Дифумаратна сіль 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(n-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну
Номер патенту: 103998
Опубліковано: 25.12.2013
Автори: Добсон Ендрю Хорнбі, Уітлок Брайан, Бернс Сюзан Елізабет, Бордман Кей Елісон







Формула / Реферат
1. Дифумарат 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)-піперидин-4-іл]окси}хіназоліну.
2. Дифумарат 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)-піперидин-4-іл]окси}хіназоліну за п. 1, де вказана сполука є кристалічною.
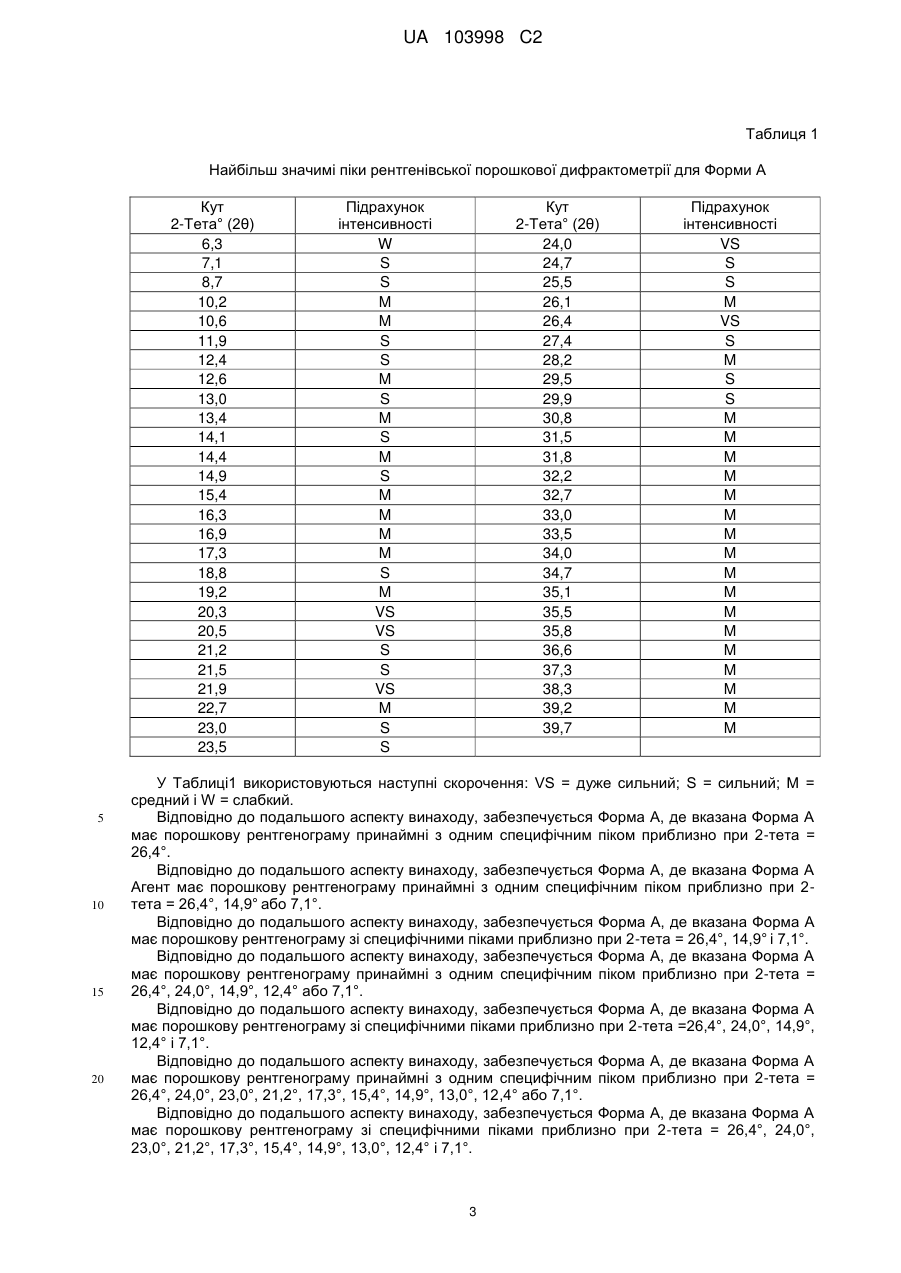
3. Кристалічний дифумарат 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну за п. 2, де вказана кристалічна сполука являє собою Форму А, що має порошкову рентгенівську дифрактограму зі специфічними піками при 2-тета = 26,4°±0,5°, 14,9°±0,5° та 7,1°±0,5°.
4. Форма А кристалічного дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну за п. 3, де вказана Форма А має порошкову рентгенівську дифрактограму зі специфічними піками при 2-тета = 26,4°±0,5°, 24,0°±0,5°, 14,9°±0,5°, 12,4°±0,5° та 7,1°±0,5°.
5. Форма А кристалічного дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну за п. 3, де вказана Форма А має порошкову рентгенівську дифрактограму зі специфічними піками при 2-тета = 26,4°±0,5°, 24,0°±0,5°, 23,0°±0,5°, 21,2°±0,5°, 17,3°±0,5°, 15,4°±0,5°, 14,9°±0,5°, 13,0°±0,5°, 12,4°±0,5° та 7,1°±0,5°.
6. Форма А кристалічного дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну за п. 3, де вказана Форма А має точку плавлення 210 °С±0,5°С.
7. Спосіб одержання Форми А дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну за п. 3, за яким здійснюють:
(і) взаємодію 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)-піперидин-4-іл]окси}хіназоліну з достатньою кількістю фумарової кислоти з утворенням дифумаратної солі;
(іі) кристалізацію дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну; і
(ііі) виділення Форми А дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну.
8. Спосіб за п. 7, де cтадію (і) способу здійснюють у суміші розчинників, що містить метанол і ізопропанол.
9. Спосіб одержання Форми А дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну за п. 3, за яким здійснюють:
(і) взаємодію розчину або суспензії 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну в етилацетаті принаймні з 1,725 молярним еквівалентом фумарової кислоти в ізопропанолі,
де придатне об'ємне співвідношення етилацетату до ізопропанолу становить від 5:1 до 1:1, наприклад від 5,1:1 до 1,9:1, і де реакцію здійснюють при температурі від 20 до 73 °С;
(іі) охолодження реакційної суміші зі Стадії (і) до 20 °С і витримування суміші при цій температурі для здійснення кристалізації Форми А дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну; і
(ііі) виділення Форми А дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну.
10. Спосіб одержання Форми А дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну за п. 3, за яким здійснюють:
(і) взаємодію розчину або суспензії 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну у воді принаймні з 2 молярними еквівалентами фумарової кислоти, де реакцію здійснюють при 85 °С;
(іі) охолодження реакційної суміші зі стадії (і) до 60 °С; і
(ііі) виділення Форми А дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну.
11. Фармацевтична композиція, яка містить дифумарат 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну відповідно до будь-якого з пунктів 1-6, у комбінації з фармацевтично прийнятним розріджувачем або носієм.
12. Фармацевтична композиція за п. 11, де дифумарат 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну являє собою Форму А дифумарату 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)-піперидин-4-іл]окси}хіназоліну.
13. Дифумарат 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)-піперидин-4-іл]окси}хіназоліну за будь-яким з пп. 1-6 для застосування як лікарського засобу.
14. Дифумарат 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)-піперидин-4-іл]окси}хіназоліну за будь-яким з пп. 1-6 для застосування для лікування злоякісного новоутворення.
Текст
Реферат: Описані дифумарат 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(Nметилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну, фармацевтична композиція, яка містить дифумарат, застосування дифумарату для лікування гіперпроліферативних розладів, таких як злоякісне новоутворення, і способи одержання дифумарату. UA 103998 C2 (12) UA 103998 C2 UA 103998 C2 5 10 15 20 25 30 35 Даний винахід стосується солі 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(Nметилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну, далі в даному винаході "Сполука (I)”, більш переважно, дифумаратної солі Сполуки (I). Вважають, що сіль є корисною для лікування або профілактики станів, опосередкованих тільки або частково шляхом передачі сигналів за допомогою erbВ рецептора, зокрема, проліферативних захворювань, таких як злоякісне новоутворення. Винахід також стосується фармацевтичної композиції, яка містить сіль, і її застосування для приготування лікарського засобу для застосування для лікування або профілактики злоякісного новоутворення, такого як рак молочної залози. Сімейство erbВ рецепторних тирозинкіназ, яке включає EGFR, erbВ2, erbВ3 і erbВ4, часто залучене в стимулювання проліферації й виживання пухлинних клітин, і, по суті, сімейство erbВ рецепторів залучене в різні епітеліальні злоякісні новоутворення (огляд в Olayioye та ін., EMBO J., 2000, 19, 3159), включаючи, наприклад, рак молочної залози (Sainsbury та ін., Brit. J. Cancer, 1988, 58, 458; Guerin та ін., Oncogene Res., 1988, 3, 21; Slamon та ін., Science, 1989, 244, 707; Klijn та ін., Рак молочної залози Res. Treat., 1994, 29, 73 і огляд в Salomon та ін., Crit. Rev. Oncol. Hematol., 1995, 19, 183), недрібноклітинний рак легенів (NSCLC), включаючи аденокарциноми (Cerny та ін., Brit. J. Cancer, 1986, 54, 265; Reubi та ін., Int. J. Cancer, 1990, 45, 269; Rusch та ін., Cancer Research, 1993, 53, 2379; Brabender та ін., Clin. Cancer Res., 2001, 7, 1850), а також інші злоякісні новоутворення легенів (Hendler та ін., Cancer Клітини, 1989, 7, 347; Ohsaki та ін., Oncol. Rep., 2000, 7, 603), рак сечового міхура (Neal та ін., Lancet, 1985, 366; Chow та ін., Clin. Cancer Res., 2001, 7, 1957, Zhau та ін., Mol Carcinog., 3, 254), рак стравоходу (Mukaida та ін., Cancer, 1991, 68, 142), рак шлунково-кишкового тракту, такий як рак ободової кишки, прямої кишки або шлунка (Bolen та ін., Oncogene Res., 1987, 1, 149; Kapitanovic та ін., Gastroenterology, 2000, 112, 1103; Ross та ін., Cancer Invest., 2001, 19, 554), рак передміхурової залози (Visakorpi та ін., Histochem. J., 1992, 24, 481; Kumar та ін., 2000, 32, 73; Scher та ін., J. Natl. Cancer Inst., 2000, 92, 1866), лейкоз (Konaka та ін., Cell, 1984, 37, 1035, Martin-subero та ін., Cancer Genet Cytogenet., 2001, 127, 174), рак яєчників (Hellstrom та ін., Cancer Res., 2001, 61, 2420), рак голови та шиї (Shiga та ін., Head Neck, 2000, 22, 599) або рак підшлункової залози (Ovotny та ін., Neoplasma, 2001, 48, 188). Таким чином, було встановлено, що інгібітор erbВ рецепторних тирозинкіназ повинен бути цінним як селективний інгібітор росту певних карцином. Різні інгібітори erbВ тирозинкіназ проявляють клінічну перевагу й різні інгібітори erbВ тирозкинкіназ були дозволені для застосування для лікування злоякісного новоутворення. Наприклад, інгібітори EGFR тирозинкіназ гефітиніб і ерлотиніб для лікування прогресуючого недрібноклітинного раку легенів, і лапатиніб, який має інгібуючу активність на erbВ2 тирозинкіназу, для застосування при метастатичному раку молочної залози. Деякі інші інгібітори EGFR і erbВ2 тирозинкіназ зараз знаходяться на стадії розробки. Сполука (I) описана в опублікованій міжнародній патентній заявці № WO2005/028469 як приклад 1 і має структуру: O N H N Cl HN O 40 45 50 O N F N Сполука (I) Сполука (I) являє собою інгібітор erbВ рецепторної тирозинкінази, зокрема, Сполука (I) є ефективним інгібітором EGFR і erbВ2 рецепторних тирозинкіназ. Отримано усе більше до- і клінічних підтверджень, які свідчать про те, що, додатково до передачі сигналів за допомогою гомодимерів EGFR і erbВ2, передача сигналів у клітинах, опосередковувана EGFR, erbВ2 і erbВ3 гетеродимерами, може бути важливим шляхом передачі онкогенних сигналів (Sergina та ін., Nature, 2007, 445, 437; Ritter та ін., Clin Cancer Res. 2007, 13, 4909; Johnston та ін., JCO, 2008, 26, 1066). Оскільки erbВ3 не має власної тирозинкіназної активності, то активація erbВ3 рецептора здійснюється тільки шляхом утворення гетеродимерних рецепторних комплексів з іншими рецепторами з кіназною активністю, 1 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 включаючи, зокрема EGFR і erbВ2. Вважають, що EGFR і erbВ2 гетеродимери, утворені з erbВ3, запускають ріст у пухлин у тих пухлинах, у яких екпресуються ці рецептори. Нами було виявлено в доклінічних дослідженнях, що Сполука (I) також інгібує опосередковану erbВ3 передачу сигналів за допомогою інгібування фосфорилування erbВ3 з наступною стимульованою лігандом EGFR/erbВ3 та/або erbВ2/erbВ3 гетеродимеризацією. Отже, Сполука (I) проявляє унікальний інгібуючий вплив на активність erbВ тирозинкінази в порівнянні з іншими інгібіторами erbВ тирозинкінази, такими як гефітиніб або ерлотиніб, які діють головним чином як інгібітори EGFR тирозинкінази. Нами були здійснені доклінічні дослідження, які підтвердили, що Сполука (I) проявляє поліпшені протипухлинні впливи в порівнянні з інгібіторами EGFR тирозинкінази, такими як гефітиніб і ерлотиніб. Не бажаючи обмежуватися будь-якою теорією, вважають, що поліпшені властивості можуть бути обумовлені інгібуванням Сполукою (I) опосередкованої erbВ3 передачі сигналів. В WO2005/028469 вказано, що сполуки, розкриті в даній заявці, можуть бути отримані у формі у фармацевтично прийнятної солі, наприклад, солі приєднання кислоти сполуки формули I, з неорганічною або органічною кислотою, такою як соляна, бромистоводнева, сірчана, трифтороцтова, лимонна або малеїнова кислота. У заявці WO2005/028469 відсутнє навіть припущення про сіль із фумаровою кислотою. Сполука (I) описана в Прикладі 1 заявки WO2005/028469 і виділена у вигляді вільної основи. У заявці WO2005/028469 відсутнє розкриття будь-якої специфічної солі Сполуки (I). Нами було виявлено, що Сполука (I) є кристалічною з деяким аморфним характером, як показано на порошковій рентгенівській дифракції XRPD на Фігурі 1. Диференціальна сканувальна калориметрія (Фігура 2A) на Сполуці (I) показує широку ендотерму з початком при 76,2 °C, яка, імовірно, обумовлена втратою розчинника, найбільш ймовірно води, з наступною ендотермою плавлення з початком при 126,2 °C. Термогравіметричний аналіз на Сполуці (I) (Фігура 2B) показує втрату ваги на 1,2 % між 25 °C і 95 °C. Сорбція динамічного випаровування (Фігура 3) показує вологопоглинання приблизно 1,9 мас. % при відносній вологості 80 %, отже, Сполука (I) є помірно гігроскопічною. Нами було виявлено, що Сполука (I) має відносно низьку власну швидкість розчинення, переважно при рН нижче 6,0 і має високу клітинну проникність. Низька розчинність і висока проникність надає можливість здійснити BCS класифікацію в Класі II для Сполуки (I). Отже, характеристики розчинення сполуки можуть бути вирішальними для контролювання абсорбції лікарського засобу й варіабельності між пацієнтами, особливо при високих дозах. Ці отримані дані разом з тими фактами, що Сполука (I) є частково аморфною і гігроскопічною, приводить до необхідності виявлення альтернативних форм Сполуки (I) з поліпшеними властивостями. Несподівано нами було виявлено, що дифумаратна сіль Сполуки (I) має сприятливі властивості в порівнянні зі Сполукою (I). Дифумарат Сполуки (I) має сприятливий профіль розчинення, проявляючи високу розчинність у воді й гарну власну швидкість розчинення. Крім того, дифумарат Сполуки (I) проявляє сприятливі властивості у твердому стані, наприклад, високу кристалічність, низьку гігроскопічність та/або сприятливі термічні властивості, такі як висока точка плавлення. Відповідно до першого аспекту даного винаходу, забезпечується дифумарат Сполуки (I). Підходяще дифумарат Сполуки (I) є кристалічним. Отже, відповідно до подальшого аспекту даного винаходу, забезпечується кристалічний дифумарат Сполуки (I). Дифумарат Сполуки (I) може існувати в сульфатованих, а також у несольватованих формах, таких як, наприклад, гідратовані форми. Мається на увазі, що винахід охоплює всі такі сольватовані й несольватовані форми дифумарату Сполуки (I). Нами було виявлено, що переважна кристалічна форма дифумарату Сполуки (I), далі в даному винаході "Форма A" характеризується тим, що вона забезпечує порошкову рентгенограму, по суті таку ж, як показано на Фігурі 4. Найбільш значимі піки Форми A представлено в таблиці 1. 2 UA 103998 C2 Таблиця 1 Найбільш значимі піки рентгенівської порошкової дифрактометрії для Форми A Кут 2-Тета° (2θ) 6,3 7,1 8,7 10,2 10,6 11,9 12,4 12,6 13,0 13,4 14,1 14,4 14,9 15,4 16,3 16,9 17,3 18,8 19,2 20,3 20,5 21,2 21,5 21,9 22,7 23,0 23,5 5 10 15 20 Підрахунок інтенсивності W S S M M S S M S M S M S M M M M S M VS VS S S VS M S S Кут 2-Тета° (2θ) 24,0 24,7 25,5 26,1 26,4 27,4 28,2 29,5 29,9 30,8 31,5 31,8 32,2 32,7 33,0 33,5 34,0 34,7 35,1 35,5 35,8 36,6 37,3 38,3 39,2 39,7 Підрахунок інтенсивності VS S S M VS S M S S M M M M M M M M M M M M M M M M M У Таблиці1 використовуються наступні скорочення: VS = дуже сильний; S = сильний; M = средний і W = слабкий. Відповідно до подальшого аспекту винаходу, забезпечується Форма A, де вказана Форма A має порошкову рентгенограму принаймні з одним специфічним піком приблизно при 2-тета = 26,4°. Відповідно до подальшого аспекту винаходу, забезпечується Форма A, де вказана Форма A Агент має порошкову рентгенограму принаймні з одним специфічним піком приблизно при 2тета = 26,4°, 14,9° або 7,1°. Відповідно до подальшого аспекту винаходу, забезпечується Форма A, де вказана Форма A має порошкову рентгенограму зі специфічними піками приблизно при 2-тета = 26,4°, 14,9° і 7,1°. Відповідно до подальшого аспекту винаходу, забезпечується Форма A, де вказана Форма A має порошкову рентгенограму принаймні з одним специфічним піком приблизно при 2-тета = 26,4°, 24,0°, 14,9°, 12,4° або 7,1°. Відповідно до подальшого аспекту винаходу, забезпечується Форма A, де вказана Форма A має порошкову рентгенограму зі специфічними піками приблизно при 2-тета =26,4°, 24,0°, 14,9°, 12,4° і 7,1°. Відповідно до подальшого аспекту винаходу, забезпечується Форма A, де вказана Форма A має порошкову рентгенограму принаймні з одним специфічним піком приблизно при 2-тета = 26,4°, 24,0°, 23,0°, 21,2°, 17,3°, 15,4°, 14,9°, 13,0°, 12,4° або 7,1°. Відповідно до подальшого аспекту винаходу, забезпечується Форма A, де вказана Форма A має порошкову рентгенограму зі специфічними піками приблизно при 2-тета = 26,4°, 24,0°, 23,0°, 21,2°, 17,3°, 15,4°, 14,9°, 13,0°, 12,4° і 7,1°. 3 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 Відповідно до іншого аспекту винаходу, забезпечується Форма A, де вказана Форма A має порошкову рентгенограму, по суті таку ж, що й порошкова рентгенограма, представлена на фігурі 4. Підходяще Форма A по суті не містить інших форм дифумарату Сполуки (I). Наприклад, принаймні 80 % дифумарату Сполуки (I) знаходиться у вигляді Форми A, переважно принаймні 90 %, більш переважно, принаймні 95 % і ще більш переважно принаймні 99 % дифумарату Сполуки (I) знаходиться у вигляді Форми A. У переважно варіанті здійснення принаймні 98 % дифумарату Сполуки (I) знаходиться у вигляді Форми A. При вказівці в даній заявці, наприклад, що 80 % дифумарату Сполуки (I) знаходиться у вигляді Форми A, стосується % за вагою дифумарату Сполуки (I). ДСК термограма для Форми A представлена на Фігурі 5 у даному винаході далі. Форма A, проявляє круту ендотерму плавлення з температурою початку розкладання приблизно 210,4 °C, що визначається шляхом аналізу диференціальної сканувальної калориметрії (ДСК), використовуючи прилад Mettler DSC820e, як описано в Прикладах. Таким чином, Форма A має точку плавлення приблизно 210 °C. Нами було виявлено, що дифумарат Сполуки (I) може існувати в інших кристалічних формах, наприклад, Формах B - Q, описаних у Прикладах у даній заявці. Відповідно до подальшого аспекту винаходу, забезпечується кристалічний дифумарат Сполуки (I), вибраний з будь-якої з Форми B - Форми Q, описаних у даній заявці. Підходяще, кожна з описаних форм кристалічного дифумарату Сполуки (I) по суті не містить інших форм дифумарату Сполуки (I). Наприклад, принаймні 80 % дифумарату Сполуки (I) знаходиться в бажаній формі, переважно принаймні 90 %, більш переважно, принаймні 95 % і ще більш переважно принаймні 99 % дифумарату Сполуки (I) знаходиться в бажаній кристалічній формі дифумарату. Кристалічні форми дифумарату Сполуки (I), описані в даній заявці, є кристалічними. Підходяще ступінь кристалічності, як визначається за допомогою даних рентгенівської порошкової дифрактометрії, становить, наприклад, більше, ніж приблизно 60 %, наприклад, більше, ніж приблизно 80 %, переважно більше, ніж приблизно 90 % і більш переважно більше, ніж приблизно 95 %. У варіантах здійснення винаходу, ступінь кристалічності, як визначається за допомогою даних рентгенівської порошкової дифрактометрії, становить більше, ніж приблизно 98 %, де % кристалічність стосується % за вагою маси сумарного зразка, який є кристалічним. У попередніх абзацах при визначенні піків порошкової рентгенограми для кристалічних форм Сполуки (I), термін "при значеннях приблизно", використовуваний у виразі “… при значеннях 2-тета приблизно =…” вказує на те, що точне положення піків (тобто наведені значення кутів 2-тета) не повинні тлумачитися як абсолютні значення, оскільки, що є очевидним для фахівця в даній галузі техніки, точне положення піків може незначно відрізнятися при вимірюванні на різних приладах, або різних зразків, або внаслідок незначних коливань використовуваних умов вимірювання. Це також вказувалося в попередніх параграфах, що Форма А дифумарату Сполуки (I) характеризується порошковими рентгенограмами "по суті" такими ж, що й порошкові рентгенограми, наведені на фігурі 4, і має головним чином найбільш значимі піки (значення кута 2-тета), наведені в таблиці 1. Слід прийняти до уваги, що використання терміну "по суті" у цьому контексті також призначене для вказівки того, що значення кутів 2-тета порошкових рентгенограм можуть незначно відрізнятися при вимірюванні на різних приладах, або різних зразків, або внаслідок незначних коливань використовуваних умов вимірювання, таким чином, піки, показані на Фігурах або наведені в Таблицях у даному винаході також на повинні тлумачитися як абсолютні значення. Щодо цього, у даній галузі техніки відомо, що може бути отримана порошкова рентгенограма, яка містить одну або декілька похибок вимірювань, які залежать від умов вимірювання (таких як використовуване устаткування або прилад). Зокрема, добре відомо, що напруга при порошковій рентгенограмі може відрізнятися залежно від умов вимірювання й приготування зразків. Наприклад, фахівці в галузі порошкової рентгенограми можуть підтвердити, що на відносну інтенсивність піків можуть впливати, наприклад, частинки розміром приблизно 30 мікрон і неодиничні формати зображення, які можуть впливати на аналіз зразків. Фахівець у даній галузі також буде враховувати той факт, що на положення відбиттів може впливати точна висота, на яку встановлюється зразок у дифрактометрі й калібрування нуля дифрактометра. Площинність поверхні зразка також може мати незначний вплив. Отже, фахівець у даній галузі техніки візьме до уваги, що дані порошкової рентгенограми, наведені в даному винаході, не повинні тлумачитися як абсолютні (для додаткової інформації див. Jenkins, R & Snyder, R.L. "Introduction to X-Ray Powder Diffractometry" John Wiley & Sons, 1996). Таким чином, слід прийняти до уваги, що кристалічні форми дифумарату Сполуки (I), описані в даному 4 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 винаході, не обмежуються кристалами, які охарактеризовані порошковою рентгенограмою, ідентичною до порошкової рентгенограми, наведеної на фігурі 1, і будь-які кристали, які характеризуються порошковою рентгенограмою, по суті такою ж, що й наведена на Фігурі 4, підпадають під обсяг даного винаходу. Фахівець в галузі порошкової рентгенограми здатний оцінити практичну ідентичність порошкових рентгенограм. У цілому, похибка вимірювання кута дифракції на порошковій рентгенограмі для кута 2-тета о становить приблизно 0,5 або менше, і такий порядок похибки вимірювання слід враховувати при аналізі порошкових рентгенограм на Фігурах 1 і 4 і при тлумаченні положень піків з посиланням на вищеописаний текст і на Таблицю 1. Точки плавлення й дані ДСК, представлені в даній заявці, визначали за допомогою приладу Mettler DSC820e, застосування якого більш докладно описане в даному винаході далі. Фахівець у даній галузі техніки візьме до уваги, що незначні коливання при вимірюванні точок плавлення за допомогою ДСК можуть відбуватися внаслідок відмінностей чистоти зразків, приготування зразків і умов вимірювань (наприклад, швидкості нагрівання). Слід прийняти до уваги, що альтернативні дані для точок плавлення можуть бути отримані при використанні устаткування інших типів або при застосуванні інших умов, що відрізняються від описаних у даному винаході далі. Таким чином, дані фігур точок плавлення й ендотерм, представлених у даній заявці, не повинні тлумачитися як абсолютні значення й такі похибки вимірювань не слід враховувати при інтерпретації даних ДСК. Звичайно, точки плавлення можуть відрізнятися на 0,5 °C або менше. Кристалічні форми дифумарату Сполуки (I), такі як Форма A відповідно до винаходу, також можуть бути охарактеризовані та/або їх можна відрізнити від інших фізичних форм, використовуючи інші підходящі аналітичні методики, наприклад, БІЧ спектроскопію або спектроскопію ядерного магнітного резонансу у твердому стані. Хімічна структура дифумарату Сполуки (I) згідно із даним винаходом може бути підтверджена за допомогою загальноприйнятих методів, наприклад, аналізу шляхом протонного ядерного магнітного резонансу (ЯМР). Синтез Сполуки (I) Сполука (I) може бути синтезована, використовуючи методи, описані в заявці WO2005/028469 або як ілюструється в прикладах, наведених у даній заявці. В WO2005/028469 описано в Прикладі 1 одержання Сполуки (I) у такий спосіб: 2-Хлор-N-метилацетамід (32 мг, 0,3 ммоль) додавали до суміші 4-(3-хлор-2-фтораніліно)-7метокси-6-[(піперидин-4-іл)окси]хіназоліну (120 мг, 0,3 ммоль), йодиду калію (16 мг, 0,1 ммоль), і карбонату калію (50 мг, 0,36 ммоль) в ацетонітрилі (5 мл). Суміш нагрівали в колбі зі зворотним холодильником протягом однієї години. Після упарювання розчинників у вакуумі, залишок ресуспендували в дихлорметані. Органічний розчин промивали водою й соляним розчином, висушували над сульфатом магнію. Після упарювання розчинників у вакуумі, залишок очищали шляхом хроматографії на силікагелі (елюент: 1 % - 2 % 7 н. метанольний аміак у дихлорметані), одержуючи Сполуку (I). Нами було виявлено, що при взаємодії 2-хлор-N-метилацетаміду безпосередньо з 4-(3хлор-2-фтораніліно)-7-метокси-6-[(піперидин-4-іл)окси]хіназоліном вдається уникнути застосування йодиду калію. Крім того, кристалізація Сполуки (I) з певних розчинників дозволяє одержати Сполуку (I) з високою чистотою. Таким чином, вважають, що новий спосіб придатний для одержання Сполуки (I) у промисловому масштабі. Отже, як подальший аспект даного винаходу, забезпечується спосіб одержання Сполуки (I), який включає: (i) взаємодію 2-хлор-N-метилацетаміду з 4-(3-хлор-2-фтораніліно)-7-метокси-6-[(піперидин4-іл)окси]хіназолін дигідрохлоридом у присутності підходящої основи; (ii) додавання розчинника, вибраного з етанолу, води й метанолу, або їх суміші, до реакційної суміші зі стадії (i) для здійснення кристалізації Сполуки (I); і (iii) виділення Сполуки (I). Взаємодію на Стадії (i) підходяще здійснюють у підходящому інертному розчиннику, такому як розчинники, описані в Процесі (c) на сторінці 30 заявки WO2005/028469. Наприклад, взаємодія може бути здійснена, використовуючи ацетонітрил як розчинник. Реакцію здійснюють у присутності підходящої основи, наприклад, однієї з основ, описаних у Процесі (c) на сторінці 30 заявки WO2005/028469, такої як триетиламін. Підходяще реакцію здійснюють при підвищеній температурі, наприклад, приблизно при 75 °C. В одному варіанті здійснення на Стадії (ii) способу розчинником є вода. Підходяще у цьому варіанті здійснення, якщо Стадію (i) здійснюють в ацетонітрилі, то об'ємне співвідношення вода:ацетонітрил становить приблизно 1:3. 5 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 В іншому варіанті здійснення винаходу на Стадії (ii) способу розчинником є етанол. Підходяще у цьому варіанті здійснення, якщо Стадію (i) здійснюють в ацетонітрилі, об'ємне співвідношення етанол:ацетонітрил становить приблизно 3,5:7. В подальшому варіанті здійснення винаходу на Стадії (ii) способу розчинником є суміш етанолу й води. Підходяще у цьому варіанті здійснення об'ємне співвідношення етанолу до води становить від приблизно 20:1 до приблизно 30:1, наприклад, приблизно 21,9:1-25:1. Якщо Стадію (i) здійснюють в ацетонітрилі, то підходяще приблизно 3,5 об'ємів етанолу й 0,15 об'ємів води додають до 7 об'ємів ацетонітрилу для здійснення кристалізації. Також слід прийняти до уваги, що при вказівці об'ємного співвідношення, наприклад, що співвідношення вода:ацетонітрил становить 1:3, означає, що 1 води додають до 3 об'ємів ацетонітрилу, присутнього у реакційній посудині, з наступним завершенням Стадії (i) способу. В одному варіанті здійснення, на Стадії (ii) способу реакційну суміш зі Стадії (i) охолоджували до приблизно 70 °C і додавали етанол. Після цього реакційну суміш охолоджували до приблизно 45 °C і додавали воду для здійснення кристалізації Сполуки (I). При необхідності, реакційну суміш можна затравлювати за допомогою Сполуки (I) для сприяння почали кристалізації. Після цього реакційну суміш охолоджували до приблизно 20 °C для завершення кристалізації. Виділення Сполуки (I) на Стадії (iii) можна здійснювати, використовуючи загальноприйняті методи, наприклад, фільтрацію й висушування Сполуки (I). 4-(3-хлор-2-фтораніліно)-7-метокси-6-[(піперидин-4-іл)окси]хіназолін дигідрохлорид, який використовували як вихідну речовину, може бути отриманий, як описано в Прикладах у даній заявці. Наприклад, шляхом додавання соляної кислоти до 6-{[(1-третбутоксикарбоніл)піперидин-4-іл]окси}-4-(3-хлор-2-фтораніліно)-7-метоксихіназоліну. Підходяще реакцію здійснюють у підходящому розчиннику, наприклад, етанолі, ацетонітрилі, тетрагідрофурані, 2-метилтетрагідрофурані, н-пропанолі, метанолі, 1-бутанолі, етилацетаті, трет-бутил ацетаті, ізопропанолі або промисловому денатурованому метиловому спирті. Переважним розчинником є етанол або більш переважно промисловий денатурований метиловий спирт. 6-{[(1-трет-бутоксикарбоніл)піперидин-4-іл]окси}-4-(3-хлор-2-фтораніліно)-7-метоксихіназолін може бути отриманий, використовуючи спосіб, описаний у Прикладі 1 заявки WO2005/028469, шляхом взаємодії 4-(3-Хлор-2-фтораніліно)-6-гідрокси-7-метоксихіназоліну із трет-Бутил (4метансульфонілокси)піперидин-1-карбоксилатом, де реакцію здійснюють у присутності фториду цезію, використовуючи DMA як розчинник й при температурі 85 °C. Цю реакцію також можна здійснювати в присутності N-метилпіролідону (NMP) у присутності підходящої основи, такої як карбонат калію. Цю реакцію підходяще здійснюють при підвищеній температурі, як описано в Прикладах у даній заявці. Проте, нами було виявлено, що здійснення реакції в присутності певних розчинників одержують продукт у хорошій формі. Крім того, вважають, що деякі із цих розчинників придатні для одержання продукту в промисловому масштабі. Отже, як подальший аспект даного винаходу, забезпечується спосіб одержання 6-{[(1-третбутоксикарбоніл)піперидин-4-іл]окси}-4-(3-хлор-2-фтораніліно)-7-метоксихіназоліну, який включає: (i) взаємодію 4-(3-хлор-2-фтораніліно)-6-гідрокси-7-метоксихіназоліну із трет-бутил (4метансульфонілокси)піперидин-1-карбоксилатом у присутності підходящої основи, де реакцію здійснюють у розчиннику, вибраному з N-метилпіролідону або спирту, вибраного з метанолу, етанолу, ізопропілового спирту, н-пропанолу й промислового денатурованого метилового спирту; і (ii) кристалізацію 6-{[(1-трет-бутоксикарбоніл)піперидин-4-іл]окси}-4-(3-хлор-2-фтораніліно)7-метоксихіназоліну. Стадію (i) цього способу здійснюють у присутності підходящої основи, наприклад, основ, описаних у заявці WO2005/028469, таких як карбонат калію. Реакцію підходяще здійснюють при підвищеній температурі, підходяще при температурі флегми. При необхідності, до розчинників, використовуваних на Стадії (i), можна додавати воду, для полегшення обробки, наприклад, для підвищення рухомості реакційної суміші. В одному варіанті здійснення, Стадію (i) реакції здійснюють у спирті, вибраному з метанолу, етанолу, ізопропілового спирту, н-пропанолу й промислового денатурованого метилового спирту, необов'язково в присутності води. В подальшому варіанті здійснення, реакцію Стадії (i) здійснюють у суміші етанолу й води. Якщо використовують суміш етанолу й води, то об'ємне співвідношення етанолу до води на Стадії (i) не є вирішальним, наприклад, об'ємне співвідношення етанолу до води може аж до приблизно 10:2 є підходящим, таке як приблизно 6 UA 103998 C2 5 10 15 10:1. Кристалізацію на Стадії (ii) способу підходяще здійснюють шляхом охолодження реакційної суміші зі Стадії (i) (наприклад, охолодження до приблизно 70 °C) і додавання води до суміші для здійснення кристалізації. Після цього продукт за допомогою загальноприйнятих методів, таких як методи, описані в Прикладах. Сполука (I) також може бути отримана відповідно до способу, проілюстрованого на схемі реакцій 1: Схема реакцій 1 Примітки для схеми реакцій 1: 1 Стадія (i): Lg являє собою підходящу групу, яка вилучається, наприклад, галоген, алкансульфонілокси або арилсульфонілокси групу, наприклад, хлор, бром, метансульфонілокси, 4-нітробензолсульфонілокси або толуол-4-сульфонілокси групу (підходяще метансульфонілокси, 4-нітробензолсульфонілокси або толуол-4-сульфонілокси 1 групу, наприклад, Lg являє собою метансульфонілокси). 1 Pg являє собою підходящу амінозахисну групу. Такі групи добре відомі, наприклад, як описано в одному з багатьох загальних посібників з даної тематики, такому як "Protective Groups 7 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 in Organic Synthesis" за редакцією Theodora Green (видавництво: John Wiley & Sons). Прикладами амінозахисних груп є ацильна група, наприклад, алканоїльна група, така як ацетил, алкоксикарбонільна група, наприклад, метоксикарбонільна, етоксикарбонільна або третбутоксикарбонільна група, арилметоксикарбонільна група, наприклад, бензилоксикарбоніл, або 1 ароїльна група, наприклад, бензоїл. Конкретним прикладом Pg є трет-бутоксикарбоніл. Реакцію підходяще здійснюють у присутності основи, наприклад, карбонату, такого як карбонат калію. Реакцію підходяще здійснюють у присутності підходящого інертного розчинника, наприклад, спирту, такого як ізопропанол. Реакцію підходяще здійснюють при підвищеній температурі, зручно при температурі флегми розчинника. 1 Стадія (ii): Захисну групу Pg видаляють, використовуючи загальноприйняті методи. 1 Наприклад, якщо Pg являє собою трет-бутоксикарбоніл, то він може бути вилучений шляхом обробки з підходящою кислотою, такою як соляна, сірчана або фосфорна кислота або трифтороцтова кислота. 2 Стадія (iii): Lg являє собою підходящу групу, яка вилучається, наприклад, галоген, такий як хлор. Реакцію підходяще здійснюють у присутності підходящої основи, такої як карбонат, органічний амін або алкоголят. Підходящі основи включають карбонат калію або триетаноламін. Реакцію підходяще здійснюють у присутності інертного розчинника, такого як ацетонітрил або спирт, такий як етанол. Реакцію підходяще здійснюють при підвищеній температурі, зручно при температурі флегми розчинника. Стадія (iv): Нітрування може бути здійснене за допомогою добре відомих методів для нітрування ароматичних кілець, наприклад, шляхом обробки 2-[4-(5-Ціано-2метоксифенокси)піперидин-1-іл]-N-метилацетаміду (4) з азотною кислотою в присутності сірчаної кислоти, використовуючи добре відомі методи для таких реакцій і як ілюструється в Прикладах. Стадія (v): Реакції відновлення, які підходять для відновлення нітрогруп до амінів, добре відомі, наприклад, шляхом відновлення в присутності підходящого відновника, такого як дитіоніт натрію. Цю реакцію підходяще здійснюють у присутності водного розчинника, наприклад, водного метанолу. Реакцію зручно здійснюють при підвищеній температурі наприклад, 40-60 °C. Альтернативно, відновлення можна здійснювати шляхом гідрування, наприклад, шляхом каталітичного гідрування за допомогою підходящого каталізатора, такого як паладій на каталізаторі вугіллі, наприклад, 10 % паладій на каталізаторі вугіллі. Гідрування зручне здійснюють у підходящому розчиннику, такому як метанол. Стадія (vi): 2-[4-(4-Аміно-5-ціано-2-метоксифенокси)піперидин-1-іл]-N-метилацетамід (6) піддають реакції з N, N-диметилформамід диметил ацеталем. Реакцію підходяще здійснюють у присутності підходящого розчинника, такого як простий ефір, наприклад, 2метилтетрагідрофуран, або ароматичного вуглеводню, такого як толуол. Реакцію підходяще здійснюють при підвищеній температурі, наприклад, приблизно при від 70 до 105 °C, підходяще приблизно при 76 °C. Стадія (vii) Реакцію підходяще здійснюють у присутності підходящої кислоти, такої як одна або декілька кислот, вибраних з оцтової, пропанової, янтарної, фумарової і лимонної кислоти. В одному прикладі кислота являє собою оцтову кислоту. Реакцію підходяще здійснюють у присутності інертного розчинника, наприклад, ароматичного вуглеводневого розчинника, такого як метоксибензол. Реакцію підходяще здійснюють при підвищеній температурі, наприклад, від приблизно 90 до приблизно 120 °C, підходяще приблизно при 90 °C. Спосіб, описаний на схемі реакцій 1, становить подальший аспект даного винаходу. Таким чином, забезпечується спосіб одержання Сполуки (I), який включає взаємодію 2-[4-(5-ціано-4{[(диметиламіно)метилен]аміно}-2-метоксифенокси)піперидин-1-іл]-N-метилацетаміду (7) з 3хлор-2-фтораніліном у присутності підходящої кислоти. Підходящими умовами реакції є умови, описані в даній заявці вище для Стадії (vii) схеми реакцій 1. Певні проміжні сполуки, представлені на схемі реакцій 1, є новими й становлять подальший аспект даного винаходу. Отже, інший аспект винаходу забезпечує сполуку, вибране з будь-якої 1 зі сполук 2, 3, 4, 5, 6 і 7 на схемі реакцій 1, або її сіль; де Pg має значення, вказані в даній заявці вище (наприклад, трет-бутоксикарбоніл). Деякі із проміжних сполук, такі як сполука (7) на схемі реакцій 1, можуть мати геометричні ізомерні центри (E- і Z- ізомери). Мається на увазі, що даний винахід охоплює всі такі геометричні ізомери і їх суміші. Синтез Форми A Дифумарату Сполуки (I) Відповідно до подальшого аспекту даного винаходу, забезпечується спосіб одержання дифумарату Сполуки (I) (Форма A), який включає: 8 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 (i) взаємодію Сполуки (I) з достатньою кількістю фумарової кислоти з утворенням дифумаратної солі; (ii) кристалізацію Форми A; і (iii) виділення Форми A. Примітки для Стадії (i) Зручно реакцію з фумаровою кислотою здійснюють у підходящому розчиннику, наприклад, вибраному з метанолу, етанолу, 1-бутанолу, 2-бутанолу й діацетонового спирту. Реакцію також можна здійснювати в суміші підходящих розчинників, наприклад, суміші, вибраній з метилетилкетону й диметилформаміду; метилетилкетону й тетрагідрофурану; метилетилкетону й метанолу; метилетилкетону й ізопропанолу; етанолу й диметилсульфоксиду; етанолу й тетрагідрофурану; етанолу й ізопропанолу; 1-бутанолу й диметилформаміду; 1-бутанолу й диметилсульфоксиду; 1-бутанолу й тетрагідрофурану; 1-бутанолу й метанолу; 1-бутанолу й ізопропанолу; етилацетату й диметилформаміду; етилацетату й метанолу; етилацетату й ізопропанолу; і метанолу й ізопропанолу. В одному варіанті здійснення, реакцію на Стадії (i) здійснюють у воді. В одному варіанті здійснення, реакцію на Стадії (i) здійснюють у суміші розчинників, що містить метилетилкетон і розчинник, вибраний з диметилформаміду, тетрагідрофурану, метанолу й ізопропанолу. В іншому варіанті здійснення, реакцію на Стадії (i) здійснюють у суміші розчинників, що містить етанол і розчинник, вибраний з диметилсульфоксиду, тетрагідрофурану й ізопропанолу. В іншому варіанті здійснення, реакцію на Стадії (i) здійснюють у суміші розчинників, що містить 1-бутанол і розчинник, вибраний з диметилформаміду, диметилсульфоксиду, тетрагідрофурану, метанолу й ізопропанолу. В іншому варіанті здійснення, реакцію на Стадії (i) здійснюють у суміші розчинників, що містить етилацетат і розчинник, вибраний з диметилформаміду, метанолу й ізопропанолу. В іншому варіанті здійснення, реакцію на Стадії (i) здійснюють у суміші розчинників, що містить етилацетат і ізопропанол. В іншому варіанті здійснення, реакцію на Стадії (i) здійснюють у суміші розчинників, що містить метанол і ізопропанол. У цих варіантах здійснення, якщо Стадію (i) реакції здійснюють у суміші розчинників, що містить метанол і ізопропанол, то об'ємне співвідношення ізопропанолу до метанолу підходяще знаходиться в діапазоні від приблизно 3,4:1 до приблизно 1,0:1, наприклад, від приблизно 1,5:1 до приблизно 1,0:1. Цю реакцію підходяще здійснюють при підвищеній температурі, наприклад, при температурі більше 60 °C, підходяще від 65 °C до температури флегми розчинника. Зручно Сполуку (I) розчиняють або диспергують в ізопропанолі й цю суміш піддають реакції з розчином або дисперсією фумарової кислоти в метанолі. У цих варіантах здійснення, якщо Стадію (i) реакції здійснюють у суміші розчинників, що містить етилацетат і ізопропанол, то об'ємне співвідношення етилацетату до ізопропанолу підходяще знаходиться в діапазоні від приблизно 5,1:1 до 1,9:1, наприклад, від приблизно 3,9:1 до 1,9:1, таке як приблизно 2,1:1. Цю реакцію підходяще здійснюють при температурі приблизно 20 до приблизно 73 °C, наприклад, приблизно при 40 °C. Зручно Сполуку (I) розчиняють або диспергують в етилацетаті й цю суміш піддають реакції з розчином або дисперсією фумарової кислоти в ізопропанолі. Альтернативно, Сполука (I) може бути попередньо розчинена в суміші етилацетату й ізопропанолу. При необхідності, ізопропанол може бути вилучений з наступним розчиненням Сполуки (I), використовуючи загальноприйняті методи, такі як дистиляція. Якщо готують розчин або дисперсію фумарової кислоти в спирті, такому як метанол або ізопропіловий спирт, то може бути необхідним нагрівати суміш для здійснення розчинення фумарової кислоти. Проте, слід уникати надлишкового нагрівання та/або витримування суміші при підвищеній температурі протягом тривалого часу для уникнення утворення складного ефіру. Як правило, на Стадії (i) способу Сполуку (I) піддають реакції принаймні з 2 молярними еквівалентами фумарової кислоти, наприклад, від приблизно 2 до приблизно 3, переважно від приблизно 2 до приблизно 2,7 молярними еквівалентами фумарової кислоти. Проте, у певних системах розчинників можна використовувати більш низькі кількості фумарової кислоти. Наприклад, якщо реакцію здійснюють у суміші етилацетату й ізопропанолу, то нами було виявлено, що дифумарат Сполуки (I) може бути отриманий, якщо молярне співвідношення фумарової кислоти до Сполуки (I) більше або дорівнює 1,725. Примітки для Стадії (ii) Кристалізація Форми A може бути здійснена за допомогою методів, відомих для кристалізації сполуки з розчину. Наприклад, шляхом викликання пересичення розчину, що 9 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 містить сіль. Пересичення можна досягати, наприклад, шляхом охолодження розчину, упарювання розчинника з розчину або шляхом додавання до розчину підходящого антирозчинника. В одному варіанті здійснення кристалізацію здійснюють шляхом охолодження розчину. Наприклад, якщо Стадію (i) реакції здійснюють у суміші метанолу й ізопропілового спирту, то реакційну суміш охолоджували до приблизно 30 °C протягом приблизно 90 хвилин і витримували при 30 °C приблизно протягом 30 хвилин. Потім реакційну суміш можна додатково охолоджувати протягом приблизно 2 годин до температури приблизно 0 °C і витримувати при цій температурі протягом періоду часу, достатнього для завершення кристалізації, наприклад, близько 1 години. Альтернативно, якщо Стадію (i) реакції здійснюють у суміші етилацетату й ізопропілового спирту, реакційну суміш охолоджували до приблизно 20 °C (наприклад, від приблизно 40 °C до приблизно 20 °C протягом приблизно 1 години). Потім реакційну суміш витримують при 20 °C протягом часу, достатнього для здійснення кристалізації. Підходяще, реакційну суміш витримують приблизно при 20 °C протягом принаймні 10 годин, наприклад, приблизно протягом 13,5 годин. В іншому варіанті здійснення, якщо Стадію (i) реакції здійснюють у суміші метанолу й ізопропілового спирту, то кристалізацію можна здійснювати шляхом видалення частини розчинника для викликання пересичення реакційної суміші, що залишився. Розчинник може бути вилучений шляхом упарювання або дистиляції. Підходяще, видаляють від приблизно 55 до 65 % за вагою розчинника, наприклад, близько 62 %. При необхідності, до суміші можна додавати додаткову кількість ізопропанолу з наступною дистиляцією приблизно такої ж ваги розчинника. Наприклад, до суміші можна додавати приблизно від 50 до 60 % додаткового ізопропанолу, де % означає % за вагою розчинника, що залишився в реакційній посудині після першої дистиляції. Після додавання ізопропанолу, аналогічну вагу розчинника видаляють шляхом дистиляції. Кристалізація може бути завершена шляхом додавання додаткової кількості ізопропанолу й охолодження суміші до приблизно 0 °C протягом приблизно 8 годин. Як правило, Форма A буде самостійно кристалізуватися на Стадії (ii) способу, але, що є очевидним для фахівця в даній галузі техніки, затравлювання за допомогою Форми A можна використовувати для сприяння кристалізації. При необхідності, затравочні кристали можуть бути приготовлені за допомогою методу, описаного вище й проілюстрованого в Прикладах для одержання дифумарату Сполуки (I) Форми A. Примітки для Стадії (iii) Будь-який підходящий метод, відомий у даній галузі техніки для виділення кристалічних матеріалів з розчину, можна використовувати на Стадії (iii) способу. Підходяще, Форму A збирають шляхом фільтрації. Після виділення Форми A, сіль можна промивати підходящим розчинником, наприклад, холодним ізопропанолом. Після виділення, Форма A може бути висушена, використовуючи загальноприйняті методи, наприклад, вакуумне сушіння. Таким чином, в одному варіанті здійснення винаходу, забезпечується спосіб одержання дифумарату Сполуки (I) (Форма A), який включає: (i) взаємодію розчину або суспензії Сполуки (I) в ізопропанолі із принаймні 2 молярними еквівалентами фумарової кислоти в метанолі, де об'ємне співвідношення ізопропанолу до метанолу становить від 3,4:1 до приблизно 1,0:1, наприклад, від приблизно 1,5:1 до приблизно 1,0:1, і де реакцію здійснюють при температурі принаймні 60 °C; (ii) кристалізацію Форми A; і (iii) виділення Форми A. Підходящими умовами для кристалізації й виділення Форми A є умови, описані в даній заявці вище. Таким чином, в іншому варіанті здійснення винаходу забезпечується спосіб одержання дифумарату Сполуки (I) (Форма A), який включає: (i) взаємодію розчину або суспензії Сполуки (I) в етилацетаті принаймні з 1,725 молярним еквівалентом фумарової кислоти в ізопропанолі (підходяще принаймні 2 молярними еквівалентами фумарової кислоти), де об'ємне співвідношення етилацетату до ізопропанолу підходяще становить від приблизно 5:1 до 1:1, наприклад, від приблизно 5,1:1 до 1,9:1, таке як приблизно 2,1:1 і де реакцію здійснюють при температурі від приблизно 20 до приблизно 73 °C (наприклад, приблизно 40 °C); (ii) охолодження реакційної суміші зі Стадії (i) до приблизно 20 °C і витримування суміші при цій температурі для здійснення кристалізації Форми A; і 10 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 (iii) виділення Форми A дифумарату Сполуки (I). Підходящими умовами для виділення Форми A є умови, описані в даній заявці вище. В іншому варіанті здійснення винаходу забезпечується спосіб одержання дифумарату Сполуки (I) (Форма A), який включає: (i) взаємодію Сполуки (I) у воді із принаймні 2 молярними еквівалентами фумарової кислоти (наприклад, принаймні 2,05, наприклад, приблизно 2,1 молярними еквівалентами фумарової кислоти), і де реакцію здійснюють приблизно при 85 °C; (ii) охолодження реакційної суміші зі Стадії (i) до приблизно 60 °C; і (iii) виділення Форма A дифумарату Сполуки (I). Підходяще на Стадії (ii) реакційну суміш повільно охолоджували до приблизно 60 °C, наприклад, при швидкості охолодження приблизно 1 °C/хвилину. При необхідності, кристалізацію Форми A можна індукувати шляхом додавання затравочних кристалів Форми A при охолодженні суміші. Підходяще затравочні кристали Форми A додають тоді, коли реакційну суміш охолоджують до приблизно 77 °C. Підходящими умовами для виділення Форми A на Стадії (iii) є умови, описані в даній заявці вище. Форми B-P кристалічного дифумарату Сполуки (I) можуть бути отримані, наприклад, за допомогою методів, описаних у Прикладах у даному описі. Фармацевтичні композиції Відповідно до подальшого аспекту винаходу, забезпечується фармацевтична композиція, яка містить дифумарат Сполуки (I) у комбінації з фармацевтично прийнятним розріджувачем або носієм. Дифумарат Сполуки (I) може використовуватися в композиції в будь-якій з форм, описаних у даній заявці, наприклад, Формі A. Композиції за винаходом можуть знаходитися у формах, які підходять для перорального застосування (наприклад, у вигляді таблеток, коржів, твердих або м'яких капсул, водних або масляних суспензій, емульсій, диспергованих порошків або гранул, сиропів або еліксирів), для місцевого введення (наприклад, у вигляді паст, мазей, гелів, водних або масляних розчинів або суспензій), для введення шляхом інгаляції (наприклад, у вигляді тонкоподрібненого порошку або рідкого аерозолю), для введення шляхом вдування (наприклад, у вигляді тонкоподрібненого порошку), або для парентерального введення (наприклад, у вигляді стерильного водного або масляного розчину для внутрішньовенного, підшкірного, внутрішньом'язового введення, або у вигляді супозиторію для ректального введення). Композиції за винаходом можуть бути отримані звичайними способами при використанні звичайних фармацевтичних наповнювачів, добре відомих у даній галузі. Таким чином, композиції, призначені для перорального введення, можуть містити, наприклад, один або декілька барвників, підсолоджувачів, ароматизаторів та/або консервантів. Наприклад, дифумарат Сполуки (I) підходяще готують у вигляді лікарського засобу у вигляді таблетки, використовуючи наступні наповнювачі: Ядро таблетки: дифумарат Сполуки (I) (наприклад, Форма A); лактоза; мікрокристалічна целюлоза; кросповідон; полівідон (PVP); і стеарат магнію Ядро таблетки може бути покрите за допомогою плівкової оболонки, такої як плівкова оболонка на основі HPMC, така оболонка необов'язково містить один або декілька барвників та/або світлозахисних засобів. Таблетки можуть бути приготовлені, використовуючи загальноприйняті методи і як ілюструється в Прикладах. При необхідності дифумарат Сполуки (I) може бути подрібнений перед приготування лікарського препарату у вигляді таблетки для забезпечення однорідного розподілу частинок дифумарату Сполуки (I) за розмірами у таблетці. Наприклад, дифумарат Сполуки (I) може бути подрібнений для забезпечення середнього розміру частинок приблизно 5 мкм. Підходящі способи подрібнення добре відомі з рівня техніки. Кількість активного компонента, яка необхідна для одержання одиничної лікарської форми в комбінації з одним або декількома наповнювачами, головним чином залежить від організму, який піддається лікування, і конкретного шляху введення. Наприклад, лікарська форма, призначена для перорального введення людині, як правило, містить, наприклад, від 0,5 мг до 0,5 г активної речовини (більш переважно від 0,5 до 200 мг, наприклад, від 1 до 30 мг) у комбінації з підходящою й прийнятною кількістю наповнювачів, яка може змінюватися від приблизно 5 до приблизно 98 % від загальної маси композиції. 11 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 Доза дифумарату Сполуки (I) для лікування або профілактики звичайно змінюється залежно від природи й тяжкості станів, віку й статі тварини або людини, і шляхи введення, і визначається відповідно до добре відомих підходів у медицині. Дифумарат Сполуки (I), який застосовується для лікування або профілактики, звичайно вводиться в добовій дозі в інтервалі, наприклад, від 0,1 мг/кг до 75 мг/кг ваги тіла пацієнта, і, при необхідності, може бути розділений на декілька прийомів. У цілому, при парентеральному введенні застосовуються більш низькі дози. Так, наприклад, для внутрішньовенного введення звичайно застосовують дозу в інтервалі, наприклад, від 0,1 мг/кг до 30 мг/кг ваги тіла. Подібним чином, для введення шляхом інгаляції застосовується доза в інтервалі, наприклад, від 0,05 мг/кг до 25 мг/кг ваги. Переважним, однак, є пероральне введення, як правило, у формі таблетки. Наприклад, дифумарат Сполуки (I) може вводитися теплокровній тварині перорально, у стандартній дозі менше, ніж 1 г на добу, але більше, ніж 1 мг. Переважно дифумарат Сполуки (I) може вводитися теплокровній тварині, у стандартній дозі менше, ніж 250 мг на добу. В іншому аспекті винаходу, дифумарат Сполуки (I) може вводитися теплокровній тварині, у стандартній дозі менше, ніж 160 мг на добу. В подальшому аспекті винаходу, дифумарат Сполуки (I) може вводитися теплокровній тварині, у стандартній дозі менше, ніж 50 мг на добу. Доза дифумарату Сполуки (I) може вводитися у вигляді однократної добової дози або у вигляді множинних фракцій загальної добової дози. Наприклад, загальна добова доза дифумарату Сполуки (I) може вводитися у вигляді двох доз, які можуть бути однаковими або різними. Проте, підходяще, кожна фракція загальної добової дози буде приблизно рівною. Як приклад, дифумарат Сполуки (I) може вводитися у вигляді однієї або декількох пероральних дозованих форм, таких як таблетка або капсула, що містять 1,5, 3,7, 14,9, 59,6 або 149 мг дифумарату Сполуки (I) (еквівалентно 1, 2,5, 10, 40 або 100 мг вільної форми Сполуки (I)). В подальшому варіанті здійснення дозу дифумарату Сполуки (I), еквівалентну 40, 80, 100, 160, 200 або 240 мг Сполуки (I), вводять два рази на добу. У переважно варіанті здійснення дозу дифумарату Сполуки (I), еквівалентну до 160 мг Сполуки (I), вводять два рази в на добу. У переважному варіанті здійснення дозу дифумарату Сполуки (I), еквівалентну до 200 мг Сполуки (I), вводять два рази в на добу. В іншому переважному варіанті здійснення дозу дифумарату Сполуки (I), еквівалентну по 240 мг Сполуки (I), вводять два рази в на добу. Біологічні дослідження Інгібуючі активності Сполуки (I) і дифумарату Сполуки (I) можуть бути виміряні в дослідженнях, описаних у заявці WO2005/028469 або як описано в Прикладах у даній заявці. Сполуки згідно із даним винаходом мають антипроліферативні властивості, такі як протиракові властивості, які, як вважають, є наслідком їх інгібуючої дії по відношенню до рецепторних тирозинкіназ сімейства erbВ, і переважно змішаним профілем erbВ2/ EGF та/або erbВ3/EGF. Таким чином, сполуки згідно із даним винаходом, як очікується, є придатними для лікування захворювань або хворобливих станів, опосередковуваних тільки erbВ рецепторними тирозинкіназами або частково опосередкованих цими тирозинкіназами, тобто сполуки можуть застосовуватися для одержання інгібуючої дії по відношенню до erbВ рецепторної тирозинкінази в теплокровної тварини, яка потребує такого лікування. Таким чином, сполуки згідно із даним винаходом забезпечують спосіб лікування злоякісних клітин, який характеризується інгібуванням однієї або декількох рецепторних тирозинкіназ сімейства erbВ. Переважно сполуки за винаходом можуть застосовуватися для одержання антипроліферативної та/або проапоптотичної та/або антиінвазивної дії, опосередкованої тільки інгібуванням erbВ рецепторної тирозинкінази, або частково опосередкованого інгібуванням цього ферменту. Більш переважно, сполуки згідно із даним винаходом, як очікується, є придатними для профілактики або лікування тих пухлин, які чутливі до інгібування однієї або декількох erbВ рецепторних тирозинкіназ, які залучені в стадії передачі сигналів, що регулюють проліферацію й виживання цих пухлинних клітин. Таким чином, сполуки згідно із даним винаходом, як очікується, є придатними для лікування псоріазу, доброякісної гіперплазії передміхурової залози (BPH), атеросклерозу й рестенозу, та/або злоякісного новоутворення шляхом забезпечення антипроліферативної дії, особливо для лікування злоякісних новоутворень, чутливих до erbВ рецепторної тирозинкінази. Такі доброякісні або злоякісні пухлини можуть виникати в будь-яких тканинах і включають несόлідні пухлини, такі як лейкоз, множинну мієлому або лімфому, а також сόлідні пухлини, наприклад, рак жовчних проток, кісток, сечового міхура, головного мозку/ЦНС, молочної залози, прямої кишки, ендометрію, шлунка, голови та шиї, печінки, легенів, нервових клітин, стравоходу, яєчників, підшлункової залози, передміхурової залози, нирок, шкіри, яєчок, щитовидної залози, матки й зовнішніх жіночих статевих органів. 12 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 Якщо наведене посилання на злоякісне новоутворення, то воно переважно стосується раку стравоходу, мієломи, печінково-клітинного раку, раку підшлункової залози, раку шийки матки, пухлини Юінга, нейробластоми, саркоми Капоши, раку яєчників, раку молочної залози, раку ободової і прямої кишки, раку передміхурової залози, раку сечового міхура, меланоми, раку легені - недрібноклітинного раку легені (NSCLC), і дрібноклітинного раку легені (SCLC), раку шлунка, раку голови та шиї, раку головного мозку, раку нирки, лімфоми й лейкозу. В одному варіанті здійснення воно стосується раку молочної залози, наприклад, позитивного за рецептором гормону раку молочної залози. В іншому варіанті здійснення посилання на злоякісне новоутворення стосується SCLC, NSCLC, раку ободової і прямої кишки, раку яєчників і / або раку молочної залози. В іншому варіанті здійснення посилання на злоякісне новоутворення стосується SCLC. В іншому варіанті здійснення посилання на злоякісне новоутворення стосується раку шлунка. Додатково, посилання стосується NSCLC. Додатково, посилання стосується раку ободової і прямої кишки. Додатково, посилання стосується раку яєчників. Додатково, більш переважно посилання стосується раку молочної залози. Додатково, більш переважно посилання стосується позитивного за рецептором гормону раку молочної залози, особливо, позитивного за рецептором гормону раку молочної залози в жінок у постклімактеричний період. В одному варіанті здійснення посилання стосується ранньої стадії не-метастатичного позитивного за рецептором гормону раку молочної залози, наприклад, ранньої стадії не-метастатичного позитивного за рецептором гормону раку молочної залози в жінок у постклімактеричний період. Крім того, посилання стосується ранньої стадії неметастатичного позитивного за рецептором естрогену та/або прогестерону раку молочної залози, особливо, ранньої стадії не-метастатичного позитивного за рецептором естрогену та/або прогестерону раку молочної залози в жінок у постклімактеричний період. Додатково, більш переважно посилання стосується метастатичного позитивного за рецептором гормону раку молочної залози, особливо, метастатичного позитивного за рецептором гормону раку молочної залози в жінок у постклімактеричний період. Крім того, посилання стосується метастатичного позитивного за рецептором естрогену та/або прогестерону раку молочної залози, особливо, метастатичного позитивного за рецептором естрогену та/або прогестерону раку молочної залози в жінок у постклімактеричний період. Крім того, посилання стосується раку сечового міхура, раку стравоходу, раку шлунка, меланомі, раку шийки матки та / або раку нирки. Додатково посилання стосується раку ендометрію, печінки, шлунка, щитовидної залози, прямої кишки та / або головного мозку. В іншому варіанті здійснення винаходу, переважно злоякісне новоутворення знаходиться на не-метастатичній стадії. В іншому варіанті здійснення винаходу, переважно злоякісне новоутворення знаходиться на метастатичній стадії. В подальшому варіанті здійснення винаходу, переважно злоякісне новоутворення знаходиться на метастатичній стадії, і більш переважно злоякісне новоутворення продукує метастази в шкіру. В подальшому варіанті здійснення винаходу, переважно злоякісне новоутворення знаходиться на метастатичній стадії, і більш переважно злоякісне новоутворення продукує метастази в лімфатичну систему. В подальшому варіанті здійснення винаходу, переважно злоякісне новоутворення знаходиться на метастатичній стадії, і більш переважно злоякісне новоутворення продукує метастази в головний мозок. Якщо наведене посилання на лікування злоякісного новоутворення, то вона переважно стосується лікування злоякісних пухлин, які експресують один або декілька рецепторів сімейства erbВ, наприклад, рецептори EFGR, erbВ2 та/або erbВ3. Протиракова дія дифумарату Сполуки (I) відповідно до винаходу може бути виміряна на основі однієї або декількох протипухлинних дій, ступені реакції-відповіді (наприклад, зменшення об'єму пухлини або зменшення пухлинного навантаження), швидкості реакції-відповіді, рівня клінічної користі (суми повної реакції-відповіді, часткової реакції-відповіді й стабільного захворювання) часу до прогресування захворювання, тривалості життя без прогресування захворювання й загального рівня виживання. Такі кінцеві точки клінічних випробувань добре відомі й описані, наприклад, у публікації FDA "Guidance for Industry Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics" у травні 2007 р. (www.fda.gov/Cber/gdlns/clintrialend.htm). Протипухлинними діями дифумарату Сполуки (I) відповідно до винаходу можуть бути, наприклад, одна або декілька дій: інгібування росту пухлини, затримка росту пухлини, регресія пухлини, стискання пухлини, збільшення проміжку часу до поновлення росту пухлини після припинення лікування або уповільнення прогресування захворювання. Застосування дифумарату Сполуки (I) також може мати сприятливий вплив для запобігання початку злоякісного новоутворення в теплокровних тварин, таких як люди. Відповідно до цього аспекту винаходу, забезпечується дифумарат Сполуки (I), для застосування як лікарський засіб. 13 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 Відповідно до подальшого аспекту винаходу, забезпечується дифумарат Сполуки (I), для застосування для одержання антипроліферативної дії в теплокровної тварини, такої як людина. Таким чином, відповідно до цього аспекту винаходу, забезпечується застосування дифумарату Сполуки (I) для приготування лікарського засобу для застосування для одержання антипроліферативної дії в теплокровної тварини, такої як людина. Відповідно до подальшої особливості цього аспекту винаходу, забезпечується спосіб забезпечення антипроліферативної дії в теплокровної тварини, такої як людина, яка потребує такого лікування, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I). Відповідно до подальшого аспекту винаходу, забезпечується дифумарат Сполуки (I), для застосування для запобігання або лікування тих пухлин, які чутливі до інгібування erbВ рецепторних тирозинкіназ, таких як комбінація EGFR і erbВ2 та/або EGFR і erbВ3, які залучені в поетапну передачу сигналів, що приводять до проліферації пухлинних клітин. Відповідно до подальшого аспекту винаходу, забезпечується застосування дифумарату Сполуки (I) для приготування лікарського засобу для застосування для запобігання або лікування тих пухлин, які чутливі до інгібування erbВ рецепторних тирозинкіназ, таких як комбінація EGFR і erbВ2 та/або EGFR і erbВ3, які залучені в поетапну передачу сигналів, що приводять до проліферації пухлинних клітин. Відповідно до подальшої особливості цього аспекту винаходу, забезпечується спосіб запобігання або лікування тих пухлин, які чутливі до інгібування однієї або декількох рецепторних тирозинкіназ сімейства erbВ, таких як комбінація EGFR і erbВ2 та/або EGFR і erbВ3, які залучені в поетапну передачу сигналів, що приводять до проліферації та/або виживання пухлинних клітин, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I). Відповідно до подальшого аспекту винаходу, забезпечується застосування дифумарату Сполуки (I) для приготування лікарського засобу для застосування для забезпечення комбінованої інгібуючої дії на EGFR і erbВ2 тирозинкінази. Відповідно до подальшої особливості цього аспекту винаходу, забезпечується спосіб забезпечення комбінованої інгібуючої дії на EGFR і erbВ2 тирозинкінази, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I). Відповідно до подальшої особливості цього аспекту винаходу, забезпечується дифумарат Сполуки (I), для застосування для забезпечення комбінованої інгібуючої дії на EGFR і erbВ2 тирозинкінази. Відповідно до подальшого аспекту винаходу, забезпечується застосування дифумарату Сполуки (I) для приготування лікарського засобу для застосування для забезпечення інгібуючої дії по відношенню до тирозинкінази на два або більше рецепторів, вибраних з EGFR, erbВ2 і erbВ3. Відповідно до подальшої особливості цього аспекту винаходу, забезпечується спосіб забезпечення інгібуючої дії по відношенню до тирозинкінази на два або більше рецепторів, вибраних з EGFR, erbВ2 і erbВ3, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I). Відповідно до подальшої особливості цього аспекту винаходу, забезпечується дифумарат Сполуки (I), для застосування для забезпечення інгібуючої дії по відношенню до тирозинкінази на два або більше рецепторів, вибраних з EGFR, erbВ2 і erbВ3. Відповідно до подальшого аспекту винаходу, забезпечується застосування дифумарату Сполуки (I) для приготування лікарського засобу для застосування для лікування стану (наприклад, пухлини), опосередкованого повністю або частково фосфорилуванням erbВ2/erbВ3 гетеродимеру. Відповідно до подальшої особливості цього аспекту винаходу, забезпечується спосіб лікування стану (наприклад, пухлини), опосередкованого повністю або частково фосфорилуванням erbВ2/erbВ3 гетеродимеру, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I). Відповідно до подальшої особливості цього аспекту винаходу, забезпечується дифумарат Сполуки (I), для застосування для лікування стану (наприклад, пухлини), опосередкованого повністю або частково фосфорилуванням erbВ2/erbВ3 гетеродимеру. Відповідно до подальшого аспекту даного винаходу, забезпечується застосування дифумарату Сполуки (I) для приготування лікарського засобу для застосування для лікування злоякісного новоутворення (наприклад, злоякісного новоутворення, вибраного з лейкозу, множинної мієломи, лімфоми, раку жовчних проток, кісток, сечового міхура, головного мозку/ЦНС, молочної залози, ободової і прямої кишки, ендометрію, шлунка, голови та шиї, 14 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 печінки, легені (переважно не-дрібноклітинного раку легені), нервових клітин, стравоходу, яєчників, підшлункової залози, передміхурової залози, нирок, шкіри, яєчок, щитовидної залози, матки й зовнішніх жіночих статевих органів і переважно злоякісного новоутворення, вибраного з раку молочної залози, шлунка, ободової і прямої кишки, голови та шиї, яєчників і легені, більш переважно раку молочної залози). Відповідно до подальшої особливості цього аспекту винаходу, забезпечується спосіб лікування злоякісного новоутворення (наприклад, злоякісного новоутворення, вибраного з лейкозу, множинної мієломи, лімфоми, раку жовчних проток, кісток, сечового міхура, головного мозку/ЦНС, молочної залози, ободової і прямої кишки, ендометрію, шлунка, голови та шиї, печінки, легені (переважно не-дрібноклітинного раку легені), нервових клітин, стравоходу, яєчників, підшлункової залози, передміхурової залози, нирок, шкіри, яєчок, щитовидної залози, матки й зовнішніх жіночих статевих органів і переважно злоякісного новоутворення, вибраного з раку молочної залози, шлунка, ободової і прямої кишки, голови та шиї, яєчників і легені, більш переважно раку молочної залози в теплокровної тварини, такої як людина, яка потребує такоголікування, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I). Відповідно до подальшого аспекту винаходу, забезпечується дифумарат Сполуки (I), для застосування для лікування злоякісного новоутворення (наприклад, вибраного з лейкозу, множинної мієломи, лімфоми, раку жовчних проток, кісток, сечового міхура, головного мозку/ЦНС, молочної залози, ободової і прямої кишки, ендометрію, шлунка, голови та шиї, печінки, легені (переважно не-дрібноклітинного раку легені), нервових клітин, стравоходу, яєчників, підшлункової залози, передміхурової залози, нирок, шкіри, яєчок, щитовидної залози, матки й зовнішніх жіночих статевих органів і переважно злоякісного новоутворення, вибраного з раку молочної залози, шлунка, ободової і прямої кишки, голови та шиї, яєчників і легені, більш переважно раку молочної залози). Як було вказано вище, доза, необхідна для терапевтичного або профілактичного лікування конкретного хворобливого стану обов'язково залежить, зокрема, від організму, який піддається лікуванню, шляху введення й тяжкості захворювання, яке піддається лікуванню. Відповідно до подальшого аспекту винаходу, забезпечується фармацевтична композиція, яка містить дифумарат Сполуки (I), у комбінації з фармацевтично прийнятним розріджувачем або носієм для застосування для лікування злоякісного новоутворення Для запобігання невизначеності, при згадуванні лікування злоякісного новоутворення мається на увазі, що воно також стосується запобігання метастаз і лікування метастаз, тобто поширення злоякісного новоутворення. Отже, Сполука (I) і дифумарат Сполуки (I) згідно із даним винаходом може застосовуватися для лікування пацієнта, у якого не виявлено метастаз для призупинення їх прояву або для збільшення періоду часу, протягом якого вони проявляються, а також для лікування пацієнта, у якого вже діагностовані метастази, для їх лікування. Крім того, лікування злоякісного новоутворення також стосується лікування первинної пухлини чи пухлин, що вже сформувалися, або первинної пухлини чи пухлин, що розвиваються. В одному варіанті здійснення винаходу лікування злоякісного новоутворення стосується запобігання метастаз. В іншому варіанті здійснення винаходу лікування злоякісного новоутворення стосується лікування метастаз. В іншому варіанті здійснення винаходу лікування злоякісного новоутворення стосується лікування первинної пухлини чи пухлин, що вже сформувалися, або первинної пухлини чи пухлин, що розвиваються. В одному варіанті здійснення лікування злоякісного новоутворення стосується ад'ювантного лікування. В іншому варіанті здійснення вказівка на лікування злоякісного новоутворення стосується неоад'ювантного лікування злоякісного новоутворення. Отже, у варіанті здійснення винаходу, дифумарат Сполуки (I) відповідно до винаходу застосовується як ад'ювантне лікування гормонально-чутливого раку молочної залози, переважно як ад'ювантне лікування позитивного за рецептором естрогену раку молочної залози в жінок у постклімактеричний період. В іншому варіанті здійснення винаходу дифумарат Сполуки (I) відповідно до винаходу застосовується як неоад'ювантне лікування гормонально-чутливого раку молочної залози, переважно як неоад'ювантне лікування чутливого до рецептора естрогену та/або прогестерону в жінок у постклімактеричний період. В іншому варіанті здійснення дифумарат Сполуки (I) застосовується для лікування прогресуючого (метастатичного) гормонально-чутливого (позитивного за рецептором естрогену та/або прогестерону) раку молочної залози, переважно прогресуючого позитивного за рецептором естрогену раку в жінок у постклімактеричний період. В подальшому варіанті здійснення дифумарат Сполуки (I) відповідно до винаходу може застосовуватися як неоад'ювантне лікування чутливого до гормону раку молочної залози в пацієнтів. В іншому варіанті здійснення дифумарат Сполуки (I) відповідно до винаходу не 15 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 застосовується як неоад'ювантне лікування. Термін "ад'ювантна терапія" стосується лікування, яке проводять після видалення первинної пухлини. Якщо злоякісне новоутворення являє собою рак молочної залози, то видалення первинної пухлини може здійснюватися, наприклад, шляхом хірургічного втручання (наприклад, секторальної резекції молочної залози або ампутації молочної залози) та/або шляхом променевої терапії. Термін "неоад'ювантна терапія" стосується лікування, які проводять перед видаленням первинної пухлини шляхом хірургічного втручання або променевої терапії. У даному винаході лікування злоякісного новоутворення також стосується запобігання злоякісного новоутворення per se. В одному варіанті здійснення винаходу, дифумарат Сполуки (I) застосовують у комбінації з ендокринним агентом, придатним для лікування раку молочної залози. Наприклад, комбінація дифумарату Сполуки (I) і ендокринного агента, вибраного з інгібітора ароматази, селективного модулятора рецептора естрогену, LHRH агоніста й понижувального регулятора рецептора естрогену. Наприклад, комбінація дифумарату Сполуки (I) і інгібітора ароматази. Наприклад, комбінація дифумарату Сполуки (I) і тамоксифену. Наприклад, комбінація дифумарату Сполуки (I) і анастрозолу. Наприклад, комбінація дифумарату Сполуки (I) і летрозолу. Наприклад, комбінація дифумарату Сполуки (I) і ексеместану. Комбінація дифумарату Сполуки (I) і ендокринної терапії переважно придатна для застосування для лікування раку молочної залози, як описано в даній заявці. Наприклад, комбінація може бути придатною для лікування метастатичного позитивного за естрогеном та/або прогестероном раку молочної залози. Альтернативно комбінація може бути придатною як ад'ювантне лікування раку молочної залози, переважно як ад'ювантне лікування позитивного за естрогеном та/або прогестероном раку молочної залози. Комбінація також може бути придатною для лікування позитивного за естрогеном та/або прогестероном раку молочної залози в пацієнтів, які раніше не одержували ендокринної терапії (наприклад, селективного модулятора рецептора естрогену, такого як тамоксифен, інгібітора ароматази, такого як анастрозол або понижувального регулятора рецептора естрогену). Отже, в одному варіанті здійснення винаходу, забезпечується спосіб лікування прогресуючого (метастатичного) позитивного за естрогеном та/або прогестероном раку молочної залози в теплокровної тварини, такої як людина, яка потребує такого лікування, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I) у комбінації з ефективною кількістю інгібітора ароматази, такого як анастрозол, де вказана тварина раніше не піддавалася ендокринній терапії, такій як, наприклад, селективний модулятор рецептора естрогену, такий як тамоксифен, інгібітор ароматази, такий як анастрозол, або понижувальний регулятор рецептора естрогену. В іншому варіанті здійснення винаходу забезпечується спосіб лікування не-метастатичного позитивного за естрогеном та/або прогестероном раку молочної залози в теплокровної тварини, такої як людина, яка потребує такого лікування, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I) у комбінації з ефективною кількістю інгібітора ароматази, такого як анастрозол, де вказана тварина раніше не піддавалася ендокринній терапії, такій як, наприклад, селективний модулятор рецептора естрогену, такий як тамоксифен, інгібітор ароматази, такий як анастрозол, або понижувальний регулятор рецептора естрогену. У цьому варіанті здійснення комбінацію підходяще вводять як ад'ювантне лікування. У двох вищеописаних варіантах здійснення лікування раку молочної залози, теплокровна тварина підходяще являє собою жінку в постклімактеричний період. Термін "постклімактеричний період" включає жінок, які знаходиться у природньому постклімактеричному періоді, і жінок, у яких менопауза була індукована, наприклад, шляхом лікування за допомогою LHRH агоніста, такого як гозерелін. Мається на увазі, що коли в даній заявці вказано, що пацієнт "раніше не піддавався ендокринній терапії", то це припускає, що лікування пацієнта за допомогою LHRH агоніста для індукування ранньої менопаузи в пацієнта не розглядається як "раніше піддавався ендокринній терапії". Отже, пацієнти, які піддавалися лікуванню за допомогою LHRH агоніста для індукування ранньої менопаузи, не виключаються з тих варіантів здійснення винаходу, які описані в даній заявці, тому що не є "лікованими за допомогою ендокринної терапії". В іншому варіанті здійснення дифумарат Сполуки (I) застосовують у комбінації з таксаном, таким як паклітаксел або доцетаксел. Ця комбінація може бути придатною для лікування раку молочної залози. Наприклад, для лікування раку молочної залози (переважно прогресуючого/метастатичного раку молочної залози), який має низьку понадекспресію erbВ2. Термін "низька понадекспресія erbВ2" стосується пухлин, які є негативними при Her2 16 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 флуоресценції in-situ гібридизації (FISH). Переважними пухлинами, які характеризуються "низькою понадекспресією erbВ2", є ті пухлини, які: (i)Her2 + шляхом імуногістохімії (IHC); та/або (ii)Her2 ++ шляхом IHC і Her2 флуоресценції in-situ гібридизації (FISH) негативними. Таким чином, у переважно варіанті здійснення винаходу, дифумарат Сполуки (I) застосовують у комбінації з таксаном, таким як паклітаксел або доцетаксел, для лікування злоякісного новоутворення з низькою понадекспресією erbВ2, вибраного з одного або декількох з: (a) раку молочної залози, який є Her2 FISH негативним; (b) раку молочної залози, який є Her2+ шляхом IHC; і (c) раку молочної залози, який є Her2++ шляхом IHC і Her2 FISH негативним. Відповідно до подальшого аспекту даного винаходу, забезпечується спосіб лікування раку молочної залози з низькою понадекспресією erbВ2 у теплокровної тварини, такої як людина, яка потребує такого лікування, який включає введення вказаній тварині ефективної кількості дифумарату Сполуки (I) у комбінації з ефективною кількістю таксану, такого як паклітаксел або доцетаксел. У даній заявці, якщо використовується термін "комбінація", то він має на увазі, що це стосується одночасного, окремого або послідовного введення. В одному аспекті винаходу, "комбінація" стосується одночасного введення. В іншому аспекті винаходу, "комбінація" стосується окремого введення. В подальшому аспекті винаходу "комбінація" стосується послідовного введення. Якщо введення є послідовним або окремим, то затримка введення другого компонента не повинна бути такою, щоб втратити позитивний вплив комбінації. Як мається на увазі, вказівки на застосування дифумарату Сполуки (I), описані в методах, застосуваннях і фармацевтичних композиціях, розкритих у даній заявці, стосується будь-якого з дифумаратів, розкритих у даній заявці, наприклад, Форми A. Позначення до фігур На Фігурі 1 представлена порошкова рентгенограма (XRPD) для вільної форми Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 2A представлена крива диференціальної сканувальної калориметрії для вільної форми Сполуки (I). На осі Х представлені температура й час, а на осі Y - потужність у мВт. Текст на фігурі означає температуру початку розкладання ендотерм і інтеграли (мДж) кривих. На Фігурі 2B представлена термогравіметрична крива для вільної форми Сполука (I). На осі Х представлені температура й час, а на осі Y - вага в мг. Текст на цьому графіку означає % втрати ваги й абсолютну втрату ваги зі зразка для явища між приблизно 30 і 80 °C. На Фігурі 3 представлена графік ізотерми сорбції динамічного випаровування для вільної форми Сполуки (I). На осі Х представлені % відносної вологості, а на осі Y - % зміни маси зразка. "сорб" стосується циклу адсорбції, а "десорб" - циклу десорбції. На Фігурі 4 представлена порошкова рентгенограма (XRPD) для Форми A дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 5 представлена крива диференціальної сканувальної калориметрії для Форми A дифумарату Сполуки (I). На осі Х представлені температура й час, а на осі Y - потужність у мВт. Текст на фігурі означає температуру початку розкладання ендотерми плавлення й інтеграл (мДж) кривої. На Фігурі 6 представлена термогравіметрична крива для Форми A дифумарату Сполуки (I). На осі Х представлені температура й час, а на осі Y - вага в мг. Текст на цьому графіку означає % втрати ваги й абсолютну втрату ваги зі зразка між приблизно 30 і 80 °C. На Фігурі 7 представлена графік ізотерми сорбції динамічного випаровування для Форми A дифумарату Сполуки (I). На осі Х представлені % відносної вологості, а на осі Y - % зміни маси зразка. "сорб" стосується циклу адсорбції, а "десорб" - циклу десорбції. На Фігурі 8 представлена порошкова рентгенограма для Форми B дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 9 представлена порошкова рентгенограма для Форми C дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 10 представлена порошкова рентгенограма для Форми D дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 11 представлена порошкова рентгенограма для Форми E дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 12 представлена порошкова рентгенограма для Форми F дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 13 представлена порошкова рентгенограма для Форми G дифумарату Сполуки (I). 17 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 14 представлена порошкова рентгенограма для Форми H дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 15 представлена порошкова рентгенограма для Форми I дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 16 представлена порошкова рентгенограма для Форми J дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 17 представлена порошкова рентгенограма для Форми K дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 18 представлена порошкова рентгенограма для Форми L дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 19 представлена порошкова рентгенограма для Форми M дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 20 представлена порошкова рентгенограма для Форми N дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 21 представлена порошкова рентгенограма для Форми O дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 22 представлена порошкова рентгенограма для Форми P дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. На Фігурі 23 представлена порошкова рентгенограма для Форми Q дифумарату Сполуки (I). На осі Х представлені значення 2-тета, а на осі Y - імпульси. Приклади Винахід надалі ілюструється за допомогою наступних прикладів, які призначені для пояснення деяких варіантів здійснення винаходу. Ці приклади не призначені й вони не повинні розглядатися як обмежуючі обсяг винаходу. Зрозуміло, що винахід може бути практично здійснений іншим чином, ніж переважно описано в даній заявці. З урахуванням відомостей, представлених у даній заявці, можливі різні модифікації й варіації даного винаходу, і, отже, вони охоплюються обсягом винаходу. У Прикладах, якщо спеціально не вказане інакше: (i) виходи представлені тільки з метою ілюстрації й необов'язково максимально досяжні; (ii) точки плавлення визначали шляхом ДСК аналізу, використовуючи прилад Mettler DSC820e ; 1 - 2 мг зразків точно зважували й аналізували у вентильованій кюветі для зразків; нагрівання здійснювали при 10 °C/хвилину від 25 °C до 325 °C; якщо спеціально не вказане інакше, точки плавлення, наведені в даній заявці, стосуються температури початку розкладання ендотерми плавлення, виміряні за допомогою ДСК; (iii) мас-спектрометричний аналіз здійснювали при енергії електронів 70 еВ методом хімічної іонізації (CI), використовуючи зонд прямого опромінення; де вказану іонізацію здійснювали шляхом електронного удару (EI), бомбардування швидкими атомами (FAB) або електророзпилення (ESP); представлені значення для m/z; як правило, тільки для іонів, які + вказані у вихідній масі; і якщо спеціально не вказане інакше, мас-іон наведений у дужках (MH) , + що стосується протонованого мас-іона; посилання на M стосується мас-іона, отриманого + шляхом втрати електрона; і посилання на M-H стосується мас-іона, отриманого шляхом втрати протона; (iv) дані ЯМР, якщо вони наведені, представлені у формі дельта-значень для основних визначальних протонів, вказаних у вигляді частинок на млн (час. на млн) відносно тетраметилсилану (TМС) як внутрішнього стандарту, визначених при 500 МГц, використовуючи оброблений дейтерієм диметилсульфоксид (ДМСО-d6) як розчинник; використані наступні скорочення: s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; br, широкий; (v) хімічні символи мають їх звичайні значення; застосовуються одиниці й символи SI; (vi) співвідношення розчинників знаходиться у вигляді об'ємних значень (об./ об.; (vii) термогравіметричний аналіз здійснювали, використовуючи устаткування Mettler TG851 [1 - 5 мг зразків точно зважували й аналізували у відкритій кюветі; нагрівання здійснювали при 10 °C/хвилину від 25 °C до 325 °C. (viii) Аналіз рентгенівської порошкової дифрактометрії здійснювали за допомогою порошкового рентгенівського дифрактометра Siemens D5000, оснащеного сцинтиляційним детектором; джерелом рентгенівських променів був Cu Kα, забезпечуючи довжину хвилі 1,54Å; дані збирали в діапазоні 2-тета 2-40, із кроком 2-тета 0,02 з 1 секундою на крок і класифікували за категоріями, ідентифікованими у Таблиці 2 нижче: 18 UA 103998 C2 Таблиця 2 % відносної інтенсивності* 25 - 100 10 - 25 3 - 10 1-3 Визначення vs (дуже сильний) s (сильний) m (середній) w (слабкий) * Відносні інтенсивності розраховували на основі дифрактограм, виміряних з фіксованими щілинами 5 10 15 20 25 30 35 40 45 [Як було вказано раніше, фахівці в галузі порошкової рентгенограми можуть підтвердити, що на відносну інтенсивність піків можуть впливати, наприклад, частинки розміром близько 30 мікронів і неодиничні формати зображення, які можуть впливати на аналіз зразків. Фахівець у даній галузі також буде враховувати той факт, що на положення відбиттів може впливати точна висота, на яку встановлюється зразок у дифрактометрі й калібрування нуля дифрактометра. Площинність поверхні зразка також може незначно впливати. Отже, представлені дані порошкової рентгенограми не повинні тлумачитися як абсолютні (для додаткової інформації див. Jenkins, R & Snyder, R.L. "Introduction to X-Ray Powder Diffractometry" John Wiley & Sons, 1996)]; (ix) сорбцію динамічного випаровування вимірювали, використовуючи SMS DVS (Surface Measurement Systems Limited, UK). Зразки аналізували при 25 °C, використовуючи газовий потік 200 кубічних сантиметрів за хвилину. Відносну вологість (ВВ) підвищували від 0 % ВВ східчасто на 10 % ВВ до 80 %ВВ з кінцевою стадією 95 %ВВ. Після цього зразки десорбували, використовуючи таку ж схему етапів ВВ, що й при сорбції; потім цю процедуру повторювали в другому циклі сорбції /десорбції. Зрівноважування на кожній стадії вологості встановлювали таким чином, щоб швидкість зміни ваги в часі (хвилина) становила 0,002 %. (x) власну швидкість розчинення вимірювали в бані для розчинення, з'єднаною із скловолоконним Уф-детектором. (xi) розчинність у воді вимірювали за допомогою ВЕРХ УФ. (xii) у прикладах, представлених нижче, вказана кількість молів і виходи стосуються сировинних матеріалів і реагентів при 100 мас. %, у такий спосіб враховується чистота використовуваних матеріалів. Приклад A Одержання 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(N-метилкарбамоїлметил)піперидин-4іл]окси}хіназоліну (Сполука (I)) 2-Хлор-N-метилацетамід (3,720 кг, 34,60 моль) і 4-(3-хлор-2-фтораніліно)-7-метокси-6[(піперидин-4-іл)окси]хіназолін дигідрохлорид (13,70 кг, 27,25 моль), розчиняли в ацетонітрилі (79,2 кг). До перемішуваної суспензії при температурі навколишнього середовища, додавали триетиламін (17,40 кг, 172,11 моль). Отриманий прозорий розчин нагрівали в колбі зі зворотним холодильником і витримували протягом 3 годин. Розчин охолоджували до 20 °C (продукт кристалізувався при 50 °C). У реактор додавали воду (54,2 кг) і суспензію перемішували додатково протягом 2 годин при 20 °C. Продукт фільтрували й промивали водою (34 кг), після цього холодним (0 °C) ацетонітрилом (13,0 кг). Продукт перекристалізовували з ацетонітрилу (94,6 кг), виділяли шляхом фільтрації й промивали холодним (0 °C) ацетонітрилом (13,2 кг). Після цього здійснювали додаткову перекристалізацію цього продукту, як описано вище, з ацетонітрилу (75,2 кг). Після цього тверду речовину висушували у вакуумі, одержуючи вказаний 1 у заголовку продукт у вигляді білої твердої речовини (6,50 кг, 50 %); H ЯМР-спектр: (CDCl3) 1,98 (m, 2H), 2,08 (m, 2H), 2,46 (-m, 2H), 2,85 (m, 2H), 2,87 (d, 3H), 3,07 (s, 2H), 4,02 (s, 3H), 4,49 (m, + 1H), 7,16 (m, 4H), 7,31 (m, 2H), 8,49 (m, 1H), 8,71 (s, 1H); Мас-спектр: MH 474. 4-(3-хлор-2-фтораніліно)-7-метокси-6-[(піперидин-4-іл)окси]хіназолін, який використовували як вихідну речовину, готували в такий спосіб: Стадія 1:6-ацетокси-4-(3-хлор-2-фтораніліно)-7-метоксихіназолін гідрохлорид 6-Ацетокси-7-метоксихіназолін-4-он (Міжнародна патентна заявка WO 96/15118, Приклад 39 у цій заявці; 21,4 кг, 89,3 моль) суспендували в толуолі (150 кг). До нього додавали Nетилдіізопропіламін (13,3 кг, 103 моль). Коричневу суспензію нагрівали до 70 °C, потім завантажували оксихлорид фосфору (36,0 кг, 228 моль). Реакційну суміш перемішували при 19 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 70 °C протягом 5 годин. Додатково додавали толуол (84,0 кг), потім додавали 3-хлор-2фторанілін (14,88 кг, 102 моль). Реакційну суміш перемішували при 70 °C протягом 2 годин, протягом цього часу осаджувалася тверда речовина. Суспензію охолоджували до 25 °C і витримували при цій температурі протягом 93 годин. Реакційну суміш фільтрували й осад на фільтрі промивали толуолом (2 × 55,5 кг). Осад додатково промивали сумішшю етанолу (24,5 кг) і води (32,0 кг) двічі, потім етанолом (50,5 кг) двічі й після цього тверду речовину висушували у вакуумі, одержуючи вказаний у заголовку продукт у вигляді бежевої твердої речовини (33,4 кг, 1 78 %); H ЯМР: 2,37 (s, 3H), 4,00 (s, 3H), 7,34 (ddd, 1H), 7,48 (s, 1H), 7,52 (ddd, 1H), 7,61 (ddd, 1H), 8,62 (s, 1H), 8,86 (s, 1H); Мас-спектр: 362,4, 364,4. Стадія 2: 4-(3-Хлор-2-фтораніліно)-6-гідрокси-7-метоксихіназолін 6-Ацетокси-4-(3-хлор-2-фтораніліно)-7-метоксихіназолін гідрохлорид зі стадії 1 (33,5 кг, 69,6 моль) суспендували в метанолі (198 кг). До перемішуваної суспензії при 25 °C додавали воду (86 кг) і гідроксид натрію (31,5 кг, 32 %). Отриманий розчин перемішували при 60 °C протягом 4,5 годин і потім охолоджували до 25 °C. Додавали оцтову кислоту (приблизно 16,0 кг) до досягнення значення рН 5,5-6,0, у цій точці продукт осаджувався з розчину. Післядодавання додаткової кількості метанолу (5,5 кг) суспензію перемішували протягом 90 хвилин. Продукт фільтрували, потім промивали за допомогою 25 % водного метанолу (39,0 кг MeОН + 17,0 кг води) і потім метанолом (55,5 кг). Неочищену тверду речовину висушували у вакуумі при 40 °C. Неочищену тверду речовину розріджували за допомогою води (145 кг) і перемішували протягом 2 годин при 65 °C. Завись охолоджували до 20 °C і фільтрували. Осад на фільтрі промивали метанолом (2 × 21,5 кг), потім висушували у вакуумі при 40 °C, одержуючи вказаний у заголовку 1 продукт у вигляді світло-коричневої твердої речовини (21,85 кг, 98 %); H ЯМР: 3,95 (s, 3H), 7,19 (s, 1H), 7,23 (dd, 1H), 7,42 (dd, 1H), 7,50 (dd, 1H), 7,64 (s, 1H), 8,32 (s, 1H), 9,43 (s, 1H), 9,67 (br.s, 1H); Мас-спектр: 320,4, 322,4. Стадія 3: 6-{[(1-трет-Бутоксикарбоніл)піперидин-4-іл]окси}-4-(3-хлор-2-фтораніліно)-7метокси хіназолін 4-(3-Хлор-2-фтораніліно)-6-гідрокси-7-метоксихіназолін зі Стадії 2 (15,591 кг, 48,44 моль), трет-Бутил (4-метансульфонілокси)піперидин-1-карбоксилат (отриманий, як описано в Chemical & Pharmaceutical Bulletin 2001, 49(7), 822-829; 16,20 кг, 57,99 моль) і карбонат калію (7,978 кг, 57,73 моль) розчиняли в N-метилпіролідиноні (114,2 кг), і суміш нагрівали до 100 °C при перемішуванні. Продовжували нагрівати при 100 °C (95 °C-105 °C) протягом 5 годин. Потім суміш охолоджували до 80 °C і загартовували шляхом додавання води (216,6 кг). Партію перемішували при 80 °C додатково протягом 60 хвилин потім охолоджували до 20 °C протягом 2 годин, протягом цього часу продукт кристалізувався. Продукт виділяли шляхом фільтрації. Продукт розчиняли в гарячому (флегма) метанолі (200 л). До цієї суміші додавали воду (20 л), що індукувало кристалізацію. Суспензію охолоджували до 0 °C і фільтрували. При здійсненні висушування у вакуумі при 50 °C одержували вказаний у заголовку продукт, 18,80 кг 1 (77 %); H ЯМР: 1,40 (s, 9H), 1,60-1,65 (m, 2H), 1,95-2,00 (m, 2H), 3,20-3,25 (m, 2H), 3,65-3,70 (m, 2H), 3,92 (s, 3H), 4,68 (m, 1H), 7,21 (s, 1H), 7,27 (dd, 1H), 7,47 (ddd, 1H), 7,51 (dd, 1H), 7,85 (s, 1H), 8,36 (s, 1H), 9,53 (s, 1H); Мас-спектр: 503,5, 505,5. Стадія 4:4-(3-хлор-2-фтораніліно)-7-метокси-6-[(піперидин-4-іл)окси]хіназолін дигідрохлорид 6-{[(1-трет-Бутоксикарбоніл)піперидин-4-іл]окси}-4-(3-хлор-2-фтораніліно)-7-метоксихіназолін зі стадії 3 (18,80 кг, 37,38 моль) суспендували в ізопропанолі (139,8 кг), і нагрівали до 40 °C при перемішуванні. Соляну кислоту (15,40 кг, ~156,3 моль) завантажували в посудину протягом 50 хвилин, що дозволяло здійснитися виділенню тепла приблизно 9 °C. При завантаженні кислоти, суспензія розчинялася, утворюючи прозорий розчин. Розчин повільно нагрівали в колбі зі зворотним холодильником приблизно протягом 90 хвилин, і потім витримували в колбі зі зворотним холодильником додатково протягом 3 годин. Продукт кристалізувався протягом цього періоду флегми. Густу суспензію охолоджували до 0 °C і фільтрували. Осад на фільтрі два рази промивали холодним (0 °C) ізопропанолом (2 × 20,6 кг). Продукт висушували у вакуумі 1 при 50 °C, одержуючи вказаний у заголовку продукт, 13,60 кг (73 %); H ЯМР: 1,53-1,64 (m, 2H), 2,00-2,05 (m, 2H), 2,64-2,72 (m, 2H), 3,00-3,07 (m, 2H), 3,92 (s, 3H), 4,60 (m, 1H), 7,20 (s, 1H), 7,26 (ddd, 1H), 7,47 (dd, 1H), 7,50 (dd, 1H), 7,82 (s, 1H), 8,34 (s, 1H), 9,56 (s, 1H); Мас-спектр: 403,2, 405,2. Приклад B Одержання 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(Nметилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну (Сполука (I)) 2-Хлор-N-метилацетамід (24,22 г, 223,1 ммоль) і 4-(3-хлор-2-фтораніліно)-7-метокси-6[(піперидин-4-іл)окси]хіназолін дигідрохлорид (86,00 г, 160,9 ммоль), розріджували в ацетонітрилі (537 мл). До перемішуваної суспензії при температурі навколишнього середовища, додавали триетиламін (101 мл, 723,9 ммоль). Реакцію нагрівали до 75 °C витримували 20 UA 103998 C2 5 10 15 20 25 30 35 40 45 протягом 5 годин. Розчин охолоджували до 70 °C і додавали етанол (268 мл). Реакцію охолоджували до 45 °C і додавали воду (9,6 мл). Додавали сполуку (I) (0,42 г) для встановлення кристалізації й потім завись охолоджували до 20 °C протягом 2 годин. Після перемішування додатково протягом 12 годин продукт виділяли шляхом фільтрації. Осад на фільтрі два рази промивали за допомогою ацетонітрил (102 мл): етанол (51 мл): вода (1,8 мл) і потім за допомогою води (153 мл). Продукт висушували у вакуумі при 60 °C, одержуючи вказану в заголовку сполуку у вигляді білої твердої речовини (45,9 г, 60 %); 1H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,76-1,87 (m, 2 H) 2,01-2,11 (m, 2 H) 2,35-2,44 (m, 2 H) 2,64 (d, J=4,74 Гц, 3 H) 2,722,80 (m, 2 H) 2,95 (s, 2 H) 3,95 (s, 3 H) 4,51-4,63 (m, 1 H) 7,23 (s, 1 H) 7,29 (td, J=8,08, 1,29 Гц, 1 H) 7,46-7,58 (m, 2 H) 7,75 (q, J=4,60 Гц, 1 H) 7,83 (s, 1 H) 8,38 (s, 1 H) 9,59 (s, 1 H) Мас-спектр: + MH 474. 4-(3-хлор-2-фтораніліно)-7-метокси-6-[(піперидин-4-іл)окси]хіназолін дигідрохлорид, який використовували як вихідну речовину, одержували в такий спосіб: Стадія 1:6-{[(1-трет-Бутоксикарбоніл)піперидин-4-іл]окси}-4-(3-хлор-2-фтораніліно)-7-метокси хіназолін 4-(3-Хлор-2-фтораніліно)-6-гідрокси-7-метоксихіназолін (отриманий, як описано на Стадії 2 Прикладу A; 60,00 г, 0,1828 моль), трет-Бутил (4-метансульфонілокси)піперидин-1-карбоксилат (88,04 г, 0,3107 моль) і карбонат калію (30,31 г, 0,2193 моль) суспендували в етанолі (584 мл) і воді (58 мл), і суміш нагрівали в колбі зі зворотним холодильником при перемішуванні. Продовжували нагрівати в колбі зі зворотним холодильником 16,5 годин. Потім суміш охолоджували до 70 °C і додавали воду (234 мл) протягом 60 хвилин. Партію перемішували при 65 °C додатково протягом 2 годин для встановлення кристалізації. Завись охолоджували до 20 °C протягом 6 годин. Продукт виділяли шляхом фільтрації. Осад на фільтрі розріджували за допомогою водного етанолу (етанол 117 мл, вода 58 мл) і потім витиснення промивали за допомогою водного етанолу (етанол 117 мл, вода 58 мл). Осад на фільтрі потім розріджували в допомогою води (175 мл) і потім витиснення промивали водою (175 мл). Продукт висушували у вакуумі при 40 °C, одержуючи вказану в заголовку сполуку (81,5 г, 84 %); 1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн 1,42 (s, 9 H) 1,601,70 (m, 2 H) 1,96-2,04 (m, 2 H) 3,23-3,30 (m, 2 H) 3,65-3,75 (m, 2 H) 3,95 (s, 3 H) 4,68-4,75 (m, 1 H) 7,24 (s, 1 H) 7,29 (t, J=8,06 Гц, 1 H) 7,49 (t, J=7,50 Гц, 1 H) 7,54 (t, J=7,19 Гц, 1 H) 7,88 (s, 1 H) 8,39 (s, 1 H) 9,57 (s, 1 H); Мас-спектр: 503,5, 505,5. Стадія 2:4-(3-хлор-2-фтораніліно)-7-метокси-6-[(піперидин-4-іл)окси]хіназолін дигідрохлорид 6-{[(1-трет-Бутоксикарбоніл)піперидин-4-іл]окси}-4-(3-хлор-2-фтораніліно)-7-метоксихіназолін (10,00 г, 0,1879 моль) суспендували в промислових денатурованих метилових спиртах (95 мл), і нагрівали до 35 °C при перемішуванні. Соляну кислоту (6,59 мл, приблизно 0,7891 моль) завантажували в посудину, дозволяючи відбутися екзотермі приблизно 5,5 °C. При завантаженні кислоти, суспензія розчинялася, утворюючи прозорий розчин. Розчин повільно нагрівали до 70 °C приблизно протягом 90 хвилин, і потім витримували при 70 °C додатково протягом 1 години. Потім реакцію охолоджували до 0 °C протягом 4 годин, протягом цього часу продукт кристалізувався. Продукт виділяли шляхом фільтрації й потім осад на фільтрі два рази промивали за допомогою промислових денатурованих метилових спиртів (2 × 14 мл). Продукт висушували у вакуумі при 50 °C, одержуючи вказаний у заголовку продукт (9,04 г, 88 %); 1H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,91-2,01 (m, 2 H) 2,27-2,35 (m, 2 H) 3,15-3,26 (m, 2 H) 3,26-3,35 (m, 2 H) 4,02 (s, 3 H) 5,07-5,15 (m, 1 H) 7,35 (td, J=8,08, 1,29 Гц, 1 H) 7,46 (s, 1 H) 7,52 (ddd, J=8,03, 5,23 Гц, 1 H) 7,63 (ddd, J=8,22, 6,76, 1,62 Гц, 1 H) 8,83 (s, 1 H) 8,91 (s, 1 H) 9,02-9,13 (m, 1 H) 9,20-9,31 (m, 1 H) 12,51 (br. s., 1 H)); Мас-спектр: 403,2, 405,2. Приклад C Одержання 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(Nметилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну (Сполука (I)) Сполуку (I) одержували відповідно до схеми, представленої нижче: 50 21 UA 103998 C2 5 10 15 20 2-[4-(5-ціано-4-{[(диметиламіно)метилен]аміно}-2-метоксифенокси)піперидин-1-іл]-Nметилацетамід (7, 7,00 г, 17,71 ммоль), суспендували в метоксибензолі (35,8 г). Завантажували оцтову кислоту (16,6 г) і до отриманого розчину додавали 3-хлор-2-фторанілін (2,71 г, 18,07 ммоль). Реакційну суміш нагрівали при 90 °C протягом 20 годин потім охолоджували до 20 °C. У реакційну суміш завантажували воду (37,04 г), і органічний шар відкидали. До отриманої водної суміші завантажували ізопропанол (39,00 г), після цього водний аміак (20,79 г, 25 %). Реакційну суміш нагрівали до 30 °C і зацьковували за допомогою Сполуки (I), яка індукує кристалізацію. Потім реакцію охолоджували до 0 °C і продукт виділяли шляхом фільтрації. Осад на фільтрі два рази промивали за допомогою суміші води (7,28 г) і ізопропанолу (4,68 г), потім висушували, 1 одержуючи Сполуку (I) (5,65 г, вихід 55 %); H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,79 (m, 2 H) 2,04 (m, 2 H) 2,38 (m, 2 H) 2,62 (d, J=4,5 Гц, 3 H) 2,74 (m, 2 H) 2,94 (s, 2 H) 3,93 (s, 3 H) 4,56 (tt, J=8,1, 3,8 Гц, 1 H) 7,21 (s, 1 H) 7,28 (m, 1 H) 7,50 (m, 2 H) 7,73 (q, J=4,5 Гц, 1 H) 7,81 (s, 1 H) 8,36 + (s, 1 H) 9,56 (br.s, 1 H); Мас-спектр: m/z (M+H) 474,2, 476,2. 2-[4-(5-ціано-4-{[(диметиламіно)метилен]аміно}-2-метоксифенокси)піперидин-1-іл]-Nметилацетамід (7), який використовували як вихідну речовину, одержували в такий спосіб: Стадія 1. Одержання трет-бутил 4-(5-ціано-2-метоксифенокси)піперидин-1-карбоксилату (2) 3-гідрокси-4-метоксибензонітрил (1, 6,00 г, 39,62 ммоль), трет-бутил (4метансульфонілокси)піперидин-1-карбоксилат (16,6 г, 59,44 ммоль) (Chemical & Pharmaceutical Bulletin 2001, 49(7), 822-829); і карбонат калію (6,71 г, 47,55 ммоль) суспендували в ізопропанолі 22 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 (78,98 г) і суміш нагрівали у колбі зі зворотним холодильником при перемішуванні. Для завершення реакції додатково додавали трет-бутил (4-метансульфонілокси)піперидин-1карбоксилат (2,08 г, 7,43 ммоль). Після цього суміш охолоджували й загартовували шляхом додавання води (100,47 г). При затравлюванні за допомогою трет-бутил 4-(5-ціано-2метоксифенокси)піперидин-1-карбоксилату (2) з наступним охолодженням до 0 °C приводило до одержання кристалічного продукту, який виділяли шляхом фільтрації. Осад на фільтрі промивали сумішшю води (8,86 г) і ізопропанолу (6,97 г), після цього водою (23,64 г) і потім 1 висушували, одержуючи вказану в заголовку сполуку (10,75 г, вихід 80 %); H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,39 (s, 9 H) 1,48 (m, 2 H) 1,88 (m, 2 H) 3,13 (m, 2 H) 3,67 (m, 2 H) 3,83 (s, 3 H) 4,56 (tt, J=8,1, 3,8 Гц, 1 H) 7,13 (d, J=8,4 Гц, 1 H) 7,42 (dd, J=8,4, 1,9 Гц, 1 H) 7,51 (d, J=1,9 + Гц, 1 H); Мас-спектр: m/z (M+H) 333,1. Стадія 2. Одержання 4-метокси-3-(піперидин-4-ілокси)бензонітрилу (3) Трет-бутил 4-(5-ціано-2-метоксифенокси)піперидин-1-карбоксилат (2, 39,31 г, 118,26 ммоль) суспендували в етанолі (155,53 г) і нагрівали до 40 °C. До цього суспензії повільно додавали HСl (46,61 г, 573,04 ммоль). Суміш нагрівали до 60 °C і витримували протягом 3 годин. Реакційну суміш охолоджували до 20 °C і завантажували затравку для початку кристалізації. Отриману тверду речовину виділяли шляхом фільтрації при 0 °C, два рази промивали етанолом (62,21 г) і потім висушували, одержуючи вказану в заголовку сполуку у вигляді гідрохлоридної 1 солі (29,84 г, вихід 77 %); H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,84 (m, 2 H) 2,09 (m, 2 H) 3,02 (ddd, J=12,7, 8,9, 3,4 Гц, 2 H) 3,20 (m, 2 H) 3,84 (s, 3 H) 4,63 (tt, J=7,7, 3,6 Гц, 1 H) 7,15 (d, J=8,5 Гц, 1 H) 7,45 (dd, J=8,5, 1,9 Гц, 1 H) 7,56 (d, J=1,9 Гц, 1 H) 9,16 (br. s, 2 H); Мас-спектр: m/z + (M+H) 233,2. Стадія 3. Одержання 2-[4-(5-ціано-2-метоксифенокси)піперидин-1-іл]-N-метилацетаміду (4) 4-Метокси-3-(піперидин-4-ілокси)бензонітрил гідрохлоридну сіль (3, 28,36 г, 95,82 ммоль), 2хлор-N-метилацетамід (12,37 г, 114,98 ммоль) і карбонат калію (33,11 г, 239,55 ммоль) суспендували в ацетонітрилі (161,36 г). Реакційну суміш нагрівали в колбі зі зворотним холодильником протягом 3 годин. Реакційну суміш охолоджували до 20 °C і завантажували воду (386,26 г). Реакцію нагрівали до 75 °C і об'єм зменшували шляхом дистиляції. При охолодженні відбувалася кристалізація. Отриману тверду речовину виділяли шляхом фільтрації, два рази промивали водою (77,25 г і 128,75 г) і потім висушували, одержуючи вказану в заголовку 1 сполуку (27,95 г, вихід 94 %); H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,68 (m, 2 H) 1,91 (m, 2 H) 2,29 (m, 2 H) 2,61 (d, J=4,7 Гц, 3 H) 2,67 (m, 2 H) 2,88 (s, 2 H) 3,83 (s, 3 H) 4,41 (tt, J=8,3, 4,0 Гц, 1 H) 7,11 (d, J=8,4 Гц, 1 H) 7,40 (dd, J=8,4, 1,9 Гц, 1 H) 7,47 (d, J=1,9 Гц, 1 H) 7,68 (q, J=4,7 Гц, 1 + H); Мас-спектр: m/z (M+H) 304,2. Стадія 4. Одержання 2-[4-(5-ціано-2-метокси-4-нітрофенокси)піперидин-1-іл]-Nметилацетаміду (5) 2-[4-(5-Ціано-2-метоксифенокси)піперидин-1-іл]-N-метилацетамід (4, 8,78 г, 26,11 ммоль) суспендували в оцтовій кислоті (22,82 г, 364,87 ммоль) і отриману реакційну суміш охолоджували до 5 °C. До неї додавали сірчану кислоту (23,64 г, 234,95 ммоль), підтримуючи температуру реакції нижче 30 °C. До отриманого розчину додавали азотну кислоту (2,40 г, 26,63 ммоль). Після цього реакційну суміш нагрівали до 35 °C і витримували протягом 3 годин. Завантажували додатково азотну кислоту (117 мг, 1,31 ммоль) і сірчану кислоту (1,31 г 13,1 ммоль) і реакційну суміш нагрівали при 35 °C протягом 30 хвилин. Розчин охолоджували до 20 °C і загартовували за допомогою водного аміаку (92,45 г 1,36 молей), що приводило до підвищення температури до 50 °C. До отриманої суспензії додавали пропіонітрил (61,58 г 1,12 молей) і воду (19 г). Реакційну суміш нагрівали до 80 °C, що приводило до одержання прозорого розчину, у якому при відстоюванні утворювалося два шари. Нижній шар видаляли. Реакційну суміш охолоджували до 20 °C, що приводило до одержання густої суспензії. Тверду речовину виділяли шляхом фільтрації, промивали пропіонітрилом (6,16 г 112,0 ммоль) і висушували, 1 одержуючи вказану в заголовку сполуку (7,44 г, вихід 82 %); H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,72 (m, 2 H) 1,97 (m, 2 H) 2,35 (m, 2 H) 2,61 (d, J=4,7 Гц, 3 H) 2,66 (m, 2 H) 2,90 (s, 2 H) 3,96 (s, 3 H) 4,73 (tt, J=8,4, 4,0 Гц, 1 H) 7,71 (q, J=4,7 Гц, 1 H) 7,82 (s, 1 H) 7,86 (s, 1 H). Мас+ спектр: m/z (M+H) 349,2 Стадія 5. Одержання 2-[4-(4-аміно-5-ціано-2-метоксифенокси)піперидин-1-іл]-Nметилацетаміду (6) 2-[4-(5-Ціано-2-метокси-4-нітрофенокси)піперидин-1-іл]-N-метилацетамід (5, 7,42 г, 19,38 ммоль) суспендували у воді (44,52 г) і метанолі (5,35 г). До нього додавали дитіоніт натрію (11,91 г, 58,15 ммоль) і отриману реакційну суміш нагрівали до 60 °C. До реакційної суміші додавали соляну кислоту (46,98 г, 463,89 ммоль)), одержуючи розчин, який витримували при 60 °C протягом 3 годин. Після цього реакційній суміші дозволяли охолонути до 20 °C. 23 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 Завантажували водний гідроксид натрію (15,51 г 182,2 ммоль), після цього 2метилтетрагідрофуран (58,0 г). Реакційну суміш нагрівали до 60 °C, у якій при відстоюванні утворювалося два шари й нижній водний шар відкидали. Об'єм реакційної суміші зменшували шляхом вакуумної перегонки й додавали метил трет-бутиловий ефір (18,54 г), одержуючи суспензію, яку охолоджували до 10 °C і потім тверду речовину збирали шляхом фільтрації. Тверду речовину промивали за допомогою 2-метилтетрагідрофурану (5,8 г) і висушували, 1 одержуючи вказану в заголовку сполуку (5,4 г, вихід 78 %); H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,62 (m, 2 H) 1,82 (m, 2 H) 2,20 (m, 2 H) 2,60 (d, J=4,7 Гц, 3 H) 2,65 (m, 2 H) 2,86 (s, 2 H) 3,72 (s, 3 H) 4,00 (tt, J=8,3, 4,0 Гц, 1 H) 5,66 (br. s, 2 H) 6,39 (s, 1 H) 6,94 (s, 1 H) 7,65 (q, J=4,7 Гц, 1 H) + Мас-спектр: m/z (M+H) 319,2. Стадія 6. Одержання 2-[4-(5-ціано-4-{[(диметиламіно)метилен]аміно}-2метоксифенокси)піперидин-1-іл]-N-метилацетаміду (7) 2-[4-(4-Аміно-5-ціано-2-метоксифенокси)піперидин-1-іл]-N-метилацетамід (6, 18,21 г, 52,05 ммоль) суспендували в 2-метилтетрагідрофурані (99,62 г). До нього додавали оцтову кислоту (162,79 мг), і N, N-диметилформамід диметил ацеталь (ДМФА-DMA) (8,63 г, 70,27 ммоль) і отриману реакційну суміш нагрівали при 76 °C протягом 16 годин. До реакційної суміші додатково додавали N, N-диметилформамід диметил ацеталь (639,41 мг, 5,20 ммоль) для завершення реакції. Реакційну суміш охолоджували до 30 °C, протягом цього часу відбувалася кристалізація. Отриману тверду речовину виділяли шляхом фільтрації, промивали за допомогою 2-метилтетрагідрофурану (14,23 г) і висушували, одержуючи вказану в заголовку 1 сполуку (19,53 г, вихід 97 %); H ЯМР (400 МГц, ДМСО-d6) δ част. на млн 1,65 (m, 2 H) 1,86 (m, 2 H) 2,24 (m, 2 H) 2,60 (d, J=4,7 Гц, 3 H) 2,66 (m, 2 H) 2,87 (s, 2 H) 2,95 (s, 3 H) 3,04 (s, 3 H) 3,81 (s, 3 H) 4,19 (tt, J=8,2, 3,8 Гц, 1 H) 6,72 (s, 1 H) 7,15 (s, 1 H) 7,67 (q, J=4,7 Гц, 1 H) 7,90 (s, 1 H); Мас+ спектр: m/z (M+H) 374,2. Біологічна активність Оцінювали активність Сполуки (I) для тестування його здатності відносно: a) інгібування активації (фосфорилування) EGFR, ErbВ2 і ErbВ3 у стимульованих лігандом клітинах; і b) інгібування базальної й стимульованої лігандом проліферації MCF-7 клітин. (a) Дослідження Сполуки (I), опосередковані лігандом Методи: KB клітини й MCF-7 клітини одержували з Американської колекції типових культур (ATCC) і загальноприйнятим способом культивували в RPMI 1640 (без фенолового червоного) + 10 % фетальної бичачої сироватки + 2 мM L-глутамін. Обробка й лізис клітин: KB клітини висівали при щільності 5000 клітин/лунку й MCF-7 клітини при щільності 4000 клітин/лунку в планшети на 96 лунок у середовищі RPMI 1640, що містить 10 % FBS. Клітини інкубували протягом 72 годин, після цього середовище заміняли на середовище без сироватки RPMI 1640 протягом 24 годин. Після цього клітини обробляли за допомогою Сполуки (I) протягом 90 хвилин при концентрації в інтервалі 0-10 мкM. Безпосередньо перед лізисом клітин, MCF-7 і KB клітини інкубували протягом 5 хвилин з лігандом (герегулин ("HRG") для MCF-7 клітин і фактор росту епідермісу ("EGF") для KB клітин) при концентрації, необхідної для підвищення фосфорилування рецептора до 90 % максимальної (ED90) для надання можливості порівняння між дослідженнями. Вимірювання p-EGFR, p-ЕrbВ2 і p-ErbВ3: p-EGFR статус KB клітин вимірювали за допомогою набору людського фосфо-EGFR Duoset ELISA (R&D systems загальний EGFR #DYC1854, p-EGFR #DYC1095). Вміст p-ErbВ2 і p-ErbВ3 для MCF-7 клітин вимірювали за допомогою набору людського фосфо-erbВ2 Duoset ELISA (R&D systems, DYC1768) і набору людського фосфо-erbВ3 Duoset ELISA (R&D systems, DYC1769) відповідно. За допомогою наборів вимірювали сумарне фосфорилування тирозину в клітинах за допомогою EGFR, ErbВ2 або ErbВ3. Дослідження здійснювали відповідно до інструкцій виробника, при цьому на лунку додавали 50 мкл лізату. Результати: Результати узагальнено в таблиці 3 24 UA 103998 C2 Таблиця 3 Активність сполуки (I) по відношенню до p-EGFR (в KB клітинах) і p-ErbВ2 і p-ErbВ3 (в MCF-7 клітинах) Сполука Сполука (I) p-EGFR Середнє геометричне IC50 (95 % CIR*) 0,004 (1,377) p-ErbВ2 Середнє геометричне IC50 (95 % CIR) 0,003 (1,817) p-ErbВ3 Середнє геометричне IC50 (95 % CIR) 0,004 (1,89) * Співвідношення довірчого інтервалу 5 10 15 20 У Таблиці 3 показано, що Сполука (I) є ефективним інгібітором фосфо-EGFR, фосфо-ErbB2 і фосфо-ErbB3 у цих клітинах. b) Сполука (I) у дослідженні базальної або HRG-стимульованої проліферації MCF-7 клітин Методи: MCF-7 клітини культивували загальноприйнятим способом в DMEM (без фенолового червоного) + 10 % фетальної бичачої сироватки + 2 мM L-глутаміну. Клітини висівали при щільності 4000 клітин на лунку в планшетах на 96 лунок в DMEM середовищу, що містить 1 % вугілля/оброблений декстраном FBS і 2 мM глутамін і дозволяли відстоюватися протягом 4 годин перед обробкою за допомогою Сполуки (I) при концентрації в інтервалі 0-3 мкM і 0-10 мкг/мл відповідно. Через дві години після обробки, клітини інкубували з 10 нг/мл HRG, концентрації, необхідної для підвищення проліферації MCF-7 клітин до 90 % максимальної (ED90). Базальні лунки не стимулювали за допомогою ліганду. Після інкубування протягом 4 днів, оцінювали життєздатність клітин, використовуючи аналіз за допомогою 3-(4,5диметилтіазол-2-іл)-5-(3-карбоксиметоксифеніл)-2-(4-сульфофеніл)-2H-тетразолію (MTS). Перед визначенням IC50 для HRG-стимульованих оброблених сполукою клітин, середнє значення базального росту при 96 годин віднімали з кожних отриманих даних, у такий спосіб оцінювали проліферацію, стимульовану за допомогою HRG-передачі сигналів. Базальні IC50 значення виражали у вигляді GI50, тобто кількість клітин у планшеті в день 0 (значення для вихідного рівня) віднімали з даних, отриманих через 96 годин. Результати: Результати узагальнено в Таблицях 4 і 5. Таблиця 4 IC50 значення для HRG-стимульованої проліферації Сполука Сполука (I) Середнє геометричне IC50 0,061 мкM 95 % CIR 2,421 25 Таблиця 5 GI50 значення базальної проліферації Сполука Сполука (I) 30 35 Середнє геометричне GI50 1,094 мкM 95 % CIR 4,423 В KB клітинах, стимуляція за допомогою EGF, який специфічно зв'язується з EGFR, викликає фосфорилування й, отже, активацію цього рецептора. Аналогічним чином, в MCF-7 клітинах HRG, який специфічно зв'язується з ErbВ3, викликає утворення його гетеродимерів з ErbВ2 і обидва рецептори стають фосфорилованими й активованими. У Таблицях 4 показано, що Сполука (I) є ефективним інгібітором HRG-стимульованої MCF-7 проліферації. Вважають, що такі впливи на проліферації обумовлюються активностями цих сполук по відношенню до ErbВ2/ErbВ3 гетеродимерів, при цьому Сполука (I) є найбільш ефективним інгібітором по відношенню до цього гетеродимеру. MCF-7 базальні дослідження характеризують ситуацію, у якій не відбувається підвищення стимуляції або активації erbВ2/erbВ3 гетеродимеризації. У Таблиці 5 показано, що навіть у таких 25 UA 103998 C2 5 10 15 20 25 30 35 40 45 50 55 60 умовах Сполука (I) інгібує MCF-7. Приклад 1 Одержання Форми A Дифумарату Сполуки (I): 2-[4-({4-[(3-хлор-2-фторфеніл)аміно]-7метоксихіназолін-6-іл}окси)піперидин-1-іл]-н-метилацетамід ди-[(2E)-бут-2-ендіоат] Форма A Розчин фумарової кислоти (2,7 г, 23,22 ммоль) у метанолі (95 мл) додавали до суміші 2-[4({4-[(3-Хлор-2-фторфеніл)аміно]-7-метоксихіназолін-6-іл}окси)піперидин-1-іл]-N-метилацетаміду (Сполука (I)) (5,62 г при 89 мас. %, 10,55 ммоль) в ізопропанолі (100 мл), підтримуючи температуру >65 °C. Суміш нагрівали в колбі зі зворотним холодильником протягом однієї години перед освітлінням. Реакційну суміш охолоджували до 30 °C протягом 90 хвилин і витримували протягом 30 хвилин для встановлення кристалізації. Реакцію охолоджували до 0 °C протягом 2 годин і витримували протягом 1 години перед виділенням шляхом фільтрації. Осад на фільтрі два рази промивали за допомогою холодного ізопропанолу (2 × 10 мл) і висушували у вакуумі при 50 °C, одержуючи вказану в заголовку сполуку у вигляді білої твердої 1 речовини (5,84 г, 78 %); H ЯМР-спектр: (ДМСО) 1,85 (m, 1H), 2,08 (m, 1H), 2,50 (m, 1H), 2,66 (d, 3H), 2,83 (m, 1H), 3,05 (s, 2H), 3,96 (s, 3H), 4,58 (m, 1H), 6,64 (s, 4H), 7,23 (s, 1H), 7,28 (m, 1H), 7,46 (ddd, 1H), 7,55 (m, 1H), 7,70 (широкий q, 1H), 7,85 (s, 1H), 8,38 (s, 1H). Приклад 2 Одержання Форми A Дифумарату Сполуки (I): 2-[4-({4-[(3-хлор-2-фторфеніл)аміно]-7метоксихіназолін-6-іл}окси)піперидин-1-іл]-н-метилацетамід ди-[(2E)-бут-2-ендіоат] Форма A Розчин фумарової кислоти (1,4 кг, 12,1 моль) у метанолі (26,6 кг) додавали до суміші 2-[4({4-[(3-хлор-2-фторфеніл)аміно]-7-метоксихіназолін-6-іл}окси)піперидин-1-іл]-N-метилацетаміду (2,93 кг, 84,8 мас. %, 5,24 моль) в ізопропанолі (39 кг), підтримуючи температуру >65 °C. Завантажували лінійне промивання метанолом (3,6 кг). Суміш нагрівали в колбі зі зворотним холодильником протягом однієї години перед освітлінням, після цього здійснювали лінійне промивання метанолом (7 кг). Реакційну суміш дистилювали при атмосферному тиску для видалення 47 кг дистилятів. Додавали ізопропанол (15,8 кг і реакційну суміш дистилювали для видалення 15,6 кг дистилятів. При дистиляції відбувалася кристалізація. Додавали ізопропанол (21 кг) і реакцію охолоджували до 0 °C протягом 8 годин і витримували протягом 1 години перед виділенням шляхом фільтрації. Осад на фільтрі промивали холодним 50:50 ізопропанол:MeОН (4 кг), після цього холодним ізопропанолом (4 кг) і висушували у вакуумі при 50 °C, одержуючи 1 вказану в заголовку сполуку у вигляді білої твердої речовини (3,64 кг, 98 %); H ЯМР-спектр: (ДМСО) 1,85 (m, 1H), 2,08 (m, 1H), 2,50 (m, 1H), 2,66 (d, 3H), 2,83 (m, 1H), 3,05 (s, 2H), 3,96 (s, 3H), 4,58 (m, 1H), 6,64 (s, 4H), 7,23 (s, 1H), 7,28 (m, 1H), 7,46 (ddd, 1H), 7,55 (m, 1H), 7,70 (широкий q, 1H), 7,85 (s, 1H), 8,38 (s, 1H). Приклад 3 Одержання Форми A Дифумарату Сполуки (I): 2-[4-({4-[(3-хлор-2-фторфеніл)аміно]-7метоксихіназолін-6-іл}окси)піперидин-1-іл]-н-метилацетамід ди-[(2E)-бут-2-ендіоат] Форма A 2-[4-({4-[(3-Хлор-2-фторфеніл)аміно]-7-метоксихіназолін-6-іл}окси)піперидин-1-іл]-Nметилацетамід (Сполука (I)) (60,19 г при 88 мас. %, 111,8 ммоль) розчиняли в етилацетаті (1550 мл). Розчин освітлювали шляхом фільтрації й фільтр промивали етилацетатом (53 мл). Розчин охолоджували до 40 °C. Після цього додавали прояснений розчин фумарової кислоти (26,60 г, 257,0 ммоль) в ізопропанолі (408 мл) протягом 1 години. Фільтр, використовуваний для посвітління розчину фумарової кислоти, потім промивали за допомогою ізопропанолу (37 мл). Після витримування протягом 1 години при 40 °C реакцію охолоджували до 20 °C протягом 1 години. Реакційну суміш витримували протягом 13,5 годин перед виділенням продукту шляхом фільтрації. Осад на фільтрі два рази промивали за допомогою етилацетату (82 мл): ізопропанолу (24 мл) і потім висушували у вакуумі при 40 °C, одержуючи вказану в заголовку 1 сполуку у вигляді білої твердої речовини (72,32 г, 90 %); H ЯМР-спектр: (ДМСО) 1,85 (m, 1H), 2,08 (m, 1H), 2,50 (m, 1H), 2,66 (d, 3H), 2,83 (m, 1H), 3,05 (s, 2H), 3,96 (s, 3H), 4,58 (m, 1H), 6,64 (s, 4H), 7,23 (s, 1H), 7,28 (m, 1H), 7,46 (ddd, 1H), 7,55 (m, 1H), 7,70 (широкий q, 1H), 7,85 (s, 1H), 8,38 (s, 1H). Приклад 4 Одержання Форми A Дифумарату Сполуки (I): 2-[4-({4-[(3-хлор-2-фторфеніл)аміно]-7метоксихіназолін-6-іл}окси)піперидин-1-іл]-н-метилацетамід ди-[(2E)-бут-2-ендіоат] Форма A 2-[4-({4-[(3-Хлор-2-фторфеніл)аміно]-7-метоксихіназолін-6-іл}окси)піперидин-1-іл]-Nметилацетамід (Сполука (I)) (2,75 г при приблизно 100 мас. %, 5,80 ммоль) розчиняли в етилацетаті (94 мл) і ізопропанолі (14 мл). Розчин дистилювали, таким чином, що збирали 25,2 мл дистилятів. Розчин охолоджували до 40 °C. Потім додавали прояснений розчин фумарової кислоти (1,38 г, 11,90 ммоль) в ізопропанолі (21 мл) протягом 1 години. Додавали затравку Форму A дифумарату Сполуки (I) (3,7 мг, 5,3 μ моль). Фільтр, використовуваний для посвітління 26 UA 103998 C2 5 10 15 20 25 30 35 40 розчину фумарової кислоти, потім промивали за допомогою ізопропанолу (2 мл). Після витримування протягом 1 години при 40 °C реакцію охолоджували до 20 °C протягом 2 годин. Реакційну суміш витримували протягом 15 годин перед виділенням продукту шляхом фільтрації. Осад на фільтрі два рази промивали за допомогою етилацетату (4,3 мл): ізопропанолу (1,2 мл) і потім висушували у вакуумі при 40 °C, одержуючи вказану в заголовку 1 сполуку у вигляді білої твердої речовини (72,32 г, 90 %); H ЯМР-спектр: (ДМСО) 1,85 (m, 1H), 2,08 (m, 1H), 2,50 (m, 1H), 2,66 (d, 3H), 2,83 (m, 1H), 3,05 (s, 2H), 3,96 (s, 3H), 4,58 (m, 1H), 6,64 (s, 4H), 7,23 (s, 1H), 7,28 (m, 1H), 7,46 (ddd, 1H), 7,55 (m, 1H), 7,70 (широкий q, 1H), 7,85 (s, 1H), 8,38 (s, 1H). Приклад 5 Одержання Форми A Дифумарату Сполуки (I): 2-[4-({4-[(3-хлор-2-фторфеніл)аміно]-7метоксихіназолін-6-іл}окси)піперидин-1-іл]-н-метилацетамід ди-[(2E)-бут-2-ендіоат] Форма A 2-[4-({4-[(3-Хлор-2-фторфеніл)аміно]-7-метоксихіназолін-6-іл}окси)піперидин-1-іл]-Nметилацетамід (Сполука (I)) (1 г, 1,86 ммоль) і фумарову кислоту (0,44 г, 3,81 ммоль) суспендували у воді (4,4 г) і нагрівали до 85 °C. Реакційну суміш охолоджували до 60 °C зі швидкістю 1 °C/хвилину й додавали затравку Форму A Сполуки (I), коли температура була 77 °C. Отриману тверду речовину виділяли шляхом фільтрації, два рази промивали ацетоном (0,70 г на промивання) і висушували у вакуумній шафі при 40 °C, одержуючи вказану в заголовку сполуку (0,89 г, вихід 68 %), 1H ЯМР (400 МГц, ДМСО-d6) d част. на млн 1,84 (m, 2 H) 2,08 (m, 2 H) 2,55 (m, 2 H) 2,63 (d, J=4,7 Гц, 3 H) 2,86 (m, 2 H) 3,12 (s, 2 H) 3,93 (s, 3 H) 4,59 (tt, J=7,8, 3,7 Гц, 1 H) 6,62 (s, 4 H) 7,21 (s, 1 H) 7,27 (td, J=8,1, 1,3 Гц, 1 H) 7,49 (m, 2 H) 7,86 (m, 2 H) 8,36 (s, 1 H) 9,63 (br. s., 1 H). Властивості Форми A Дифумарату Сполуки (I) Форма A дифумарату Сполуки (I) являє собою рідкотекучий порошок. Порошкова рентгенограма дифумарату Сполуки (I) (Фігура 4) вказує на те, що матеріал є кристалічним. Найбільш значимі піки з картини XRPD Форми A описані в даній заявці вище й перераховано в Таблиці 1. При диференціальній сканувальній калориметрії представлена єдина ендотерма плавлення з початком при 210,4 °C (Фігура 5). При термогравіметричному аналізі не було виявлено вимірюваних втрат ваги (Фігура 6). При сорбції динамічного випаровування було встановлено, що сполука негігроскопічно абсорбує 90 % вивільнення через 45 хвилин (Таблиця 7), що вказує на придатність цих дозованих форм для застосування в PK дослідженнях на собаках. Таблиця 7 % розчинення Сполуки (I) і Форми A Дифумарату Сполуки (I) у цитратному буфері рН 4,5. Сполука Сполука (I) Форма A дифумарату Сполуки (I) 25 % розчинення через 45 хвилин 93,6 100,3 Характеристики композицій у вигляді таблеток, які перорально вводили собакам, порівнювали із внутрішньовенним введенням у дозі 20 мг сполуки (I), що містить розчиненого 4 -1 мг.мл і доведеного до об'єму за допомогою 25 мас./ об. % гідроксипропіл-бета-циклодекстрину (Hp-beta-cd) у воді для ін'єкцій рН доводили до 4 за допомогою 1M HСl. Результати дослідження на собаках представлено в таблиці 8. 30 Таблиця 8 Результати фармакокінетичних параметрів для Сполуки (I) і Форми A дифумарату Сполуки (I), які вводили перорально, і Сполуки (I), яку вводили внутрішньовенно самцям собак породи бігль (n=4, середнє значення СКВ) Параметр Cmax (мкмоль/л) Tmax (год.) Період напіввиведення (год.) AUC(0-48) (мкмоль.год./л) Cl (мл/хв/кг) Hbf (мл/хв) Vss (л/кг) Біодоступність (0-48ч) (%) Дослідження 0383KD - Препарат сполуки (I) /Шлях (Доза) 1 Дифумарат Форма A 1 ВВ (20 мг) Вільна форма (100 мг) (еквівалентно до 100 мг вільної форми) 9,0±1,1 14,0±1,7 32,6±3,6 0,2±0,0 1,6±0,8 1,1±0,3 5,3±0,2 8,2±0,6 16,9±4,0 33,4±7,7 1,9±0,5 39 0,6±0,1 103,9±25,3 63,5±7,0 190,3±46,9 112,5±8,1 1 дозовані у вигляді таблеток прямого пресування; Cmax = Пік концентрації в плазмі 28
ДивитисяДодаткова інформація
Назва патенту англійськоюFumarate salt of 4-(3-chloro-2-fluoroanilino)-7-methoxy-6-{[1- (n-methylcarbamoylmethyl)piperidin-4-yl]oxy}quinazoline
Автори російськоюBoardman, Kay, Alison, Burns, Susan, Elizabeth, Dobson, Andrew, Hornby, Whitlock, Brian
МПК / Мітки
МПК: A61K 31/517, A61P 35/00
Мітки: дифумаратна, сіль, 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(n-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну
Код посилання
<a href="https://ua.patents.su/52-103998-difumaratna-sil-4-3-khlor-2-ftoranilino-7-metoksi-6-1-n-metilkarbamolmetilpiperidin-4-iloksikhinazolinu.html" target="_blank" rel="follow" title="База патентів України">Дифумаратна сіль 4-(3-хлор-2-фтораніліно)-7-метокси-6-{[1-(n-метилкарбамоїлметил)піперидин-4-іл]окси}хіназоліну</a>
Кристалічна малеатна сіль хіназоліну (варіанти), корисна як антиангіогенний агент

Номер патенту: 91332
Опубліковано: 26.07.2010
Автор: Маккейб Джеймс
МПК: C07D 403/12, A61P 35/00, A61K 31/505
Мітки: агент, корисна, кристалічна, хіназоліну, варіанти, малеатна, сіль, антиангіогенний
Формула / Реферат:
1. Малеатна сіль 4-((4-фтор-2-метил-1Н-індол-5-іл)окси)-6-метокси-7-(3-(піролідин-1-іл)пропокси)хіназоліну у вигляді кристалічної форми (Форма А), де згадана сіль має рентгенограму на порошку з принаймні одним окремим піком при 2-тета приблизно 21,5° або при 2-тета приблизно 16,4°.2. Малеатна сіль 4-((4-фтор-2-метил-1Н-індол-5-іл)окси)-6-метокси-7-(3-(піролідин-1-іл)пропокси)хіназоліну у вигляді кристалічної форми (Форма А) за п. 1,...
Кристалічний моногідрохлорид 2,5-діон-3-[(1-метил-1н-індол-3-іл)-4-[1-(піперидин-2-ілметил)піперидин-4-іл]-1н-індол-3-іл]-1н-піролу

Номер патенту: 78801
Опубліковано: 25.04.2007
Автори: Фоул Маргарет Мері, Рутцель-Іденс Сюзан Марія, Буш Джулі Кей
МПК: C07D 401/14
Мітки: 2,5-діон-3-[(1-метил-1н-індол-3-іл)-4-[1-(піперидин-2-ілметил)піперидин-4-іл]-1н-індол-3-іл]-1н-піролу, кристалічний, моногідрохлорид
Формула / Реферат:
1. Кристалічний моногідрохлорид 2,5-діон-3-[(1-метил-1Н-індол-3-іл)-4-[1-(тридин-2-ілметил)піперидин-4-іл]-1Н-індол-3-іл]-1Н-піролу, його гідрат або його суміші.2. Кристалічний моногідрохлорид 2,5-діон-3-[(1-метил-1Н-індол-3-іл)-4-[1-(піридин-2-ілметил)піперидин-4-іл]-1Н-індол-3-іл]-1Н-піролу, його гідрат або його суміші, порошкова рентгенограма якого включає такі піки: 6,8±0,1°, 10,9±0,1°, 14,2±0,1° і 1б,6±0,1° при
Сукцинат біфеніл-2-ілкарбамінової кислоти 1-[2-(2-хлор-4-{[(r)-2-гідрокси-2-(8-гідроксі-2-оксо-1,2-дигідрохінолін-5-іл)етиламіно]метил]}-5-метоксифенілкарбамоїл)етил]піперидин-4-іл-естеру та його застосування д

Номер патенту: 100364
Опубліковано: 25.12.2012
Автори: Чудасама Решма, Кіндон Лінда Джейн, Мале Франк Патрік, Кеннеді Ендрю
МПК: C07D 401/12, A61K 31/4709
Мітки: біфеніл-2-ілкарбамінової, кислоти, сукцинат, 1-[2-(2-хлор-4-{[(r)-2-гідрокси-2-(8-гідроксі-2-оксо-1,2-дигідрохінолін-5-іл)етиламіно]метил]}-5-метоксифенілкарбамоїл)етил]піперидин-4-іл-естеру, застосування
Формула / Реферат:
1. Сукцинат біфеніл-2-ілкарбамінової кислоти 1-[2-(2-хлор-4-{[(R)-2-гідрокси-2-(8-гідроксі-2-оксо-1,2-дигідрохінолін-5-іл)етиламіно]метил}-5-метоксифенілкарбамоїл)етил]піперидин-4-іл-естеру або його сольват, який є у кристалічній формі 1, що характеризується рентгенодифрактограмою порошку, що має піки дифракції при значеннях 2θ 5,0±0,3 та 10,0±0,3.2. Сіль за п. 1, де сіль характеризується: а) даними диференційної...
Натрієва сіль 2-гідрокси-3-(8-метокси-2-метилхінолін-4-ілтіо)пропанової кислоти, що має антиоксидантну, нейропротекторну активність

Номер патенту: 49819
Опубліковано: 11.05.2010
Автори: Бражко Олександр Анатолійович, Омельянчик Людмила Олександрівна, Генчева Вікторія Іванівна, Завгородній Михайло Петрович
МПК: C07D 215/00
Мітки: активність, сіль, нейропротекторну, антиоксидантну, натрієва, 2-гідрокси-3-(8-метокси-2-метилхінолін-4-ілтіо)пропанової, кислоти, має
Формула / Реферат:
Натрієва сіль 2-гідрокси-3-(8-метокси-2-метилхінолін-4-ілтіо)пропанової кислоти формули:,що має антиоксидантну, нейропротекторну активність.
D-(+)-глюкозиламонієва сіль 4-хлор-n-(4′-карбоксифеніл) антранілової кислоти, яка виявляє протизапальну, анальгетичну активність

Номер патенту: 55973
Опубліковано: 15.04.2003
Автори: Брунь Лідія Володимирівна, Яковлева Лариса Василівна, Павлій Олег Олександрович, Зупанець Ігор Альбертович, Ісаєв Сергій Григорович
МПК: A61K 31/196, A61K 31/7004, A61P 29/00, C07C 229/58
Мітки: яка, антранілової, анальгетичну, активність, 4-хлор-n-(4'-карбоксифеніл, кислоти, протизапальну, d-(+)-глюкозиламонієва, сіль, виявляє
Формула / Реферат:
D-(+)-глюкозиламонієва сіль 4-хлор-N-(4’-карбоксифеніл) антранілової кислоти загальної формули,яка виявляє протизапальну, анальгетичну активність.
Попередній патент: Спосіб стереоселективного ферментативного гідролізу естеру 5-метил-3-нітрометилгексанової кислоти
Наступний патент: Діоксид титану з покриттям та його застосування в композиціях
Випадковий патент: Спосіб вмикання електричних машин змінного струму в мережу