами амінокислот, переважно замикання дисульфідних зв'язків з вільних сульфгідрильних груп, утворення амідних зв'язків або утворення стабільних ароматичних чи аліфатичних гідразонів здійснюють пропусканням сумішей реагентів через повторно скручений продукт, який зв'язаний з твердою фазою. 33. Спосіб за п. 24, який відрізняється тим, що тверду фазу вибирають з діоксиду кремнію, целюлози або з полімерів, переважно із полістирольної смоли, поперечнозшитої з дивінілбензолом, хлортритильної смоли, з модифікованих, переважно карбоксильованих, частинок меламіну або модифікованого, переважно карбоксильованого, полімерного носія на основі полівінілового спирту. 34. Спосіб за п. 24 і/або 33, який відрізняється тим, що матеріал смоли містить феромагнітні частинки, що дозволяє застосовувати технології магнітної сепарації. 35. Спосіб за будь-яким з пп. 20-27, який відрізняється тим, що моно- або олігомерні амінокислоти містять імідазольні бічні ланцюги, переважно не менше шести залишків гістидину, двох або більше залишків гістидину, більш переважно 6-10 залишків гістидину. 36. Спосіб за пп. 23 і 35, який відрізняється тим, що моно- або олігомерні амінокислоти, що використовують, подовжують на N-кінці короткими амінокислотними послідовностями, що забезпечують ділянку розпізнавання для специфічної протеази. 37. Спосіб за будь-яким з пп. 23, 35 і/або 36, який відрізняється тим, що модифіковані або немодифіковані за п. 35 моно- чи олігомерні амінокислоти подовжують на С-кінці на одну або більше амінокислот, що дозволяє проводити визначення системами детектування, такими як біотиніловані амінокислоти, які приєднують, щоб мати можливість визначати за допомогою авідиноподібних взаємодій. 38. Спосіб за будь-яким з пп. 28 і/або 29 і/або 3537, який відрізняється тим, що конкурентний хелатний реагент містить структурні фрагменти, що містять метал-хелатні фрагменти, які мають електронні пари для координаційних фрагментів, одержаних з імідазолілу, N-метилімідазолілу, імінодіоцтової кислоти, нітрилотриоцтової кислоти, етилендіамінтетраоцтової кислоти, амінопурину, фенантроліну, біпіридилу, терпіридинілу, триазациклононану або тетраазациклододекану. 39. Спосіб за будь-яким з пп. 20-26, який відрізняється тим, що хелатний ліганд містить структурні фрагменти, що мають електронні пари для координаційних зв'язків, такі як трифенілфосфін, 6амінопурин, фталоціанін. 40. Спосіб за п. 39, який відрізняється тим, що ліганд являє собою 5-аміно-1,10-фенантролін або амінотерпіридин чи триазациклононан або тетраазациклододекан чи їхні похідні. 41. Спосіб за будь-яким з пп. 20-40, який відрізняється тим, що хелатні групи бічних ланцюгів амінокислот вибирають з імідазоло, амінофрагментів, гідроксильного, карбоксильного, тіолового фрагментів, фрагментів нітрилотриоцтової кислоти, фрагментів імінодіоцтової кислоти, фенантролінового фрагмента, піридинового, біпіридинового, терпіридинового, триазациклононанового, тетраазациклододеканового або пуринового фрагментів чи похідних перелічених фрагментів, які здатні утворювати комплекси з металом за будь-яким з пп. 20-26. 42. Спосіб за будь-яким з пп. 20-41, який відрізняється тим, що повністю автоматизований, сумісний з роботами для синтезу і при якому поділ рідини та твердої фази під час циклів синтезу здійснюють відомими способами, переважно просіванням, поділом за розміром частинок, центрифугуванням або технологією магнітної сепарації. Хімічний синтез пептидів добре вивчений. По суті можна розрізняти два різні методи. Синтез у розчині часто займає дуже багато часу і тому його не використовують для наукових досліджень, тоді як синтез на твердому носії робить можливою швидку оптимізацію реакційних циклів. Методи, що підходять для твердофазового синтезу пептидів (SPPS), засновані на технології синтезу Мерифілда [Merrfield, R.B., J. Amer. Chem. Soc.85, 1963, 2149], відповідно до якої синтезують пептиди з певними послідовностями на нерозчинному твердому носії, перевага якої полягає у тому, що відокремлення розчинних реагентів легко можна здійснювати простою фільтрацією. SPPS був дуже 9 швидко вдосконалений у вигляді варіантів (позначених в описі як синтез за протоколом типу Мерифілда) і спосіб включає наступні стадії: 1. На першій стадії добирають відповідну матрицю твердого носія (смолу), до якого амінокислотне похідне ковалентно приєднують по С-кінцю. За звичай першу С-кінцеву амінокислоту синтезованої послідовності приєднують до твердого носія за допомогою лінкера. Аміногрупи та функціональні групи бічного ланцюга першої амінокислоти захищають захисними групами, відомими з рівня техніки, звичайно сумісними з Fmoc- або Вос-хімією. 2. На другій стадії групу, що захищає N-кінець першої амінокислоти, вибірково видаляють. 3. На 3-ій стадії наступне амінокислотне похідне, відповідне 2-ій амінокислоті у синтезованій послідовності, приєднують до вільної кінцевої аміногрупи першої амінокислоти, тоді як вона сама має аміногрупу та функціональну групу бічного ланцюга, захищені підхожими захисними групами. 4. На 4-ій стадії групу, що захищає N-кінець, видаляють з тільки що приєднаного амінокислотного похідного і повторюють стадію 3 для наступної амінокислоти. Таким чином, твердофазовий синтез пептидів включає циклічний процес, за допомогою якого певна послідовність амінокислот може бути створена на твердому носії. Наприкінці процесу пептид відщеплюють від твердого носія, наприклад, обробкою трифтороцтовою кислотою (включаючи різні вбирачі). Одночасно захисні групи, які можуть бути присутні на бічних ланцюгах амінокислот, захищаючи імідазол-, меркапто-, карбокси-, аміно-, спирт- або інші функціонально підхожі групи, можуть бути видалені. Це призводить до кінцевого пептиду, який часто потребує очищення на відповідному хроматографічному обладнанні, за звичай системі РХ/МС, яка дозволяє одночасно поділяти та аналізувати компоненти. Описані та інші стадії розглядають детально у стандартних посібниках з синтезу пептидів. Тоді як синтез невеличких пептидів (аж до 30членних) звичайно не становить якихось проблем з SPPS-методиками, існують обмеження стосовно пептидів розміром від 40 до 120 (або навіть більше) амінокислот. Їх властивості значно більше відповідають терміну «невеличкий білок», ніж терміну «великий пептид»: a) Конструйовані пептиди зазначеного розміру звичайно мають не тільки первинну структуру (послідовність), а також вторинну структуру (наприклад, спіраль, бета-шар) і третинну структуру (наприклад, лейцин-„замок”, з'єднання доменів дисульфідним містечковим зв'язком). Тоді як різні варіанти методу SPPS призначені виключно для побудови первинної структури, вони не передбачають жодного інструменту, щоб контролювати внутрішньомолекулярне утворення вторинної і третинної структур підхожим способом. Відсутність контрольних механізмів фолдінгу більш крупних пептидів ускладнюється ще тим фактом, що одночасне відокремлення всіх продуктів від смоли і видалення захисних груп приводить до дуже високої локальної концентрації продукту. За описаних умов відбувається не тільки внутрішньомолекулярний фолдінг. 86023 10 У багатьох випадках невеличкі білки, відщеплені від смоли, прагнуть до взаємодії один з одним, а не до внутрішньомолекулярного скручування. Подібні проблеми мають місце, коли ліофілізовані фракції, що містять очищений продукт, елюйовані із системи РХ, розбавляються розчинником. Таким чином, внутрішньомолекулярний фолдінг і міжмолекулярні взаємодії конкурують, і не існує жодного способу, щоб адекватно контролювати описаний процес. Це часто призводить до утворення функціонально непотрібних мультимолекулярних агрегатів, які не виявляють жодної бажаної біологічної активності. b) У випадку синтезу великих пептидів виявляється переважним використовувати відповідно модифіковані (захищені, частково захищені або незахищені) пептидні фрагменти замість окремих амінокислотних залишків для кожного циклу синтезу. Звичайно невеличкі фрагменти сполучають або лігуванням або методами конденсації фрагментів для того, щоб мінімізувати кількість стадій синтезу Це обумовлене тим фактом, що цикли звичайно не дають кількісних виходів, а надто багато циклів часто закінчується неприйнятним зниженням виходу. Крім того, проблем з конденсацією окремих амінокислот легко можна запобігти за допомогою конденсації цілого фрагмента замість окремої амінокислоти. У випадку конденсації фрагментів виникає проблема, обумовлена необхідністю застосовувати більш тривалі періоди взаємодії під час кожного циклу конденсації. Низька реакційна швидкість обмежена дифузією і обумовлена відсутністю дифузії фіксованого пептидного ланцюга та субоптимальною дифузійною поведінкою фрагментів у порах навколишньої смоли. Тривалий час реакції часто приводить до значного ступеня рацемізації по С-кінцевій амінокислоті конденсованих фрагментів, що визначає необхідність підтримувати час реакції настільки коротким, наскільки є можливим. Найкращим способом подолання описаної проблеми є конденсація у розчині. Класичний SPPS не забезпечує спосіб доставки обох учасників реакції у розчині і можливість повторно приєднувати їх під час наступної стадії циклу при повторенні цієї процедури на кожній стадії синтезу. с) Часто буває важко контролювати необхідну густину початкових залишків на полімерному носії, а навантаження смоли першою амінокислотою часто відбувається разом з рацемізацією. Хоча більшість попередньо навантажених смол постачається комерційно, комерційні смоли часто не охоплюють всі навантаження, а найкращу величину для даної задачі пептидного синтезу нерідко виявляється важко підібрати. Спорідненість до металу до цього часу знаходить дише незначне застосування у синтезі пептидів, що документально підтверджують у публікації Comely et al. [Journal of the Chemical Society, 2001, 2525-2531]. У згаданій роботі утворюють комплекс амінокислоти з хромом, використовуючи ароматичні електро-пі-системи, наприклад, яка присутня у бічному ланцюгу модифікованого фенілаланіну. Такі комплекси утворюються у розчині і попередньо одержаний комплекс з хромом потім прикріп 11 люють до твердої фази та успішно проводять один цикл синтезу. Хоча принципова придатність прикріплення комплексу з металом до твердої основи була продемонстрована, процедура відрізняється від наступних аспектів винаходу, що пропонується. Comely et al. спочатку приєднують іон металу до розчинної частини системи, а потім прикріплюють комплекс до твердої фази на другій стадії, тоді як у пропонованому винаході спочатку здійснюють приєднання іонів металу до твердої фази, а потім прикріпляють зростаючий пептидний ланцюг до твердої фази. Це усуває один з недоліків системи, що пропонується Comely, тобто в одержаному хромвмісному комплексі на твердій фазі координаційний зв'язок між бічними ланцюгами амінокислоти і хромом виявляється сильніше, ніж зв'язок між хромом і твердим носієм. Таким чином, елюювання комплексу конкурентами елюює пептидний комплекс завжди з хромом, приєднаним до пептиду у стехіометричних кількостях. Це не придатне ані для аналізу та поділу, ані для передбачуваного застосування пептиду. Утворений комплекс даного винаходу характеризується сильним хелатоутворенням іона металу у твердій фазі і легко оборотним утворенням комплексу з пептидним ланцюгом. Процес завершують елююванням пептиду без значних кількостей іонів металу, приєднаних до нього. Comely et al. використовують дуже незвичайні комплекси, утворені ароматичними системами, тоді як у даному винаході застосовуються утворюючі комплекси фрагменти, які координаційно зв'язують іон металу через атоми N, Р, S або О. Згадані атоми можуть бути частиною стандартних органічних груп. Такі групи можна створювати і оптимізувати для хелатоутворення та міцності сполучення, використовуючи всю різноманітність методів органічної хімії, і не обмежуватися ароматичними системами. Таким чином, принцип винаходу, що пропонується, передбачає більш значну гнучкість і пристосовність, ніж система Comely. Система, що пропонується Comely, є невигідною у тому сенсі, що вона потребує застосування атмосфери інертного газу, щоб захистити деякі з реагентів, що використовуються. Навіть за таких умов вихід не перевищує 90% на кожну стадію, це означає, що не можна очікувати, що більш довгі пептиди, ніж максимально декамери, будуть одержані з необхідним виходом. Даний винахід і використовувана хімія комплексів є сумісними із стандартною fmoc-хімією і не потребують застосування спеціальної атмосфери або обладнання. Comely застосовує жорсткі умови, які є повільними і деструктивними відносно до смоли, щоб звільнити пептид (48 годин окислення утворюючого основу полімеру на повітрі при білому світлі у ДХМ). У той час як жодного іншого застосування комплексів металів досі не знайдено у галузі хімічного синтезу пептидів, спорідненість металів відома у галузі рекомбінантного одержання біомолекул. Проте його використовують виключно у хроматографічному методі як інструмент для очищення біологічних молекул з неочищених біологічних сумішей або біологічних рідин в одну стадію. Є патенти щодо таких стратегій, які використовують 86023 12 похідні агарози, що містять імінодіацетат та нітрилотриацетат (ЕР-А-253303; US-A-4877830). Гранульована агароза, придатна для хроматографічного очищення рекомбінантних продуктів, постачається комерційно. Крім того, пептиди, що містять бічні ланцюги із спорідненістю до металу, конструювали з наміром використовувати комплекси перехідного металу, фіксовані бічним ланцюгом, як люмінесцентні мічені реагенти для даного пептиду (наприклад, WO-A-9603651A1; ЕРА-А-0178450). Всі згадані описи документально підтверджують придатність способів з використанням хелату металу для аналізу і/або очищення біомолекул. Однак вони не містять в собі суті винаходу та його застосування, які описуються нижче. Технічна проблема, яку необхідно вирішити, полягає у створенні підхожого способу твердофазового синтезу, очищення і рефолдінгу/деагрегації невеличких та великих пептидів, включаючи пептиди понад 120 амінокислот. Щоб розв'язати проблеми SPPS, пов'язані з великими пептидами, пептиди слід оборотно приєднувати до твердого носія. Інший аспект винаходу полягає у повторному скручуванні та відокремленні синтезованих пептидів, що мають небажані, неправильно скручені структури і/або міжмолекулярні агрегати. Згадані проблеми можна вирішити згідно із способом пункту 1. Пропонується застосування активованої твердої фази, що містить твердий носій, металхелативні ліганди, ковалентно зв'язані з твердим носієм, іони металу Мn+ з n=1-3, координаційно зв'язані з метал-хелативними лігандами, причому вказана активована тверда фаза забезпечує координаційні ділянки для координаційного і оборотного приєднання якірної частини пептиду, для твердофазового синтезу пептидів або очищення пептидів. Активовану тверду фазу ще називають «смола із спорідненістю до металу». У переважному втіленні пептид являє собою «зростаючий пептид» і його піддають процедурам подовження пептиду. Переважно зростаючий пептид складається, щонайменше, з однієї амінокислоти. В іншому переважному втіленні моно- або олігомерні амінокислоти приєднують до карбокси- або амінокінця (Сабо N-кінець) «зростаючого пептиду» за протоколом послідовних реакцій типу протоколу Мерифілда, переважно на основі fmoc-хімії. В іншому переважному втіленні моноабо олігомерні амінокислоти містять природні і/або неприродні амінокислоти. Крім того, легко можна обрати відповідно захищені амінокислотні похідні або олігомерні фрагменти, приєднувані у кожному циклі за протоколом послідовних реакцій типу протоколу Мерифілда. Переважний твердий носій є носієм на основі діоксиду кремнію, скла або целюлози чи полімеру, обраного з групи, що складається із полістирольних смол, меламінових смол і смол на основі полівінілового спирту. Інші придатні носії даного винаходу являють собою, наприклад, полістироли, що мають функціональні групи, в яких вказані функціональні групи 13 86023 можуть бути ковалентно зв'язані з підхожими метал-хелативними лігандам. Прикладами таких функціональних груп є групи хлортритилу, аміно, гетероциклічного азоту, карбокси, гідрокси, меркапто і вінілу. Для твердофазового синтезу пептидів вільні карбоксильні групи або інші реакційноздатні групи твердого носія повинні бути захищені таким чином, щоб вони не могли заважати процедурам подовження пептиду. У переважному втіленні твердий носій містить феромагнітні частки. В іншому переважному втіленні поділ рідкої і активованої твердої фази під час циклів синтезу здійснюють, наприклад, просіванням, поділом за розміром, центрифугуванням або за способом магнітної сепарації. Реакційноздатні функціональні групи можна вводити у твердий носій в результаті реакції із попередньо одержаними фрагментами твердого носія або, у випадку полімерів, також в результаті співполімеризації з відповідно модифікованими співполімерами. У переважному втіленні кожний ліганд, утворюючий комплекс з металом, містить, щонайменше, один атом азоту, кисню, фосфору або сірки, який здатний утворити координаційний зв'язок метал-ліганд. В іншому переважному втіленні кожний металхелативний ліганд містить, щонайменше, одну 14 функціональну групу, обрану з групи, що складається з аміно, гетероциклічного азоту, карбокси, гідроксилу і меркапто. Метал-хелативні ліганди ковалентно з'єднані з твердим носієм або безпосередньо або через групи лінкера. Підхожі лінкерні групи являють собою, наприклад, групи аміно, карбокси, метилену, окси, метилендіокси, поліметилендіокси, етилендіокси і поліетилендіокси. Для твердофазового синтезу пептидів вільні карбоксильні групи металхелативних лігандів повинні бути захищені таким чином, щоб вони не могли заважати процедурам подовження пептиду. На Фіг.1a зображений приклад твердого носія, модифікованого 5-аміно-1,10-фенантроліном. Фахівці у даній галузі легко зможуть ідентифікувати численні органічні фрагменти, які здатні координаційно зв'язувати іони металу, наприклад, з бази даних, яку можна використовувати, щоб знайти підхожі метал-хелативні фрагменти для метал-хелативних лігандів згідно з даним винаходом. В рамках даного винаходу існуючі відомості можна використовувати, щоб ідентифікувати підхожі фрагменти, які відповідають сфері дії винаходу. Приклад бази даних, що містить відповідні відомості, наведений з показовими даними для груп, придатних для здійснення даного винаходу, у таблиці 1 нижче. Таблиця 1 Константи зв'язування комплексів показових комбінацій ліганд/іон металу Стадія зв'язування Ліганд m 1 2 Імідазол 1 2 3 4 1-Метилімідазол 1 2 3 4 Імінодіоцтова кислота 1 2 3 Нітрилотриоцтова кислота 1 2 Етилендіамінтетраоцтова ки1 слота Етилендіамін 1 2 3 1,3-Діамінопропан 1 2 3 Транс-1,2-діаміноциклогексан 1 2 3 1,4,7-Триазациклононан 1 2 рК Мn2+ 3 1,25 2,3 3,23 Fe2+ 4 1,31 3,04 4,72 7,82 5,8 10,1 Со2+ 5 2,44 4,34 5,76 6,7 2,29 4,25 5,32 6,7 6,97 12,2 7,27 10,44 11,1 8,9 11,98 14,3 10,38 14,33 16,5 11,51 16,32 18,4 12,7 17,43 18,8 10,45 14,24 16,5 2,6 4,5 5,21 4,13 7,32 9,12 5,5 10,1 13,4 7,3 13,44 17,51 6,31 10,6 12,9 7,9 14,3 20 13 10,49 19,6 5,69 10,64 13 1,34 2,08 3,08 2,94 5,33 5,8 6,37 11,74 15,22 11,2 Ni2+ 6 3,01 5,53 7,5 8,8 3,05 5,95 7,61 9,13 8,3 14,7 Cu2+ 7 4,21 7,72 10,57 12,6 4,22 7,76 10,65 12,86 10,56 16,3 Zn2+ 8 2,55 4,89 7,17 9,18 2,38 4,92 6,6 9,21 7,15 12,4 9,7 16,74 11,09 20,8 15,6 27,7 6,37 11,98 14,1 11,5 15 86023 16 Продовження таблиці 1 1 1,4,7,10Тетраазациклододекан Піридин 2,2'-Дипіридил 2,2'; 6', 2'' - Терпіридин 2,4,6-Три-(2-піридил)-1, 3,5триазин 4-Гідроксипіридин-2,6дикарбонова кислота 1,10-Фенантролін 2-Метил-1,10-фенантролін 2 1 3 4 5 14,1 6 16,4 7 24,6 8 16,2 1 2 3 4 1 2 3 1 2 1 0,24 0,7 0,9 1,22 1,8 1,88 3 3,3 1,05 1,45 2,6 4,2 7,9 17,2 7,1 20,7 12,3 5,8 11,3 16 9,5 18,6 7,04 13,86 20,16 10,7 21,8 2,54 4,38 5,7 6,03 8,12 13,63 17 12,3 19,1 7,1 13,7 19,68 5,1 10 13,9 8,4 16,2 8,7 16,8 24,4 5,95 11,8 16,7 9,2 17,3 9,13 15,84 21,03 7,4 13,8 16,95 12,2 22,1 6,38 12,08 17,1 4,96 9,36 12,7 1 2 1 2 3 1 2 3 4,44 6,7 4,09 7,52 10,2 3 5,5 7,9 Значення рК при 25°С. Значення рК визначали як негативний десятковий логарифм константи К зв'язування комплексу. У кожному стовпчику подане значення рК для кожної послідовної стадії зв'язування іона металу або його комплексу MLm-1 з додатковою молекулою ліганду L (Me Lm-1+LÛMLm, K=c (MLm)/(c(M Lm-1) * с (L)). Узято з: A.E. Martell, R.M. Smith, RJ. Motekaitis, „Database 46 NIST Critically Selected Stability Constants of Metal Complexes, Version 7", Texas A&M University, College Station, TX. Таким чином, дані, що стосуються утворення комплексу іонів різних металів з лігандними молекулами, відомі та можуть бути знайдені в основній літературі та базах даних. Дані у таблиці 1 узяті з бази даних Мартелла (Martell) та співробітників (2003) і являють собою показовий огляд щодо взаємодії відповідних іонів металів з лігандами. Такі бази даних можна використовувати, щоб приєднувати іон металу до твердої фази. Згадані ліганди також можуть функціонувати у бічному ланцюзі одиночних або олігомерних, природних чи неприродних амінокислот як хелативні групи, які зв'язують ненасичений іон металу, як описано нижче. В іншому переважному втіленні кожний металхелативний ліганд, ковалентно зв'язаний з твердим носієм, містить, щонайменше, один фрагмент, обраний з групи, що складається з трифенілфосфінових фрагментів, амінопуринових фрагментів, переважно 6-амінопуринових фрагментів, фталоціанінових фрагментів, 1, 10-фенантролінових фрагментів, переважно 5-аміно-1,10фенантролінових фрагментів, терпіридинових фрагментів, переважно 4'-аміно[2, 2'; 6', 2'']терпіридинових фрагментів, триазациклононанових фрагментів, переважно [1,4,7]триазациклононанових фрагментів, і тетраазациклододеканілових фрагментів, переважно [1,4,7,10]тетраазациклододеканових фрагментів. Переваж 5,85 11,15 21 4,2 7,9 10,8 5,12 9,63 13,3 6 но метал Мn+ обирають з групи, що складається з Mn2+, Cu2+, Ni2+, Co2+, Zn2+, Mg2+, Са2+, Fe2+, Fe3+ та іонів лантаніду, особливо переважними Мn+ є Cu2+, Ni2+, Co2+ і Zn2+. На Фіг.1b показаний приклад активованої твердої фази, що містить твердий носій, ковалентно зв'язаний з 5-аміно-1,10-фенантроліновим лігандом, утворюючим комплекс з металом, та іони зв'язаного хелатом металу, Сu2+. На Фіг.1с показаний приклад приєднаного пептиду, що містить пептидний залишок і якірну частину олігогістидинового фрагмента, в якому вказана якірна частина ковалентно приєднана до активованої твердої фази, що включає твердий носій, ковалентно зв'язаний 5-аміно-1,10фенантроліновим метал-хелативним лігандом, та іони зв'язаного хелатом Сu2+. Покривний шар прикріплених пептидів і прикріплених (моно- або олігомерних) амінокислот, останні називають вихідною ланкою для твердофазового синтезу пептидів, на поверхні активованого твердого носія дуже легко можна контролювати. За умови, що координаційні ділянки активованої твердої фази рівномірно розподілені по поверхні, густину приєднаних пептидів і вихідних ланок (покривний шар) можна коректувати для певних задач синтезу, очищення або рефолдінгу пептидів і визначати за кількістю вихідних ланок, доданих до смоли під час 1-ої стадії приєднання. Покривний шар поверхні можна розрахувати з відомої величини ділянки поверхні та кількості пептидів (визначених на моль) і/або діаметрів. Оскільки реакції комплексоутворення звичайно є швидкими, приєднання може бути завершено за присутності органічного або водного розчинника протягом декількох хвилин. Пропонується спосіб, при якому якірну частину пептиду, яка координаційно зв'язана з координаційними ділянками активованої твердої фази, що 17 містить твердий носій, метал-хелативні ліганди, ковалентно зв'язані з твердим носієм, іони металу Мn+ з n=1-3 у вказаних метал-хелативних лігандах, відокремлюють від згаданої активованої твердої фази доданням конкурентного ліганду. Згідно з винаходом, конкурентний реагент можна додавати до прикріпленого пептиду для того, щоб конкурентно відокремити якірну частину пептиду від активованої твердої фази. Переважно конкурентний реагент додають до суміші реагентів стадії зв'язування за протоколом реакцій типу протоколу Мерифілда. Підхожий конкурентний хелативний реагент має приблизно таку ж або більш слабку спорідненість до вільних координаційних ділянок на активованій твердій фазі як кожний індивідуальний хелативний іон металу фрагмент якірної частини пептиду до вказаних координаційних ділянок. У переважному втіленні конкурентний реагент є розчинним у суміші реагентів стадії зв'язування і не взаємодіє з інгредієнтами з сумішей реагентів. На Фіг.2а показаний принцип відокремлення з прикладом, у якому якірна частина пептиду є олігогістидиновим залишком. На відміну від відщеплення, повторне приєднання якірної частини пептиду до активованої твердої фази виявляється можливим в результаті розбавлення суміші, що містить активовану тверду фазу, конкурентний реагент та не приєднаний пептид. У переважному втіленні в межах протоколу реакцій типу протоколу Мерифілда повторне приєднання проводять перед наступними стадіями промивання. На Фіг.2b показаний принцип повторного приєднання з прикладом, у якому якірна частина пептиду являє собою олігогістидиновий залишок. Приклад впливу концентрацій конкурентного реагенту на зв'язування даного пептиду з активованою твердою фазою показаний на Фіг.3а, приклад, коли відщеплення зростаючими концентраціями конкурента застосовують для елюювання координаційно зв'язаного пептиду. Описаний процес є оборотним. В іншому способі, що пропонується, конкурентний ліганд містить, щонайменше, один фрагмент, здатний утворювати комплекс з іонами металів, переважно азотвмісний фрагмент, обраний з групи, що складається з імідазолу, N-метилімідазолу, амінопурину, фенантроліну, біпіридину, герпіридину, триазациклононану і тетраазациклододекану, фрагментів імінодіоцтової кислоти, фрагментів нітрилотриоцтової кислоти і фрагментів етилендіамінтетраоцтової кислоти. В іншому переважному втіленні конкурентні ліганди містять структурні фрагменти, що мають електронні пари для координаційних зв'язків, такі як трифенілфосфінові фрагменти, 6-амінопуринові фрагменти або фталоціанінові фрагменти. Конкретними прикладами конкурентних лігандів є глутатіон, етилендіамінтетраоцтова кислота, імідазол, N-метилімідазол, фенантроліни, переважно 5-аміно-1,10-фенантроліни, амінотерпіридини, триазациклононани і тетраазациклододекани. 86023 18 У переважному втіленні вказаного способу пептид являє собою «зростаючий пептид», зв'язаний через якірну частину з іоном металу, який приєднаний до метал-хелативного ліганду, зв'язаного з твердим носієм, і піддається процедурам подовження пептиду. Термін «зростаючий пептид» має відношення до послідовного створення пептидного ланцюга звичайно з використанням амінокислоти із захищеними N-кінцем і бічним ланцюгом, наприклад, як у випадку всіх схем пептидного синтезу із застосуванням fmoc. Переважно моно- або олігомерні амінокислоти приєднують на С- або N-кінці «зростаючого пептиду» за протоколом послідовних реакцій типу протоколу Мерифілда. Як згадувалося вище, зростаючий пептид, що містить, щонайменше, одну моно- або олігомерну амінокислоту, ковалентно зв'язану з іоном металу якірної частини, розглядають як вихідну ланку для твердофазового синтезу пептидів. Прикладом такої вихідної ланки є єдиний залишок гліцину, ковалентно зв'язаний через його карбоксильну групу з фрагментом, утворюючим комплекс з іоном металу. Переважно вихідна ланка містить, щонайменше, один бічний ланцюг і/або С-кінцеву модифікацію, яка здатна координувати іони металу активованої твердої фази. Вказані амінокислоти можуть бути природними або неприродними. Кожна прикріплена моно- або олігомерна амінокислота, що використовується як вихідна ланка для твердофазового синтезу пептидів, повинна бути сумісною з протоколами пептидного синтезу. Переважно якірна частина пептиду містить, щонайменше, один фрагмент, утворюючий комплекс з іоном металу, кожний вказаний фрагмент включає в себе, щонайменше, одну групу, що містить азот, кисень, фосфор або сірку, яка здатна координувати іони металу активованої твердої фази. У переважному втіленні кожний фрагмент, утворюючий комплекс з іоном металу, містить від 1 до 10 згаданих груп, що містять азот, кисень, фосфор або сірку. Більш переважно кожний фрагмент, утворюючий комплекс з іоном металу, містить від 1 до 10 груп, що містять аміно, гетероциклічний азот, аза, карбокси, сірку і фосфор. Утворюючі комплекс з іоном металу фрагменти якірної частини можуть бути модифіковані за умови, що одержане похідне є сумісним із способами пептидного синтезу. На Фіг.1с, 2а і 2b показаний олігогістидиновий фрагмент, амінокінець якого захищений відповідно до Fmoc-хімії. В іншому втіленні винаходу координаційний зв'язок метал-хелативних лігандів з іонами металу активованої твердої фази є більш сильним, ніж координаційний зв'язок якірної частини пептиду з вказаними іонами металу. Більш переважну азотвмісну групу, здатну координувати іони металу активованої твердої фази, обирають з групи, що складається з аміно, гідроксилу, карбоксилу, меркапто, імідазолілу, Nметилімідазолілу, амінопіринілу, фрагментів фенантролілу, фрагментів піридилу, фрагментів біпі 19 ридилу, терпіридинілових фрагментів, триазациклононанонілових фрагментів, тетраазациклододеканілових.фрагментів, фрагментів імінодіоцтової кислоти, фрагментів нітрилотриоцтової кислоти і фрагментів етилендіамінтетраоцтової кислоти. Особливо переважний вказаний фрагмент містить від 2 до 10 природних або неприродних амінокислот. Підхожі бічні ланцюги вказаної амінокислоти, що містять фрагменти, які утворюють комплекс з іоном металу, переважно можна обрати з імідазоло-, аміно-, гідроксильного, карбоксильного, меркапто-, фенантролінового, піридинового, біпіридинового, терпіридинового, триазациклононанового, тетраазациклододеканового і пуринового фрагментів та їх похідних, які ще здатні утворювати комплекси з металом. Підхожі органічні фрагменти, здатні утворювати координаційні зв'язки з іонами металів, можна знайти в базах даних, які ілюстративно показані у таблиці 1. Для здійснення процесів подовження пептиду вільні карбоксильні групи повинні бути відповідно захищені. Переважні моно- або олігомерні амінокислоти якірної частини містять можливо захищені на Nкінці імідазольні бічні ланцюги. В іншому переважному втіленні вказані моно- або олігомерні амінокислоти являють собою олігогістидин або коротку (1-6 залишків) послідовність неприродних амінокислот, що несуть фенантролінові фрагменти у своїх бічних ланцюгах, щонайменше, з однією додатковою амінокислотою, причому додаткова амінокислота не взаємодіє з твердим носієм, наприклад, гліцин. Переважно вказані олігогістидинові фрагменти містять, щонайменше, 2 залишки гістидину; більш переважно 6-10 залишків гістидину. В іншому переважному втіленні вказані олігогістидинові фрагменти містять послідовність, щонайменше, з 2 послідовних залишків L- або Dгістидину, більш переважно 6-10 залишків L- або D-гістидину. В іншому переважному втіленні вказані моноабо олігомерні амінокислоти якірної частини містять, щонайменше, один 5-аміно-1, 10фенантроліновий фрагмент. Виявилося, що можна повторно відщеплювати від активованої твердої фази і повторно приєднувати до активованої твердої фази зростаючий пептидний ланцюг. У переважному втіленні відщеплення здійснюють під час стадії конденсації синтезу пептидів, тоді як повторне приєднання індукують розбавленням реакційної суміші до стадій промивання, що передує подальшому видаленню захисту. Використовуючи описані стадії, виявилося, що можна застосовувати «перехідний» рідкофазовий синтез і використовувати його переваги особливо для синтезу великих пептидів способами конденсації фрагментів. Переважно вказану якірну частину пептидного ланцюга поміщають на С-кінці і/або, щонайменше, в одному бічному ланцюзі амінокислот пептиду. В іншому переважному втіленні вказану якірну частину пептиду можна подовжувати певними групами і/або послідовностями природних і/або неприродних амінокислот, що робить можливою подальшу обробку та детектування пептидів. 86023 20 У переважному втіленні, щонайменше, одну амінокислоту якірної частини на С-кінці пептиду подовжують однією або більше амінокислотами, що дозволяє проводити визначення системами виявлення. У переважному втіленні до зростаючого пептиду додають послідовність, яка забезпечує пряме розпізнавання кінцевого пептидного ланцюга антитілами. Прикладом такої tag-послідовності є myctag. Крім того, додання біотинілованої амінокислоти (наприклад, біотинілований D-, L- лізин), переважно на С-кінці олігомерної вихідної ланки, дозволяє проводити пряме кількісне визначення кінцевого продукту, засноване на специфічній взаємодії між біотином і авідин-подібними молекулами. В іншому переважному втіленні якірну частину пептиду подовжують на N-кінці амінокислотною послідовністю, забезпечуючи ділянку розпізнавання для специфічної протеази. У ще одному переважному втіленні вихідну ланку подовжують короткою амінокислотною послідовністю, яка кодує ділянки розпізнавання для протеази. Це робить можливим селективне видалення вихідної ланки після процесу синтезу та процесу скручування. У переважному втіленні пептид повторно приєднують до активованої твердої фази розбавленням реакційної суміші у ході послідовних реакцій типу протоколу Мерифілда, що містить конкурентний ліганд. Підхожі конкурентні ліганди мають однакову спорідненість до активованої твердої фази (смола із спорідненістю до металу) у порівнянні із спорідненістю окремих залишків переважно олігомерних азотвмісних груп, що утворюють комплекс з іонами металу фрагментів якірної частини пептиду (вихідна ланка), який необхідно відокремити. У випадку олігогістидину у вигляді якірної частини пептиду відповідні конкурентні ліганди являють собою імідазол (у водних розчинниках) або N-метилімідазол (в органічних розчинниках). Відокремлення здійснюють доданням великого надлишку конкурентного ліганду (за звичай 102-106 молярного надлишку конкурентного ліганду відносно до приєднаного ліганду) у порівнянні з приєднаним пептидом у розчиннику. Наприклад, у водних розчинниках концентрація від 100 до 250мМ імідазолу (конкурентний ліганд) виявляється придатною для того, щоб досягти повного відщеплення. За таких же умов повторного приєднання досягають розведенням розчинника, що містить конкурентний ліганд, переважно у 10-20 разів. Це призводить до зниження концентрації конкурентного ліганду і забезпечує повторне приєднання полідентатної якірної частини пептиду до носія. Молярне співвідношення конкурентного ліганду до якірних частин можна легко визначити для пар якірної частини і активованої твердої фази у відповідному розчиннику хроматографічними способами, елююючи якірну частину з смоли із спорідненістю до металу. В іншому переважному втіленні приєднання якірної частини до активованої твердої фази при синтезі пептиду (наприклад, олігогістидину) здійс 21 нюють за присутності розбавленого лужного або нейтрального розчину вихідної ланки. Крім того, пропонується спосіб очищення можливо захищених пептидів, що містять якірну частину; вказану якірну частину координаційно зв'язують з координаційними ділянками активованої твердої фази, що містить твердий носій, металхелативні ліганди, ковалентно зв'язані з твердим носієм, іони металу Мn+ з n=1-3 у вказаних металхелативних лігандах; промивають, щоб змити домішки, такі як залишки захисних груп та вбирачі або небажані побічні продукти пептидного синтезу. В іншому переважному втіленні винаходу використовують хелатні групи, приєднані на кінцевих стадіях синтезу до пептиду (на амінокінці ланцюга, наприклад, похідне Gly-фенантроліну). У згаданому випадку очищення неочищеного продукту від забруднюючих побічних продуктів здійснюють методом хроматографії, використовуючи координаційне приєднання пептиду до активованої твердої фази. Описаний метод можна застосовувати, використовуючи звичайне блокування кінця вільних незв'язаних аміногруп у кожному циклі синтезу. При застосуванні описаного методу очищення неочищенного продукту можна успішно провести за одну хроматографічну стадію. Відщеплення можна здійснити доданням підхожого конкурентного реагенту, який описаний вище, або збільшенням кислотності розчинника до критичного ступеня, переважно, щонайменше, до величини pH нижче 6, більш переважно до pH 5 або нижче, особливо у водному розчині, що є іншим переважним способом відщеплення приєднаного пептиду від смоли із спорідненістю до металу. Пропонується інший спосіб, заснований на вищезазначеному координаційному і оборотному приєднанні пептиду до активованої твердої фази. Спосіб рефолдінгу неправильно скручених структур і/або деагрегації міжмолекулярних агрегатів можливо захищеного пептиду, у якому якірна частина пептиду координаційно та оборотно приєднана до активованої твердої фази, і повторне створення правильно скрученої пептидної структури, включає стадії: a) обробки пептиду, щонайменше, одним хаотропним або денатурувальним реагентом, і b) подальшої обробки рядом розчинників, щоб поступово знизити хаотропність і забезпечити відповідні умови рефолдінгу та повторного створення вторинної і третинної структур. У переважному втіленні хаотропний або денатурувальний реагент обирають з групи, що складається з сечовини, детергентів, таких як додецилсульфат натрію, високі концентрації солей і меркаптоетанол або їх суміші у підхожому розчиннику. Замість ковалентного приєднання до відповідної смоли, у даному винаході передбачена можливість відщеплення ланцюга зростаючого пептиду під час стадії конденсації, хоча його можна приєднувати знов перед наступними стадіями. Крім того, той же самий принцип оборотного прикріплення пептиду до активованої твердої фази можна використовувати, щоб контролювати фолдінг очищеного продукту. З цією метою продукт очищають і по 86023 22 вторно приєднують до активованої твердої фази, при цьому співвідношення молекул приєднаного пептиду (визначене у молях) до ділянки поверхні смоли із спорідненістю до металу обирають таким чином, щоб приєднані пептиди не могли взаємодіяти один з одним. Це сприяє внутрішньомолекулярному фолдінгу, а не міжмолекулярним взаємодіям (агрегації). Очищений і приєднаний продукт потім обробляють рядом розчинників, які роблять можливим поступовий фолдінг молекули і підтримують правильну внутрішньомолекулярну конфігурацію пептидних ланцюгів. Стадії фолдінгу можуть завершуватися утворенням, наприклад, дисульфідних зв'язків у правильному положенні. Наприкінці описаної процедури стабілізований продукт вивільняють з активованої твердої фази доданням конкурентного ліганду або збільшенням кислотності розчинника. Рефолдінг приєднаного пептиду проводять згідно з протоколом рефолдінгу. За вказаним протоколом продукт розчиняють у підхожому розчиннику. За звичай найбільш переважним розчинником є елюційний розчинник з РХхроматографічної системи, хоча продукт можна ліофілізувати і перерозчинити у підхожому розчиннику. З одержаного розчину продукт повторно координаційно приєднують до активованої твердої фази. Процедуру можна здійснювати, використовуючи смоли на основі незахищеної імінодіоцтової кислоти або тринітрилооцтових кислот. Це виявляється можливим, оскільки звичайно для рефолдінгу не застосовують реагенти, які активують вільні карбоксильні групи. Віддають перевагу дуже низькій щільності приєднання (покривний шар поверхні), щоб уникнути того, аби молекули продукту вступали у близький контакт одна з одною. Після завершення приєднання ряд розчинників поступово пропускають через смолу. В принципі, перші розчинники є високо денатурувальними і/або хаотропними для того, щоб привести всі молекули продукту у порівнянний стан фолдінгу. Поступово наступні стадії і розчинники наближають фізіологічні умови з тим, щоб підтримати молекулярний фолдінг. Наприкінці процедури молекули продукту опиняються в необхідному, переважно водному, кінцевому розчиннику, можливо скріплені, наприклад, дисульфідним зв'язком, і можуть вивільнятися з активованої твердої фази. В іншому переважному втіленні вторинну і третинну структури пептиду підтримують ковалентними зв'язками між реакційноздатними бічними групами ланцюгів вказаного пептиду в результаті обробки пептиду відповідними реагентами, що включає утворення вказаних ковалентних зв'язків до відщеплення пептиду від активованої твердої фази. Переважно ковалентні зв'язки, утворені між реакційноздатними бічними ланцюгами повторно скручених пептидів, є дисульфідними зв'язками, амідними зв'язками або стабільними ароматичними чи аліфатичними гідразонами. Відповідні реагенти для утворення, наприклад, дисульфідних місточкових зв'язків, можна обрати з різних окислювально-відновних реагентів, таких як йод, фероціанати, кисень і перекиси. Також можна 23 обрати інші реагенти, які підходять для утворення ковалентних зв'язків у загальноприйнятих реакціях у розчині. Пропонується спосіб, при якому вторинну і третинну структури правильно скручених пептидів фіксують утворенням ковалентних зв'язків між бічними ланцюгами амінокислот, переважно утворенням дисульфідних зв'язків з вільних меркаптогруп, 86023 24 утворенням амідних зв'язків, або утворення стабільних ароматичних чи аліфатичних гідразонів досягають пропусканням сумішей реагентів через повторно скручений продукт, який приєднують до активованої твердої фази. Крім того, пропонується спеціальний спосіб синтезу пептиду з послідовністю: з X = d-аланін і ßAla = бета-аланін, що включає утворення дисульфідного зв'язку між залишками цистеїну з одержанням таким чином: . у якій X ßAla є такими, як описано вище. Спеціальний спосіб синтезу пептиду з послідовністю (І), яку синтезують на активованій твердій фазі, використовуючи описаний вище спосіб послідовних реакцій за протоколом типу Мерифілда, включає, щонайменше, одну стадію очищення та рефолдінгу і утворення дисульфідного зв'язку між залишками цистеїну. Такий же спосіб застосовують відносно похідного пептиду з амінокислотними делеціями або амінокислотними заміщеннями, оскільки ще два залишки цистеїну є на відстані, яка припускає утворення дисульфідного зв'язку. Крім того, пропонуються пептиди, що містять якірну частину, яка переважно складається з неприродних амінокислот, для координаційного та оборотного приєднання пептиду до поверхні активованої твердої фази, якірна частина яких, розміщена на N- або С-кінці і/або у бічному ланцюзі пептиду, містить, щонайменше, один фрагмент, що утворює комплекс з іоном металу, кожний вказаний фрагмент, що містить, щонайменше, одну азотвмісну групу, за винятком пептидів, у яких вказаний фрагмент, утворюючий комплекс з іоном металу, є амінокислотною послідовністю з 2-5 залишків гістидину. Переважно групи, що утворюють комплекс з іоном металу, переважно азотвмісні групи пропонованих пептидів, фіксують на N- чи С-кінці або частині бічного ланцюга, щонайменше, однієї амінокислоти, і їх обирають з групи, що складається з аміно, гідроксильного, карбоксильного, меркапто, імідазольного, N-метилімідазольного, амінопуринільного фрагментів, фенантролільних фрагментів, піридильних фрагментів, біпіридильних фрагментів, терпіридинільних фрагментів, триазациклононанонільних фрагментів і тетраазациклододеканільних фрагментів або їх комбінацій. В іншому переважному втіленні амінокислотний фрагмент містить окремі або олігомерні природні чи неприродні амінокислоти. У переважному втіленні якірна частина містить, щонайменше, один імідазолільний фрагмент. Більш переважно, якірна частина пептиду містить від 1 до 10 імідазолільних фрагментів. В іншому переважному втіленні амінокислота містить, щонайменше, два, більш переважно від 6 до 10 фрагментів гістидинілу. У переважному втіленні амінокислотний фрагмент містить, щонайменше, один фрагмент імідазолілу. Більш переважно, якірна частина містить від 1 до 10 фрагментів імідазолілу. Інша перевага винаходу полягає у тому, що смоли із спорідненістю до металу можна повторно використовувати після синтезу пептидів. Повторне застосування може бути здійснене в результаті видалення катіона і повторного завантаження смоли тим же самим або іншим підхожим катіоном. Оскільки міцність зв'язування різних катіонів відрізняється одна від одної, спосіб модифікації процедур приєднання та повторного приєднання і стабільності одержаних комплексів добирають для іншого катіона як місточкового ліганду між поверхнею смоли і вихідною ланкою синтезу. Винахід надалі ілюструється наступними прикладами, що не обмежують винахід. Приклади: Скорочення Речовини ДМФА N,N-диметилформамід (чистий для синтезу пептидів) Дхм Дихлорметан (чистий для пептидного синтезу) HOBT 1-Гідроксибензотриазол (безводний) ДБУ 1,8-Діазабіцикло[5,4,0]ундек-7-ен 98% РуВОР Гексафторфосфат бензотриазол1-іл-окси-трис-піролідінофосфонію DIPEA, DIEA N-етилдіізопропіламін 98+% TIS Триізопропілсилан 99% MeOH Метанол ВЕРХ (чистий для градієнта) TFE 2,2,2-Трифторетанол 99,8% EDT 1,2-Етандитіол HCL Хлористоводнева кислота NaOH Розчин гідроксиду натрію ТГФ Тетрагідрофуран ДМСО-d6 Диметилсульфоксид, дейтерований ш широкий (сигнал) с синглет д дуплет 25 т триплет кв квадруплет п квінтет μ мультиплет TFA Трифтороцтова кислота NMI N-метилімідазол Ni-NTA Смола Superflow (Novagen) Приклад 1: Синтез Fmoc-Gly-His4-GIy-OMe [TAG1] 2ммоль першої амінокислоти і 4ммоль DIPEA розчиняли в 10мл сухого ДХМ. Одержаний розчин додавали до 1,0г сухої 2хлортритилхлоридної смоли (200-400 меш) і суміш залишали для взаємодії протягом 60 хвилин на обертовій мішалці. Наприкінці реакції смолу двічі залишали для взаємодії з 20мл суміші ДХМ/МеОН/DIPEA протягом 3 хвилин, двічі промивали 20мл ДХМ і двічі ДМФА. Смолу потім двічі обробляли 20мл суміші піперидин/ДМФА (1:3) протягом 3/20 хвилин і промивали 6 разів 20мл ДМФА. 5ммоль амінокислоти, 7,5ммоль HOBT і 10ммоль DIPEA розчиняли у 15мл ДМФА. Через 5 хвилин додавали 5ммоль РуВОР та 10ммоль DIPEA і одержаний розчин вливали в смолу. Після 60 хвилин перемішування на обертовій мішалці смолу промивали 6 разів 20мл ДМФА, двічі обробляли 20мл суміші піперидин/ДМФА (1:3) протягом 20/20 хвилин і промивали 6 разів 20мл ДМФА. У випадку N-кінцевої амінокислоти смолу не обробляли сумішшю піперидин/ДМФА. Після приєднання останньої амінокислоти смолу промивали 6 разів 20мл ДМФА, двічі 20мл ДХМ, а потім залишали для взаємодії з 50мл суміші TFE/ДХМ (2/8) протягом 60 хвилин. Смолу відфільтровували, розчинники видаляли у вакуумі, а неочищений пептидний фрагмент використовували без подальшого очищення. Неочищений пептид, 10ммоль С1-НОВТ, 10ммоль DIEA і 20ммоль DIC розчиняли у мінімальній кількості сухого ДХМ. Розчин залишали для взаємодії протягом 10 хвилин і додавали розчин 10ммоль DIEA та 10ммоль Gly-OMe. Після 2 годин перемішування на обертовій мішалці додавали 100мл ДХМ, а розчин екстрагували 3 рази порціями по 200мл бісульфату натрію (1-2моль/л), 3 рази порціями по 200мл концентрованого хлориду натрію і 3 рази порціями по 200мл бікарбонату натрію (1-2моль/л). Органічний шар висушували над сульфатом натрію, розчинник видаляли, а неочищений пептид використовували без подальшого очищення. Неочищений пептид розчиняли у 50мл суміші TFA/TIS/H2O (95/2,5/2,5) і залишали для взаємодії протягом 60 хвилин на обертовій мішалці. TFA видаляли при одночасному випаровуванні з ДХМ. Неочищений пептид розчиняли у ДМСО і 1000мкл очищали на системі РХ-МС Gilson Nebula LCMS System, використовуючи колонку Kromasil RP С18. Лінійний градієнт складав від 5% водної TFA (0,1%) до 50% ацетонітрилу (що містив 0,085% TFA) протягом 30 хвилин. Швидкість потоку складала 20мл/хв., і оптичну густину контролювали при 214нм. Приклад 2: Синтез Fmoc-Gly-His6-Gly-OMe [TAG3] 86023 26 2ммоль першої амінокислоти і 4ммоль DIPEA розчиняли у 10мл сухого ДХМ. Одержаний розчин додавали до 1,0г сухої 2-хлортритилхлоридної смоли (200-400 меш) і суміш залишали для взаємодії протягом 60 хвилин на обертовій мішалці. Наприкінці реакції смолу двічі залишали для взаємодії з 20мл суміші ДХМ/МеОН/DIPEA протягом 3 хвилин, двічі промивали 20мл ДХМ і двічі ДМФА. Смолу потім двічі обробляли 20мл суміші піперидин/ДМФА (1:3) протягом 3/20 хвилин і промивали 6 разів 20мл ДМФА. 5ммоль амінокислоти, 7,5ммоль HOBT і 10ммоль DIPEA розчиняли у 15мл ДМФА. Через 5 хвилин додавали 5ммоль РуВОР і 10ммоль DIPEA і одержаний розчин вливали в смолу. Після 60 хвилин перемішування на обертовій мішалці смолу промивали 6 разів 20мл ДМФА, двічі обробляли 20мл суміші піперидин/ДМФА (1:3) протягом 20/20 хвилин і промивали 6 разів 20мл ДМФА. У випадку N-кінцевої амінокислоти смолу не обробляли сумішшю піперидин/ДМФА. Після приєднання останньої амінокислоти смолу промивали 6 разів 20мл ДМФА, двічі 20мл ДХМ, а потім залишали для взаємодії з 50мл суміші TFE/ДХМ (2/8) протягом 60 хвилин. Смолу відфільтровували, розчинники видаляли у вакуумі, а неочищений пептидний фрагмент використовували без подальшого очищення. Неочищений пептид, 10ммоль С1-НОВТ, 10ммоль DIEA і 20ммоль DIC розчиняли у мінімальній кількості сухого ДХМ. Розчин залишали для взаємодії протягом 10 хвилин і додавали розчин 10ммоль DIEA та 10ммоль Gly-OMe. Після 2 годин перемішування на обертовій мішалці додавали 100мл ДХМ, а розчин екстрагували 3 рази порціями по 200мл бісульфату натрію (1-2моль/л), 3 рази порціями по 200мл концентрованого хлориду натрію і 3 рази порціями по 200мл бікарбонату натрію (1-2моль/л). Органічний шар висушували над сульфатом натрію, розчинник видаляли, а неочищений пептид використовували без подальшого очищення. Неочищений пептид розчиняли у 50мл суміші TFA/TIS/H2O (95/2,5/2,5) і залишали для взаємодії протягом 60 хвилин на обертовій мішалці. TFA видаляли при одночасному випаровуванні з ДХМ. Неочищений пептид розчиняли у МеОН і 1000мкл очищали на системі РХ-МС Gilson Nebula LCMS System, використовуючи колонку Kromasil RP С18. Лінійний градієнт складав від 5% водної TFA (0,1%) до 50% ацетонітрилу (що містив 0,085% TFA) протягом 50 хвилин. Швидкість потоку складала 20мл/хв., і оптичну густину контролювали при 214нм. Приклад 3: Синтез Fmoc-GIy-His3-Gly-His3-Gly-OMe [TAG4] 2ммоль першої амінокислоти і 4ммоль DIPEA розчиняли у 10мл сухого ДХМ. Одержаний розчин додавали до 1,0г сухої 2-хлортритилхлоридної смоли (200-400 меш) і суміш залишали для взаємодії протягом 60 хвилин на обертовій мішалці. Наприкінці реакції смолу двічі залишали для взаємодії з 20мл суміші ДХМ/МеОН/DIPEA протягом 3 хвилин, двічі промивали 20мл ДХМ і двічі ДМФА. Смолу потім двічі обробляли 20мл суміші піпери 27 дин/ДМФА (1:3) протягом 3/20 хвилин і промивали 6 разів 20мл ДМФА. 5ммоль амінокислоти, 7,5ммоль HOBT і 10ммоль DIPEA розчиняли у 15мл ДМФА. Через 5 хвилин додавали 5ммоль РуВОР та 10ммоль DIPEA і одержаний розчин вливали в смолу. Після 60 хвилин перемішування на обертовій мішалці смолу промивали 6 разів 20мл ДМФА, двічі обробляли 20мл суміші піперидин/ДМФА (1:3) протягом 20/20 хвилин і промивали 6 разів 20мл ДМФА. У випадку N-кінцевої амінокислоти смолу не обробляли сумішшю піперидин/ДМФА. Після приєднання останньої амінокислоти смолу промивали 6 разів 20мл ДМФА, двічі 20мл ДХМ, а потім залишали для взаємодії з 50мл суміші TFE/ДХМ (2/8) протягом 60 хвилин. Смолу відфільтровували, розчинники видаляли у вакуумі, а неочищений пептидний фрагмент використовували без подальшого очищення. Неочищений пептид, 10ммоль С1-НОВТ, 10ммоль DIEA і 20ммоль DIC розчиняли у мінімальній кількості сухого ДХМ. Розчин залишали для взаємодії протягом 10 хвилин і додавали розчин 10ммоль DIEA та 10ммоль Gly-OMe. Після 2 годин перемішування на обертовій мішалці додавали 100мл ДХМ, а розчин екстрагували 3 рази порціями по 200мл бісульфату натрію (1-2 моль/л), 3 рази порціями по 200мл концентрованого хлориду натрію і З рази порціями по 200мл бікарбонату натрію (1-2 моль/л). Органічний шар висушували над сульфатом натрію, розчинник видаляли, а неочищений пептид використовували без подальшого очищення. Неочищений пептид розчиняли у 50мл суміші TFA/TIS/H2O (95/2,5/2,5) і залишали для взаємодії протягом 60 хвилин на обертовій мішалці. TFA видаляли одночасним випаровуванням з ДХМ. Неочищений пептид розчиняли у ДМСО і 1000мкл очищали на системі РХ-МС Gilson Nebula LCMS System, використовуючи колонку Kromasil RP С18. Лінійний градієнт складав від 5% водної TFA (0,1%) до 50% ацетонітрилу (що містив 0,085% TFA) протягом 30 хвилин. Швидкість потоку складала 20мл/хв., і оптичну густину контролювали при 214нм. Приклад 4: Синтез Fmoc-СІу-5-аміно-1,10-фенантроліну [TAG5] 2мл DIEA, 6ммоль C1-HOBt і 12ммоль DIC додавали до розчину 2,56ммоль Fmoc-Gly-OH у мінімальній кількості ДМФА. Одержаний розчин перемішували на обертовій мішалці протягом 5 хвилин і додавали розчин 0,5г 5-аміно-1,10-фенантроліну у мінімальній кількості ДМФА. Суміш інкубували протягом 12 годин і розбавляли 5-кратним обсягом етилацетату. Розчин три рази промивали бікарбонатом натрію. Осади неочищеного продукту відфільтровували і очищали методом колонкової хроматографії або РХ-МС. Неочищений пептид розчиняли у ДМСО і 1000мкл очищали на системі РХ-МС Gilson Nebula LCMS System, використовуючи колонку Kromasil RP С18. Лінійний градієнт складав від 5% водної TFA (0,1%) до 50% ацетонітрилу (що містив 0,085% TFA) протягом 50 хвилин. 86023 28 Швидкість потоку складала 20мл/хв., і оптичну густину контролювали при 214нм. Приклад 5: Синтез (Біс-трет-бутоксикарбонілметил-аміно)оцтової кислоти [TAG8b] a) Синтез бензилового ефіру (біс-третбутоксикарбонілметил-аміно)-оцтової кислоти [TAG8a] 20ммоль п-тозилату бензилового ефіру гліцину, 40ммоль трет-бутилового ефіру бромоцтової кислоти і 60ммоль DIEA розчиняли у 35мл сухого ДМФА. Реакційну суміш перемішували на обертовій мішалці протягом 4 днів. Солі, що випали в осад, відфільтровували, а розчин розчиняли у 250мл етилацетату. Органічний шар екстрагували наступними розчинами: 2´200мл 1N NaOH, 3´1N NaOH/сольовий розчин 1:1. Розчин висушували над Na2SO4, розчинник видаляли перегонкою у вакуумі, а неочищений продукт очищали колонковою флеш-хроматографією (етилацетат/гексан 1:4). 1 H-ЯМР (400МГц, ДМСО-d6)млн. д. 7,42-7,29 (м, 5Н), 5,10 (с, 2Н), 3,60 (с, 2Н), 3,50 (с, 4Н), 1,38 (с, 18Н); РХ-МС (ESI): (М+Н)+ = 394. b) Синтез (біс-трет-бутоксикарбонілметиламіно)-оцтової кислоти [TAG8b] 17,5ммоль бензилового ефіру (біс-третбутоксикарбонілметил-аміно)-оцтової кислоти розчиняли у 75мл ТГФ і додавали паладієвий каталізатор, 10% вугілля, 0,70г. Реакційну судину декілька разів заповнювали воднем, а реакційну суміш перемішували в атмосфері водню доти, поки не припинялося споживання водню. Каталізатор відфільтровували через целіт, а розчинник видаляли у вакуумі. 1 H-ЯМР (400МГц, ДМСО-d6)млн. д. 12,2 (ш с, 1Н), 3,46 (с, 2H), 3,44 (с, 4Н), 1,40 (с, 18Н); РХ-МС (ESI): (М+Н)+=304. Приклад 6: Синтез 4-(біс-трет-бутоксикарбонілметил-карбамоїл)-масляної кислоти [TAG10b] a) Синтез бензолового ефіру 4-(біс-третбутоксикарбонілметил-карбамоїл)-масляної кислоти [TAG10a] 24ммоль монобензилового ефіру пентандіової кислоти, 30ммоль оксалілхлориду і дві краплі ДМФА розчиняли у 20мл сухого ДХМ. Реакційну суміш кип'ятили із зворотним холодильником доти, поки не припинялося виділення газу. Розчин випаровували з 10мл толуолу. Неочищений продукт розчиняли у ДХМ і додавали крапля з краплею до розчину 12,0ммоль трет-бутилового ефіру (третбутоксикарбонілметил-аміно)-оцтової кислоти і 26,4 DIEA у ДХМ при 0°С. Реакційну суміш перемішували протягом 1 години при 0°С та протягом ночі при кімнатній температурі. Розчинник видаляли у вакуумі, а залишок розчиняли у 150мл діетилового ефіру. Органічний шар екстрагували наступними розчинами: 100мл 1N HCl, 100мл напівконцентрованого розчину бікарбонату натрію і 100мл сольового розчину. Розчин висушували над сульфатом натрію, а розчинник видаляли у вакуумі. Неочищений продукт очищали колонковою хроматографією на діоксиді кремнію (етилацетат/гексан 1:1). 29 1 H-ЯМР (400МГц, ДМСО-d6)млн. д. 7,4-7,2 (м, 5Н), 5,08 (с, 2Н), 4,11 (с, 2Н), 3,91 (с, 2Н), 2,38 (т, J=7,4Гц, 2Н), 2,28 (т, J=7,3Гц, 2Н), 1,75 (п, J=7,4Гц, 2Н), 1,41 (с, 9Н), 1,39 (с, 9Н); РХ-МС (ESI): (М+Н)+ =450. b) Синтез 4-(біс-трет-бутоксикарбонілметилкарбамоїл)-масляної кислоти [TAG 10b] 2ммоль бензилового ефіру 4-(біс-третбутоксикарбонілметил-карбамоїл)-масляної кислоти розчиняли у 20мл ТГФ і додавали паладієвий каталізатор, 10% вугілля, 0,09г. Реакційну судину декілька разів заповнювали воднем, а реакційну суміш перемішували в атмосфері водню доти, поки не припинялося споживання водню. Каталізатор відфільтровували через целіт, а розчинник видаляли у вакуумі. 1 H-ЯМР (400МГц, ДМСО-d6)млн. д. 12,1 (ш с, 1H), 4,11 (с, 2Н), 3,91 (с, 2Н), 2,30-2,17 (м, 4Н), 1,68 (п, J=7,2Гц, 2Н), 1,42 (с, 9Н), 1,39 (с, 9Н); РХ-МС (ESI): (M+Na)+ =382. Приклад 7: Синтез 4-([1,10]-фенантролін-5-іл-карбамоїл)масляної кислоти [TAG12b] а) Синтез метилового ефіру 4-([1,10]фенантролін-5-іл-карбамоїл)-масляної кислоти [TAG 2а] В атмосфері аргону 5ммоль 5-аміно-[1,10]фенантроліну розчиняли у 25мл сухого ДМФА. Додавали 6,5ммоль DIEA і крапля за краплею додавали 5,5ммоль метилового ефіру 4хлоркарбоніл-масляної кислоти. Після перемішування протягом 1 години розчинник і надлишок основи видаляли перегонкою у вакуумі. Неочищений продукт очищали методом колонкової хроматографії з оберненою фазою (колонка Merck Lobar RP18, елюент: ацетонітрил/бікарбонат амонію, 5% мас, градієнт: від 20% до 40%). 1 H-ЯМР (400МГц, ДМСО-d6)млн. д. 10,11 (с, 1Н), 9,13 (дд, J=1,6, 4,3Гц, 1Н), 9,03 (дд, J=1,7, 4,3Гц, 1H), 8,60 (дд, J=1,6, 8,4Гц, 1H), 8,44 (дд, J=1,7, 8,2Гц, 1H), 8,17 (с, 1H), 7,82 (дд, J=4,2, 8,4Гц, 1H), 7,74 (дд, J=4,3, 8,1Гц, 1H), 3,63 (с, 3Н), 2,59 (т, J=7,2Гц, 2Н), 2,46 (т, J=7,4Гц, 2Н), 1,95 (п, J=7,3Гц, 2Н); РХ-МС (ESI): (М+Н)+ =324. b) Синтез 4-([1,10]-фенантролін-5-ілкарбамоїл)-масляної кислоти [TAG12b] 1,5ммоль метилового ефіру 4-([1,10]фенантролін-5-іл-карбамоїл)-масляної кислоти розчиняли у 15мл 1,4-діоксану і 15мл дистильованої води. Додавали 1,5мл 1N розчину гідроксиду калію і розчин кип'ятили протягом 1 години. Розчинник видаляли перегонкою у вакуумі, а неочищений продукт очищали методом колонкової хроматографії з оберненою фазою (колонка Merck Lobar RP18, елюент: ацетонітрил/трифтороцтова кислота, 1% мас, градієнт: від 10% до 20%). 1 H-ЯМР (400МГц, CD3OD) у вигляді солі К: млн. д. 9,11 (дд, J=1,6, 4,3Гц, 1H), 9,04 (дд, J=1,6, 4,4Гц, 1H), 8,65 (дд, J=1,5, 8,4Гц, 1H), 8,41 (дд, J=1,6, 8,1Гц, 1Н), 8,14 (с, 1Н), 7,82 (дд, J=4,4, 8,4Гц, 1H), 7,74 (дд, J=4,4, 8,1Гц, 1H), 2,64 (т, J=7,6Гц, 2H), 2,36 (т, J=7,2Гц, 2Н), 2,10 (кв, J=7,5Гц, 2Н)); РХ-МС (ESI): (M+H)+ =310. Крім того, мітки [TAGs], що містять похідні інших дикарбонових кислот окрім глутарової кисло 86023 30 ти, можна одержувати аналогічним чином, використовуючи описані процедури. Приклад 8: Синтез [5-([1,10]-фенантролін-5-іл-карбамоїл)пентаноїл-аміно]-оцтової кислоти [TAG15] В атмосфері аргону 1ммоль смоли Ванга (Wang), на яку завантажений Fmoc-Gly-OH (0,7ммоль/г), суспендували у суміші піперидин/ДМФА 1:3 і опромінювали у мікрохвильовій печі (система "Discovery" від СЕМ) три рази протягом 30 секунд. Смолу промивали три рази ДМФА і три рази ДХМ. Смолу суспендували у 15мл ДХМ і додавали 9ммоль DIEA та 10ммоль адіпоїлхлорид. Після перемішування суспензії протягом 30 хвилин на обертовій мішалці реакційний розчин відфільтровували, а смолу промивали двічі ДХМ і один раз ДМФА. Смолу суспендували у 15мл ДМФА і додавали розчин 3ммоль 5-аміно-1,10-фенантроліну та 5ммоль DIEA у 20мл ДМФА. Після перемішування на обертовій мішалці суспензії протягом ночі смолу промивали шість разів ДМФА, а продукт відокремлювали від смоли суспендуванням у суміші TFA/TIS/H2O, 95:2,5:2,5. Смолу відфільтровували, а фільтрат декілька разів випаровували одночасно з хлороформом, щоб одержати продукт. Описаний спосіб також аналогічним чином можна застосовувати для інших пептидів, зв'язаних із смолою, та інших активованих похідних дикарбонових кислот. Приклад 9: 5-аміно-1,10-фенантролін/2хлортритилхлоридна смола 2ммоль 5-аміно-1,10-фенантролін суспендували в атмосфері інертного газу у 50мл сухого ДМФА. До одержаної суміші додавали 5г 2хлортритилхлоридної смоли (200-400 меш) і смолу інкубували при кімнатній температурі протягом 2 годин. Смолу відфільтровували і промивали ДМФА доти, поки не видаляли надлишок фенантроліну. Щоб блокувати групи, які не прореагували на смолі, останню інкубували три рази з 10мл суміші ДХМ/МеОН/DIEA (80:15:5) протягом 10 хвилин, а потім промивали 3 рази 10мл ДМФА. Смолу можна зберігати під ДМФА при кімнатній температурі. Щоб завантажити смолу іонами Ni2+, її інкубували протягом 30 хвилин з насиченим розчином NiCl2 у ДМФА. Смолу промивали ДМФА доти, поки більше не виявляли жодного NiCl2. Після 5 додаткових промивок ДМФА застосовували наступні процедури: 4´5' ДМФА/N-метилімідазол (0,25моль/л) 4´15' ДМФА/N-метилімідазол (0,25моль/л) 1´12 годин ДМФА/N-метилімідазол (0,25моль/л) 2´5' ДМФА 1´1' ізопропанол 1´15' ізопропанол 1´5' ДМФА 1´30' ДМФА 1´1' ізопропанол 1´15' ізопропанол 1´5' ДМФА 1´30' ДМФА 1´1' ізопропанол 1´15' ізопропанол 31 1´5' ДМФА 1´30' ДМФА. Приклад 10: 5-Аміно-1,10-фенантролін/Novasyn-ТG-карбокси-смола 5г смоли Novasyn TG зрівноважували 50мл сухого ДМФА протягом 30 хвилин. Додавали 10ммоль DIEА і 10ммоль HOBt. Після 5 хвилинної інкубації додавали 2г 5-аміно-1,10-фенантроліну та 10ммоль Pybop, а також 10ммоль DIEA і суміш перемішували протягом ночі на обертовій мішалці. На наступний ранок смолу промивали 6 разів 50мл ДМФА кожного разу. Для завантаження смоли нікелем додавали 10мл насиченого розчину хлориду нікелю у ДМФА і перемішували на обертовій мішалці протягом 30 хвилин. Після описаної стадії супернатант видаляли, а смолу промивали порціями по 50мл ДМФА доти, поки не можна було виявити жодного видимого фарбування супернатанта. Адсорбований надлишок іонів нікелю видаляли наступними процедурами промивання: 4´5' ДМФА/N-метилімідазол (0,25моль/л) 4´15' ДМФА/N-метилімідазол (0,25моль/л) 1´12 годин ДМФА/N-метилімідазол (0,25моль/л) 2´5' ДМФА 1´1' ізопропанол 1´15' ізопропанол 1´5' ДМФА 1´30' ДМФА 1´1' ізопропанол 1´15' ізопропанол 1´5' ДМФА 1´30' ДМФА 1´1' ізопропанол 1´15' ізопропанол 1´5' ДМФА 1´30' ДМФА. Приклад 11: Синтез 1, 4, 7-триазациклононан/2хлортритилхлоридної смоли 2,5г 2-хлортритилхлоридної смоли (200-400 меш) суспендували у 40мл сухого ДМФА. Додавали 500мг тригідрохлориду 1,4,7триазациклононану і 2мл діізопропілетиламіну. Смолу інкубували при кімнатній температурі протягом 8 годин. Смолу фільтрували і промивали ДМФА шість разів. Щоб блокувати групи, які не прореагували на смолі, останню інкубували двічі з 20мл суміші ДХМ/МеОН/DIEA (80:15:5) протягом 30 хвилин, а потім промивали 3 рази 20мл ДМФА. Завантаження смоли нікелем можна проводити згідно з прикладом 10. Смолу зберігали під ДМФА у холодильнику. Приклад 12: Завантаження смоли мітками (завантажувальні експерименти) 1мл відповідної смоли промивали 3 рази ДМФА (чистий для пептидного синтезу). 1мг мітки розчиняли у 1мл ДМФА і додавали 1 краплю DIEA. 50мкл одержаного розчину розбавляли 950мкл води і аналізували методом ВЕРХ. Залишок суміші додавали до промитої смоли і інкубували протягом 30 хвилин. Щоб перевірити приєднання міток до 86023 32 смоли, 50мкл розчину розбавляли 950мкл води і аналізували методом ВЕРХ. Приклад 12а: Завантаження TAG1 на Ni-фенантролін-2СlТrt-смолу При ВЕРХ супернатантної рідини більше не виявляли жодного визначуваного TAG1. Приклад 12b: Завантаження TAG3 на Niфенантролін-2Сl-Тrt-смолу При ВЕРХ супернатантної рідини більше не виявляли жодного визначуваного TAG3. Сліди, що залишилися, з подібним часом утримування являли собою мінорні домішки, які не були зв'язані. Проведений експеримент також продемонстрував потенціал способу для очищення продуктів від забруднення. Приклад 12с: Завантаження TAG4 на Niфенантролін-2Сl-Тrt-смолу При ВЕРХ супернатантної рідини більше не виявляли жодного визначуваного TAG3. Приклад 12d: Завантаження TAG3 на NovasynТG смолу При ВЕРХ супернатантної рідини більше не виявляли жодного визначуваного TAG3. Приклад 13: Елюювання TAG3 із смол (завантажувальні експерименти) 10мл насиченої смоли 3 рази промивали 20мл суміші 1 і смолу поділяли на 10 порцій. Супернатантну рідину видаляли, а смоли інкубували протягом 5 хвилин з наступними розчинами: (TAG3_01) 6мг глутатіону (окисленого) у 2мл суміші 1 (5ммоль/л) (TAG3_02) 3мг глутатіону (відновленого) у 2мл суміші 1 (5ммоль/л) (TAG3_03) 2мл 1N HCl (TAG3_04) 2мл 0,1N HCl (TAG3_05) 0,68мг імідазолу у 2мл суміші 1 (5ммоль/л) (TAG3_06) 1,36мг імідазолу у 2мл суміші 1 (10ммоль/л) (TAG3_07) 2,72мг імідазолу у 2мл суміші 1 (20ммоль/л) (TAG3_08) 0,82мг N-метилімідазолу у 2мл суміші 1 (5ммоль/л) (TAG3_09) 1,64мг N-метилімідазолу у 2мл суміші 1 (10ммоль/л) (TAG3_10) 3,28мг N-метилімідазолу у 2мл суміші 1 (20ммоль/л). Що стосується експериментів з елюювання більш високими концентраціями імідазолу та Nметилімідазолу, дослідження проводили, використовуючи смоли з пробірок (TAG3-07) і (TAG3-10). Вказані смоли інкубували із зростаючими концентраціями елюційних реагентів (100ммоль, 200ммоль, 500ммоль), супернатант аналізували, а смолу інкубували протягом 5 хвилин з наступною концентрацією елюційного реагенту. Результати: Елюювання міток може бути легко здійснене за допомогою хлористоводневої кислоти. Концентрації понад 100ммоль/л імідазолу або Nметилімідазолу також елюювали ефективно. Приклад 14: 33 Зв'язування та елюювання TAG3 на Ni-NTAсмолу (FPLС-(рідинна хроматографія високого розрізнення) експерименти) Пептид: TAG 3 (Fmoc-(His)6-OMe, M=1191,25г/моль), чистий для ВЕРХ Смола: Ni-NTA SuperFlow (місткість приблизно 5мкмоль/мл) Колонка: Biorad, 2,0мл Обладнання для FPLC: Akta Pharmacia a) Елюювання N-метилімідазолом Вводили 1,1мг (0,92мкмоль) TAG3 у 0,75мл елюенту А Елюент А: 2,2,2-трифторетанол/Н2O/діізопропілетиламін/N-метилімідазол, 1:1:+ 1%: + 5мМ Елюент В: TFE/H2O/DIEA/NMI, 1:1:+1%:+500мМ Швидкість: 1мл/хв Програма: Введення (5мл А) ® Промивка (10мл А) ® Градієнт: елюювання (0% В ® 100% В для 10мл) ® Збір (5мл В) ® Регенерація (8мл А). Певну кількість TAG3 зв'язували та елюювали при концентраціях N-метилімідазолу 100-125мМ. Таку FPLC-процедуру також можна використовувати для визначення порогових концентрацій, вище яких приєднана молекула ефективно конкурентно витісняється хелативним реагентом. Нижче порогової концентрації приєднана молекула залишається ще зв'язаною із смолою. У завантажувальній процедурі розбавлення суміші до точки нижче порогової концентрації призводить до повторного зв'язування із смолою змитої приєднаної молекули. b) Елюювання хлористоводневою кислотою 2,4мг TAG3 (2,0мкмоль) у 0,75мл елюенту А вводили в FPLC-прилад Akta. Здійснювали наступну програму: Елюент A1: TFE/H2O/DIEA/NMI, 1:1:+1%:+5мМ Елюент В: TFE/H2O/NMI, 1:1:+5мМ Елюент А2: TFE/H2O/HCl, 1:1:+10мМ Швидкість: 0,5-1мл/хв. Програма: Введення (2,5мл А1) ® Промивка (10мл А1) ® Градієнт: Видалення DIEA (0% В ® 100% В для 10мл) ® потім «Стадія-градієнта» елюентом А2 (10мМ НСl: 20%, 40%, 60%, 70%). TAG3 елюювали при 60% елюенту А2. Приклад 15: Цикл синтезу з Fmoc-Gly Конденсація TAG3 з Fmoc-Gly на TG-смолі 1мл TG-смоли, в який вводили TAG3, промивали 3 рази 5мл ДМФА. Щоб видалити захист Nкінця, додавали 5мл 20% піперидину і смолу інкубували протягом 30 хвилин. Далі смолу промивали 6 разів ДМФА і додавали попередньо активований розчин 1ммоль Fmoc-Gly-OH, 2ммоль DIEA, 2ммоль HOBt і 2ммоль DIC у мінімальній кількості ДМФА. Через 60 хвилин смолу промивали 6 разів порціями ДМФА по 5мл і 6 разів порціями води по 2мл. Для відокремлення пептиду від смоли додавали 1мл 1N HCl, а смолу інкубували протягом 30 хвилин. Результат: Відщеплений пептид аналізували методом РХ-МС на колонці (218MS5415) масспектрометра Vydac С18. Використовували градієнт від 5 до 95% В протягом періоду 30 хвилин і 86023 34 швидкість потоку 1мл/хв (А = вода, 0,1% TFA; В = ацетонітрил, 0,085% TFA). Жодних виділених речовин не визначали. Тільки пептид, присутній у суміші, показав очікувану масу (М = 1247, М2+ = 625). Окрім HOBt не змогли виявити жодних інших суттєвих домішок. Приклад 16: Синтез TAG5-G-A-Fmoc на Ni-NTA-смолі 2,65мг TAG5 зв'язували з Ni-NTA-смолою у колонці Omnifit, 1,7мл. Смолу переносили у мікрохвильову реакційну судину і промивали 6 разів 5мл ДМФА кожного разу. Супернатанти відкидали, а смолу інкубували з 5мл 25% піперидину у ДМФА, піддаючи дії мікрохвильових імпульсів, 3´30сек, при 50В кожний у мікрохвильовій печі Discovery (СЕМ GmbH). Смолу промивали 6 разів ДМФА. Одержували розчин 1мМ необхідної амінокислоти (відповідно захищеної) у мінімальній кількості розчинника (ДМФА). До одержаного розчину додавали 2ммоль DIEА, 2ммоль HOBt і 2ммоль DIC. Після 5 хвилинної інкубації одержаний розчин додавали до смоли, позбавленої захисту, і піддавали дії мікрохвильових імпульсів, 3´30сек, при 50В кожний. Після приєднання останньої амінокислоти кінцеву Fmoc-групу не видаляли. Смолу промивали 5 разів водою (pH понад 7). Нарешті 2мл 1N HCl додавали до смоли. Смолу інкубували протягом 30 хвилин, супернатант видаляли і зберігали. Супернатант використовували безпосередньо для аналітичної РХ-МС. Смолу збирали для рециркуляції. При РХ-МС встановили точну масу пептиду в єдиному визначуваному пептидному піку. Непептидними домішками були HOBt і HOBtH2O. Приклад 17: Очищення імуномера IL-2 Очищення імуномера IL-2 (DIM03A) на Ni-NTA у суміші 2,2,2-трифторетанол/Н2О/діізопропілетиламін/N-метилімідазол Пептид: який вказаний у прикладі 19 (М=6237г/моль), неочищений продукт після розщеплення смоли; Смола: Ni-NTA SuperFlow 76,7мг (12,2мкмоль) у 5мл елюенту А, введені у 5 фракціях, див. підписи до Фіг.3а-с. Колонка: Biorad, 2,0мл Елюент A: TFE/H2O/DIEA/NMI, 1:1:+1% + 5мМ Елюент В: TFE/H2O/DIEA/NMI, 1:1:+1%+500мМ Швидкість: 1мл/хв. Програма: Введення (5мл А) ® Промивка (10млА) ® Градієнт: елюювання (0% В ® 100% В у межах 10мл) ® Збір (5мл В) ® Регенерація (8мл А). Необхідний продукт елюювали в одному піку приблизно при 125мМ NMI. Ідентичність підтверджували аналітичною РХ-МС. Приклад 18: Постсинтетичні модифікації пептиду Утворення дисульфідного місточкового зв'язку у пептиді H2N-HHHHGC(Acm) GNGGGC(Trt) GSGN-COOH Acm і Trt є стандартними захисними групами для SH-групи на залишках цистеїну. Пептид синтезували звичайним способом синтезу пептиду і очищали на системі РХ/МС до чистоти понад 95%. 35 86023 36 1мл Ni-NTA-смоли, яку завантажували пептидом, промивали 3 рази 2мл МеОН. Протягом 2 годин додавали 50мкл розчину йоду (0,5моль/л), а суміш інкубували протягом додаткових 2 годин при кімнатній температурі. Смолу промивали 6 разів МеОН, 3 рази водою (pH>7), а потім інкубували з 2мл НСl (1N). Смолу відфільтровували і розчин аналізували у системі РХ/МС. Одержано єдиний пік продукту, який показав точну масу. Приклад 19: Синтез Інтерлейкін-2-імуномера, використовуючи стадії рефолдінгу способу, показаного в описі Хімічна формула речовини, яку синтезували: X = D-аланін bAla = бета-аланін інші літери = стандартний амінокислотний код. Загальна стратегія: Синтез проводили на 2-хлортритилхлоридній смолі (200-400 меш) із ступенем заміщення 0,2ммоль/г. Перші сім амінокислот конденсували у вигляді фрагмента. Приєднання наступних амінокислот досягали за допомогою одиночної, подвійної або потрійної конденсації при 10-кратному надлишку амінокислот та PyBOP/HOBT/DIPEA як конденсуючих добавок. Видалення захисту з Nкінця зростаючого пептидного ланцюга проводили подвійною обробкою сумішшю піперидин/ДМФА (1/3). У складних випадках проводили третю обробку сумішшю ДБУ/піперидин/ДМФА (2/2/96). Кількість застосованих циклів конденсації і зняття захисту показана у наступній схемі (інформація щодо складної конденсації/зняття захисту, одержана при перевірці кожного подовження методом ВЕРХ-МС). 1,2,3 =кількість стадій конденсації та видалення захисту X = D-аланін В = b-аланін. Протокол 1: Синтез першого пептидного фрагмента 2ммоль першої амінокислоти і 4ммоль DIPEA розчиняли у 10мл сухого ДХМ. Одержаний розчин додавали до 1,0г сухої 2-хлортритилхлоридної смоли (200-400 меш) і суміш залишали для взаємодії протягом 60 хвилин на обертовій мішалці. Наприкінці реакції смолу двічі залишали для взаємодії з 20мл суміші ДХМ/МеОН/DIPEA протягом 3 хвилин, двічі промивали 20мл ДХМ і двічі ДМФА. Смолу потім двічі обробляли 20мл суміші піперидин/ ДМФА (1:3) протягом 3/20 хвилин і промивали 6 разів 20мл ДМФА. Приєднання амінокислот 2-7 досягали за наступним способом: 5ммоль амінокислоти, 7,5ммоль HOBT і 10ммоль DIPEA розчиняли у 15мл ДМФА. Через 5 хвилин додавали 5ммоль, РуВОР та 10ммоль DIPEA і одержаний розчин вливали в смолу. Після 60 хвилин перемішування на обертовій мішалці смолу промивали 6 разів 20мл ДМФА, двічі обробляли 20мл суміші піперидин/ДМФА (1:3) протягом 20/20 хвилин і промивали 6 разів 20мл ДМФА. У випадку Nкінцевої амінокислоти смолу не обробляли сумішшю піперидин/ДМФА. Протокол 2: Відщеплення пептидного фрагмента Після приєднання останньої амінокислоти смолу промивали 6 разів 20мл ДМФА, двічі 20мл ДХМ, а потім залишали для взаємодії з 50мл TFE/ДХМ (2/8) протягом 60 хвилин. Смолу відфільтровували, розчинник видаляли у вакуумі, а неочищений пептидний фрагмент використовували без подальшого очищення. Протокол 3: Повторне приєднання пептидного фрагмента 1ммоль фрагмента і 2ммоль DIPEA розчиняли у 50мл сухого ДХМ. Одержаний розчин додавали до 5,0г сухої 2-хлортритилхлоридної смоли (200400 меш) і суміш залишали для взаємодії протягом 12 годин на обертовій мішалці. Наприкінці реакції смолу двічі залишали для взаємодії з 50мл суміші ДХМ/МеОН/DIPEA протягом 3 хвилин, двічі промивали 50мл ДХМ і двічі 50мл ДМФА. Протокол 4: Конденсація амінокислот 8-57 Висушену смолу залишали набрякати у 50мл суміші піперидин/ДМФА (1/3) протягом 30 хвилин, обробляли 30мл суміші піперидин/ДМФА (1/3) протягом 20 хвилин, обробляли 30мл суміші ДБУ/піперидин/ДМФА (2/2/96) протягом 20 хвилин і промивали 6 разів 30мл ДМФА. 10ммоль амінокислоти, 15ммоль HOBT і 20ммоль DIPEA розчиняли у 30мл ДМФА. Через 5 хвилин додавали 10ммоль РуВОР та 20ммоль DIPEA і одержаний розчин вливали в смолу. Після 60 хвилин перемішування на обертовій мішалці смолу промивали двічі 30мл ДМФА і конденсацію повторювали один раз або (у складних випадках) двічі протягом 60 хвилин. Смолу потім промивали 6 разів 30мл ДМФА. Смолу можна використовувати безпосередньо для приєднання наступної амінокислоти або після 2 промивок 30мл ДХМ висушувати у вакуумі і зберігати при -80°С. 37 Протокол 5: Відщеплення та видалення захисту Після конденсації останньої амінокислоти Nкінцеву захисну групу видаляли подвійною обробкою 30мл суміші піперидин/ДМФА (1/3) протягом 20 хвилин і обробкою сумішшю ДБУ/піперидин/ДМФА (2/2/96) протягом 20 хвилин. Смолу промивали 6 разів 30мл ДМФА, двічі 30мл ДХМ і обробляли 50мл суміші TFE/ДХМ (2/8) протягом 180 хвилин. Після фільтрування розчинник видаляли у вакуумі, а захисну групу видаляли обробкою 30мл суміші TFA/TIS/EDT/вода (94/1/2,5/2,5) протягом 180 хвилин в атмосфері інертного газу. Одержаний розчин вливали у 300мл холодного ефіру, осад розчиняли в ацетонітрилі, а пептид очищали методом ВЕРХ-ОФ (Kromasil 100 С4 10мкм, 250´4,6мм) і зібрані фракції безпосередньо використовували для процедури рефолдінгу. Альтернативно неочищений продукт імуномера IL-2 можна очищати ефективно на афінній колонці із спорідненістю до металу: Неочищений продукт IL-2, 76,7мг (12,2мкмоль) вводили приблизно у 5мл елюенту А за 5 циклів (також див. Фіг.3а-с). Колонка: Omnifit, 1,7мл Елюент A: TFE/H2O/DIEA/NMI, 1:1: +1%: +5мМ Елюент В: TFE/H2O/DIEA/NMI, 1:1:+1%:+500мМ Швидкість: 1мл/хв Програма: Введення (5мл А) ® Промивка (16,5мл А) ® Градієнт: елюювання (0% В ® 100% В у межах 10мл) ® Збір (5мл В) ® Регенерація (8мл А). Протокол 6: Процедура рефолдінгу Відповідні елюйовані фракції розбавляли до кінцевого обсягу 20мл, використовуючи точно такий розчинник, який присутній в елюйованій фракції. Одержаний розчин очищеного продукту інкубували протягом 30 хвилин при кімнатній температурі з 10мл Ni-NTA Superflow (Qiagen) при незначному перемішуванні у хімічній склянці. Частки Superflow упаковували у порожню колонку FPLC і прикріпляли до приладу FPLC. Використовуючи хроматографічну програму цього приладу, розчинник замінювали за 10хв. Потім градієнт водою протягом інших 10хв. Використовували градієнт, щоб замінити розчинник сумішшю вода/трифторетанол (1/1). Під час 24 годинного періоду розчинник, що використовувався, окисляли барботуванням кисню через резервуарну судину і при використанні атмосфери кисню у судині, щоб закрити дисульфідні місточкові зв'язки. Під час насичення киснем підтримували постійну швидкість 0,1мл/хв протягом ночі по всій колонці, тоді як елюат постійно рециркулював у резервуарній судині. Наприкінці 24 годинного періоду кінцевий продукт, повторно скручений і ущільнений в результаті утворення дисульфідного зв'язку, елюювали застосуванням забуференого фосфатом фізіологічного розчину (PBS, pH 7,2), що містив 150мМ імідазолу, або застосуванням 0,1Μ ацетатного буфера (pH 4,5). Приклад 20: 86023 38 Конденсація (біс-трет-бутоксикарбонілметиламіно)-оцтової кислоти [TAG8b] з N-кінцем пептиду 1ммоль пептиду з послідовністю WETGLRLAPL одержували на 2-хлортритилхлоридній смолі (200-400 меш), дотримуючись стандартних процедур, які описані у прикладі 19, протокол 1. Для конденсації Tag8b до N-кінця пептиду аналогічним чином застосовували процедуру конденсації амінокислот. 5ммоль Tag8b, 7,5ммоль HOBT і 10ммоль DIPEA розчиняли у 15мл ДМФА. Через 5 хвилин додавали 5ммоль РуВОР та 10ммоль DIPEA і одержаний розчин вливали в смолу. Після 60 хвилин перемішування на обертовій мішалці смолу промивали 6 разів 20мл ДМФА, двічі 30мл ДХМ. Відщеплення і зняття захисту пептиду здійснювали, дотримуючись стандартних процедур: смолу обробляли 50мл суміші TFE/ДХМ (2/8) протягом 180 хвилин. Після фільтрування розчинник видаляли у вакуумі, а захисні групи видаляли обробкою 30мл суміші TFA/TIS/вода (95/2,5/2,5) протягом 180 хвилин в атмосфері інертного газу. Одержаний розчин вливали у 300мл холодного ефіру, осад розчиняли в ацетонітрилі, а пептид очищали методом ВЕРХ-ОФ (Kromasil 100 С4 10мкм, 250´4,6мм). На стадії видалення захисту групи третбутилових ефірів Tag відщеплювали з тим, щоб одержати пептид Tag8-WETGLRLAPL. PX-MC(ESI): (M+H)+:1331. Приклад 21: Конденсація 4-(біс-трет-бутоксикарбонілметилкарбамоїл)-масляної кислоти [TAG10b] з Nкінцем пептиду 1ммоль пептиду з послідовністю GDQYIQQAHRSHI був одержаний на 2хлортритилхлоридній смолі (200-400 меш), дотримуючись стандартних процедур, які описуються у прикладі 19, протокол 1. Для конденсації Tag10b до N-кінця пептиду аналогічним чином застосовували процедуру конденсації амінокислот. 5ммоль Tag10b, 7,5ммоль HOBT і 10ммоль DIPEA розчиняли у 15мл ДМФА. Через 5 хвилин додавали 5ммоль РуВОР та 10ммоль DIPEA і одержаний розчин вливали в смолу. Після 60 хвилин перемішування на обертовій мішалці смолу промивали 6 разів 20мл ДМФА, двічі 30мл ДХМ. Відщеплення і зняття захисту пептиду здійснювали, дотримуючись стандартних процедур: смолу обробляли 50мл суміші TFE/ДХМ (2/8) протягом 180 хвилин. Після фільтрування розчинник видаляли у вакуумі, а захисні групи видаляли обробкою 30мл суміші TFA/TIS/вода (95/2,5/2,5) протягом 180 хвилин в атмосфері інертного газу. Одержаний розчин вливали у 300мл холодного ефіру, осад розчиняли в ацетонітрилі, а пептид очищали методом ВЕРХ-ОФ (Kromasil 100 С4 10мкм, 250´4,6мм). На стадії видалення захисту групи третбутилових ефірів Tag відщеплювали з тим, щоб одержати пептид TaglO-GDQYIQQAHRSHI. РХ-МС (ESI): (М+Н)+: 1783. 39 86023 40 41 86023 42 43 86023 44 45 Комп’ютерна верстка Л.Литвиненко 86023 Підписне 46 Тираж 28 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюSolid phase peptide synthesis and use of activated solid phase in the process
Автори англійськоюRybka Andreas, Frank Hans-Georg, Bracht Franz-Peter, Haberl Udo
Назва патенту російськоюСпособ твердофазного синтеза пептидов и применение активированной твердой фазы в способе
Автори російськоюРибка Андреас, Франк Ханс-Георг, Брахт Франц-Петер, Хаберл Удо
МПК / Мітки
МПК: C07K 1/22, C07K 1/04, C07K 14/55
Мітки: пептидів, способи, спосіб, синтезу, застосування, твердої, твердофазного, активованої, фазі
Код посилання
<a href="https://ua.patents.su/23-86023-sposib-tverdofaznogo-sintezu-peptidiv-ta-zastosuvannya-aktivovano-tverdo-fazi-v-sposobi.html" target="_blank" rel="follow" title="База патентів України">Спосіб твердофазного синтезу пептидів та застосування активованої твердої фази в способі</a>
Спосіб автоматичного керування процесом осадження твердої фази

Номер патенту: 65879
Опубліковано: 15.04.2004
Автори: Шпильовий Леонід Вікторович, Білецький Володимир Стефанович
МПК: B01D 21/00
Мітки: керування, фазі, автоматичного, твердої, осадження, спосіб, процесом
Формула / Реферат:
Спосіб автоматичного керування процесом осадження твердої фази шляхом вимірювання вагової витрати твердої фази у вихідній пульпі та концентрації іонів водню та зміни витрати флокулянта, який відрізняється тим, що додатково вимірюють питому поверхню частинок твердої фази, а витрату флокулянта змінюють в залежності від загальної поверхні твердої фази з корекцією за величиною концентрації іонів водню в рідкій фазі пульпи.
Спосіб автоматичного контролю параметрів твердої фази пульпи

Номер патенту: 27086
Опубліковано: 10.10.2007
Автори: Поркуян Ольга Вікторівна, Моркун Володимир Станіславович
МПК: G01N 29/02
Мітки: фазі, твердої, автоматичного, пульпи, спосіб, контролю, параметрів
Формула / Реферат:
Спосіб автоматичного контролю параметрів твердої фази пульпи, який включає випромінювання ультразвукових коливань у потік пульпи, який проходить через вимірювальну камеру, приймання коливань, що пройшли фіксовану відстань крізь потік пульпи, вимірювання інтенсивності прийнятих коливань та визначення з урахуванням її величини контрольованих параметрів, який відрізняється тим, що на стінку вимірювальної камери наносять металеву плівку,...
Спосіб твердофазного синтезу альдегідів, кетонів, оксимів, амінів і гідроксамової кислоти

Номер патенту: 67733
Опубліковано: 15.07.2004
Автори: Мортон Джордж К., Мейсон Хелен Дж., Сальвіно Джозеф М., Лябодіньєр Річард Ф.
МПК: C07C 47/20, C07D 311/82, C07D 307/33, C07C 47/52, C07C 209/00, C07C 271/00, C07C 45/00, C07B 61/00, C07C 239/00, C07C 317/44, C07D 313/00, C07C 49/213, C07C 209/42, C07C 269/00, C07C 49/327, C07C 315/00, C07C 45/45, C07C 259/00, C07D 311/80, C07C 45/61, C07D 311/68, C07C 45/51, C08F 12/00, C08F 8/00, C07D 209/42, C07D 311/86
Мітки: кетонів, гідроксамової, твердофазного, спосіб, амінів, синтезу, альдегідів, оксимів, кислоти
Формула / Реферат:
1. Спосіб одержання кетону формули,в якійRc і Ra незалежно є аліфатичною або ароматичною групою, що включаєа) взаємодію N-алкілованої гідроксамової кислоти на полімерній смолі формули:,в якій є твердим носієм, L відсутній або є зв'язувальною групою, a Ra і Rb незалежно є аліфатичною або арильною групою з металоорганічним реагентом формули RcM, в якій Rc є аліфатичним або арильним аніоном і Μ є...
Спосіб автоматичного контролю параметрів твердої фази рудної суспензії

Номер патенту: 27846
Опубліковано: 12.11.2007
Автори: Моркун Володимир Станіславович, Подгородецький Микола Сергійович, Поркуян Ольга Вікторівна
МПК: G01N 29/02
Мітки: фазі, контролю, суспензії, твердої, параметрів, автоматичного, рудної, спосіб
Формула / Реферат:
Спосіб автоматичного контролю параметрів твердої фази рудної суспензії, що включає формування потоку суспензії рудного матеріалу у вимірювальній камері, періодичний вплив на потік суспензії ультразвуковими коливаннями, формування високочастотних об'ємних ультразвукових хвиль у потоці суспензії рудного матеріалу та поверхневих ультразвукових хвиль, вимірювання інтенсивності високочастотних об'ємних ультразвукових хвиль і поверхневих...
Спосіб очистки твердої фази суспензії

Номер патенту: 55912
Опубліковано: 15.04.2003
Автори: Стринадко Мирослав Танасійович, Тимочко Богдан Михайлович, Божок Аркадій Михайлович
МПК: B04B 1/00
Мітки: очистки, спосіб, суспензії, твердої, фазі
Формула / Реферат:
Спосіб очистки твердої фази суспензії, що включає попередній розгін ротора центрифуги, подачу суспензії в ротор центрифуги до заповнення його робочого об'єму, холостий хід, відділення рідкої фази суспензії під дією поля відцентрових сил в процесі основного розгону при максимальній частоті обертання ротора, ущільнення осаду, подачу дози промивної води, промивку й вивантаження осаду, який відрізняється тим, що в ньому одночасно вимірюється...
Попередній патент: Спосіб виготовлення багатошарової плити з перегородками
Наступний патент: Трициклічні дельта-опіоїдні модулятори
Випадковий патент: Похідні 6-дифторметил-5,6-дигідро-2н-[1,4]оксазин-3-аміну