Спосіб хроматографічного визначення сумарного жирнокислотного складу біологічних рідин
Номер патенту: 94373
Опубліковано: 26.04.2011
Автори: Цяпець Сергій Васильович, Ростока-Резнікова Мар'яна Василівна, Аріповскій Алєксандр Вікторовіч, Глазкова Галіна Пєтровна, Колесник Павло Олегович, Веждел Марія Іванівна, Кірсанова Марина Петрівна




Формула / Реферат
Спосіб хроматографічного визначення сумарного жирнокислотного складу біологічних рідин, який включає відділення ліпідів від матричного матеріалу, який відрізняється тим, що рідкий зразок біологічного матеріалу, вибраний з групи: кров, сироватка, суспензія клітин, гомогенізат тканини, сушать для одержання зразка безводного біологічного матеріалу в апараті сублімаційного сушіння або в ротаційному вакуумному концентраторі типу "Savant SpeedVac", при цьому аліквоту зразка досліджуваного рідкого біологічного матеріалу в кількості 20-500 мкл зневоднюють безпосередньо в пробірці для дериватизації, а ліпіди висушеного зразка біологічного матеріалу у цій же пробірці перетворюють у відповідні метилові ефіри нагріванням з розчином 0,5 N метилату натрію в метанолі з розрахунку 150 мкл розчину метилату натрію на 100 мкл крові або 15 % гомогенізату, а присутні вільні жирні кислоти нагрівають з 10 % метанольним розчином трифтористого бору з розрахунку 0,8-1,0 мл на 100 мкл крові або 15 % гомогенізату, причому після закінчення метилювання спиртовий розчин зразка біологічного матеріалу розбавляють дистильованою водою вдвічі, після чого екстрагують одержані метилові ефіри жирних кислот в гептанову неполярну фазу, при цьому дещо уповільнене розшарування водно-спиртової та неполярної гептанової фази на стадії екстракції ефірів з реакційної суміші прискорюють додаванням краплі пропілового спирту або короткочасним (1-2 хв.) центрифугуванням в низькооборотній центрифузі, після чого екстраговані в гептанову фазу метилові ефіри жирних кислот визначають кількісно методом капілярної газової хроматографії на колонці з полярною іммобілізованою рідкою фазою.
Текст
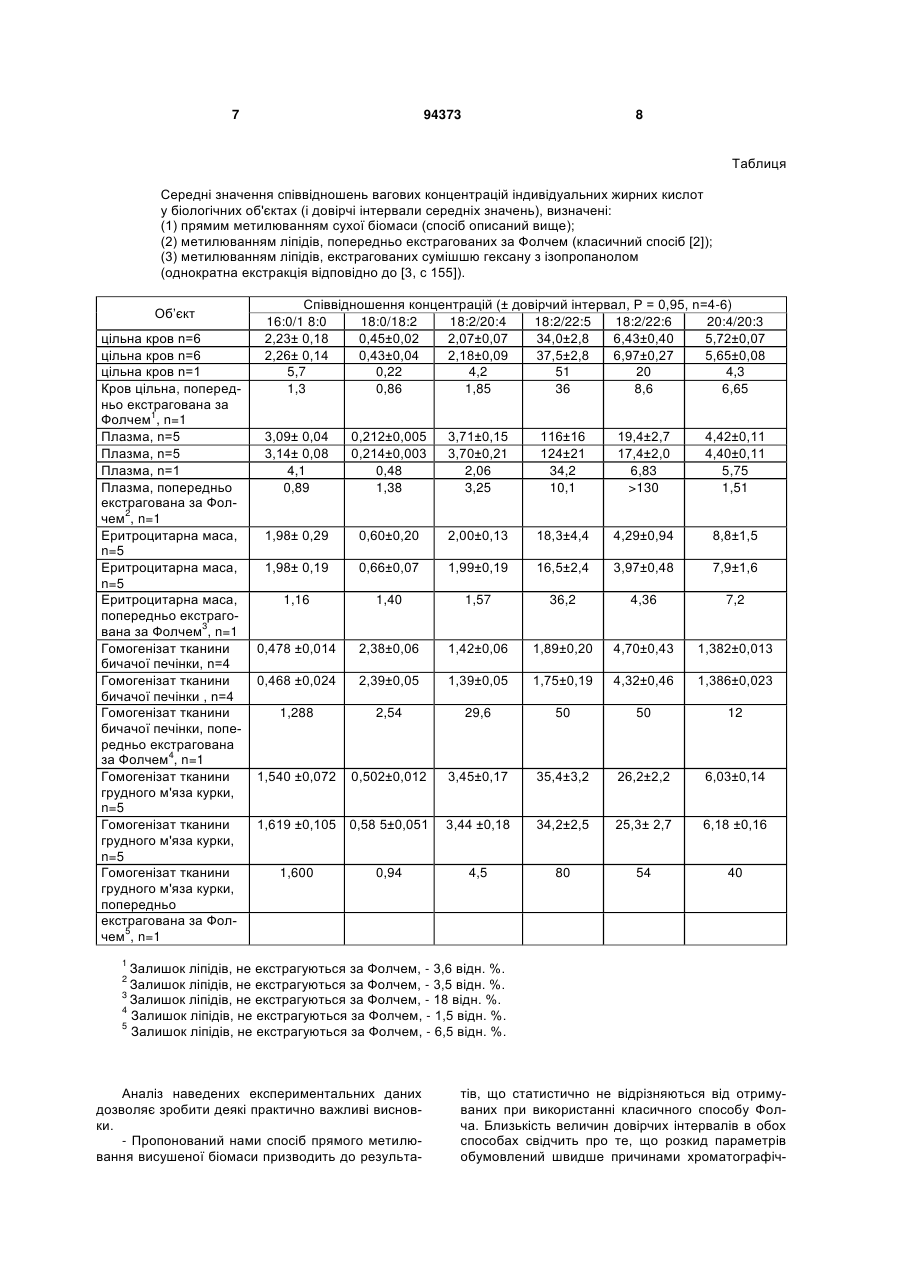
Спосіб хроматографічного визначення сумарного жирнокислотного складу біологічних рідин, який включає відділення ліпідів від матричного матеріалу, який відрізняється тим, що рідкий зра C2 2 (11) 1 3 В даний час дослідження жирнокислотного складу біологічних об'єктів у всіх випадках починається з екстракційного вилучення суми ліпідів та концентрування екстракту. Сучасні варіанти цього способу (наприклад, екстракція вуглекислотою в надкритичних умовах [1]) представляють безсумнівний інтерес для хімічної технології та біотехнології, однак як спосіб лабораторного дослідження вважаються малоперспективними. В ході лабораторних досліджень відділення ліпідів від матричного матеріалу в усіх випадках, що нам відомі, проводиться за способом Фолча. Отже, саме цей спосіб слід вважати прототипом за відсутністю спрощених безекстракційних варіантів. Суть загальновідомого і надзвичайно популярного способа Фолча полягає в дворазовій екстракції рідкого зразка або гомогенізату великими (зазвичай 6- або 10-кратними) обсягами суміші хлороформу з метанолом, 2:1, і промиванням об'єднаних екстрактів ізотонічним розчином хлоридів натрію або калію [2]. Інколи зустрічаються посилання на можливість використання суміші спирту з вуглеводнем [3]. Даний спосіб має ряд суттєвих недоліків: вихід ліпідів при їх екстракції за способом Фолча в оптимальних умовах складає 90-95 %, однак величина ступеня вилучення відчутно залежить, наприклад, від ефективності гомогенізації вихідного матеріалу. Фрагменти жирних кислот, що хімічно іммобілізовані на клітинних мембранах або ковалентно зв'язані в молекулах гліколіпідів і ліпопротеїдів, не можуть бути вилучені в ході екстракції за Фолчем; зрозуміло, що переведенню в метилові ефіри вони вже не піддаються і не можуть бути визначені за допомогою газової хроматографії. Також, як правило, трифазні суміші "водний метанол - хлороформ - дрібнодисперсний твердий матеріал" після екстракції зразка за Фолчем розшаровуються дуже погано, потребують тривалого (іноді багатогодинного) настоювання, фільтрування або ефективного центрифугування. Такі процедури є вкрай трудомісткими й тривалими, і до того ж не піддаються автоматизації чи суттєвому спрощенню. Операції з великими обсягами розбавлених розчинів ліпідів в органічних розчинниках завжди супроводжуються частковим окисленням і фотодеструкцією поліненасичених сполук. Використання ж низьких температур, інертних атмосфер і попередньо дегазованих розчинників надзвичайно ускладнює препаративні роботи. В основу винаходу поставлена задача створити варіант хроматографічного визначення сумарного жирнокислотного складу зразка, що був би вільний від перерахованих вище недоліків прототипу (крайня трудомісткість, необхідність тривалих маніпуляцій з розбавленими розчинами, неповнота екстракції ліпідів). Отже, запропоновано спосіб хроматографічного визначення сумарного жирнокислотного складу біологічних рідин, який включає відділення ліпідів від матричного матеріалу, який відрізняється тим, що рідкий зразок біологічного матеріалу, а саме кров, сироватка, суспензія клітин, гомогенізат тка 94373 4 нини, сушать для одержання зразка безводного біологічного матеріалу в апараті сублімаційного сушіння або в ротаційному вакуумному концентраторі типу "Savant SpeedVac", при цьому аліквоту зразка досліджуваного рідкого біологічного матеріалу в кількості 20-500 мкл зневоднюють безпосередньо в пробірці для дериватизації, а ліпіди висушеного зразка біологічного матеріалу у цій же пробірці перетворюють у відповідні метилові ефіри нагріванням з розчином 0,5 N метилату натрію в метанолі з розрахунку 150 мкл розчину метилату натрію на 100 мкл крові або 15 % гомогенізату, а присутні вільні жирні кислоти нагрівають з 10 % метанольним розчином трифтористого бору з розрахунку 0,8-1,0 мл на 100 мкл крові або 15 % гомогенізату, причому після закінчення метилювання спиртовий розчин зразка біологічного матеріалу розбавляють дистильованою водою вдвічі, після чого екстрагують одержані метилові ефіри жирних кислот в гептанову неполярну фазу, при цьому дещо уповільнене розшарування водно-спиртової та неполярної гептанової фази на стадії екстракції ефірів з реакційної суміші прискорюють додаванням краплі пропілового спирту або короткочасним (1-2 хв.) центрифугуванням в низькооборотній центрифузі, після чого екстраговані в гептанову фазу метилові ефіри жирних кислот визначають кількісно способом капілярної газової хроматографії на колонці з полярною іммобілізованою рідкою фазою. Основна перевага пропонованого нами способу полягає в принциповому спрощенні та прискоренні підготовки біологічного зразка для хроматографічного аналізу. Продуктивність сушильних установок типу "SpeedVac" дуже висока і визначається тільки числом гнізд в каруселі центрифуги (те ж саме справедливо стосовно препаративних приладів для сублімаційного сушіння); таким чином, кілька десятків зразків можуть бути висушені, прометильовані і повністю підготовлені до аналізу протягом порівняно короткого часу (2,5-3 години). Здається істотною і та обставина, що процес підготовки зразка за цим способом полягає у виконанні послідовності нескладних операцій, що не вимагають високої кваліфікації персоналу та легко автоматизуються - на відміну від повсюдно використовуваної в даний час схеми "дворазова екстракція за Фолчем - промивання - концентрування дериватизація". Одже, як бачимо, даний спосіб може бути використаний у великомасштабних дослідженнях та є зручним для використання. Зразки цільної крові, плазми, еритроцитарної маси надані Серпуховською станцією переливання крові. Гомогенізати (м'язової і печінкової тканини) отримані за допомогою стандартного поршневого гомогенізатора. Спосіб здійснюється наступним чином. Приклад У скляні пробірки для роботи під тиском вміщують по 100 мкл досліджуваних зразків рідких біологічних матеріалів, додають по 100 мкл розчину внутрішнього стандарту в метанолі (маргаринова кислота, 200-600 мкг/мл), поміщають пробірки до гнізд карусельної центрифуги установки "Savant SpeedVac" і сушать зразки у вакуумі при кімнатній 5 температурі до повітряно-сухого стану (30-60 хв.). До ідентичного результату приводить використання сублімаційноі сушарки «Іней 3-2» (у цьому випадку спиртовий розчин внутрішнього стандарту додавався до зразка вже після її ліофілізації). Дериватизацію проводять відповідно до рекомендацій [3]. До висушеного залишку біологічного матеріалу додають 150 мкл 0,5 N розчину метилату натрію в метанолі і нагрівають 2-3 хв. в настільному термоблоці при температурі 60-65 ºС, після чого додають 1,0 мл 10 % метанольного розчину трифториду бору, щільно загвинчують кришку пробірки і нагрівають суміш протягом 40 хв. при 75ºС. До ідентичного результату приводить сапоніфікація сухого зразка дією 150 мкл 0,5 N розчину їдкого натру (замість метилату натрію) у метанолі і подальше нагрівання з 1 мл суміші «2N метанольний розчин сухого хлористого водню - бензол - 2,2диметоксипропан», із співвідношенням 100:20:5. Температурні умови лужної сапоніфікації і кислого метилювання в цьому випадку ідентичні наведеним вище. Після закінчення метилювання в охолоджені пробірки вносять по 1,0 мл чистого нгептану, 1,0 мл води, екстрагують метилові ефіри в вуглеводневу фазу. До верхньої фази додають 10 мкл н-пропанолу і домагаються повного розшарування емульсії з використанням низькооборотної центрифуги установки "Savant SpeedVac" (1-2 хв.). Екстраговані в гептанову фазу метилові ефіри визначають способом газової хроматографії. Умови хроматографування Використовують аналітичний газовий хроматограф «Варіан 3900» (США) і кварцову капілярну колонку з іммобілізованою нерухомою фазою «Супелковакс-10» (15 м 0,25 мм 0,3 мкм) виробництва «СУПЕЛКО», Швейцарія. Введення зразка (2 мкл) - без поділу потоку газу-носія (гелій), перехід до режиму поділу потоку - через 12-30 секунд, залежно від концентрації досліджуваних речовин. Температурна програма аналізу - від 90 ºС (0,5хв.) 94373 6 до 240 ºС (5 хв.) зі швидкістю 6 С/хв.. Детектор полум'яно-іонізаційний (260 ºС), реєстрація сигналу - комп'ютерна програма «Мультіхром-1, 5х» виробництва ЗАТ «Амперсенд», РФ. Кількісне визначення - за способом внутрішнього стандарту (з попереднім обчисленням відповідних калібрувальних коефіцієнтів з хроматограми модельної суміші жирних кислот з стандартною маргариновою кислотою). Результати Дослідники рідко використовують абсолютні значення концентрацій певних ЖК у медикодіагностичних цілях: величини абсолютних концентрацій сильно залежать від ступеня розбавлення зразка крові консервантом і антикоагулянтом, від величин препаративних втрат зразка на різних стадіях роботи, від похибок при відборі аліквот, визначенні калібрувальних коефіцієнтів та інших факторів . Крім того, в ході фракціонування біоматеріалу (наприклад, отримання еритроцитарної або тромбоцитарної маси, ліпопротеїнів високої та низької щільності і т.д.) визначення сухої ваги фракцій, як правило, виявляється вельми скрутним. З наведених причин для побудови біомедичних кореляцій частіше використовуються відносні величини: відносини концентрацій суми насичених кислот до суми поліненасичених, відносини сумарних концентрацій ώ3- і ώ6-кислот, відношення концентрацій кислот C20:4ώ6 / C20:5ώ3 та ін. Подібний підхід дозволяє звести до мінімуму похибки препаративних та аналітичних маніпуляцій, забезпечує значно кращу міжлабораторну відтворюваність результатів. Тому для порівняльної оцінки метрологічних характеристик двох обговорюваних аналітичних способів в таблиці наведено величини середніх значень і довірчих інтервалів (при 95 % надійності) ряду безрозмірних відносин концентрацій індивідуальних жирних кислот у біологічних зразках різної природи. 7 94373 8 Таблиця Середні значення співвідношень вагових концентрацій індивідуальних жирних кислот у біологічних об'єктах (і довірчі інтервали середніх значень), визначені: (1) прямим метилюванням сухої біомаси (спосіб описаний вище); (2) метилюванням ліпідів, попередньо екстрагованих за Фолчем (класичний спосіб [2]); (3) метилюванням ліпідів, екстрагованих сумішшю гексану з ізопропанолом (однократна екстракція відповідно до [3, с 155]). Об’єкт цільна кров n=6 цільна кров n=6 цільна кров n=1 Кров цільна, попередньо екстрагована за Фолчем1, n=1 Плазма, n=5 Плазма, n=5 Плазма, n=1 Плазма, попередньо екстрагована за Фолчем2, n=1 Еритроцитарна маса, n=5 Еритроцитарна маса, n=5 Еритроцитарна маса, попередньо екстрагована за Фолчем3, n=1 Гомогенізат тканини бичачої печінки, n=4 Гомогенізат тканини бичачої печінки , n=4 Гомогенізат тканини бичачої печінки, попередньо екстрагована за Фолчем4, n=1 Гомогенізат тканини грудного м'яза курки, n=5 Гомогенізат тканини грудного м'яза курки, n=5 Гомогенізат тканини грудного м'яза курки, попередньо екстрагована за Фолчем5, n=1 Співвідношення концентрацій (± довірчий інтервал, Р = 0,95, n=4-6) 16:0/1 8:0 18:0/18:2 18:2/20:4 18:2/22:5 18:2/22:6 20:4/20:3 2,23± 0,18 0,45±0,02 2,07±0,07 34,0±2,8 6,43±0,40 5,72±0,07 2,26± 0,14 0,43±0,04 2,18±0,09 37,5±2,8 6,97±0,27 5,65±0,08 5,7 0,22 4,2 51 20 4,3 1,3 0,86 1,85 36 8,6 6,65 3,09± 0,04 3,14± 0,08 4,1 0,89 0,212±0,005 0,214±0,003 0,48 1,38 3,71±0,15 3,70±0,21 2,06 3,25 116±16 124±21 34,2 10,1 19,4±2,7 17,4±2,0 6,83 >130 4,42±0,11 4,40±0,11 5,75 1,51 1,98± 0,29 0,60±0,20 2,00±0,13 18,3±4,4 4,29±0,94 8,8±1,5 1,98± 0,19 0,66±0,07 1,99±0,19 16,5±2,4 3,97±0,48 7,9±1,6 1,16 1,40 1,57 36,2 4,36 7,2 0,478 ±0,014 2,38±0,06 1,42±0,06 1,89±0,20 4,70±0,43 1,382±0,013 0,468 ±0,024 2,39±0,05 1,39±0,05 1,75±0,19 4,32±0,46 1,386±0,023 1,288 2,54 29,6 50 50 12 1,540 ±0,072 0,502±0,012 3,45±0,17 35,4±3,2 26,2±2,2 6,03±0,14 1,619 ±0,105 0,58 5±0,051 3,44 ±0,18 34,2±2,5 25,3± 2,7 6,18 ±0,16 4,5 80 54 40 1,600 0,94 1 Залишок ліпідів, не екстрагуються за Фолчем, - 3,6 відн. %. Залишок ліпідів, не екстрагуються за Фолчем, - 3,5 відн. %. 3 Залишок ліпідів, не екстрагуються за Фолчем, - 18 відн. %. 4 Залишок ліпідів, не екстрагуються за Фолчем, - 1,5 відн. %. 5 Залишок ліпідів, не екстрагуються за Фолчем, - 6,5 відн. %. 2 Аналіз наведених експериментальних даних дозволяє зробити деякі практично важливі висновки. - Пропонований нами спосіб прямого метилювання висушеної біомаси призводить до результа тів, що статистично не відрізняються від отримуваних при використанні класичного способу Фолча. Близькість величин довірчих інтервалів в обох способах свідчить про те, що розкид параметрів обумовлений швидше причинами хроматографіч 9 94373 ної природи (дискримінація компонентів у випарнику, неточності інтегрування малих піків та інше), ніж факторами, пов'язаними з особливостями хімічної підготовки зразків. - Ліпіди, що екстрагуються за Фолчем, характеризуються істотно іншим жирнокислотним складом у порівнянні з ліпідами того самого зразка, що за Фолчем, однак, не екстрагуються. Це слід враховувати при плануванні тонких ліпідних досліджень: «ідентичність» результатів (табл.), отриманих способами 1 і 2, обумовлена лише порівняльно незначним вмістом (кілька відсотків від суми ліпідів) не виділеного залишку. - Одноразова екстракція біологічного матеріалу сумішшю спирту з вуглеводнем (спосіб 3) не може бути рекомендована для визначення жирнокислотного складу зразків. Комп’ютерна верстка Л.Литвиненко 10 Винахід може бути використаний для масштабних скринінгових досліджень жирнокислотного спектру біологічних субстратів у клінічних та біохімічних лабораторіях, що значно спрощує підготовку зразка до проведення аналізів і сприятиме оптимізації лабораторної діагностики. Джерела інформації: 1. Под ред. Северина С.Е., Соловьевой Г.А. Практикум по биохимии. Изд. 2, издательство Московского Университета, 1989. С. 68-70. 2. Folch J., Lees M., Sloane-Stanley G.H. A simple method for isolation and purification of total lipids from animal tissues. J.Biol.Chem. 1957, 226:497-509. - прототип. 3. Knapp D.R. Handbook of analytical derivatization reactions. A Wiley-Interscience Publication, John Wiley&Sons Inc. New York, 1979. P. 154, с. 155, 164-167. Підписне Тираж 23 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for chromatographic determination of total fat-acid composition of biological liquids
Автори англійськоюArchipovskyi Aleksandr Viktorovych, Kolesnyk Pavlo Olehovych, Vezhdel Maria Ivanivna, Rostoka-Rieznykova Mariyana Vasylivna, Kirsanova Maryna Petrivna, Tsiapets Serhii Vasyovych, Hlazkova Halyna Petrivna
Назва патенту російськоюСпособ хроматографического определения суммарного жирнокислотного состава биологических жидкостей
Автори російськоюАриповский Александр Викторович, Колесник Павел Олегович, Веждел Мария Ивановна, Ростока-Резникова Марьяна Васильевна, Кирсанова Марина Петровна, Цяпец Сергей Василиевич, Глазкова Галина Петровна
МПК / Мітки
МПК: G01N 30/00, G01N 33/48
Мітки: сумарного, рідин, хроматографічного, спосіб, жирнокислотного, складу, біологічних, визначення
Код посилання
<a href="https://ua.patents.su/5-94373-sposib-khromatografichnogo-viznachennya-sumarnogo-zhirnokislotnogo-skladu-biologichnikh-ridin.html" target="_blank" rel="follow" title="База патентів України">Спосіб хроматографічного визначення сумарного жирнокислотного складу біологічних рідин</a>
Спосіб хроматографічного визначення сумарного жирнокислотного складу біологічних рідин

Номер патенту: 58675
Опубліковано: 26.04.2011
Автори: Глазкова Галіна Пєтровна, Цяпець Сергій Васильович, Аріповскій Алєксандр Вікторовіч, Колесник Павло Олегович, Ростока-Резнікова Мар'яна Василівна, Веждел Марія Іванівна, Кірсанова Марина Петрівна
МПК: G01N 33/48, G01N 30/00
Мітки: жирнокислотного, складу, біологічних, визначення, спосіб, сумарного, хроматографічного, рідин
Формула / Реферат:
Спосіб хроматографічного визначення сумарного жирнокислотного складу біологічних рідин, який включає відділення ліпідів від матричного матеріалу, який відрізняється тим, що рідкий зразок біологічного матеріалу, а саме кров, сироватку, суспензію клітин, гомогенізат тканини, сушать для одержання зразка безводного біологічного матеріалу в апараті сублімаційного сушіння або в ротаційному вакуумному концентраторі типу "Savant SpeedVac",...
Спосіб визначення жирнокислотного складу ліпідів листя топінамбура

Номер патенту: 56471
Опубліковано: 10.01.2011
Автори: Середа Петро Іванович, Максютіна Ніна Павлівна, Брюзгіна Тетяна Семенівна, Цимбаліста Юлія Андріївна
МПК: G01N 33/68
Мітки: спосіб, топінамбура, листя, ліпідів, жирнокислотного, складу, визначення
Формула / Реферат:
Спосіб визначення жирнокислотного складу ліпідів листя топінамбура, що включає дослідження порушень обміну речовин, який відрізняється тим, що визначають жирнокислотний склад ліпідів листя топінамбура за допомогою газорідинної хроматографії, визначають вміст пальмітинової, ліноленової та арахідонової вищих жирних кислот, порівнюють з контролем сироватки і розраховують їх вміст в процентах.
Спосіб визначення жирнокислотного складу ліпідів

Номер патенту: 10045
Опубліковано: 30.09.1996
Автори: Слушняк Сергій Григорович, Данилик Богдана Богданівна, Рівіс Йосип Федорович
МПК: G01N 30/02, G01N 33/06
Мітки: ліпідів, спосіб, визначення, складу, жирнокислотного
Формула / Реферат:
Спосіб визначення жирнокислотного складу ліпідів, який включає прямий метаноліз зразка, концентрування отриманої суміші, обробку концентрату розчинниками, очистку метилових ефірів жирних кислот і газорідинну хроматографію очищених метилових ефірів жирних кислот, який відрізняється тим, що обробку концентрату розчинниками і очистку метилових ефірів жирних кислот проводять одночасно за допомогою гексану і метилату натрію.
Спосіб визначення жирнокислотного складу ліпідного комплексу кореня соняшника

Номер патенту: 56472
Опубліковано: 10.01.2011
Автори: Брюзгіна Тетяна Семенівна, Максютіна Ніна Павлівна, Цимбаліста Юлія Андріївна, Середа Петро Іванович
МПК: G01N 33/68
Мітки: визначення, складу, кореня, спосіб, комплексу, ліпідного, соняшника, жирнокислотного
Формула / Реферат:
Спосіб визначення жирнокислотного складу ліпідного комплексу кореня соняшника, що включає дослідження порушень обміну речовин, який відрізняється тим, що визначають жирнокислотний склад ліпідів кореня соняшника за допомогою газорідинної хроматографії, визначають вміст лінолевої та арахідонової вищих жирних кислот і розраховують їх в процентах.
Спосіб визначення жирнокислотного стану ліпідного складу комплексу кипрію (іван-чаю)

Номер патенту: 56496
Опубліковано: 10.01.2011
Автори: Середа Петро Іванович, Абдудеийх Зеад Хельмі, Максютіна Ніна Павлівна, Брюзгіна Тетяна Семенівна
МПК: G01N 33/60
Мітки: жирнокислотного, комплексу, визначення, кипрію, стану, спосіб, ліпідного, складу, іван-чаю
Формула / Реферат:
Спосіб визначення жирнокислотного складу ліпідного комплексу Кипрію (Іван-чаю) шляхом дослідження порушень обміну речовин, який відрізняється тим, що визначають жирнокислотний склад ліпідів Іван-чаю за допомогою газорідинної хроматографії, виявляють вміст пальмітинової, лінолевої та арахідонової вищих жирних кислот, порівнюють з контрольними показниками плазми крові і розраховують їх в процентах.
Попередній патент: Спосіб склеювання солом`яних частинок
Наступний патент: Технологія виконання земляних робіт при капітальному ремонті лінійної частини магістральних трубопроводів і комплекс технологічного обладнання для її здійснення
Випадковий патент: Спосіб попередження зіткнень на залізничному переїзді