Спосіб одержання енантіомерів о-диметилтрамадолу
Номер патенту: 45342
Опубліковано: 15.04.2002
Автори: Янсен Пітер, Граудумс Іварс, Штрассбургер Вольфганг, Фрідеріхс Ельмар, Бушманн Хельмут, Вінтер Вернер




Формула / Реферат
1. Способ получения энантиомеров О-диметилтрамадола, согласно которому трамадол преобразуют в основание, осаждают кислотой в присутствии органического растворителя и высвобождают один из энантиомеров в виде свободного основания, отличающийся тем, что рацемический раствор (-) и (+)-энантиомеров трамадола преобразуют в основание, осаждают (-)-энантиомер трамадола из маточного раствора L-(+)-винной кислотой и отделяют осадок от маточного раствора, высвобождают (-)-энантиомер трамадола из осадка в виде свободного основания (-)-энантиомера трамадола, основание (-)-энантиомера трамадола преобразуют в (-)-энантиомер O-диметилтрамадола посредством диизобутилалюминийгидрида, высвобождают из маточного раствора (+)-энантиомер трамадола в виде свободного основания (+)-энантиомера трамадола и преобразуют основание (+)-энантиомера трамадола в (+)-энантиомер O-диметилтрамадола посредством диизобутилалюминийгидрида.
2. Способ по п.1, отличающийся тем, что упомянутый рацемический раствор получают преобразованием рацемической соли трамадола в соответствующее основание.
3. Способ по п.2, отличающийся тем, что в качестве рацемической соли трамадола используют рацемический гидрохлорид трамадола.
4. Способ по п.1, отличающийся тем, что раствор осаждают в присутствии органического растворителя.
5. Способ по п.4, отличающийся тем, что в качестве упомянутого органического растворителя используют алифатический С1-5 спирт.
6. Способ по п.1, отличающийся тем, что (-)-энантиомер трамадола получают кристаллизацией L-(+)-тартрата (-)-трамадола.
7. Способ по п.1, отличающийся тем, что основание энантиомера трамадола преобразуют в другую соль, отличную от тартрата, и затем высвобождают соответствующий энантиомер в виде свободного основания из этой другой соли, до преобразования свободного основания посредством диизобутилалюминийгидрида.
8. Способ по п.7, отличающийся тем, что свободное основание энантиомера трамадола преобразуют в гидрохлорид.
Текст
1. Способ получения энантиомеров Одиметилтрамадола, согласно которому трамадол преобразуют в основание, осаждают кислотой в присутствии органического растворителя и высвобождают один из энантиомеров в виде свободного основания, отличающийся тем, что рацемический раствор (-) и (+)-энантиомеров трамадола преобразуют в основание, осаждают (-)энантиомер трамадола из маточного раствора L(+)-винной кислотой и отделяют осадок от маточного раствора, высвобождают (-)-энантиомер трамадола из осадка в виде свободного основания ()-энантиомера трамадола, основание (-)энантиомера трамадола преобразуют в (-)энантиомер O-диметилтрамадола посредством диизобутилалюминийгидрида, высвобождают из C2 2 45342 1 3 45342 4 зали исследования, оба энантиомера трамадола и Целесообразно в качестве рацемической соли два энантиомера метаболитного трамадола иметрамадола использовать рацемический гидрохлоют болеутоляющее действие (J. Pharmacol. Exptl. рид трамадола. Ther. 260, 275 (1992); Arzneim.-Forschimg 38, 877 Преобразование основания трамадола в гид(1988)). рохлорид может быть осуществлено посредством Получение О-днметилтрамадола как рацемаконцентрированной хлористоводородной кислоты та или в форме энантиомеров известно из патента или газообразного хлорида водорода в органичеWO 93/04675. В описанном способе Оском растворителе, например ацетоне, диоксане, диметилтрамадол получают преобразованием диэтиловом эфире и/или диизопропиловом эфире, трамадола в свободное основание, например, с или триметилхлорсиланом/водой в растворителе, помощью реакции с использованием сильного например 2-бутаноне. основания типа гидрид натрия или калия в присутПредпочтительно раствор осаждают в присутствии тиофенола и диэтиленгликоля, с нагревом ствии органического растворителя. Причем в кадо дефлегмации. Реакцию проводят в течении честве упомянутого органического растворителя, часа, с последующим охлаждением, а затем тушат может быть использован алифатический С 1-5 смешивая с водой. После этого смесь осаждают спирт. кислотой в присутствии органического раствориЦелесообразно (-)-энантиомер трамадола потеля, например, этилового эфира, высвобождают лучать кристаллизацией L-(+)-тартрата (-)в виде свободного основания, а затем выделяют с трамадола. использованием галогенированного органического При этом предпочтительно основание энанрастворителя, например, метилен хлорида. Затем тиомера трамадола преобразуют в другую соль, экстракт высушивают, а раствор выпаривают для отличную от тартрата, и затем высвобождают сополучения конечного О-диметилтрамадол продукответствующий энантиомер в виде свободного та, который затем может быть легко дистиллирооснования из этой другой соли, до преобразоваван, и преобразуют в соответствующую соль, нания свободного основания посредством диизобупример, обработкой кислотным раствором (НСl тилалюминийгидрида этанолом), и перекристализуют из раствора смеПричем свободное основание энантиомер шанного с органическим растворителем. трамадола преобразуют в гидрохлорид. Однако, при использовании данного способа, При использовании способа согласно изобреО-диметилтрамадол может быть получен только в тению, энантиомеры О-диметилтрамадола могут неудовлетворительных количествах, при чем в быть получены экономично и экологически благокачестве реагентов используют вредные для здоприятным способом, и в больших количествах. ровья компоненты, сильные основания типа гидТолько энантиомерная форма винной кислоты, а рид натрия или калия в присутствии тиофенола и именно недорогая L-(+)-винная кислота, является диэтиленгликоля. необходимой для разделения рацемата соли траВ основу настоящего изобретения поставлена мадола. С помощью L-(+)-винной кислоты в прозадача разработать способ получения энантиомедукте может быть получено более чем 85% отноров О-диметилтрамадола, с помощью которого сительно используемого рацемата энантиомеров энантиомеры трамадола могут быть получены с трамадола с чистотой более чем 98%. Маточный высокой степенью чистоты, стабильно большой раствор может быть повторно использован в пропроизводительностью и экологически благоприятцессе разделения рацемата после освобождения ным способом. основания трамадола. Расщепление метилового Поставленная задача решается тем, что раэфира дает энантиомеры О-диметилтрамадола в цемический раствор (-) и (+)-энантиомеров трамаколичестве более 95%. дола преобразуют в основание, осаждают (-)Использование О-диметилтрамадола в сочеэнантиомер трамадола из маточного раствора Lтании с кодеином, оксикодоном, гидрокодоном или (+)-винной кислотой и отделяют осадок от маточацетаминофеном для лечения проявления боли ного раствора, высвобождают (-)-энантиомер траописано в WO 93/04675. Теперь установлено, что мадола из осадка в виде свободного основания (даже О-диметилтрамадол сам по себе или в соче)- энантиомера трамадола, основание (-)тании с трамадолом обладает высоким болеутоэнантиомера трамадола преобразуют в (-)- энанляющим воздействием. тиомер О-диметилтрамадола посредством диизоТаким образом, настоящее изобретение отнобутилалюминийгидрида, высвобождают из маточсится также к использованию Оного раствора (+)-энантиомер трамадола в виде диметилтрамадола в качестве основания и/или свободного основания (+)-энантиомера трамадола соли в форме рацемата или энантиомера, оти преобразуют основание (+)-энантиомера трамадельно или в сочетании с трамадолом в качестве дола в (+)-энантиомер 0основания и/или соли в форме рацемата или дииетилтрамадола посредством диизобутилалюэнантиомера, как болеутоляющий активный комминийгидрида (ОГОАН). Расщепление эфира мепонент в лекарстве. тила с помощью DIBAH обычно проводится в ароПредпочтительно использовать (+)матическом углеводороде, например толуоле, при энантиомер О-диметилтрамадола. температуре между 60 и 130°С. В дополнение к основанию и/или по крайней Причем упомянутый рацемический раствор мере одной соли О-диметилтрамадола отдельно может быть получен преобразованием рацемичеили в сочетании с основанием трамадола и/или по ской соли трамадола в соответствующее основакрайней мере одной соли трамадола, эти анальгение. тики согласно изобретению содержат носители, 5 45342 6 наполнители, растворители, разбавители, красического материала при комнатной температуре в тели и/или связующие вещества. Выбор этих вакууме (60мбар) было получено 2050г (49% отновспомогательных веществ и их количества для сительно общего количества использованных раиспользования зависит от того, будет лекарство цемических оснований) (1S, 2S)-2применяться орально, внутривенно, трансбукдиметиламинометил-1-(3-метокси-фенил)- циклокально, интраперитонеально, подкожно, внутригексанол-L-(+)-тартрата с точкой плавления 173 мышечно, интраназально или местно, например, 175°С (удельное вращение: [a]RTD = -12,2° (с = на коже, слизистой оболочке или в глазу. Препа1,01; метанол)). раты в форме таблеток, драже, капсул, гранул, Этап 3: высвобождение основания из соли Lкапель, соков и сиропов пригодны для орального (+)-винной кислоты. применения. Растворы, суспензии, или сухие пре2050г (4,95моль) (1S, 2S)-2параты в виде готовых форм и аэрозоли пригодны диметиламинометил-1-(3-метокси-фенил)для парентерального или местного применения и циклогексанол L-(+)-тартрата, полученного на этаингаляции. Составы, которые по изобретению для пе 2, растворили в 4000мл воды и обработали использования в виде порошков или в повязок, 900г измельченного льда. При перемешивании желательно с добавлением веществ,. которые добавили по каплям 1000мл 36-38%-ного (технипромотируют проникновение через кожу, примеческого) едкого натра. Затем смесь экстрагированяют для введения через кожу. Формы препарали с помощью 2500мл дихлорометана, и после тов, которые применяются орально или через коразделения фаз вновь экстрагировали с помощью жу, способны выделять составы по изобретению дополнительных 500мл дихлорометана. Смесь постепенно, распределено во времени. органических фаз была высушена посредством Количество активного компонента, который сульфата натрия. После удаления растворителя назначают пациенту, изменяется в зависимости от дистилляцией было получено 1280г (99% теоревеса пациента, от способа применения, от услотически) (1S, 2S)-2-диметиламинометил-1-(3вий и от степени тяжести болезни. Назначают метокси-фенил)-циклогексанола в виде сиропа. обычно от 5 до 500мг/кг по крайней мере одного из Этап 4: преобразование (1S, 2S)-2вышеупомянутых составов. диметиламинометил-1-(3-метокси-фенил)Пример 1 циклогексанола в гидрохлорид (-1). (-)-(1S, 2S)-2-диметиламинометил-1 -(31280г (4,86моль) основания, полученного на метокси-фенил)-циклогексанол-гидрохлорид (-1) этапе 3, растворили в 16л 2-бутанона и обработали 88мл (4,9моль) воды и 621мл (532г; 4,9моль) триметшгхлорсилана при перемешивании. Смесь перемешивали в течение 3 часов при комнатной температуре и оставили стоять в течение 24 часов при 4°С для кристаллизации. Осажденные твердые частицы отфильтровали отсасыванием, промыли в 5000мл 2-бутанона при 4°С и высушили до Этап 1: высвобождение рацематического оспостоянного веса при 90°С в вакууме (60мбар). нования. Было получено 1390г гидрохлорида (-1) (95 % 3кг (10моль) (1RS, 2К8)-2теоретически относительно использованного осдиметиламинометил-1-(3-метокси-фенил)нования из этапа 3, и 92% относительно содержациклогексанол-гидрохлорида (1) были суспендиния энантиомеров использованного рацемата из рованы в 4800мл воды и обработаны измельченэтапа 1), в виде бесцветных кристаллов. ным льдом массой 1,6кг. При перемешивании по Точка плавления: 172 - 173°С каплям было добавлено 1300мл 36 - 38%-ного Удельное вращение: [a]RTD = -29,6° (технического) едкого натра. Затем смесь экстра(с = 1,00; метанол)). гировали с помощью 7000мл дихлорометана, и Этап 5: преобразование гидрохлорида(-1) в (после разделения фаз была вновь экстрагирована )-(1S, 2S)-3-(2-диметиламинометил-1 -гидроксис помощью дополнительных 2000мл дихлоромециклогексил)-фенол-гидрохлорид тана. Смесь органических фаз была высушена посредством сульфата натрия. После удаления растворителя посредством дистилляции было получено 2630г (99% теоретически) (1RS, 2RS)-2диметиламинометил-1-(3-метокси-фенил)циклогексанола в виде сиропа. Этап 2: осаждение с помощью L-(+)-винной Основание было вьщелено из гидрохлокислоты. рида (-1) посредством дихлорометана или раство2630г (10моль) основания, полученного на ра гидрохлорида натрия при условиях, приведенэтапе 1, растворили в 2400мл этанола и обрабоных для этапа 1. После высушивания раствора тали раствором, состоящим из 1500г (10моль) Lдихлорометан был отогнан в вакууме. 208,1г (+)- винной кислоты и 11200мл этанола. Смесь (0,79моль) полученного основания, растворенного перемешивали в течение двух часов при комнатв 360мл толуола, добавили по каплям при комнатной температуре и оставили на 24 часа при 4°С ной температуре к 1,6л 20%-ного раствора диизодля кристаллизации. Осажденные кристаллы бутилалюминийгидрида (1,58моль) в толуоле. Запрофильтровали отсасыванием и промыли 6400 тем смесь нагревали орошением в течение 11 мл этанола при 4°С. После высыхания кристалли 7 45342 8 часов, и после охлаждения до комнатной темпеЭтап 2: преобразование (1R, 2R)-2ратуры ее охладили далее почти до 0°С с помодиметиламинометил-1-(3-метокси-фенил)щью льда/поваренной соли. Затем по каплям доциклогексанол в гидрохлорид (+1). бавили 450мл этанола таким образом, чтобы 1340г (5,09моль) основания, полученного на внутренняя температура не превышала 15°С. Поэтапе 1, растворили в 17,5л 2-бутанона и обрабосле завершения добавления, смесь размешивали тали с помощью 105мл (5,8моль) воды и 670мл в течение дальнейших 15 минут и разбавили с (573г; 5,3моль) триметилхлорсилана при переме1000мл толуола. 450мл смеси этанол/вода (1 : 1) шивании. Смесь перемешивали в течение 3 часов добавили по каплям при охлаждении смеси во при комнатной температуре и оставили отстаильду/поваренной соли; после того, как добавление ваться в течение 24 часов при этой же температубыло завершено, смесь размешивали в течение ре для кристаллизации. Осажденные твердые одного часа при комнатной температуре. Осажчастицы отфильтровали отсасыванием, промыли денную гидроокись алюминия отфильтровали отпосредством 5000мл 2-бутанона и просушили до сасыванием и перемешали с пятью частями по постоянного веса при 90°С в вакууме (60мбар). объему этилацетата при 60°С для последующей Было получено 1350г гидрохлорида (+1) (88% теоэкстракции. После отфильтровывания снова при ретически относительно использованного основаотсасывании, смешанные органические фазы быния из этапа 1, и 89% относительно содержания ли высушили с помощью сульфата натрия и сконэнантиомеров использованного из Примера 1) в центрированы при 60°С в роторном испарителе. виде бесцветных кристаллов. Было получено 193г (98% теоретически) основаТочка плавления: 171 - 172°С ния; основание выкристаллизовано как твердое Удельное вращение: [a]RTD = +29,6° тело с точкой плавления 139 - 142°С. (с = 1,00; метанол)). Полученный полуфабрикат растворили в Этап 3: преобразование гидрохлорида (+1) в 1,93л ацетона и обработали с помощью 65мл кон(+)-(1R, 2R)-3-(2-диметиламинометил-1 -гидроксицентрированной хлористоводородной кислоты. циклогексил)-фенол-гидрохлорид После того, как началась кристаллизация, продукт размешивали в течение одного часа при охлаждении в ледяной ванне перед тем, как осадок был отфильтрован отсасыванием. Осадок был дважды промыт ацетоном и диэтиловым эфиром, и кристаллический материал затем высушили" до постоянного веса при 70°С в вакууме, создаваемом (+)-энантиомер О-диметилтрамадола был масляным насосом. Было получено 216,8г (96% получен как гвдрохлорид с вы ходом 96%, считая теоретически) бесцветных кристаллов. от гидрохлорида (+1), при условиях, данных в Точка плавления: 247 -248°С Примере 1, этап 5. RT Удельное вращение: [a] D = -35,2° Точка плавления: 247 -248°С (с = 1,00; метанол)). Удельное вращение: [a]RTD = +35,4° Пример 2 (с = 1,00; метанол)). (+)-(1R, 2R) - 2 - диметиламинометил - 1 - (3 Фармакологические исследования Испытание метокси - фенил) -циклогексанол- гидрохлорид анальгетика с использованием теста "корчи" на (+1) мышах. Этап 1: высвобождение основания из маточного раствора из осадка L-(+)-винной кислоты. Этаноловый маточный раствор и фаза промывки осадка L-(+)-винной кислоты (пример 1, этап 2) были объединены. После удаления растворителя дистилляцией, остаток (2080г) растворили в 2500мл воды и обработали 900г измельченного льда. При перемешивании по каплям добавили 1000мл 36 - 38%-ного (технического) едкого натра. Затем смесь экстрагировали с помощью 2700мл дихлорометана, и после разделения фаз снова экстрагировали с помощью дополнительных 600мл дихлорометана. Смесь органических фаз высушили посредством сульфата натрия. После удаления растворителя посредством дистилляции было получено 1340г (99% теоретически) (1R, 2R)-2-диметиламинометил-1(3-метокси-фенил)-циклогексанола в виде сиропа. Было проведено испытание эффективности анальгетика при использовании теста на боль у мышей (теста "корчи"), вызываемой фенилхиноном (модифицированного по I.C. Hendershot, J. Forsaith, J. Pharmacol. Exp. Ther. 125, 237 - 240 (1959)). Для этой цели использовались мыши NMRI мужского пола весом 25 - 30г. Для каждой дозы вещества использовались группы из 10 животных. Через 10минут после внутривенного введения составов по изобретению, каждой мыши было введено внутрибрюшинно 0,3мл 0,02% водного раствора фенилхинона (фенилбензохинон производства Sigma, Deisenhofen; раствор подготовлен с добавлением 5% этанола и хранился в водной ванне при 45°С). Животных поместили индивидуально в клетки для наблюдения, и подсчитывали количество движений вытягивания, вызванных болью (так называемые реакции корчи = расправление тела с вытягиванием задних конечностей), в течение 5 - 20 минут после введения фенилхинона посредством нажатия кнопки счетчика. Значения ED50 были рассчитаны с точностью 95% регрессионным анализом (программа расчета Martens EDV Service, Eckental) уменьшения 9 45342 10 реакций корчи в зависимости от дозы в сравнении рассчитано как среднее число предварительного с контрольными группами, которые были исследоисследования. Измерение боли было сделано ваны параллельно и которые были обработаны через 20, 40 и 60 минут после внутривенного вветолько фенилхиноном. дения. Когда время ожидания боли было увеличеИспытание анальгетика с использованием но, максимальное время воздействия было огратеста движений хвоста у крыс. ничено 12 секундами и увеличение скрытого Болеутоляющая эффективность составов по периода на более, чем 150% относительно предизобретению была исследована тестом на тепловарительного среднего значения расценивалось вое излучение (движение хвоста) на крысах, при как болеутоляющее воздействие. Чтобы опредеиспользовании методики D'Amour и Smith (J. лить зависимость от дозировки, составы вводиPharm. Exp. Ther. 72, 74 - 79 (1941). Для этой цели лись в увеличиваемых логарифмически с коэфиспользовались крысы женского пола Sprague фициентом от 3 до 5 дозах, которые включает в Dawley с весом 120 - 160г. Животных поместили себя начальную и максимальную эффективную индивидуально в клетки для наблюдения, а оснодозу каждый раз. Значения ЕD50 были определены вания их хвостовых частей были подвержены для большого количества обработанных анальгесфокусированному тепловому излучению от электиком животных методом Litchfield и Wilcoxon (J. трической лампы (Rhema Analgesiemeter для Pharm.Exp. Ther. 96, 99 - ИЗ, (1949)). ED50 было крыс). Интенсивность лампы была отрегулирована рассчитано для эффективных максимальных 20 таким образом, чтобы время от включения лампы минутах после внутривенного введения вещества. до рывка хвостовой части (время ожидания боли) Энантиомеры О-диметилтрамадола по собыло 3 - 6 секунд для необработанных животных. гласно изобретению проявили явный болеутоПеред введением составов по изобретению жиляющий эффект в тесте "корчи" на мышах и в тесвотные были дважды исследованы в течение пяти те движений хвоста на крысах. Полученные минут, и среднее значение этих измерений было результаты приведены в следующей таблице. Таблица Испытание анальгетика при использовании теста "корчи" на мышах и тесте движения хвоста на крысах ED50 (мг/кг, внутривенно) в тесте ED50 (мг/кг, внутривенно) в Состав "корчи" тесте движения хвоста (+)-энантиорем О-диметилтрамадол0,489 1,01 гидрохлорида (-)-энантиорем О-диметилтрамадол6,60 > 10,0 гидрохлорида Для сравнения: рацемический тра3,59 6,47 мадолгидрохлорид ДП «Український інститут промислов ої в ласності» (Укрпатент) вул. Сім’ї Хохлов их, 15, м. Київ , 04119, Україна (044) 456 – 20 – 90 ТОВ “Міжнародний науков ий коміт ет” вул. Артема, 77, м. Київ , 04050, Україна (044) 216 – 32 – 71
ДивитисяДодаткова інформація
Автори англійськоюBushman Helmut, Strassburger Wolfgang
Автори російськоюБушманн Хельмут, Штрассбургер Вольфганг
МПК / Мітки
МПК: C07B 57/00, A61P 25/04, C07C 217/74, A61K 31/135, C07C 213/00, C07C 215/00
Мітки: о-диметилтрамадолу, енантіомерів, спосіб, одержання
Код посилання
<a href="https://ua.patents.su/5-45342-sposib-oderzhannya-enantiomeriv-o-dimetiltramadolu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання енантіомерів о-диметилтрамадолу</a>
Спосіб одержання похідних азепіна або їх солей, або енантіомерів

Номер патенту: 8041
Опубліковано: 26.12.1995
Автори: Карл-Гейнц Вебер, Армін Фюгнер, Герхард Вальтер, Клаус Шнейдер
МПК: C07D 513/04, C07D 223/00, A61K 31/55, A61P 7/02, C07D 487/04, C07D 267/00, C07D 498/04, A61P 43/00, A61P 37/08, C07D 281/00
Мітки: азепіна, спосіб, солей, похідних, одержання, енантіомерів
Формула / Реферат:
Способ получения производных азепина общей формулыгде R1 = Н; 7-Сl, R2 = Н при Х = СН2, О, S;R1 = Н, 6-Сl, 6-СН3, R2 = Н, 12-Сl при Х = О;R1 = H, R2 =12-Cl при X = O, S;R1 = 6 - ОСН3, R2 = Н при Х = S,или их солей, отличающийся тем, что соединение общей формулыгде R1, R2, Х имеют указанные значения, подвергают взаимодействию с бромцианом в среде тетрагидрофурана с последующим...
Спосіб біокаталітичного одержання одного з енантіомерів хіральної 2-арилалканової кислоти

Номер патенту: 27817
Опубліковано: 16.10.2000
Автори: ЕВАНС Крістофер Томас, ВІЗДОМ Річард Ентоні, СТЕЙБЛЕР Пітер Джозеф, Карганіко Джермано
МПК: C07D 213/55, C12P 17/10, C07D 401/04, C12P 7/40, C12P 41/00
Мітки: кислоти, одного, енантіомерів, спосіб, хіральної, біокаталітичного, 2-арилалканової, одержання
Текст:
...новых поступлений 348917. Ниже приведены следующие характеристики депозитированного микроорганизма ENZA-13: Ферментативное кутильтивирование: Глюкоза -ve Аэробное культивирование: D-Глюкоза +ve D-Галактоза +ve L-Сорбоза +ve D-Глюкозамин +ve D-Рибоза +ve D-Ксилоза +ve L-Арабиноза +ve D-Арабиноза -ve L-Рамноза +ve Сахароза +ve Мальтоза +ve aa -Трегалоза +ve Метил а -Глюкозид +ve Целлобиоза +ve Салицин +ve Арбутин +ve Мелибиоза +ve Лактоза...
Похідні 1,4-дигідропіридину у формі рацемату або енантіомерів, 2-хлор-3-ціанобензальдегід як проміжний продукт для їх одержання та фармацевтична композиція, що має антагоністичну активність по відношенню до кат

Номер патенту: 37211
Опубліковано: 15.05.2001
Автори: Вольфганг Хартвіг, Маартен де Йонге, Цан Гао, Тойніс Шуурман, Бернхард Шмідт, Бодо Юнге, Рудольф Шохе-Лооп, Генріх Мейєр
МПК: A61P 25/28, A61K 31/455, C07D 401/12, C07D 211/90
Мітки: відношенню, 1,4-дигідропіридину, похідні, композиція, проміжний, рацемату, має, форми, антагоністичну, одержання, активність, продукт, енантіомерів, фармацевтична, 2-хлор-3-ціанобензальдегід, кат
Формула / Реферат:
1. Производные 1,4-дигидропиридина общей формулы (I):где указанные заместители имеют следующие значения: R1 R2 R3 R4 Рацемат/ Энантиомер (Н3С)2-НС- Η 2-F -(СН2)2-ОСН3 ...
Похідні тетрагідроізохіноліну і їх фармацевтично прийнятні солі у вигляді індивідуальних енантіомерів, рацематів або інших сумішей енантіомерів

Номер патенту: 42730
Опубліковано: 15.11.2001
Автори: Джонстон Девід Нормен, Крю Ендрю Філіп Остін, Сарджент Брюс Джеремі
МПК: C07D 217/04, C07D 405/08, A61K 31/472, C07D 409/08, A61P 21/00, A61K 31/47, C07D 217/14, A61P 25/28, C07D 401/08
Мітки: рацематів, фармацевтично, солі, вигляді, тетрагідроізохіноліну, похідні, прийнятні, енантіомерів, індивідуальних, інших, сумішей
Формула / Реферат:
1. Производные тетрагидроизохинолина общей формулы I:в которойR1 - водород или алкил с 1-3 атомами углерода,R2 - алкил с 1-3 атомами углерода,Е - алкиленовая цепь, содержащая 2 или 3 атома углерода,G - насыщенная или ненасыщенная алициклическая группа, содержащая 3-8 атомов углерода, алкил с 1-6 атомами углерода, незамещенный или замещенный алкилом с 1-3 атомами углерода или циклоалкилом с 3-7 атомами...
Спосіб одержання (-)-[[4-(1,4,5,6-тетрагідро-4-метил-6-оксо-3-піридазиніл)феніл]гідразоно]пропандинітрилу, проміжні сполуки, спосіб одержання оптично чистого (-)-6-(4-амінофеніл)-5-метилпіридазин-3(2н)ону, опти
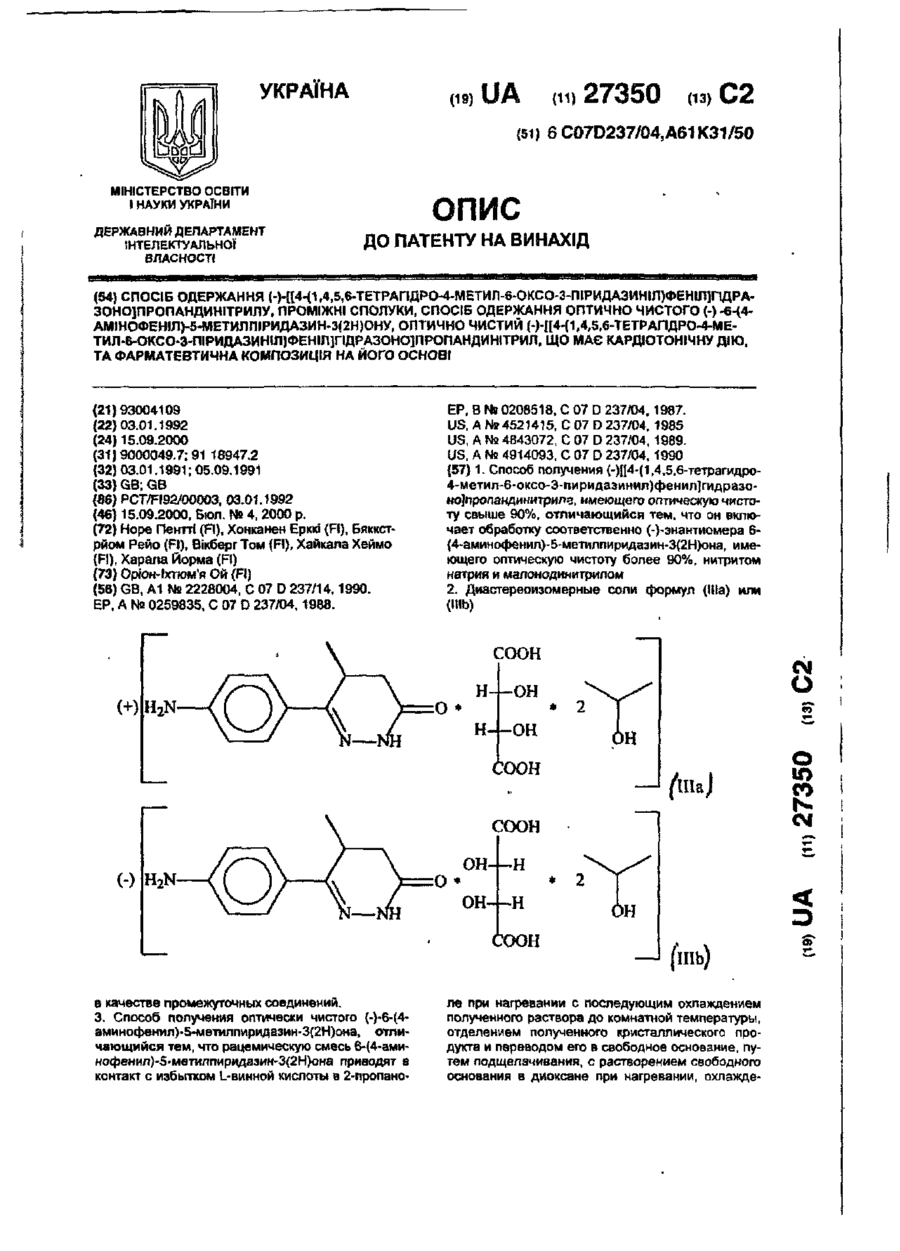
Номер патенту: 27350
Опубліковано: 15.09.2000
Автори: Бяккстрйом Рейо, Вікберг Том, ХОНКАНЕН Ерккі, Хайкала Хеймо, Норе Пентті, Харала Йорма
МПК: A61P 9/10, A61P 9/08, A61K 31/50, C07D 241/08, A61P 9/12, C07B 57/00, C07D 237/04
Мітки: оптично, 4-(1,4,5,6-тетрагідро-4-метил-6-оксо-3-піридазиніл)феніл]гідразоно]пропандинітрилу, опти, спосіб, сполуки, 6-(4-амінофеніл)-5-метилпіридазин-3(2н)ону, одержання, проміжні, чистого
Текст:
...Великобритании 2228004. Было показано, что соединение (f) является сильнодействующим при лечении застойной сердечной недостаточности и обладает значительной способностью в связывании кальция с тропонином. Наши дальнейшие исследования в настоящее время неожиданно выявили, что кардиотоническэя активность преимущественно относится к оптически активному {-) энантиомеру этого соединения. Кроме того, было установлено, что растворимость в воде (-)...
Попередній патент: Спосіб купірування синдрому відміни внаслідок вживання психоактивних речовин
Наступний патент: Спосіб розділення рацемату трамадолу та спосіб розділення рацемічної солі трамадолу на (-) і (+)-енантіомери
Випадковий патент: Спосіб закупорки вина в скляну тару