Спосіб гідразинолізу нітрогуанідину
Номер патенту: 73053
Опубліковано: 16.05.2005
Автори: Панасюк Олександр Григорович, Шестозуб Анатолій Борисович


Формула / Реферат
1. Спосіб гідразинолізу нітрогуанідину у середовищі спирту (етилового або метилового), який включає взаємодію нітрогуанідину з еквімолярною кількістю гідразингідрату при температурі кипіння впродовж 75÷80 хвилин з наступним охолодженням до кімнатної температури або при кімнатній температурі протягом 6 діб в інертній атмосфері, з виділенням біс(аміногуанідиній)-1,6-динітробігуанідину (І) або біс(аміногуанідиній)-1,6-динітро-2(аміногуаніл)бігуанідину (II) фільтруванням, витримку фільтрату протягом 12 годин при температурі 0 - 20°С та його фільтрування з виділенням іншої сполуки (І чи ІІ), розведення спиртових фільтратів водою та підкислення концентрованою азотною кислотою, їх взаємодію з надлишком бензальдегіду протягом 1 години, витримку при 0°С протягом декількох годин, потім відфільтровування утвореної суміші бензальазину та дибензальдиаміногуанідину нітрату (III), виділення бензальазину із суміші ефіром, наступну перекристалізацію (III), впарювання водноспиртових маточників під вакуумом на ¾ об’єму, охолодження залишку, осадження бензальаміногуанідиній нітрату (IV), який відрізняється тим, що гідразиноліз нітрогуанідину проводять у середовищі спирту ROH (R = n-С3Н7, і-С3H7) з концентрацією води в спирті 5,00 - 0,01 % тривалість процесу при кипінні розчину із зворотнім охолодженням становить 70 - 150 хвилин, а тривалість процесу при кімнатній температурі - 7200 - 14400 хвилин в атмосфері повітря або інертного газу при стехіометричному співвідношенні ν(G)/ ν(HГ) = 0,95 - 1,15, переробку спиртових фільтратів після виділення сполук (І) та (II) проводять наступним чином: спиртовий фільтрат за необхідності впарюють під вакуумом, залишок обробляють кислотою НХ при температурі -20÷50°С на повітрі або в інертному середовищі в стехіометричному співвідношенні ν(HX)/ ν(HT) = 0,5÷0,8, можливо виділяючи при цьому NH4X, потім виділяють суміш солей АГ·Х та ДАГ·НХ у вигляді концентрованих водних, водноспиртових розчинів або в твердому стані або обробляють залишок кислотою при температурі -10÷30°С в стехіометричному співвідношенні ν(HAn)/ ν(HГ) = 0,5÷0,8, з наступною обробкою отриманої суміші солей стехіометричною кількістю солі MX за схемою:
АГ·НАn+МХ↔АГ·НХ+МАn
ДАГ·НАn + MX↔ДАГ·НХ + МАn,
де НАn = НСl, HBr, HClO4, НNO3, 1/2H2SO4;
М = Ag+, Na+, K+, NH4+, Li+, 1/2Рb+, l/2Ca++, l/2Ba++, та виділенням суміші солей АГ·НХ та ДАГ·НХ, як вказано вище;
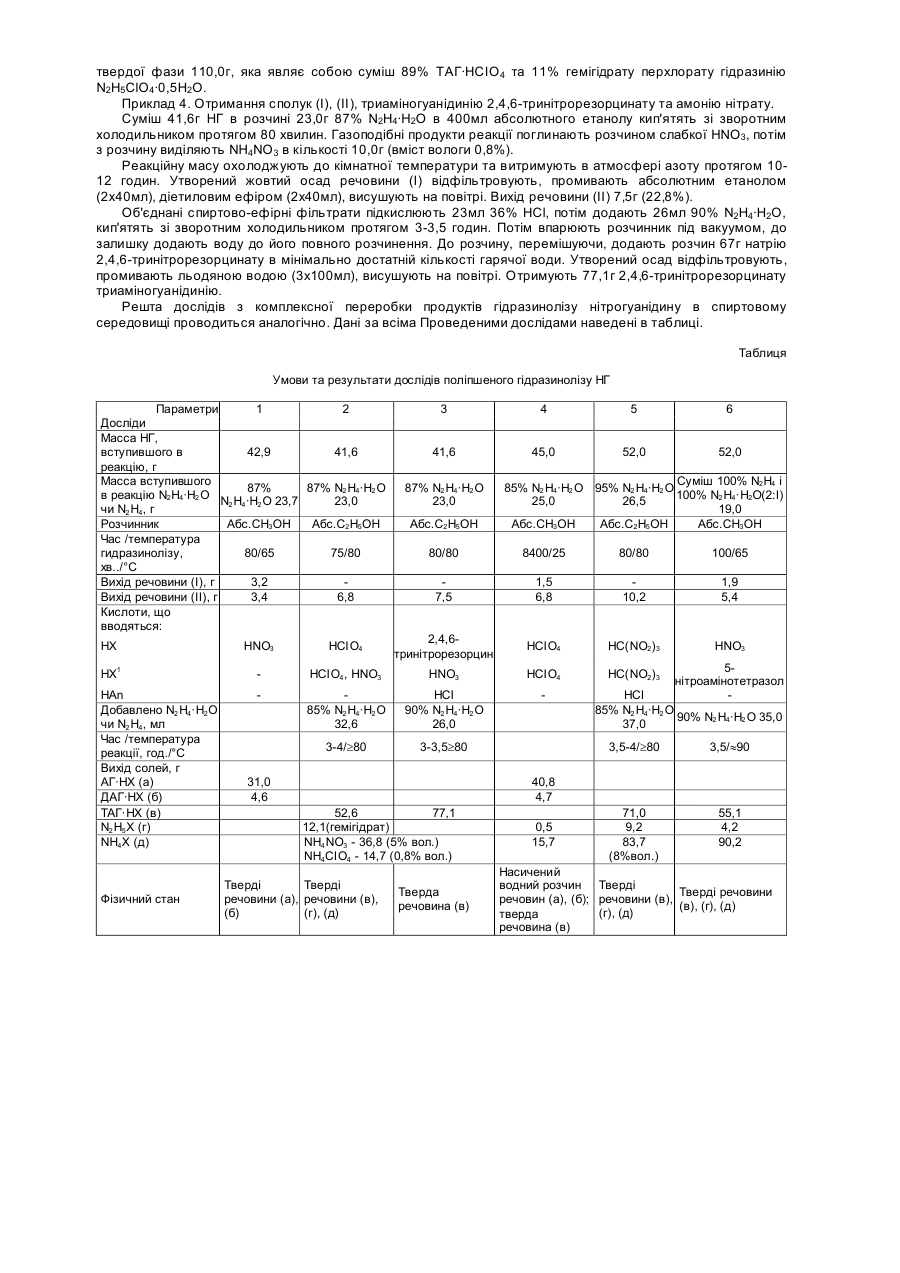
НХ = HClO4, НNО3, НС(NO2)3, HN(NO2)2, НN3,
 ,
, 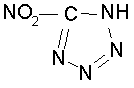 ,
,  ;
;
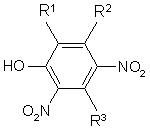
R1=Η, NO2, NH2, R2=R3 = HO, H, СН3, СН3О.
2. Спосіб за п. 1, який відрізняється тим, що як G, крім гідразингідрату N2H4·Н2О, застосовують безводний гідразин N2H4 або їх суміш, причому ν(N2H4·H2O)+ ν(N2H4)= ν(G).
3. Спосіб за п. 1, який відрізняється тим, що гази, які утворюються при проведенні гідразинолізу нітрогуанідину, пропускають крізь розчин кислоти НХ1, отримуючи при цьому солі NH4X1, причому
НХ1 = НСlO4, HNO3, HC(NO2)3, HN(NO2)2,
 ,
, 
4. Спосіб за п. 1, який відрізняється тим, що розчин суміші солей АГ·НХ та ДАГ·НХ піддають взаємодії з реагентом G в стехіометричному співвідношенні ν(G)/ ν(HT) = 1,0÷1,5 при температурі 60-100°С протягом 3-5 годин, потім реакційну масу, за необхідності, впарюють повністю або частково під вакуумом та виділяють суміш солей N2H5X та [C(NHNH2)3]X, молярна концентрація останньої в суміші становить 80-97 %.
5. Спосіб за п. 4, який відрізняється тим, що в разі наявності суміші солей АГ·НАn та ДАГ·НАn у розчині реакційну масу після взаємодії з реагентом G вводять у взаємодію зі стехіометричною кількістю солі MX, видаляючи побічні сполуки МАn відомими способами.
6. Спосіб за п. 4, який відрізняється тим, що гази, які утворюються при взаємодії розчинів суміші солей АГ·НХ та ДАГ·НХ або АГ·НАn та ДАГ·НАn з реагентом G, абсорбують розчином кислоти НХ1, виділяючи солі ΝΗ4Χ1.
Текст
Винахід стосується області загальної та органічної хімії, зокрема, хімії похідних гуанідину. Похідні гуанідину та інші енерговмістні речовини, одержані за даним винаходом, можуть бути використані як індивідуальні вибухові речовини (ВР), можливі компоненти промислових ВР, в тому числі водовмісних (ВВР) та емульсійних (ЕВР), а також як компоненти деяких сумішевих твердих ракетних палив (ТРП) чи унітарних рідких ракетних палив (РРП). Найбільш близьким за технічною суттю та результатом, що досягається, є спосіб гідразинолізу нітрогуанідину (НГ) у середовищі спирту (етилового або метилового) взаємодією НГ з еквімолярною кількістю гідразин-гідрату при температурі кипіння впродовж 75¸80 хвилин з наступним охолодженням до кімнатної температури або при кімнатній температурі протягом 6 діб в інертній атмосфері, з виділенням біс(аміногуанідиній)-1,6-динітро-бігуанідину (І) або біс(аміногуанідиній)-1,6-динітро-2(аміногуаніл)бігуанідину (ІІ) фільтруванням, витримку фільтрату протягом 12 годин при температурі 0¸20°С та його фільтрування з виділенням іншої сполуки (І чи II), розведення спиртових фільтратів водою та підкислення концентрованою азотною кислотою, їх взаємодію з надлишком бензальдегіду протягом 1 години, витримку при 0°С протягом декількох годин, відфільтровування утвореної суміші бензальазину та дибензальдиаміногуанідину нітрату (ІІІ), виділення бензальазину із суміші ефіром, наступну перекристалізацію (ІІІ), впарювання водноспиртових маточників під вакуумом на ¾ об'єму, охолодження залишку, осадження бензальаміногуанідиній нітрату (IV) з утворенням 1,6-динітробігуанідину (І) та 1,6-динітро2(аміногуаніл)бігуанідину (ІІ) у вигляді їх аміногуанідиніевих солей [R.A.Henry, S.Skolmk, G.B.L. Smith. The hydrazinolysis of nitroguanidine in alcoholic systems. Preparation and reactions of 1,6-dinitrobiguanidsne / JACS. 1953. - P.955-962]. Сполуки (І) та (II) можна використати як напівпродукти виробництва компонентів твердого ракетного палива (ТРП), бризантних та ініціюючих вибухових речовин (БВР та ІВР). Недоліком способу є те, що вихід високоенергетичних речовин (І) та (II) невисокий, значна частина нітрогуанідину (41,8¸62,2%) переходить в аміногуанідин, 8-12% - в диаміногуанідин для розділення яких автори осаджували кожну органічну основу в вигляді нітратів бензіліденових похідних послідовною взаємодією з бензальдегідом та азотною кислотою. Селективне виділення отриманих нітратів гідразонів достатньо важка процедура, що також є недоліком вказаного способу. Крім цього, отримані нітрати гідразонів є мало перспективними з точки зору їх енергоємності. Задача винаходу - вдосконалення способу гідразинолізу нітрогуанідину в спиртовому середовищі так, щоб усі продукти гідразинолізу НГ та побічні продукти могли бути застосовані в якості високоенергетичних речовин як компоненти твердих або унітарних ракетних палив, БВР чи ІВР. Поставлена задача вирішується тим, що в відомому способі гідразинолізу нітрогуанідину (НГ) у середовищі спирту (етилового або метилового) взаємодією НГ з еквімолярною кількістю гідразин-гідрату при температурі кипіння впродовж 75¸80 хвилин з наступним охолодженням до кімнатної температури або при кімнатній температурі протягом 6 діб в інертній атмосфері, з виділенням біс(аміногуанідиній)-1,6динітро-бігуанідину (І) або біс(аміногуанідиній)-1,6-динітро-2(аміногуаніл)бігуанідину (II) фільтруванням, витримку фільтрату протягом 12 годин при температурі 0¸20°С та його фільтрування з виділенням іншої сполуки (І чи ІІ), розведення спиртових фільтратів водою та підкислення концентрованою азотною кислотою, їх взаємодію з надлишком бензальдегіду протягом 1 години, витримку при 0°С протягом декількох годин, відфільтровування утвореної суміші бензальазину та дибензальдиаміногуанідину нітрату (ІІІ), виділення бензальазииу із суміші ефіром, наступну перекристалізацію (III), впарювання водноспиртових маточників під вакуумом на ¾ об'єму, охолодження задишку, осадження бензальаміногуанідиній нітрату (IV), згідно винаходу, відрізняється тим, що гідразиноліз НГ проводять також у середовищі спирту ROH (R=n-С3H7, і-С3 Н7) з концентрацією вода в спирті 5,00¸0,01%, тривалість процесу при кипінні розчину із зворотним холодильником дорівнює 70¸150 хвилин, а тривалість процесу при кімнатній температурі дорівнює 7200-14400 хвилин в атмосфері повітря або інертного газу при стехіометричному співвідношенні v(G)/n(HГ)=0,95¸1,15, переробку спиртових фільтратів після виділення сполук (І) та (ІІ) проводять наступним чином: Спиртовий фільтрат за необхідності впарюють під вакуумом, залишок обробляють кислотою НХ при температурі -20¸50°С на повітрі або в інертному середовищі в стехіометричному співвідношенні n(ΗΧ)/n(ΗΓ)=0,5¸0,8, можливо виділяючи при цьому NН4X, потім виділяють суміш солей АГ·НХ та ДАГ·НХ у вигляді концентрованих водних, водноспиртових розчинів або в твердому стані, або обробляють залишок кислотою при температурі -10¸30°С в стехіометричному співвідношенні n(ΗΑn)/n(ΗΓ)=0,5¸0,8, з наступною обробкою отриманої суміші солей стехіометричною кількістю солі MX за схемою: АГ·НАn+MX«АГ·НХ+МАn, ДАГ·НАn+MX«ДАГ·НХ+МАn, де НАn=НСI, НВr, НСlО4, HNO3,1/2H2SO4; Μ=Ag+, Na+, К+, NН4+, Li+, 1/2Pb++, 1/2Ca++, 1/2Ва++, та виділенням суміші солей АГ·НХ та ДАГ·НХ, як вказано вище; При цьому в якості G, крім гідразингідрату N2 H4·Н2О, застосовують безводний гідразин N2H4 або їх суміш, причому n(Ν2Η4·Η2Ο)+n(N2H4)=n(G), a гази, котрі утворюються при проведенні гідразинолізу НГ, за необхідності, пропускають крізь розчин кислоти НХ1, отримуючи при цьому солі NH4X1, розчинуміші солей АГ·НХ та ДАГ·НХ чи ΑΓ·ΗΑn та ДАГ·HAn піддають взаємодії з реагентом G в стехіометричному співвідношенні n(G)/n(HГ)=1,0¸1,5 при температурі 60¸100°С протягом 3¸5 годин. В разі наявності суміші солей АГ·НАn та ДАГ·HAn в розчині реакційну масу вводять зі стехіометричною кількістю солі MX. Потім реакційну масу, за необхідності, впарюють повністю або частково під вакуумом та виділяють суміш солей N2 H5X та [C(NHNH2)3 ]X, молярна концентрація останньої в суміші становить 80¸97%. Гази, які утворюються при взаємодії реагента G з сумішшю солей АГ·НХ та ДАГ·НХ чи ΑΓ·ΗΑn та ДАГ·HAn, за необхідності, абсорбують розчином кислоти НХ1, виділяючи солі ΝΗ4Χ1· Істотною відмінністю винаходу в порівнянні з прототипом є: - значне розширення технології переробки побічних продуктів гідразинолізу НГ. Заміна складної технології селективного виділення АГ та ДАГ у вигляді їх бензіліденових похідних нейтралізацією суміші основ кислотами з можливим наступним гідразинолізом суміші солей та виділенням солей триаміногуанідину; - трансформація побічних продуктів гідразинолізу НГ в енергоємні солі складу Β·ΗΧ(Β=АГ, ДАГ, ТАГ); - розширення часу процесу гідразинолізу НГ та вибору розчинників, що веде до розширення можливостей способу. Приклад 1. Отримання ди(аміногуанідиній)-1,6-динітробігуанідину (І), ди(аміногуанідиній)-1,6-динітро-2(аміногуаніл)-бігуанідину (II), бензальаміногуанідиній нітрату (III) та дибензальдиаміногуанідиній нітрату (VI) (прототип). До розчину 23,7г 87% гідразин-гідрату в 400мл абсолютного метанолу прибавляють 42,9г нітрогуанідину. Отриману суміш кип'ятять зі зворотним холодильником протягом 80 хвилин, потім швидко охолоджують до кімнатної температури. Яскраво-жовтий осад, що утворився, відфільтровують, промивають диетиловим ефіром (3х50мл), висушують на повітрі. Це речовина (І), вихід 3,4г (9,3%) з температурою плавлення 166-167°С (з розкл.). Знайдено, %: С 13,71; Η 5,00; Ν 62,65 та 62,90. Для 2CH6N4·C2H6N8O4 обчислено, %: С 13,56; Η 5,12; Ν 63,26. Фільтрати витримують при температурі нижче нуля (за Цельсієм) протягом 12 годин та виділяють 3,2г (9,8%) речовини (II) з температурою правлення 145-147°С (з розкл.). (Перекристалізація з метанолу не призводить до зміни Тпл). Знайдено, %: С 15,17; Η 5,18 та 5,13; Ν 64,36. Для C3H8 N10О4·2CH6N4 обчислено, %: С 15,15; Η 5,09; Ν 63,62. Об'єднані спиртові розчини розводять 400мл води та підкислюють 40мл концентрованої азотної кислоти, після чого добавляють 40г С6Н5СНО та перемішують протягом 1 години, потім охолоджують до 0°С та витримують кілька годин. Осад, що випав, відфільтровують, промивають ефіром для видалення бензальазину та надлишку бензальдегіду, потім промивають водою (видалення кислоти). Вихід речовини (IV) 9,9г (7,3%), Тпл=158-163°С. Після перекристалізації з 95% етанолу отримана речовина з Тпл=195°С. Концентрування водно-спиртових фільтратів на роторному впарювачеві під вакуумом до об'єму 200мл та послідуюче охолодження призводить до утворення осаду речовини (III), вихід 51,0г (56,7%), Тпл=160-161°С. Приклад 2. Отримання сполук (І), (II), а також аміногуанідинію нітрату та диаміногуанідинію нітрату. До розчину 23,7г 87% гідразин-гідрату в 400мл абсолютного метанолу прибавляють 42,9г нітрогуанідину. Отриману суміш кип'ятять зі зворотним холодильником протягом 80 хвилин, потім швидко охолоджують до кімнатної температури. Яскраво-жовтий осад, що утворився, відфільтровують, промивають діетиловим ефіром (3х50мл), висушують на повітрі. Це речовина (І), вихід 3,4г (9,3%) з температурою плавлення 166-167°С (з розкл.). Знайдено, %: С 13,71; Η 5,00; Ν 62,65 та 62,90. Для 2СН6N4·C2H6N8O4 обчислено, %: С 13,56; Η 5,12; Ν 63,26. Фільтрати витримують при температурі нижче нуля (за Цельсієм) протягом 12 годин та виділяють 3,2г (9,8%) речовини (II) з температурою плавлення 145-147°С (з розкл.). (Перекристалізація з метанолу не призводить до зміни Тпл). Знайдено, %: С 15,17; Η 5,18 та 5,13; N 64,36. Для C3H8 N10O4·2CH6N4 обчислено, %: С 15,15; Η 5,09; Ν 63,62. Об'єднані спиртові розчини впарюють під вакуумом на роторному впарювачеві до об'єму 30¸40мл, після чого нейтралізують 23мл 53% ΗΝΟ3. Отриманий розчин висушують під вакуумом, отримують 35,6г суміші солей (87% АГ·НNO3 та 13% ДАГ·НNO3). Приклад 3. Отримання сполук (І), (II), а також триаміногуанідинію перхлорату, гідразинію перхлорату та амонію перхлорату. Суспензію 41,6г НГ в розчині 23,0г 87% Ν2Η4·Н2О в 400мл абсолютного етанолу кип'ятять зі зворотним холодильником протягом 75 хвилин. Аміак, що виділяється, барботують крізь охолоджену до -5¸0°С 70% НСIO4, при цьому утворюється 14,1г NH4CIO4, який відфільтровують, промивають льодяною водою до рН=7. Після закінчення реакції реакційну масу охолоджують до кімнатної температури та витримують без доступу повітря протягом 12 годин. Утворений жовтий осад відфільтровують, промивають абсолютним етанолом (2x50мл), діетиловим ефіром (2x50мл), висушують на повітрі. Вихід речовини (ll) 6,8г (20,7%) з температурою плавлення 144-147°С (з розкл.). Об'єднані спиртово-ефірні фільтрати підкислюють 49мл 69% НСIO4. Випавший осад (0,5г) NH4CIO4 відфільтровують, промивають спиртом до рН=7, висушують. До спиртово-ефірного фільтрату добавляють 32,6мл 85% N2H4·Н2О, реакційну масу кип'ятять зі зворотним холодильником протягом 3-4 годин. і Газоподібні продукти, що виділялись, барботують крізь водний розчин HNO3, потім з розчину виділяють 36,8г NH4NO3, який містить 5% води. Потім впарюють розчинник під вакуумом, висушують залишок. Вихід твердої фази 110,0г, яка являє собою суміш 89% ТАГ·НСIО4 та 11% гемігідрату перхлорату гідразинію N2H5ClO4·0,5H2O. Приклад 4. Отримання сполук (І), (II), триаміногуанідинію 2,4,6-тринітрорезорцинату та амонію нітрату. Суміш 41,6г НГ в розчині 23,0г 87% N2H4·Н2О в 400мл абсолютного етанолу кип'ятять зі зворотним холодильником протягом 80 хвилин. Газоподібні продукти реакції поглинають розчином слабкої HNO3, потім з розчину виділяють NH4NO3 в кількості 10,0г (вміст вологи 0,8%). Реакційну масу охолоджують до кімнатної температури та витримують в атмосфері азоту протягом 1012 годин. Утворений жовтий осад речовини (І) відфільтровують, промивають абсолютним етанолом (2x40мл), діетиловим ефіром (2x40мл), висушують на повітрі. Вихід речовини (II) 7,5г (22,8%). Об'єднані спиртово-ефірні фільтрати підкислюють 23мл 36% НСl, потім додають 26мл 90% Ν2Η4·Н2О, кип'ятять зі зворотним холодильником протягом 3-3,5 годин. Потім впарюють розчинник під вакуумом, до залишку додають воду до його повного розчинення. До розчину, перемішуючи, додають розчин 67г натрію 2,4,6-тринітрорезорцинату в мінімально достатній кількості гарячої води. Утворений осад відфільтровують, промивають льодяною водою (3х100мл), висушують на повітрі. Отримують 77,1г 2,4,6-тринітрорезорцинату триаміногуанідинію. Решта дослідів з комплексної переробки продуктів гідразинолізу нітрогуанідину в спиртовому середовищі проводиться аналогічно. Дані за всіма Проведеними дослідами наведені в таблиці. Таблиця Умови та результати дослідів поліпшеного гідразинолізу НГ Параметри 1 2 Досліди Масса НГ, вступившого в 42,9 41,6 реакцію, г Масса вступившого 87% 87% Ν2 Η4·Η2 Ο в реакцію N2H4·Н2 О Ν2 Η4·Η2 Ο 23,7 23,0 чи N2 H4, г Розчинник Абс.СН3ОН Абс.С2Н5ОН Час /температура гидразинолізу, 80/65 75/80 хв../°С Вихід речовини (І), г 3,2 Вихід речовини (ІІ), г 3,4 6,8 Кислоти, що вводяться: 3 4 5 6 41,6 45,0 52,0 52,0 87% Ν2 Η4·Η2 Ο 23,0 Абс.С2Н5ОН Суміш 100% Ν2Η4 і 85% Ν2 Η4·Η2 Ο 95% Ν2 Η4·Η2 Ο 100% Ν2 Η4·Η2Ο(2:Ι) 25,0 26,5 19,0 Абс.СН3ОН Абс.С2Н5ОН Абс.СН3ОН 80/80 8400/25 80/80 100/65 7,5 1,5 6,8 10,2 1,9 5,4 ΗΝΟ3 НХ ΗΝΟ3 НСIO4 2,4,6тринітрорезорцин НСIO4 HC(NO2)3 НХ1 НСIO4, ΗΝΟ3 ΗΝΟ3 НСIO4 HC(NO2)3 НАn Добавлено N2 H4·H2О чи N2 H4, мл Час /температура реакції, год./°С Вихід солей, г АГ·НХ (а) ДАГ·НХ (б) ТАГ·НХ (в) N2 H5X (г) NH4Х (д) 85% Ν2 Η4·Η2 Ο 32,6 НСI 90% Ν2 Η4·Η2 Ο 26,0 3-4/³80 3-3,5³80 Фізичний стан 31,0 4,6 5нітроамінотетразол НСI 85% Ν2 Η4·Η2 Ο 90% Ν2 Η4·Η2 Ο 35,0 37,0 3,5-4/³80 3,5/»90 71,0 9,2 83,7 (8%вол.) 55,1 4,2 90,2 40,8 4,7 52,6 77,1 12,1(гемігідрат) NH4 NO3 - 36,8 (5% вол.) NH4CIO4 - 14,7 (0,8% вол.) Тверді Тверді речовини (а), речовини (в), (б) (г), (д) Тверда речовина (в) 0,5 15,7 Насичений водний розчин Тверді Тверді речовини речовин (а), (б); речовини (в), (в), (г), (д) (г), (д) тверда речовина (в)
ДивитисяДодаткова інформація
Назва патенту англійськоюA method for hydrazinolysis of nitroguanidine
Автори англійськоюPanasiuk Oleksandr Hryhorovych, Shestozub Anatolii Borysovych
Назва патенту російськоюСпособ гидразинолиза нитрогуанидина
Автори російськоюПанасюк Александр Григорьевич, Панасюк Олександр Григорович, Шестозуб Анатоллй Борисович
МПК / Мітки
МПК: C07C 279/00, C07C 279/02
Мітки: нітрогуанідину, спосіб, гідразинолізу
Код посилання
<a href="https://ua.patents.su/3-73053-sposib-gidrazinolizu-nitroguanidinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб гідразинолізу нітрогуанідину</a>
Спосіб одержання похідних заміщеного 2-нітрогуанідину

Номер патенту: 69387
Опубліковано: 15.09.2004
Автор: Майєнфіш Петер
МПК: C07D 261/10, C07D 413/14, C07D 401/14, C07D 417/14, C07D 277/32, C07D 405/14, C07D 261/08, C07D 213/89, C07D 277/20, C07D 277/28, C07D 251/08, C07D 213/40, C07D 213/61, C07D 307/14
Мітки: похідних, одержання, 2-нітрогуанідину, спосіб, заміщеного
Формула / Реферат:
1. Спосіб одержання заміщеного 2-нітрогуанідину формули, (I)та, у відповідних випадках, його E/Z-ізомерів, сумішей E/Z-ізомерів та/або таутомерів, відповідно, у вільній формі або у формі солей, де R1 є воднем або С1-С4-алкілом,R2 є воднем, С1-С6-алкілом, С3-С6-циклоалкілом або радикалом -СН2В, Неt є незаміщеним або - залежно від здатності...
Спосіб одержання похідних нітрогуанідину

Номер патенту: 64781
Опубліковано: 15.03.2004
Автори: Щепанскі Генрі, Рапольд Томас, Майєнфіш Петер
МПК: C07D 277/32, C07D 307/14, C07D 213/89, C07D 413/06, C07D 213/61
Мітки: спосіб, одержання, похідних, нітрогуанідину
Формула / Реферат:
1. Спосіб одержання сполуки формули Ів якій R1 являє собою водень або С1-С4-алкіл; R2 являє собою водень, С1-С6-алкіл, C2-C6-алкеніл, С2-С6-алкініл, С3-С6-циклоалкіл або радикал -СН2В;А являє собою незаміщений ароматичний або неароматичний, моноциклічний або біциклічний гетероциклічний радикал або, залежно від можливостей заміщення...
Азотнокислі й хлорнокислі солі похідних 1,2,4-триазолу та спосіб їх одержання

Номер патенту: 64855
Опубліковано: 15.03.2004
Автори: Шестозуб Анатолій Борисович, Панасюк Олександр Григорович
МПК: C07C 243/00, C07C 279/28, C07D 249/04, C07C 241/00
Мітки: спосіб, солі, азотнокислі, хлорнокислі, похідних, 1,2,4-триазолу, одержання
Формула / Реферат:
1. Хлорнокислі й азотнокислі солі гідразинопохідних 1,2,4-триазолу (s-триазолу), заміщених в положеннях 3, 4, 5, загальної формули:де R1=Н, СН3, С2Н5, NH2; R2=Н, CH3, NH2; R3=Н, NH2, NHNH2;Х=NO3-,ClO4-; n=1, 2.2. Спосіб одержання солей за п. 1, що включає гідразиноліз вихідної сполуки - ціангуанідину (субстрату), циклізацію інтермедіату при...
Спосіб отримання порошкового модифікатора іржі

Номер патенту: 64785
Опубліковано: 15.03.2004
Автор: Науменко Олександр Олександрович
МПК: C09D 197/00, C09D 05/02
Мітки: отримання, спосіб, модифікатора, іржі, порошкового
Формула / Реферат:
1. Спосіб отримання порошкового модифікатора іржі, що передбачає сушку гідролізного лігніну, подрібнення та модифікацію, який відрізняється тим, що у кульковому млині послідовно виконують подрібнення гідролізного лігніну протягом 5 годин, після чого здійснюють нейтралізацію залишкової сульфатної кислоти 25% водним розчином аміаку протягом 2 годин, потім додають 2% за масою моноетаноламіну і проводять модифікацію лігніну протягом 2...
Спосіб отримання твердих розчинів gete-cu2te

Номер патенту: 62892
Опубліковано: 15.12.2003
Автори: Борик Віктор Васильович, Фреїк Дмитро Михайлович, Іванишин Ірина Мирославівна, Шперун Володимир Михайлович
МПК: C30B 11/02
Мітки: отримання, gete-cu2te, твердих, спосіб, розчинів
Формула / Реферат:
Спосіб отримання твердих розчинів GeTe-Cu2Te, який полягає в тому, що вихідні речовини сплавляють у кварцових вакуумованих ампулах при температурі 1470 К протягом 5 годин з подальшим охолодженням на повітрі, після чого отриманий сплав піддають відпалу у атмосфері аргону при температурі 820 К протягом 600 год., який відрізняється тим, що як вихідну речовину використовують сплав (GeTe)1-x(Cu2Te)x складу х=0,025.
Попередній патент: Пристрій для поперечного накатування зубів зірочок
Наступний патент: Установка для одержання тепла
Випадковий патент: Установка для сепарації та сушіння дрібнозернистих сипучих матеріалів