Похідні піридопіримідину, їх одержання, їх застосування в терапії
Номер патенту: 95223
Опубліковано: 25.07.2011
Автори: Перро П'єр, Казелла П'єр, Жегам Самір, Буррі Бернар







Формула / Реферат
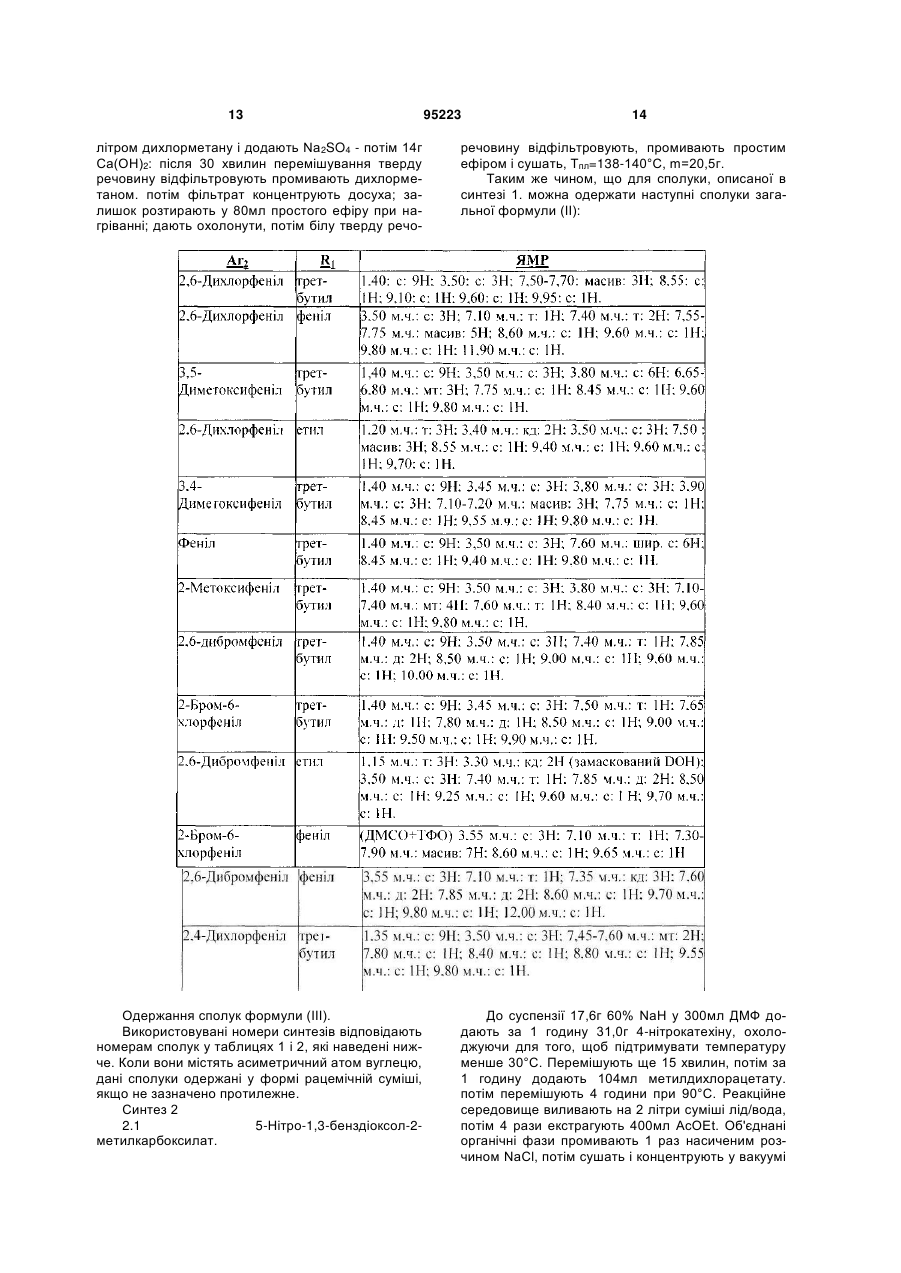
1. Сполука, що відповідає формулі (І):
 , (I)
, (I)
в якій:
- R1 вибраний із групи, яка складається з (С1-С6)алкілу, (С3-С7)циклоалкілу, CH2COR4, фенілу або фенілу, заміщеного гідроксилом і/або галогеном, і/або (С1-С6)алкілом;
- R4 означає гідроксильну групу, (С1-С4)алкоксигрупу, аміногрупу, (С1-С4)алкіламіногрупу, ді(С1-С4)алкіламіногрупу;
- Аr1 означає радикал, вибраний із:
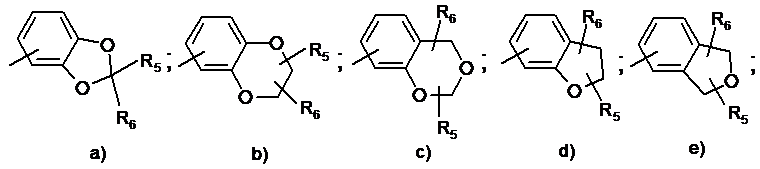
- R5 означає ціаногрупу, гідроксі(С1-С4)алкіл, (С1-С6)алкоксі(С1-С6)алкіл або групу (CH2)nNR7R8, CO2R7, CONHNR7R8, CONR7R8, CONR8OR9, (CH2)nNR7COR8, (CH2)nNR7COOR8;
- R6 означає атом водню, (С1-С4)алкіл або одне із значень R5; або
- R5 і R6, такі як визначені перед цим, пов'язані разом з утворенням циклу, від чотиричленного до семичленного, що містить від 0 до 2 гетероатомів, вибраних із N і О, причому зазначений цикл, що складається із 4-7 ланок, може бути заміщений одним або декількома замісниками, незалежно вибраними з наступних груп: галоген, (С1-С4)алкіл, галогенований (С1-С4)алкіл, гідроксі(С1-С4)алкіл, (С1-С4)алкоксі(С1-С4)алкіл, (CH2)mNR7R8, трет-бутоксикарбоніл;
- R7 і R8 означають, кожен незалежно один від іншого, замісник, вибраний із наступних: Н, (С1-С4)алкіл, (С1-С4)алкіл-ОН, (С3-С7)циклоалкіл, (С3-С7)циклоалкіл-NH2, (С1-С4)алкіл-(С3-С7)циклоалкіл, C(=NH)NH2, SО2(С1-С6)алкіл, SО2-феніл, R8 може також означати трет-бутоксикарбонільну групу або бензилоксикарбонільну групу; або
- R7 і R8 разом з атомом азоту, з яким вони зв'язані, утворюють радикал азетидиніл, піролідиніл, піперидиніл, піперазиніл або морфолініл, причому зазначений радикал є незаміщеним або заміщеним, один або декілька разів, (С1-С6)алкілом, (С1-С4)алкіл-ОН, СОО(С1-С6)алкілом, F;
- R9 означає атом водню або (С1-С4)алкіл;
- Аr2 означає фенільну групу, незаміщену або заміщену від 1 до 5 разів однаковими або різними замісниками, вибраними з атома галогену, (С1-С4)алкільної групи, трифторметильної групи або (С1-С4)алкоксигрупи;
- n означає 1, 2 або 3;
- m означає 0, 1, 2 або 3.
2. Сполука формули (І) за п. 1, яка відрізняється тим, що:
- R1 означає трет-бутил, етил або феніл; і/або
- Аr означає радикал, вибраний із:
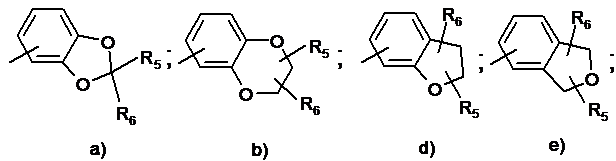 і/або
і/або
- R5 означає групу (CH2)nNR7R8, CONHNR7R8, CONR7R8, гідроксі(С1-С4)алкіл або (CH2)nNR7COR8; і/або
- R6 означає атом водню, метил або групу (CH2)nNR7R8, або гідроксиметил; і/або
- Аr2 означає фенільну групу, заміщену 1-2 замісниками, незалежно вибраними з галогену, (С1-С4)алкілу, (С1-С4)алкоксигрупи;
- n, R7 і R8 такі, як визначені перед цим для сполук формули (І);
у формі основи або солі приєднання з кислотою, а також у формі гідрату або сольвату.
3. Сполука за будь-яким із попередніх пунктів, яка відрізняється тим, що R5 вибраний із (CH2)nNR7R8, CONR7R8 і (CH2)nNR7COR8.
4. Сполука за будь-яким із попередніх пунктів, яка відрізняється тим, що вона знаходиться у формі:
нехіральній або
рацемічній, або
збагаченій одним стереоізомером, або
збагаченій одним енантіомером;
і тим, що вона може бути сольватована або гідратована, і тим, що вона може бути перетворена у сіль.
5. Проміжний продукт синтезу для одержання продуктів згідно з будь-яким із пп. 1-4, який відповідає наступній загальній формулі:
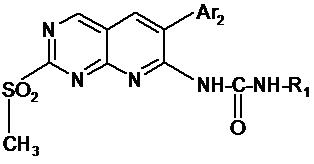 , (ІІ)
, (ІІ)
в якій R1 і Аr2 такі, як визначено в одному з пп. 1-4.
6. Проміжний продукт синтезу для одержання продуктів згідно з будь-яким із пп. 1-5, який відповідає наступній загальній формулі:
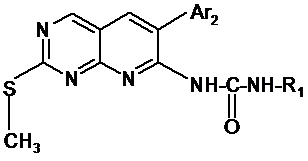 ,
,
в якій R1 і Аr2 такі, як визначено в одному з пп. 1-5.
7. Проміжний продукт синтезу за п. 5 або п. 6, який відрізняється тим, що замісник Аr2 вибраний із наступних: феніл, 2-метоксифеніл, 2,6-дихлорфеніл, 3,5-диметоксифеніл, 3,4-диметоксифеніл, 2,6-дибромфеніл, 2-бром-6-хлорфеніл, 2,4-дихлорфеніл і 3,5-дихлорфеніл.
8. Проміжний продукт синтезу за п. 5 або п. 6, який відрізняється тим, що замісник R1 вибраний із наступних: етил, трет-бутил і феніл.
9. Спосіб одержання сполуки формули (І) за будь-яким із пп. 1-3, який відрізняється тим, що вводять у реакцію зі сполукою формули:
, (ІІ)
в якій R1 і Ar2 такі, як визначені для сполуки формули (І),
амін формули
Ar'1NH2, (III)
в якій Аr'1 являє собою Аr1, такий як визначений для (І);
за необхідності перетворюють групу Аr'1 сполуки, одержаної таким чином, у групу Аr1.
10. Спосіб одержання сполуки формули (І) за будь-яким із пп. 1-3, який відрізняється тим, що вводять у реакцію:
і) сполуку формули:
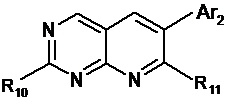 , (IIb)
, (IIb)
в якій Ar2 має значення, визначені для сполуки формули (І);
R10 означає групу, що видаляється, таку як (а) галоген, зокрема Сl або Вr, або (b) aлкіл-S(O)m-, в якій m=0, 1 або 2; в якій R11 означає NHC(=R12)-NH-R1, з R12=O або S; і
іі) амін формули
Ar'1NH2, (III)
в якій Аr'1 являє собою Аr1, такий як визначений для сполуки формули (І);
за необхідності перетворюють групу Аr'1 сполуки, одержаної таким чином, у групу Аr1;
в якому:
(a) коли R10 являє собою галоген або алкіл-S(О)m- з m=2, реакцію здійснюють у розчиннику, переважно полярному:
(і) наприклад, тетрагідрофурані, диметилсульфоксиді або етанолі, у відомих випадках, у присутності слідової кількості кислоти, такої як соляна кислота; або
(іі) у диметилсульфоксиді в присутності сильної основи, такої як t-BuOK;
при температурі, що знаходиться в інтервалі від кімнатної температури до температури кипіння розчинника;
(b) коли R10 являє собою алкіл-S(О)m- з m=0 або 1, реакцію здійснюють з
Ar'1NH2 (III)
у розплавленому стані при 200 °С;
у разі необхідності, аміногрупи, присутні в групі Аr'1 сполуки (III), заздалегідь перетворюють у сольові або захищають.
11. Лікарський засіб, який відрізняється тим, що він містить сполуку формули (І) за будь-яким із пп. 1-2 або її сіль приєднання з фармацевтично прийнятною кислотою або гідратом, або сольват сполуки формули (І).
12. Фармацевтична композиція, яка відрізняється тим, що містить сполуку формули (І) за будь-яким із пп. 1-3 або фармацевтично прийнятну сіль, гідрат або сольват згаданої сполуки, а також принаймні один фармацевтично прийнятний ексципієнт.
13. Фармацевтична композиція за п. 12, яка відрізняється тим, що вона додатково містить один (або декілька) інший(их) цитостатично(их) активний(их) компонент (компонентів).
14. Застосування сполуки формули (І) за будь-яким із пп. 1-3 для одержання лікарського засобу, призначеного для лікування і профілактики захворювань, що викликаються або загострюються проліферацією клітин.
15. Застосування за п. 14 для профілактики і лікування лейкозу, первинних і метастатичних солідних пухлин, карцином і раку.
Текст
1. Сполука, що відповідає формулі (І): Ar2 N 2 (19) 1 ; 3 95223 - R9 означає атом водню або (С1-С4)алкіл; - Аr2 означає фенільну групу, незаміщену або заміщену від 1 до 5 разів однаковими або різними замісниками, вибраними з атома галогену, (С1С4)алкільної групи, трифторметильної групи або (С1-С4)алкоксигрупи; - n означає 1, 2 або 3; - m означає 0, 1, 2 або 3. 2. Сполука формули (І) за п. 1, яка відрізняється тим, що: - R1 означає трет-бутил, етил або феніл; і/або - Аr означає радикал, вибраний із: R6 O O R5 ; O O R6 a) R5 ; R6 ; O O R5 d) b) R6 N N R5 e) O , (ІІ) в якій R1 і Аr2 такі, як визначено в одному з пп. 1-4. 6. Проміжний продукт синтезу для одержання продуктів згідно з будь-яким із пп. 1-5, який відповідає наступній загальній формулі: Ar2 N S CH3 N N Ar2 N N N NH-C-NH-R1 ; NH-C-NH-R1 CH3 наступних: феніл, 2-метоксифеніл, 2,6дихлорфеніл, 3,5-диметоксифеніл, 3,4диметоксифеніл, 2,6-дибромфеніл, 2-бром-6хлорфеніл, 2,4-дихлорфеніл і 3,5-дихлорфеніл. 8. Проміжний продукт синтезу за п. 5 або п. 6, який відрізняється тим, що замісник R1 вибраний із наступних: етил, трет-бутил і феніл. 9. Спосіб одержання сполуки формули (І) за будьяким із пп. 1-3, який відрізняється тим, що вводять у реакцію зі сполукою формули: SO2 і/або - R5 означає групу (CH2)nNR7R8, CONHNR7R8, CONR7R8, гідроксі(С1-С4)алкіл або (CH2)nNR7COR8; і/або - R6 означає атом водню, метил або групу (CH2)nNR7R8, або гідроксиметил; і/або - Аr2 означає фенільну групу, заміщену 1-2 замісниками, незалежно вибраними з галогену, (С1С4)алкілу, (С1-С4)алкоксигрупи; - n, R7 і R8 такі, як визначені перед цим для сполук формули (І); у формі основи або солі приєднання з кислотою, а також у формі гідрату або сольвату. 3. Сполука за будь-яким із попередніх пунктів, яка відрізняється тим, що R5 вибраний із (CH2)nNR7R8, CONR7R8 і (CH2)nNR7COR8. 4. Сполука за будь-яким із попередніх пунктів, яка відрізняється тим, що вона знаходиться у формі: нехіральній або рацемічній, або збагаченій одним стереоізомером, або збагаченій одним енантіомером; і тим, що вона може бути сольватована або гідратована, і тим, що вона може бути перетворена у сіль. 5. Проміжний продукт синтезу для одержання продуктів згідно з будь-яким із пп. 1-4, який відповідає наступній загальній формулі: Ar2 N SO2 4 NH-C-NH-R1 O , в якій R1 і Аr2 такі, як визначено в одному з пп. 1-5. 7. Проміжний продукт синтезу за п. 5 або п. 6, який відрізняється тим, що замісник Аr2 вибраний із O CH3 , (ІІ) в якій R1 і Ar2 такі, як визначені для сполуки формули (І), амін формули Ar'1NH2, (III) в якій Аr'1 являє собою Аr1, такий як визначений для (І); за необхідності перетворюють групу Аr'1 сполуки, одержаної таким чином, у групу Аr1. 10. Спосіб одержання сполуки формули (І) за будь-яким із пп. 1-3, який відрізняється тим, що вводять у реакцію: і) сполуку формули: Ar2 N R10 N N R11 , (IIb) в якій Ar2 має значення, визначені для сполуки формули (І); R10 означає групу, що видаляється, таку як (а) галоген, зокрема Сl або Вr, або (b) aлкіл-S(O)m-, в якій m=0, 1 або 2; в якій R11 означає NHC(=R12)NH-R1, з R12=O або S; і іі) амін формули Ar'1NH2, (III) в якій Аr'1 являє собою Аr1, такий як визначений для сполуки формули (І); за необхідності перетворюють групу Аr'1 сполуки, одержаної таким чином, у групу Аr1; в якому: (a) коли R10 являє собою галоген або алкіл-S(О)mз m=2, реакцію здійснюють у розчиннику, переважно полярному: (і) наприклад, тетрагідрофурані, диметилсульфоксиді або етанолі, у відомих випадках, у присутності слідової кількості кислоти, такої як соляна кислота; або (іі) у диметилсульфоксиді в присутності сильної основи, такої як t-BuOK; при температурі, що знаходиться в інтервалі від кімнатної температури до температури кипіння розчинника; (b) коли R10 являє собою алкіл-S(О)m- з m=0 або 1, реакцію здійснюють з Ar'1NH2 (III) у розплавленому стані при 200 °С; у разі необхідності, аміногрупи, присутні в групі Аr'1 сполуки (III), заздалегідь перетворюють у сольові або захищають. 5 95223 6 11. Лікарський засіб, який відрізняється тим, що він містить сполуку формули (І) за будь-яким із пп. 1-2 або її сіль приєднання з фармацевтично прийнятною кислотою або гідратом, або сольват сполуки формули (І). 12. Фармацевтична композиція, яка відрізняється тим, що містить сполуку формули (І) за будь-яким із пп. 1-3 або фармацевтично прийнятну сіль, гідрат або сольват згаданої сполуки, а також принаймні один фармацевтично прийнятний ексципієнт. 13. Фармацевтична композиція за п. 12, яка відрізняється тим, що вона додатково містить один (або декілька) інший(их) цитостатично(их) активний(их) компонент (компонентів). 14. Застосування сполуки формули (І) за будьяким із пп. 1-3 для одержання лікарського засобу, призначеного для лікування і профілактики захворювань, що викликаються або загострюються проліферацією клітин. 15. Застосування за п. 14 для профілактики і лікування лейкозу, первинних і метастатичних солідних пухлин, карцином і раку. Об’єктом даного винаходу є похідні піридо[2,3d]піримідину. їхнє одержання та їхнє застосування в терапії. Сполуки, що є похідними піридо[2,3d]піримідину. описані в патентних заявках WO 01/55147 і WO 03/000011 і в патентах ЕР-В-790997 і US 5733913. Ці сполуки є потенційно придатними для лікування порушень проліферації клітин. Згідно з першим аспектом, об’єктом даного винаходу с сполуки, що відповідають формулі (І): сі(С1-С4)алкіл, (С1-С4)алкоксі(С1-С4)алкіл. (CH2)mNR7R8, трет-бутоксикарбоніл; - R7 і R8 означають, кожен незалежно один від іншого, замісник, вибраний із таких: Н, (С1С4)алкіл, (С1-С4)алкіл-ОН, (С3-С7)циклоалкіл, (С3С7)циклоалкіл-NН2, (С1-С4)алкіл-(С3-С7)циклоалкіл, C(=NH)NH2, SО2(С1-С6)алкіл, SО2-феніл, R8 може також означати трет-бутоксикарбонільну групу або бензилоксикарбонільну групу; - або R7 і R8 разом з атомом азоту, з яким вони зв'язані, утворюють радикал азетидиніл, піролідиніл, піперидиніл, піперазиніл або морфолініл, причому зазначений радикал є незаміщеним або заміщеним, один або декілька разів, (С1 С6)алкілом. (С1-С4)алкіл-ОН. СОО(С1-С6)алкілом, F; - R9 означає атом водню або (С1-С4)алкіл; - Аr2 означає фенільну групу, незаміщену або заміщену від 1 до 5 разів однаковими або різними замісниками, вибраними з атома галогену. (С1С4)алкільної групи, трифторметильної ірупи або (С1-С4)алкоксиірупи; - n означає 1, 2 або 3: - m означає 0, 1, 2 або 3. Сполуки формули (І) можуть містити один або декілька асиметричних атомів вуглецю. Отже, вони можуть існувати у формі енантіомерів або діастереоізомерів. Згадані енантіомери, діастереоізомери, а також їхні суміші, включаючи рацемічні суміші, є частиною винаходу. Сполуки формули (І) можуть існувати у формі основ або солей приєднання з кислотами. Коли сполуки формули (І) містять вільні кислотні групи, наприклад, карбоксильну, сульфонову. фосфонову. згадані групи можуть бути перетворені в сіль за допомогою основ з одержанням солей приєднання. Такі солі приєднання становлять частину винаходу. Переважно, солі приєднання з кислотами або основами одержують, відповідно, із фармацевтично прийнятими кислотами або основами, але солі з іншими кислотами або основами, що використовуються, наприклад, для очищення або виділення сполук формули (І), також є частиною винаходу. Так само, сполуки формули (І) можуть існувати у формі гідратів або сольватів, а саме, у формі асоціатів або комбінацій з однією або декількома молекулами води або з розчинником. Такі гідрати або сольвати також с частиною винаходу. в якій: - R1 вибраний із групи, яка складається з (С1С6)алкілу. (С3-С7)циклоалкілу. CH2COR4, фенілу або фенілу, заміщеного гідроксилом і/або галогеном і/або (С1-С6)алкілом; - R4 означає гідроксильну групу, (С1С4)алкоксигрупу. аміногрупу. (С1С4)алкіламіногрупу, ді(С1-С4)алкіламіногрупу; - Аг1 означає радикал, вибраний із: - R5 означає ціаногрупу, гідроксі(С1-С4)алкіл. (С1-С6)алкоксі(С1-С6)алкіл або групу (CH2)nNR7R8, CO2R7 CONHNR7R8, CONR7R8. CONR8OR9. (CH2)nNR7CORx, (CH2)nNR7COOR8: - R6 означає атом водню, (С1-С4)алкіл або одне зі значень R^; - або. R5 і R6, такі як визначені перед цим. пов’язані разом з утворенням циклу, від чотирьохчленного до семичленною, що містить від 0 до 2 гетероатомів, вибраних із N і О. причому зазначений цикл, що складається із 4-7 ланцюгів, може бути заміщений одним або декількома замісниками, незалежно вибраними з таких груп: галоген. (С1-С4)алкіл, галогенований (С1-С4)алкіл. гідрок 7 У рамках даного винаходу мають на увазі: - атом галогену: фтор, хлор, бром або йод; - алкільна група: насичена аліфатична група, пряма або розгалужена. Як приклад можна назвати такі групи: метил, н-пропіл, н-бутил, н-пентил, нгексил, н-гептил, 1-метилетил, 1-метилпропіл, 2метилпропіл, 1,1-диметил, 1-метилбутил, 2метилбутил, 3-метилбутил, 1,1-диметилпропіл, 1,2-диметилпропіл, 2,2-диметилпропіл, 1метилпентил, 2-метилпентил, 3-метилпентил, 4метилпентил, 1,1-диметилбутил, 1,2диметилбутил, 1,3-диметилбутил, 2,2диметилбутил, 2,3-диметилбутил, 3,3диметилбутил, 1,1,2-триметилпропіл, 1,2,2триметилпропіл, 1-етил-1-метилпропіл, 1-етил-2метилпропіл, 1-етилбутил, 2-етилбутил, 1метилгексил, 2-метилгексил, 3-метилгексил, 4метилгексил, 5-метилгексил, 1,1-диметилпентил, 1,2-диметилпентил, 1,3-диметилпентил, 1,4диметилпентил, 2,2-диметилпентил, 2,3диметилпентил, 2,4-диметилпентил, 3,3диметилпентил, 3,4-диметилпентил, 4.4диметилпентил, 1,1,2-триметилбутил, 1,1,3триметилбутил, 1,2,2-триметилбутил, 1,2,3триметилбутил, 1,3,3-триметилбутил, 2,2,3триметилбутил, 2,3,3-триметилбутил, 1,1,2,2тетраметилпропіл, 1-етилпентил, 2-етилпентил, 3етилпентил, 1-етил-1-метилбутил, 1-етил-2метилбутил, 1-етил-3-метилбутил, 2-етил-1метилбутил, 2-етил-2метилбутил, 2-етил-3метилбутил, 1-пропілбутил, 1-(1-метилетил)бутил, 1-(1-метилетил)-2-метилпропіл: - циклоалкільна група: циклопропіл, циклобутил, циклопентил, циклогексил. Циклогептил, біцикло[2,2,1]гептил, циклооктил, біцикло[2,2,2]октил, біцикло[3,2,1]октил, адамантил. Зі сполук формули (І), предметів винаходу, можна назвати переважні сполуки, які визначаються таким чином: - R1 означає трет-бутил. етил або феніл: - і/або - Аr, означає радикал, вибраний із: - і/або R5 означає групу (CH2)nNR7R8. CONHNR7R8, CONR7R8, гідроксі(С1-С4)алкіл або (CH2)nNR7COR8; - і/або R6 означає атом водню, метил або групу (CH2)nNR7R8 або гідроксиметил; - і/або Аr2 означає арильну групу, заміщену 1-2 замісниками, незалежно вибраними з галогену, (С1-С4)алкілу, (С1-С4)алкоксигрупи; - n, m, R7 і R8 такі як визначені перед цим для сполук формули (І): у формі основи або солі приєднання кислоти, а також у формі гідрату або сольвату. 95223 8 Продукти згідно з винаходом, переважно, будуть мати замісник R5, вибраний із (CH2)nNR7R8. CONR7R8 і (CH2)nNR7COR8. Продукт згідно з винаходом може знаходитися в нехіральній або рацемічній, або збагаченій одним стереоізомером, або збагаченій одним енантіомером формі; може знаходитися в сольватованій або гідратованій формі, і може бути перетворений у сіль. Відповідно до другого аспекту, винахід стосується проміжних продуктів синтезу, що використовуються при одержанні продуктів згідно з його першим аспектом, причому зазначені проміжні продукти відповідають такій загальній формулі: в якій R1 і Аr2 такі, як визначені перед цим. Відповідно до третього аспекту, винахід стосується одержання проміжних продуктів згідно з його першим і його другим аспектами, що відповідають такій загальній формулі: в якій R1 і Аr2 такі, як визначені перед цим. Відповідно до четвертого аспекту, винахід стосується одержання проміжних продуктів згідно з його першим, його другим і його третім аспектами, що відповідають такій загальній формулі: в якій Аг2 такий, як визначений перед цим. Проміжні продукти синтезу згідно з другим, третім і четвертим аспектами винаходу, містять замісник Аr2, вибраний із наступних: феніл, 2метоксифеніл, 2,6-дихлорфеніл, 3,5диметоксифеніл, 3,4-диметоксифеніл, 2,6дибромфеніл, 2-бром-6-хлорфеніл, 2,4дихлорфеніл і 3,5-дихлорфеніл. Проміжні продукти синтезу згідно з другим і третім аспектами винаходу містять замісник Аr1. вибраний із наступних: етил, трет-бутил і феніл. Відповідно до винаходу, сполуки формули (І) можна одержати згідно зі способом, який відрізняється тим. що вводять у реакцію: і) сполуку формули: в якій R10 означає групу, що видаляється, таку як (а) галоген, зокрема. СІ або Вr. або (b) алкілS(О)m- з m=0, 1 або 2: R,, означає NHC(=R12)-NHR1, з R12=O або S:i іі) амін формули Ar'1NH2 (Ill), в якій Аr'1 являє собою Аr1, такий як визначений для (І), або попередника Аr1: у випадку необхідності перетворює 9 95223 10 групу Аr'1 сполуки, одержаної таким чином, у групу Аr1. Коли R10 являє собою галоген або алкіл-S(О)m з m=2, реакцію здійснюють у розчиннику, переважно, полярному: (і) наприклад, тетрагідрофурані. диметилсульфоксиді або етанолі, у відомих випадках, у присутності слідової кількості кислоти, такої як соляна кислота: або (іі) у диметилсульфоксиді у присутності сильної основи, такої як t-BuOK: при температурі, що знаходиться в інтервалі від кімнатної температури до температури кипіння розчинника. Коли R10 являє собою алкіл-S(О)m- з m=1, можна здійснити реакцію з Ar'1NH2 (III) у розплаві, переважно, при температурі, близькій до 200°С, без каталізатора. За необхідності, аміногрупи, присутні в групі Аr'1 сполуки (III), заздалегідь перетворюють у сольові або захищають. Під попередником Аr1 мають на увазі групу a), b), c), d) або e), таку як визначену вище для (І), в якій замісники R5 і/або R6 є такими, як визначені вище для (І), або є попередниками R5 і/або R6. Сполуки формули (II) одержують, дотримуючись методики, описаної в європейському патенті 790997 і американському патенті US 5733913, як наведено на схемі 1. що наведена нижче: м-ПХБК: мета-перхлорбензойна кислота. Аміни формули (III) відомі або одержані відомими способами, виходячи з відповідних нітропохідних Ar'1NO2 (IV). відновленням або (і) у кислому середовищі в присутності металу, такого як залізо або цинк у порошкоподібному стані, або (іі) воднем у присутності каталізатора, такого як Pd/C. Сполуки формули (IV) відомі або одержані відомими способами. Так, 5-нітро-1,3-бенздіоксоли, монозаміщені у положенні 2 групою Я5=метоксикарбоніл, можуть бути одержані дією метилдихлорацетату на 4нітрокатехін(4-нітробензол-1,2-діол). 5-Нітро-1,3-бенздіоксоли, гем-дизаміщені в положенні 2, можуть бути одержані, згідно з Pharmazie, 2003, 58 (1), 13-17, дією етилдиброммалонату на 4-нітрокатехін(4-нітробензол-1,2діол). 7-Нітро-2,3-дигідро-1,4-бенздіоксини, заміщені в положенні 2 або 3 групами R5 і R6, можуть бути одержані згідно з методом, описаним у міжнародній заявці на патент WO 01/021577, виходячи з 4нітрокатехіну або за допомогою відомих хімічних перетворень. 1,3-Дигідро-2-бензофуран-5-амін описаний у J. Med. Chem., 1978, 2J, 965-978; 4Н-1,3-бенздіоксин6-амін описаний у J. Org. Chem.. 1994. 59 (4). 754757; 4Н-1,3-бенздіоксин-7-амін описаний у Chimie Therapeutique. 1972. 7,443-449. Діють відомими методами, такими як описані в березневому номері Advanced Organic Chemistry, 5th Edition. 2005, ISBN 0471585890. для того, щоб перетворити групу R5 і/або R6 сполук формули (IV). відповідно, у замісники R5 і/або R6. що є бажаними для сполук формули (І). Так само, можна перетворити групи R5 і/або R6 сполук формули (І) для того, щоб одержати нові сполуки формули (І), що несуть бажані замісники R5 і/або R6. Так. група R5=CO2Me дозволяє одержати сполуки формули (IV) або (І), в яких R5 являє собою групу СО2Н. CN. СН2OH, CONR7R8, CONHNR7R8, CONR8OR9, CH2NR7R8. методами, відомими фахівцеві. Виходячи зі сполуки формули (IV) або (І), що містить групу R5=(CH2)nOH з n=1, 2 або 3, можна одержати сполуку формули (IV) або (І), в якій R5=метилхлориду, дією метилхлориду, потім сполуку формули (IV) або (І), в якій R5=-CH2NR7R8, дією HNR7R8. при цьому R7 і R8 такі, як визначені для сполук формули (І). Сполуку згідно з винаходом одержують у рацемічній формі; потім можна одержати оптично чисті ізомери, використовуючи методи поділу, відомі фахівцеві, такі як кристалізація внаслідок утворення солей з хіральними агентами. Можна також одержати сполуку згідно з винаходом в оптично чистій формі, використовуючи методи асиметричного або стереоспецифічного синтезу, або хроматографічні методики із застосуванням хіральної фази. Крім того, продукти згідно з винаходом можуть бути розділені через утворення діастереоізомерів, їхній поділ, потім розщеплення фармако 11 логічно застосовного діастереоізомера з утворенням його енантіомерно чистого продукту. Ферментативні методики також можуть бути застосовані. Можуть бути використані відомі додаткові методики поділу. Вони включають у себе методики, опубліковані в книзі: Enantiomers, Racemates and Resolutions. John Wiley and Sons, New York (1981). Починаючи з одержання проміжних продуктів синтезу, сполуки згідно з винаходом можуть бути також одержані у формі, збагаченій одним стереоізомером. Так, поділ енантіомерів амінів формули (III) або нітрованих попередників (IV) може бути здійснене одним із зазначених методів. Наступні приклади описують одержання деяких проміжних продуктів і сполук згідно з винаходом. Наведені приклади не є такими, що обмежують і служать тільки для ілюстрації даного винаходу. У прикладах використовують такі скорочення: Тпл: температура плавлення Вос: трет-бутоксикарбоніл ВОР: гексафторфосфат бензотриазол-1ілокситрис(диметиламіно)фосфонію (БОФ) ТГФ (THF): тетрагідрофуран ТК (ТА): температура кімнатна ДХМ (DCM): дихлорметан МеОН: метанол ДЦКДІ (DCCI): дициклогексилкарбодиімід ДІПЕА (DIPEA): діізопропілетиламін KHSO4/K2SO4: 5% розчин KHSO4/K2SO4 Z: бензилоксикарбоніл Спектри ядерного магніїною резонансу (ЯМР) протонів зареєстровані при 200МГц або при 250МЕц у DMSO-d6. якщо не зазначено протилежне. Сигнал DMSO-d6 знаходиться при 2,5м.ч. і служить стандартом порівняння. При інтерпретації спектрів використовують наступні скорочення: с синглет. d: дублет, t: триплет, m: масив, ml: мультиплет, se: широкий синглет, dd: дублет дублетів, qd: квартет, qt: квінтет. Одержання сполуки формули (II) Синтез 1 N(трет-Бутил)-N'-[6-(2,6-дихлорфеніл)-2(метилсульфоніл)піридо[2,3-d]піримидин-7іл]карбамід. 1.1.4-Аміно-2-(метилтіо)піримідин-5етилкарбоксилат. До суспензії 50,7г 4-хлор-2(метилтіо)піримідин-5-етилкарбоксилату в 400мл ЕtOН за 20 хвилин додають, підтримуючи температуру при 20°С. 140мл 20% розчину NH4OH. Після 20 годин перемішування при кімнатній температурі реакційне середовище концентрують у вакуумі майже досуха, потім залишок витягують у 350мл води, перемішують 20 хвилин, фільтрують, промивають 3×60мл водою, потім сушать у вакуумі у присутності Р2О5. Одержують білу тверду речовину. Tпл=134-135°C. m=39,9г. 1.2 [4-Аміно-2-(метилтіо)піримідин-5іл]метанол. До 39,68г складного ефіру, одержаного на попередній стадії, розчиненого в 1л ТГФ, за 45 хвилин додають 210мл розчину 1М LiAlH4 у ТГФ. підтримуючи температуру менше 30°С. Перемішують ще 1 годину, потім знижують температуру до 5°С і 95223 12 послідовно, по краплях, додають 9мл води. 6,5мл 5н. гідроксиду натрію, потім 32мл води. Після 10 хвилин перемішування тверду речовину відфільтровують, потім промивають ТГФ. Фільтрат концентрують у вакуумі досуха, потім залишок знову розчиняють у 600мл толуолу при кип’ятінні, швидко фільтрують у гарячому стані, щоб видалити невелику кількість нерозчинної речовини, і дають фільтрату охолонути протягом ночі. Одержані білі кристали відфільтровують. промивають невеликою кількісно толуолу, потім простого ефіру і сушать. Tпл=124-127°C. m=23,9г. 1.3 4-Аміно-2-(метилтіо)піримідин-5карбальдегід. До суспензії 23,8г спирту, одержаного на попередній стадії, у 1600мл хлороформу за 2 хвилини додають 79.5 г активного МnО2 і перемішують протягом 1 ночі при кімнатній температурі: тверду речовину відфільтровують, промивають 3×75мл СНСl3 і фільтрат концентрують у вакуумі досуха; білий твердий залишок витягують простим ефіром, фільтрують, сушать, Тпл=184-186°С, m=21,05г. 1.4 6-(2,6-Дихлорфеніл)-2(метилтіо)піридо[2,3-d]піримідин-7-амін. До 21г альдегіду, одержаного на попередній стадії, розчиненого у 240мл ДМФ (DMF) та охолодженого до 5°С, за 5 хвилин додають 5,47г 60% NaH, потім за 20 хвилин, невеликими порціями, 29,05г 2,6-дихлорфенілацетонітрилу. Перемішування продовжують протягом 30 хвилин при 5°С, потім протягом ночі при кімнатній температурі. Реакційне середовище охолоджують до 5°С і додають 65мл насиченого розчину NH4CІ. потім 500мл суміші вода/лід; утворюється червоний осад, який відфільтровують, промивають 2 рази водою, максимально зневоднюють, промивають простим ефіром. 100мл хлороформу, потім знову простим ефіром; після сушіння одержують бежеву тверду речовину, Тпл=250-253°С, m=29,92 г. Фази простого ефіру і хлороформу, що утворилися при промиванні, концентрують досуха, витягують невеликою кількістю хлороформу, до якого додають простий ефір: одержують другу порцію 3,15г, мзагальна=33,07г. 1.5 N-(mpem-Бутил)-N'-[6-(2,6-діхлорфеніп)-2(метилтіо)піридо[2,3-d]піримідин-7-іл]карбамід. До розчину 29,9г аміну, одержаного вище, у 300мл ДМФ за 10 хвилин додають, підтримуючи температуру менше 25°С, 4г 60% NaH; перемішають ще 20 хвилин, потім за 20 хвилин додають 12,2мл трет-бутилізоціанату. потім перемішують протягом ночі. Реакційне середовище повільно виливають на 800мл суміші вода/лід+100мл 6N НСl: осад, що утворився, відфільтровують, промивають простим ефіром і сушать. Одержують бежевого кольору тверду речовину. Тпл=195-196°С (розкл.), m=26,5г. 1.6 N(трет-Бутил)-N'-[6-(2,6-дихлорфеніл)-2(метилсульфоніл)піридо[2,3-d]піримідин-7іл]карбамід. До розчину 21,95г карбаміду, одержаного вище, у 300мл хлороформу за 25 хвилин додають. підтримуючи температуру менше 25°С, 27г метаперхлорбензойної кислоти. Утворюється осад. Через 2 години реакційне середовище розводять 1 13 95223 14 літром дихлорметану і додають Na2SO4 - потім 14г Са(ОН)2: після 30 хвилин перемішування тверду речовину відфільтровують промивають дихлорметаном. потім фільтрат концентрують досуха; залишок розтирають у 80мл простого ефіру при нагріванні; дають охолонути, потім білу тверду речо речовину відфільтровують, промивають простим ефіром і сушать, Тпл=138-140°С, m=20,5г. Таким же чином, що для сполуки, описаної в синтезі 1. можна одержати наступні сполуки загальної формули (II): Одержання сполук формули (III). Використовувані номери синтезів відповідають номерам сполук у таблицях 1 і 2, які наведені нижче. Коли вони містять асиметричний атом вуглецю, дані сполуки одержані у формі рацемічній суміші, якщо не зазначено протилежне. Синтез 2 2.1 5-Нітро-1,3-бенздіоксол-2метилкарбоксилат. До суспензії 17,6г 60% NaH у 300мл ДМФ додають за 1 годину 31,0г 4-нітрокатехіну, охолоджуючи для того, щоб підтримувати температуру менше 30°С. Перемішують ще 15 хвилин, потім за 1 годину додають 104мл метилдихлорацетату. потім перемішують 4 години при 90°С. Реакційне середовище виливають на 2 літри суміші лід/вода, потім 4 рази екстрагують 400мл AcOEt. Об'єднані органічні фази промивають 1 раз насиченим розчином NaCl, потім сушать і концентрують у вакуумі 15 (випарювання ДМФ). Залишок витягують у суміш AcOEt/H2O і доводять до рН=8,6 за допомогою Na2CO3; органічну фазу дскантують. промивають насиченим NaHCO> H2O, 5% KHSO4/K2SO4, H2O. насиченим NaCl, потім сушать і упарюють у вакуумі: одержують напівтвердий залишок, який витягують, потім розтирають у гептані з одержанням твердої речовини, m=27,7г, Тпл=90-92°С. 2.2 5-Аміно-1,3-бенздіоксол-2метилкарбоксилат. До 900мл складною ефіру, одержаного на попередній стадії, розчиненого в 30мл ТГФ додають 3.92 порошкоподібною цинку і. після охолодження до -5°С, за 30 хвилин додають 4мл оцтової кислоти. розведеної 4мл ТГФ. потім дозволяють температурі знову піднятися. Через 1,5 години фільтрують, промивають тверду речовину невеликою кількісно ТГФ і метанолу. Фільтрат розводять AcOEt і промивають Н2О, насиченим NaHCO2, H2O, насиченим NaCl; після сушіння і концентрування у вакуумі одержують жовтий віск, що ідентифікується методом ЯМР, m=800мг. Синтез 3 3.1 5-Нітро-1,3-бенздіоксол-2-карбоксамід. До 1,12г складногометилового ефіру, одержаного в синтезі 2.1, доливають 20мл розчину 2М аміаку в метанолі. Через 25 хвилин концентрують у вакуумі, витягують твердий залишок в Et2O, фільтрують і сушать, m=0,99г, Тпл=202-207°С. 3.2 5-Аміно-1,3-бенздіоксол-2-карбоксамід. До 0,98г аміду, одержаного в синтезі 3.1, у 35мл ТГФ додають 4,57г порошкоподібного цинку; після охолодження до -5°С. за 30 хвилин додають 5мл оцтової кислоти, розведеної 5мл ТГФ. По закінченні додавання дозволяють температурі знову піднятися. Через 1 годину тверду речовину відфільтровують, промивають невеликою кількістю ТГФ. метанолу, AcOEt. Фільтрат розводять AcOEt, додають туди воду і доводять до рН=6 насиченим NaHCO3. Осад, що утворився, видаляють фільтруванням, фільтрат декантують, потім органічну фазу промивають насиченим NНСО3, Н2О, насиченим NaCl, сушать і упарюють, одержують віск, який затвердіє при охолодженні. m=0,63г. Синтез 4 4.1 5-Нітро-(1,3-бенздіоксол-2-іл)метанол. До 5,02г складного метилового ефіру, одержаного в синтезі 2.1, розчиненого в 25мл ТГФ, при 5°С за 1 годину 15 хвилин додають 22,3мл розчину 1М LіАlН4 у ТГФ: через 20 хвилин після закінчення додавання, по краплях, додають 20мл AcOEt, потім 9мл 1н. NaOH: осад, що утворився виділяють фільтруванням, промивають AcOEt. фільтрат розводять AcOEt і промивають Н2О, 5% KHSO4/K2SO4, Н2О. насиченим NaCl; після сушіння і концентрування у вакуумі одержують віск, який кристалізується. m=2,74г, Тпл=80-82°С. На наступній стадії нітропохідне. одержане в синтезі 4.1. відновлюють згідно з методиками, описаними вище, з одержанням аміну формули (III) синтезу 4.2. Синтез 5 5.1 (5-Нітро-1,3-бенздіоксол-2іл)метилсульфонат 95223 16 До 4,12г спирту, одержаного на стадії 4.1, розчиненого в 30мл СН2СІ2, при 5°С додають 3мл триетиламіну, потім за 15 хвилин 1,85г мезилхлориду. Через 15 хвилин прибирають льодяну баню. Через 55 хвилин реакційне середовище розводять СН2СІ2 і водою; органічну фазу декантують, промивають Н2О, насиченим NaCl, сушать, упарюють. Після розтирання в гептані одержують тверду речовину каштанового кольору. m=5,20г, Тпл, 112115°С. 5.2 [(5-Нітро-1,3-бенздіоксол-2іл)метил]діетиламін До 2,91г мезилату. одержаного на попередній стадії, у 18мл ДМФ додають 2,19г діетиламіну і нагрівають до 80°С. Через 15 годин додають 0,73г діетиламіну, потім через 8 годин ще 0,73г. Через 48 годин, загалом, реакційне середовище розводять AcOEt, промивають водою, потім насиченим NaCl; після сушіння AcOEt упарюють, потім залишок витягують у 40мл Еt2О+10мл AcOEt і екстрагують 2 рази 60мл 0,2 н. НСl: кислі фази змішують, приводять у контакт з AcOEt і доводять до рН=9 за допомогою 10н. NaOH; органічну фазу декантують. промивають водою, потім насиченим NaCl, потім сушать і упарюють. Одержують масло, m=1,55г. 5.3 [(5-Аміно-1,3-бенздіоксол-2іл)метил]діетиламін До 1,93г нітросполуки, одержаної на попередній стадії, розчиненої в 70мл ТЕФ. додають 7,45г порошкоподібного цинку, потім при -5°С за 25 хвилин додають 7,6мл AcOEt, потім продовжують перемішування при температурі в інтервалі від 0 до 5°С. Через 1,5 години тверду речовину відфільтровують, промивають ТГФ і невеликою кількістю метанолу: фільтра і розводять AcOEt+H2O і доводять до рН=9 за допомогою 10н. NaOH; осад, що утворився видаляють фільтруванням: фільтрат декантують: органічну фазу промивають водою, насиченим NaCl, сушать, упарюють, одержують чорне масло. m=1,75г. Синтез 6 6.1 1-Метил-4-((5-нітро-1,3-бенздіоксол-2іл)метил)піперазин Реакція згідно з методикою 5.2, виділений продукт у формі дихлоргідрату. 6.2 2-((Метилпіперазин-1-іл)метил)-36енздіоксол-5-амін Реакція за методикою 5.3. Синтез 7 7.1 5-Нітро-1,3-бенздіоксол-2-карбонова кислота До 12г складного ефіру, одержаного в синтезі 2.1, у 12мл метанолу за 30 хвилин додають 1,5мл 5н. гідроксиду натрію. Через 35 хвилин після закінчення додавання реакційне середовище розводять AcOEt і НгО і доводять до рН=2 за допомогою 2 н. НСl; органічну фазу декантують, промивають водою і насиченим NaCl. потім сушать, потім упарюють, одержують 1,25 г масла. 7.2 N,N-Диметил-5-нітро-1,3-бенздіоксол-2карбоксамід До 0,84г кислоти, одержаної на попередній стадії, розчиненій у 15мл дихлорметану, додають 0,36 хлоргідрату диметиламіну, 0,77мл ДІПЕА (DIPEA), потім 0,91г ДЦКДІ (DCCI). Після 3 годин 17 перемішування при кімнатній температурі реакційне середовище фільтрують, потім фільтрат розводять СН2СІ2. який послідовно промивають насиченим розчином NHCO3. Н2О. 5% KHSO4/K2SO4. Н2О, насиченим NaCl. після сушіння розчинник випарюють, потім залишок хроматографують на діоксиді кремнію сумішшю дихлорметан/метанол 99/1. Одержують 0,5г твердого продукту з Тпл=109°С. Синтез 9 9.1 5-Нітро-1,3-бенздіоксол-2-карбонітрил До 45мл ДМФ, охолодженого до 5°С, повільно додають 3,7мл РОСІ3- Після 30 хвилин перемішування при 5°С додають за 1 раз 1,67г аміду, одержаного на стадії 3.1. Після 3 годин перемішування при кімнатній температурі реакційну суміш виливають на 250мл суміші вода/лід. Осад, що утворився, фільтрують, промивають водою, потім сушать. m=1,32г, Тпл=105-110°С. 9.2 5-Аміно-1,3-бенздіоксол-2-карбонітрил Відновлення NO2 групи продукту, одержаного на стадії 9.1, у NH2 здійснюють згідно з методикою, описаною перед цим, застосовуючи суміш Zn/AcOH. Синтез 11 11.1 5-Нітро-1,3-бенздіоксол-2,2діетилкарбоксилат Дану сполуку одержують згідно з методикою, описаною в Pharmazi 2003 58 (1) 13-17. 11.2 5-Нітро-1,3-бенздіоксол-2,2дикарбоксамід До 14мл розчину 2М аміаку в метанолі за 1 раз додають 1,24г складного діефіру, одержаного на попередній стадії. Після 30 хвилин перемішування реакційне середовище концентрують досуха, потім витягують залишок у простий ефір, фільтрують і сушать, m=1,01г, Тпл=231-233°С. Синтез 12 12.1 (5-Нітро-1,3-бенздіоксол-2,2дііл)диметанол До 1,87г складного діефіру, одержаного на стадії 11.1, у 60мл ТГФ при кімнатній температурі за 1 годину додають 1,50г NaBH4; через 25 хвилин після закінчення додавання розводять 250мл AcOEt. потім 5мл метанолу, потім 40мл води по краплях. Органічну фазу декантують, промивають водою, 5% розчином KHSO4/K2SO4, потім Н2О, насиченим розчином NaCl. Після сушіння і випарювання залишок хроматографують на діоксиді кремнію сумішшю хлороформ/метанол (98/2). одержують 0,64г масла, яке тужавіє в масі. Тпл=111113°С. Синтез 13 13.1 3-(2-Метил-5-нітро-1,3-бенздіоксол-2іл)етилпропаноат До суспензії 15,51г 4-нітрокатехіну в 22,10г етилацетоацетату додають при 70°С за 15 хвилин 22,4г фосфорного аніідриду. Через 1 годину 45 хвилин реакційну суміш охолоджують. потім екстрагують 4×150мл теплим толуолом. Толуольні фази об'єднують, промивають Н2О, 1 н. NaOH, H2O, 5% KHSO4/K2SO4. Н2О, насиченим розчином NaCl. Після сушіння і випарювання продукт очищають хроматографією на діоксиді кремнію, елююючи хлороформом, одержують 2,44г твердої речовини, Тпл=76-78°С. 95223 18 13.2 3-(2-Метил-5-нітро-1,3-бенздіоксол-2іл)пропан-1-ол До 2,33г складного ефіру, одержаного вище, розчиненого в 40мл ТГФ. додають при -5°С за 45 хвилин 8мл 1 М LiAlH4 у ТГФ. Через 35 хвилин по краплях додають 8мл етилацетату, потім 1мл води, потім 1мл 1 н. гідроксиду натрію. Тверду речовину виділяють фільтруванням; фільтрат розводять AcOEt, промивають Н2О, 5% KHSO4/K2SO4, Н2О, насиченим розчином NaCl; після сушіння органічну фазу концентрують у вакуумі; одержують масло m=1,90г. 13.3 3-(2-Метил-5-нітро-1,3-бенздіоксол-2іл)пропілметансульфонат До 1,89г спирту, одержаного перед цим на стадії 13.2, у 40мл дихлорметану при 5°С додають 1,01г триетиламіну. потім 1,14г мезилхлориду за 20 хвилин. По закінченні додавання льодяну баню прибирають і продовжують перемішування 1 годину; реакційне середовище розводять СН2СІ2 і промивають Н2О, потім насиченим розчином NaCl: після сушіння розчинник випарюють; одержують 2,46г воску, який при охолодженні стає твердим. 13.4 N,N-Діетил-3-(2-Метил-5-нітро-1,3бенздіоксол-2-іл)пропан-1-амін. До 1,21г мезилату в 20мл ДМФ додають 0,87г діетиламіну, потім нагрівають до 80°С. Через 8,5 годин додають 0,44г діетиламіну і продовжують нагрівання протягом 14 годин. Реакційне середовище концентрують у вакуумі, залишок знову розчиняють у AcOEt і доводять до рН=9,5 за допомогою 1 н. NaOH, органічну фазу декантують, промивають Н2О, насиченим розчином NaCl, потім сушать, після випарювання одержують неочищений продукт, який знову розчиняють у 20мл AcOEt плюс 20мл Et2O. потім 2 рази екстрагують 50мл 0,5 н. НСІ; 2 водні фази змішують, приводять у контакт із AcOEt і доводять до рН=9 за допомогою 10 н. NaOH; органічну фазу декантують. повторно промивають Н2О, потім насиченим розчином NaCl, сушать, упарюють, одержують 0,66г масла. 13.5 Замісника NO2 продукту, одержаного на стадії 13.4, відновлюють у NH2 способом, описаним перед цим. використовуючи суміш Zn/AcOH для одержання відновленого продукту 13.5. 13.6 До 2,00 продукту, одержаного на стадії 13.3, у 20мл ДМФ додають 0,85г азиду натрію і перемішують 5 днів при кімнатній температурі, потім екстрагують простим ефіром і промивають органічну фазу водою, потім насиченим розчином NaCl. Одержують масло, m=1.50г. ЯМР відповідає. 13.7 Суміш 1,49г продукту, одержаного на стадії 13.6, 1,71г трифенілфосфіну і 0,12г води у 25мл ТГФ перемішують протягом 24 годин. Потім реак 19 ційне середовище екстрагують AcOEt і промивають водою. Одержаний неочищений продукт розчиняють у суміші AcOEt/Et2O ra екстрагують водним розчином 1 н. НСl. Кислу водну фазу приводять у контакт з AcOEt і доводять до рН 9 за допомогою 10 н. NaOH. Органічну фазу виділяють, промивають водою, потім насиченим розчином NaCl. Органічну фазу концентрують при зниженому тиску з одержанням 1,10г масла. 13.8 У 10мл дихлорметану (ДХМ) (DCM) розчиняють 1,09г аміну, одержаного на стадії 13.7, потім додають 0,20г триетиламіну, потім 1,18г Вос 2О. Через 5 годин реакційне середовище розводять ДХМ, потім промивають, послідовно, 5% розчином KHSO4/K2SO4, водою, насиченим розчином NaCl. Після сушіння і випарювання при зниженому тиску органічну фазу виділяють, одержують 1,36г масла. 13.9 (попередник прикладу 55) Продукт, одержаний на стадії 13.8, у кількості 1,5г обробляють Zn/AcOH, як описано на стадії 2.2. щоб відновити групу NO2 у NН2. Одержують 1,13г воску. Синтез 14 2-(2-Метоксиметил)-5-нітро-1,3-бенздіоксол У 25мл ТГФ розчиняють 1,45г гідроксиметилу. одержаного на стадії 4.1; після охолодження до 5°С невеликими порціями додають 353мг 60% NaH; через 30 хвилин додають 0,92мл метилйодиду, потім перемішують протягом однієї ночі при кімнатній температурі; додають 1мл метилйодиду і продовжують перемішування протягом 5 годин; до реакційного середовища додають 30мл насиченого розчину NH4Cl. потім воду і етилацетат: органічну фазу декантують, повторно промивають Н2О. потім насиченим NaCl, сушать і упарюють. Неочищений продукт хроматографують на діоксиді кремнію, елююючи сумішшю СНСІ3/гептан 9/1. Одержують 595мг масла, яке ідентифікують методом ЯМР. Синтез 16 16.1 трет-Бутил-2-((5-нітро-1,3-бенздіоксол-2іл)карбоніл)гідразинкарбоксилат Суміш 900мг складного ефіру (стадія 2.1) і 2,114г трет-бутилкарбазату в 40мл метанолу нагрівають при 60°С протягом 60 годин; додають 600мг трет-бутилкарбазату і гріють ще 3 години. Метанол випарюють, залишок витягують етилацетатом і промивають водою. 0,2 н. соляною кислотою, насиченим розчином бікарбонату натрію, водою і насиченим розчином NaCl. Одержують віск, який тужавіє в масі, m=1,27г. 16.2 трет-Бутил-2-((5-аміно-1,3-бенздіоксол-2іл)карбоніл)гідразинкарбоксилат До 1,30г продукту, такого як одержаний на попередній стадії, розчиненого в 25мл ТГФ. додають 3,92г порошкоподібного цинку, потім при 5°С за 30 хвилин 4,8г оцтової кислоти; прибирають льодяну баню і продовжують перемішування 2 години; тверду речовину відфільтровують, промивають невеликою кількістю ТГФ, потім AcOEt: додають до 95223 20 фільтрату воду і доводять до рН 6,5 15% розчином Na2СО3; AcOEt декантують. промивають насиченим NaHCO3, H2O, насиченим NaCl, сушать і упарюють. Одержують очікувану сполуку у формі чорного масла, m=1,12г. Синтез 21 21.1 До 2,14г мезилату, одержаного на стадії 5.1, розчиненого в 17мл ДМФ, додають 1,51 г азиду натрію і нагрівають при 70°С протягом 3 годин. Реакційне середовище екстрагують AcOEt. який промивають водою, потім насиченим розчином NaCl. Одержують масло. m=1,71г. 21.2 До 1,7г продукту, одержаного на стадії 21.1, розчиненого в 20мл AcOEt. невеликими порціями додають 3,41г трифенілфосфіну, потім, через 10 хвилин. 2,34мл води і нагрівають до 60°С. Через 1 годину реакційне середовище упарюють досуха, потім витягують у Et2O. Нерозчинні речовини видаляють, потім додають надлишок насиченого розчину НСІ у простому ефірі. Тверду речовину, що утворилася, відфільтровують, промивають простим ефіром, потім сушать з одержанням очікуваного продукту у формі хлоргідрату. Відповідний амін одержують вивільненням із хлоргідрату. 21.3 Продукт, одержаний на стадії 21.2, може бути розділений на два його енантіомери: До 2г аміну, одержаного на стадії 21.2, розчиненого в 70мл води і 7мл діоксану при 70°С. додають 1,97г (S)(+) кислоти. Потім реакційному середовищу дають повільно повернутися до 30°С при перемішуванні магнітною мішалкою. Одержаний осад відфільтровують, розчиняють у 40мл води і 4мл діоксану при 70°С. потім дають повільно повернутися до 30°С при перемішуванні. Тверду речовину, яка утворюється при охолодженні, відфільтровують і сушать, m=0,49г. Одержаний таким чином продукт витягують 20мл води і 100мл AcOEt, потім доводять до рН=9,5 додаванням 1 н. NaOH. Суміш декантують, органічну фазу виділяють, промивають декілька разів водою, потім насиченим розчином NaHCO3, водою, і нарешті насиченим розчином NaCl. Органічну фазу збирають, сушать і розчинник випарюють при зниженому тиску. Одержують 0,27г воску, який поступово твердне. []D=+94,7° при 25°С; С=0,5 (МеОН); оптична чистота: хіральна ВЕРХ: 96/4 (обертальна здатність енантіомера, очищеного до 100%=102°). 21.4 До продукту, одержаного на стадії 21.2, у 30мл ДХМ додають 1,49мл триетиламіну. потім невеликими порціями 2,53г Вос2О. Через 1 годину реакційне середовище промивають 5% KHSO4/K2SO4, H2O. насиченим розчином NaCl. Після сушіння органічну фазу концентрують досуха, потім залишок розтирають у гептані; одержують 2г твердої речовини. 21.5 До 480мг 60% NaH у 20мл ТГФ при 5°С за 30 хвилин додають 1,79г продукту, одержаного на стадії 21.4, розчиненого в 15мл ТЕФ. Суміш пере 21 мішують 45 хвилин при кімнатній температурі, потім за 10 хвилин додають 1,2мл йодметану. Через 3 години реакційне середовище доливають до 60мл насиченого водного розчину лимонної кислоти, потім екстрагують етилацетатом. Органічну фазу виділяють, промивають водою, потім насиченим розчином NaCl, сушать і упарюють досуха. Одержаний неочищений продукт очищають імпульсною хроматографією з градієнтом дихлорметану в циклогексані. Одержують білу тверду речовину: m=1,4г. 21.6 Продукт, одержаний на стадії 21.5, відновлюють згідно зі звичайною методикою за допомогою Zn/AcOII. Синтез 22 22.1 Суміш 11,65г 4-N-Z-піперидону, 6,35г метилортоформіату і 40мг паратолуолсульфокислоти нагрівають у колбі Кляйзена (Claisen) при 60°С протягом 1 години, потім при 70°С протягом 1 години, не заважаючи перегонці метилформіату. Залишок розводять АсОЕt+Н2О і додають декілька крапель 1 н. NaOH, щоб довести до рН 7. Органічну фазу декантують, виділяють, промивають водою, потім насиченим розчином NaCl сушать, упарюють. Одержують 14г незабарвленого масла. 22.2 Суміш 16,09г кеталю, одержаного на стадії 22.1, 10,80г 4-нітрокатехіну і 60мг паратолуолсульфокислоти нагрівають у 120мл толуолу з повільною перегонкою толуолу. Через 4 години 30 хвилин реакційне середовище розводять толуолом, охолоджують; фільтруванням відділяють нерозчинну речовину, промивають фільтрат 1 н. NaOH, H2O, 5% розчином KHSO4/K2SO4, H2O, насиченим розчином NaCl. Після сушіння і випарювання неочищений продукт хроматографують на діоксиді кремнію, елююючи сумішшю CHCІ3/AcOEt 98/2. Одержують 0,65г очікуваного продукту. 22.3 До 0,60г продукту, одержаного вище, у 5мл трифтороцтовій кислоті додають 0,5мл тіоанізолу. Через 3 години реакційне середовище концентрують у вакуумі: залишок витягують СН2СІ2 з водою і доводять до рН 0 за допомогою 1 н. NaOH. Після декантації органічну фазу знову промивають водою, потім насиченим розчином NaCl. Органічну фазу сушать і упарюють. Неочищений продукт збирають і хроматографують на діоксиді кремнію, елююючи сумішшю CHCl3/MeOH/NH4OH 95/5/0,1: одержують 100мг очікуваного твердого продукту. 22.4 Протягом 1 години 95мг аміну, одержаного на стадії 22.3, обробляють 98мг Вос2О і 20мг триети 95223 22 ламіну в 3мл дихлорметану. Після реакції і звичайних обробок одержують білу тверду речовину, m=130мг. 22.5 Групу NO2 відновлюють в амін за допомогою Zn/AcOH відповідно до вже описаної методики. Синтез 23 23.1 До 985мг продукту стадії 4.1, розчиненого у 25мл ДХМ. додають 0,46мл піридину. Потім, при 5°С за 20 хвилин додають 0,84мл ангідриду трифтороцтової кислоти, розчиненого в 3мл ДХМ. Через 1 годину при 5°С реакційне середовище промивають сумішшю води з льодом, потім насиченим розчином NaCl. Органічну фазу сушать і концентрують у вакуумі. Одержують 1,42г твердої речовини. 23.2 До 1,15г продукту, одержаного на стадії 23.1, розчиненого в 10мл ДХМ+0,5мл ДМФ, додаюіь 75мг діетаноламіну. Після перемішування протягом ночі, реакційне середовище розводять 100мл ДХМ, промивають водою, промивають насиченим розчином NaCl, сушать і концентрують при зниженому тиску. Залишок очищають імпульсною хроматографією з градієнтом метанолу від 0% до 15% у хлороформі. Одержують 730мг твердої речовини. 23.3 Групу NО2 відновлюють в амін за допомогою Zn/AcOH. як описано перед цим. Виходячи з 720мг продукту, одержаного на стадії 23.2, одержують 400мг очікуваного продукту у формі смолоподібної речовини. Синтез 24 24.1 Продукт одержують згідно з методикою, описаною в Org. Lett., 2001, 3(9), 1399-1402. Синтез 24.2 До 2,73г продукту, одержаного на стадії 24.1, у 30мл ДХМ при 5°С за 2 хвилини додають 5мл 69% азотної кислоти. Через 2.5 години перемішування реакційне середовище розводять Et2O; органічну фазу промивають 2 рази Н2О, 2 рази охолодженим 7% розчином Na2СО3 1 раз водою, 1 раз 5% розчином KHSO4/K2SO4. 1 раз водою, потім 1 раз насиченим розчином NaCl. Після сушіння і випарювання одержують білу тверду речовину, m=3,20г. Синтез 24.3 При -5°С протягом 1 години 3,63г продукту, одержаного за методикою, описаною вище, у 80мл ТГФ обробляють 532мг LiAlH4. Після звичайної обробки виділяють 2,60г продукту у формі густого масла. 23 95223 Синтез 24.4 24 Синтез 24.7 Спирт, одержаний на стадії 24.3, у кількості 2,59г обробляють мезилхлоридом за методикою, описаною в синтезі 13.3, з одержанням 3,52г мезилату. Ідентифікація методом ЯМР. Синтез 24.5 Продукт, одержаний на стадії 24.4, у кількості 3,51г обробляють 1,97г азиду натрію за методикою, описаною в синтезі 13.6. Одержують 2,60г очікуваного продукту. Синтез 24.6 Продукт, одержаний на стадії 24.5, у кількості 2,59г обробляють 4,90г трифенілфосфіну і 2мл води за методикою, описаною в синтезі 13.7. Одержують 2г очікуваною продукту у формі масла. Продукт, одержаний на стадії 24.6, у кількості 1,99г обробляють ВОС2О за методикою, описаною в синтезі 13.8. Одержують 2,41г твердої речовини. Синтез 24.8 Продукт, одержаний на стадії 24.7, обробляють Zn/AcOH згідно зі звичайним способом відновлення нітрогрупи в аміногрупу. На основі 0,93г вихідного продукту одержують 0,84г очікуваного продукту у формі воску. Сполуки формули (III) і проміжні продукти формули (IV), що є похідними бенздіоксолу, охарактеризовані в таблиці, що наводиться нижче. Перетворення сполуки формули(ІУ) у сполуку формули (III) було здійснене відповідно до синтезу 5.3 для таких сполук: 7.2. 9.1. 11.1. 12.1, 13.4 і 14.1. ТАБЛИЦЯ 1 Синтези сполук формули (III) 25 95223 26 27 Синтез 18 18.1 Хлоргідрині ((7-нітро-2,3-дигідро-1,4бетдіоксин-2-іл)(метил)діетиламіну 95223 28 До 1,50г (7-нітро-2,3-дигідро-1,4-бенздіоксин-2іл)метансульфонату, описаного в міжнародній заявці на патент WO 01/021577, у 30мл ДМФ дода 29 95223 ють 800мкл діетиламіну і нагрівають до 80°С, потім з інтервалом у 12 годин 3 рази додають по 800мкл діетиламіну. Через 48 годин реакційне середовище упарюють досуха: залишок витягують 100мл AcOЕt плюс 5мл 4 н. NaOH; органічну фазу декантують. промивають водою, потім насиченим NaCl; після сушіння і випарювання AcOEt залишок витягують 20мл AcOEt плюс 50мл 0,5 н. НСІ, перемішують і потім декантують: водну фазу упарюють досуха і залишок розтирають у простому ефірі, фільтрують і сушать, m=0,95г, Тпл=192-194°С. 18.2 ((7-аміно-2,3-дигідро-1,4-бенздиоксин-2іл)метил)діетиламін До суспензії 0,94г нітропохідного. одержаного на попередній стадії, у 40мл ТГФ додають 3,05г порошкоподібного цинку. Після охолодження до 30 0°С, за 20 хвилин додають 3,1мл оцтової кислоти; через 10 хвилин льодяну баню прибирають і продовжують перемішування протягом 2 годин. Тверду речовину відфільтровують, промивають ТГФ. потім невеликою кількістю метанолу. Фільтрат упарюють досуха, витягують етилацетатом, додають воду і доводять до рН=8 за допомогою 10 н. NaOH; осад, що утворився виділяють фільтруванням, промивають AcOEt; фільтрат декантують. промивають насиченим NaCl. сушать і упарюють; одержують масло каштанового кольору, m=0,51 і. Сполуки формули (III) і проміжні продукти формули (IV), що с похідними бенздіоксину, одержані у формі рацемічній суміші та охарактеризовані в таблиці 2, наведеній нижче. ТАБЛИЦЯ 2 Синтез сполук формули (III) Номери сполук, наведених у прикладах, відповідають номерам, наданим у Таблиці 3. ідо наведена нижче, яка ілюструє хімічні структури і фізичні властивості деяких сполук згідно з винаходом. Коли вони містять асиметричний атом вуглецю, дані сполуки одержані у формі рацемічної суміші. ПРИКЛАД 1 Сполука №2 5-((7-(((трет-Бутиламіно)карбоніл)аміно)-6-(2,6дихлорфетл)піридо[2,3-d]піримідин-2-іл)аміно-1,3бенздіоксол-2-метилкарбоксилат Суміш 0,97г сполуки синтезу 2,2 і 1,40г сполуки синтезу 1 у 15мл ТГФ кип’ятять у колбі зі зворотним холодильником. Через 6 годин реакційне середовище концентрують у вакуумі. Продукт очищають хроматографією на діоксиді кремнію, елююючи сумішшю AcOEt/толуол (2/3), потім по 31 вторною хроматографією, елююючи сумішшю СНСІ3/МеОН 98/2, одержують 0,45г жовтої твердої речовини, ідентифікованої методом мас+ спектрометрії. МН =583. ПРИКЛАД 2 Сполука №4 5-((7-(((трет-Бутиламіно)карбоніл)аміно)-6-(2,6дихлорфеніл)піридо[2,3-d]піримідин-2-іл)аміно-1,3бенздіоксол-2-карбонова кислота До 0,25г складного ефіру, одержаного в прикладі, наведеному вище, у 10мл метанолу додають 0,3мл 2 н. гідроксиду натрію. Через 1 годину 10 хвилин перемішування при кімнатній температурі реакційне середовище розводять AcOEt+H2O і доводять до рН=4 за допомогою 1 н. НСl. Органічну фазу декантують, промивають Н2О, потім насиченим NaCl, сушать і упарюють. Твердий жовтий залишок витягують простим ефіром, розтирають, фільтрують і сушать. Одержують 205 мг продукту, ідентифікованого методом мас-спектрометрії. + МH =569. ПРИКЛАД 3 Сполука №3 5-((7-(((трет-Бутиламіно)карбоніл)аміно)-6-(2,6дихлорфеніл)тридо[2,3-d]піримідин-2-іл)аміно-1,3бенздіоксол-2-карбоксамід Суміш 0,62г сполуки синтезу 3.2 і 1,40г сульфопохідного синтезу 1 у 20мл ТГФ кип'ятять у колбі зі зворотним холодильником. Через 3 години реакційне середовище концентрують у вакуумі, потім залишок хроматографують на діоксиді кремнію, елююючи сумішшю СНСІ3/МеОН 97/3об./об. Одержують 315мг жовтої твердої речовини, іден+ тифікованої методом мас-спектрометрії. МН =568. ПРИКЛАД 4 Сполука №9 N-(трет-Бутил)-N'-(6-(2,6-дихлорфеніл)-2-((2гідроксиметил)-1,3-бенздіоксол-5іл)аміно)піридо[2,3-d]піримідин-7-іл)карбамід До суміші 350мг сполуки синтезу 4.2 і 937мг сполуки синтезу 1 у 15мл ЕtOН додають 43мкл концентрованої НСl і гріють при 55°С протягом 6 годин. Реакційне середовище концентрують у вакуумі, потім залишок хроматографують на діоксиді кремнію, одержують 490мг жовтої твердої речовини, ідентифікованої методом мас-спектрометрії, + МН =555. ПРИКЛАД 5 Сполука №6 5-((7-(((трет-Бутиламіно)карбоніл)аміно)-6-(2,6дихлорфеніл)піридо[2,3-d]піримідин-2-іл)аміно-1,3бенздіоксол-2-карбонітрил До 1,00г сполуки синтезу 1 і 450мг сполуки синтезу 9.2 в 15мл ЕtOН додають 50мкл концентрованої НСl і несильно нагрівають зі зворотним холодильником. Через 1,5 години реакційне середовище концентрують у вакуумі. Залииюк хроматографують на діоксиді кремнію, елююючи сумішшю CHCl3/AcOEt (90/10; об./об.). Одержують 465мг жовтої твердої речовини, ідентифікованої + методом мас-спектрометрії. МН =550. ПРИКЛАД 6 Сполука №12 95223 32 N-(2-((Амінометил)-1,3-бенздіоксол-5-іл)аміно)6-(2,6-дихлорфеніл)піридо[2,3-d]піримідин-7-іл)-N'(трет-бутил)карбамід До 530мг нітрильного похідного попереднього прикладу, розчиненого у 25мл ТГФ. при 10°С за 30 хвилин додають 1,9мл розчину 1 М LiAlН4 у ТГФ. Через 15 хвилин після закінчення додавання, доливають 1,5мл AcOEt. потім 10мл насиченого розчину NH4CІ, потім дозволяють температурі піднятися. Після розведення AcOEt. промивають водою, потім насиченим NaCl. Після сушіння, органічну фазу концентрують у вакуумі і залишок хроматографують на діоксиді кремнію, елююючи сумішшю СНСІ3/МеОН (95/5об./об.). Одержують 165мг жовтої твердої речовини, ідентифікованої методом + мас-спектрометрії. МН =554. ПРИКЛАД 7 Сполука № 19 трет-Бутілкарбоксилат 2-((5-((7-((третбутиламіно)(карбоніл)аміно)-6-(2,6дихлорфеніл)піридо[2,3-d]піримідин-2-іл)аміно-1,3бенздіоксол-2-іл)карбоніл)гідразину До 0,645г продукту, одержаного в синтезі 1, і 0,63г продукту, одержаного в синтезі 16.2, у 25мл ЕtOН додають 0,025мл концентрованої НСІ і перемішують 5 годин при 70°С. Реакційне середовище упарюють і залишок витягують СНСІ3, який промивають водою, насиченим розчином NaHCO3. водою, насиченим NaCl, потім сушать і упарюють у вакуумі; залишок хроматографують на діоксиді кремнію. Одержують 450мг жовтої твердої речовини, ідентифікованої методом мас-спектрометрії. + МН =683. ПРИКЛАД 8 Сполука №20 N-(трет-Бутил)-N'-(6-(2,6-дихлорфеніл)-2-((2гідразинкарбоніл)-1,3-бенздіоксол-5іл)аміно)піридо[2,3-d]піримідин-7-іл)карбамід Сполуку попереднього прикладу у кількості 360мг перемішують 45 хвилин у суміші 4мл СН2СІ2 і 14мл ТФО (TFA): реакційне середовище упарюють у вакуумі; залишок витягують СНСІ3. який промивають Н2О, 15% водним розчином Na2CO3. Н2О. насиченим розчином NaCl; після сушіння СНСІ3 випарюють у вакуумі, потім залишок розтирають у простому ефірі, відфільтровують. Одер+ жують 225мг жовтої твердої речовини. МН =583. ПРИКЛАД 9 Сполука №34 При кімнатній температурі 431мг сполуку №33 перемішують 30 хвилин у 5мл ДХМ і 5мл ТФО. Після випарювання, залишок витягують сумішшю хлороформ/вода і доводять до рН=9 додаванням 15% водного розчину Na2CO3. Органічну фазу декантують. промивають водою, потім насиченим розчином NaCl, сушать і концентрують при зниженому тиску. Збирають 116мг твердої речовини. + [М+Н] =568. ПРИКЛАД 10 Сполука №35 Стадія 1 33 Стадію здійснюють згідно з методикою, описаною в J. Organometall. Chem., 1996,507, 1-21. Стадія 2 Стадію здійснюють згідно J. Med. Chem. 1988. 31. 84-91. Стадія 3 До 2,91г продукту, одержаного на попередній стадії, у 40мл метанолу додають при 8°С за 35 хвилин 800мг боргідриду нагрію. Через 50 хвилин реакційне середовище виливають у 150мл вода/лід плюс 400мл етилацетату: після 5 хвилин перемішування проводять декантацію і промивають органічну фазу 5% розчином KHSO4/K2SO4, водою, насиченим розчином NaCl; після сушіння і випарювання органічної фази виділяють тверду речовину коричневого кольору; m=2,25г. Стадія 4 До 2,24г попереднього спирту в 60мл ДХМ послідовно додають 1,43г триетиламіну. потім за 25 хвилин 1,67г метансульфонілхлориду. Через 45 хвилин реакційне середовище розводять 100мл ДХМ; промивають 2 рази сумішшю води з льодом і 1 раз насиченим розчином NaCl, органічну фазу сушать і концентрують у вакуумі. Одержують тверду речовину каштанового кольору, m=3,01г. Стадія 5 Суміш 3,0г продукту, одержаного на стадії 4, і 1,82г азиду натрію нагрівають протягом 7,5 годин у 20мл ДМФ при 65°С. Потім реакційне середовище виливають у 75мл суміші води з льодом і 300мл простого ефіру. Органічну фазу виділяють, промивають декілька разів водою, потім насиченим розчином NaCl. Потім органічну фазу сушать і концентрують у вакуумі з одержанням твердої речовини каштанового кольору. m=2,20г. Стадія 6 До 2,19г продукту, одержаного на стадії 5, розчиненого в 50мл етилацеїату. за 15 хвилин додають 4,33г грифенілфосфіну, потім, через 10 хвилин. 1,8мл води за 2 хвилини. Реакційне середовище перемішують протягом 1 години 40 хвилин при 60°С. потім розводять 150мл етилацетату. Одержаний таким чином розчин промивають 2 рази водою, 1 раз насиченим розчином NaCl, сушать, упарюють. Залишок після випарювання 95223 34 розчиняють у суміші 50мл етилацетату і 50мл діетилового ефіру та екстрагують 2 рази 50мл 1 н. НСІ. Кислі водні фази об’єднують і екстрагують сумішшю 25мл етилацетату і 25мл діетилового ефіру, потім приводять у контакт із 300мл етилацетату і доводять рН до 9 за допомогою 10 н. NaOH. Після декантації органічну фазу промивають водою, насиченим розчином NaHCO3, водою, потім насиченим розчином NaCl. Залишкову органічну фазу сушать, потім концентрують у вакуумі. Одержують масло. m=1,40г. Стадія 7 До 1,39г аміну, одержаного на стадії 6, у 35мл ДХМ при 5°С протягом 10 хвилин додають 0,35г триетиламіну, потім 1,75г Вос2О. Після перемішування протягом 1 ночі при кімнатній температурі реакційне середовище розводять 150мл ДХМ. промивають водою, 5% розчином KHSO4/K2SO4, водою, потім насиченим розчином NaCl. Після сушіння і випарювання ДХМ одержану тверду речовину розчиняють у мінімальній кількості діетилового ефіру потім додають гептан до повного осадження. Одержують тверду речовину: m=1,90г. ЯМР: 1.30 м.ч.: с: 9Н; 3.40-3.55 м.ч.: мт: 2Н; 6.55 м.ч.: т: 1Н; 7,00 м.ч.: т: 1Н; 7.15-7.30 м.ч.: мг: 2Н; 7,55 м.ч.: д: 1Н. Стадія 8 До 0,60г продукту, одержаного на стадії 7, у 25мл ТГФ додають 1,96г порошкоподібного цинку, потім при -3°С протягом 25 хвилин 2мл АсОН. По закінченні додавання реакційному середовищу дають повернутися до кімнатної температури. Через 1 годину 15 хвилин реакційне середовище фільтрують, фільтрат розводять 150мл етилацетату і 30мл води. рН доводять до 9 додаванням 15% розчину Na2CO3. Після декантації органічну фазу виділяють, промивають насиченим розчином NaHCO3, водою, потім насиченим розчином NaCl. Органічну фазу виділяють, сушать, потім концентрують при зниженому тиску. Одержують 0,53г в'язкого жовтого масла. ЯМР: 1,35 м.ч.: с: 9Н; 3.30 м.ч.: мт: 2Н: 4,80 м.ч.: с: 2Н; 6,05 м.ч.: т: 1Н; 6,10-6.25 м.ч.: мт: 2Н; 6,50 м.ч.: т: 1Н: 7,10 м.ч.: т: 1Н. Стадія 9 Зв’язування. для одержання сполуки 35. за стандартних умов, таких як описані перед цим. ПРИКЛАДИ Сполука №45 До 1г сполуки 12, розчиненої у 15мл метанолу і 2 мл ДМФ, додають 1,035мл ДІПЕА. потім 0,675г формамідинілсульфокислоти (H2N-C(=NH)-SO3H). Після перемішування протягом ночі при 25°С до реакційного середовища додають воду, осад, що утворився, відфільтровують, промивають водою, потім сушать при зниженому тиску. Неочищений продукт піддають імпульсній хроматографії на си 35 лікагелі з градієнтом МеОН. від 10 до 50%. у ДХМ. Одержують 170мг твердої речовини, перетвореної в сіль у співвідношенні моль/моль H2SO4. MC (MS): + МН =596. ПРИКЛАД 12 Сполука №39 Синтез 39.1 До 555мг сполуки 9 (приклад 4) у 10мл ДХМ при 5°С додають 0,11мл піридину, потім за 15 хвилин 0,20мл ангідриду трифтороцгової кислоти розведених у 2мл ДХМ. Через 45 хвилин реакційне середовище розводять 50мл ДХМ. промивають послідовно сумішшю води з льодом, водою, 5% розчином KHSO4/K2SO4. водою, потім насиченим розчином NaCl. Органічну фазу потім сушать і концентрують при зниженому тиску. Одержують 593мг твердої речовини. -1 ЯМР Н: 1.40 м.ч.: с: 9Н: 4.65 м.ч.: шир. с: 2Н: 6.50 м.ч.: т: 1Н: 6.90 м.ч.: д: 1Н: 7.40 м.ч.: шир. д: 1Н; 7.50-7.70 м.ч.: масив: 3Н: 8.00 м.ч.: шир. с: 1Н; 8.05 м.ч.: с: 1Н; 8.20 м.ч.: шир. с: 1Н: 9.00 м.ч.: с: 1Н: 10.15 м.ч.: с: 1Н: 10.70 м.ч.: с: 1Н. Синтез 39.2 До 579мг сполуки одержаної на стадії 39.1, у 10мл ДХМ додають 0,17мл морфоліну. Реакційне середовище перемішують 3 години 30 хвилин при кімнатній температурі, потім систему упарюють при зниженому тиску. Залишок очищають імпульсною хроматографією на силікагелі з градієнтом AcOEt. від 0 до 20%. у ДХМ. Збирають 300мг жов+ того порошку. МС: МН =624. ПРИКЛАД 13 Сполуки №№40-43 Сполуки з 40 по 43 одержують таким самим чином, що сполуку 39. описану в прикладі 12, використовуючи сполуку, одержану на стадії 39.1 і замінюючи морфолін стадії 39.2, відповідно, третбутиламіном. 1-БОК-піперазином (БОК (для тексту)=Вос (для хімічних формул і схем реакції)), циклопропіламіном. цис-2.6-диметилпіперидином. Приклад 14 Сполука №44 Сполуку №44 одержують видаленням захисної групи зі сполуки 41, одержаної в прикладі 13, способом, описаним перед цим. що використовує ТФО. ПРИКЛАД 15 Сполуки №№46-48 Сполуку 12 вводять у реакцію з кислотою в активованій формі, наприклад, ангідриду, хлориду кислоти, кислота + зв’язувальний агент, наприклад. ДЦКДІ, БОФ. Так. сполуки з 46 по 48 одержують у результаті реакції між сполукою 12 і, відповідно, оцтовим ангідридом. бензоїлхлоридом і циклопропанкарбонілхлоридом. ПРИКЛАД 16 Сполука №49 Охолоджують до 5°С 444мг сполуки 12 у 12мл ацетонітрилу. Додають 0,14мл триетиламіну, потім 0,075мл метансульфонілхлориду. Через 40 хвилин перемішування при 25°С реакційне середовище витягують AcOEt. Органічну фазу промивають во 95223 36 дою, промивають насиченим розчином NaCl, сушать і упарюють при зниженому тиску. Залишок очищають імпульсною хроматографією на силікагелі з градієнтом від 0 до 5% метанолу в хлороформі. Одержують 220мг твердої речовини. МС: + МН =632. ПРИКЛАД 17 Сполука №50 До 284мг сполуки 4 (приклад 2) у 5мл ДМФ додають 40мг трет-бутиламіну, 71г діізопропіламіну і 176мг тетрафторборату О-(1Н-бензотриазол-ііл)-N,N,N',N'-тетраметилуронію (ТБТУ) (TBTU). Реакційне середовище перемішують 1 годину при 25°С потім розводять AcOEt. Органічну фазу послідовно промивають водою, насиченим розчином NaHCO3, водою, потім насиченим розчином NaCl. Органічну фазу сушать, упарюють при зниженому тиску і залишок очищають імпульсною хроматографією на силікагелі з градієнтом від 0 до 5% метанолу в ДХМ. Одержують 196мг жовтої твердої + речовини. МС: МН =624. ПРИКЛАД 18 Сполуки №№51-54 Сполуки з 51 по 54 одержують таким самим чином, що сполуку 50, виходячи зі сполуки 4 і замінюючи трет-бутиламін. відповідно, на циклопропіламін, піролі дин, N-ізопропілметиламін і метиламін. ПРИКЛАД 19 Сполука №62 У 25мл абсолютного спирту, що містить 0,02мл концентрованої НСІ. при 70°С протягом 3 годин перемішають 819мг сполуки, описаної в синтезі 1, і 0,314мг 6-аміно-2,3дигідробензо[b]фурану, який може бути одержаний згідно Еur. J. Med. Chem. Chimica Therapeutica, 1977. Vol. 12, 231-235. Після охолодження осад відфільтровують, промивають теплим МеОН. потім Et20. Одержують 637мг жовтої твердої речови+ ни, яка плавиться при 197°С. МС: МН =523. ПРИКЛАД 20 Сполука №69 Стадія 1 До 3г продукту, одержаного на стадії 1.4, у 25мл ДМФ за 10 хвилин додають 432мг 60% NaH. Через 30 хвилин при перемішуванні додають 1,40г етилізоціанатацетату за 10 хвилин, потім реакційне середовище залишають на 3.5 години при перемішуванні при кімнатній температурі. Реакційне середовище екстрагують AcOEt, послідовно промивають водою, 5% розчином KHSO4/K2SO4. водою і насиченим розчином NaCl. Органічну фазу сушать, концентрують у вакуумі та одержаний неочищений продукт очищають хроматографією на силікагелі, елюент: CHCl3/AcOEt 85/15об./об. з одержанням 1,52г очікуваного продукту. 37 95223 38 Стадія 2 Продукт, одержаний на стадії 1, окисляють метахлорпербензойною кислотою (МХГІБК) (МСРВА) згідно зі способом, описаним у синтезі 1.6, Одержують 900мг очікуваного продукту у формі бежевої твердої речовини. Стадія 3 При 65°С протягом 6 годин у присутності 15мл ЕtOН і 0,06мл концентрованої НСl нагрівають 798мг продукту стадії 2 і 295мг 1,3-дигідро-2бензофуран-5-аміну. Реакційне середовище розводять СНСl3 і водою, потім доводять водну фазу до рН=9 додаванням насиченого розчину NaHCO3. Після декантації органічну фазу виділяють, промивають водою, потім насиченим розчином NaCl, сушать і концентрують при зниженому тиску. Продукт перекристалізовують з AcOEt. Одержують 0,67г жовтої твердої речовини. Стадія 4 Протягом 5 годин 0,66г складного ефіру, одержаного на стадії 3, у 25мл етанолу і 2мл ДМФ обробляють 1,5мл 2 н. NaOH. Реакційне середовище розводять СНСІ3 потім доводять до рН=4 додаванням 1 н. НСІ і декантують. Органічну фазу виділяють, промивають водою, потім насиченим розчином NaCl, сушать і концентрують при зниженому тиску з одержанням 0,61г жовтого порошку. Стадія 5 Протягом 1 години 15 хвилин у 2,5мл ДМФ перемішують 105мг продукту, одержаного на стадії 4, 16мг трет-бутиламіну. 28мг ДІПЕА (діізопропілетиламін) і 70мг ТБТУ. Потім реакційне середовище екстрагують СНСІ3, органічну фазу послідовно промивають водою. 5% розчином KHSO4/K2SO4. насиченим розчином NaCl, потім сушать і упарюють при зниженому тиску. Неочищений продукт очищають хроматографією на діоксиді кремнію, елююючи сумішшю СНСІ3/МеОН, 94/6об./об. Одержують 90мг очікуваного продукту у формі жовтої + твердої речовини. МС: МН =580. ПРИКЛАД 21 Сполуки 31 і 32 Сполука 12, одержана в прикладі 6, може бути об'єктом для поділу її енантіомерів хроматографією на хіральній нерухомій фазі таким чином: Техніка Berger Prep SFC Детектування УФ 230нм Нерухома фаза Chralpack AD-H 250×21мм (5мкм)) Рухома фаза 60%/40% СО2/(Етанол+0,5% ізопропіламіну) Витрата 50мл/хвилину Тиск 100 бар Кількість введень Багато з 350 введень по 28мг Виходячи з 10г рацемічної суміші, після поділу одержують 4,147г правообертального оптичного ізомеру (сполука 31) і 4.077г лівообертальною оптичного ізомеру (сполука 32). Таблиці 3 і 4. наведені нижче, ілюструють хімічну будову і фізичні властивості деяких прикладів згідно з винаходом. У згаданих таблицях Me. Et. iPr і tBu означають, відповідно, метильну, етильну. ізопропільну і трет-бутильну групи, і Вос означає трет-бутоксикарбонільну групу. Якщо не зазначено протилежне, продукти, що містять асиметричний атом вуглецю, одержані у формі рацемічної суміші. 39 95223 ТАБЛИЦЯ 3 40 41 95223 42 43 95223 44 45 95223 46 47 95223 48 49 95223 50 51 95223 52 53 95223 54 55 95223 56 57 Сполука 34 є об’єктом поділу на два її оптичних ізомери хіральною хроматографією. Правообертальний оптичний ізомер: []D=+72,l°; t=25°C: 00,5 (МеОН). Лівообертальний оптичний ізомер:[]D=-73.0°; t=25°C: С=0,5 (МеОН). Так само, використовуючи класичні методики комбінаційної хімії і застосовуючи способи одержання, описані в даному винаході, були одержані бібліотеки продуктів згідно з винаходом. Будова 95223 58 одержаних продуктів, а також їхні характеристики наведені тут. Аналізи РХ/МС, були здійснені на приладі Micromass, модель LCT, з'єднаному з приладом моделі HP 1100. Відносний вміст сполук вимірювали детектором із лінійкою діодів G1315A в області довжин хвиль від 200 до 600нм і за допомогою випарного детектора розсіяння світла (ВДРС) (DEDL) Sedex, модель 65. Мас-спектри реєстрували в діапазоні від 180 до 800 (M/z). Дані аналізува 59 95223 ли за допомогою програмного забезпечення Micromass MassLynx. Поділ проводили на колонці Hypersil BDS С18 (50×4,6мм), розмір частинок: 3мкм. Градієнтне елюювання від 5 до 90% ацетонітрилу, що містить 0,05% (об./об.) трифтороцто 60 вої кислоти (ТФО). у воді, що містить 0,05% (об./об.) ТФО, за 3,5 хвилини при витраті 1мл/хвилину. Загальна тривалість процедури, включаючи відновлення рівноваги колонки,становить 7 хвилин. ТАБЛИЦЯ 5
ДивитисяДодаткова інформація
Назва патенту англійськоюPyrido-pyrido pyrimidine derivatives, preparation thereof, therapeutic use thereof
Автори англійськоюBourrie Bernard, Casellas Pierre, Gegam Samir, Perreaut Pierre
Назва патенту російськоюПроизводные пиридопиримидина, их получение, их применение в терапии
Автори російськоюБурри Бернар, Казелла Пьер, Жегам Самир, Перро Пьер
МПК / Мітки
МПК: A61K 31/519, A61P 35/02, C07D 417/04, A61P 35/00
Мітки: піридопіримідину, терапії, застосування, одержання, похідні
Код посилання
<a href="https://ua.patents.su/33-95223-pokhidni-piridopirimidinu-kh-oderzhannya-kh-zastosuvannya-v-terapi.html" target="_blank" rel="follow" title="База патентів України">Похідні піридопіримідину, їх одержання, їх застосування в терапії</a>
Похідні 4-арилморфолін-3-ону, спосіб їх одержання і їх застосування в терапії

Номер патенту: 86082
Опубліковано: 25.03.2009
Автори: ЕМОН-АЛЬТ Ксав'є, Проієтто Вінченцо
МПК: C07D 413/14, C07D 265/10, C07D 413/06, A61K 31/537
Мітки: терапії, 4-арилморфолін-3-ону, одержання, спосіб, похідні, застосування
Формула / Реферат:
1. Сполука формули (І):, (І)в якійАr означає феніл, моно- або двозаміщений атомом галогену;R1 означає феніл, незаміщений або заміщений один або два рази одним або двома замісниками, незалежно вибраними з атома галогену, (С1-С4)алкілу або (С1-С4)алкокси;R2 означає:піридил;феніл, незаміщений або заміщений один або два рази одним або двома замісниками, незалежно вибраними з атома...
Похідні піридо[2,3-d]піримідину, їх одержання, їх застосування у терапії

Номер патенту: 92021
Опубліковано: 27.09.2010
Автори: Перро П'єр, Мюно Клод, Жегам Самір, Казелла П'єр, Буррі Бернар
МПК: A61K 31/519, A61P 35/00, C07D 471/04
Мітки: терапії, застосування, одержання, похідні, піридо[2,3-d]піримідину
Формула / Реферат:
1. Сполука формули (І), в якій Аr2 і R1 є відповідно: 2,6-дихлорофеніл та трет-бутил або 2-бромо-6-хлорфеніл та трет-бутил; або 2,6-дихлорофеніл та етил; або 2,6-дибромофеніл та трет-бутил; або 2,6-дибромофеніл та етил; або...
Похідні піридоіндолону, заміщені у положенні 6, їх одержання і застосування у терапії

Номер патенту: 85709
Опубліковано: 25.02.2009
Автори: Вермют Камілль-Жорж, Чіапетті Паола, Жегам Самір, Сазелла П'єр, Мюно Іветт, Буррі Бернар, Дерок Жан-Марі
МПК: A61K 31/437, A61K 31/407, C07D 471/04
Мітки: застосування, заміщені, піридоіндолону, одержання, положенні, похідні, терапії
Формула / Реферат:
1. Сполука, яка відповідає формулі:, (I)де:R1 являє собою атом водню, (С1-С4)-алкіл, групу -(СН2)mОН, групу -(CH2)mCN, групу -(CH2)mNR9R10;R2 являє собою атом водню або (С1-С4)-алкіл;R3 являє собою феніл, заміщений радикалами R6, R7, R8;R4 являє собою:групу
Похідні 5-піридиніл-1-азабіцикло[3.2.1]октану, їх одержання і їх застосування у терапії

Номер патенту: 92917
Опубліковано: 27.12.2010
Автори: Ваше Жюльєн, Галлі Фредерік, Леклерк Оділь, Локхед Алістер
МПК: C07D 471/18, A61K 31/439, A61P 25/00
Мітки: терапії, 5-піридиніл-1-азабіцикло[3.2.1]октану, одержання, застосування, похідні
Формула / Реферат:
1. Сполука загальної формули (І), (I) в якій R означає групу, вибрану з піразолілу, імідазолілу, триазолілу, оксазолілу, оксадіазолілу, тіазолілу, ізотіазолілу, тіадіазолілу, тетразолілу, причому вказана група, можливо, заміщена однією або декількома групами, вибраними з галогенів, (С1-С6)алкілу, (С1-С6)алкокси,...
Похідні тетрагідроізохінолілсульфонамідів, їх одержання й застосування в терапії

Номер патенту: 84771
Опубліковано: 25.11.2008
Автори: Хімінес Баргуено Марія Долорес, Діас Мартін Хуан Антоніо
МПК: A61P 3/04, A61K 31/472, C07D 401/12, A61K 31/5377, C07D 405/06, A61K 31/496, C07D 217/04, A61K 31/4725, C07D 217/02, A61P 25/00, C07D 409/06, A61P 3/10, C07D 217/06
Мітки: похідні, застосування, терапії, тетрагідроізохінолілсульфонамідів, одержання
Формула / Реферат:
1. Сполука формули І:, Іде:n може приймати значення від 1 до 6;-(С)n- являє собою С1-6-алкіліден, за необхідності заміщений замісниками в числі від 1 до 4, вибраними з атома галогену, гідрокси, нітро, ціано, аміно, С1-3-моноалкіламіно, С2-6-діалкіламіно або С1-3-алкокси;R1 являє собою:атом водню;С1-6-алкіл;R2 являс...
Попередній патент: Побутовий прилад з електроприводом
Наступний патент: Антацидна фармацевтична композиція у порошковій формі, фармацевтичний засіб, що її вміщує, та спосіб її одержання
Випадковий патент: Композиція інгредієнтів бальзаму-настойки "назар стодоля"