Пептид, який має активність антагоніста бомбезину, фармацевтична композиція на його основі і спосіб лікування раку у ссавців








Текст
1. Пептид, имеющий формулу (1): D-Tpi-Gln^-Ala-Val-Gly-His-Leu-esj-Tac-NI^. X-A'-A'-Tnp-Ala-Vat-Gly-His-Leu-esi-A9-©.. 9. Пептид по п. 4, имеющий формулу: 8. Пептид по п. 3, имеющий формулу: где X представляет собой водород, простую связь, D-Phe-Gln^-Ala-Val-Gly-His-Leu-esi-DMTac-NHz. связывающую альфа-аминогруппу А с гамма-карбоксильной частью на 3-пропионильной группе А2, если 10. Пептид по п. 1, где X представляет собой А2 является Glu(-); или группу формулы R'CO-, где R1 R'CO-; А1 представляет собой пептидную связь, обозначает СНз-группу; А1 представляет собой D-, L- связывающую ацильную часть R1CO- с альфаили DL-аминокислот-ный остаток, выбранный из аминочастью Аг; А2 представляет собой Gin или группы, включающей в себя Phe, Hca, p-HI-Phe, pGlu, His; a Q представляет собой NH2. Nat, Pal, Tpi, незамещенный Тгр; либо пептидную связь, 11. Пептид по п. 1-, где X представляет собой во связывающую ацильную часть R'-CO- с альфа-а- дород, А1 представляет собой Нса. Аг представ миночастью А , при условии, чтоХ = R1 -СО-; А2 ляет собой Gin; А9 представляет собой Тас. представляет собой Gin, Glu[-], Glu(Y), или His, где j-J 12. Пептид по п. 11, имеющий следующую форму означает простую связь, если X является простой лу: связью, а А3 является Gtu(-]. причем Hca-Gln-Trp-Ala-Vai-Gly-His-Leu-gsJ-Tac-NHz связь [-] связывает гамма-карбоксипьную часть или 3пропионильную часть Аг с альфа-аминогруппой A1, a 13. Пептид, имеющий формулу (I): X-A'Y представляет собой ОСНз-группу; Leu-QSi представляет собой редуцированную форму A^Trp-Afa-Vel-Gly-Hts-Leu-Esj-A'-Q, Leu. где вместо -СНг- присутствует С=О, так что связь указанной -СНг- части с альфа-аминогруппой соседнего где X представляет собой водород; А А9 - остатка является псевдо-пелтидной связью; представляет собой ненатуральную аминокисА* представляет собой Тас, МТас или DMTac; Q лоту, выбранную из группы, включающей в себя представляет собой NHs, или его фармацевтически L-. D-Pat, L- или D-Tpi; А представляет собой Gin; приемлемые кислоты или соли, обладающие Leu-gsf представляет собой редуцированную форактивностью антагониста бомбезина. му Leu, где вместо -СНг-присутствует С=О, так что 2. Пептид по п. 1, где X представляет собой водород, связь указанной -СНг- части с альфа-аминогрупили R'CO-, где R1 является СНз-группой, Af пой соседнего А* • остатка является псевдопептидной связью; А* представляет собой Су»; 34456 Q представляет собой или его фармацевтически приемлемые кислоты или соли, обладающие активностью антагониста бомбезина. 14 Фармацевтическая композиция, обладающая антагонистической активностью по отношению к бомбеэину, включающая в себя.активный агент и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного агента она содержит фармацевтически эффективное ко личество пептида формулы (1) или его терапевтически приемлемую аддитивную соль или комплекс. 15. Способ лечения рака у млекопитающих, заключающийся в том, что указанному млекопитающему вводят эффективную дозу активного агента, отличающийся тем, что в качестве активного агента вводят пептид формулы (1) или его терапевтически приемлемую кислоту или соль. Изобретение откосится к новым пептидам, которые влияют на рост раковых опухолей у человека. Более конкретно, изобретение относится к антагонистам бомбезина, которые представляют собой у69- псевдопептиды, содержащие Тас-, МТас- или ОМТас- остаток е С-концевом положении; к их солям и фармацевтическим композициям, а также к способам синтеза указанных пептидов и к способам их использования Эти пептиды являются антагонистами по отношению к бомбезину или бомбеэинподобным пептидам. Изобретение относится к полипептидным соединениям, которые обладают антагонистической активностью по отношению к бомбезину или бомбезинподобным пептидам, таким как гастрин высвобождающий пептид (GRP). нейромедин С и т. п (называемый далее бомбеэин-внтагонистической активностью) и которые могут быть использованы, например, для лечения злокачественных опухолей у теплокровных животных, включая человека. Изобретение относится к новым полипептидным соединениям и способам их получения, а также к новым фармацевтическим композициям, содержащим указанные полипептидные соединения, и к способам изготовления лекарственных препаратов, способных продуцировать бомбезинантагонистический эффект при их введении теплокровным животным, включая человека. Бомбезин представляет собой амид тетрадекапептида, который впервые был выделен из кожи лягушки Bombtna bornbina (краснобрюхая жерлянка) (Anastasi, Erspamer and Bucci, Experientia, 1971. 27, 166). Известно, что указанный бомбезин является сильным митогеном для фибробластных. клеток мышей Swiss ЗТЗ (Rozengurt and Sinnett-Smith, Proc. Nat. Acad. Sci. USA, 1983, 80, 2936) и стимулирует секрецию амилазы из ацинуса поджелудочной железы морских свинок (Jensen, Jones. Folkers and Gardner, Nature, 1984, 309, 61). Кроме того, известно, что бомбезинподобные пептиды продуцируются и секретируются клетками мелкоклеточного рака легких (SCL С) человека (Moody, Pert, Gazdar, Garney and Minna, Science, 1981, 214, 1246). Известно также, что экзогенно добавленные бромезинподобные пептиды могут стимулировать sn vrtro рост клеток SCLC человека (Gamey, Cuttrta, Moody and Minna, Canser Research, 1987, 47,821) и что моноклональное антитело, которое является специфическим к С-концевой области бомбезина и GRP, может блокировать связывание GRP с его рецептором и предупреждать рост SCLC-клеток человека как in vitro, так и in vivo (Cuttita, Carney, Mulshine. Moody. Fedorko, Fischier and Minna, Nature, 1985, 316, 823). GRP (гастрин-высвобождающий пептид), который обладает бомбезинподобными свойствами, является широко распространенным амидированным пептидом, содержащим 27 аминокислот, выделенных из кишки свиньи (McDonald, Jornvall, Nilsson, Vagne, Ghatei, Bloom and Mutt, Biochem. Biophys. Res. Commun., 1979, 90, 227). где С-концевая аминокислотная последовательность является почти идентичной С-концеаой последовательности бомбезина. Нейромидин С представляет собой амидированный декалептид, который имеет структуру, идентичную десяти последним аминокислотным в С-концевой области СРР, и который был выделен из тонкой кишки собаки (Reeve, Walsh, Chew, Clark, Hawke and Shively, J. Biol. Chem., 1983, 258, 5582). GRP стимулирует ряд биологических реакций, включая высвобождение гастрина в большом круге кровообращения, а в фибробластных мышиных клетках ЗТЗ и в клетках мелкоклеточного рака легких (SCLC), он также играет роль фактора роста Поэтому было высказано предположение, что GRP играет непосредственную патофизиологическую роль в развитии SCLC посредством аутокрмнного механизма роста Бомбезин, нейромедин С и С-концевой нонапептид GRP имеют следующую структуру: Бомбезин: pGlu-Gln-Arg-Leu-Gly-Asn-Gln-TrpAla-Val-Gly-His-Leu-Met-NH2. Нейромедин С: H-G!y-Asn-His-Trp-Ala-ValGly-His-Leu-Met-NH2. С-концевой нонапептид GRP: Asn-Hts-TrpAla-Vai-Gly-His-Leu-Met-NHs. В результате исследования других бомбезинподобных пептидов, происходящих от земноводных, из кожи лягушки, обитающей в Папуа-Новой Гвинеи, удалось выделить литорин, который представляет собой нонапептид (pGJu-GIn-Trp-AlaVal-Gly-Hts-Leu-Met-NH2 и который, как оказалось, является исключительно сильным аналогом бомбезинз (Yasukara et al, Chem. Pharm. Bud., 1979, 27, 492). Исследования аналогов бомбезина показали, что сегмент, состоящий по меньшей мере из 9 аминокислотных остатков в 6-14 положениях, обладают полным спектром бомбезиновой активности. Было охарактеризовано несколько видов антагонистов бомбезина. Субстанция Р (Arg-Pro-LyePro-Gin-Gen-Phe-Phe-Gly-Leu-Met-NH2), которая имеет очень слабую гомологию с аминокислотной 34456 последовательностью бомбезина, не способна ингибировать связывание бомбезина и бомбеэинподобных пептидов, однако, как было обнаружено, аналоги субстанции Р, модифицированные путем замены нескольких L-аминокислот D-аминокислотами, например, такие, как fD-Arg1, О-Ргог, D-Trp7 . Leu11), субстанция Р и (D-Arg\ D-Phe5, D-Trp79, LBU'1). субстанция Р (Moody и др, Fed Proceedings, 1987. 46. 2201), могут блокировать секрецию бомбезина в клетках ацинуса поджелудочной железы и подавлять ростстимулирующее действие бомбезина в клетках Swiss ЗТЭ Было также установлено, что два типа антагонистов бомбезина, происходящих от бомбезина. например (D-Phe6, DPne12). бомбезин и [Leu13 -psj-Leu'*], бомбеэин (Coy и др., J. Biol. Chem , 1988, 263, 5056 и пептиды. 1989, 10, 587), являются сильными in vitro и in vivo ингибиторами бомбеэинового ответа. Другой тип антагонистов бомбезина, обнаруженный Hetmbrook и др. , (Bio. Chem., 1989, 264, 11258), представляет собой N-anenm-GRP (20-26) и его аналоги, которые образуются от GRP (20-27) -аналогов путем делеции С-концевого метионинового остатка Coy {J. Bio). Chem., 264, 1989, 25, 14691)1 показал, что некоторые короткоцепочечные антагонисты бомбезина, полученные на основе последовательности литорина, например, (ОPhe® Leu13 -psi-Phe16] бомбезин (6-14) и [D-Phefl, Leu1 -pjsi-Leu ] бомбезин (6-14), обладают гораздо более высокой активностью, чем их соответствующие родственные пептиды [Leul3-ES)-Leu14] бомбезин. Линейные (нециклические) бомбезиноаые аналоги GRP и бомбезина земноводных, имеющие, но необязательно, в СНг-NH непептидную связь, описаны в заявке на патент PCTWO 90/03980 (а родственные аналоги описаны в WO 91/02746). Как указывается в этой заявке, эти аналоги, которые действуют как ингибиторы натуральных бомбезиновых пептидов, имеют следующую формулу: R R где Ri R2 = Н; А0 может быть делетирован: из многих возможных аминокислот в каждом положении: А1 может представлять собой D-Phe, D-Trp или DNal. Аг может представлять собой Gin: А4 может представлять собой Ala; А5 может представлять собой Val; Ae может представлять собой Gly; A7 может представлять собой His, a W представляет собой: R Z Z О Iа IіI2 I II II -N-€H-ft -CH-C-V где R4=CHj-NH; в некоторых случаях Ту может представлять собой идентифицирующую боковую цепь Leu, т. е. - СНгОН{СН3)г; Ъ может представлять собой идентифицирующую боковую цепь Cys или Met, т. в. - СНг-SH или (СН8)г- S-CH3; V = = N(R6)R7. где Re, R? И RB могут представлять собой Н, a Ri и R? могут представлять совой If ими COEi. где Ei может быть С,С,-о-апкилом Линейные пептидные аналоги бомбезина пписаны также в ЕР 0309297 Эги пептиды огут иметь С - концевой Met - остаток и [СН? - NH] псевдопептидную связь между С - концом и примыкающим к нему остатком Изобретение относится к новым полипептидам, которые являются сильными антагонистами бомбезина, к способам их синтеза и к новым лекарственным средствам, включая фармацевтические композиции, содержащие указанные попипептиды, и к использованию этих композиций в качестве фармацевтически активных агентов А. Синтетические пептиды. Бопее конкретно, один из вариантов осуществления изобретения относится к псевдопептидам, обладающим сильной бомбезин-антагонистической активностью, и имеющим формулу I' Х-А1 -Аг -Trp-Ala-Val-Gly-His-Leu-Bsj-A9 -Q (І) где X представляет собой водород; простую связь, связывающую альфа-аминогруппу А с гамма-карбоксильной частью на 3-пропионильной группе Аг. если А2 является Glu, или фулпу формулы R'CO-. где R1 выбирают из группы, включающей в себя а) водород, СгСю-апкил, фенил, фенил-Сг Сю-алкил. Р-Н1-фенил, р-НІ-фенил-Сі-Сю-апкил, нафтзлин-СгСю-алкил, индолип, индолил- СгСюалкил, пиридил. пиридил-Сі-Сю-алкил, тиенил, тиенил-СгСю-алкил. циклогексил, или циклогексил-Сі-Сю-алкил, где Н1. например, является F, СІ, Вг, ОН. СНз или ОСНз; Ь) R' где R представляет собой водород, Ci-do алкил, фенил, или фенил-Сі-Сю-алкил; R3 представляет собой водород, или С і-С кг алкил: с) R4 - О, где R4 представляет собой Сі-Сюалкил. Фенил, или фенил-Сі-Сю-алкил; А представляет собой D-, L- или OL-aминокислотный остаток, выбранный из группы, включающей в себя Phe, p-HI-Phe, pGlu, Nal, Pal, Tpi незамещенный Тгр или Тгр, замещенный в бензольном кольце одним или несколькими заместителями, выбранными из F, CI. Вг, NH? или Сі-Сз-алкила; либо пептидную связь, связывающую аципьную часть R1CO с альфааминочастью А2; А2 представляет собой Gfn, Glu{-J . Glu (Y), или His, где [ } означает простую связь, если X является простой связью, а Аг является Glu, причем эта связь связывает гамма-карбоксильную часть или 3-пропионильную часть А2 с альфа-аминогруппой A1, a Y представляет собой: (а) - OR5, где R является водородом. Сі-Сгалкихюм или фенилом; К или(Ь) , где Я является водородом R 34456 или СгСз-алкилом, a R7 является водородом, CiСз-алкилом, или -NHCONH2; Leu;j5£j представляет собой редуцированную форму Leu, где вместо СНг- присутствует часть С-О, так, что связь указанной -СНг- части с альфа-аминогруппой соседнего А9 - остатка является псевдопептидной связью: А9 представляет собой Тас, МТас, или DMTac; Q представляет собой NH2 или -OQ1, где Q1 является водородом, Ci-Cto - алкилом, фенилом, или фенил - Сі-do - алкилом, и, кроме того, изобретение относится к фармацевтически приемлемым кислотам или солям описанных псевдопептидоэ Если А = Тас, МТас или DMTac, то у А имеется 5-членное гетероциклическое кольцо. Это кольцо в основном образуется путем окисления боковой цепи остатка А9 на определенной стадии синтеза нонапептида формулы 1 (проводимого предпочтительно с использованием формальдегида или ацетальдегида). в результате чего происходит циклизация боковой цепи с альфа-аминогруппой остатка А9. Идентичность полученного циклизованного остатка А9 зависит от идентичности оригинала (т. е., неоксидированного А9) и от оксиданта. Так, например, если -CH2-SH-rpynna Cys циклизуется с альфа-аминогруппой Cys9, связанной посредством Leu-psi-псевдопептидной связью, в реакции с формальдегидом, то образующееся кольцо имеет структуру формулы НА (ниже показана как -Leue-psi-Tac -NHz-фрагмент нонапептида формулы I)'. S • 2 | Iа • •. N H -C H - CH - N ----- CH-CO-NH Эти псевдопептиды обладают более высокой биологической активностью и большей стабильностью, чем их нециклические варианты, где А9 является Cys или Ren. Тем не менее в некоторых предпочтительных вариантах осуществления изобретения в том случае, если в пептидах формупы ( А9 является нецихпизованным, а X, А2 и Q являются такими, как они определены выше, то указанный А9 - остаток представляет собой Cys или Ren, а А' представляет сдбой не встречающуюся в природе аминокислоту, выбранную из Lили D-Pal, L- или D - Трі или Нса. В некоторых предпочтительных вариантах X представляет собой R CO, R' представляет собой водород или Сі-Сю -алкил (предпочтительно метил); А1 я D - Сра, D - Nal, D - Rhe, D- или D-Tpi или D-Trp; A2 = Gin; A9 * Тас или DMTac, a'Q* NH2. Однако в другом предпочтительном варианте изобретения, если А1 является пептидной связью, связывающей ацильиую часть R1 - СО-группы с альфа-аминогруппой остатка А2, то А2 является Gtn или His; А9 является Тас, М-Тас, или DM-Tac, в Q является Nhfe В предпочтительной форме указанных пептидов X является Нса, Нпа, Раа, Мрр, Нрр или Naa; А2 является Сіп; а А является Тас. В. Способы синтеза. Антагонисты бомбезина формулы I могут быть синтезированы путем твердофазного синтеза. В первой схеме синтеза все аминокислоты последовательно присоединяются друг к другу после того, как С-концевой остаток будет связан с твердофазным полимерным носителем. Затем после связываний аминокислотных остатков с полимером осуществляют реакцию боковой цепи С-концевого остатка с оксидантом, в результате которой образуется 5-членное гетероциклическое кольцо псевдопепгида. Полученный псевдопелтид подвергают HF-обработке для его отделения от твердофазного полимерного носителя. Эта реакция позволяет также удалить защитные группы боковой цепи. Во второй схеме синтеза псевдопептиды формулы I могут быть получены в виде двух фрагментов, которые синтезируются путем твердофазного синтеза, либо путем синтеза в жидкой фазе. Трипептид, содержащий С-конец, связывают с олигопептидом, в результате чего получают полный антагонист бомбезина формулы I. 5-членное гетероциклическое кольцо образуют при помощи реакции боковой цепи А с оксидантом. Затем пептид подвергают HF-обработке, в результате которой удаляются все защитные группы боковой цепи аминокислотного остатка, а пептид отщепляется от полимерного носителя. С Лекарственные средства. Псевдопептиды формулы 1 могут быть использованы в фармацевтических композициях, предназначенных для лечения раковых или других заболеваний у млекопитающих путем введения этим млекопитающим эффективной дозы лсевдопептида или его терапевтически приемлемой кислоты ипи сопи в сочетании с фармацевтически приемлемым носителем. Указанные псевдопептиды в сочетании с фармацевтически приемлемым носителем могут быть введены в дозе, составляющей от около 1 до 1000 мг на 1 кг веса тепа в день. Эти композиции могут быть введены парентерально, внутривенно, подкожно, внутримышечно, интраназально, с помощью легочных аэрозолей и инъекций замедленного всасывания. На фиг. 1 изображен график, иллюстрирующий величину объема опухоли рака молочной железы мыши МХТ в зависимости от введения не* которых антагонистов бомбезина и полученный исходя из данных, представленных в табл. 7 примера 7; на фиг. 2 • график, иллюстрирующий величину объема опухоли SCLC у "голых^ мышей в зависимости от введения некоторых антагонистов бомбезина и полученный на основе данных, представленных в табл. 9 примера 8; на фиг. 3 - график, иллюстрирующий величину объема опухоли поджелудочной железы (MIA РАСА-2) у "голых" мышей в зависимости от введения некоторых антагонистов бомбезина и полученный на основе данных, представленных в табл. 10 примера 9; на фиг. 4 - график, иллюстрирующий величину объема опухоли поджелудочной железы CAPAN-2 у "голых" мышей в зависимости от введения некоторых антагонистов бомбезина и полученный на основе данных, представленных в табл. 11 примера 10. Описание предпочтительных вариантов осуществления настоящего изобретения. 34456 А. Синтетические пептиды. 1 Номенклатура. В данном описании для удобства используемые аминокислоты, пептиды и их производные обозначаются в соответствии со стандартными аббревиатурами, обычно принятыми в пептидной химии и рекомендованными Комиссией по биохимической номенклатуре Международного союза по теоретической и прикладной химии (IUPAC IUB) [European J. Biochem., 1984, 138,9-37]. Аббревиатуры для отдельных аминокислотных остатков образованы на основе тривиальных названий аминокислот, например, Ala означает эланин, Cys - цистеин, Gin - гпутамин, Glu - глутаминовая кислота, pGlu - пироглутаминовая кислота, Gly - глицин, His - гистидин. Leu - лейцин, Phe - фенилаланин, Тгр - триптофан и Val - валин. Cfu может иметь функциональные группы, связанные с его гамма-карбоксильной боковой цепью. [-J или Y является такими, как они были определены выше, Dpa представляет собой 2,3-диаминопропионовую кислоту. В том случае, если аминокислотный остаток имеет изомерные формы, то, если нет каких-либо указаний, подразумевается L-форма, в противном же случае перед названием аминокислоты имеются D- или DL. Используемые в данном описании редко встречающиеся аминокислоты имеют следующие обозначения: Сра • п-хлорфенилаланин Dpa • 2,3-диаминопропионовая кислота PGlu - пироглутаминовая кислота Na( - 3-(2-нафтил)-аланин Pal • 3-(3-пиридил)-аланин Реп • леницилламин Tpt • 2,3,4,9-тетрагидро-1Н-пиридо-[3,4-Ь] индол-3-карбоновая кислота Тас - тиазолидин-4-карбоновая кислота DMTac - 5,5-диметил-тиазолидин-4-карбоновая кислота МТас • 2-метип-тиаэолидин-4-кэр6оновая кислота Аналоги аминокислот имеют следующие обозначения: Нса - гидрокоричная кислота или дес-аминофенилалаиин Нпа - З-гидрокси-2-нафтойная кислота Нрр - 3-{4-гидроксифенил)пролионовая кислота Мрр - Э-(4-метоксифенил)пропионовая кислота Naa - нафтилуксусная кислота Раа - фенилуксуснвя кислота Кроме того, были использованы следующие обозначения: АС - ацил Ас - ацетип АсОН - уксусная кислота ВНА - бензидриламин Вое - трет-бутоксикзрбонил "' (ВОС)гО - ди-трет-бутипдикарбонат Вот • бекзилоксиметил But - бутил Вг\ • бензил DSA * альбумин бычьей сыворотки DIC - 1,3-диизопролилкарбодиимид DM ЕМ • модифицированная по способу Дульбекко. среда Игла DMF(flM)- диметилформамид Е\ - этил EDTA - этипендиаминтетрауксусная кислота FCBS - окопоплодная сыворотка теленка Fmoc - фяуоренилметилоксикарбомил For - формил HITES - среда RPMI 1640 + 108 М гидрокортизона. 5 мкл/мл бычьего инсулина. 10 мкг/мл трамсферрина человека. 10 е М р-эстрадиола и 3 * 1 0 MNa2SeO3 НОВІ - 1-гидроксибензотриаэол НР1.С (ВРЖХ) - высокораэрешающая жидкостная хроматография Leu - psi редуцированная форма Leu, где вместо - СНг - присутствует С = О - часть, так, что связь указанной - СНг - части с аминочастью остатка А9 является псевдопептидной связью Me - метил MeCN - ацетонитрил МеОН - метиловый спирт PBS - фосфатно-буферный раствор TEA - тризтиламин TFA - трифтороуксусная кислота. В данном описании запись пептидных пос, ледовательностей проводится в соответствии с общепринятой нормой, т. е., N-концевая аминокислота находится слева, а С-концевая аминокислота находится справа. Однако при этом следует отметить, что в описании, если это не оговорено особо, стандартная нумерация аминокислотных остатков во фрагментарном пептиде не соблюдается и не соответствует их истинным положениям в полном тетрадекапептидном антагонисте бомбезина. Если следовать стандартной нумерации, то остатки в нонапептиде срормулы I должны быть пронумерованы от 6 до 14, а ядро Тгр - Ala - Val • Gly -His Leu - антагонистов бомбезина должно быть пронумеровано А 8 - А9 - А10 - А11- А -А13. Вместо этого во избежание путаницы пептидные антагонисты бомбезина, используемые в данном описании, были пронумерованы следующим образом: N- концевой аминокислотный остаток (или остаток аналога) был обозначен А1; С-концевой аминокислотный остаток (или остаток аналога) был обозначен А9; а промежуточные остатки были пронумерованы по порядку от А5 (остаток, примыкающий к N-концевому остатку А1) до А8 (остаток, примыкающий к С-концевому остатку А9). 2. Предпочтительные варианты осуществления изобретения. В предпочтительных вариантах своего осуществления настоящее изобретение относится к пептидным антагонистам бомбезина, имеющим формулу I: X - А1 - А2 - Тгр - Ala - Val - Gly • His-Leu - psi - A9 - Q I где X, A1, A2, Leu - e£i, А* и Q являются такими, как они были определены выше. Указанные псевдопептиды, антагонисты бомбезина, отличаются своей аминокислотной последовательностью, особенно в остатках А1, А2 и А9, а также наличием псевдопептидной связи Между А8 и А8 и необязательно наличием 5-членного гетероциклического кольца у А8, Кольцевая 34456 9. - 0-Pa!-Gtn-Trp-Ata-Val-Gly-Hi5-Leu-B£i Tac-NH2 10. - D-Trp-Gln-Trp-Ala-Val-Gly-His-Leu-psi Tac-NH2 11. - Ac-D;Trp-G)n-Trp-Ala- Val-Gly-His-LeuRSJ -Tac-NHa 12. - Tpi-Gin-Trp-Ala-Val-Gly-His-Leu-esj-TacNH2 13. - D-Tpi-Gln-Trp-Ala-Va!-Gly-His-Leu-E£i • Tac-NHa 14. - Hca-Gln-Trp~Afa-Val-Gly-His-Leu-2£i Tac-NH2 15. - D-Phe-His-Trp-Ala-Val-Gly-His-Leii-esi • Tac-NH2 16. - D-Phe-Glu(OMe)-Trp-Ala-Val-Gly-HisLeu- esi -Tac-NHa 17. - D-Phe-GluH -Тф-Ala-Vat-Gly-His-Leu- С5І •Tac-NHj 18. - D-Phe-Gln-Trp-Ala-Val-Gty-His-Leu-ESi DMTac-NH2 19. - Ac-D-Phe-Gln-Trp-Ala-Vaf-Gty-His-LeuCSJ -DMTac-NHa 20. - D-Cpa-Gln-Trp-Ala-Val-Gly-His-Leu-csJ DMTac-NH2 21.- Трі-ОІп-Тф-АІа-Уаі-Оіу-Ніз-Ьеи- esi DMTac-NH2 22. - D-Tpi-Gln-Trp-Ala-Val-GIy-His-Leu-cai DMTac-NHz Особенно предпочтительными вариантами являются следующие пептиды 2. - D-Phe-Gin-Trp-Ala-Gly-His-Leu-BSi-TacNH2 13. - 0-ТрІ-вІп-Тф-АІа-Уа!-СІу-Ніз-іеи-еа • Tac-NH2 18. - D-Pbe-Gln-Trp-Ala-Val-Gly-His-Leu-pjsi OMTac-NHz В. Методы синтеза 1. Обзор Псевдопептиды формулы I могут быть получены любым способом, обычно используемым в пептидной химии Такие способы описаны, например, в книге М. Bodanszky, Principees of Peptide Synthesis, Springer-Verfag, Heidelberg, 1984. Все псевдопептиды формулы I могут быть получены в соответствии с техникой твердофазного синтеза. Этот способ является наиболее предпочтительным для получения указанных псевдепептидов и их промежуточных пептидов. Техника твердофазного синіеза описана в руководстве J. М. Stewart and J. D. Young, Solid Phase Peptide Synthesis, Pierce Chem. Co., Rocrtord, П., 1984 (2oe изд.), и в образе G. Barany и др., Imt J. Peptide Protein Res. , 30, 705 - 739, 1987. Дополнительная стадия окисления С-концевого остатка А9, проводимая для циклизации конкретной боковой цепи этого остатка с его альфа-аминогруппой, может быть осуществлена в одной из нескольких ступеней синтеза пептидов формулы I. Для получения псевдопептидных антагонистов бомбезина формулы I могут быть использованы по крайней мере две схемы синтеза. В первой схеме все аминокислоты последовательно присоединяются друг к другу, начиная от С-концевого остатка А9, который предварительно присоединяют к твердофазному носителю. Во второй схеме сначала на полимерном носителе синтезируют трипептид: Затем этот трипептид присое струхіура опредепяется идентичностью остатка А и соединением, используемым для его окисления Так например, если используется формальдегид, а А представляет собой Cys, то полученное кольцо имеет структуру Тас формулы НА (показанную ниже в виде Leu -pjj -Тас9 - Q - фрагмента пептида формулы I, где Q = NHa): н сг \н H-CONH ПА NH- CH-CH -N 2 ---------------------С 2 І СН -CHtCH ) 2 3 2 Если используется ацетальдегид, а А является Cys, то полученное 5-членное гетероциклическое кольцо имеет структуру МТас формулы MB (показанную ниже в виде Leu8 £si -МТас - Q фрагмента пептида формулы I, где Q - NH2): СН НС' з і NH-CH-CH -N ---- CH-CO-NH ИВ снг-сн(снз)г Если испопьзуется формальдегид, а А является Реп, то полученное кольцо имеет структуру DMTac формулы НС (показанную ниже в виде Leue-esi - DMTac9 - Q - фрагмента нонапептида формулы 1, где Q = NHa): - ••NH- CH- CH - N ----CH-CO-NH IIC сн2-сн(снз)2 В описанных предпочтительных вариантах осуществления изобретения желательно, чтобы Х= Н или Ас, А1 = D-Phe, А2 = Gin и Q = NH2. Ниже представлены наиболее предпочтительные псевдопептидные антагонисты бомбеэина данного изобретения: Пептид - Структура 1. - D-pGlu-Gln-Trp-Afa-Val-Gty-His-Leu-pjy Tac-NHa 2. - D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-BSi Тас-МНг 3. - D-Phe-Gln-Trp-Ata-Val-Gfy-His-Leu-csi Tac-NHa 4. • Ac-D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu'Csi Tac-NH2. 5. • D-Cpa-GJn-Trp-Ala-Gty-His-Leu-Djgi-TacNHa 6. - D-Cpa-Gln-Trp-AJa-Val-Gly-His-Leu-esj Tac-NHa 7. - D-Nat-Glh-Tn>AJa-Val-Gty-His-Leu-eai • Tac-NHa 8. - Pal-G1n-Tn>A!a-Val-Gly-Htfl-Leu-B2i-TacNH2 6 34456 диняют it олигопептиду, несущему все остальные аминокислоты, необходимые для образования нужного пептида. В обеих схемах синтез начинают с С-концевого остатка Ав, к которому последовательно добавляют соответствующие аминокислоты вплоть до N-концевого остатка. 1 (а) Первая схема. 8 качестве полимерного твердофазного носителя для синтеза псевдопептидов может быть использована бензгидриламиновая (ВНА) смола или хлорометилироваиная полистироловая смола, сшитая (на 1%) с дивинилбензолом, причем указанные две смолы являются коммерчески доступными. С-концевой остаток А9 присоединяют к одной из указанных смол посредством его карбоксильной группы. Для предупреждения реакции между двумя указанными остатками, перед тем как присоединить этот остаток (А8) к смоле-носителю, альфа-аминофулпу каждого А9- остатка блокируют с помощью химической защитной группы. Защитной группой для альфааминогруппы Ая-остатка, и каждого последовательно присоединяемого остатка, может быть либо 9 • флуоренилметилоксикарбонильная (Fmoc) или трет-бутоксикарбонильная (Вое) группа. Fmoc предпочтительно использовать при связывании Сконцевого остатка А9 с остатками до А2, поскольку реакция удаления Вое также способствует удалению защитной группы боковой цепи от С-концевого А9 остатка. В процессе указанных реакций присоединения функциональные группы боковых цепей некоторых аминокислотных остатков могут быть также защищены от нежелательных химических реакций путем их связывания с химической защитной группой (подходящей группой для Аа является But) до осуществления реакции присоединения После присоединения С-концевого Fmoc» А9(Ви1)-остатка к твердому носителю альфааминогруппу этого остатка подвергают разблокированию, а затем к нему присоединяют Fmoc-LeuСНО, образуя тем самым псевдопептидную связь. Остальнье остатки, А5, А4, А3 и А2, защищенные предпочтительно ос-группой, и А1, защищенный предпочтительно Вос-группой, постадийно добавляют в обратном порядке (А7, Ав и т. п.), в результате чего получают нужный псевдопептид. Если в указанном псевдопептиде присутствует His или Clu то в процессе реакций их присоединения или реакций присоединения других остатков боковые цели указанных остатков могут оказаться незащищенными от нежелательных химических воздействий. В соответствии с этим перед осуществлением реакции присоединения этих аминокислот их боковые цепи могут быть связаны с химической защитной группой. После того как все аминокислотные остатки будут присоединены к ВНА-смопе, Вос-группа у Nконца псевдопептида может быть удалена. 5-членное гетероциклическое кольцо у Тэс, МТас или DMTac может быть образовано с помощью реакции - СН2 - SH-части Cys (или -C(CH3)zSH Pen) и вторичной аминогруппы редуцированной связи, соединяющей остатки А9 и А9 (т. е. альфа-аминогруппы А8), с оксидантом, таким, как формальдегид или ацетальдегид. Промежуточный псевдопептид, еще несущий защитные группы боковых цепей, подвергают HF-обработке в целях его отщепления от твердофазного носителя, а также в целях удаления защитных групп боковой цепи. 1, Вторая схема. Во второй схеме в соответствии с процедурой твердофазного синтеза конструируют три ептид с использованием ВНА-смолы в качестве носителя, в результате чего попучают следующий защищенный трипептид на носителе' Boc-His (Bom)7-Leue -BSi -Cys(But)9-BHA-CMona Вое' - группу и But - группу удаляют, а - СНгSH-часть Cys подвергают циклизации с вторичным амином редуцированной связи, соединяющей остаток А с остатком А9, в результате чего получают His(Bom)7 -Leu -ESi-Tac -ВНА-смолу После HF-обработки, получают свободный трипептид: His7 -Leue -psI-Tac9 -NH2. В соответствии со стандартным методом твердофазного синтеза, на GlyОСНг -смоле постадийно конструируют пептид на носителе: "Вое A' -A8 -Trp-Ala-Val-G1y-OCH2 -смола", после чего эту конструкцию обрабатывают 95% метанолом, содержащим 1% KCN (в течение 12 ч), в результате чего Вос-олигопептид отщепляется. После конструирования этих двух фрагментов . Вос-олигопептид присоединяют к свободному His7Leu8- esj -Тасв-МНг-трипептиду с использованием ВОР-реагента. Затем Вос-грулпу удаляют и получают нужный пептидНиже перед подробным описанием синтеза антагонистов бомбезина формупы 1 приводится несколько общих процедур осуществления синтеза, характерных для одной или обеих из указанных схем. 2. Общие процедуры полипептидного синтеза. Процедура 1. Получение некоторых синте тических остатков для реагентов. a) L и D-Tpi: 2,04 г (10 мМ) L-Trp растворяли в 25 мл кипящей воды, содержащей 2,1 г лимон ной кислоты. Затем добавляли 0,5 мл 40%-ного водного формальдегида, после чего сразу начали образовываться твердые вещества. Полученную смесь охлаждали в ледяной бане, а осадок со бирали, промывапи холодной водой, осушали воз духом, а затем осушали при комнатной температу ре, в результате чего получали 2,t4 г твердого ве щества (99%), т. пл. (с разложением) около 310*С. D -изомер, который получали тем же спо собом из D-Trp, также имел т. пл. (разлож.) около 310-С. b) L - и D-Boc-Tpi, К размешанной суспензии из 10,8 г (50 мМ)-Трі В 250 мл 0,2 н. NaOH и 7,5 мл триэтиламина добавляли 10 г Di-трет-бутипдикарбоната, полученную смесь размешивали 4 ч, а за тем, размешивая, добавляли еще 10 г дикарбо ната и после повторного 3-часового размешива ния добавляли еще 10 г дикарбоната. После этого смесь размешивали в течение ночи и экстрагиро вали (2 • 100 мл) эфиром, а эфирный слой отде ляли и отбрасывали. К водному слою добавляли лимонную кислоту (в целях доведения рН до 3-5). Твердый остаток собирали, промывали водой и осушали воздухом в течение ночи. Твердые вещества суспендировали в 100 мл тетрагидрофурана. При этом почти все твердые вещества растворялись. Нерастворимые вещества удаляли путем фильтрации, а ТГФ удаляли в вакууме. Остаток растирали с эфиром и по* 34456 пуча л и 9.20 г (или 58%) нужного материала. Этот материал имел такую же т. пл.. что и исходный материал, но отличался от исходного материала по своей растворимости. ТСХ нэ силикагеле проводили с использованием смеси СНС1Э . МеОН ; : НОАС- 65 15 0. 5 Повторяя аналогичную процедуру с использованием 2.55 г - ТрІ, получали 2,22 г или 59% Вос-Тр. c) Fmooleu-CHO. Сложный метиловый эфир Fmoc-лейцина (35 г. 134 мМ) в безводном толуоле (250 мл) в атмосфере азота охлаждали с использованием смеси сухого льда и ацетона, после чего в течение 30 мин добавляли 150 мл 25% гидрида ди-иэобутипалюминия в толуоле. После этого смесь размешивали 20 мин в бане из сухого льда/ацетона, а затем осторожно добавля ли метанол (15 мл). Полученную смесь выливали в 1000 мл ледяной водь!, размешивали и фильтро вали Затем толуол отделяли, а водную фазу сно ва экстрагировали эфиром (3x300 мл). Толуол и эфирные экстракты объединяли и осушали суль фатом натрия Полученное масляное вещество быстро пропускали через колонку с силикагелем (3x50 см) в 1500 мл 15% ЕЮАс/ петрола. И на конец получали Fmoc-Leu-CHO s виде твердого вещества (27,6 г). d) Синтетические аминокислоты или анало ги аминокислот, вводимые в псевдопептиды дан ного изобретения, являются в основном коммер чески доступными. Так, например, Нса постав ляется фирмой Aldrich Co., 1001 St. Paul Avenue, Milwaukee, W1 53233 Вое- или Fmoc-защищенные аминокислоты поставляются фирмой Advanced Chem. Tech., 5609 Fern Valfey Road, Louivitle. KV 40228; или Bachem California, 3132 Kashiwa Street, Torrance, CA 90505. , Процедура 2. Получение смопы и присоединение А -остатка. ВНА-смолу получали путем обработки 10% TEA в СНгСІ2 (для нейтрализации) (два раза по 3 мин), а после обработки ее 6 раз промывали метиленхлоридом. Fmoc-A9 (But) - остаток связывали со смолой путем добавления 1,35 мМ Fmoc-AB (But) и 1.5Q мМ 1-гидрокси-бензотриазола (НОВІ) в ДМФ с последующим перемешиванием полученной смеси в течение 3 мин. Затем смесь перемешивали путем встряхивания в течение 60 мин при комнатной температуре. Полученную Fmoc-A9 (Bul)-BHA-cмолу промывали метиленхлоридом, метанолом (каждый раз дважды) и три раза снова метиленхлоридом, после чего эту смолу анализировали с помощью теста Keiser (Anal. Biochem. 34, 595 (1970). 8 случае, если обнаружилось неполное связывание, описанную процедуру повторяли. Процедура 3. Образование псеедопептидяой связи. Отщепление (т. ©. разблокирование) Fmoc группу от А9 осуществляли путем добавления 50% пиридина в ДМФ с последующим 30-минутным размешиванием, промыванием ДМФ (6x1 мин), а затем (і) добавлением Fmoc-Leu-CHO (3 экз) в ДМФ, содержащей 1% АсОН; (Іі) добавлением NaBH3CN(3,5 экв. ) в ДМФ, и размешиванием (путем встряхивания) в течение 60 мин. Полученную смесь последовательно промывали 50% МеОН (3x1 мин); 100% МеОН (3x1 мин) и ДМФ (3x1 мин). Процедура 4. Присоединение аминокислот и образование пептидной связи. 8 качестве защитной группы для альфааминогруппы А9-остатка и каждого последовательно присоединяемого аминокислотного остатка могут быть использованы либо 9-флуоренилметилоксикарбонил (Етос)-группы. либо трет-бутилоксикарбонил (Вос)-группа. При связывании А9 с последующими аминокислотными остатками вплоть до N-концевого аминокислотного остатка предпочтительной является Fmoc-группа, поскольку реакция удаления Вое позволяет одновременно удапить защитную группу боковой цепи А9-осхатка. A) Использование Fmoc-аминокислоты. Для введения Fmoc-аминохислоты в Fmoc* промежуточный пептид осуществляли следующие процедуры. (1) Fmoc - промежуточный пептид подверга ли разблокированию и нейтрализации, а затем промывали метиленхлоридом (3x1 мин) и ДМФ (3x1 мин). (2) Fmoc - аминокислоту присоединяли к разблокированному промежуточному пептиду пу тем (і) добавления Fmoc-аминокислоты (З экв.) и HOBt (3,3 экв.) в ДМФ (3 мин) к промежуточному . пептиду; (іі) добавления 3 экв. OIC (в виде 20%-ного раствора в СНгСЬ); и 90-минутного встряхивания полученной смеси. (3) Затем смесь промывали этанолом (3x1 мин) и диметилформамидом (3x1 мин). Для присоединения других остатков только что присоединенную Fmoc-аминокислоту подвергали разблокированию 50% пиперидином в течение 30 мин, а затем промывали диметилформамидом (6x1 мин) Последующие процедуры присоединения осуществляли в соответствии с описанием в стадии (2). Каждый раз после присоединения новой аминокислоты к смоле или пептиду проводили тест Kaizer и в случае обнаружения неполного связывания реакцию повторяли. Для присоединения Fmoc-Gly и Fmoc-GIn стадию (2) описанной процедуры модифицировали следующим образом: к смеси ДМФ - раствора Fmoc - аминокислоты (3,0 экв) и HOBt (3,3 экв.) добавляли (в течение 15 мин при 0'С и в течение 15 мин при комнатной температуре) 3 экв. 1С (в виде 20%-ного раствора в СНгС12). Затем реакционную смесь добавляли к пептидной смоле и в случае Fmoc-Gly размешивали 1 ч, а в случае FmocGtn размешивали 2 ч. B) Использование Вос-аминокислоты. Для введения Вос-аминокислоты в Fmocпромежуточный пептид осуществляли следующие процедуры: (1) Вос-rpynny удаляли пучем добавления 50% TFA в СН2С1г; (2) а затем добавления 50% TFA в СИ2СІ2, содержащего 5% меркаптоэтанола и 5% анизола в течение 25 мин и (3) промывания метиленхлоридом (2x1 мин), метанолом (2x1 мин) и диметилформамидом (3x1 мин); (4) присоединение Вос-аминокислотного ос татка осуществляли путем добавления 3 экви валентов Вос-аминокислоты и 3,3 эквивалентов HOBt в ДМФ и последующего размешивания в те 34456 чемие 3 мин Затем добавляли 3 эквивалента 20% дииэопропилкарбодиимида в 01ЬС1; и размешивали путем встряхиваний в течение 90 мин После этого реакционную смесь промывали метанолом (3x1 мин), а затем метиленхлоридом (3x1 мин) (5) Вос-группу удаляли (т е осуществляли разблокирование) путем промывания продукта, полученного в стадии (5), 50% 1FA в СН?СІ?. содержащего 5% меркаптоэтанопа (5 мин) и 5% анизола (25 мин) Затем промывали в СН?СЬ (2x1 мин), МеОН (2x1 мин) и ДМФ (3x1 мин) При этом следует иметь ввиду, что удапение Вос-группы влечет за собой также удаление защитной Bul-группы. соответствующим образом связанной с боковой цепью Ая - остатка Поэтому удаление Вос-группы следует проводить лишь непосредственно перед циклизацией А - остатка. Процедура 5 Образование 5-членного гетероциклического кольца Формирование кольцевой структуры формулы НА осуществляли следующим образом промежуточный пептид, имеющий разблокированный Cys* - остаток, в смеси 50% АсОН. 3,7% НСНО, и ДМФ (1 1 8) размешивали 3 мин при комнатной температуре, э затем фильтровали Попученный продукт промывали диметилформамидом, метанолом и метиленхлоридом (каждым по 3 раза). Формирование кольцевой структуры формулы ІІ8 осуществляли следующим образом: промежуточный пептид, имеющий разблокированный Cys - остаток, в смеси 50% АсОН, 10% СНЭСНО и ДМФ ( 1 5 0, 58) размешивали 10 мин при комнатной температуре, а затем фильтровали Полученный продукт промывали диметипформэмидом, метанолом и метиленхлоридом (каждым по 3 раза) Формирование кольцевой структуры формулы НС осуществляли следующим образом' промежуточный пептид, имеющий разблокированный Реп"* - остаток в смеси 50% АсОН,'3,7% НСНО и ДМФ (1 1 8) размешивали 10 мин при комнатной температуре, а затем фильтровали Полученный продукт промывали диметилформамидом, метанолом и метиленхлоридом (каждым по 3 раза) Процедура 6 Отделение пептида от смолы После присоединения всех нужных аминокислотных остатков промежуточный пептид на ног.иієле обрабатывали жидким фтороводородом в присутствии анизола для тог о чтобы полученный попипепгид отделить от носителя В результате этой реакции также удаляются защитные группы боковых цепей В случае использования ВНА-смолы попученный промежуточный пептид имеет Q = NH?. Если X не явпяется И, то X - группу помещают в N-конец либо путем введения аминокислоты, уже несущей X - группу (например, АсO-Phe). либо путем реакции разблокированной альфа-аминогруппы у N-концевого А1-остатка промежуточного псевдопептида с соответствующим реагентом Так. например, после отделения от смолы, используемой о качестве твердофазного носителя попный пептид может быть подвергнут реакции с КОС с получением I іроиудура 7 Очистка пептидов Псевдопептиды формулы I очищали тайным образом с помощью обрпщенно-фазоаой высокораэрешающей жидкостной хроматографии (ВРЖХ) на колонке системы Ramin (Rfwun Inc. Со , Wohum. Ma), состоящей из трек на псов Hamin Rabbit HP, HPLC, контролируемых компьютером (Apple Macintosh Plus), дозатором (Pheocyne) и монитором переменного УФ-иэпучения (Knauer Model 87) Неочищенные пептиды (10 АО мг) загружали в копонку Dynamax Marco (Лі,2*250 мм), упакованную сферическим Сіа-сипикаїемем (размер пор - ЗООА, размер частиц - \J мкм) (Ramin Inc Co ) и эпюировали линейным градиентом, используя систему растворителей, состоящую из (А) 0,1% TFA, и (В) 0.1% 1FA в 70%-ном водном ацетонитриле при скорости потока 2,0 мл/мин Все фракции анализировали из чистоту и - время удерживания с помощью аналитической ВРЖХ, описанной ниже. Качество и характеристики элюированил неочищенного и очищенного пептида определяли с помощью аналитической ВРЖХ, ислопьлуя уста новку для жидкостной хроматографии (модели Hewlett) Packard 1090), снабженную диодным матричным детектором, установленным на 220 и 280 нм, и обращенно-фаэовой W-Рогех-Сів-колонкой (4,6x250 мм) (размер пор ЗООА. размер частиц 5 мкм) Скорость потока систем растворителей (А) и (В), описанных выше, поддерживали на уровне 1,2 мл/мин, а разделение осуществляли при комнатной температуре. В большинстве случаев псеедопептидные антагонисты бомбезина очищали с помоіиью повторной хроматографии на той же самой колонке, но с небольшими изменениями в условиях градиентного элюироеания Как показала аналитическая ВЭЖХ, полученные гомогенно очищенные пеп тиды имени чистоту бопее 97% Если необходимо. то может быть осуществлен аминокислотньїй анализ псевдопептидов формулы I с использованием аминокислотного анализатора Beckman (530 и образцов, гидролизованных в течение 20 ч при t тО'С о герметично запаянных, обезгаженных пробирках с 4 М метансульфоновой кислотой, содержащей 0,2% 3-2(2-аминоэтил)-индол С Синтетические промежуточные пептиды 1 Защитные группы боковой цепи В твердофазном синтезе обычно блокируют функциональные группы реактивных боковых групп различных аминокислотных частей или пептидных фрагментов. Функциональные группы боковых цепей могут быть защищены во избежание нежелательных химических реакций, происходящих в этих цепях Поэтому обычно продуцируют промежуточный пептид, который включает в себя каждый из аминокислотных остатков, локализованных в нужной последовательности в пептидной цепи, с боковыми защитными группами, связанными с соответствующими остатками При выборе конкретной защитной группы боковой цепи, которая может быть использована в синтезе пептидов, обычно руководствуются следующими правилами (а) предпочтительно, чтобы защитная группа сохраняла свои защитные свойства в условиях реакции присоединения, ф) защитная (руппа должна быть стабильной по отно 34456 жить гидрохлорид, гидробромид, сульфат, фосфат, фумарат, глюхонаі. таннат, малеат, ацетат, цитрат, бензоат, сукцинат. альгинат. памоат. малат, аскорбат, тертрат и т. п. В основном фармацевтические композиции содержат псевдопептиды формулы І в сочетании со стандартными фармацевтически приемлемыми носителями, из которых наиболее подходящим является физиологический раствор, ХОТИ могут быть испопьзованы и другие известные носители Лечение с испопьзованием указанных фармацевтических композиций может быть осуществлено тем же способом, которым обычно проводят клиническое лечение с использованием других агонистов и антагонистов LHRH или аналогов соматостатинв. Так, например, антагонисты бомбезина формулы I могут быть введены внутривенно, подкожно, внутримышечно, интраназально, с помощью легочных аэрозолей или с использованием форм замедленного всасывания (например, микрокапсулы, микрогранулы или цилиндрические стержнеобразные имплантаты), изготовленных из биопогически разлагаемого полимера (такого, как DL-лактидкогликолид). Изобретение включает в себя также и другие способы введения композиций, например путем капельного вливания, ели-вания с использованием инфуэионного насоса и введения с использованием лекарственных форм пролонгированного высвобождения, например микрокапсул и т. п. Эффективные дозы пептидов формулы I могут варьироваться в зависимости от способа введения и от вида млекопитающего, подвергающегося лечению. При парентеральном введении доза вводимых пептидов может составлять от около 1 до 1000 мг на 1 кг массы тела в день. Этот диапазон доз является предпочтительным, однако в некоторых случаях могут быть использованы и более высокие дозы. Физиологический раствор, содержащий указанный пептид, может быть введен в таком количесгве. которое обеспечивало бы дозу около 0,01 - 0,20 мг/кг массы тела в день, за исключением тех случаев, когда используются формы замедленного всасывания, где количество инъецируемого средства рассчитывается примерно на 15-30 дней или более. В приведенных примерах для идентификации промежуточных пептидов на определенных стадиях синтеза иа юльзуются три кодовых обозначения. Пептид под кодом "1/01/Аи представляет собой промежуточный пептид для пептида "01", полученного в примере 1, который был синтезирован в стадии А синтеза. Аналогично коды "2/08/В" и "4/19/С" относятся к промежуточным пептидам для пептидов "08я и "19", полученных в примерах 2 и 4, которые были синтезированы в стадиях В и С соответственно. Пример 1. Пептид Й 1. D-pGtu-Gln-Trp-Ala-Val-Gly-His-Leu-gsiTac-NH2 2. D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu- es|Tac-NHz 4. Ac-D-Phe-Gln-Trp-Ala-Va!-G1y-His-Leu-esiTac-NHj 5. D-Cpa-Gln^-Aia-Vat-Gly-His-Leu-Bsj-TacNH2 шению к связывающему реагенту и предпочтительно стабильной в условиях реакции присоединения, выбранных для удаленип эльфа-аминозащитной группы н каждой стадии синтеза; (с) защитная группа боковой цепи должна быть удаляемой после завершения синтеза нужной аминокислотной последовательности, причем в таких реакционных условиях, которые не приводили бы к нежелательным изменениям в пептидной цепи. Защитные группы боковой цепи связывают с аминокислотными остаткзми постадийным способом, хорошо известным специалистам. Подходящими боковыми защитными группами для Н является Вот, а для Glu и Cys является But. 2. Синтетические промежуточные пептиды. При этом следует отметить, что в объем изобретения входят промежуточные пептиды на соответствующих стадиях синтеза. Ниже проиллюстрированы промежуточные пептиды 2 на трех стадиях. Стадия А: после присоединения всех аминокислот и смолы: Boc-0-Phe-Gln-Trp-Ala*Val-G!y-His(Bom)-LeuEsi -СуБ(Єи!ї-ВНА-смола Г1/02/А") Стадия В. после удаления - концевой Восгруппы: D-PhG-Gln-Trp-Ala-Val-G)y-Hi5Ala-Val-Gly-His-Leu-Bsi-Tac-NH2. Смесь из 15 мг промежуточного пептида 2/17/1 и 15 мг НОВІ в 0,8 ДМФ при 0е С добавляли к 50 мкл 25% диизопропилкарбодиимида в СНгС12 и размешивали 2 ч при 0° С. В результате образовывалась простая связь пептида 17, связывающая альфа-аминогруппу остатка D-Phe1 с гаммакарбоксильной частью на 3-пропионипьной части остатка Glu2: D-Phe-Glul-J-Trp-Ala-Val-Giy-His-Leu-Bsi-TacЗатем реакционную смесь подвергали очистке с помощью ВРЖХ в соответствии с процедурой 7. 8 табл 2 приводятся времена удерживания для указанных пептидов. Пример 3. Пептид N 3. D-Phe-Gtn-Trp-Ala-Val-Gly-His-Leu-esi-MTac6. D-Cpa-Gln-Trp-Ala-Vat-Gly-His-LeU'Esj-MTac Эти попипептиды могут быть синтезированы иэ общего промежуточного пептида 1-4 (Fmoc-TrpAla-Va!-Gly-His(Bom)-Leu-Bsi-Pen(But)-BHA-cMona). Этот промежуточный пептид постадийно конструировали в соответствии со стандартными методами твердофазного синтеза (описанного в примерах 1 и 2 с использованием бенэгидриламиновой (ВНА) смолы. Так, например, 1,0 г ВНА-смолы (0,55 мМ NHa/r), полученной в соответствии с процедурой 2. обрабатывали 10 мл 10% TEA в CH2CI2 (нейтрализация) (два раза по 3 мин) и б раз промывали 10 мл метилекхлорида, после чего в течение 3 мин смешивали с 1.6 мМ Fmoc-Pen (But) и 1,8 мМ 1-гидроксибенэотриазопом (HOBt) в ДМФ. Затем добавляли 20% 1,3-диизопропилкарбодиимид (DLC) (1,6 мМ) в СН2С1г. Смесь размешивали в шейкере в течение 90 мин при комнатной температуре. Полученный пептид "Fmoc-Pen (But)-BHAсмола" промывали метиленхлоридом, метанолом (каждым по 2 раза) и снова метиленхлоридом (три раза), а затем подвергали тесту Kaizer. Отщепление Fmoc-группы (разблокирование) от пептида "Fmoc-Pen(But)-BHA-CMOna" осуществляли в соответствии с процедурой 4А. Присоединение Fmoc-Leu-CHO осуществляли в соответствии с процедурой 3. Аэ (But)-BHA промывали два раза диметилформамидом. Затем добавляли 1,6 мМ Fmoc-Leu-CHO в ДМФ, содержащем 1% АсОН, а после этого 1,8 мМ NaBH3CN в ДМФ. Реакционную смесь размешивали 60 мин в шейкере, а затем два раза промывали 50% метанолом в воде, два раза 100% метанолом, три раза метиленхлоридом (каждый раз по 1 мин). После удаления Fmoc-группы из пептида Tmoc-Leu-p_si-Pen (ВиО-ВНА-смола" и нейтрализации осуществляли присоединение Fmoc-His (Bom) в соответствии с процедурой 4. Присоединение Fmoc-Gty осуществляли, как описано в процедуре 4. Для этого к ДМФ-раствору (0*С), содержащему 1,5 мМ Fmoc-Gly и 1,65 мМ HOBt. добавляют 20% 1,3*диизопропипкарбодиимид (1,5 мМ) в CH2CI2 и размешивали 15 мин NH2 Указанные пептидные антагонисты бомбезика могут быть синтезированы иэ общего промежуточного пептида 1 - 3 (т. е., Fmoc-Gln-TrpAla-Val-Gly-His(Bom)-Leu-fisi-Cys(But)-BHA-CMona). Этот пептид был получен исходя из ВНА-смолы (1,0 г. 0,55 мМ NHs/r) путем последовательных реакций присоединения, как описано в примере 1, начиная с Fmoc-Cys(But) с последующим присоединением Fmoc-teu*CHO (с NaBH3CN); FmocHis(Bom); и т. п. в соответствии с процедурой, описанной в примере 1 Fmoc-Gln присоединяли к N-концу в соответствии с процедурой 4 и получали промежуточный пептид 1-3. После конечного присоединения Boc-D-Phe или Boc-D-Cpa к промежуточному пептиду 1-3, проводимого в соответствии с процедурой 4, получали промежуточные пептиды "3/3/А", или "З/б/А" соответственно. Удаление Вос-группы осуществляли с использованием 50% TFA в СНгСІг. содержащем 5% меркаптоэтаноп и 5% анизол, в соответствии с процедурой 4. На этой стадии боковую Cys-rpynny промежуточного пептида 3/3/В подвергали циклизации путем окисления с образованием 5-членного гетероциклического кольца (показанного в формуле ИВ), как описано в процедуре 5. Для этого к указанному пептиду добавляли 10 мл смеси 50% АсОН, 10 СНэСНО и ДМФ (1,5: 0,5: 8). Реакционную смесь размешивали в шейкере в течение 10 мин, а затем промывали водой, диметилформамидом и метиленхлоридом (по 3 раза каждым), в результате чего получали промежуточный пептид 3/3/С D-Phe-Trp-Ata-Va(-Gly-His-Leu-esi-MTacВНА-смола Затем промежуточные пептиды 3/3/С и 3/6/С отщепляли от смолы-носителя, как описано в процедуре б, т. е. путем обработки {1 ч. при 0е С) фтороводородом (5 мл) и анизолом (0,25 мл). В этой процедуре также отщеплялась от His защитная Во-rpynna В результате получали конечные нонапептиды Э и 6. 13 34456 зтипэцетатом, экстрагировали 70-80%-ной уксусной кислотой и лиофипиювали, в результате чего получапи неочищенную нонапептидную смолу Реакционную СМРСЬ очищали и получапи пептидный антагонист бомбеэина N 18 Аналогичные стадии удаления защитных групп циклизации разблокированного А9-осгатка и отщепления пептида от ВНА-смолы осуществляли с использованием промежуточных пептидов 4/19/А 4/20/А, 4/21/А и 4 /22 /А. в результате чего получали пептиды 19, 20, 21 и 22 Очистку проводили с помощью ВРЖХ в соответствии с процед/рой 7, используя систему растворителей, состоящую из (A) 0,1%TFA и (В) 1% TFA в 70% ацетонигрила Как показала аналитическая ВРЖХ, очищенные пептиды имели чистоту свыше 97% Времена удерживания для этих пептидов представлены в табп 4 Альтернативно А9-остаток может быть циклизован в растворе путем добавления 25 мг свободного пептида, содержащего структуру -Leu - p_si - Pen-NHj, к 25 мг HOBt в 0.8 мл ледяной уксусной кислоты, смешанной с 100 мкл 10%-ного формальдегида, с последующим размешиванием в течение 30 мин при 0" С В результате этой процедуры получапи пептид, имеющий 5-членную кольцевую структуру Leu - pjsi- DMTac-NH? Пример 5. Pal-Gln-Trp-Ala-Val-Gly-His-Leu- gsi -Cys-NHz D-Pal Gln-Trp-Ala-Val-Gly-His-Leu- p_sj -CysNH2 Tpi-Gln-T'p-Ala-Val-Gty-His-Leu - QS\ -Cys-NH2 D-Tpi-Gin-Trp-AJa-Vat-Glv-His-Leu-pst-CvsNHS Hea-Gln-Trp-Ala-Val-Gly-His-Leu-esj-Cys-NH;. Эти пептиды могут быть синтезированы из промежуточного пептида "Fmoc-Gln-Trp-Ala-ValG(y-His (Born)-Leu- QSI -Cys (But)-BHA-cMona" Fmoc-Leu- p_si -Cys(But)-BHA-cMony получали следующим образом 1,0 г ВНА-смопы (0,55 мМ Н^/г) последовательно связывали с Fmoc-Cys (But) и Fmoc-Leu-CHO в соответствии с процедурами 2 и 3 (см выше) В результате последовательного присоединения Fmoc-His (Pom), Fmoc-Gly, Fmoc-Val, Fmoc-Ala и Fmoc-Trp и Fmoc-GIn получали промежуточный пептид 1-5 (Fmoc-G)n-Tfp-Ala-Val-Gly-His(Bom)-Leu-psi-Cvs (ВЫ)-ВНА-смола). 150 мг - аликвоту этого (общего для всех пептидов данного примера) промежуточного пептида 1-5 подвергали дополнительным реакциям присоединения, проводимым в соответствии с процедурой 4, в резупьтзте чего получали пептиды на смоле-носителе После присоединения Boc-Pal к промежуточному пептиду 1-5 получали пептид Boc-Pal-GlnТгр-Ala-Va! Gly-His (Bom)-Leu- ESI -Cys (But)-BHAсмола ("5/08/А") После присоединения Boc-D-Pal к промежуточному пептиду 1-5 попучали пептид Boc-D-PalGln-Trp-Ala-Val-Gly-HtsiBom)-Leu-ESi-Cys(But)-BHAсмола ("5"09/А") После присоединений Boc-Tpi к промежуточному пептиду 1-5 попучали пептид Boc-Tpi-GlnTrp-Ala-Val-Gly-His(Bom)-Leu-esi-Cys(But)-BHAсмола ("5/12/А*) при охлаждении и 15 мин при комнатной температуре, после чего осадок отфильтровывали, добавляли к смоле и раэмешивапи 60 мин в шейчере Затем таким же образом последовательно присоединяли аминокислотные остатки Fmoc-Val, Гтос-А!а и Fmoc-Trp, в результате чего получали 1,9 t промежуточного пептида 1-4, т е защищенного пептида на НОСИ ТРЛЄ, имеющего структуру Fmoc-Trp- Ala- Val-Gly-Ht
ДивитисяДодаткова інформація
Назва патенту англійськоюPeptide bombesin antagonist, pharmaceutical composition based thereon and method for cancer treatment
Автори англійськоюSchally Andrew, Cai Ren Zhi
Назва патенту російськоюПептид-антагонист бомбезина, фармацевтическая композиция на его основе и способ лечения рака
Автори російськоюСкелли Эндрю, Кей Рен Жи
МПК / Мітки
МПК: C07K 7/08, A61K 38/04, C07K 7/06, A61P 35/00
Мітки: має, активність, основі, композиція, антагоніста, лікування, ссавців, спосіб, бомбезину, пептид, раку, фармацевтична
Код посилання
<a href="https://ua.patents.su/31-34456-peptid-yakijj-maeh-aktivnist-antagonista-bombezinu-farmacevtichna-kompoziciya-na-jjogo-osnovi-i-sposib-likuvannya-raku-u-ssavciv.html" target="_blank" rel="follow" title="База патентів України">Пептид, який має активність антагоніста бомбезину, фармацевтична композиція на його основі і спосіб лікування раку у ссавців</a>
Похідне тетрагідробензимідазолу або його фармацевтично прийнятна сіль, що проявляють активність антагоніста 5-нт3-рецептора, та фармацевтична композиція на його основі

Номер патенту: 27290
Опубліковано: 15.09.2000
Автори: Дзунйа Охморі, Акіра Матсухіса, Такесі Сузукі, Токуо Коіде, Кейдзі Міята, Ісао Янагісава, Мітсуакі Охта
МПК: C01B 7/00, C07D 235/06, C07D 403/12, A61K 31/40, A61K 31/415, C07D 403/08
Мітки: проявляють, сіль, фармацевтична, прийнятна, основі, похідне, тетрагідробензимідазолу, антагоніста, композиція, фармацевтично, активність, 5-нт3-рецептора
Текст:
...%: C 71,77; H 6,13; N 15,13. Масс-спектр (Е1): m/z; 358 (M+). Примеp 17. Гидрохлорид N-[(4,5,6,7- тетрагидробензимидазол-5-ил)карбонил]-фенотиазина Физико-химические свойства: Т.пл. 268 – 270оС. Элементный анализ для C20H17N3О × HCl × 0,5 х х х H2O: Рассчитано, %: C 61,14; H 4,87; N 10,69; Cl 9,02. Найдено, %: C 61,15; H 4,64; N 10,60; Cl 8,59. Масс-спектр (Е1): m/z; 347 (M+, сво бодное соединение). Пример 18....
Спосіб отримання речовини, яка має протипухлинну активність, речовина, яка отримана цим способом та фармацевтична композиція на її основі

Номер патенту: 26321
Опубліковано: 30.08.1999
Автор: Д'арріго Клаудіо
МПК: C07G 17/00, A61K 36/18, A61P 35/00, A61K 35/00
Мітки: протипухлинну, має, отримання, яка, композиція, фармацевтична, основі, отримана, речовини, речовина, активність, цим, способом, спосіб
Формула / Реферат:
1. Способ получения вещества, обладающего противоопухолевой активностью, включающий экстракцию растительного сырья спиртом, упаривание, извлечения, очистку и сушку, отличающийся тем, что в качестве растительного сырья используют незрелые плоды растений семейства Pittosporacea, экстракцию спиртом проводят методом мацерации с получением спиртового экстракта, полученный спиртовой экстракт встряхивают с хлороформом с получением...
Засіб для лікування порушення сну, фармацевтична композиція на його основі

Номер патенту: 26717
Опубліковано: 12.11.1999
Автор: Фло Майкл Едвард
МПК: A61P 25/20, A61K 31/40, C07D 209/14, A61K 31/404, A61K 31/403
Мітки: порушення, основі, фармацевтична, засіб, композиція, лікування, сну
Формула / Реферат:
1. Применение производных мелатонинагде R1 представляет водород C1-C4-алкил или C1-C4-алкоксигруппу;R2 представляет водород или C1-C4-алкил;R3 представляет водород C1-C4-алкил, фенил или замещенный фенил;R4 представляет водород, галогенацетил, C1-C5-алканоил, бензоил или бензоил, замещенный галогеном или метилом;R5 и R6 каждый независимо представляет водород или галоген;R7 представляет водород...
1,2-заміщені 1н-імідазо[4,5-с]хінолін-4-аміни, проміжні сполуки для їх синтезу, антивірусна фармацевтична композиція, спосіб лікування інфікованих ссавців, спосіб індукування біосинтезу інтерферону у ссавців, с

Номер патенту: 32546
Опубліковано: 15.02.2001
Автори: Ліндстрьом Кайл Дж., Крукс Штефен Л., Герстер Джон Ф.
МПК: C07D 471/04, A61K 31/4353
Мітки: композиція, біосинтезу, лікування, фармацевтична, проміжні, сполуки, 1н-імідазо[4,5-с]хінолін-4-аміни, 1,2-заміщені, інтерферону, ссавців, синтезу, індукування, спосіб, інфікованих, антивірусна
Текст:
...от одного до четырех атомов углерода, - галогена и - алкила с нормальной или разветвленной цепью, содержащего от одного до четырех атомов углерода, применяемые как антивирусные препараты или стимуляторы продукции интерферона, или антибпастомные препараты 19. 1, 2-замещённые 1Н-имидазо(4,5-с] хинолин-4амины по п. 18, отличающиеся тем, что Rs содержит от двух до десяти атомов углерода. 20. 1, 2-замещённые 1Н-имидазо14.5-с]...
Похідні 3-циклоалкілпропен-2-аміду, їх таутомерні форми та їх солі, що мають протизапальну активність, спосіб їх одержання та фармацевтична композиція на їх основі
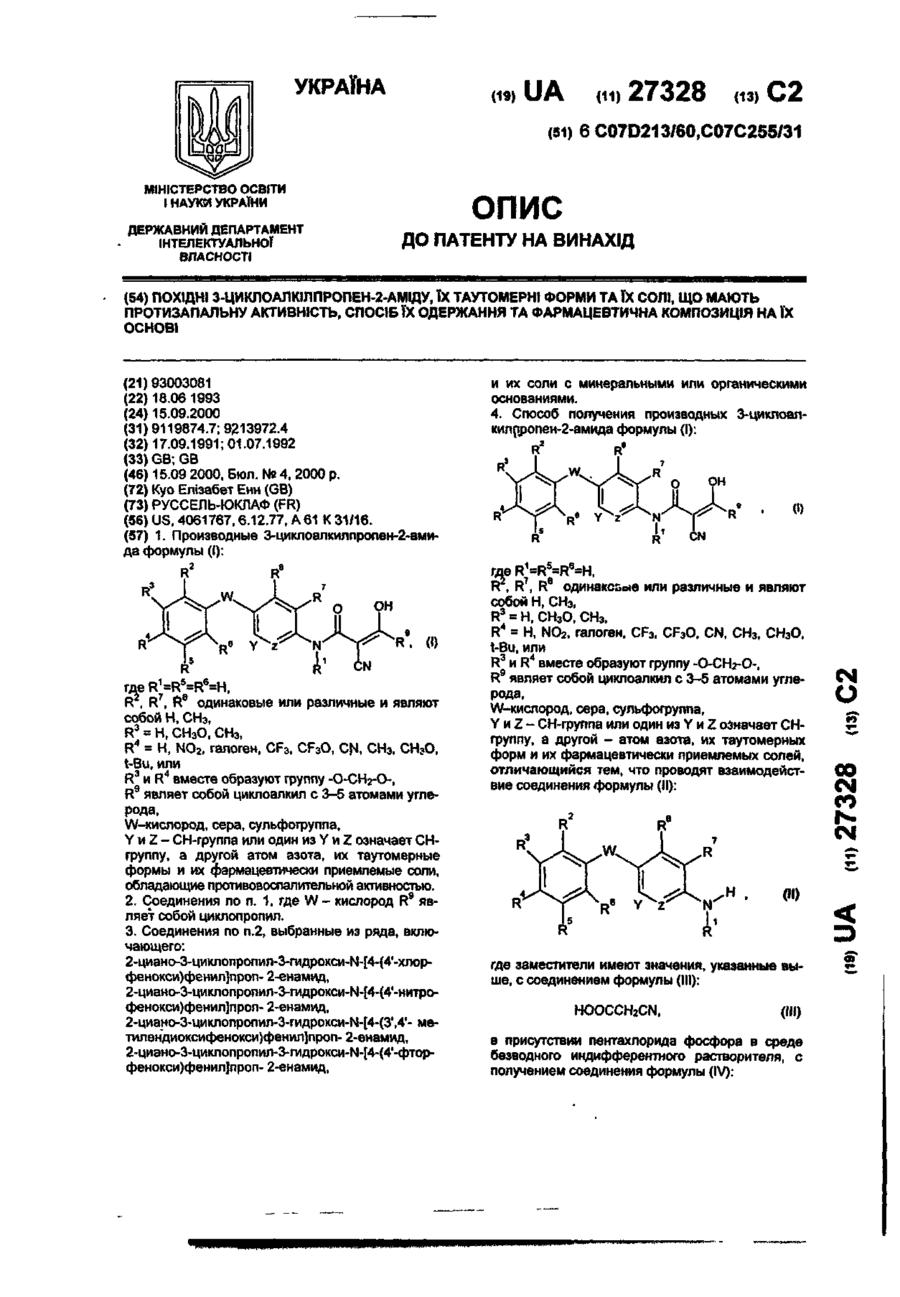
Номер патенту: 27328
Опубліковано: 15.09.2000
Автор: Куо Елізабет Енн
МПК: C07C 255/31
Мітки: солі, активність, протизапальну, похідні, одержання, фармацевтична, форми, таутомерні, спосіб, композиція, 3-циклоалкілпропен-2-аміду, мають, основі
Текст:
...фармакологическими свойствами. В частности, они проявляют противовоспалительное действие Они ингибируют, с одной стороны, воспалительные явления, вызванные раздражающими агентами, а с другой стороны, тормозят реакции запаздывающей суперчувствительности, препятствуя активации иммунных клеток специфическим антигеном. Эти свойства иллюстрированы ниже в опытной части. Эти свойства позволяют использовать новые производные...