Спосіб захисту рослин від фітопатогенів
Номер патенту: 29649
Опубліковано: 15.11.2000
Автори: Хірофумі Ошіта, Шігео Ямамото, Тошіро Като, Юджі Фунакі, Шізуя Танака





















Формула / Реферат
1. Способ защиты растений от фитопатогенов путем воздействия соединением триазола, отличающийся тем, что на поверхности растения или почвы, в которой растут или будут выращиваться растения создают защитный слой, содержащий соединения триазола, представленного формулой I:
(I)
где R1 - это атом водорода, С1-С4 алкил, С3-С4 алкенил или 2-пропиниловая группа, R2 - зто C1-C6 алкил, циклопропил или 1-метилцклопропиловая группа, R3, которая может быть такой же или отличающейся, представляет собой атом галогена, выбранный из группы, состоящей из хлора, брома или фтора, С1-С4, алкил, галогензамещенной С1-С3 алкил, С1-С4 алкокси-, фенокси-, фенил-, циано- или нитро-группу, n- зто целое число от 0 до 3, или соли указанного соединения при величине дозирования от 0,02 до 5 кг на гектар, посредством чего растения защищают от фитопатогенов.
2. Способ по п. 1, отличающийся тем, что защитный слой формируют в виде пленки.
3. Способ по п. 1, отличающийся тем, что упомянутый защитный слой формируют путем нанесения микрогранул на поверхность растения.
Текст
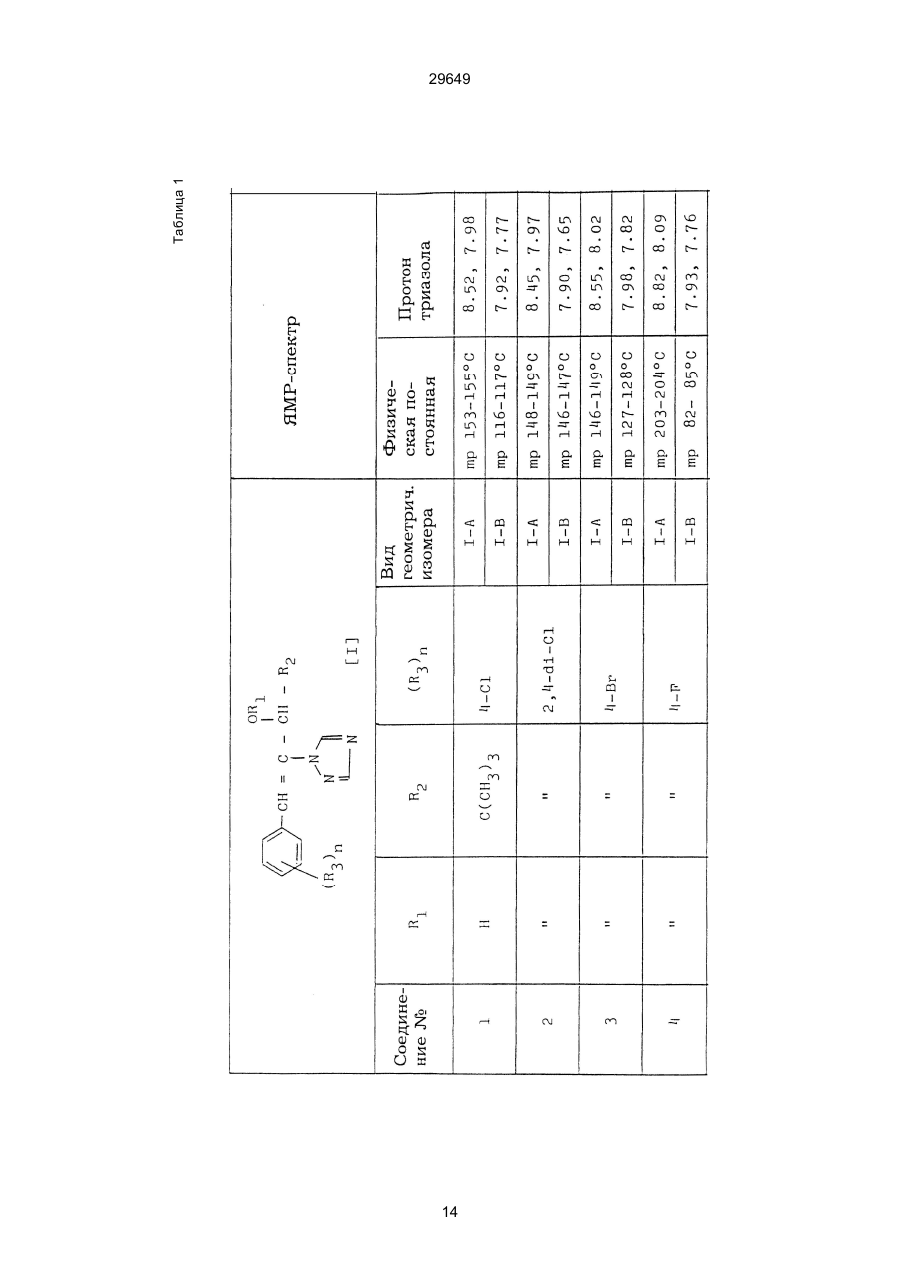
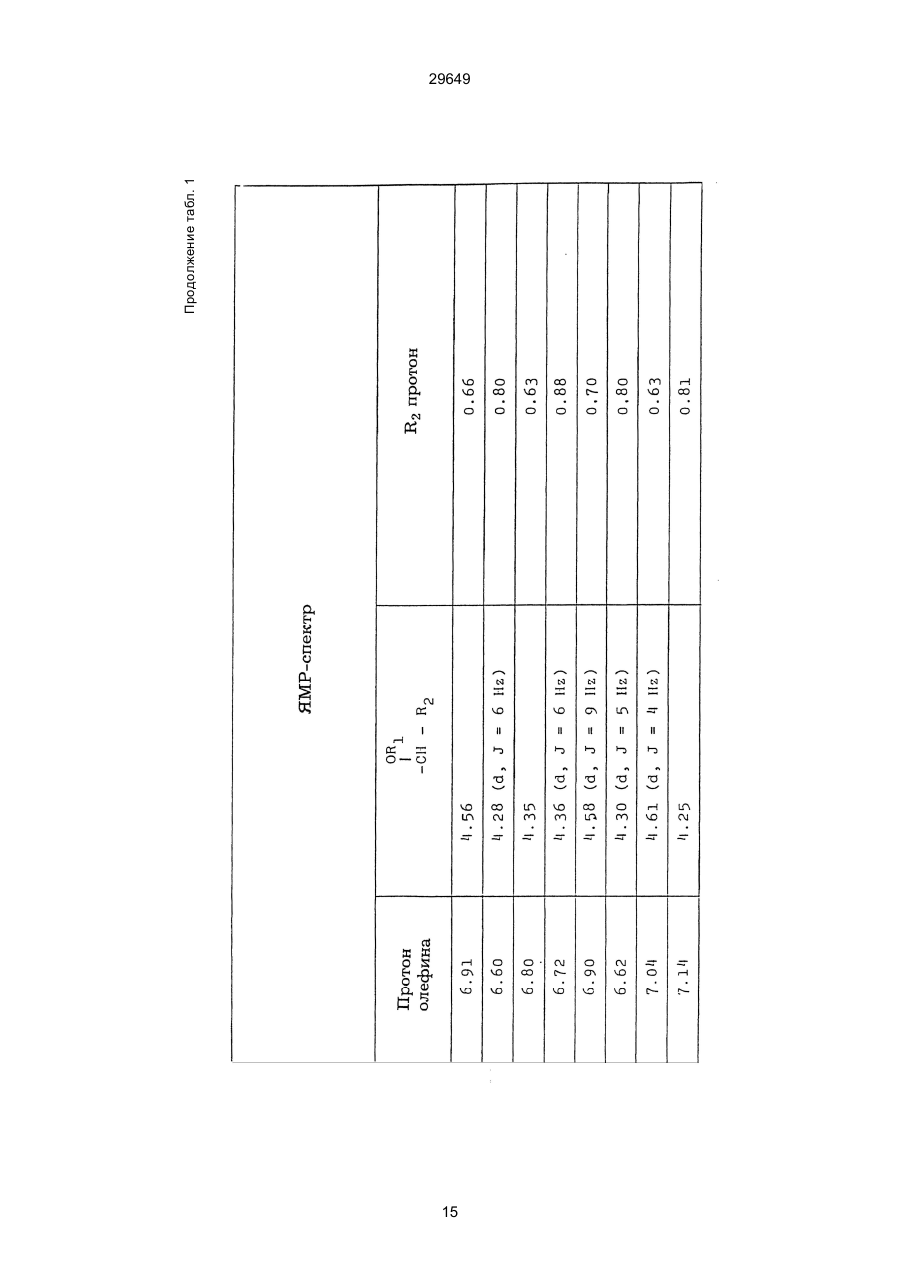
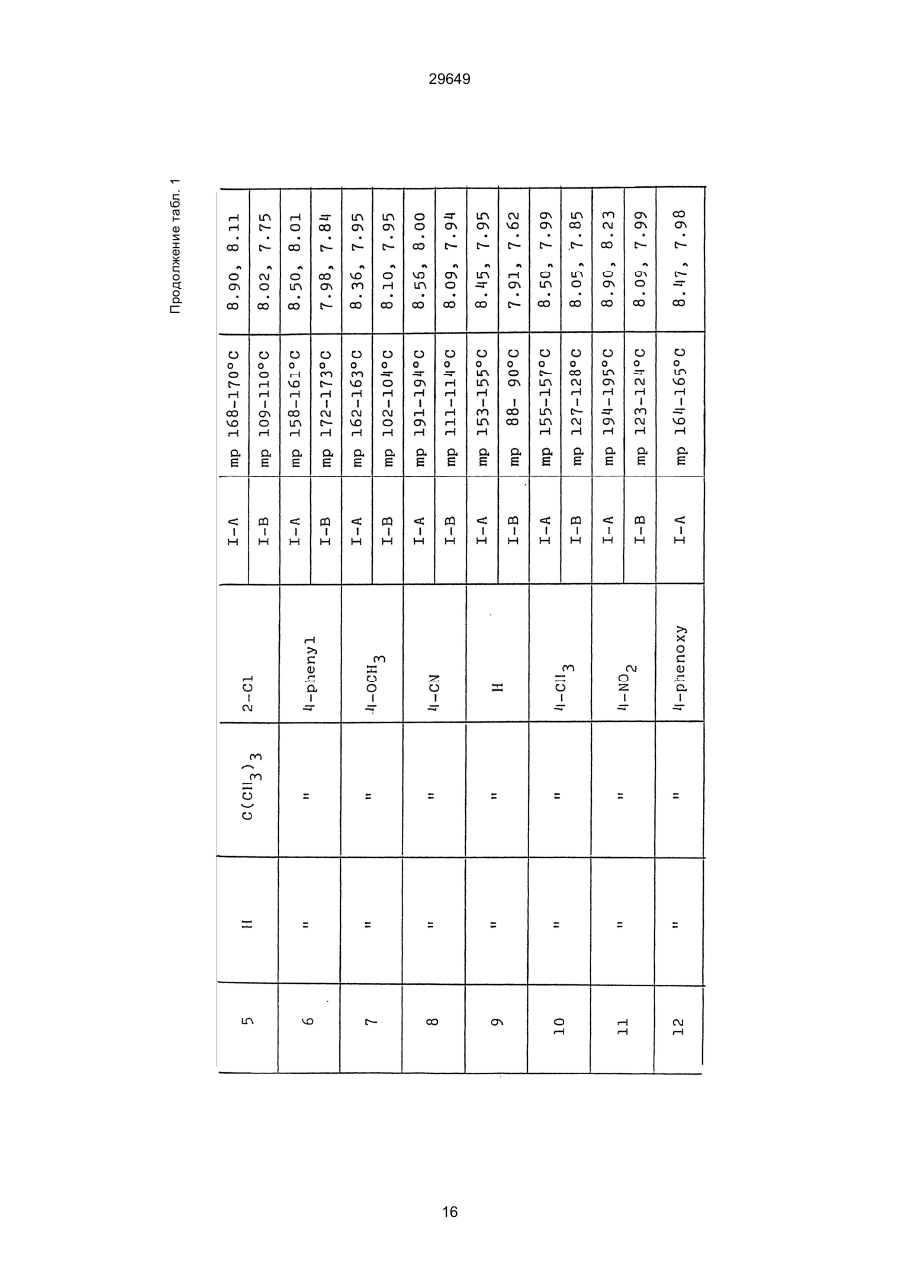
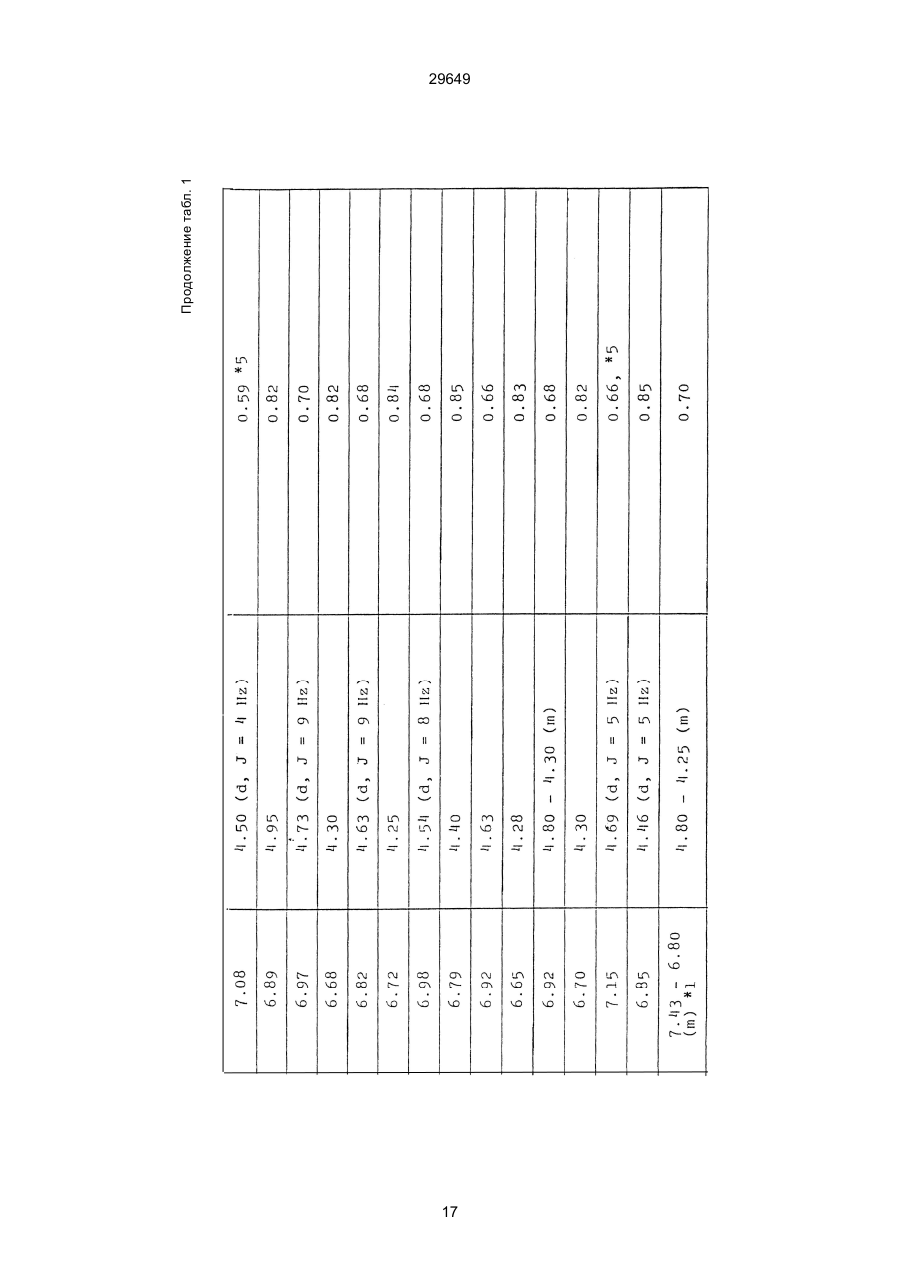
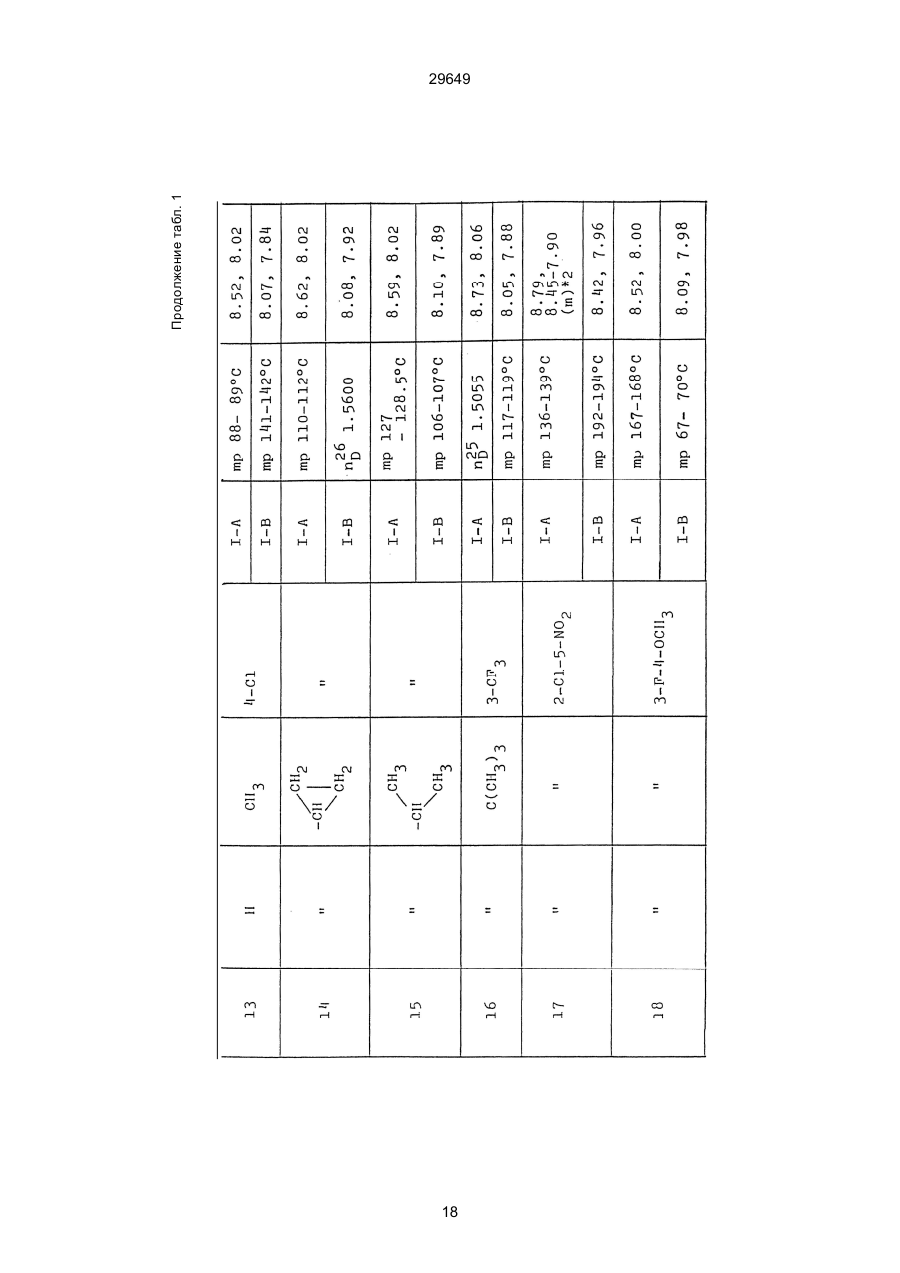
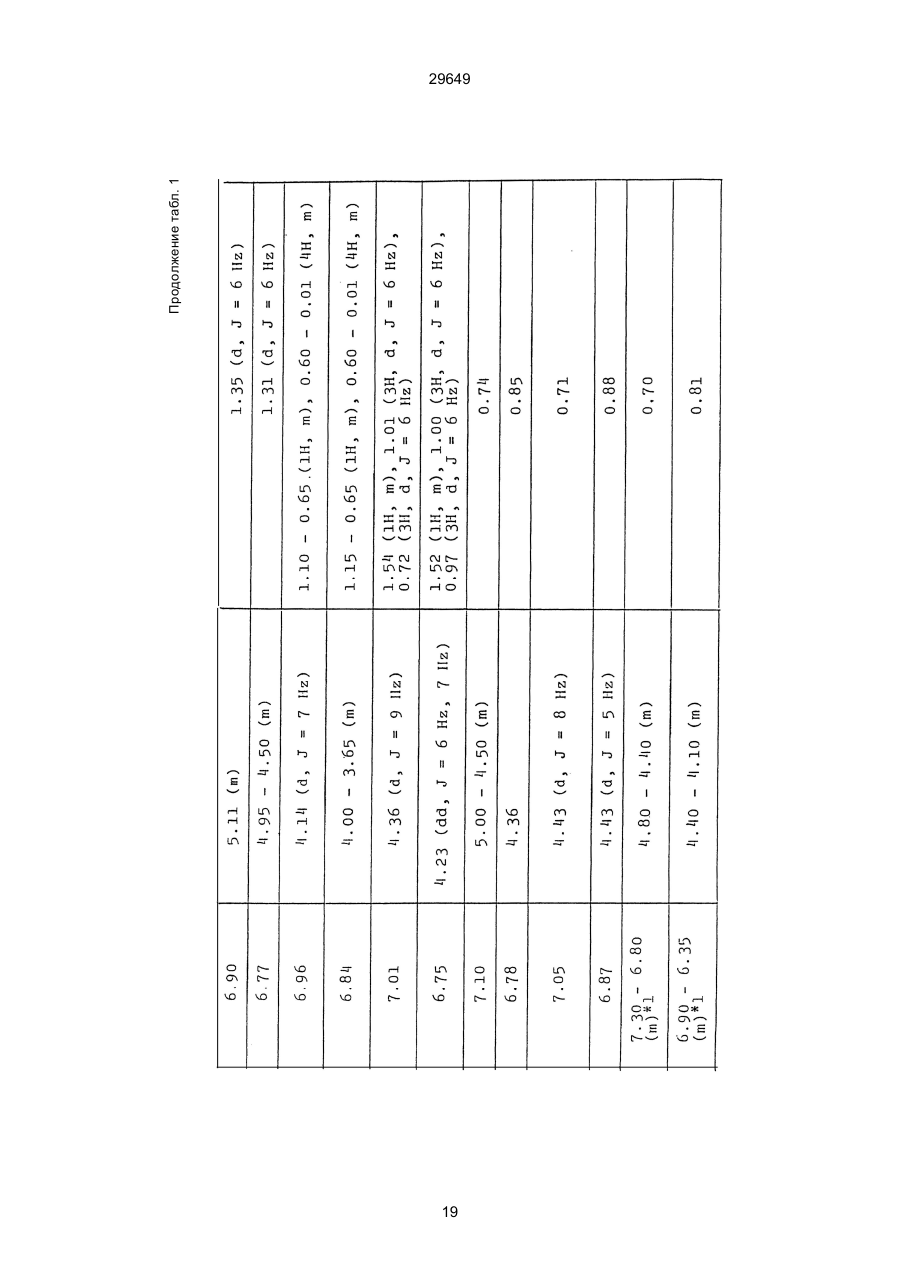
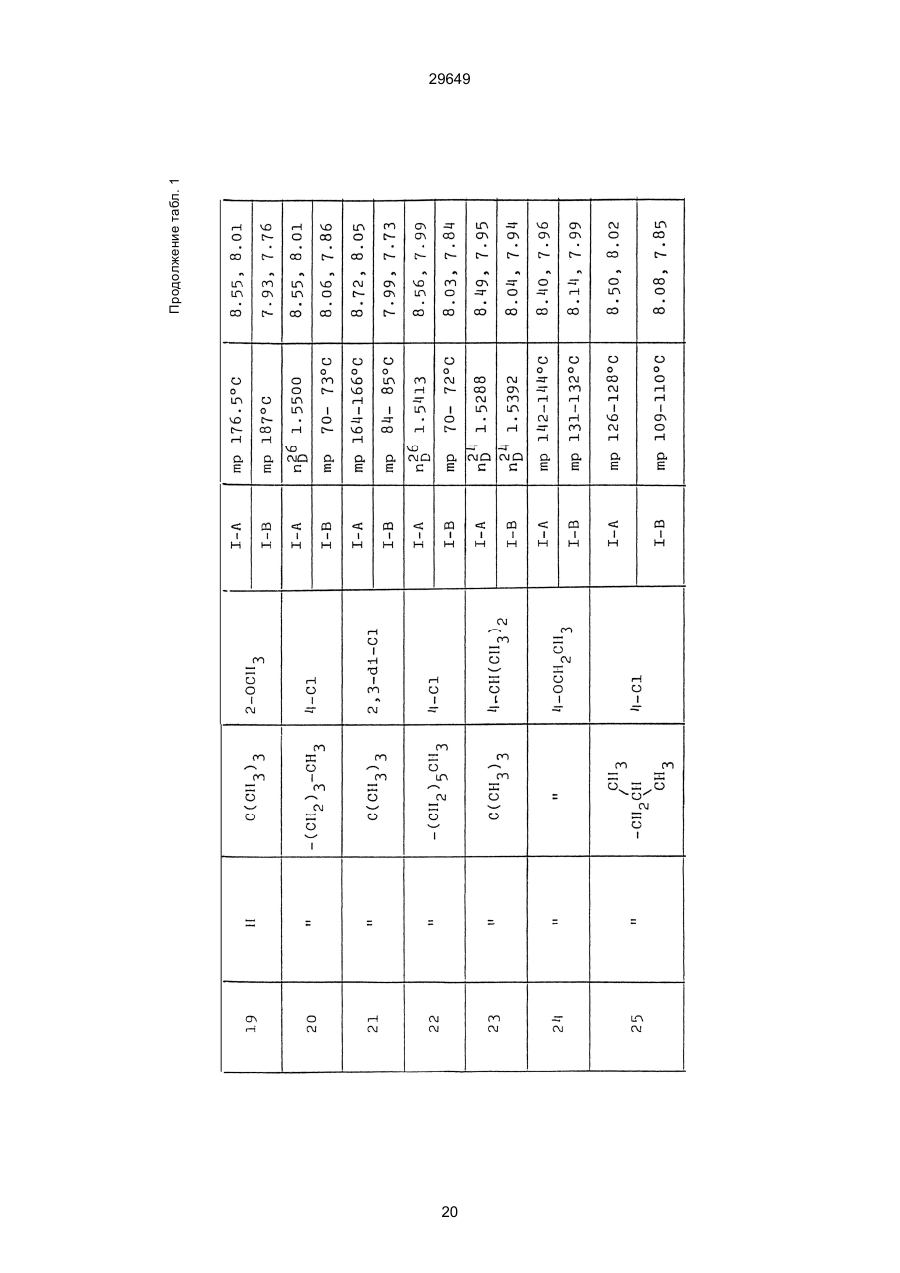
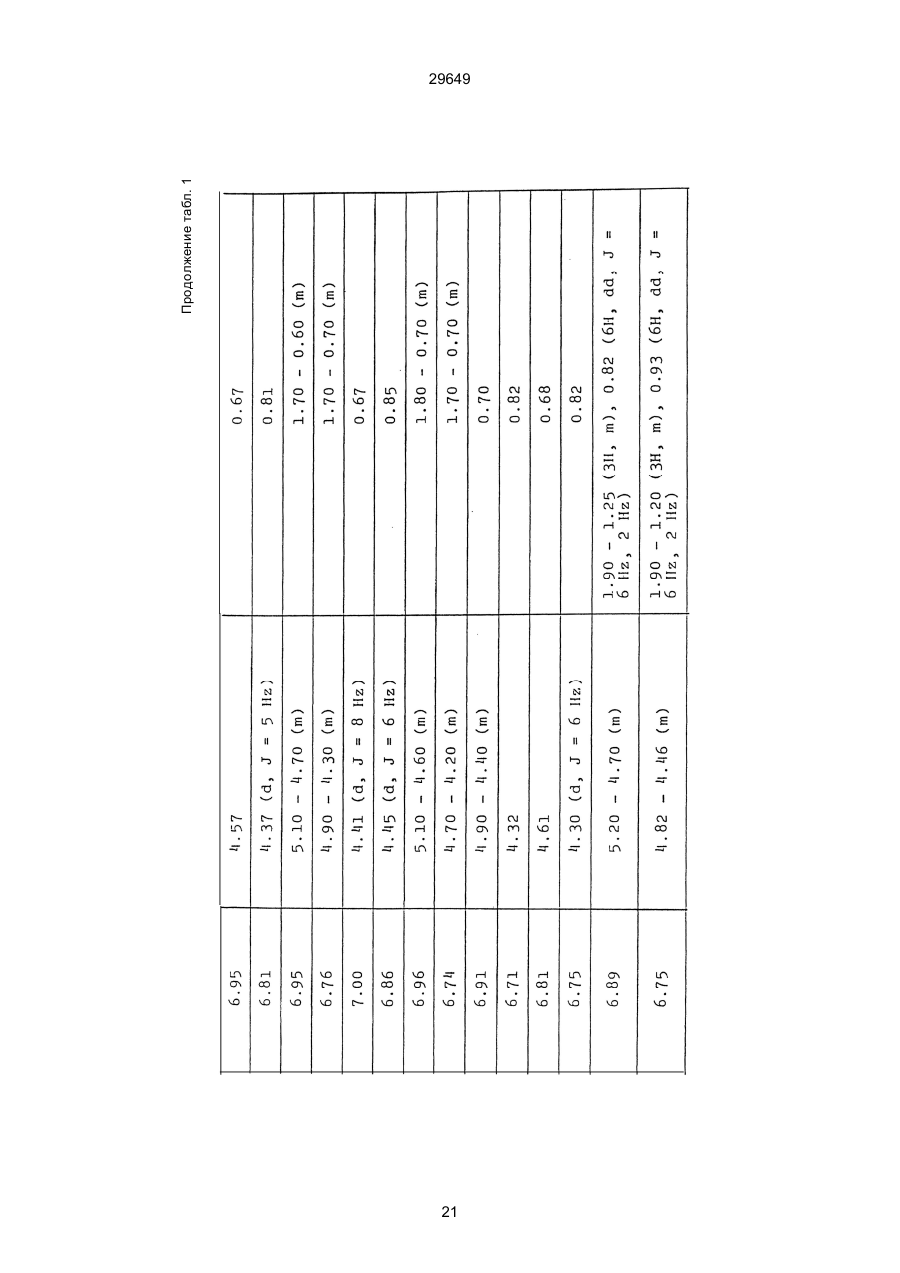
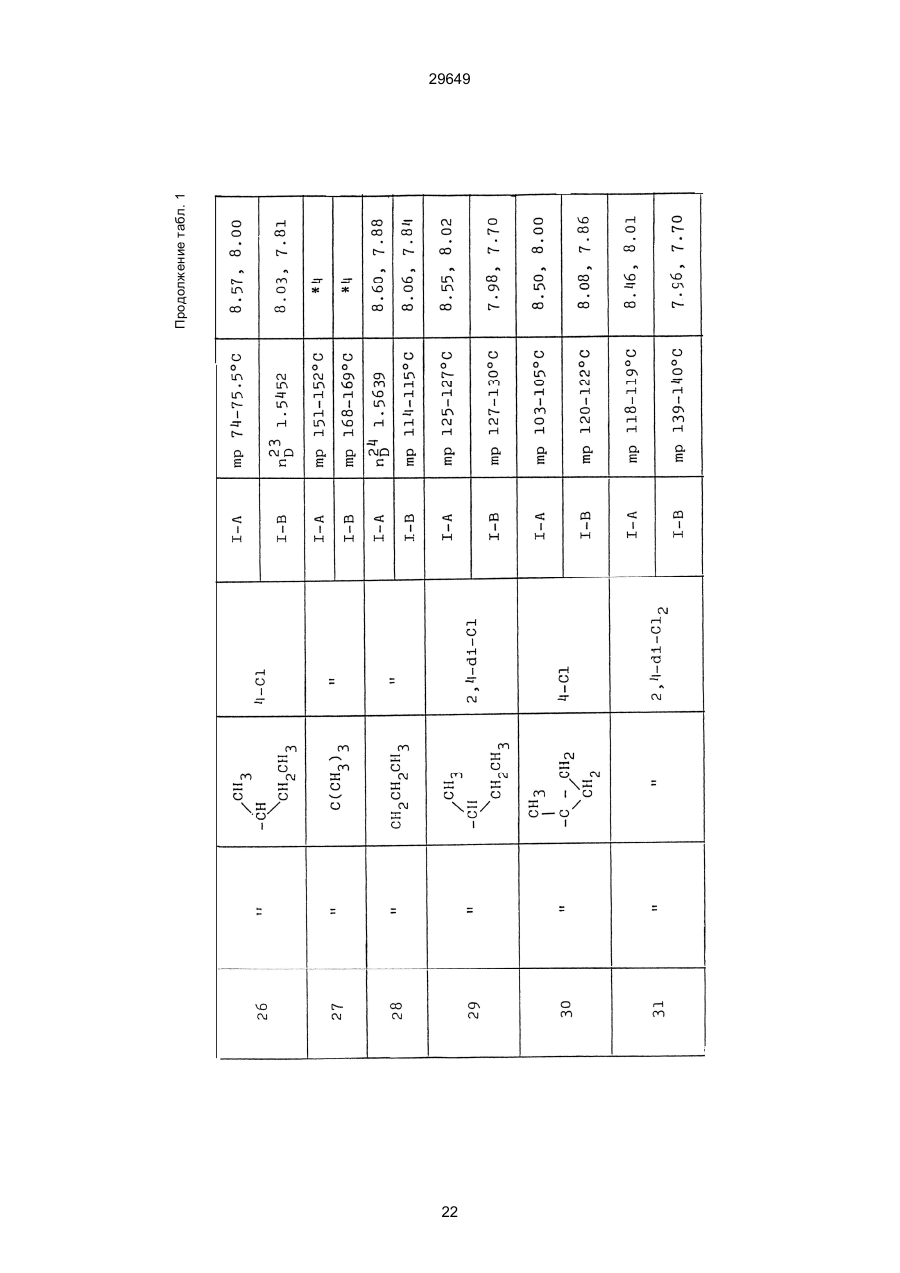
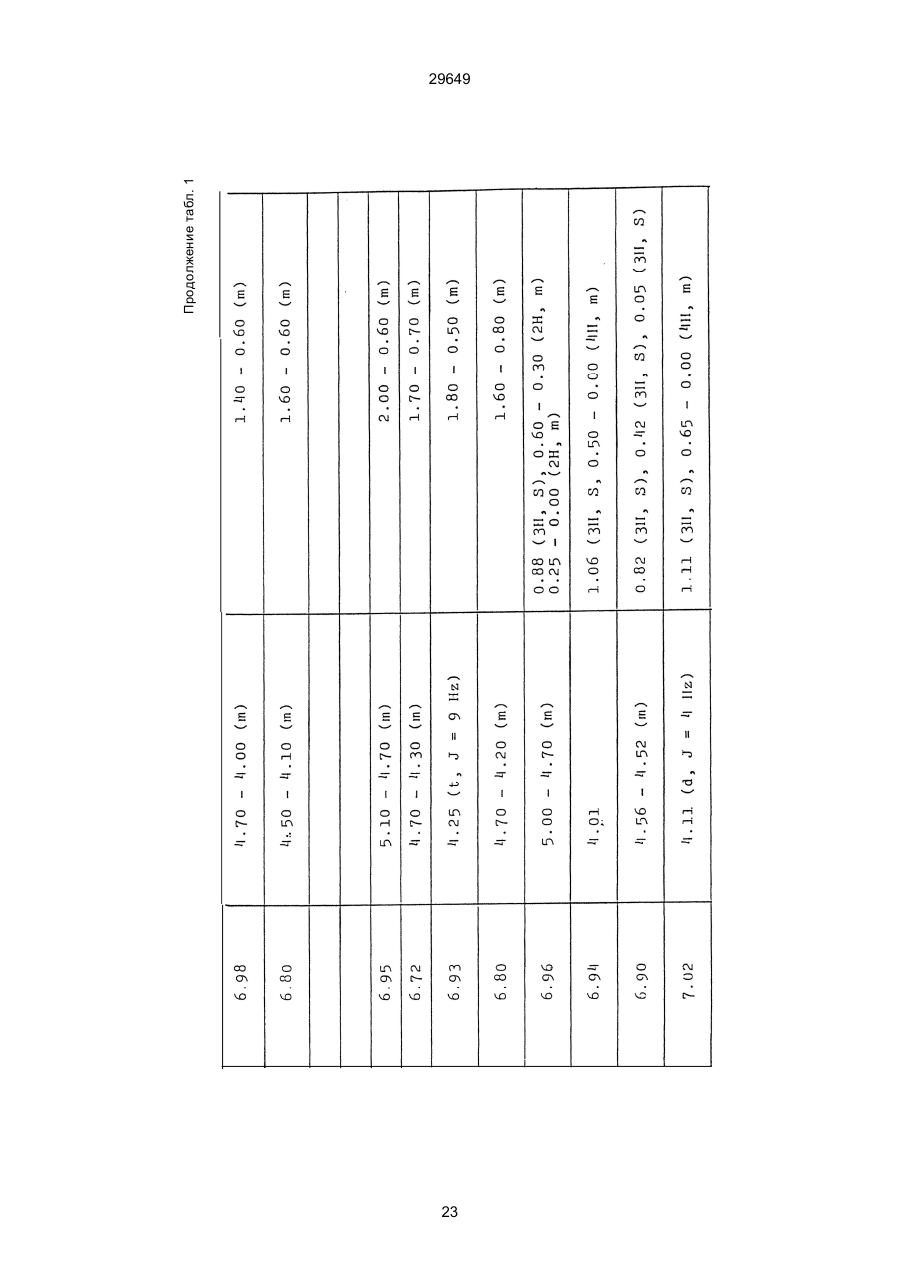
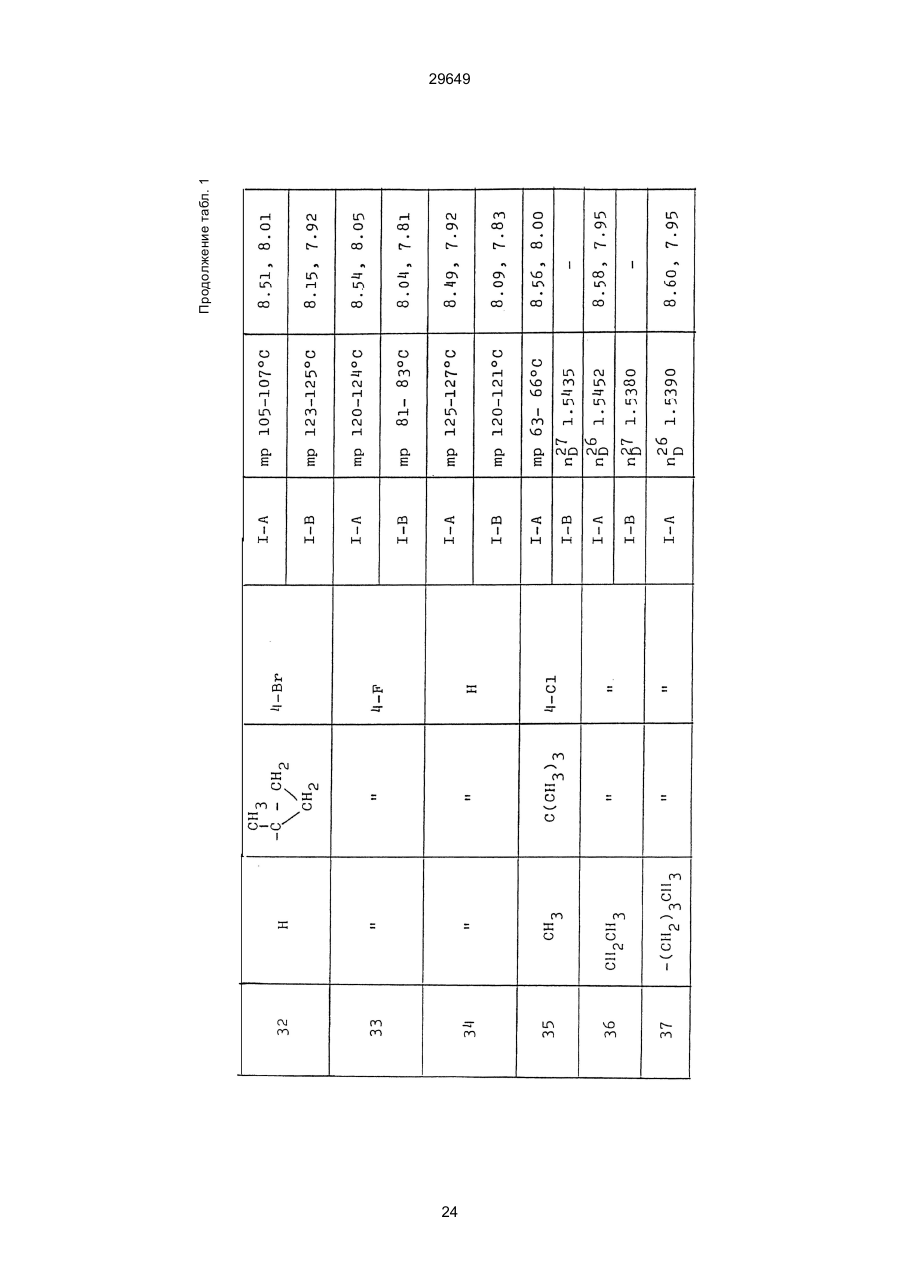
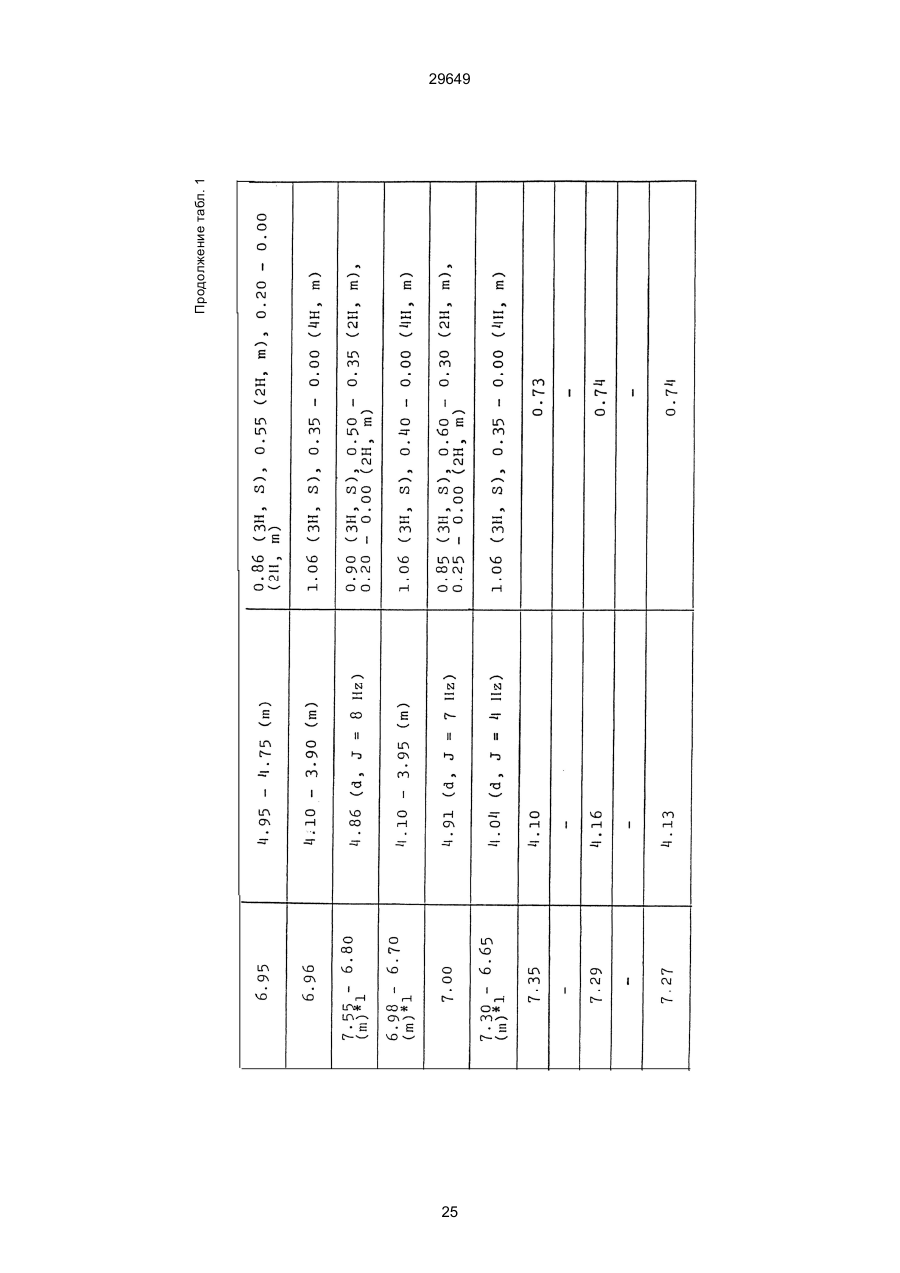
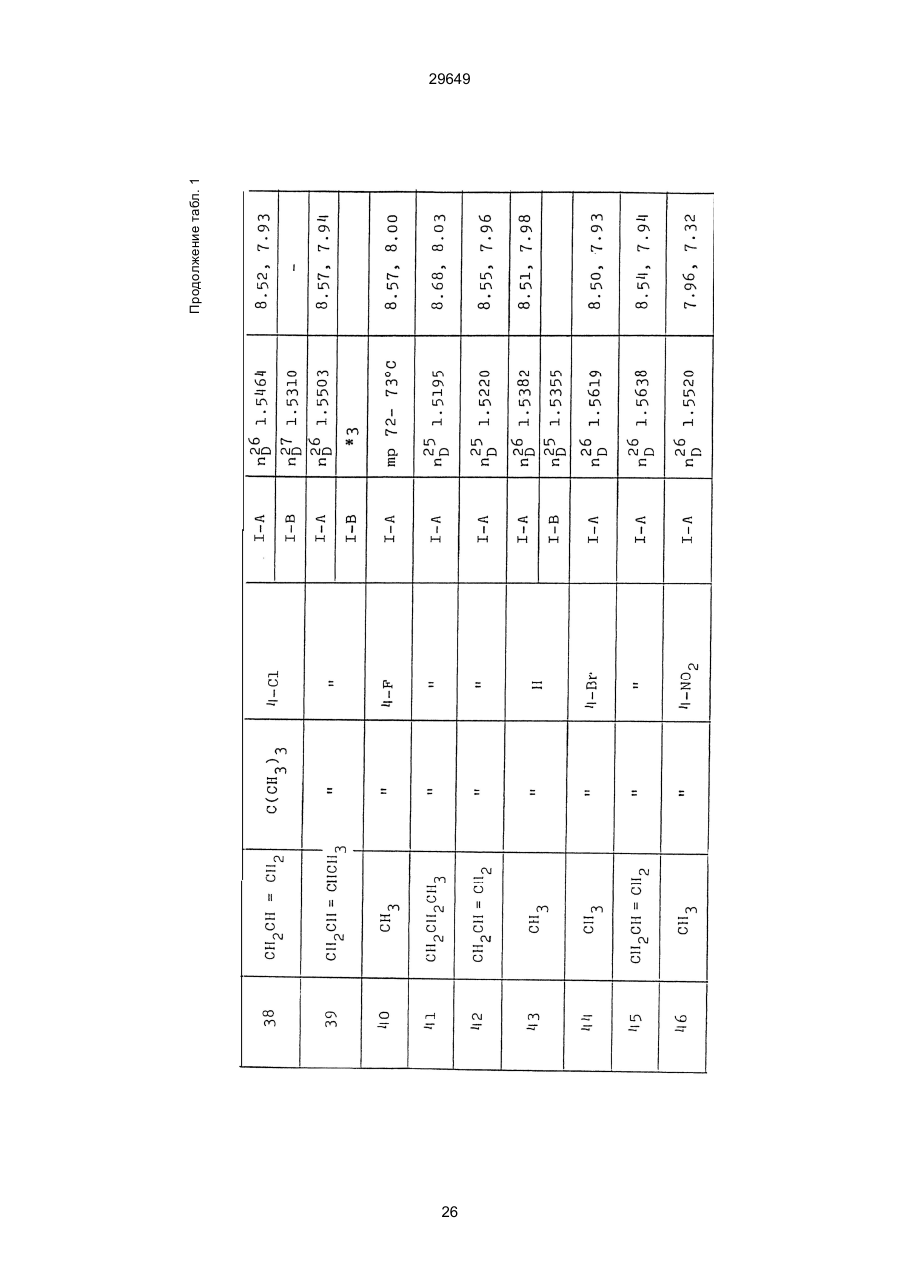
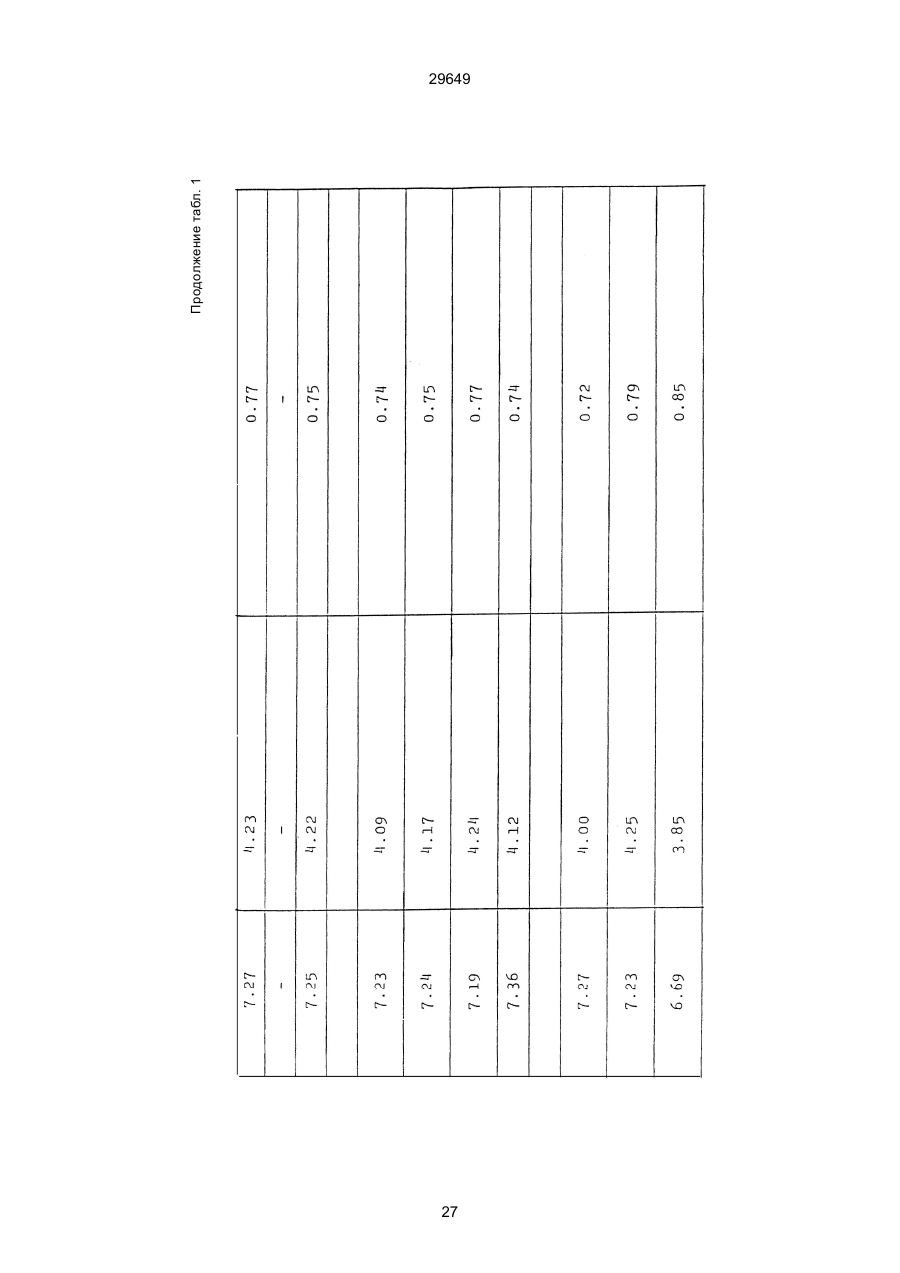
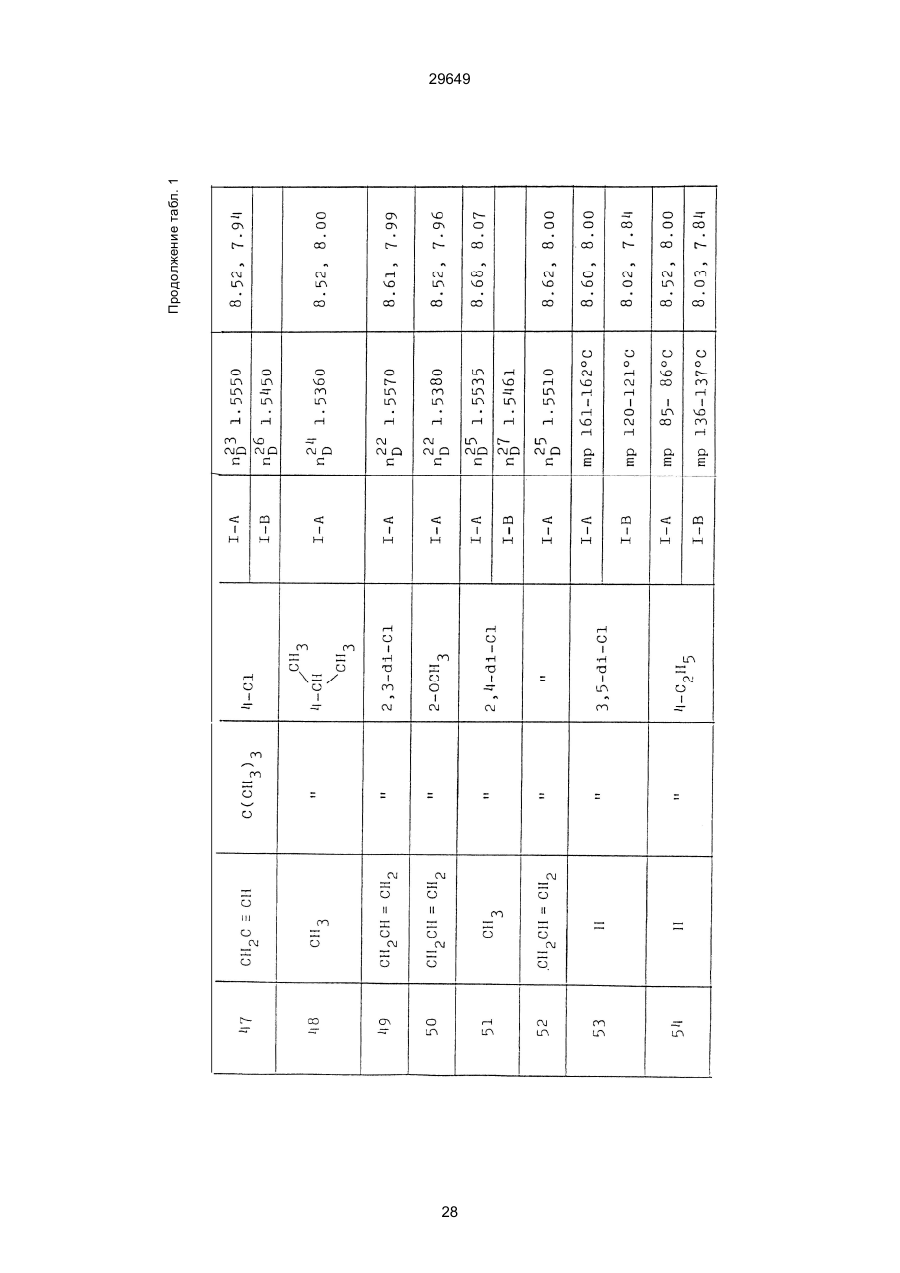
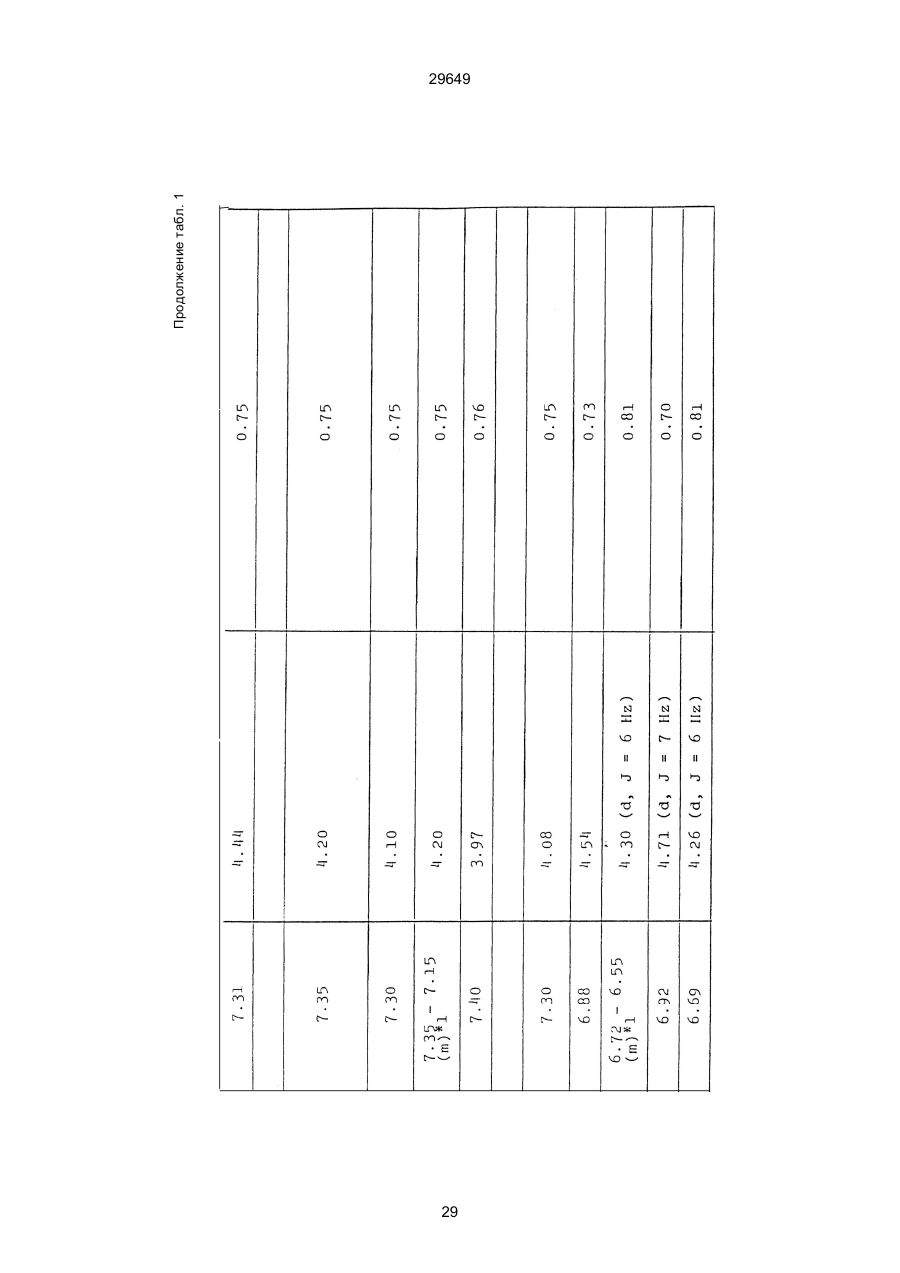
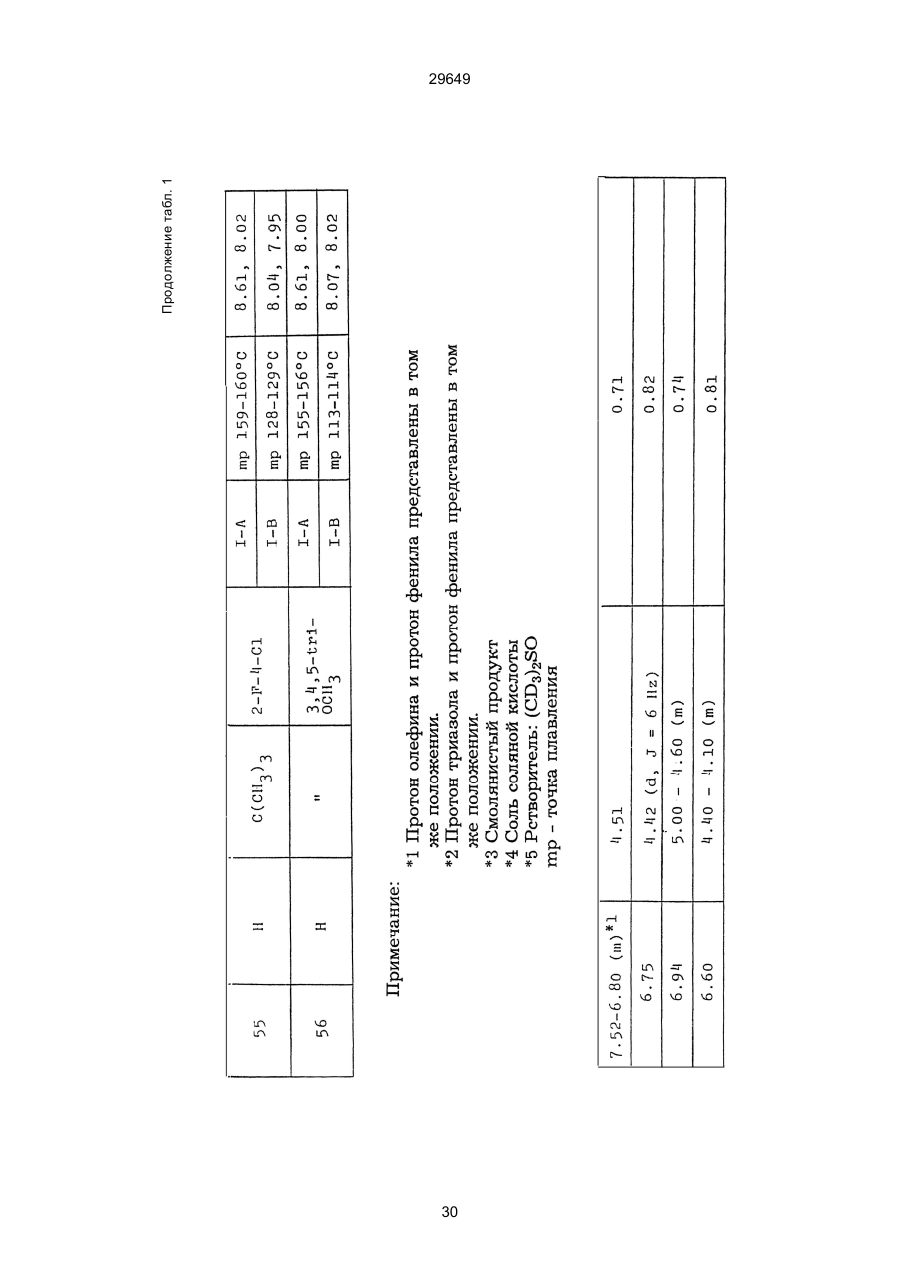
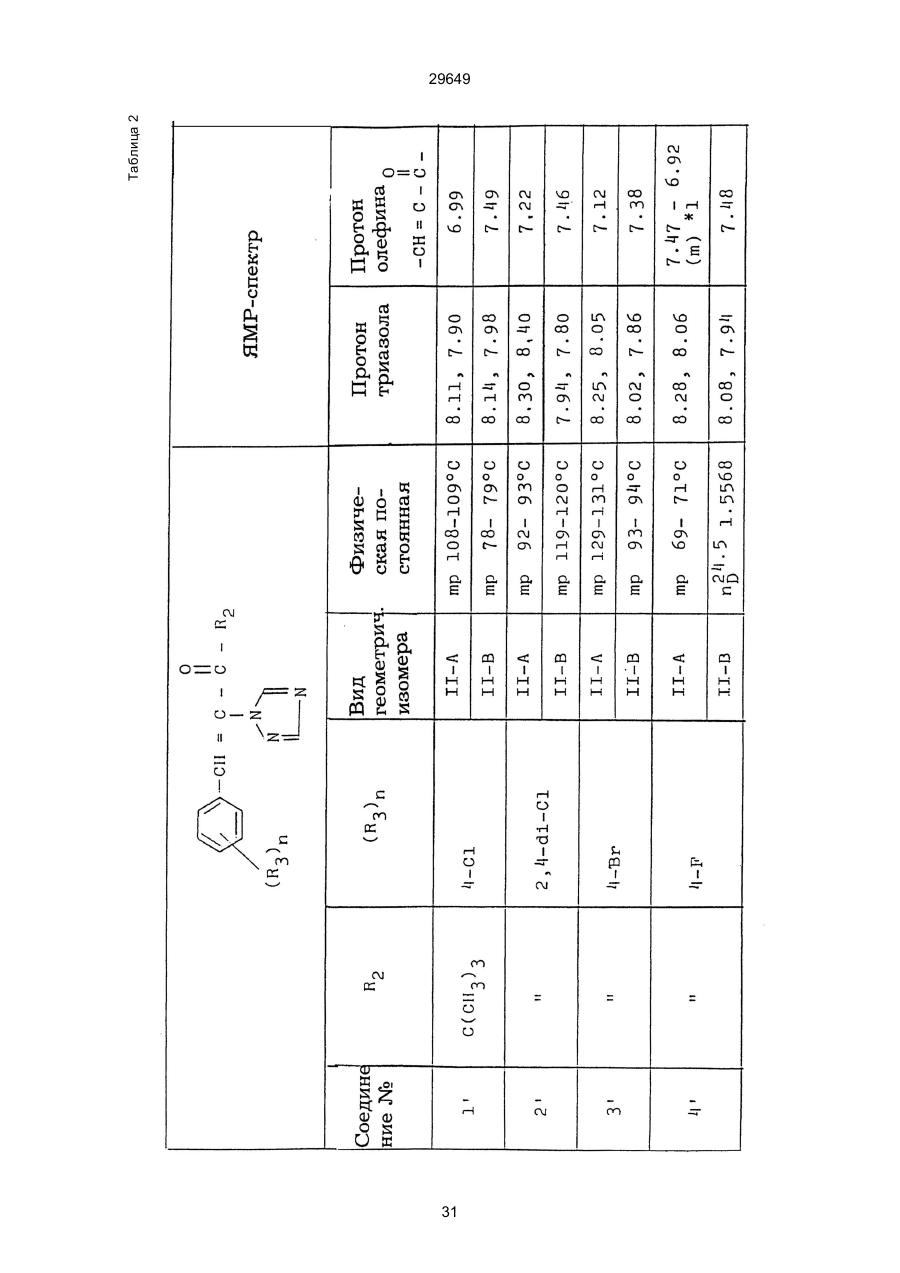
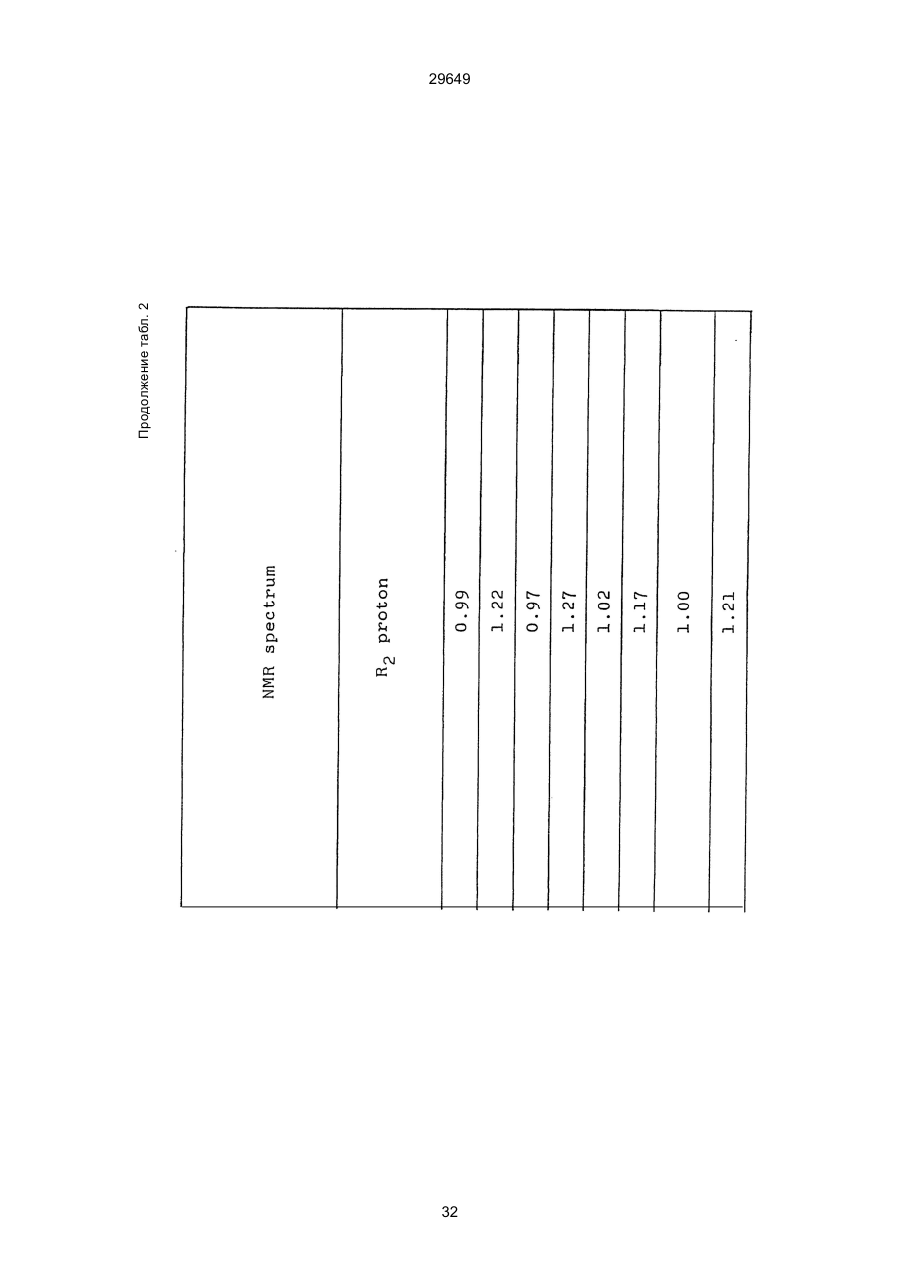
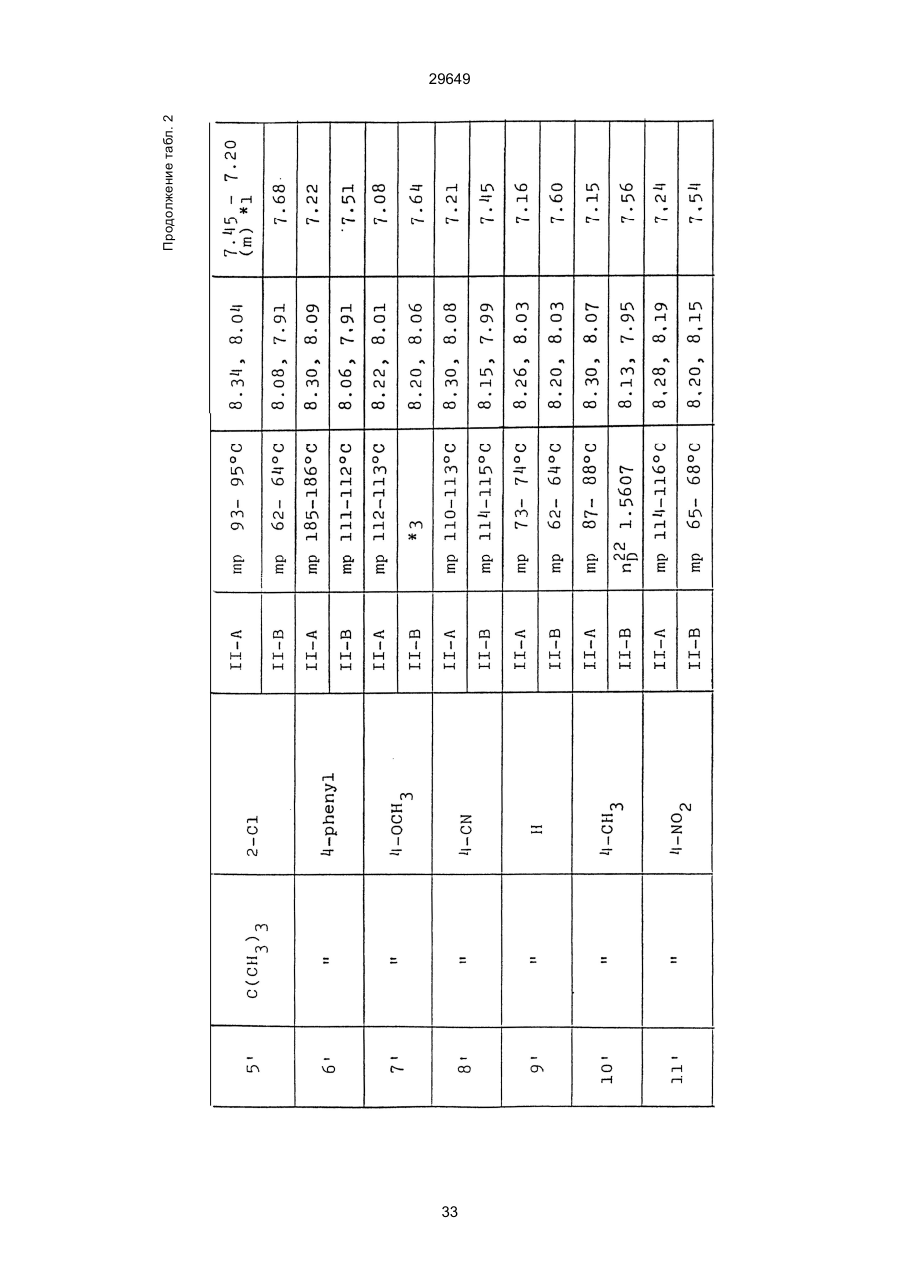
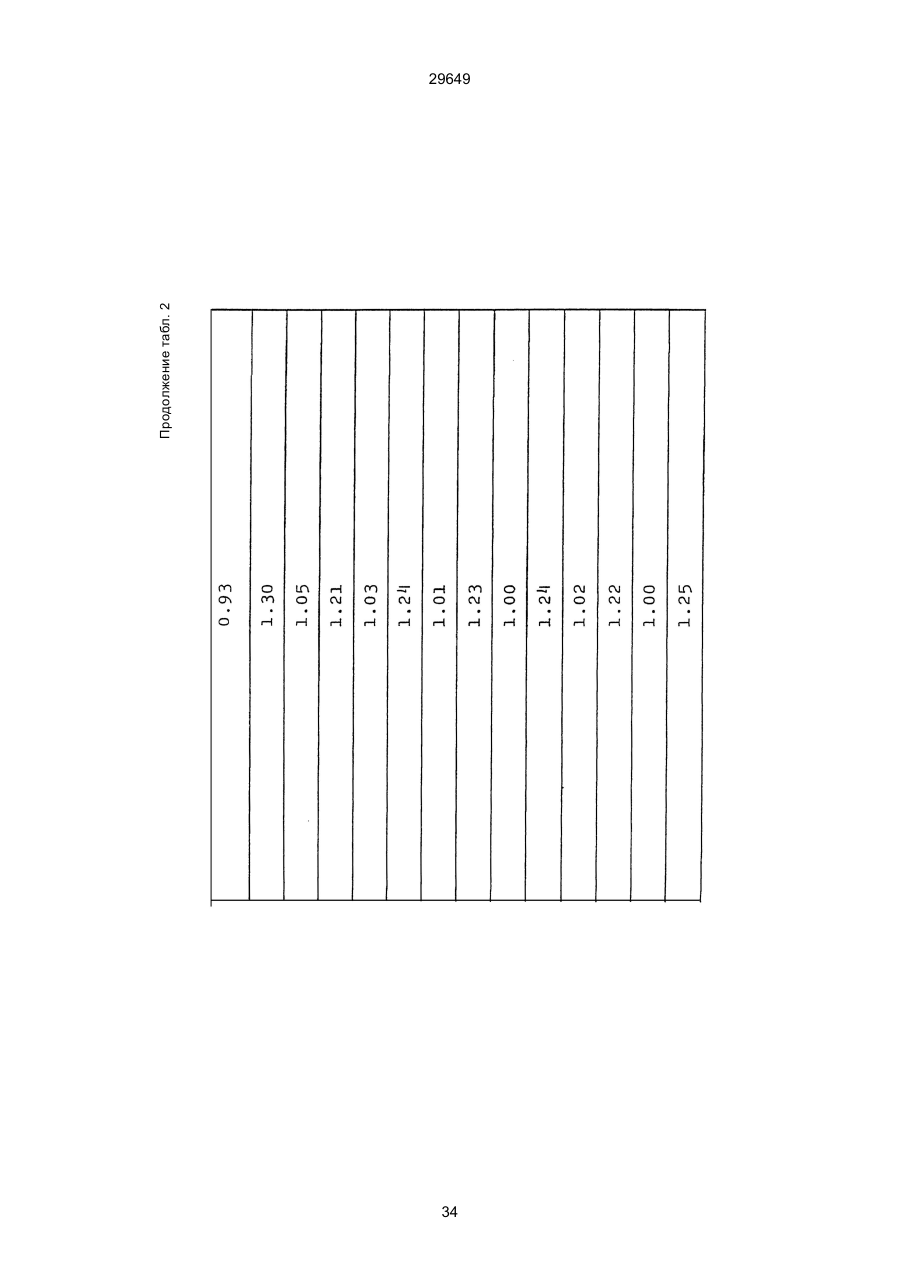
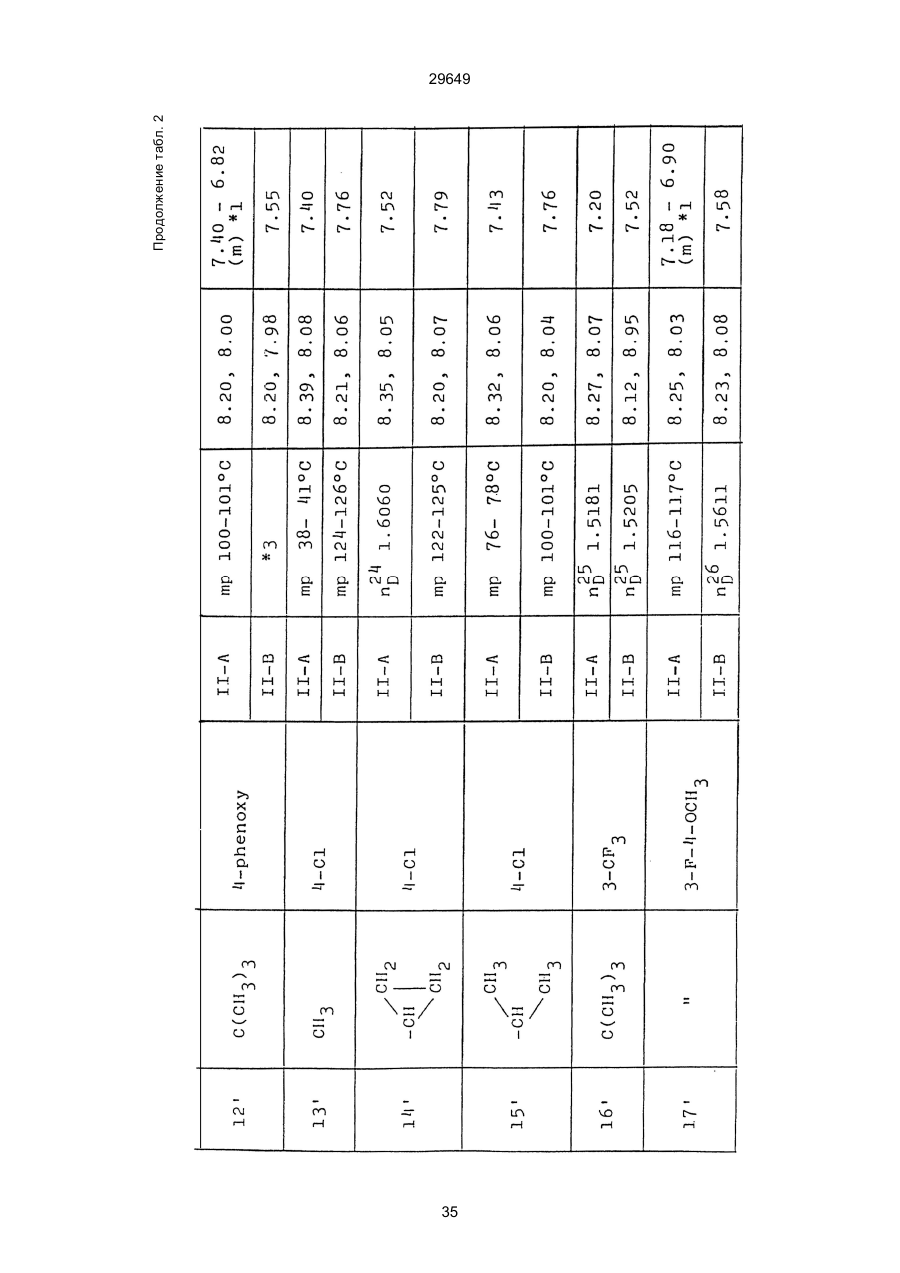
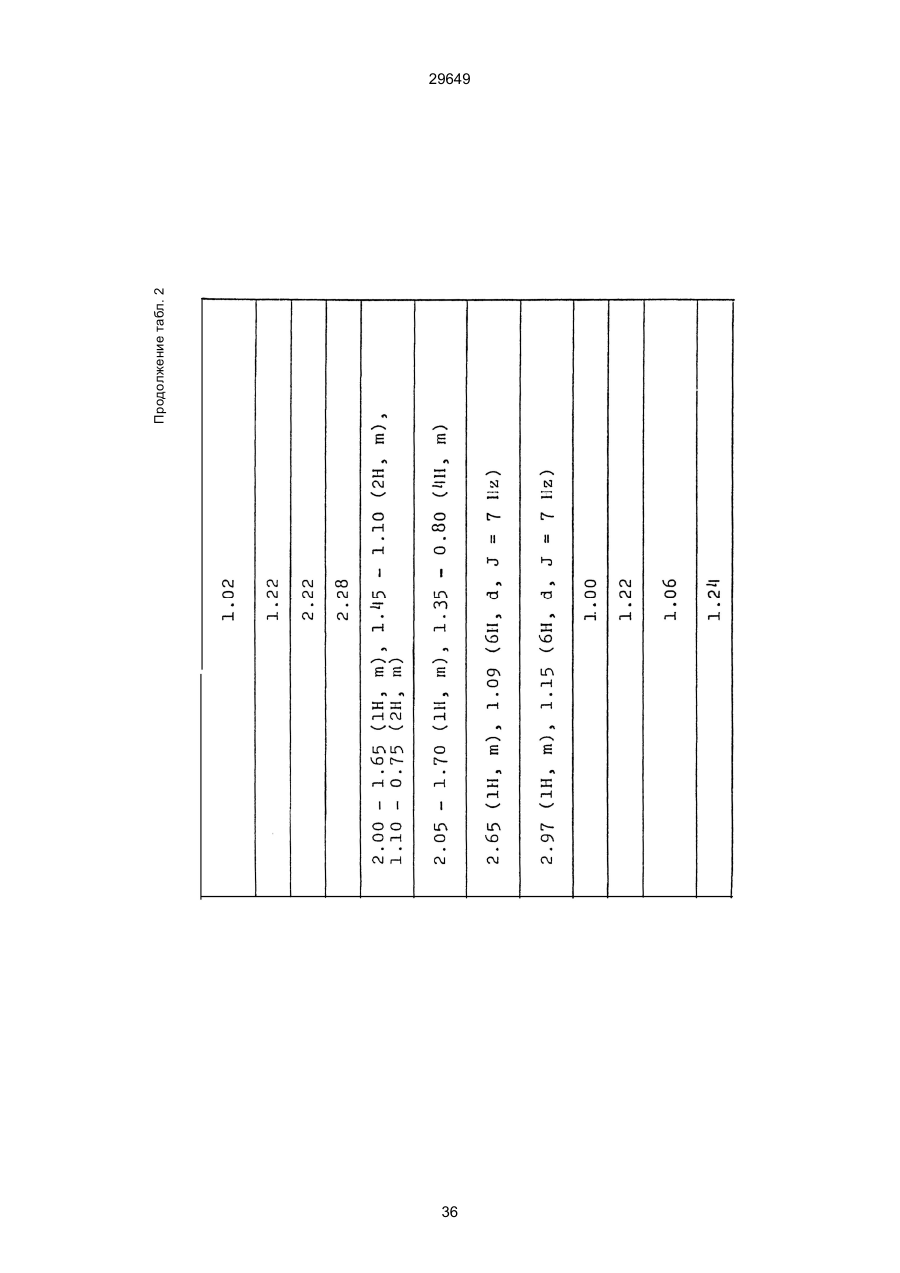
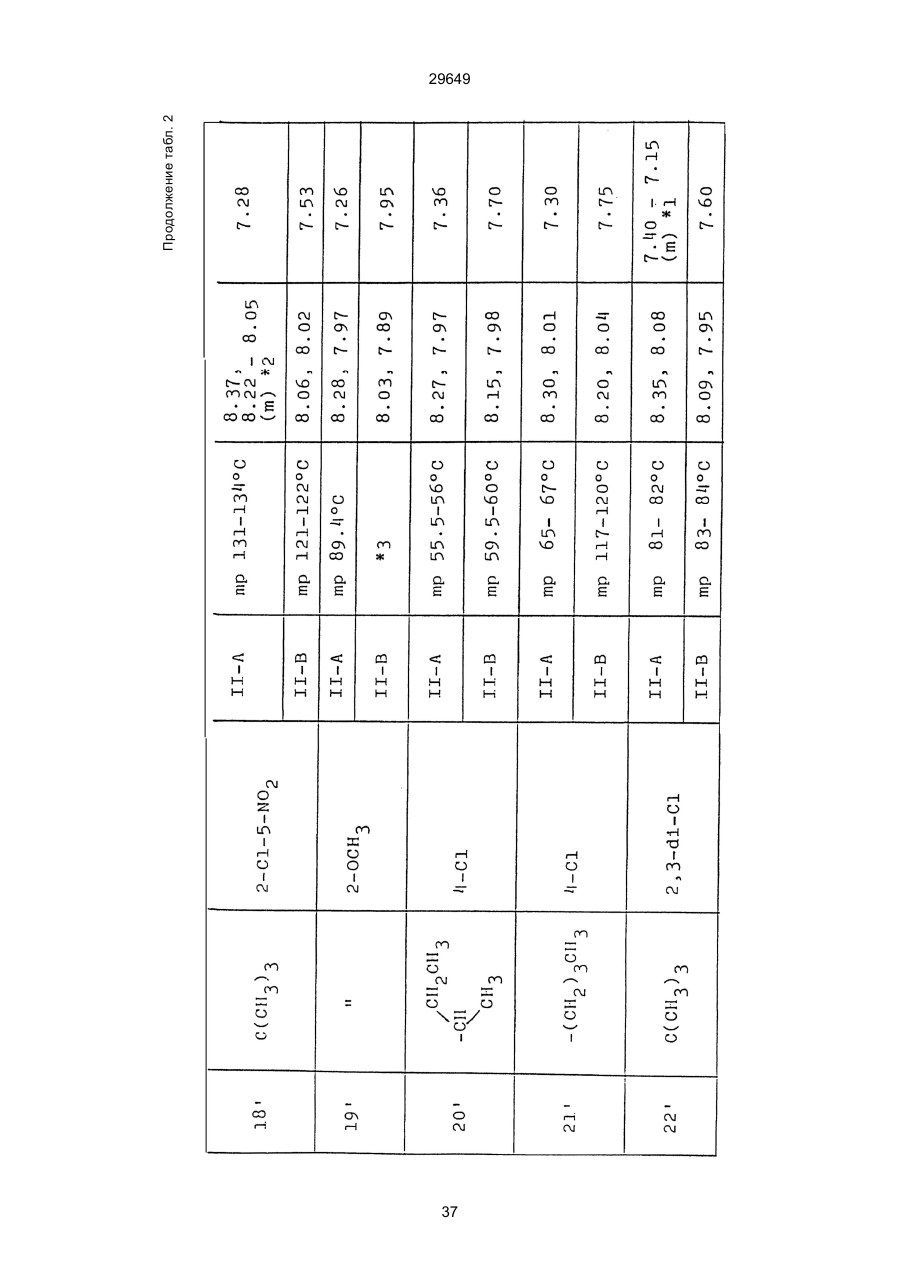
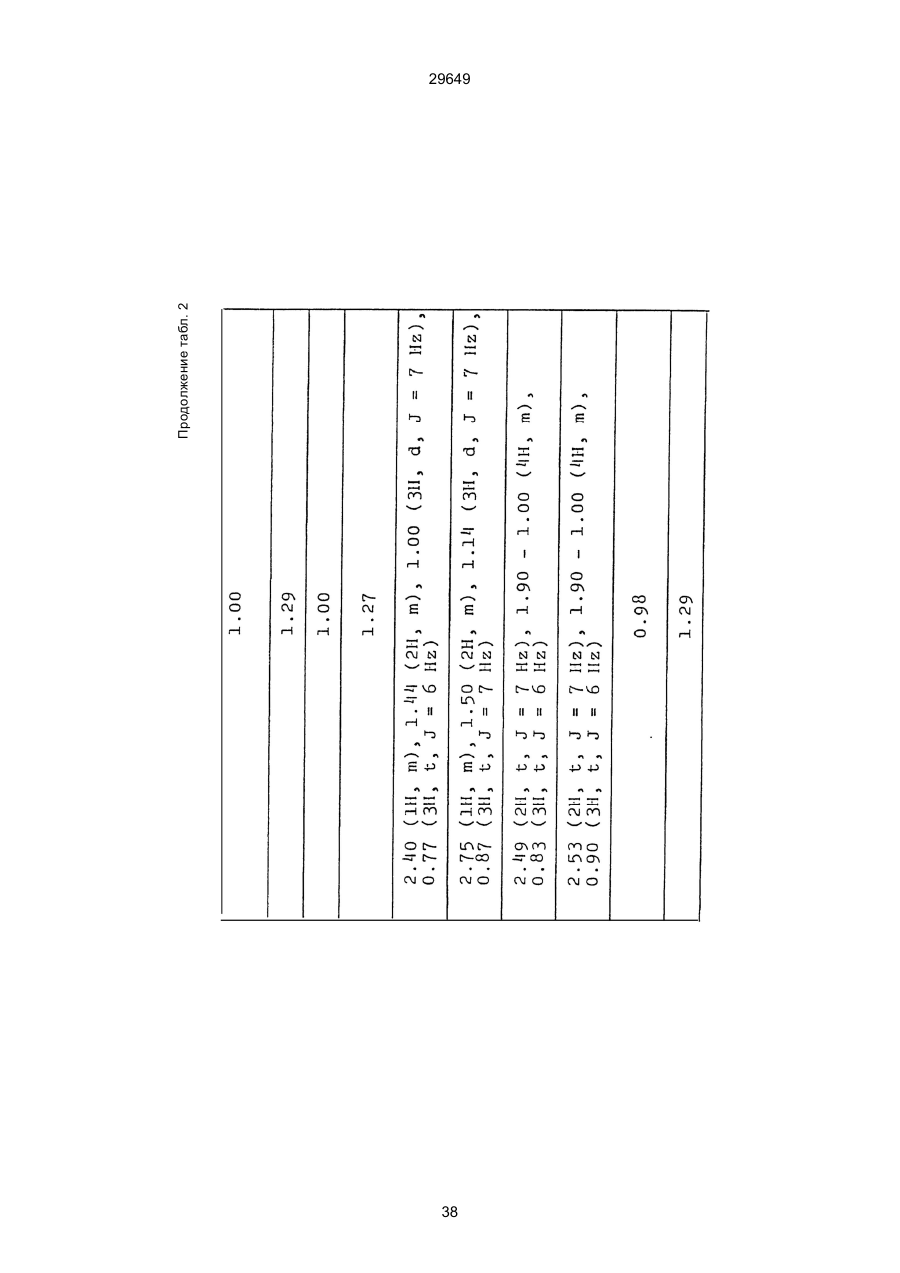
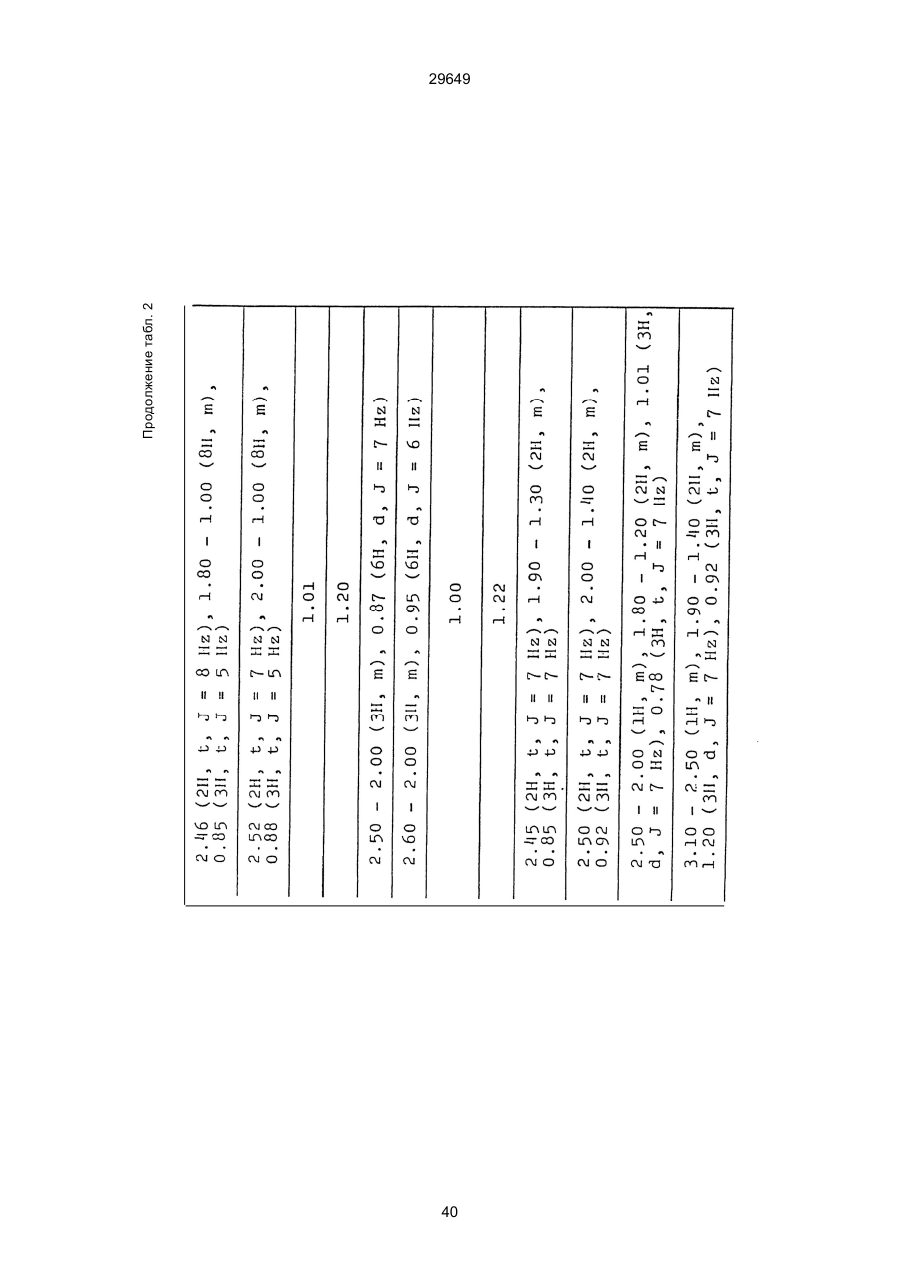
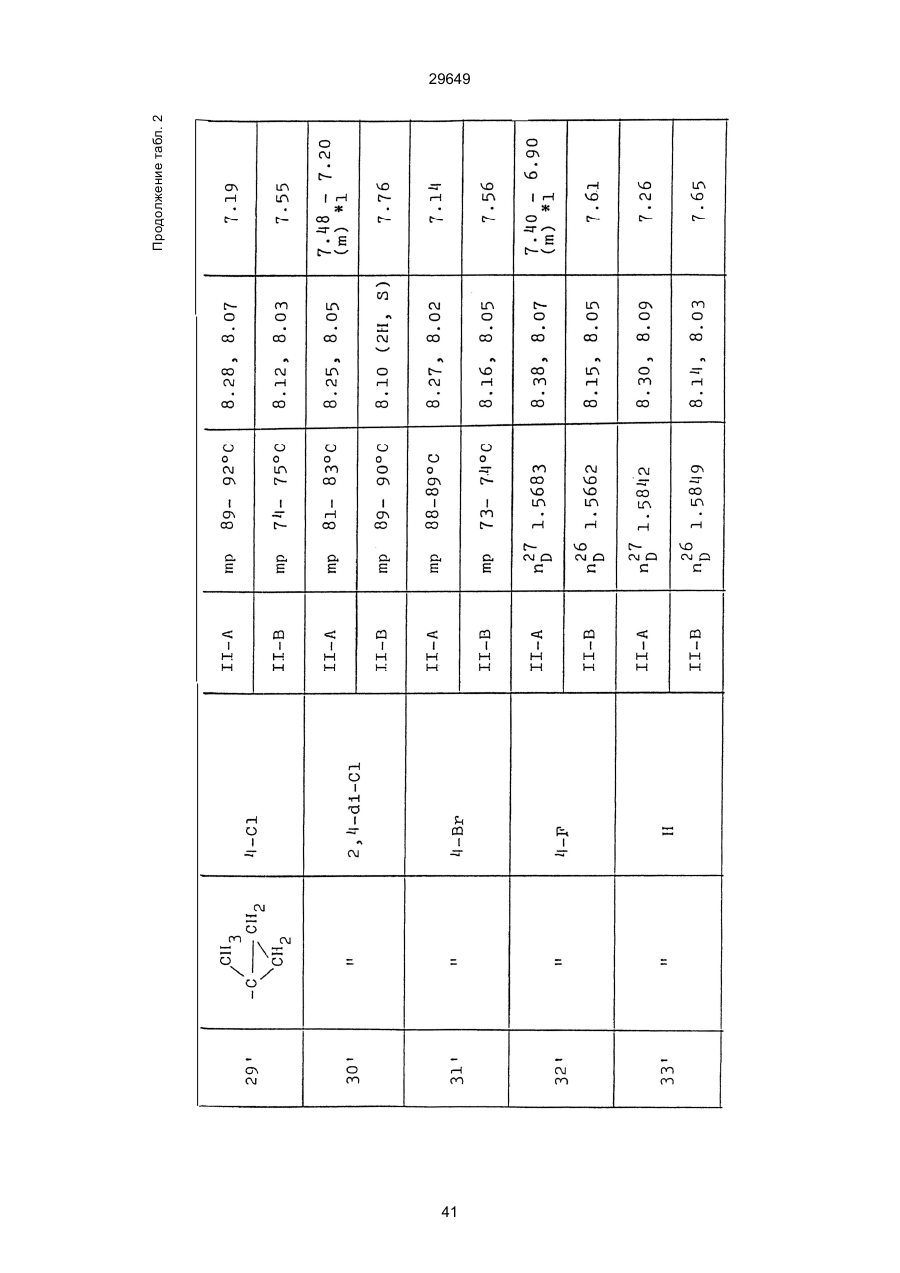
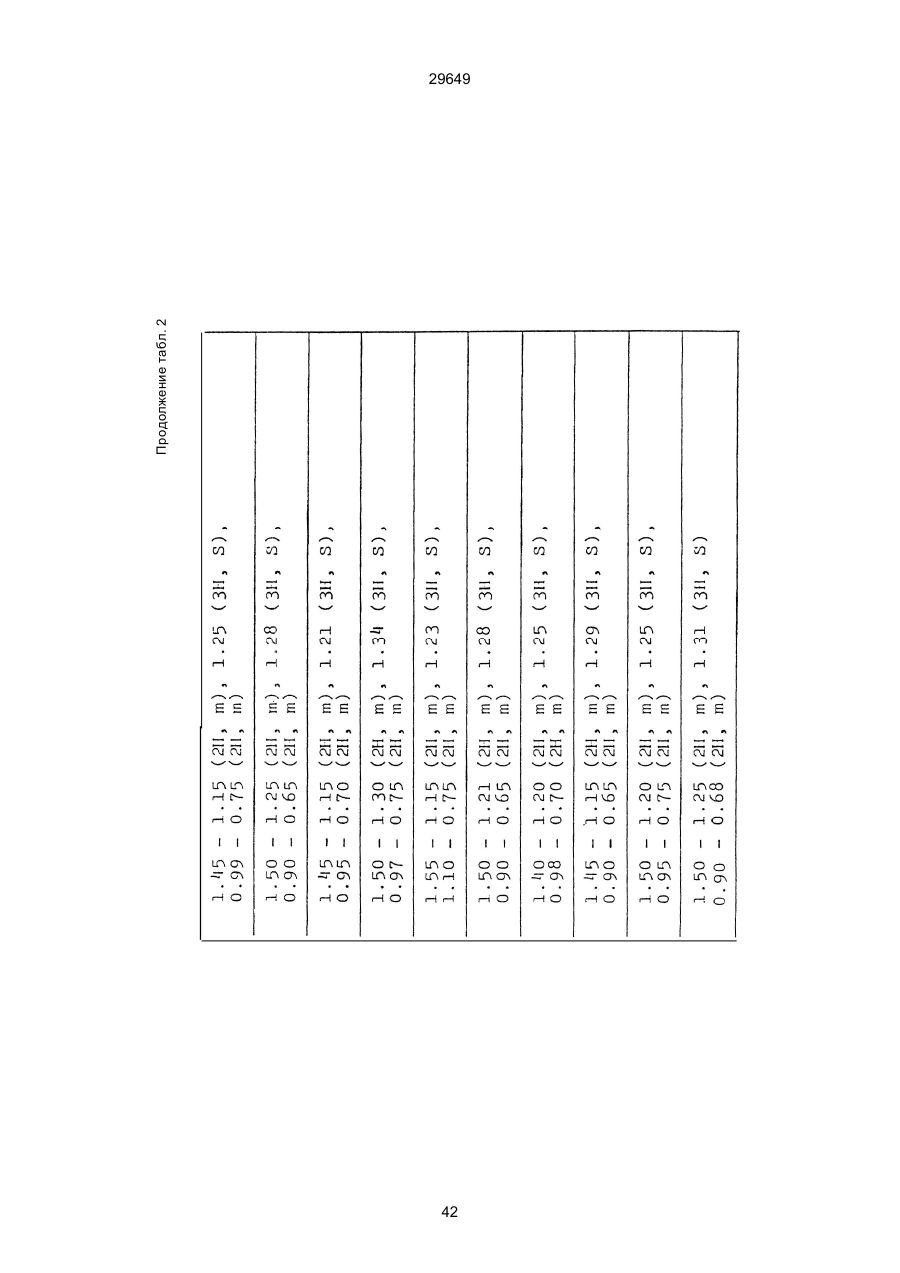
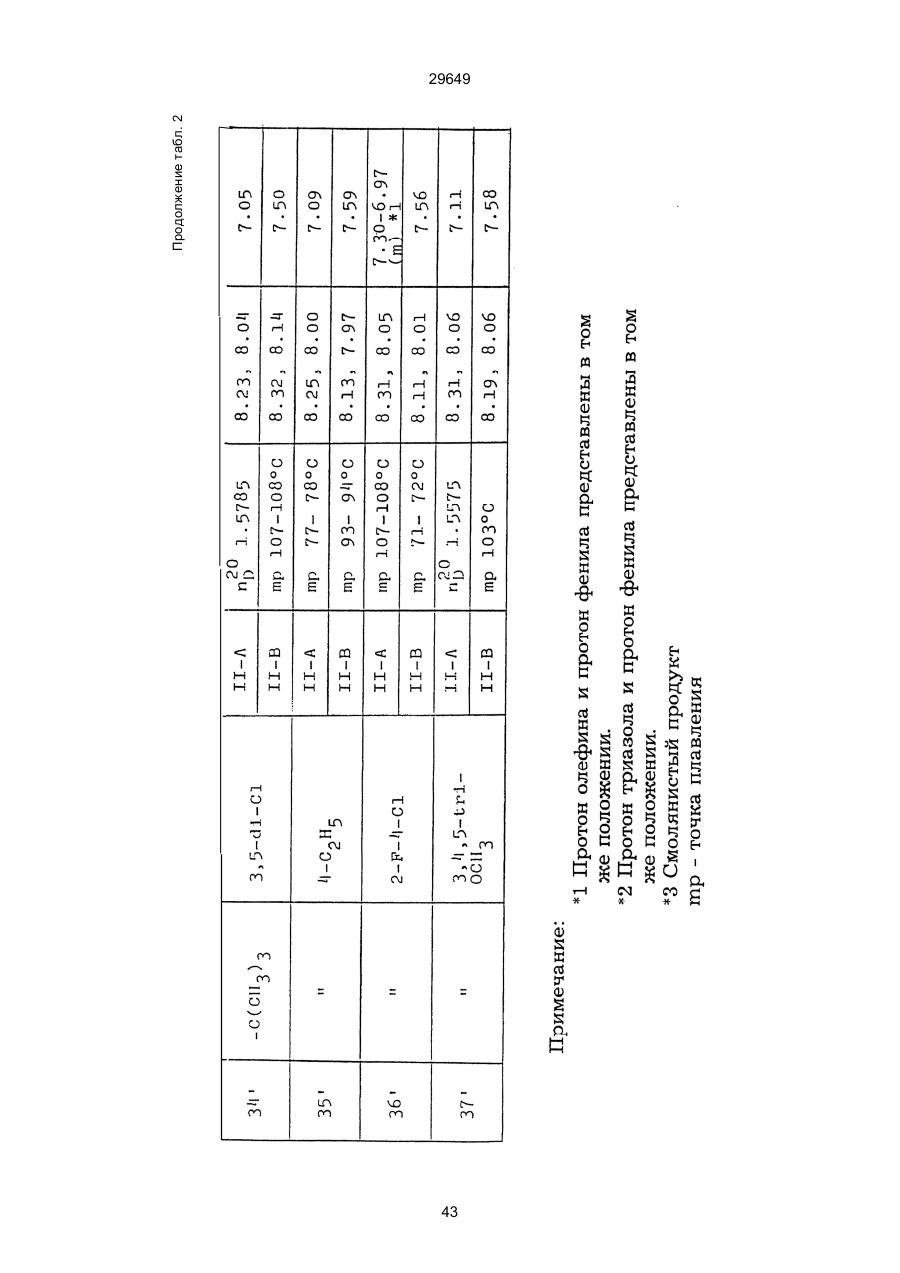
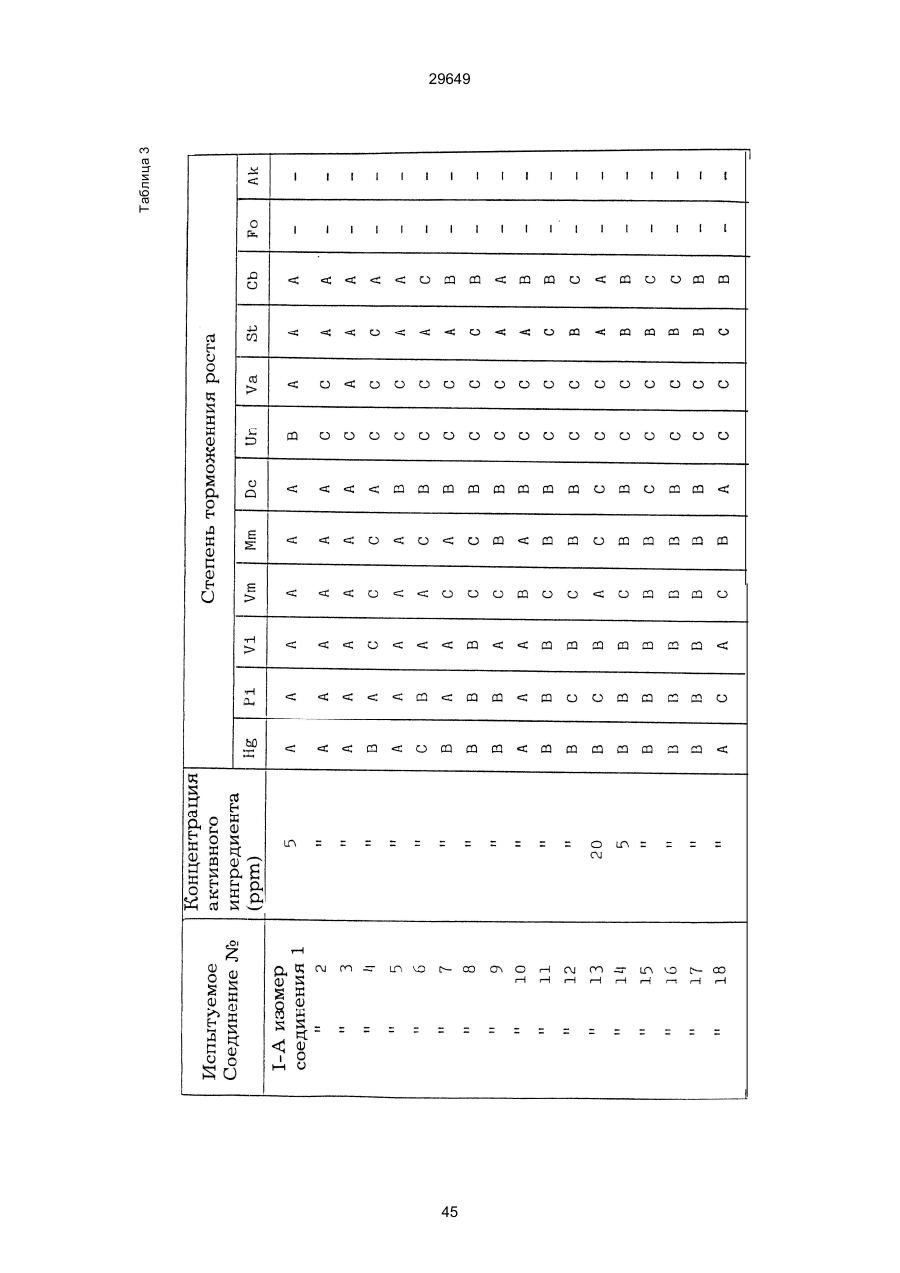
1. Способ защиты растений от фитопатогенов путем воздействия соединением триазола, отличающийся тем, что на поверхности растения или почвы, в которой растут или будут выращиваться растения создают защитный слой, содержащий соединения триазола, представленного формулой I: где R1 - это атом водорода, С1-С4 алкил, С3-С4 алкенил или 2-пропиниловая группа, R2 - зто C1-C6 алкил, циклопропил или 1-метилцклопропиловая группа, R3, которая может быть такой же или отличающейся, представляет собой атом галогена, выбранный из группы, состоящей из хлора, брома или фтора, С1-С4, алкил, галогензамещенной С1С3 алкил, С1-С4 алкокси-, фенокси-, фенил-, цианоили нитро-группу, n- зто целое число от 0 до 3, или соли указанного соединения при величине дозирования от 0,02 до 5 кг на гектар, посредством чего растения защищают от фитопатогенов. 2. Способ по п. 1, отличающийся тем, что защитный слой формируют в виде пленки. 3. Способ по п. 1, отличающийся тем, что упомянутый защитный слой формируют путем нанесения микрогранул на поверхность растения. (11) UA изомеров, который проявляет более сильную активность. Известен способ защиты растений от фитопатогенов, заключающийся в использовании соединений триазола (см. патент Великобритании № 2004276, МКИ C07D231/52). Соединение, полученное этим способом обладает способностью подавлять различные заболевания растений. Известен также способ защиты растений от фитопатогенов, заключающийся в использовании соединений триазола (см. патент США № 4086351, МКИ С07D231/52). Соединения, полученные этим способом имеют другие структурные характеристики и обладают более высокой фунгицидной активностью. Данный способ выбран в качестве прототипа. Эти соединения, однако, недостаточно эффективны для борьбы с различными фитопатогенами. В основу изобретения поставлена задача в способе защиты растений от фитопатогенов провести такие усовершенствования, с помощью ко (19) Настоящее изобретение относится к области химии и может быть использовано в сельском хозяйстве, садоводстве для борьбы с заболеваниями и вредителями растений. Известно большое количество синтетических органических соединений, которые применяются для борьбы с заболеваниями и вредителями сельскохозяйственных и садоводческих культур. Однако остается значительное количество проблем, требующих своего решения. Эти проблемы иногда решают за счет создания новых пестицидов, но можно решать, исследуя обычные пестициды для установления подходящих форм или способов применения агрохимикатов. Существует немало синтетических органических соединений, которые могут присутствовать в виде геометрических или оптических изомеров с различной биологической активностью. В настоящее время серьезное значение приобретает проблема загрязнения окружающей среды в области сельского хозяйства и садоводства, и важно уменьшить эту проблему, используя один из двух 29649 (13) A (I) 29649 торых обеспечить эффективную борьбу с фитопатогенами. Поставленная задача решается тем, что в способе защиты растений от фитопатогенов путем воздействия соединением триазола, согласно изобретению на поверхности растения или почвы, в которой растут или будут выращиваться растения, создают защитный слой, содержащий соединения триазола, представленного формулой I: , группа; R3, которая может быть такой же или отличающейся, представляет собой атом галогена, выбранный из группы, состоящей из хлора, брома или фтора, С1-С4 алкил, галогензамещенной С1-С3 алкил, С1-С4 алкокси-, фенокси-, фенил-, цианоили нитро-группу; n - это целое число от 0 до 3; или соли указанного соединения при величине дозирования от 0,02 до 5 кг на гектар, посредством чего растения защищают от фитопатогенов. Защитный слой наносят в виде пленки. Упомянутый защитный слой формируют путем нанесения микрогранул на поверхность растения. Предложенный способ осуществляют следующим образом. Соединение триазола по формуле (I) получают восстановлением соединения триазола формулы (II), получая соединение триазола формулы (І), в котором R1 - это атом водорода, и последующей этерификацией полученного соединения: (I) где R1 - это атом водорода, С1-С4 алкил, С3-С4 алкенил или 2-пропиниловая группа; R3 - это С1-С6 алкил, циклопропил или 1-метилциклопропиловая (II) можно осуществлять путем применения соединения триазола формулы (І) таким образом, что защитньй слой, содержащий соединение триазола формулы (І) на нем, может быть сформирован на поверхности растения и/или почвы. Термин "защитный слой" здесь и далее обозначает покрытие или пленку, или слой, образованный микрогранулами, прилипшими к поверхности растения или почвы. Термин "пленка или покрытие" может быть образован применением триазольного соединения вместе с поверхностно-активным веществом, имеющим хорошо сбалансированную величину HLB и произвольно соответствующий носитель. Слой, сформированный микрогранулами, может быть образован применением триазольного соединения, содержащего связующее, сформированное в микрогранулы на растение и/или почву таким образом, что микрогранулы прилипают на поверхность растения и/или почвы. В качестве поверхностно-активного вещества (ПАВ) для предложенного способа может быть проиллюстрировано поверхностно-активное вещество, имеющее величину HLB от 8 до 18. Может быть использовано любое ПАВ, имеющее упомянутую величину HLB. (I) (R1: Заместители иные, чем Н) где R1, R2, R3 и n - такие же, как определены выше. Далее один из двух геометрических изомеров соединения триазола (II), олефиновый протон которого проявляется при повышенном магнитном поле на ЯМР-спектре в дейтерохлороформе, определяется как изомер II-А, а другой изомер, олефиновый протон которого появляется при пониженном магнитном поле на ЯМР-спектре в дейтерохлороформе, определяется как изомер ІІ-В. Кроме того, соединение (І), в котором R1 - это атом водорода, полученное восстановлением изомера ІІ-А, определяется как изомер І'-А; соединение (І), в котором R1 - это иная, нежели атом водорода, замещающая группа из числа вышеуказанных, полученная этерификацией изомера І'-А, определяется как изомер І''-А; а изомеры І'-А и І"-А имеют обобщающее определение как изомер І-А. Соответствующие соединения, полученные из изомера ІІ-В, аналогичным образом определяются как изомер І'-В, изомер І"-В и изомер I-В, соответственно. Настоящее изобретение относится к изомеру І-А и к изомеру ІІ-А, который является промежуточным соединением для изомера І-А. В результате интенсивных исследований создателям настоящего изобретения удалось установить, что эффективную борьбу с фитопатогенами 2 29649 вая гниль персиков (Sclerotinia cinerea), антракноз винограда (Elsinoe ampelina), гниль спелого винограда (Glomerella cingulata), серая гниль винограда (Botrytis cinerea), мучнистая роса винограда (Uncinula necator), ржавчина винограда (Phakopsora ampelopsidis), ржавчина верхушек овса (Puccinia coronata), мучнистая роса ячменя (Erysiphe graminis), пятнистость листьев ячменя (Rhynchosporium secalis), полосатость ячменя (Helmithosporium gramineum), рыхлая головня ячменя (Ustilago nuda), твердая головня ячменя (Ustilago hordei), снежная гниль ячменя (Typhula incarnata), стеблевая ржавчина ячменя (Puccinia graminis), листовая ржавчина пшеницы (Puccinia recondita), рыхлая ржавчина пшеницы (Ustilago tritici), твердая головня пшеницы (Tilletia caries), пятнистость листьев пшеницы (Septoria tritici), пятнистость колосков пшеницы (Septoria nodorum), желтая ржавчина пшеницы (Puccinia striiformis), стеблевая ржавчина пшеницы (Puccinia graminis), мучнистая роса пшеницы (Erysiphe graminis), мучнистая роса огурцов (Sphaerotheca fuliginea), серая гниль огурцов (Botrytis cinerea), гуммозная стеблевая гниль огурцов (Mycosphaerella melonis), гниль семянки огурцов (Sclerotinia sclerotiorum), антракноз огурцов (Colletotrichum lagenarium), лиственная гниль томатов (Cladosporium fulvum), настоящая мучнистая роса томатов (Erysiphe cichoracearum), ранняя гниль томатов (Altenaria solani), серая гниль баклажан (Botrytis cinerea), мутовчатое увядание баклажан (Vertcillium alboatrum), мучнистая роса баклажан (Erysiphe cichoracearum), мучнистая роса душистого перца (Leveillula taurica), серая гниль земляники (Botrytis cinerea), настоящая мучнистая роса земляники (Sphaerotheca humuli), коричневая пятнистость табака (Alternaria longipes), мучнистая роса табака (Erysiphe cichoracearum), пятнистость листьев свеклы (Cercospora beticola), пятнистость листьев земляного ореха (Cercospora personata), коричневая пятнистость листьев горошка (Cerospora arachidicola) и тому подобное. Более конкретно способы получения соединений настоящего изобретения будут описаны ниже. Способ А Восстановление триазольного соединения II В качестве связующе формирующих материалов могут быть приведены следующие ПАВ: производные целлюлозы, такие как карбоксиметилцеллюлоза, гидроксипропилцеллюлоза, поли(мет)акриловая кислота, как, например, карбоновый виниловый полимер. Микрогранулы могут быть получены путем смешивания расплава триазольного соединения формулы (І) со сложным эфиром полиглицериновой жирной кислоты, такой как эфир полиглицерина, представленной формулой , где n - целое число 2 или более, с жирной кислотой, как например, лауриновой кислотой, пальмитиновой кислотой, стеариновой кислотой, бегеновой кислотой, олеиновой кислотой, линолевой кислотой и им подобным кислотам, добавляя связующе-образующее вещество к полученному, перемешивая полученную смесь до конца, и каплеобразно добавляя полученную смесь на алюминиевый диск, вращающийся со скоростью 1500 об/мин и более, получая микрогранулы, имеющие размер частиц от 30 до 80 мешей (число отверстий на линейный дюйм). В качестве заболеваний, против которых соединения настоящего изобретения (1-А) (изомер) обладает превосходной защитной активностью, можно привести пирикуляроз риса (Pyricularia oryzae), гниль оболочки риса (Pellicularia sasakii), рак яблок (Valsa mali), цветочная гниль яблок (Sclerotinia mali), мучнистая роса яблок (Podospaera leucotricha), парша яблони (Venturia inaequalis), пятнистость яблок (Mycosphaerella pomi), пятнистость листьев у яблони (Alternaria mali), черная пятнистость груши (Alternaria kikuchiana), мучнистая роса груши (Phyllactinia ругі), ржавчина груши (Gymnosporangium haraeanum), парша груши (Venturia nashicola), меланоз цитрусовых (Diaporthe citri), парша цитрусовых (Elsinoe fawcetti), обычная зеленая плесень цитрусових плодов (Penicillium digitatum), синяя плесень апельсинов (Penicillium italicum), коричне где R2, R3 и n имеют указанные ранее значения. Изомер І'-А получают, восстанавливая изомер ІІ-А в подходящем растворителе металлгидридным комплексом (например, литийалюминийгидридом, натрийборгидридом) или алкоголятом алюминия (например, изопропилатом алюминия). II-А изомер, подлежащий восстановлению, можно получить в чистом виде, например, осуществляя фракционную кристаллизацию или хромато графируя на колонке смесь геометрических изомеров триазольного соединения (II), полученного в соответствии со следующей схемой реакции. Изомер ІІ-А можно также легко получить с хорошим выходом, например, облучая смесь ультрафиолетовыми лучами для проведения фотоизомеризации. Более детальные разъяснения будут приведены далее со ссылками на способы С и Д. 3 29649 где R2, R3 и n имеют указанные ранее значения. Растворитель, который можно использовать при восстановлении металгидридным комплексом, включает, например, простые эфиры (например, диэтиловый эфир, тетрагидрофуран) и спирты (например, метанол, этанол, изопропанол). Если в качестве металгидридного комплекса используют натрийборгидрид, то реакцию осуществляют, добавляя в растворитель 1 моль II-А изомера и от 0,25 до 2 молей натрийборгидрида. Температуру реакции поддерживают, предпочтительно, в интервале от 0°С до комнатной температуры. Используемые растворители включают, например, простые эфиры (например, диэтиловый эфир, тетрагидрофуран) и спирты (например, этанол, метанол, изопропанол). Если в качестве металгидридного комплекса используют литийалюминийгидрид, то реакцию осуществляют, растворяя литийалюминийгидрид в количестве от 0,25 до 0,8 моля в расчете на II-А изомер в растворителе, и, добавляя полученный раствор к раствору изомера в том же самом растворителе. Температуру реакции поддерживают, предпочтительно, в интервале от -60°С до 70°С. Используемый растворитель включает простые эфиры (например, диэтиловый эфир, тетрагидрофуран). После завершения реакции воду или разбавленную водой кислоту добавляют к реакционному раствору, и после нейтрализации щелочью, в случае необходимости, высадившиеся кристаллы собирают фильтрованием или экстрагированием органическим растворителем, умеренно растворимым в воде. Последующую обработку проводят обычными способами. Если в качестве восстанавливающего агента используют изопропилат алюминия, то предпочтительно использовать такие растворители как спирты (например, изопропанол) или ароматические углеводороды (например, бензол). Обычно дают возможность 1 молю II-А изомера взаимодействовать с 1-2 молями изопропилата алюминия при температуре между комнатной температурой и 100°С. Полученное соединение алюминия разлагают разбавленной серной кислотой или водным раствором гидроокиси натрия, после чего экстрагируют органическим растворителем, умеренно растворимым в воде. Последующую обработку проводят обычными способами. Соли І'-А изомера относятся к солям, полученным из растительно- и физиологически приемлемых кислот, например, галоидоводородных кислот (например, бромистоводородной кислоты, хлористоводородной кислоты, йодистоводородной кислоты), карбоновых кислот (например, уксусной кислоты, трихлоруксусной кислоты, малеиновой кислоты, янтарной кислоты), сульфокислоты (например, паратолуолсульфокислоты, метаносульфокислоты), азотной кислоты, серной кислоты и фосфорной кислоты. В случае необходимости эти соли получают обычными способами. Способ В Этерификация І'-А изомера где R1, R2, R3 и n имеют указанные ранее значения. Соединения настоящего изобретения, І"-А изомеры, получают при взаимодействии І'-А изомера с реакционноспособным С1-С4-алкильным, 4 29649 Пример 1 Синтез І'-А изомера 1-(4-хлорфенил)-4,4-диметил-2-(1,2,4-триазол-1-ил)-1-пентен-3-ола (Соединение № 1) по способу А. II-А-изомер (2,9 г 0,01 моля; т. пл. 108-109°С) 1-(4-хлорфенил)-4,4-диметил-2(1,2,4-триазол-1-ил)1-пентен-3-она (Соединение № 1') растворили в метаноле (50 мл). К этому добавили натрийбромид (0,38 г, 0,01 моля), причем температуру реакции поддерживали при 20°С или менее, охлаждая льдом. Реакционную смесь выдерживали при температуре 20°С в течение 3 часов, а затем разложили, добавив воду (100 мл) и уксусную кислоту (1 мл). Органический слой экстрагировали этилацетатом (100 мл), полученный экстракт промыли 5% водным раствором бикарбоната натрия (50 мл) и высушили над безводным сульфатом натрия. Затем растворитель удалили при пониженном давлении, и полученный остаток перекристал- лизовали из изопропанола до получения 2,0 г (выход 69%) І'-А изомера, точка плавления которого 153155°С. Элементный анализ и полученный спектр ЯМР этого соединения приведены ниже. Элементный анализ: С% Н% N% Cl% Рассчитано для С15Н18N3ОСІ 61,74 6,23 14,40 12,15 Найдено 61,82 6,33 14,38 12,15 Спектр ЯМР: 8,52 (1Н, s, триазольный протон) 7,98 (1Н, s, триазольный протон) 7,30 (4H, s, фенильный протон) 6,91 (1H, s, олефиновый протон) 4,56 (2Н, широкий синглет, гидроксильный протон и протон метина, содержащего ОН группу) 0,66 (9Н, s, бутиловый протон). Сравнительный пример 1 Синтез І'-В изомера 1-(4-хлорфенил)-4,4-диметил-2-(1,2,4-триазол-1-ил)-1-пентен-3-ола (Соединение № 1) ІІ-В изомер (2,9 г, 0,01 моля; т. пл. 78°-79°С 1-(4-хлорфенил)-4,4-диметил-2-(1,2,;-триазол-1-ил)1-пернтен-3-она (Соединение № 1') растворили в метаноле (50 мл). Этот изомер оставили реагировать с натрийборгидридом, а затем обработали таким же способом, что и в примере 1. Полученный остаток перекристаллизовывали из смеси 1:10 четыреххлористого углерода и n-гексана до получения 2,2 г (выход 76%) І'-В изомера (т. пл. 116°-117°С) соединения № 1. Элементный анализ и спектр ЯМР этого соединения приведены далее. Элементный анализ: С% Н% N% Cl% Рассчитано для С15Н18N3ОСІ 61,74 6,23 14,40 12,15 Найдено 61,80 6,25 14,52 12,09 Спектр ЯМР: 7,92 (1H, s, триазольный протон) 7,77 (1H, s, триазольный протон) 7,05 (2H, d, фенильный протон, J=9 Гц;) 6,58 (2Н, d, фенильный протон, J=9 Гц) 6,66 (1H,s, олефиновый протон) 4,28 (1H, d, метиновый протон, несущий ОН группу, J=6 Гц) 3,21 (1H,d, гидроксильный протон, J=6 Гц) 0,80 (9Н, s, бутиловый протон). С3-С4-алкенильным или 2-пропильным производным в подходящем растворителе в присутствии основания. Реакционноспособные производные включают, например, алкил-алкенил- или алкинилгалоиды (например, метилйодид, аллилбромид, пропаргилбромид), сульфаты (например, диметилсульфат, диэтилсульфат) и сульфонаты (например, паратолуолсульфонат, нафталинсульфонат). Растворитель включает, например, общие инертные органические растворители, например, диэтиловый эфир, тетрагидрофуран, диоксан, бензол, толуол, ксилол и диметилформамид. Эту реакцию можно проводить в присутствии воды, используя катализатор переноса фазы, известный как акселератор реакции (например, триэтилбензиламмонийхлорид, триметилбензиламмонийбромид). Основания включают, например, подходящие сильные основания (например, гидриды щелочных металлов, такие как натрийгидрид, амиды щелочных металлов, например, натрийамид), карбонаты (например, карбонат натрия, карбонат калия) и гидроокиси щелочных металлов (например, гидроокись калия, гидроокись натрия). Эту реакцию осуществляют, смешивая І'-А изомер, реакционноспособное С1-С4-алкил, С3-С4алкенил или 2-пропинил-производное и основание, предпочтительно, в эквимолярном соотношении в подходящем растворителе. Реакцию проводят в интервале температур от 0° до 100°С, и предпочтительно, от 20° до 60°С. Иногда предпочтительно, во-первых, вызывать реакцию І'-А изомера с подходящим сильным основанием (например, гидриды щелочных металлов, амиды щелочных металлов) в инертном растворителе, а затем полученную соль щелочного металла подвергнуть взаимодействию с реакционноспособным С1-С4алкил, С3-С4-алкенил, или 2-пропинил-произ- водным. В некоторых случаях для выделения соединений настоящего изобретения І"-А может быть желательным следующий путь: Реакционную смесь выделяют из растворителя путем выпаривания, воду и органический растворитель, умеренно растворимый в воде, добавляют к остатку, органический слой после экстрагирования выделяют и затем очищают обычными способами. Соли І"-А изомера относятся к тем солям, которые получены с помощью физиологически приемлемых кислот, таких как, например, галоидоводородные кислоты (например, бромистоводородная кислота, хлористоводородная кислота, йодистоводородная кислота), карбоновые кислоты (например, уксусная кислота, трихлоруксусная кислота, малеиновая кислота, янтарная кислота), сульфоновые кислоты (например, паратолуолсульфокислота, метансульфоновая кислота), азотная кислота, серная кислота и фосфорная кислота. В случае необходимости, эти кислоты получают обычными способами. Более подробно настоящее изобретение будет проиллюстрировано со ссылкой на следующие примеры. Если нет других указаний, то ЯМР спектр в примерах указывают значениями d для дейтерохлороформа как растворителя и тетраметилсилана в качестве внутреннего стандарта. 5 29649 № 1) растворили в диметилформамиде (20 см3) и к нему добавили 65% натрийгидрида в масле (0,26 г). После перемешивания в течение 1 часа при комнатной температуре реакционную смесь охладили до 10°С и добавили метилйодид (1 г). После выстаивания при комнатной температуре в течение 20 часов растворитель удалили при пониженном давлении, а полученный остаток экстрагировали, добавив ледяную воду (100 г) и хлороформ (100 см3). Органический слой высушили над безводным сульфатом магния, и растворитель удалили при пониженном давлении. Полученный маслянистый неочищенный продукт очистили на хроматографической колонке на силикагеле (ацетон:n-гексан=1:10) и далее перекристаллизовали из смеси четыреххлористого углерода и n-гексана (1:2) до получения 1,6 г указанного в заглавии соединения (т. пл. 63°-66°С). Сравнительный пример 3 Синтез І'-В изомера 1-пара-хлорфенил-4,4диметил-3-метокси-2-(1,2,4-триазол-1-ил)-1-пентена (Соединение № 35) І'-В изомер (2 г) 1-(4-хлорфенил)-4,4-диметил2-(1,2,4-триазол)-1-ил)-1-пентен-3-ола (Соединение № 1) растворили в диметилформамиде (20 см3) и к нему добавили 65% натрийгидрида (0,2 г). После перемешивания при комнатной температуре в течение 1 часа реакционную смесь охладили до 10°С и добавили 1 г метилйодида. Реакционную смесь выдерживали при 10°С в течение 1 часа, а затем оставили выстаиваться при комнатной температуре в течение 16 часов. Диметилформамид удалили при пониженном давлении, и полученный остаток экстрагировали, добавив ледяную воду (100 г) и хлороформ (100 см3). Органический слой высушили над безводным сульфатом магния, и растворитель удалили при пониженном давлении. Полученный в результате неочищенный продукт очистили на хроматографической колонке на силикагеле (ацетон:n-гексан=1:10) до получения 1,0 г соединения, указанного в заглавии, в виде масляного продукта. Показатель преломления n27D=1,5435. Результаты элементного анализа: С% Н% N% Cl% Рассчитано 62,90 6,60 13,77 11,50 для С16Н20N3СІО Найдено 62,84 6,59 13,74 11,59 Соединения настоящего изобретения (І-А изомеры), полученные по способам А и В приведены в табл. 1. Для сравнения рядом приведены данные для І-В изомеров. Если нет других указаний, спектры ЯМР в таблице приведены в значениях d для СDСІ3 как растворителя и тетраметилсилана в качестве внутреннего эталона. І'-изомер и І"-изомер обычно называют І-А изомером, І'-В изомер также обычно называют І-В изомером. Это общее обозначение также используется в тестовых примерах, приводимых далее. Далее приводится объяснение для получения ІІ-А изомера триазольного соединения (II), которое Пример 2 Синтез І'-А изомера 3-(4-хлорфенил)-1-(1-метилциклопропан)-2-(1,2,4-триазол-1-ил)-2-пропен1-ола (Соединение № З0) по способу А І'-А изомер (2,9 г 0,01 моля; т. пл. 89°-92°С) 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4триазол-1-ил)-2-пропен-1-она (Соединение № 29'), охарактеризованный по спектру ЯМР, описанный далее, растворили в метаноле (50 мл). Натрийборгидрид (0,38 г, 0,01 моля) добавили к этому, причем температуру реакции поддерживали 20°С или ниже, охлаждая льдом. Температуру реакции поддерживали при 20°С в течение 3 часов, а затем разлагали, добавив воду (100 мл) и уксусную кислоту (2 мл). Органический слой экстрагировали хлороформом (100 мл), экстракт промыли 5% водным раствором бикарбоната натрия (50 мл) и высушили над безводным сульфатом магния. Затем растворитель удалили при пониженном давлении, а полученный осадок перекристаллизовали из смеси четыреххлористого углерода и n-гексана (1:1) 5 мл до получения 2,4 г (выход 85%) указанного в заглавии соединения. ЯМР спектр исходного материала ІІ-А изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4триазол-1-ил)-2-пропен-1-она следующий: 8,28 (1Н, s, триазольный протон) 8,07 (1Н, s, триазольный протон) 7,32 (4Н, s, фенильный протон) 7,19 (1Н, s, олефиновый протон) 1,45-1,15 (2Н, m, метиленовый протон циклопропильной группы) 1,25 (3Н, s, метильный протон) 0,99-0,75 (2Н, m, метиленовый протон циклопропильной группы). Сравнительный пример 2 Синтез І'-В изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-2-пропен1-ола (Соединение № З0) І'-В изомер (2 г, 0,007 моля; т. пл. 74°-75°С) 3-(4-хлорфенил)-(1-метилциклопропил)-2-(1,2,4триазол-1-ил)-2-пропен-1-она (Соединение № 29'), характеризующийся спектром ЯМР, приведенным далее, восстановили таким же способом, что и в примере 2, натрийбромгидридом (0,27 г, 0,007 моля) в метаноле (50 мл). Таким образом, получили 1,7 г (выход 85%) соединения, указанного в заглавии. Спектр ЯМР исходного материала, II-В изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2(1,2,4-триазол-1-ил)-2-пропен-1-она: 8,12 (2Н, s, триазольный протон) 8,03 (1Н, s, триазольный протон) 7,55 (1Н, s, олефиновый протон) 7,21 (2Н, d, фенильный протон, J=8 Гц) 6,81 (2Н, d, фенильный протон, J=8 Гц) 1,50-1,25 (2Н, m, метиленовый протон циклопропильной группы) 1,28 (3Н, s, метильный протон) 0,90-0,65 (2Н, m, метиленовий протон циклопропильной группы). Пример 3 Синтез І'-А изомера 1-(4-хлорфенил)-4,4-диметил-3-метокси-2-(1,2,4-триазол-1-ил)-1-пентен (Соединение № 35) по способу В І'-изомер (2 г) 1-пара-хлорфенил-4,4-диметил2-(1,2,4-триазол-1-ил)-1-пентен-3-ола (Соединение 6 29649 является исходным материалом для І'-А изомера триазольного соединения (І). Способ С Изомеризация ІІ-В изомера или смеси ІІ-В и ІІ-А изомеров триазольного соединения (II): где R2, R3 и n имеют указанные ранее значения. ІІ-А изомер можно получить, облучая ІІ-В изомер или смесь II-В и ІІ-А изомеров УФ-лампами или ксеноновыми лампами, или экспериментально флюоресцентными лампами, или солнечными лучами в растворителе, инертном по отношению к этим лучам. В качестве обычно используемого растворителя можно указать, например, спирты (например, метанол, этанол, пропанол); простые эфиры (например, тетрагидрофуран, диоксан), кетоны (например, ацетон, метилэтилкетон, метилизобутилкетон), алифатические углеводороды (например,гексан, циклогексан, петролейный эфир) и ароматические углеводороды (например, бензол, толуол, ксилол). Реакции можно проводить при температурах, при которых обычно протекает фотоизомеризация, однако предпочтительными на практике являются температуры между 0°С и 100°С. Естественно, реакцию можно проводить, добавляя сенсибилизатор, используемый в обычных фотореакциях, например, фенилкетоны, такие, как ацетофенон и пропиофенон, однако преимущество это дает небольшое. Далее будет проиллюстрирован способ получения триазольного соединения, представленного формулой (II). Способ D Получение смеси геометрических изомеров триазольного соединения (II) и каждого из изомеров (ІІ-В и ІІ-А) где R2, R3 и n имеют указанные ранее значения. Триазольное соединение (II) получают, подвергая взаимодействию 1 моль кетона формулы (IV) с 1-2 молями бензальдегида формулы (III) в подходящем растворителе в присутствии основного катализатора. Основные катализаторы включают, например, гидроокиси щелочных и щелочноземельных металлов (например, гидроокись натрия, гидроокись калия, гидроокись кальция), алкоголята щелочных металлов (например, метилат натрия, этилат натрия, метилат калия), карбонаты (например, карбонат натрия, карбонат калия), ацетаты (например, ацетат натрия, ацетат калия), вторичные амины (например, диэтиламин, дипропиламин, пирролидин, пиперидин, морфолин) и третичные амины (например, триэтиламин, трибутиламин, пиридин, пиколин, диметиланилин) и его используют в количествах от 0,01 моля до 10,0 молей. Растворители включают, например, спирты (например, метанол, этанол), ароматические углеводороды (например, бензол, толуол, ксилол), простые эфиры (например, диэтиловый эфир, тетрагидрофуран, диоксан), воду и их сме си. Реакцию проводят в интервале температур от 0°С до температуры кипения растворителя. Если в качестве основного катализатора используют ацетаты (например, ацетат натрия, ацетат калия), карбонаты (например, карбонат натрия, карбонат калия) или третичные амины, то в качестве растворителя для реакции можно использовать ледяную уксусную кислоту или уксусный ангидрид. Полученное таким образом триазольное, соединение (II) является смесью двух геометрических изомеров, например, изомера ІІ-А и изомера ІІ-В, причем каждый из изомеров можно выделить хроматографированием на колонке или фракционной кристаллизацией. Смесь геометрических изомеров обычно содержит большее количество изомера ІІ-В, нежели изомера II-А. Все изомеры ІІ-А кетонового соединения представляют собой, естественно, новые соединения, а из ІІ-В изомеров новыми соединениями являются такие, в которых R2 является І-метилциклопропильной группой. Далее способы С и D будут проиллюстрированы более подробно со ссылкой на следующие примеры. Пример 4 7 29649 Синтез 1-(4-хлорфенил)-4,4-диметил-2-(1,2,4триазол)-1-ил)-1-пентен-3-она (Соединение № 1') по способу D. a-(1,2,4-триазол-1-ил)пинаколон (50 г), безводный карбонат калия (41 г) уксусный ангидрид (200 мл) и 4-хлорбензальдегид (46,3 г) смешали, и полученную смесь нагревали при температуре 90°С в течение 12 часов при перемешивании. После охлаждения реакционного раствора выпавший осадок отфильтровали. Полученный фильтрат по каплям добавляли к теплой воде (500 мл) при температуре 60°С для разложения уксусного ангидрида, а затем понемногу добавили карбонат калия для подщелачивания раствора. Полученный маслянистый продукт экстрагировали этилацетатом (500 мл), и органический слой высушили над безводным сульфатом натрия, и затем сконцентрировали при пониженном давлении. Одну каплю полученного остатка растворили в ацетоне, ацетоновый раствор проанализировали с помощью газовой хроматографии в условиях, описанных далее. Затем было найдено, что время удерживания для пика, соответствующего ІІ-А изомеру, составляет 300 сек, а время, соответствующее пику II-В изомера, составляет 360 сек. Отношение обоих изомеров, полученное при расчете площадей пиков составило 19,8/61,2, то есть, около 1/3. Условия проведения газово-хроматографического анализа были следующими: Аппаратура: Газовый хроматограф Ниппон Денши 20К, снабженный детектором FID Колонка: Стеклянная колонка длиной 1 м Жидкая фаза 5% ХЕ-6 О Носитель Хромосорб W Температура (колонки): 200°С Температура (ввода): 240°С Газ-носитель: газообразный азот, 1кг/см2 Остаток растворили в бензоле (100 мл) Полученный раствор пропустили через колонку, заполненную силикагелем с размерами частиц 100200 межей (1,2 кг), и провели хроматографирование смесью н-гексан/ацетон (10:1) в качестве проявляющего растворителя. Фракции, соответствующие каждому изомеру, перекристаллизовывали из четыреххлористого углерода, в результате чего получили 36 г (выход 41,6%) чистого II-В изомера), т. пл. 78-79°С (и 10 г), выход 11,5% чистого II-А изомера (т. пл. 108-109°С) Проявляющий растворитель-смесь н-гексан/ацетон (10/3) пропускали далее через колонку, в результате чего выделили 8 г a-(1,2,4-триазол-1ил)пинаколона. Элементные анализы в спектре ЯМР каждого изомера приведены далее. Спектр ЯМР получали в дейтерохлороформе в качестве растворителя, и химические сдвиги выражали в величинах с тетраметилсиланом в качестве внутреннего стандарта. ІІ-А изомер 1-(4-хлорфенил)-4,4-диметил-2(1,2,4-триазол-1-ил)-1-пентен-3-она (Соединение № 1'). Элементный анализ: С% Н% N% Cl% Рассчитано для 62,17 5,58 14,50 12,23 С15Н16N3ОСІ Найдено 62,32 5,60 14,41 12,20 8,11 (1Н, s, триазольный протон) 7,00 (1Н, s, триазольный протон) 7,15 (4Н, s, фенильный протон) 6,99 (1Н, s, олефиновый протон) 0,99 (9Н, s, бутильный протон). II-В-изомер-1-(4-хлорфенил)-4,4-диметил-2(1,2,4-триазол-1-ил)-пентен-3-она (Соединение № 1'). Элементный анализ: С% Н% N% Cl% Найдено 62,35 5,59 14,38 12,18 ЯМР спектр: 8,14 (1Н, s, триазольный протон) 7,98 (1Н, s, триазольный протон) 7,22 (2Н, d, фенильный протон, J=8 Гц) 6,73 (2Н, d, фенильный протон, J=8 Гц) 7,49 (1Н, s, олефиновый протон) 1,22 (9Н, s, бутильный протон). Пример 5 Синтез ІІ-А изомера 1-(4-хлорфенил)-4,4-диметил-2-(1,2,4-триазол-1-ил)1-пентен-3-она по способу С. ІІ-В-изомер (8,0 г) 1-(4-хлорфенил)-4,4-диметил-2-(1,2,4-триазол-1-ил)-1-пентен-3-она, полученный в примере 4, растворили в ацетоне (500 мл) и изолировали при температуре 45°С под действием источника ультрафиолетового излучения, снабженного 500 Вт ртутной лампой высокого давления. В ходе реакции время от времени отбирали следовые количества реакционного раствора и с помощью газовой хроматографии в тех же условиях, что и в примере 4, отделяли соотношение изомеров (ІІ-В изомер) ІІ-А-изомер Получили следующие результаты: Время /мин Отношение изомеров (ІІ-В/ІІ-А) 0 100/0 20 10/90 60 6/94 120 6/94 Спустя 2,5 часа реакционный раствор перенесли в 500 мл колбу в форме баклажана и удалили ацетон при пониженном давлении, в результате чего получили 7,9 г кристаллов. Полученные кристаллы перекристаллизовали из четыреххлористого углерода, в результате чего получили 6,2 г (выход 78%) кристаллов (т. пл. 108-109°С). Это соединение растворили в ацетоне и хроматографировали в описанных ранее условиях, но при этом не наблюдали пика, соответствующего ІІ-В изомеру. Пример 6 Синтез ІІ-А изомера из смеси геометрических изомеров 1-(4-хлорфенил)-4,4-диметил-2-(1,2,4триазол-1-ил)-1-пентен-3-она (Соединение № 1') Реакционную смесь (10 г), содержащую ІІ-А и ІІ-В изомеры в соотношении 1 к 3, полученную в примере 4, облучали ультрафиолетовыми лучами в тех же условиях, что и в примере 5. После 1,5 часа с помощью газовой хроматографии снова измеряли соотношение ІІ-А изомера и ІІ-В изомера, и было обнаружено, что это отношение составило около 19 к 1. После выпаривания растворителя полученные кристаллы перекристаллизовывали из четыреххлористого углерода до получения 5,1 г ІІ-А изомера. Пример 7 ЯМР спектр: 8 29649 Смесь 1,2,4-триазола (18,3 г), безводного карбоната калия (37 г) иацетонитрила (250 мл) кипятили с обратным холодильником в течение 1 часа, а затем охладили до 60°С. Неочищенный 1-(1метилциклопропил)-2-бромэтан-1-он (53 г), полученный ранее, добавили к этой смеси за 2 часа, после чего перемешивание продолжали в течение ночи. Выпавшие из реакционного раствора осадки удалили фильтрованием, полученный фильтрат сконцентрировали при пониженном давлении. Полученный остаток экстрагировали, добавив 100 мл воды и 300 мл хлороформа, и органический слой высушили над безводным сульфатом магния и сконцентрировали при пониженном давлении. Полученный маслянистый остаток перекристаллизовали из 100 мл петролейного эфира и получили 27 г 1-(1-метилциклопропил)-2-(1,2,4-триазол-1ил)-этан-1-она (выход 57% в расчете на метил 1метилциклопропилкетон, т. пл. 57-60°С. Пример 8 Синтез II-А изомера 3-(4-хлорфенил)-1-(1метилциклопропил)-2-(1,2,4-триазол-1-ил)-2-пропен-1-она из ІІ-В изомера по способу С II-В изомер 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-2-пропен-1-он (4 г), полученный в примере 7, растворили в 500 мл ацетона и провели изомеризацию при температуре 45°С в течение 2 часов с помощью источника ультразвукового излучения, снабженного 500 Вт ртутной лампой высокого давления. Отношение ІІ-А изомера к II-В изомеру измеряли с помощью газовой хроматографии таким же образом, как и в примере 7. Было найдено, что это отношение составило 81,2 к 18,1. Реакционный раствор сконцентрировали при пониженном давлении до получения 3,9 г кристаллов. Эти кристаллы перекристаллизовали из четыреххлористого углерода до получения 2,8 г (выход 70%) ІІ-А изомера. Пример 9 Синтез ІІ-А изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-2-пропен1-она из смеси геометрических изомеров Реакционную смесь (3 г), содержащую II-А и ІІ-В изомеры (II-А/II-В=1/3), полученную в примере 7, облучали ультрафиолетовыми лучами в течение 1,5 часов в тех же условиях, что и в примере 8. После этого измерили отношение II-А изомера к ІІ-В изомеру с помощью газовой хроматографии. Было обнаружено, что это отношение изменилось с 1/3 до 7/3. После удаления растворителя выпариванием, полученные кристаллы перекристаллизовывали из четыреххлористого углерода до получения 1,5 г II-А изомера. Пример 10 Синтез II-В изомера 1-(4-хлорфенил)-2-(1,2,4триазол-1-ил)-1-гептан-3-она (Соединение № 22) по способу D К смеси 2-гексанона (50 г) и метанола (300 мл) добавили 80 г брома при температуре 0°С, и полученную смесь выдерживали при 10°С в течение 2 часов. К этому добавили 200 мл воды и 50 г концентрированной серной кислоты, и после перемешивания в течение 16 часов добавили еще 500 мл воды. Реакционную смесь перенесли в делительную воронку и экстрагировали 500 мл эфира. Органический слой промыли 5% водным раствором карбоната калия и высушили над хлори (А) Синтез 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-2-пропен-1-она (Соединение № 29') по способу С 1-(1-метилциклопропил)-2-(1,2,4-триазол-1ил)этан-1-он (10 г, 0,06 моля), 4-хлорбензальдегид (9 г, 0,06 моля), безводный карбонат калия (8 г, 0.06 моля) и уксусный ангидрид (100 мл) смешали, и полученную смесь нагревали до 100°С в течение 6 часов при перемешивании. Образовавшийся в реакции осадок удалили фильтрованием, и полученный фильтрат сконцентрировали при пониженном давлении до получения маслянистого продукта. Этот маслянистый продукт экстрагировали хлороформом (300 мл), и полученный экстракт промыли водой, насыщенной бикарбонатом натрия (300 мл). Органический слой высушили над безводным сульфатом натрия и сконцентрировали при пониженном давлении. Одну каплю полученного остатка растворили в ацетоне, и ацетоновый раствор подвергли газовой хроматографии в условиях, описанных далее. Пик, соответствующий ІІ-А изомеру, как было обнаружено, имеет время удерживания 250 сек, а соответствующий пик ІІ-В изомера характеризуется временем удерживания 300 сек. Отношение обоих изомеров составило 19,1/63,5, то есть около 1/3, что было рассчитано из процентных соотношений каждой из площадей. Условия для газовой хроматографии были следующими: Аппаратура: Газовый хроматограф Ниппон Денши 20К, снабженный детектором FID Колонка: Стеклянная колонка длиной 1 м Жидкая фаза 5% ХЕ-60 Носитель Хромосорб W Температура (колонки): 181°С Температура (ввода): 240°С Газ-носитель: газообразный азот, 1кг/см2. Полученный остаток растворили в бензоле (100 мл). Полученный раствор пропустили через колонку, заполненную силикагелем (300 г) с размерами зерна 100-200 мешей, и хроматографирование провели смесью n-гексан/ацетон (10:1) в качестве проявляющего растворителя. Фракции, соответствующие каждому из изомеров, перекристаллизовали из четыреххлористого углерода до разделения двух указанных геометрических изомеров. ЯМР спектры каждого из изомеров приведены в табл. 2. II-А изомер: 1,7 г (выход 10%) II-В изомер: 6,7 г (выход 38%). (В) Синтез исходного материала, 1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-этан-1-она К смеси метил 1-метилциклопропилкетона (28 г, хорошо известное соединение, описанное в Bull. Soc. Chim. Fr., 1708 (1960)), хлората калия (5,8 г) и воды (70 мл) добавили 28 г брома при температуре от 40 до 50°С за 4 часа при интенсивном перемешивании, а затем реакционный раствор перемешивали в течение 2 часов при комнатной температуре. После этого реакционный раствор экстрагировали двумя порциями эфира по 200 мл, а затем органический слой высушили над хлористым кальцием и сконцентрировали при пониженном давлении до получения 53 г неочищенного продукта 1-(1-метилциклопропил)-1бромэтан-1-она. 9 29649 стым кальцием. Затем растворитель удалили при пониженном давлении до получения 89 г неочищенного 1-бром-гексанола в виде маслянистого продукта. Смесь триазола (35 г), безводного карбоната калия (69 г) и ацетонитрила (300 мл) кипятили с обратным холодильником в течение 1 часа и оставили охлаждаться до 50°С. Неочищенный 1бром-2-гексанон (89 г), полученный ранее, по каплям добавляли к полученной смеси, которую затем перемешивали при комнатной температуре в течение 16 часов. Осадок в реакционном растворе удалили фильтрованием, и растворитель удалили при пониженном давлении. К полученному таким образом остатку добавили 200 мл воды и 200 мл хлороформа, и полученную смесь перенесли в делительную воронку, после чего экстрагировали. Органический слой высушили над безводным сульфатом магния, растворитель удалили при пониженном давлении до получения 77 г неочищенного 1-(1,2,4-триазолил)-2-гексанон (20 г), безводный карбонат калия (20 г), парахлорбензальдегид (20 г) и уксусный ангидрид (200 мл) перемешивали и нагревали при 90°С в течение 5 часов. Затем реакционную смесь сконцентрировали при пониженном давлении, полученный остаток растворили в этилацетате (500 мл) и переместили в делительную воронку. Раствор этилацетата промыли водой, насыщенной карбонатом калия (200 мл), и выделили органический слой. Затем растворитель удалили из органического слоя при пониженном давлении, остаток ввели в колонку с силикагелем (0,5 кг силикагеля с размером 100-200 мешей) и провели хроматографирование, используя смесь n-гексан/ацетон (10:1) в качестве проявляющего растворителя. Таким образом, получили 3,7 г ІІ-В изомера (т. плавления 117-120°С) 1-(4-хлорфенил)-2-(1,2,4-триазол-1-ил)-1-гептен-3-она и 9 г 1(4-хлорфенил)-2-(1,2,4-триазол-1-ил)-3-ацетокси1,3-гептадиена (т. пл. 112-113°С). К полученному 1-(4-хлорфенил)-2-(1,2,4-триазол-1-ил)-3-ацетокси-1,3-гептадиену (9 г) добавили концентрированную соляную кислоту (100 мл), и полученную смесь нагревали при температуре 50°С в течение 2 часов, после чего ее вылили в ледяную воду (500 мл). Водный раствор нейтрализовали карбонатом калия и экстрагировали этилацетатом (300 мл). Органический слой высушили над безводным сульфатом магния, и растворитель удалили выпариванием. Кристаллический остаток перекристаллизовали из смеси четыреххлористый углерод/н-гексан (1:1), в результате чего получили 6 г II-В изомера 1-(4-хлофенил)-2(1,2,4-триазол-1-ил)-гептан-3-она. Пример 11 Синтез 1-А изомера 1-(2,4-дихлорфенил)-2(1,2,4-триазол-1-ил)-4,4-диметил-1-пентен-3-ола (Соединение № 2) Первая стадия (конденсация) Способ D Смесь a-(1,2,4-триазол-1-ил)-пинаколона (200 г), 2,4-дихлорбензальдегида (220 г) и уксусного ангидрида (700 см3) нагревали до 50°С, и к этому добавили 255 г триэтиламина. В течение 7 часов выдерживали температуру 70°С, после чего уксусный ангидрид удалили при пониженном давлении. К остатку добавили 3 литра воды, и образовавшиеся кристаллы отфильтровали, промыли водой и высушили. Полученный неочищенный продукт перекристаллизовали из этанола (600 см3) до получения 304 г ІІ-В изомера 1-(2,4дихлорфенил)-2-(1,2,4-триазол-1-ил)-4,4-диметил1-пентен-3-она (Соединение № 2'). Вторая стадия (фотоизомеризация) Способ С ІІ-В изомер (300 г) Соединения № 2', полученного на первой стадии, растворили в 2 литрах ацетона и подвергли изомеризации при 30°С в течение 26 часов под действием источника ультрафиолетового излучения, снабженного 500 Вт ртутной лампой высокого давления. Затем растворитель удалили при пониженном давлении до получения 300 г масляного продукта. С помощью газовой хроматографии было найдено, что этот продукт представляет собой смесь, состоящую на 75% из ІІ-А изомера соединения № 2 и на 25% из ІІ-В изомера того же соединения. Этот продукт передали на следующую стадию, не разделяя изомеры. Третья стадия (восстановление) Способ А 300 г смеси геометрических изомеров Соединения № 2', полученного на второй стадии, суспендировали в метаноле (1 кг), и к этому добавили боргидрид натрия (38 г) по частям, охлаждая реакционную смесь до 10°С. После перемешивания при комнатной температуре в течение 1 часа реакционный раствор сконцентрировали при пониженном давлении. Полученный осадок экстрагировали, добавив 10% водный раствор уксусной кислоты (2 литра) и этилацетат (3 литра). Выделенный органический слой промыли 5% водным раствором карбоната калия (1 литр) и высушили над безводным сульфатом магния (100 г). После удаления фильтрованием высушивающего агента растворитель удалили при пониженном давлении, в результате чего получили 280 г неочищенного продукта в виде кристаллов. Этот продукт представлял собой смесь I-А и І-В изомеров Соединения № 2 (отношение смеси: І-А/І-В=75/25). Неочищенный продукт (280 г) перекристаллизовали из четыреххлористого углерода (600 см3), в результате чего получили 209 г указанного в заглавии соединения (I-А изомер Соединения № 2). Маточный раствор от перекристаллизации сконцентрировали до половинного объема, в результате чего получили 25 г І-В изомера Соединения № 2 в виде вторичных кристаллов. ІІ-А изомеры кетонного соединения (II), полученные способами С и D, приведены в табл. 2 наряду с ІІ-В изомерами. ЯМР спектры в таблице в таком же виде, что и в табл. 1. Согласно изобретению на поверхности растения или почвы, в которой растут или будут выращиваться растения создают защитный слой, содержащий описанные выше соединения триазола, представленного упомянутой формулой (І). Величина дозирования составляет от 0,02 до 5 кг на гектар. Защитный слой формируют в виде пленки на поверхности растения или почвы или формируют путем нанесения микрогранул на поверхность растения. При практическом применении соединений настоящего изобретения, полученных таким образом, они могут использоваться сами по себе без других компонентов или в смеси с носителями для 10 29649 облегчения их применения в качестве фунгицидов, гербицидов и регуляторов роста растений. Обычно используемые препараты включают, например, дусты, смачиваемые порошки, масляные распылительные рецептуры, эмульгируемые концентраты, таблетки, гранулы, мелкие гранулы, аэрозоли и способные к истечению препараты. Описанные выше препараты обычно содержат 0,1-95,0% вес активного ингредиента (включая другие ингредиенты смеси). Подходящее количество применяемого активного ингредиента обычно составляет 2-500 г на 10 ар, а концентрация применяемого активного ингредиента, предпочтительно, лежит в интервале 0,001-1,0%. Однако, поскольку количество и концентрация зависят от формы препарата, времени применения, устройств для применения, заболеваний культур и их природы, эти значения могут быть уменьшены или увеличены относительно указанных интервалов. При получении фунгицида, гербицида и регулятора роста растений по настоящему изобретению, примешиваются подходящие твердые носители или жидкие носители. В качестве примеров твердых носителей можно указать, например, неорганические соединения (например, глины каолиновой группы, монтморилонитной группы и аттапульгитной группы, тальк, слюда, пирофиллит, пемза, вермикуллит, гипс, карбонат кальция, доломит, диатомовая земля, известь, апатит, цеолит, ангидрид кремниевой кислоты, синтетический силикат кальция), растительные органические соединения (например, порошкообразная соя, табак, порошкообразный грецкий орех, мука, порошкообразная древесина, крахмал, кристаллическая целлюлоза), синтетические или природные высокомолекулярные соединения (например, кумапроновые смолы, петролейные смолы, алкидные смолы, поливинил хлорид, полиалкилен гликоль, кетонные смолы, эфирные смолы, копал, дамаровые смолы), а также воски (например, карнубский воск, пчелиный воск) и мочевина. В качестве примеров жидких носителей можно привести парафиновые или нафтеновые углеводороды (например, керосин, минеральное масло, белое масло), ароматические углеводороды (например, бензол, толуол, ксилол, этилбензол, кумол, метилнафталин), галогенированные углеводороды (например, четыреххлористый углерод, хлороформ, трихлорэтилен, монохлорбензол, о-хлортолуол), эфиры (например, диоксан, тетрагидрофуран), кетоны (например, ацетон, метил этил кетон, диизобутил кетон, циклогексан, ацетофенон, изофорон), сложные эфиры (например, этил ацетат, амил ацетат, ацетат этилен гликоля, ацетат диэтилен гликоля, дибутил малеат, диэтил сукцинат), спирты (например, метанол, н-гексанол, этилен гликоль, диэтилен гликоль, циклогексанол, бензиловый спирт), простые эфиры спиртов (например, этиловый эфир этилен гликоля, фениловый эфир этилен гликоля, этиловый эфир диэтилен гликоля, бутиловый эфир диэтилен гликоля), полярные растворители (например, диметилформамид, диметилсульфоксид) и вода. Поскольку поверхностно-активные вещества применяют для эмульгирования, диспергирования, смачивания, распределения по поверхности, связывания, регулирования дизинтеграции, ста билизации активного ингредиента, улучшение текучести и антикоррозитности, любое из неионного, анионного, катионного и амфотерного поверхностно-активного вещества может быть использовано до значения HLB Rrs от 8 до 18, но обычно используют неионные и/или анионные вещества. В качестве подходящих неионных поверхностноактивных агентов могут использоваться соединения, полученные полимеризацией окиси этилена и высшего спирта (например, лаурилового спирта, стеарилового спирта, олеилового спирта), окиси этилена и алкилфенола (например, изооксилфенола, нонилфенола), окиси этилена и алкилнафтола (например, бутилнафтола, октилнафтола), окиси этилена и высшей жирной кислоты (например, пальмитиновой кислоты, стеариновой кислоты, олеиновой кислоты), окиси этилена и моноили ди-алкил фосфата (например, стеарил фосфата, дилаурил фосфата) или окиси этилена и амина (например, додециламина, амида стеариновой кислоты), эфиры высших жирных кислот и полиатомного спирта (например, сорбитан), а также соединения, полученные полимеризацией указанных эфиров и окиси этилена, а также полимеры на основе окиси этилена и окиси пропилена. Подходящими анионными поверхностно-активными агентами могут служить соли алкил сульфата (например, натрий лаурил сульфат, аминовые соли олеил сульфата), алкилсульфонаты (например, натриевая соль диоксил сульфосукцината, натрий 2-этилгексенсульфонат), а также арилсульфонаты (например, натрий изопропилнафталинсульфонат, натрий метиленбиснафталинсульфонат, натрий лигносульфонат, натрий додецилбензолсульфонат). Кроме того, микротонкие гранулы предложенного соединения могут содержать высокомолекулярные соединения и другие вспомогательные агенты с целью усиления технологических характеристик и биологической активности. Такие высокомолекулярные соединения включают, например, казеин, желатин, альбумин, клей, альгинат натрия, карбоксиметил целлюлозу, метил целлюлозу, гидроксиэтил целлюлозу и поливиниловый спирт. Указанные выше носители и вспомогательные агенты обычно применяются сами по себе или в комбинации в соответствии с предполагаемым использованием, принимая во внимание формы препарата и технику применения. Содержание активного ингредиента в дустах обычно составляет 1-25% вес., а остаток составляет твердый носитель. Что касается смачиваемых порошков, то содержание в них активного ингредиента обычно составляет 25-90 вес. частей. Остаток представляет собой твердый носитель и дисперсионно-смачивающий агент и, если необходимо, защитный коллоид, тиксотропный агент и антипенный агент добавляют в композицию. В гранулах содержание активного ингредиента обычно составляет 1-35 вес. Частей, и большую часть остатка составляет твердый носитель. Активный ингредиент однородно смешивают с твердым носителем или его однородно фиксируют или адсорбируют на поверхности твердого носителя. Такие частицы имеют диаметр 0,2-1,5 мм. 11 29649 метиленурацил, гербициды ряда пиридиновых солей, такие как 1,1'-диметил-4,4'-бипиридиний хлорид, фосфорсодержащие гербициды, такие как N(фосфонометил)глицин, N,N-бис(фосфонометил)глицин, 0-этил 0-(2-нитро-5-метилфенил) Nсек-бутил фосфороамидотиоат S-(2-метил-1-пиперидилкарбонилметил) 0,0-ди-н-пропилдимиофосфат и S-(2-метил-1-пиперидилкарбонилметил) 0,0дифенилтиофосфат, гербициды толуидиновой серии, такие как a,a,a-трифтор-2,6-динитро-N,Nдипропил-п-толуидин, 5-трет. бутил-3-(2,4-дихлор5-изопропоксифенил)-1,3,4-оксадиозолин-2-он, 3изопропил-(1Н)-2,1,3-бензотиадиазин-(ЗН)-он-2,2диоксид, a-(b-нафтокси)пропионанилид, 4-(2,4-дихлорбензоил)-1,3-диметилпиразол-5-ил п-толуолсульфонат, 3-(метоксикарбониламино)фенил-3метилфенилкарбамат, 4-амино-3-метил-6-фенил1,2,4-триазин и т. д. Соединения настоящего изобретения могут применяться в смеси с другими инсектицидами, что не понижает регулирующего эффекта каждого активного ингредиента смеси. В качестве таких инсектицидов могут использоваться фосфор-органические инсектициды, такие как 0,0-диметил-0(4-нитро-3-метилфенил)фосфортиоат, 0-(4-цианофенил) 0,0-диметилфосфоротиоат, 0-(4-цианофенил) 0-этилфенилфосфонотиоат, 0,0-диметил S(N-метилкарбамоилметил)фосфородитиоат, 2-метокси-4Н-1,3,2-бензодиоксафосфорин-2-сульфид и 0,0-диметил S-(1-этоксикарбонил-1-фенилметил)фосфородитиоат, и такие инсектициды пиретроидной серии, как a-циано-3-феноксибензил-2-(4хлорфенил)изовалерат, 3-феноксибензил 2,2-диметил-3-(2,2-дихлорвинил)циклопропанкарбоксилат и a-цино-З-феноксибензил 2,2-диметил-3-(2,2дибромвинил)циклопропанкарбоксилат. Следовательно, в одно и то же время могут контролироваться одно или более заболеваний и насекомых и, кроме этого, можно ожидать синергетического эффекта из-за смешивания. Далее полезность соединений настоящего изобретения в качестве фунгицидов, гербицидов и регуляторов роста растений для сельского хозяйства и садоводства будет более подробно проиллюстрирована на предоставленных ниже испытательных примерах и примерах получения. Испытательный пример 1 Фунгицидный эффект Среда, содержащая 5 г полипептона, 20 г солодового экстракта, 20 г сахарозы и 20 г агарагара на 1 литр воды была превращена в раствор нагреванием. Водный разбавленный раствор эмульгируемого концентрата каждого испытуемого концентрата был добавлен к нему так, что концентрация испытуемого состава в этой среде была предопределена. После тщательного перемешивания среды эта среда была разлита в стеклянную чашку Петри, чтобы сделать пластину агарагара. После того, как агар-агар затвердел, он был инокулирован мицелиевой структурой или споровой суспензией испытуемого грибка. Название испытуемого грибка и период выращивания от изенокуляции до наблюдения будут показаны ниже. Температура выращивания была 20°С для Venturia inaequalis и 28°С для других грибков. Что касается эмульгируемых концентратов, то содержание активного ингредиента в них составляет обычно 5-30 вес. частей, причем эмульгатор занимает 5-20 вес. частей, а остаток представляет собой жидкий носитель. Если необходимо, добавляют противоплесневые агенты. Кроме этого, соединения настоящего изобретения могут использоваться в смеси с другими фунгицидами, гербицидами и регуляторами роста растений без уменьшения регулирующего эффекта каждого активного ингредиента смеси. В качестве фунгицидов могут использоваться N-(3,5дихлорфенил)-1,2-диметилциклопропан-1,2-дикарбоксимид, S-н-бутил S-п-трет.-бутилбензилдитиокарбонимидат, 0,0-диметил 0-(2,6-дихлор-4-метилфенил)фосфортиоат, метил-1-бутилкарбамоил-1H-бензилмидазол-2-ил-карбамат, N-трихлорметио-4-циклогексен-1,2-дикарбоксимид, цис-N(1,1,2,2-тетрахлорэтилтио)-4-циклогексен-1,2-дикарбоксимид, полиоксин, стрептомицин, цинк этилен-бис(дитиокарбамат), цинк диметилтиокарбамат, марганец этилен-бис(дитиокарбамат), бис(N,N-диметилтиокарбамоил)дисульфид, тетрахлоризофталонитрил, 8-гидроксихинолин, додецилгуанидин ацетат, 5,6-дигидро-2-метил-1,4-оксатиин-3-карбоксанилид, N'-дихлорфторметилтиоN,N-диметил-N'-фенилсульфамид, 1-(4-хлорфенокси)-3;3-диметил-1-(1,2,4-триазол-1-ил)-2-бутаон, 1,2-бис(3-метоксикарбонил-2-тиоуреидо), бензол, метил N-(2,6-диметилфенил)-N-метоксиацетил-2метилглюцинат, алюминий этилфосфит и т. д. В качестве гербицидов могут использоваться также гербициды фенокси-ряда, как 2,4-дихлорфеноксиуксусная кислота, 2-метил-4-хлорфеноксиуксусная кислота, 2-метил-4-хлорфеноксимасляная кислота и 2-метил-4-хлорфеноксиуксусная кислота (включая сложные эфиры и соли), гербициды серии дифениловых эфиров, такие как 2,4дихлорфенил-4-нитрофениловый эфир, 2,4,6-трихлорфенил 4'-нитрофениловый эфир, 2-хлор-4трифторметилфенил, 3'-этокси-4'-нитрофениловый эфир, 2,4-дихлорфенил, 4'-нитpo-3'-мeтoкcифeнилoвый эфир и 2,4-дихлорфенил 3'-метоксикарбонил-4'-нитрофениловый эфир, гербициды триазиновой серии, такие как 2-хлор-4,6-бисэтиламино-1,3,5-триазин, 2-хлор-4-этиламино-6-изопропиламино-1,3,5-триазин, 2-метилтио-4,6-бисэтиламино-1,3,5-триазин и 2-метилтио-4,6-бисизопропиламино-1,3,5-триазин, гербициды мочевинной серии, такие как 3-(3,4-дихлорфенил)-1,1диметилмочевина, 3-(3,4-дихлорфенил)-1-метокси-1-метилмочевина, 1-(a,a,-диметилбензил)-3-птолилмочевина и 1-(2-безотиазолил)-1,3-диметилмочевина, гербициды карбаматной серии, такие как изопропил N-(3-хлорфенил)-карбамат и метил N-(3,4-дихлорфенил)карбамат, гербициды тиокарбаматной серии такие как S-(4-хлорфензил)N,Nдиэтилтиокарбамат и S-этил N,N-гексаметиленэтиолкарбамат, гербициды анилидной серии, такие как 3,4-дихлорпропионанилид, 2-xлop-N-мeтoкcимeтил-2',6'-диэтилицeтaнилид, 2-xлop-2',6'-диэтил-N-(бутоксиметил)ацетанилид, 2-xлop-2',6'-диэтил-N-(н-пропоксиэтил)ацетанилид и N-хлорацетил-N-(2,6-диэтилфенил)глицин этиловый эфир, гербициды урацильной серии, такие как 5-бром-3сек-бутил-6-метилурацил и 3-циклогексил-5,6-три 12 29649 Наименование грибка Сокра- Период щенное выращинаимевания нование обработали капусту в соотношении 30 кг/га. Эти поверхностно обработанные микрогранулы становятся липкими и остались на прежнем месте на поверхности листьев. Защитное действие сохранялось в течение по крайней мере 30 дней после обработки. Пример состава 1 (Смачиваемый порошок) Тридцать частей I-А изомера каждого из представленных соединений 1-52, 45 частей диатомовой почвы, 20 частей белой сажи, 3 части смачивающего агента, имеющего HLB значение от 8 до 18, и 2 части диспергирующего агента (лигносульфонат кальция) хорошо перемешали до порошкообразного состояния, чтобы получить смачиваемый порошок, содержащий 30% активного ингредиента. Пример состава 2 (Микротонкие гранулы) Один килограмм пента(тетра)глицерида стеариновой кислоты нагрели до 85°С для плавления в реакционном сосуде, 30 г каждого І-А изомера представленного триазольного соединения, приведенного в табл. 1, были добавлены туда при помешивании. После получения гомогенной смеси, 200 г Карбпола № 934 (производимого фирмой the В. F. Goodrich Co., Ltd.) добавили в дальнейшем при помешивании в течение 15 минут, выдерживая при той же самой температуре. Полученный состав был скраплен капля за каплей на алюминиевый диск, вращающийся со скоростью 15000 об/мин, чтобы получить микротонкие гранулы, имеющие размер от 30 до 80 мешей. Helminthosporium gramineum Hg 6 дней Penicillium italicum Pi 6 дней Venturia inaequalis Vi 7 дней Valsa mali Vm 4 дня Micosphaerella melonis Mm 4 дня Diaporthe citri Dс 6 дней Ustilago nuda Un 6 дней Verticillium albo-atrum Va 7 дней Septoria tritici St 7 дней Cercospora beticola Cb 7 дней Fusarium oxisporium f. sp. lycopersici Fo 4 дня Alternaria kikuchiana Ac 4 дня Степень торможения роста испытуемых соединений была определена в четырех рейтингах А, В, С и D: А Степень торможения роста 100% 90% или больше B C 89-50% D 40% или меньше Как показано в табл. 3, было определено, что I-А изомер предложенного изобретения имеет явно широкий антимикробный спектр, так же как и необыкновенно высокую активность по сравнению с І--В, II-А, ІІ-В изомерами. Испытательный пример 2 Микротонкими гранулами, полученными в соответствии с примером состава 2, поверхностно 13 Таблица 1 29649 14 Продолжение табл. 1 29649 15 Продолжение табл. 1 29649 16 Продолжение табл. 1 29649 17 Продолжение табл. 1 29649 18 Продолжение табл. 1 29649 19 Продолжение табл. 1 29649 20 Продолжение табл. 1 29649 21 Продолжение табл. 1 29649 22 Продолжение табл. 1 29649 23 Продолжение табл. 1 29649 24 Продолжение табл. 1 29649 25 Продолжение табл. 1 29649 26 Продолжение табл. 1 29649 27 Продолжение табл. 1 29649 28 Продолжение табл. 1 29649 29 Продолжение табл. 1 29649 30
ДивитисяДодаткова інформація
Назва патенту англійськоюA method for protection of plants against phytopathogens
Автори англійськоюYugi Funaki, Hirofumi Oshita, Shigeo Yamamoto, Shizuya Tanaka, Toshiro Kato
Назва патенту російськоюСпособ защиты растений от фитопатогенов
Автори російськоюЮджи Фунаки, Хирофуми Ошита, Шигео Ямамото, Шизуя Танака, Тоширо Като
МПК / Мітки
МПК: A01P 7/04, C07D 311/52, A01N 43/56
Мітки: фітопатогенів, захисту, рослин, спосіб
Код посилання
<a href="https://ua.patents.su/52-29649-sposib-zakhistu-roslin-vid-fitopatogeniv.html" target="_blank" rel="follow" title="База патентів України">Спосіб захисту рослин від фітопатогенів</a>
Спосіб одержання твердого препарату для захисту культурних рослин і твердий препарат для захисту культурних рослин

Номер патенту: 27522
Опубліковано: 15.09.2000
Автори: Уедлок Девід Джон, де Лінд ван Вейнгарден Герхард
МПК: A01N 53/00, A01N 25/12, A01N 25/14, A01N 25/10, A01N 25/34
Мітки: культурних, препарату, твердого, твердий, рослин, одержання, препарат, захисту, спосіб
Текст:
..., где после растворения водой требуе тся (и предпочти тельно) быстрое ди спергиро вание . В па тен те США 4801460 вкратце указывае тся , что процесс экструзи и может быть также адап тиро ван и для по 27522 лучения препар а то в с бы стрым высвобождением активного ингредиента; однако, в указанной работе рассматривается лишь возможность изменения типа и количества сомономеров. используемых для получения поливинилпирролидонового полимера...
Спосіб захисту рослин кукурудзи від фітотоксичної дії хлорацетанілідних гербицидів

Номер патенту: 4907
Опубліковано: 28.12.1994
Автор: Ханс Мозер
Мітки: дії, рослин, хлорацетанілідних, спосіб, гербіцидів, фітотоксичної, кукурудзи, захисту
Формула / Реферат:
Формула изобретенияСпособ защиты растений кукурузы от фитотоксического действия хлорацетанилидиных гербицидов формулы где R - метил, или этил, путем обработки почвы после посева антидотом, отличающийся тем, что, с целью повышения степени защиты, в качестве антидота используют 4-дихлорацетил-2,3-дигидро-3-метил-1,4-бензоксазин в дозе 0,25-1,5 кг/га.
Спосіб захисту рослин проса, кукурудзи, рису від ушкодження гербицидами класу ацетанілідів

Номер патенту: 4905
Опубліковано: 28.12.1994
Автори: Ханс-Георг Бруннер, Курт Бурдеска, Гуглієльмо Кабас, Вернер Фьорі
МПК: C07D 239/52, A01N 25/32, C07F 9/6512, C07D 239/42, C07D 239/47, C07D 239/30
Мітки: гербицидами, класу, спосіб, проса, рису, ацетанілідів, ушкодження, рослин, кукурудзи, захисту
Формула / Реферат:
Способ защиты растений проса, кукурузы, риса от повреждения гербицидами класса ацетанилидов, включающий использование антидота, отличающийся тем, что, с целью повышения эффективности защиты, в качестве антидота используют соединение общей формулыгде при R1 и R2 - хлор каждый и n = 1 R - водород, 4-метил, 4-хлор, 4-метоксил, а при n = 2R-...
Спосіб захисту від ураження культурних рослин фітопатогенними мікроорганізмами з групи fungi imperfecti

Номер патенту: 12327
Опубліковано: 25.12.1996
Автори: Роберт Ніфелер, Хайде Д Мен, Роберт Джон Віл'ямс, Теодор Штауб
МПК: A01N 43/36
Мітки: фітопатогенними, ураження, imperfecti, fungi, рослин, культурних, мікроорганізмами, групи, захисту, спосіб
Формула / Реферат:
(57) Способ защиты от поражения культурных растений фитопатогенными микроорганизмами из группы Fungi imperfecti, включающий обработку растений или посевного материала препаратом биологически активного вещества на носителе, отличающийся тем, что в качестве биологически активного вещества используют 3-циано-4-(2,3-дихлорфенил)-пиррол, причем обработку растений проводят в дозе 0,05-5 кг активного вещества на 1 га.
Фунгіцидна композиція для захисту рослин

Номер патенту: 25956
Опубліковано: 26.02.1999
Автори: Шандор Анд'яр, Тамаш Детре, Хорст Лір, Андраш Сєго, Марлієш Штрумр, Каталін Мармарошіне Келлнер, Ференц Віран'ї, Д'юла Орош, Дітер Цанке, Йожеф Шош, Дьондьвер Надьне Хед'ї, Бріта Леннер, Тібор Ершек, Лайош Рейте, Аттіла Молнар, Ласло Хорнок, Ержебет Шюслер
МПК: A01N 47/18, A01N 43/78, A01N 43/84, A01N 47/38, A01N 43/76, A01P 3/00, A01N 37/44, A01N 43/80, A01N 47/34
Мітки: фунгіцидна, композиція, рослин, захисту
Формула / Реферат:
Фунгицидная композиция для защиты растений в форме смачиваемого порошка или эмульгируемого концентрата, включающая активный ингредиент - смесь биологически активных соединений, и целевые добавки, отличающаяся тем, что в качестве смеси биологически активных соединений она содержит смесь следующих компонентов:а - компонент, выбранный из группы фунгицидов: металаксил, беналаксил, оксадиксил, ZAB 149202 F,б - фунгицид из группы...
Попередній патент: Спосіб лікування хімічних опіків стравоходу
Наступний патент: Електричний з’єднувач
Випадковий патент: Спосіб сухого гасіння коксу