Спосіб стереоселективного ферментативного гідролізу естеру 5-метил-3-нітрометилгексанової кислоти
Номер патенту: 103997
Опубліковано: 25.12.2013
Автори: Осль Доріс, де Соуца Домінік, Рітхорст Вандер, Сальхенеггер Йорг, Бергер Андреас, Цепек Фердінанд, Ремлер Петер, Лушніг Даніель, Шваб Хельмут, Альберт Мартін

























Формула / Реферат
1. Спосіб стереоселективного ферментативного гідролізу естеру 5-метил-3-нітрометилгексанової кислоти (VIII), у якому рацемічний естер 5-метил-3-нітрометилгексанової кислоти (VIII)
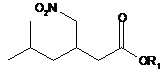 (VIII)
(VIII)
контактує з ферментом з одержанням
(R)-енантіомеру естеру 5-метил-3-нітрометилгексанової кислоти (VIII) та (S)-енантіомеру солі 5-метил-3-нітрометилгексанової кислоти, де R1 являє собою алкільну, арильну або арилалкільну групу, а фермент вибраний з групи, яка складається з естерази з печінки борова, ліпази А з Candida Antarctica, естерази з печінки свині (ICR-123) та естерази EstC з Burkholderia gladioli.
2. Спосіб за п. 1, де фермент являє собою естеразу EstC з Burkholderia gladioli.
3. Спосіб за п. 1 або 2, де конверсія складає від приблизно 40 % до приблизно 50 %.
4. Спосіб за будь-яким з пп. 1-3, де енантіомерний залишок (eн) естеру 5-метил-3-нітрометилгексанової кислоти (VIII), що залишився, або утвореної солі 5-метил-3-нітрометилгексанової кислоти (IX), при конверсії 50 % є вищим за 80 %.
5. Спосіб за п. 4, де енантіомерний залишок (ен) естеру 5-метил-3-нітрометилгексанової кислоти (VIII), що залишився, або утвореної солі 5-метил-3-нітрометилгексанової кислоти (IX), при конверсії 50 % є вищим за 95 %.
6. Спосіб за будь-яким з пп. 1-5, де стереоселективний ферментативний гідроліз проводять у водній системі, яка містить метанол.
7. Спосіб за будь-яким з пп. 1-6, де стереоселективний ферментативний гідроліз проводять у водному розчині при рН у межах від приблизно 5 до приблизно 11.
8. Спосіб за будь-яким з пп. 1-7, де енантіомер естеру (R)-5-метил-3-нітрометилгексанової кислоти (VIII) та солі (S)-5-метил-3-нітрометилгексанової кислоти розділяють та (S)-енантіомер 5-метил-3-нітрометил-гексанової кислоти додатково вступає в реакцію до утворення 3-амінометил-5-метилгексанової кислоти.
9. Спосіб за будь-яким з пп. 1-6, де естер 5-метил-3-нітрометилгексанової кислоти піддають ферментативному гідролізу при рН у межах від приблизно 8 до приблизно 14 для одержання солі 5-метил-3-нітрометилгексанової кислоти.
10. Спосіб одержання 3-амінометил-5-метилгексанової кислоти шляхом одержання солі 5-метил-3-нітрометилгексанової кислоти способом за будь-яким з пп. 1-9 та її наступного відновлення.
11. Спосіб за п. 10, де сіль 5-метил-3-нітрометилгексанової кислоти відновлюють при рН у межах від приблизно 8 до приблизно 14.
Текст
Реферат: Даний винахід стосується способів з одержанням (R)-енантіомеру естеру 5-метил-3нітрометилгексанової кислоти (VIII) та (S)-енантіомеру солі 5-метил-3-нітрометилгексанової кислоти, а також способу одержання 3-амінометил-5-метилгексанової кислоти. UA 103997 C2 5 10 15 20 Галузь винаходу Даний винахід стосується способу стереоселективного ферментативного гідролізу естеру 5метил-3-нітрометил-гексанової кислоти. Також описані спосіб одержання естеру 5-метил-3нітрометил-гексанової кислоти, а також способи одержання солі 5-метил-3-нітрометилгексанової кислоти та 3-(амінометил)-5-метилгексанової кислоти. Також описані (S)-5-метил-3нітрометил-гексанова кислота у енантіонасиченій формі або енантіочистій формі або (R)-5метил-3-нітрометил-гексанова кислота у енантіонасиченій формі або енантіочистій формі, а також їх солі, естер (S)-5-метил-3-нітрометил-гексанової кислоти у енантіонасиченій формі або енантіочистій формі або естер (R)-5-метил-3-нітрометил-гексанової кислоти у енантіонасиченій формі або енантіочистій формі та сполука, а саме у рацемічній формі, енантіонасиченій формі або енантіочистій формі. Рівень техніки (S)-3-(амінометил)-5-метилгексанова кислота (прегабалін, сполука (І); фігура 1) була у перший раз описана в ЕР-А-641330 і у даний час присутня на ринку під торговою назвою Lyrica® як агент для протиконвульсивної терапії. У ЕР-А-641330 описаний шлях синтезу цієї сполуки. Однак, описаний спосіб одержання цієї сполуки є тривалим (> 10 стадій), має малу ефективність та використовує пірофорні або реагенти, що дорого коштують, такі як відповідно бутил літій та (+)-4метил-5-феніл-2-оксазолідон, що обмежує його застосування на промисловому рівні. 25 30 35 Фігура 1. Структура прегабаліну (І) У роботі Hoekstra M. S. et al., Org. Proc. & Res. Dev. 1997, 1, 26-38 описані деякі підходи до одержання прегабаліну. Два способи, які мають зокрема економічний інтерес, описані в EP-А828704 та EP-A-830338, відповідно. В патентній заявці ´704 3-ізобутилглутарова кислота, одержана з ізовалеральдегіду та етилціаноацетату, слугує як ключова проміжна сполука, яку трансформують через відповідний циклічний ангідрид до аміду, який може бути розщеплений у класичний спосіб за допомогою енантіочистого фенілетиламіну як агента розщеплення (Схема 1). Амід потім піддають реакції деградації Гоффмана, що приводить до одержання (S)прегабаліну. Покращання та варіації цього способу були описані у WO 2006/122255, WO 2006/122258, WO 2006/122259, WO 2006/136087, WO 2007/035789, WO/035790 та WO/139933. 1 UA 103997 C2 5 10 15 Схема 1: Синтез прегабаліну (І) згідно з ЕР-А-828704. В ЕР-А-830338 одержують рацемічну 3-(амінометил)-5-метилгексанову кислоту і рацемат розщеплюють за допомогою (S)-мигдальної кислоти як хірального агента розщеплення. Рацемічний вихідний матеріал одержують у п’ять стадій з ізовалеральдегіду та діетилмалонату. Розщеплення рацемату у кінці робить синтез таким, що дорого коштує, а неефективним тому, що небажаний ізомер потрібно вилучати протягом усього процесу (Схема 2). Варіація цього процесу з розщепленням перед відновленням ціано-групи описана в WO 2007/143152. Обидва процеси страждають від недоліків, таким як довгий період синтезу та низький загальний вихід. Схема 2: Синтез прегабаліну (І) згідно з ЕР-А-830338. Асиметричний синтез проміжної сполуки, який веде одержання прегабаліну, включає гомогенне каталітичне гідрування з хіральними лігандами на основі фосфіну, описаний в WO 2001/550090 та WO 2005/087370. Вихідний матеріал одержують у три стадії, які включають застосування окису вуглецю, який являє собою небезпечний реагент та Pd, який являє собою каталізатор, що дорого коштує. 20 Схема 3: Синтез прегабаліну (І) згідно з WO 2001/55090 та WO 2005/087370. У WO 2006/110783 була описана конверсія хірального діалкілового естеру 2-(3-метил-1 2 UA 103997 C2 5 10 15 20 25 30 35 40 45 нітрометил-бутил)-малонової кислоти у прегабалін з використанням стратегії відновленнядекарбоксилювання. Послідовність реакцій наслідує послідовність реакцій, яку застосовували для синтезу, наприклад, баклофену (Ooi, T.; Fujioka, S.; Maruoka, K. J. Am. Soc., 2005, 127, 119125). Процеси очищення, які ведуть до одержання прегабаліну, чистого від деяких домішків, пов’язаних з процесом, описані в WO 2006/122255 WO 2006/121557. Усі з вищеописаних способів застосовують хіральні допоміжні агенти, каталізатори чи добавки. Такі сполуки зазвичай важко вилучити та вони присутні у кінцевому продукті у небажаних кількостях. Ферментативні кінетичні розщеплення прегабалінових попередників, які містять нітрил (сполуки (ІІ) та (ІІІ), (Фігура 2) описані в WO 2005/100580 та WO 2005/00904. У цих двох способах описані синтези прегабаліну, недоліком яких є використання ціаніду натрію, поводження з яким може бути проблематичним на промисловому рівні через проблеми безпеки. У WO 2007/143113 описане ферментативне кінетичне розщеплення через гідроліз або етерифікацію чотирьох субстратів ((IV та (V); R = H та Et, відповідно. Однак, не наводяться ніякі експериментальні деталі, такі як селективність та виходи. Фігура 2: Структури сполук, які були піддані ферментативному розщепленню. Синтез рацемічного прегабаліну описаний в Andruszkiewicz, R; Silverman, R. B., Synthesis 1989, 953-955. Синтез починається з етилового естеру (Е)-5метил-гекс-2-енової кислоти, який конвертують в етиловий естер 5-метил-нітрометил-гексанової кислоти шляхом кон’югованого додавання нітрометру. Цю сполуку конвертують в рацемічний прегабалін шляхом каталітичного гідрування з наступним омиленням. Схема 4: Синтез рацемічного прегабаліну (І) за способом Andruszkiewicz et al. Нещодавно був описаний ферментативний гідроліз етилового ефіру 5-метил-3-нітрометилгексанової кислоти, одержаного як описано у Andruszkiewicz et al. (Felluga, F. et al. Tetrahedron Assymetry 2008, 19, 945-955, опубл. online 6 травня 2008 р.). В описаному способі використовують конкретний фермент, а саме – Novozyme 435, який приводить до одержання енантіомерно насиченого етилового естеру (S)-5-метил-3-нітрометил-гексанової кислоти та енантіомерно насиченого етилового естеру (R)-5-метил-3-нітрометил-гексанової кислоти. Хороші селективності можуть бути одержані лише тоді, коли конверсії становлять нижче 30% або вище 60%, відповідно, що значно обмежує вихід. Для одержання прегабаліну конверсії треба зупиняти при 95%. Для цілей даного винаходу термін “енантіомерно насичений” означає енатіомерне співвідношення R/S або S/R більше ніж 75/25, що відповідає значенню “ен” > 50%. Можуть бути використані будь-які придатні умови для проведення стереоселективного ферментативного гідролізу. Вони будуть залежати від вибраного ферменту. Краще, коли реакцію проводять у такий спосіб, щоб “ен” енантіомеру естеру 5-метил-3-нітрометил-гексанової кислоти (VIII), що залишився, або “ен” утвореної солі 5-метил-3-нітрометил-гексанової кислоти (ІХ) становили 50% або більше, краще – 80% або більше, а найкраще – 90% або більше. Стереоселективний ферментативний гідроліз можна проводити у водній системі, такій як розчин, суспензія чи емульсія. Реакційна суміш може містити одну або множину фаз, наприклад, бути двох- чи трьохфазовою системою. Приклади таких двох- чи трьохфазових систем описані, наприклад, на сторінці 30, рядок 14-33 у WO 2006/000904. У кращому варіанті втілення реакцію проводять у водному розчиннику, такому як вода або суміш води та органічного розчинника, такого як етанол, який є сумісним з нею. Кращим водним розчинником є вода. Оскільки, естер 5-метил-3-нітрометил-гексанової кислоти (VIII) є лише слабо розчинним у воді, реакційна система є зазичай гетерогенною. Несподівано було виявлено, що стереоселективність ферментативного гідролізу естеру 5метил-3-нітрометил-гексанової кислоти (VIII) можна з користю підсилити шляхом використання метанолу як спільного розчинника у водній системі згідно з даним винаходом. Ферментативний гідроліз, здійснений з ферментом Estc з Burkoholderia gladioli в присутності метанолу дав несподіване підвищення стереоселективності ферментативного гідролізу естеру 5-метил-3нітрометил-гексанової кислоти (VIII) та показав енантіомерний надлишок (ен) естеру 5-метил-3нітрометил-гексанової кислоти (VIII), що залишився, до 98%; див. Приклад 8b. Результатом ферментативного гідролізу, здійсненого у відсутності метанолу, був енантіомерний надлишок (ен) приблизно 88% або нижче; див. Приклад 8а. Таким чином, в іншому кращому варіанті втілення стереоселективний ферментативний гідроліз проводять у водній системі, яка містить метанол. Краще, коли водна система являє собою водний розчин. Краще, коли метанол міститься у водній системі у концентрації від приблизно 0,01% до приблизно 5% [об./об.], краще – у концентрації від приблизно 1% до 6 UA 103997 C2 5 10 15 20 25 30 35 40 45 50 55 60 приблизно 3,5% [об./об.], а ще краще - у концентрації від приблизно 1,5% до приблизно 2,5% [об./об.], а найкращою концентрацією є приблизно 2,5% [об./об.]. Краще, коли стереоселективний ферментативний гідроліз проводять у буферній суміші води та метанолу. В кращому варіанті втілення фермент, який використовують у комбінації з метанолом, являє собою естеразу EstC з Burkholderia gladioli, або естеразу, яка містить амінокислотну послідовність, принаймні на 50% ідентичну амінокислотній послідовності EstC з Burkholderia gladioli, краще – ідентичній принаймні на 60%, ще краще – ідентичній принаймні на 70%, а ще краще – ідентичній принаймні на < 75%, ще краще – ідентичній принаймні на 80%, ще краще – ідентичній принаймні на 90%, та ще краще – ідентичній принаймні на 95%, ще краще – ідентичній принаймні на 97%, а ще краще – ідентичній принаймні на 99%, а найкраще – повність ідентичній амінокислотній послідовності EstC з Burkholderia gladioli. Ідентичність амінокислотної послідовності, на яку тут є посилання, визначають як ступінь ідентичності між двома послідовностями, яка показує відхилення першої послідовності від другої. Ідентичність можна зручно визначити за допомогою комп’ютерної програми в цій галузі, такої як GAP, яка забезпечується в програмному пакеті GCG (Program Manual for the Wisconsin Package, Version 8, August 1994, Genetics Computer Group, 575 Science Drive, Madison, Wisconsin, USA 53711) (Needleman, S. B., and Wunsch, C. D., (1970), Journal of Molecular Biology, 48, 443-453. GAP використовують з нижчевказаними налаштуваннями для порівняння поліпептидної послідовності: штраф за утворення GAP 3,0 та штраф за розширення GAP 0,1, зріла частина амінокислотної послідовності естерази згідно з винаходом показує принаймні 50% ступінь ідентичності амінокислотній послідовності EstC з Burkholderia gladioli, краще – принаймні 60% ідентичності, ще краще – принаймні 70% ідентичності, а ще краще – принаймні < 75% ідентичності, ще краще – принаймні 80% ідентичності, ще краще – принаймні 90% ідентичності, та ще краще – принаймні 95% ідентичності, ще краще – принаймні 97% ідентичності, а ще краще – принаймні 99% ідентичності амінокислотній послідовності EstC, одержаній з Burkholderia gladioli, від позиції 1 до 298 (в нумерації BASBPN). Відповідно, ідентичність буде визначатись як кількість ідентичних залишків, розділена на 298. Даний винахід стосується способу стереоселективного ферментативного гідролізу хіральних естерів, які є субстратами EstC з Burkholderia gladioli, в присутності метанолу, де хіральні естери мають хіральний або прохіральний центр в кислотній групі біля карбонільної групи. Краще, коли хіральний центр знаходиться в α, β або γ позиції до атома вуглецю карбонільної групи, краще – коли в α або β позиції. Кислотна група хірального естеру може бути С 3-15 алкілом, лінійним або розгалуженим, необов’язково заміщеним одним чи більше –CN, -галогеном, -NO2, -N3, -OH, -SH, -NH2, -NHR, -NR2, -OR або –SR, де R являє собою С1-6 алкіл або С1-6 алканоїл; С6-10 арил або заміщений арил, ненасичений або насичений гетероарил або заміщенй гетероарил, який містить один або більше гетероатомів. Спиртова група ROH може бути вибрана з-поміж R = C1-6 лінійного або розгалуженого алкілу; краще – з MeOH, EtOH, 2-пропанолу або бутанолу; або С1-10 арилу або заміщеного арилу. Стереоселективний ферментативний гідроліз хіральних естерів в присутності метанолу можна проводити, використовуючи EstC з Burkholderia gladioli або естеразу, яка містить амінокислотну послідовність, що показує принаймні 50% ідентичність амінокислотній послідовності EstC, одержаній з Burkholderia gladioli, краще – принаймні 60% ідентичність, ще краще – принаймні 70% ідентичність, а ще краще – принаймні < 75% ідентичність, ще краще – принаймні 80% ідентичність, ще краще – принаймні 90% ідентичність, та ще краще – принаймні 95% ідентичність, ще краще – принаймні 97% ідентичність, ще краще – принаймні 99% ідентичність, а найкраще – повну ідентичність амінокислотній послідовності EstC з Burkholderia gladioli. Стереоселективний ферментативний гідроліз хіральних естерів в присутності метанолу можна проводити при будь-яких умовах, як тут описано, для процесу стереоселективного ферментативного гідролізу естеру 5-метил-3-нітрометил-гексанової кислоти. Стереоселективний ферментативний гідроліз можна проводити при будь-якому придатному значенні рН. Краще вибирати, щоби значення рН знаходилось в межах від приблизно 5 до приблизно 11, ще краще – від приблизно 6 до приблизно 9,5. Значення рН можна регулювати, наприклад, додаванням основи, такої як неорганічна чи органічна основа. Приклад органічних основ являють собою триетиламін, диізопропілетиламін, триоетиламін. Краще додавати неорганічну основу, таку як гідроксид амонію, лужного або лужноземельного металу (наприклад, NH4OH, NaOH, KOH, LiOH) або карбонати амонію, лужного або лужноземельного металу (наприклад, Na2CO3, K2CO3 або Li2CO3). Основу можна додавати у розчині, краще – у водному розчині. Концентрація цього розчину може варіювати від насичення до високого 7 UA 103997 C2 5 10 15 20 25 30 35 40 45 50 55 60 ступеню розведення (наприклад, приблизно 0,01М). Краще, коли концентрація основи варіює від приблизно 5М до приблизно 10М. При потребі для регулювання рН реакційне середовище може містити буфер. Придатні буфери включають фосфати амонію, лужного або лужноземельного металу (наприклад, фосфат амонію, фосфат калію та фосфат натрію) або ацетати амонію, лужного або лужноземельного металу (наприклад, ацетат амонію та ацетат кальцію) або буфери з рКа від приблизно 5 до приблизно 10. Температура, при якій можна проводити стереоселективний ферментативний гідроліз може варіювати у широких межах. Наприклад, температура може варіювати від приблизно 0 °С до приблизно 70 °С. В кращому варіанті втілення температура реакції становить від приблизно 5 °С до приблизно 30 °С. Для того, щоб одержати високий надлишок бажаного енантіомеру краще буде зупинити реакцію після досягнення відповідної конверсії. Якщо реакцію проводити до її завершення, то одержаною буде відповідна рацемічна сіль 5-метил-3-нітрометил-гексанової кислоти (ІХ). Найбільш придатна ступінь конверсії буде залежати від вибраного ферменту та може бути визначена фахівцем у даній галузі. Якщо використовують EstВ з Burkholderia gladioli, реакцію краще зупиняти при конверсії від приблизно 50% до приблизно 70%. Краще реакцію зупиняти при конверсії від приблизно 50% до приблизно 55%. Реакцію можна зупинити шляхом додавання органічного розчинника. Краще додавати сумісний з водою органічний розчинник, такий як етилацетат. Реакцію можна також зупинити за допомогою стандартного методу, відомого фахівцю у даній галузі, такого як підвищення температури, додавання кислоти чи основи і т.п. Якщо використовують EstС з Burkholderia gladioli, реакцію краще зупиняти при конверсії від приблизно 40% до приблизно 50%. Краще реакцію зупиняти при конверсії від приблизно 45% до приблизно 50%. Реакцію можна зупинити шляхом додавання органічного розчинника. Краще додавати сумісний з водою органічний розчинник, такий як етилацетат. Якщо використовують Candida Antarctica B, реакцію краще зупиняти при конверсії від приблизно 40% до приблизно 50%. Краще реакцію зупиняти при конверсії від приблизно 45% до приблизно 50%. Реакцію можна зупинити шляхом додавання органічного розчинника. Краще додавати сумісний з водою органічний розчинник, такий як етилацетат. Краще, коли значення рН буде вищим за 7,4. Величину конверсії можна визначити за допомогою будь-якого придатного методу, такого як кількість вживаної основи або вимірювань за допомогою ВЕРХ. Після або під час стереоселективного ферментативного гідролізу енантіомер естеру 5метил-3-нітрометил-гексанової кислоти (VIII), що не вступив у реакцію, (наприклад, естер (S)-5метил-3-нітрометил-гексанової кислоти S-(VIII)) та одержаний енантіомер солі 5-метил-3нітрометил-гексанової кислоти (ІХ) (наприклад, сіль (R)-5-метил-3-нітрометил-гексанової кислоти R-(IX)) можна відділити, використовуючи методики, відомі фахівцю у даній галузі. Наприклад, енантіомер естеру 5-метил-3-нітрометил-гексанової кислоти (VIII), що не вступив у реакцію, можна вилучити з реакційної суміші за допомогою однієї чи більше екстракцій органічним розчинником, який не змішується з водою, таким як етилацетат або гептан, таким чином, що одержаний енантіомер солі 5-метил-3-нітрометил-гексанової кислоти (ІХ) залишається у водному шарі. Необов’язково небажаний енантіомер (наприклад, у випадку прегабаліну – R-енантіомер) може бути підданий процесу рацемізації та знову введений у процес стереоселективного ферментативного гідролізу. Хоча стереоселективний ферментативний гідроліз можна використати у цілому ряді процесів, він особливо добре підходить для приготування енантіонасиченої чи енантіочистої 3(амінометил)-5-метилгексанової кислоти (І), зокрема прегабаліну. Схема 5 показує повну схему реакції одержання (S)-3-(амінометил)-5-метилгексанової кислоти (І), де використовується заявлений стереоселективний ферментативний гідроліз (реакція (g)). Як можна побачити на схемі реакції, вихідний матеріал – рацемічний естер рац(VIII) 5-метил-3-нітрометил-гексанової кислоти можна одержати різними шляхами синтезу. Крім того, кінцевий продукт заявленої реакції, а саме – бажаний енантіомер естеру 5-метил-3нітрометил-гексанової кислоти (VIII) можна піддавати подальшій переробці до одержання бажаного енантіомеру 3-(амінометил)-5-метилгексанової кислоти (І), використовуючи різні шляхи синтезу. Ці реакції, які наводяться для ілюстрації і не являються вичерпними, будуть описані далі. Для простоти реакції базуються на одному або більше варіантах енантіомерів. Однак, зрозуміло, що схему реакції можна застосувати і до іншого енантіомеру. Крім того, хоча всі проміжні сполуки показані на Схемі 5, зрозуміло, що не всі вони повинні бути вилучені до 8 UA 103997 C2 того як вони будуть далі приймати участь в реакції. 5 Схема 5 Загальний вигляд реакцій, які обговорюються у даній заявці, використовуючи (R)селективний фермент 10 Схема 6 Загальний вигляд реакцій, які обговорюються у даній заявці, використовуючи (S)селективний фермент Процеси, показані на Схемі 5 та Схемі 6, є швидкими, економічними та простими і забезпечують одержання прегабаліну з високим виходом та високої оптичної чистоти. Кращий процес містить стадії a) та b) для одержання сполуки VIII. Один переважний процес одержання сполуки І включає стадії послідовно проведення реакцій g), h), i) або g), j), k), відповідно. Додатковою перевагою є раннє розділення енантіомерів. У рівні техніки процеси, такі як описані, наприклад, в WO 2008/007145 або US-A-5637767, розділення енантіомерів має місце на стадії рацемічного прегабаліну. Одна головна перевага даного винаходу полягає в тому, що для останньої стадії потрібна лише половина кількості каталізатору на основі перехідного металу, який дорого коштує, тому, що небажаний енантіомер відділяється на більш ранній стадії і, таким чином, не піддається відновленню. Перевагою процесу є те, що для приготування бажаного енантіомеру 3-(амінометил)-5метилгексанової кислоти (І) не потрібно хіральних додаткових речовин. Такі речовини призводять до появи домішок у кінцевому продукті. Для цілей даного опису сполука вважається рацемічною, якщо вона включає два можливі енантіомери у співвідношенні приблизно 50:50. Сполука вважається “в основному енантіочистою” або “еннатіонасиченою”, якщо вона містить приблизно 90% або більше лише одного енатіомеру. 15 20 25 9 UA 103997 C2 5 Для цілей даного опису сполука вважається “енантіомерно чистою”, якщо вміст одного енантіомеру становить приблизно 95% або вище, краще – приблизно 98% або вище, а ще краще – приблизно 99% або вище. Для цілей даного опису сполука вважається “в основному вільною” від домішок, якщо відповідна домішка присутня у кількості приблизно 3% або менше, краще – 1% або менше, а ще краще – приблизно 0,1% або менше. Реакція (а) 10 15 20 25 30 35 У реакції (а) 3-метилбутиральдегід (VI) конвертується в естер 5-метил-гекс-2-енової кислоти (VII). Хвиляста лінія у формулі (VII) показує, що подвійна лінія може мати цис- або трансорієнтацію. Для цієї реакції можуть бути вибрані різні шляхи синтезу. В одному способі 3-метилбутиральдегід (VI) може бути підданий реакції Віттіг-Хорнера. Одна реакція такого типу, зокрема, була у центрі недавньої патентної заявки WO 2003/062185, яка включена шляхом посилання. Згідно цієї заявки реакція Віттіг-Хорнера 3метилбутиральдегіду (VI) та придатного фосфонату (RO)2P(=O)-CH2-COOR1 (де R являє собою аліфатичну С1-3 групу та R1 є таким, як визначений вище) здійснюють у воді при точній температурі в присутності карбонату лужного иеталу. Вихід продукту завдяки цьому процесу становить приблизно 90%. Недоліком процесу, описаного в WO 2003/062185, є використання такого доволі дорогого фосфонату як С2-синтон. В альтернативному та кращому варіанті втілення 3-метилбутиральдегіду (VI) може вступати в реакцію з моноалкілмалонатом HOOC-CH2-COOR1 для одержання естеру 5-метил-гекс-2енової кислоти (VII). Реакцію можна проводити в присутності розчинника, або краще – без розчинника. При потребі можна додати каталітичну кількість однієї чи більше основ. Наприклад, як першу основу можна використати піперидин в каталітичних кількостях (наприклад, < 0,05 екв. відносно 1 екв. альдегіду VI), а піридин може бути використаний як друга основа у кількості від приблизно 1,0 до приблизно 5,0 еквівалентів відносно 1 еквіваленту альдегіду VI. Реакцію зазвичай проводять при температурі від 50 °С до 100 °С. Інші умови для такої конверсії, які також можуть бути застосовані до даного винаходу, описані в: Gazz. Chim. Ital. 1953, 83, 1043-1045; або J. Am. Chem. Soc. 1948, 70, 2601; або Tetrahedron 2006, 82, 476-482. Естер 5-метил-гекс-2-енової кислоти (VII) може бути виділений чи підданий подальшій переробці без очищення. Краще, коли естер 5-метил-гекс-2-енової кислоти (VII) піддають очищенню шляхом екстракції кислотою перед конверсією в естер 5-метил-3-нітрометилгексанової кислоти (VIIІ). Реакція (b) 40 45 50 В цій реакції R1 є таким, як визначений вище. Естер 5-метил-гекс-2-енової кислоти (VII) можна конвертувати в естер 5-метил-3-нітрометил-гексанової кислоти (VIII) шляхом додавання нітрометану. Краще, коли використовують від приблизно 1 до приблизно 5 еквівалентів нітрометану CH3NO2, ще краще від приблизно 1,5 до приблизно 2,5 екв. нітрометану відносно 1 екв. естеру 5-метил-гекс-2-енової кислоти (VII). Реакцію (b) можна проводити в присутності розчинника, або краще – без розчинника. Якщо використовують розчинник, його можна вибрати з будь-якого протонного або апротонного органічного розчинника. Кращими органічними розчинниками є СН 2Cl2, ацетонітрил, етанол, метанол або тетрагідрофуран. Реакцію (b) можна проводити при різних температурах, наприклад, при температурі від 10 UA 103997 C2 5 10 15 приблизно 0 °С до приблизно 100 °С, краще - при температурі від приблизно 40 °С до приблизно при 60 °С. При потребі реакцію (b) можна необов´язково проводити в присутності основи. Можна використати будь-яку придатну основу, наскільки вона буде здатна до тривалого депротонування кислого протону нітрометильної групи. Основа може бути органічною основою, такою як триалкіламін (де алкільна група краще містить від 1 до 4 атомів вуглецю), алкоксид (такий як метоксид натрію або трет-бутоксид натрію), сильні органічні основи, такі як 1,8диазабіцикло[5,4,0]ундец-7-ен (ДБУ) або N,N,N'N'-тетраметилгуанідин (ТМГ), або неорганічною основою, такою як карбонат амонію, лужного або лужноземельного металу, гідроксид амонію, лужного або лужноземельного металу, або гідрокарбонат амонію, лужного або лужноземельного металу. Краще, коли конверсію проводять в присутності сильної органічної основи, такої як 1,8-диазабіцикло[5,4,0]ундец-7-ену (ДБУ) або N,N,N'N'-тетраметилгуанідину (ТМГ). Кількість основи не є конкретно обмеженою. Але її зазвичай додають в субстехіометричних кількостях. Наприклад, використовують від приблизно 0,1 до приблизно 0,5 екв. основи відносно 1 екв. естеру 5-метил-гекс-2-енової кислоти (VII). Естер 5-метил-3-нітрометил-гексанової кислоти (VIІI) у реакції (b) зазвичай одержують з виходом, який перевищує приблизно 80%, більш звично - з виходом, який перевищує приблизно 90%. Реакція (с) 20 25 30 35 3-метилбутиральдегід (VI) можна конвертувати в диестер 2-(3-метил-бутиліден)-малонової кислоти (ХІ) шляхом реакції конденсації Кньовенагеля з диалкілмалонатом R1OOC-CH2-COOR2. В цій реакції R1 та R2 можуть бути однакові або різні і можуть приймати значення, вказані для R1 вище. Краще, коли реакцію (с) проводять в присутності основи, такої як ди-n-пропіламін. Краще, коли використовують стехіометричну кількість або невеликий надлишок диалкілмалонату (від приблизно 1,0 до 1,5 екв.) відносно 1 екв. 3-метилбутиральдегіду (VI). Також краще використовувати стехіометричну або субстехіометричну кількості аміну (приблизно 1,0 екв. чи менше) відносно 1 екв. 3-метилбутиральдегіду (VI). Синтез диестеру (ХІ) 2-(3-метил-бутиліден)малонової кислоти з використанням реакції конденсації Кньовенагеля, описаної, наприклад, в ЕР-А-830338. При потребі диестер 2-(3-метил-бутиліден)-малонової кислоти (ХІ), одержаний в цій реакції, можна очистити за допомогою методів, відомих фахівцю у даній галузі, перед тим, як він буде залучений в подальшій реакції. Однак, диестер 2-(3-метил-бутиліден)-малонової кислоти (ХІ) краще використовувати у подальшому процесі без очищення. Реакція (d) 40 45 В цій реакції R1 та R2 є такими як визначені вище. Диестер 2-(3-метил-бутилдіден)-малонової кислоти (ХІ) може вступати в реакцію до естера 5-метил-гекс-2-енової кислоти (VIII) шляхом декарбоксилування. Декарбоксилування краще проводити при температурі у межах від приблизно 100 °С до приблизно 180 °С в придатному полярному апротонному розчиннику (такому як ДМСО або НМП). Необов’язково можна додавати сіль (таку як NaCl або LiCl) для прискорення декарбоксилування. Інші умови реакції, такі як різна температура, розчинник або добавки також прийнятні. Приклади таких умов з використанням інших субстратів, описані в Tetrahedron 1990, 46, 3929-3940. 11 UA 103997 C2 Реакція (е) 5 10 15 20 25 30 35 40 45 В цій реакції R1 та R2 є такими як визначені вище. Диестер 2-(3-метил-бутиліден)-малонової кислоти (ХІ) можна конвертувати в диестер 2-(3метил-1-нітрометил-бутил)-малонової кислоти (ХІІ) шляхом додавання нітрометану. Краще, коли використовують від приблизно 1 до приблизно 5 еквівалентів нітрометану CH3NO2, ще краще – від приблизно 1,5 до приблизно 2,5 екв. нітрометану відносно 1 екв. диестеру 2-(3-метил-бутилдіден)-малонової кислоти (ХІ). Реакцію (е) можна проводити в присутності розчинника, або краще - без розчинника. Якщо використовують розчинник, його можна вибрати з групи, яка складається з будь-якого протонного або апротонного органічного розчинника. Кращими органічними розчинниками є СН2Cl2, ацетонітрил, етанол, метанол або тетрагідрофуран. Реакцію (е) можна проводити при різних температурах, наприклад, при температурі від приблизно 0 °С до приблизно 100 °С, краще - при температурі 40 °С до приблизно 60 °С. Реакцію (е) можна необов’язково проводити в присутності основи. Можна використати будьяку придатну основу, здатну до депротонування кислого протону нітрометану. Основа може бути органічною основою, такою як триалкіламін (де алкільна група краще містить від 1 до 4 атомів вуглецю), алкоксид (такий як метоксид натрію або трет-бутоксид натрію), сильною органічною основою, такою як 1,8-диазабіцикло[5,4,0]ундец-7-ен (ДБУ) або N,N,N'N'тетраметилгуанідин (ТМГ), або неорганічною основою, такою як карбонат амонію, лужного або лужноземельного металу, гідроксид амонію, лужного або лужноземельного металу, або гідрокарбонат амонію, лужного або лужноземельного металу. Краще, коли конверсію проводять в присутності сильної органічної основи, такої як 1,8-диазабіцикло[5,4,0]ундец-7-ену (ДБУ) або N,N,N'N'-тетраметилгуанідину (ТМГ). Кількість основи не є конкретно обмеженою. Але її зазвичай додають в субстехіометричних кількостях. Наприклад, використовують від приблизно 0,1 до приблизно 0,5 екв. основи відносно 1 екв. диестеру 2-(3-метил-бутиліден)-малонової кислоти (ХІ). Типові умови додавання нітрометану, які можна застосувати в реакції (е), описані в J. Am. Chem. Soc. 1950, 72, 2537-2542; Synthesis 1972, 44-45; J. Med. Chem. 1993, 36, 1041-1047 або Chem. Pharm.Bull. 1995, 43, 1125-1131. Вихід реакції (е) зазвичай становить вище, ніж приблизно 90%, краще - вище приблизно 95%. Реакція (f) В цій реакції R1 та R2 є такими як визначені вище. Диестер 2-(3-метил-1-нітрометил-бутил)-малонової кислоти (ХІІ) можна конвертувати в естер 5-метил-3-нітрометил-гексанової кислоти (VIIІ) шляхом декарбоксилування. Декарбоксилування краще проводити при температурі у межах від приблизно 100 °С до приблизно 200 °С в придатному полярному апротонному розчиннику (такому як ДМСО або ДМФ). Для збільшення виходу необов’язково можна додавати сіль, таку як NaCl. Така реакція описана, наприклад, в WO 2006/110783. 12 UA 103997 C2 5 10 Реакція (g) Реакція (g) є одним зі способів стереоселективного ферментативного гідролізу, описаного вище. Однак, слід розуміти, що реакцію (g) можна також застосовувати до іншого енатіомеру. В стереоселективному ферментативному гідролізі естер (VIIІ) 5-метил-3-нітрометилгексанової кислоти контактують з ферментом. Продукти реакції будуть різнитись в залежності від вибраного ферменту. В одному способі рацемічний естер 5-метил-3-нітрометил-гексанової кислоти (VIIІ) одержують, використовуючи реакції a) та b), описані вище. В одному способі рацемічний естер 5-метил-3-нітрометил-гексанової кислоти (VIIІ) конвертують в суміш естеру (S)-5-метил-3-нітрометил-гексанової кислоти S-(VIIІ) та сіль (R)-5метил-3-нітрометил-гексанової кислоти R-(IX). В способі рацемічний естер 5-метил-3-нітрометил-гексанової кислоти (VIIІ) конвертують в суміш естеру (S)-5-метил-3-нітрометил-гексанової кислоти S-(VIIІ) та сіль (R)-5-метил-3нітрометил-гексанової кислоти R-(IX) шляхом ферментативного гідролізу при рН 8-14. 15 20 25 30 35 40 45 50 55 В способі рацемічний естер 5-метил-3-нітрометил-гексанової кислоти (VIIІ) конвертують в суміш естеру (R)-5-метил-3-нітрометил-гексанової кислоти R-(VIIІ) та сіль (S)-5-метил-3нітрометил-гексанової кислоти S-(IX). Катіон М* солі може являти собою будь-який придатний катіон, такий як катіон лужного чи лужноземельного металу. Його зазвичай визначають за умовами, в яких проводять реакцію, і він буде, зокрема, відповідати катіону основи, яку зазвичай використовують. Фермент можна використовувати у формі сирого лізату або в очищеній формі. Альтернативно, фермент може бути у формі цілих мікробних клітин, пермебілізованих мікробних клітин, екстрактів мікробних клітин, частково очищених ферментів, очищених ферментів та т.п. Краще, коли фермент використовують у формі сирого лізату чи ліофілізату. Альтернативно, фермент може бути іммобілізованим та використовуватися у такому вигляді. Методики іммобілізації відомі фахівцю у даній галузі. Придатні тверді носії включають, ® наприклад, полімерні матриці, такі як альгінат кальцію, поліакриламід, Eupergit та інші ® полімерні матеріали, а також неорганічні матриці, такі як Celite . Методики з іммобілізацією є кращими тому, що фермент та продукт можна легко розділити. Крім того, іммобілізований фермент можна рециркулювати та використовувати повторно, що робить процес більш економічним. Інші методики, такі як використання зшитих ферментних агрегатів (CLEAs) або зшитих ферментних кристалів (CLEСs) також можуть бути використані у даному винаході. Енантіомерний надлишок (ен) естеру 5-метил-3-нітрометил-гексанової кислоти (VIII), що залишився, або утвореної солі 5-метил-3-нітрометил-гексанової кислоти (ІХ) при конверсії 50% був вищим за 80% у кожному випадку. В залежності від умов реакції (конверсія, температура, рН) можуть бути досягнуті значення “ен” естеру 5-метил-3-нітрометил-гексанової кислоти (VIII), що залишився, до 99%. Умови проведення стереоселективного ферментативного гідролізу будуть зазвичай залежати від вибраного ферменту. Краще, коли реакцію проводять у такий спосіб, щоб “ен” енантіомеру естеру 5-метил-3-нітрометил-гексанової кислоти (VIII), що залишився, або „ен” утвореної солі 5-метил-3-нітрометил-гексанової кислоти (ІХ) становили 50% або більше, краще – 80% або більше, а найкраще – 90% або більше. Стереоселективний ферментативний гідроліз можна проводити у водній системі, такій як розчин, суспензія чи емульсія. Реакційна суміш може містити одну або множину фаз, наприклад, бути двох- чи трьохфазовою системою. Приклади таких двох- чи трьохфазових систем описані, наприклад, на сторінці 30, рядок 14-33 у WO 2006/000904. У кращому варіанті втілення реакцію проводять у водному розчиннику, такому як вода або суміш води та органічного розчинника, такого як етанол, який є сумісним з нею. Кращим водним розчинником є вода. Оскільки, естер 5-метил-3-нітрометил-гексанової кислоти (VIII) є лише слабо розчинним у воді, реакційна система є зазичай гетерогенною. Як описано вище, стереоселективність ферментативного гідролізу естеру 5-метил-3нітрометил-гексанової кислоти (VIII) можна з користю підсилити шляхом використання метанолу як спільного розчинника у водній системі згідно з даним винаходом. 13 UA 103997 C2 5 10 15 20 25 30 35 40 45 50 55 60 Таким чином, в іншому кращому варіанті втілення стереоселективний ферментативний гідроліз проводять у водній системі, яка містить метанол. Краще, коли водна система являє собою водний розчин. Краще, коли метанол міститься у водній системі у концентрації від приблизно 0,01% до приблизно 5% [об./об.], краще – у концентрації від приблизно 1% до приблизно 3,5% [об./об.], а ще краще - у концентрації від приблизно 1,5% до приблизно 2,5% [об./об.], а найкращою концентрацією є приблизно 2,5% [об./об.]. Краще, коли стереоселективний ферментативний гідроліз проводять у буферній суміші води та етанолу. В кращому варіанті втілення фермент, який використовують у комбінації з метанолом згідно з цим винаходом, являє собою естеразу EstC з Burkholderia gladioli, або естеразу, яка містить амінокислотну послідовність, принаймні на 50% ідентичну амінокислотній послідовності EstC з Burkholderia gladioli, краще – ідентичній принаймні на 60%, ще краще – ідентичній принаймні на 70%, а ще краще – ідентичній принаймні на < 75%, ще краще – ідентичній принаймні на 80%, ще краще – ідентичній принаймні на 90%, та ще краще – ідентичній принаймні на 95%, ще краще – ідентичній принаймні на 97%, а ще краще – ідентичній принаймні на 99%, а найкраще – повність ідентичній амінокислотній послідовності EstC з Burkholderia gladioli. Ідентичність амінокислотної послідовності, на яку тут є посилання, визначають як ступінь ідентичності між двома послідовностями, яка показує відхилення першої послідовності від другої. Ідентичність можна зручно визначити за допомогою комп’ютерної програми в цій галузі, такої як GAP, яка забезпечується в програмному пакеті GCG (Program Manual for the Wisconsin Package, Version 8, August 1994, Genetics Computer Group, 575 Science Drive, Madison, Wisconsin, USA 53711) (Needleman, S. B., and Wunsch, C. D., (1970), Journal of Molecular Biology, 48, 443-453. GAP використовують з нижчевказаними налаштуваннями для порівняння поліпептидної послідовності: штраф за утворення GAP 3,0 та штраф за розширення GAP 0,1, зріла частина амінокислотної послідовності естерази згідно з винаходом показує принаймні 50% ступінь ідентичності амінокислотній послідовності EstC з Burkholderia gladioli, краще – принаймні 60% ідентичність, ще краще – принаймні 70% ідентичність, а ще краще – принаймні 99%; E/Z 6/1; вихід 94%). 15 1 20 25 30 35 40 45 50 Н-ЯМР головного ізомеру (CDCl3, 300 МГц) δ (ч.на млн.) = 0,91 (d, 2xCH3, 6H, J 7,0 Гц), 1,27 (t, CH3, 3H, J 7,1 Гц), 1,73 (m, CH, 1H), 2,06 (bt, CH2, 2H, J 7,0 Гц), 4,16 (q, CH2, 2H, 7,1 Гц), 5,78 (bd, CH, 1H, J 15,6 Гц), 6,92 (bt, CH, 1H, J 15,6 Гц, 7,7 Гц та 7,4 Гц). 13 C-ЯМР головного ізомеру (CDCl3, 75,47 МГц) δ (ч.на млн.) = 14,4; 22,4; 27,9; 41,6; 60,2; 122,4; 148,4; 166,8. Приклад 6: Синтез етилового естеру 5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) Етиловий естер 5-метил-гекс-2-енової кислоти (VII, R1 = етил) (123,9 г; 0,79 моль) розчиняли в 112 мл (1,98 моль) нітрометану. До цього розчину додавали 36 мл ДБУ (0,24 моль). Реакційну суміш нагрівали до 60 °С і перемішували при цій температурі протягом 150 хвилин. Аналіз методом ГХ показав повне споживання вихідного матеріалу. Додавали метил трет-бутиловий етер (100 мл) і органічний шар промивали 200 мл 2М водної HCl. Водний шар двічі екстрагували 50 мл метил трет-бутиловим етером. Органічні шари комбінували і промивали 50 мл насиченого водного NaHCO3. Розчинник видаляли при зниженому тиску для одержання 172,4 г в основному чистого γ-нітро естеру VIII (чистота, визначена за допомогою ГХ, >97%; вихід 98%). 1 Н-ЯМР (CDCl3, 300 МГц) δ (ч.на млн.) = 0,78 (t, 2xCH3, 6H, J 7,0 Гц), 1,12 (t, CH3, 2H, J 7,0 Гц), 1,13 (t, CH3, 3H, J 7,0 Гц), 1,52 (m, CH, 1H), 2,29 (d, CH2, 2H, J 6,6 Гц), 2,54 (m, CH, 1H), 4,01 (q, CH2, 2H, J 7,2 Гц), 4,29 (dd, CH2, 1H, J 5,7 Гц та 12,4 Гц), 4,36 (dd, CH2, 1H, J 6,8 Гц та 12,4 Гц), 13 C-ЯМР (CDCl3, 75,47 МГц) δ (ч.на млн.) = 14,2; 22,3; 22,6; 25,1; 36,1; 40,6; 60,8; 78,9; 171,6. Приклад 7: Синтез етилового естеру (S)-5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) та натрієвої солі (R)-5-метил-3-нітрометил-гексанової кислоти (ІХ). 100 г етилового естеру 5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) додали до водного розчину EstB (500 мл клітинного екстракту; ~ 5 г загальної концентрації протеїнів). При температурі 25 °С рН підтримували на рівні 7,0 шляхом безперервного додавання 5М водного NaOH. Після 55% конверсії (відповідає споживанню 50,6 мл NaOH) реакцію зупинили шляхом додавання 100 мл етилацетату. Додавали 100 мл 5М водного NaOH і шари розділяли. Водний шар один раз промивали 100 мл етилацетату. Комбіновані органічні шари концентрували при зниженому тиску для одержання 43 г етилового естеру (S)-5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил; ен = 98%). 20 UA 103997 C2 5 10 15 20 25 30 35 40 45 50 55 60 Приклад 8: Синтез етилового естеру (R)-5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) та натрієвої солі (S)-5-метил-3-нітрометил-гексанової кислоти (ІХ). 100 г етилового естеру 5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) додавали до водного розчину EstС (250 мл клітинного екстракту; ~ 10 г загальної концентрації протеїнів). При температурі від 5 °С до 10 °С рН підтримували на рівні 9,0 шляхом безперервного додавання 5М водного NaOH. Після 45% конверсії (відповідає споживанню 41,4 мл NaOH) реакцію зупинили шляхом додавання 100 мл етилацетату. Додавали 100 мл 5М водного NaOH і шари розділяли. Водний шар один раз промивали 100 мл етилацетату. Комбіновані водні шари фільтрували та концентрували при зниженому тиску для одержання приблизно 200 мл натрієвої солі (S)-5-метил-3-нітрометил-гексанової кислоти у воді (ен = 92%). Приклад 8а: Синтез натрієвої солі (S)-5-метил-3-нітрометил-гексанової кислоти (ІХ). В хімічному стакані в 100 мл натрієвого фосфатного буферу (1 мМ, рН 7,2) розчиняли/суспендували 100 мг EstC (ліофілізованої). Значення рН впало до рН ~ 6,8, а потім його встановили на рівні рН = 7,4 за допомогою водного NaOH (0,1 М). Потім додавали 200 мг етилового естеру 5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) і рН підтримували на рівні 7,4 шляхом безперервного додавання водного NaOH (0,1 М). Після 45% конверсії (відповідає споживанню 4,0 мл 0,1 М NaOH) реакцію зупинили шляхом додавання 10 мл етилацетату. Шари розділяли і водний шар екстрагували один раз 10 мл етилацетату. Потім водний шар концентрували для одержання сполуки, вказаної у назві, з ен 88%. Приклад 8b: Синтез натрієвої солі (S)-5-метил-3-нітрометил-гексанової кислоти (ІХ). В хімічному стакані в 100 мл калієвого фосфатного буферу (1 мМ, рН 7,2) розчиняли/суспендували 100 мг EstC (ліофілізованої). Значення рН впало до рН ~ 6,8, а потім його встановили на рівні рН = 7,4 за допомогою водного NaOH (0,1 М). Потім додавали 250 мкл метанолу та 200 мг етилового естеру 5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) і рН підтримували на рівні 7,4 шляхом безперервного додавання водного NaOH (0,1 М). Після 45% конверсії (відповідає споживанню 4,0 мл 0,1 М NaOH) реакцію зупинили шляхом додавання 10 мл етилацетату. Шари розділяли і водний шар екстрагували один раз 10 мл етилацетату. Потім водний шар концентрували для одержання сполуки, вказаної у назві, з ен 98%. Приклад 9: Синтез (S)-3-амінометил-5-метил-гексанової кислоти (І, прегабаліну). 150 г етилового естеру (S)-5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил; аналіз: 97,2%) суспендували в 300 мл Н2О. Додали KОН (90,1 г; аналіз: 2,05 екв.). Спочатку мутна реакційна суміш стала прозорою, що вказувало на те, що реакція була близька до завершення. Після завершення конверсії (як визначено за допомогою ВЕРХ) реакційну суміш перемістили в реактор гідрування. Додали 90,0 г водної суспензії нікелевого каталізатора Ренея. При тиску водню 12 бар та температурі 45 °С суміш перемішували до повної конверсії, що визначено за допомогою ВЕРХ, що дало 88,0 г прегабаліну у водному розчині. Розчин фільтрували і потім концентрували до приблизно 270 г при зниженому тиску і додавали 400 мл 2-пропанолу. При температурі 45 °С додавали оцтову кислоту до досягнення рН 7,0. Прегабалін почав кристалізуватись. Через приблизно 60 хвилин реакційну суміш охолоджували до 10 °С. Перемішування продовжували пртягом 1 години, а потім продукт виділили шляхом фільтрування. Коржик на фільтрі промивали 90 мл суміші холодних Н 2О / 2пропанолу у співвідношенні 1:1. Після висушування одержали 75 г в основному чистого (чистота 99,6%) прегабаліну (вихід: 70%). Частину прегабаліну рекристалізували, як описано в WO 2006/000904, для підвищення чистоти від 99,6% до 99,9%. Аналітичні дані відповідали тим, які наводяться в літературі. Приклад 10: Синтез (S)-3-амінометил-5-метил-гексанової кислоти (І, прегабаліну). 150 г етилового естеру (S)-5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил; аналіз: 97,2%) суспендували в 300 мл Н2О. Додали KОН (90,1 г; аналіз: 86,1%, 2,05 екв.). Спочатку мутна реакційна суміш стала прозорою, що вказувало на те, що реакція була близька до завершення. Після завершення конверсії (як визначено за допомогою ВЕРХ) додали NH4форміат та Pd/C. Реакційну суміш перемішували до того, як почали спостерігати завершення конверсії. Розчин фільтрували, а потім концентрували до приблизно 250 г при зниженому тиску і додали 400 мл 2-пропанолу. При температурі 45 °С додавали оцтову кислоту до досягнення рН 7,0. Прегабалін почав кристалізуватись. Через приблизно 60 хвилин реакційну суміш охолоджували до 10 °С. Перемішування продовжували пртягом 1 години, а потім продукт виділили шляхом фільтрування. Коржик на фільтрі промивали 90 мл суміші холодних Н 2О / 2пропанолу у співвідношенні 1:1. Після висушування одержали 65 г в основному чистого (чистота 97,9%) прегабаліну (вихід: 61%). Приклад 11: динатрієва сіль (S)-5-метил-3-нітрометил-гексанової кислоти (ІХ – динатрієва сіль) 21 UA 103997 C2 5 830 мг етилового естеру (S)-5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) суспендували в 0,8 мл Н2О. Додали 900 мг 50% водного KОН. Після 5 год. при 25 °С конверсія була завершена (визначено за допомогою ВЕРХ). При зниженому тиску розчинник вилучили для одержання твердого залишку, який складався зі сполуки, вказаної у назві, та невеликої кількості KОН. 13 10 15 20 25 30 35 40 45 C-ЯМР (D2О, 75,47 МГц) δ (ч.на млн.) = 21,9; 22,8; 26,0; 33,9; 40,9; 41,6; 123,5; 181,7. Приклад 12: мононатрієва сіль (S)-5-метил-3-нітрометил-гексанової кислоти (ІХ мононатрієва сіль) 830 мг етилового естеру (S)-5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) суспендували в 0,8 мл Н 2О. Додали 900 мг 50% водного KОН. Після 5 год. при 25 °С конверсія була завершена (визначено за допомогою ВЕРХ). Додали 4,2 мл 1 М водної HCl і розчинник вилучили при зниженому тиску для одержання твердого залишку, який складався зі сполуки, вказаної у назві, та KCl. 13 C-ЯМР (D2О, 75,47 МГц) δ (ч.на млн.) = 23,0; 23,3; 25,2; 32,5; 36,7; 41,0; 79,9; 173,8. Приклад 13: Синтез (S)-3-амінометил-5-метил-гексанової кислоти (І, прегабаліну). Для відновлення з використанням Ренея-Нікель 5,0 г етилового естеру (S)-5-метил-3нітрометил-гексанової кислоти (VIII, R1 = етил) розчинили в 10 мл етанолу та 0,4 мл води і додали 3,0 г водної суспензії Ренея-Ni. Реакційну суміш перемішували при 40 °С під тиском 4 бар водню. Після завершення конверсії вихідного матеріалу реакційну суміш фільтрували і розчинник вилучали під зниженим тиском для одержання 3,02 г сирого лактону Х як маслянистого залишку, який кристалізувався при зберіганні. 30 мл 6N водної HCl додавали до сирого лактону і реакційну суміш нагрівали до кип’ятіння зі зворотним холодильником. Після 4 год. реакційну суміш концентрували під зниженим тиском для одержання 4 г маслянистого залишку. Додавали воду (5 мл) і значення рН встановили на рівні 6 шляхом додавання 50% водного KОН. Суміш нагрівали до 50 °С і повільно охолоджували до 10 °С. Утворені кристали видаляли фільтрацією. Концентрація маточного розчину дала можливість ще додатково зібрати кристали з виходом 2,5 г прегабаліну (72%). Приклад 14: Синтез (S)-3-амінометил-5-метил-гексанової кислоти (І, прегабаліну). Етиловий естер (S)-5-метил-3-нітрометил-гексанової кислоти (VIII, R1 = етил) (10,4 г) розчинили в 160 мл МеОН. Додали 4 г 10% Pd/C та 29 г форміату амонію. Через кілька хвилини спостерігали екзотермічну реакцію. Через 30 хвилин аналіз методом ВЕРХ показав повну конверсію. Каталізатор видалили фільтрацією. Фільтрат концентрували при зниженому тиску до об’єму приблизно 20 мл. Додали воду (20 мл) і розчин знову концентрували при зниженому тиску до приблизно 20 мл. Потім додали 50 мл 6N водної HCl і суміш кип’ятили зі зворотним холодильником протягом 6 годин. Після завершення конверсії реакційну суміш концентрували при зниженому тиску до об’єму приблизно 20 мл. Додавали воду (20 мл) та 2-пропанол (40 мл) і реакційну суміш нагрівали при 45 °С. Додавали KОН, поки не було досягнуто рН 7. Продукт почав кристалізуватись. Через 60 хвилин реакційну суміш охолодили до 10 °С. Перемішування продовжували протягом 1 години, а потім продукт виділили шляхом фільтрації. Коржик на фільтрі промивали 90 мл сумішшю холодних Н2О / 2-пропанолу, взятих у співвідношенні 1:1. Після висушування одержали 5,9 г в основному чистого (чистота 98,0%) прегабаліну (вихід: 22 UA 103997 C2 5 10 15 20 25 30 35 40 45 50 55 60 81%). Частину прегабаліну рекристалізували, як описано в WO 2006/000904, для підвищення чистоти від 98,0% до 99,9%. Приклад 15: Синтез (S)-3-амінометил-5-метил-гексанової кислоти (І, прегабаліну). 3-метилбутиральдегід (100 мл; 0,91 моль, сполука VI) додавали до 180 г (1,36 моль) моноетилмалонату в 260 мл (3,1 моль) піридину. До цього розчину додали 0,23 мл (0,25 моль %) піперидину. Реакційну суміш нагрівали до 80 °С і перемішували при цій температурі протягом 90 хвилин. Аналіз методом ГХ показав повне споживання вихідного матеріалу. Додавали метил-трет-бутиловий естер (300 мл) і органічний шар промивали три рази 200 мл 2М водної H2SO4, потім двічі 150 мл 0,5М водної NaHCO3. Основну частину розчинника вилучили при зниженому тиску і додали нітрометан (120 мл). До цього розчину додали 40 мл ДБУ. Реакційну суміш нагрівали при 60 °С та перемішували при цій температурі протягом 150 хвилин. Аналіз методом ГХ показав повне споживання вихідного матеріалу. Додавали метил-трет-бутиловий естер (100 мл) і органічний шар промивали 200 мл 2М водної HCl. Водний шар екстрагували двічі 50 мл метил-трет-бутиловий етеру. Органічні шари комбінували та промивали 50 мл насиченого водного NaHCO3. ДБУ відновлювали з водного шару шляхом додавання 50% водного NaOH і екстракції метил-трет-бутиловим етеромом (вихід відновлення після двох екстракцій – кожної зі 100 мл метил-трет-бутилового етеру становив 75%; рН водного шару > 12). Розчинник органічного шару вилучали при зниженому тиску для одержання 188,2 г в основному чистого γ-нітро естеру VIII (чистота, визначена методом ГХ > 95%; метил-третбутиловий етер < 5%). Естер може бути трансформований в прегабалін, як описано в Прикладах 7, 8 та 9. Приклад 16: Швидкий скринінг придатних ферментів, використовуючи комерційно доступні ферменти. Скринінг ферментів проводили як описано в статі M. Ivancic et al., J. of Biotechnology 2007, 129, 109-122, повний зміст якої включено шляхом посилання для всіх цілей. Всі ферменти були одержані від Сігма-Одріч (Sigma-Aldrich) (St. Louis, MO), Флука (Fluka) (Buchs, Switzerland), Амано (Amano) (Nagoya, Japan), Ново Нордіск Novo Nordisk (Bagswaerd, Denmark), БайоКаталітікс/Кодексис (BioCatalytics/Codexis) або від Технічного Університету Граца (Technical University of Graz). Для аналізу комерційно доступних естераз або ліпаз використовували швидкий скринінг, який базувався на зсуві значення рН. Аналіз проводився у два етапи: (і) активні ферменти були ідентифіковані; (іі) активні ферменти аналізували стосовно їх активності по відношенню до R- та S-енантіомерам етилового естеру 5-метил-3-нітрометил-гексанової кислоти. Розчини індивідуальних ферментів поміщали на фільтрувальні папірці та висушували при 30 °С протягом 30 хвилин. Висушені фільтрувальні папірці просочували скринінговим розчином, -1 який включав Triton X 100 (0,6%), фенольний червоний (2 гЛ ), буфер Трис-HCl рН = 7,5 та 50 мМ рацемічного етилового естеру 5-метил-3-нітрометил-гексанової кислоти. Гідроліз естеру визначали шляхом візуального моніторингу за зміною кольору з червоного на жовтий при падінні рН, викликаного вивільненою кислотою. Позитивні зразки, які показували активність естерази, відбирали та піддавали подальшому аналізу шляхом розміщення їх на фільтрувальному папері, який висушували та потім просочували R та S скринінговими розчинами, які містили як субстрати замість рацемічних чисті енантіомери. Активність ферментів встановлювали шляхом моніторингу на основі часу, необхідного для зміни кольору рН індикатора. Вибрані ферменти піддавали подальшому аналізу шляхом проведення ферментативного гідролізу на препаративній шкалі, використовуючи 200 мг рацемічного субстрату в 5 мл буферу Трис-HCl при рН = 7,5. Ферментний препарат додавали у достатній кількості для того, щоб час реакції становив менше 24 годин. Конверсію визначали шляхом вимірювання кількості спожитого 1М водного NaOH. При споживанні, яке відповідає 50% конверсії, реакцію зупиняли шляхом додавання 5 мл етилацетату. Шари розділяли і органічний шар аналізували шляхом хіральної ГХ. Приклад 17: Рекомбінантна експресія EstC від Burkholderia gladioli в E. coli. 377 г клітин E. coli, які піддають надекспресії EstC від Burkholderia gladioli, суспендували в 830 мл 200 мМ розчину фосфату натрію/цитрату (рН=7,0) та двічі піддавали гомогенізації. ® Суспензію клітин розбавляли 1:2 Sepipur CL930, що дало 4000 ч. на млн. флокулянта. Активність естерази вологих клітин визначали, використовуючи р-нітрофеніл ацетат як субстрат. Естерази каталізують гідроліз р-нітрофеніл ацетат в р-нітрофенол та оцтову кислоту. Активність ферменту визначають шляхом вимірювання збільшення абсорбції р-нітрофенолу 23 UA 103997 C2 5 (жовтий, 404 нм) в залежності від часу. Для вологих клітин була визначена активність 826 Од./г. Після центрифугування одержали прозорий сирий лізат з питомою активністю 158 Од./г. Розбавлений сирий лізат концентрували, використовуючи систему ультрафільтрації від Pall Corporation (Centramate) з відрізком мембрани 50 kDa. Фактор концентрації становив 8, що дало ретентат з питомою активністю 855 Од./мл та пермеат з питомою активністю 6,3 Од./мл. Ліофілізаційний залишок становив 23,3 г та мав питому активність 9125 Од./г. Загальний вихід становив 68,3%. ФОРМУЛА ВИНАХОДУ 10 1. Спосіб стереоселективного ферментативного гідролізу естеру 5-метил-3нітрометилгексанової кислоти (VIII), у якому рацемічний естер 5-метил-3-нітрометилгексанової кислоти (VIII) O2N O OR1 15 20 25 30 35 40 (VIII) контактує з ферментом з одержанням (R)-енантіомеру естеру 5-метил-3-нітрометилгексанової кислоти (VIII) та (S)-енантіомеру солі 5метил-3-нітрометилгексанової кислоти, де R1 являє собою алкільну, арильну або арилалкільну групу, а фермент вибраний з групи, яка складається з естерази з печінки борова, ліпази А з Candida Antarctica, естерази з печінки свині (ICR-123) та естерази EstC з Burkholderia gladioli. 2. Спосіб за п. 1, де фермент являє собою естеразу EstC з Burkholderia gladioli. 3. Спосіб за п. 1 або 2, де конверсія складає від приблизно 40 % до приблизно 50 %. 4. Спосіб за будь-яким з пп. 1-3, де енантіомерний залишок (eн) естеру 5-метил-3нітрометилгексанової кислоти (VIII), що залишився, або утвореної солі 5-метил-3нітрометилгексанової кислоти (IX), при конверсії 50 % є вищим за 80 %. 5. Спосіб за п. 4, де енантіомерний залишок (ен) естеру 5-метил-3-нітрометилгексанової кислоти (VIII), що залишився, або утвореної солі 5-метил-3-нітрометилгексанової кислоти (IX), при конверсії 50 % є вищим за 95 %. 6. Спосіб за будь-яким з пп. 1-5, де стереоселективний ферментативний гідроліз проводять у водній системі, яка містить метанол. 7. Спосіб за будь-яким з пп. 1-6, де стереоселективний ферментативний гідроліз проводять у водному розчині при рН у межах від приблизно 5 до приблизно 11. 8. Спосіб за будь-яким з пп. 1-7, де енантіомер естеру (R)-5-метил-3-нітрометилгексанової кислоти (VIII) та солі (S)-5-метил-3-нітрометилгексанової кислоти розділяють та (S)-енантіомер 5-метил-3-нітрометил-гексанової кислоти додатково вступає в реакцію до утворення 3амінометил-5-метилгексанової кислоти. 9. Спосіб за будь-яким з пп. 1-6, де естер 5-метил-3-нітрометилгексанової кислоти піддають ферментативному гідролізу при рН у межах від приблизно 8 до приблизно 14 для одержання солі 5-метил-3-нітрометилгексанової кислоти. 10. Спосіб одержання 3-амінометил-5-метилгексанової кислоти шляхом одержання солі 5метил-3-нітрометилгексанової кислоти способом за будь-яким з пп. 1-9 та її наступного відновлення. 11. Спосіб за п. 10, де сіль 5-метил-3-нітрометилгексанової кислоти відновлюють при рН у межах від приблизно 8 до приблизно 14. Комп’ютерна верстка С. Чулій Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 24
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the stereoselective enzymatic hydrolysis of 5-methyl-3-nitromethyl-hexanoic acid ester
Автори російськоюAlbert, Martin, Zepeck, Ferdinand, Berger, Andreas, Riethorst, Waander, Schwab, Helmut, Luschnig, Daniel, Remler, Peter, Salchenegger, Joerg, Osl, Doris, de Souza, Dominic
МПК / Мітки
МПК: C12P 7/62, C07C 205/51, C12P 41/00, C12P 7/40, C07C 229/08, C12P 13/00
Мітки: гідролізу, 5-метил-3-нітрометилгексанової, спосіб, естеру, ферментативного, кислоти, стереоселективного
Код посилання
<a href="https://ua.patents.su/26-103997-sposib-stereoselektivnogo-fermentativnogo-gidrolizu-esteru-5-metil-3-nitrometilgeksanovo-kisloti.html" target="_blank" rel="follow" title="База патентів України">Спосіб стереоселективного ферментативного гідролізу естеру 5-метил-3-нітрометилгексанової кислоти</a>
Застосування суміші етилового естеру ейкозапентаєнової кислоти та етилового естеру докозагексаєнової кислоти для лікування серцевої недостатності

Номер патенту: 78693
Опубліковано: 25.04.2007
Автори: Граната Франческо, Пампарана Франко, Страгліотто Едуардо Ліно
МПК: A61K 31/23, A61P 9/04
Мітки: застосування, ейкозапентаєнової, докозагексаєнової, естеру, суміші, недостатності, серцевої, етилового, лікування, кислоти
Формула / Реферат:
1. Застосування суміші есенційних жирних кислот, що включає етиловий естер ейкозапентаєнової кислоти (ЕПК) і етиловий естер докозагексаєнової кислоти (ДГК), при виготовленні медикаменту, корисного для профілактики й лікування серцевої хвороби, вибраної з серцевої недостатності та застійної серцевої недостатності, як хронічної, так і гострої, у пацієнта, що цього потребує.2. Застосування за пунктом 1, в якому медикамент призначений для...
Спосіб одержання 4′-[[4-метил-6-(1-метил-1н-бензімідазол-2-іл)-2-пропіл-1н-бензімідазол-1-іл]метил]біфеніл-2-карбонової кислоти (телмісартан)

Номер патенту: 99140
Опубліковано: 25.07.2012
Автори: Стах Ян, Цінібулк Йозеф, Яррах Камаль, Стрелєц Іво, Радл Станіслав
МПК: C07D 235/18
Мітки: одержання, 4'-[[4-метил-6-(1-метил-1н-бензімідазол-2-іл)-2-пропіл-1н-бензімідазол-1-іл]метил]біфеніл-2-карбонової, спосіб, телмісартан, кислоти
Формула / Реферат:
1. Спосіб одержання кристалічної форми А телмісартану (І), (I) який відрізняється тим, що карбонову кислоту загальної формули R1COOH, де R1 є атом водню або С1-С4алкіл, додають до розчину калієвої солі телмісартану (VII) (VII)в спирті формули R2OH із...
Спосіб одержання пропінілового естеру (r)(+)-2-[4-(5-хлор-3-фторпіридин-2-ілокси)фенокси]пропіонової кислоти

Номер патенту: 64718
Опубліковано: 15.03.2004
Автори: Зайферт Готфрід, Урвілер Бернхард, Стінг Андреа Рольф
МПК: A01P 13/00, C07D 213/61, C07D 213/643
Мітки: r)(+)-2-[4-(5-хлор-3-фторпіридин-2-ілокси)фенокси]пропіонової, кислоти, естеру, пропінілового, одержання, спосіб
Формула / Реферат:
1. Спосіб одержання пропінілового естеру (R)(+)-2-[4-(5-хлор-3-фторпіридин-2-ілокси)фенокси]пропіонової кислоти формули (І) , (I)який відрізняється тим, що сполуку формули (II) перетворюють , (II)в інертному органічному розчиннику разом з М2СО3, де М являє собою натрій чи...
Спосіб отримання та/або очищення клавуланової кислоти або фармацевтично прийнятної солі або її естеру

Номер патенту: 59343
Опубліковано: 15.09.2003
Автор: Цапудер Егідій
МПК: C12P 17/18, C07D 503/00, C12P 17/10
Мітки: фармацевтично, прийнятної, спосіб, клавуланової, кислоти, солі, естеру, отримання, очищення
Формула / Реферат:
1. Спосіб отримання та/або очищення клавуланової кислоти або її фармацевтично придатної солі чи естеру, що включає видалення твердих речовин з ферментаційного середовища, що містить клавуланову кислоту, мікрофільтруванням з одержанням першого фільтрату, що має рН 5,8-6,2, подальшого видалення твердих речовин з першого фільтрату ультрафільтруванням з утворенням другого фільтрату, концентрування другого фільтрату - видаленням води та обробку...
Сукцинат біфеніл-2-ілкарбамінової кислоти 1-[2-(2-хлор-4-{[(r)-2-гідрокси-2-(8-гідроксі-2-оксо-1,2-дигідрохінолін-5-іл)етиламіно]метил]}-5-метоксифенілкарбамоїл)етил]піперидин-4-іл-естеру та його застосування д

Номер патенту: 100364
Опубліковано: 25.12.2012
Автори: Кеннеді Ендрю, Мале Франк Патрік, Кіндон Лінда Джейн, Чудасама Решма
МПК: C07D 401/12, A61K 31/4709
Мітки: кислоти, застосування, сукцинат, біфеніл-2-ілкарбамінової, 1-[2-(2-хлор-4-{[(r)-2-гідрокси-2-(8-гідроксі-2-оксо-1,2-дигідрохінолін-5-іл)етиламіно]метил]}-5-метоксифенілкарбамоїл)етил]піперидин-4-іл-естеру
Формула / Реферат:
1. Сукцинат біфеніл-2-ілкарбамінової кислоти 1-[2-(2-хлор-4-{[(R)-2-гідрокси-2-(8-гідроксі-2-оксо-1,2-дигідрохінолін-5-іл)етиламіно]метил}-5-метоксифенілкарбамоїл)етил]піперидин-4-іл-естеру або його сольват, який є у кристалічній формі 1, що характеризується рентгенодифрактограмою порошку, що має піки дифракції при значеннях 2θ 5,0±0,3 та 10,0±0,3.2. Сіль за п. 1, де сіль характеризується: а) даними диференційної...
Попередній патент: Гідробромід івабрадину
Наступний патент: Дифумаратна сіль 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(n-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну
Випадковий патент: Багатошпиндельна свердлильна головка