Спосіб одержання піразолу і його похідних





Формула / Реферат
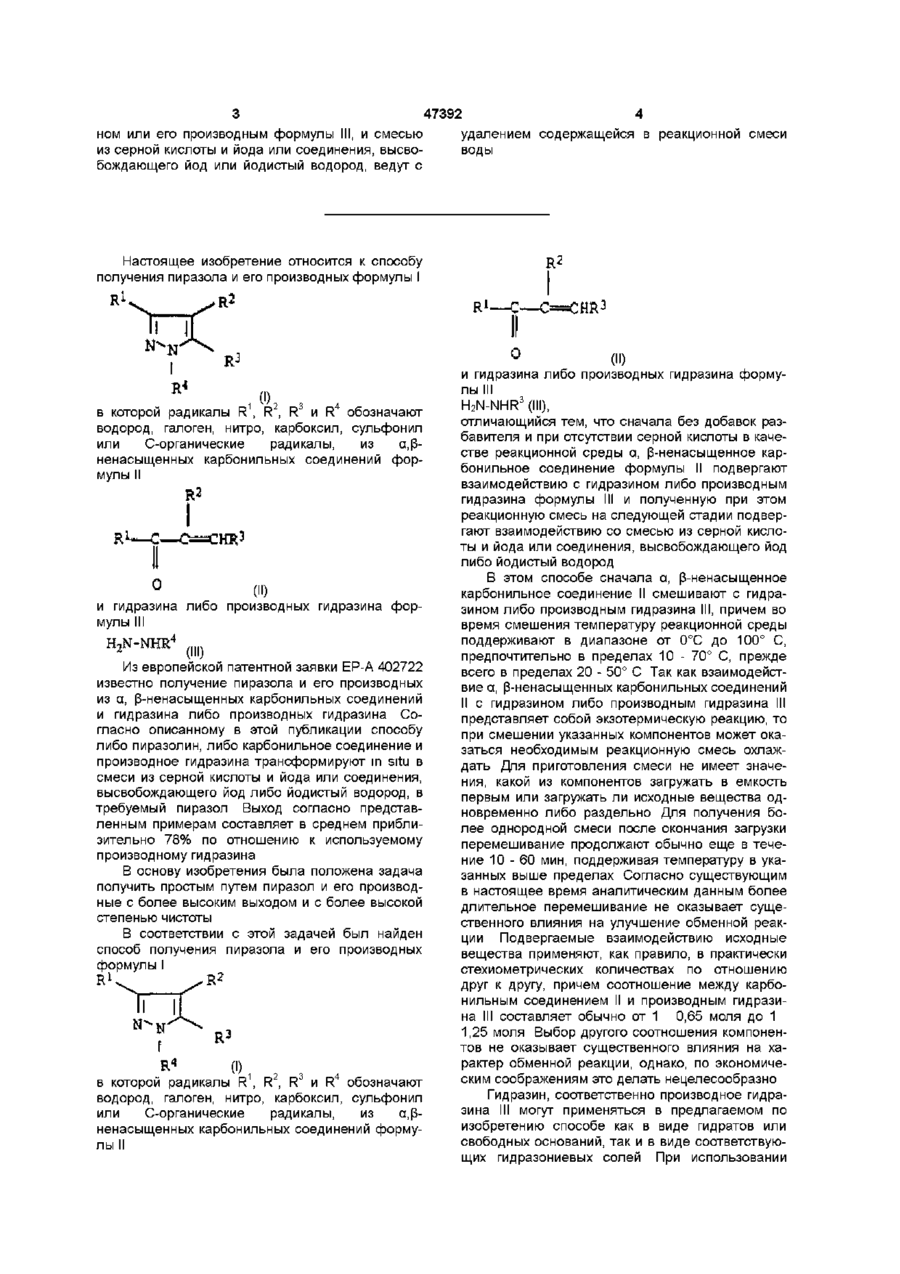
1. Способ получения пиразола и его производных формулы (І)
(I),
в которой радикалы R1 - R4 обозначают водород или алкильную группу, линейную или разветвленную, предпочтительно С1-С10-алкильную группу, более предпочтительно С1-С8-алкильную группу и наиболее предпочтительно С1-С6-алкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, циклоалкил, бициклоалкил, арил и гетероарил; циклоалкильную группу или бициклоалкильную группу, предпочтительно С3-С8-циклоалкильную группу или С6-С10-бициклоалкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил; арильную или гетероарильную группу, такую как фенил, нафтил и пиридил, причем эти группы могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил;
из -ненасыщенного карбонильного соединения формулы (II)
(II),
где R1-R3 имеют указанные значения, и гидразина или его производных формулы (III)
H2N-NHR4 (ІІІ),
где R4 имеет указанные значения,
с использованием серной кислоты и йода или соединения, высвобождающего йод или йодистый водород, отличающийся тем, что сначала ведут взаимодействие -ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III без добавления растворителя и в отсутствие серной кислоты и полученную при этом реакционную смесь на последующей стадии подвергают взаимодействию со смесью из серной кислоты и йода или соединения, высвобождающего йод или йодистый водород.
2. Способ по п. 1, отличающийся тем, что взаимодействие -ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III осуществляют при 0-100 °С.
3. Способ по п. 1, отличающийся тем, что используют 40-99 мас. %-ную серную кислоту.
4. Способ по п. 1, отличающийся тем, что йод или соединение, высвобождающее йод или йодистый водород, используют в количестве 0,01-10,0 мол. % по отношению к гидразину или его производному формулы III.
5. Способ по п. 1, отличающийся тем, что реакционную смесь, полученную взаимодействием -ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III, подвергают взаимодействию со смесью из серной кислоты и йода или соединения, высвобождающего йод или йодистый водород, при 50-250 °С.
6. Способ по п. 1, отличающийся тем, что стадию взаимодействия между реакционной смесью, полученной взаимодействием -ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III, и смесью из серной кислоты и йода или соединения, высвобождающего йод или йодистый водород, ведут с удалением содержащейся в реакционной смеси воды.
Текст
1 Способ получения пиразола и его производных формулы (I) 1()' 4 в которой радикалы R - R обозначают водород или алкильную группу, линейную или разветвленную, предпочтительно СгСю-алкильную группу, более предпочтительно Ci-Cs-алкильную группу и наиболее предпочтительно Сі-Сє-алкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, циклоалкил, бициклоалкил, арил и гетероарил, циклоалкильную группу или би циклоалкил ьную группу, предпочтительно Сз-Csциклоалкильную группу или Сб-Сюбициклоалкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил, арильную или гетероарильную группу, такую как фенил, нафтил и пиридил, причем эти группы могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил, из а, р -ненасыщенного карбонильного соедине ния формулы где R1-R3 имеют указанные значения, и гидразина или его производных формулы (III) H 2 N-NHR i (lll), где R4 имеет указанные значения, с использованием серной кислоты и йода или соединения, высвобождающего йод или йодистый водород, отличающийся тем, что сначала ведут взаимодействие а, р-ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III без добавления растворителя и в отсутствие серной кислоты и полученную при этом реакционную смесь на последующей стадии подвергают взаимодействию со смесью из серной кислоты и йода или соединения, высвобождающего йод или йодистый водород 2 Способ по п 1, отличающийся тем, что взаимодействие а, р -ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III осуществляют при 0-100 °С 3 Способ по п 1, отличающийся тем, что используют 40-99 мае %-ную серную кислоту 4 Способ по п 1, отличающийся тем, что йод или соединение, высвобождающее йод или йодистый водород, используют в количестве 0,01-10,0 мол % по отношению к гидразину или его производному формулы III 5 Способ по п 1, отличающийся тем, что реакционную смесь, полученную взаимодействием а, р-ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III, подвергают взаимодействию со смесью из серной кислоты и йода или соединения, высвобождающего йод или йодистый водород, при 50-250 °С 6 Способ по п 1, отличающийся тем, что стадию взаимодействия между реакционной смесью, полученной взаимодействием а, р-ненасыщенного карбонильного соединения формулы II с гидрази О го З 47392 ном или его производным формулы III, и смесью удалением содержащейся в реакционной смеси из серной кислоты и йода или соединения, высвоводы бождающего йод или йодистый водород, ведут с Настоящее изобретение относится к способу получения пиразола и его производных формулы I 0 в которой радикалы R1, R2, R3 и R4 обозначают водород, галоген, нитро, карбоксил, сульфонил или С-органические радикалы, из а,рненасыщенных карбонильных соединений формулы II 0 (ID и гидразина либо производных гидразина формулы III Hл2 N-NHR 4 ,. m (III) Из европейской патентной заявки ЕР-А 402722 известно получение пиразола и его производных из а, р-ненасыщенных карбонильных соединений и гидразина либо производных гидразина Согласно описанному в этой публикации способу либо пиразолин, либо карбонильное соединение и производное гидразина трансформируют in situ в смеси из серной кислоты и йода или соединения, высвобождающего йод либо йодистый водород, в требуемый пиразол Выход согласно представленным примерам составляет в среднем приблизительно 78% по отношению к используемому производному гидразина В основу изобретения была положена задача получить простым путем пиразол и его производные с более высоким выходом и с более высокой степенью чистоты В соответствии с этой задачей был найден способ получения пиразола и его производных формулы I 1 (О в которой радикалы R1, R2, R3 и R4 обозначают водород, галоген, нитро, карбоксил, сульфонил или С-органические радикалы, из а,рненасыщенных карбонильных соединений формулы II (И) и гидразина либо производных гидразина формулы III H2N-NHR3 (III), отличающийся тем, что сначала без добавок разбавителя и при отсутствии серной кислоты в качестве реакционной среды а, р-ненасыщенное карбонильное соединение формулы II подвергают взаимодействию с гидразином либо производным гидразина формулы III и полученную при этом реакционную смесь на следующей стадии подвергают взаимодействию со смесью из серной кислоты и йода или соединения, высвобождающего йод либо йодистый водород В этом способе сначала а, р-ненасыщенное карбонильное соединение II смешивают с гидразином либо производным гидразина III, причем во время смешения температуру реакционной среды поддерживают в диапазоне от 0°С до 100° С, предпочтительно в пределах 10 - 70° С, прежде всего в пределах 20 - 50° С Так как взаимодействие а, р-ненасыщенных карбонильных соединений II с гидразином либо производным гидразина III представляет собой экзотермическую реакцию, то при смешении указанных компонентов может оказаться необходимым реакционную смесь охлаждать Для приготовления смеси не имеет значения, какой из компонентов загружать в емкость первым или загружать ли исходные вещества одновременно либо раздельно Для получения более однородной смеси после окончания загрузки перемешивание продолжают обычно еще в течение 1 0 - 6 0 мин, поддерживая температуру в указанных выше пределах Согласно существующим в настоящее время аналитическим данным более длительное перемешивание не оказывает существенного влияния на улучшение обменной реакции Подвергаемые взаимодействию исходные вещества применяют, как правило, в практически стехи о метрических количествах по отношению друг к другу, причем соотношение между карбонильным соединением II и производным гидразина III составляет обычно от 1 0,65 моля до 1 1,25 моля Выбор другого соотношения компонентов не оказывает существенного влияния на характер обменной реакции, однако, по экономическим соображениям это делать нецелесообразно Гидразин, соответственно производное гидразина III могут применяться в предлагаемом по изобретению способе как в виде гидратов или свободных оснований, так и в виде соответствующих гидразониевых солей При использовании 47392 солеи, не растворяющихся в реакционной среде, вследствие недостаточно тщательного перемешивания могут иметь место потери в выходе продукта По этой причине предпочтительное применение находят гидраты или свободные основания производных гидразина III Предлагаемый способ основан на том принципе, что сначала взаимодействием гидразинов с а, [3-ненасыщенными карбонильными соединениями образуют соответствующие пиразолины и производные продукты Последующая переработка, проводимая методом дистилляции, после удаления реакционной воды позволяет получать лишь весьма невысокий выход, поскольку наряду с пиразолинами имеются также побочные продукты, которые образуются в результате присоединения пиразолинов к исходным карбонильным соединениям и которые в свою очередь могут образовывать с гидразинами гидразоны и азины Полученную описанным выше путем реакционную смесь без последующей переработки подвергают затем взаимодействию со смесью из серной кислоты и йода либо соединения, высвобождающего йод или йодистый водород При этом работают обычно аналогично тому, как это указано в европейской заявке ЕР-А 402722, а именно смесь из серной кислоты и йода, соответственно соединения, высвобождающего йод или йодистый водород, нагревают до температуры в пределах 50 - 250° С, предпочтительно 70 - 200° С, преимущественно 100 - 180° С, и при этой температуре подвергают ее взаимодействию со смесью из первой стадии Наряду с этим возможен и другой вариант, а именно полученную на первой стадии реакционную смесь вводить при более низких температурах, например, при 10 - 30°С, в смесь из серной кислоты и йода либо соединения, высвобождающего йод или йодистый водород Однако по технологическим причинам целесообразно осуществлять это введение при более высокой температуре, так как в процессе смешения образуются соли и перемешивание обеих смесей может ухудшаться Выбор температуры для введения одной смеси в другую согласно имеющимся данным не оказывает никакого влияния на выход продукта Осуществление этой второй стадии основано, повидимому, на том же принципе, по которому проводят обменную реакцию, описанную в европейской патентной заявке ЕР-А 402722 В соответствии с этим принципом, как правило, применяют серную кислоту в концентрации не менее чем 30 масс % Обычно применяют 40 - 99 масс %-ную серную кислоту, предпочтительно 45 95 масс %-ную серную кислоту Количество используемого в указанной реакции иода, соответственно соединения, высвобождающего йод либо йодистый водород, составляет, как правило, 0,01 - 10 мол %, предпочтительно 0,05 - 5 мол %, прежде всего 0,1 - 2 мол % по отношению к гидразину, соответственно к производному гидразина III Наряду с йодом и йодистым водородом в качестве соединений, высвобождающих йод либо йодистый водород, пригодны также, например, иодиды щелочных и щелочноземельных металлов, такие, как йодид лития, йодид натрия, йодид калия, йодид цезия, йодид магния и йодид кальция, а также йодиды других металлов, в принципе могут применяться все йодистые или йодистоводородные соединения, способные в соответствующих условиях реакции высвобождать йод либо йодистый водород К ним относятся, например, такие другие неорганические йодистые соединения, как гипоиодиды, йодиты, йодаты и периодаты щелочных, щелочноземельных или других металлов, или органические йодистые соединения, например, алкилиодиды, как метилиодид Было найдено, что для дегидрирования, соответственно окисления йодида до йода оптимальная температура для проведения соответствующей реакции зависит от концентрации серной кислоты Чем ниже эта концентрация серной кислоты, тем выше температура, необходимая для осуществления реакции Поэтому для того, чтобы температуру реакции поддерживать в низком диапазоне, рекомендуется во время проведения реакции отгонять реакционную воду, равно как и воду, наличие которой обусловлено использованием гидратов Удаленная из смеси вода содержит большую часть введенного йодида в виде йода и йодистого водорода, который после восстановления, соответственно после нейтрализации, например, с помощью гидросульфита натрия, может быть возвращен После завершения реакции реакционной смеси дают остыть, и при этом производное пиразола кристаллизуется в основном в виде соли серной кислоты Для выделения пиразола реакционную смесь нейтрализуют, после чего эту нейтральную смесь экстрагируют с помощью инертного органического, не смешиваемого с водой растворителя Затем органическую фазу по обычной методике сушат и разделяют Таким путем получают сырые пиразолы со степенью чистоты порядка 85 - 90%, которую однократной перегонкой можно довести до 99% Предлагаемый согласно изобретению способ пригоден для получения пиразола и его производных общей формулы I R4 (О, в которой радикалы R1, R , R3 и R4 независимо друг от друга обозначают водород, галоген, нитро, карбоксил, сульфонил или С-органические радикалы При этом под галогеном имеются в виду в первую очередь фтор, хлор и бром В качестве соответствующих С-органических радикалов следует назвать алкильные группы, которые могут быть линейными либо разветвленными, например, С-і-С-ю алкил, предпочтительно С-і-Сз алкил, прежде всего С-і-Сб алкил, причем эти радикалы в свою оче 47392 редь могут быть разорваны гетероатомами, такими, как азот, кислород и сера, и могут нести заместители из следующей группы нитро, карбоксил, сульфонил, галоген, циклоалкил, бициклоалкил, арил и гетарил, циклоалкильные группы либо бициклоалкильные группы, например, Сз-Cs циклоалкил либо СбС1 бициклоалкил, причем эти радикалы в свою -0 очередь могут быть разорваны гетероатомами, такими, как азот, кислород и сера, и могут нести заместители из следующей группы нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетарил, арильные группы либо гетарильные группы, такие, как фенил, нафтил и пиридил, причем эти радикалы в свою очередь могут нести заместители из следующей группы нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетарил Предлагаемый согласно изобретению способ пригоден в первую очередь для получения производных пиразола, в которых по крайней мере один из радикалов R1, R2, R3 и R4 обозначает не водород Пиразол и его производные находят применение, например, в качестве побочных продуктов, используемых для получения обладающих фармакологическим действием соединений, действующих веществ в средствах защиты растений или же для изготовления красителей Примеры 1 Получение 3-метилпиразола 1 а Трансформирование кротонового альдегида и гидрата гидразина К 115г (2,3 моля) гидрата гидразина добавляли 169,1г (2,415 моля) кротонового альдегида, причем температуру охлаждением поддерживали на уровне 30°С После введения добавки смесь продолжали перемешивать еще в течение ЗОмин при температуре 25 - 30° С 1 6 Трансформирование в 3-метилпиразол Смесь из 720,8г (5,06 молей) 68,8%-ной серной кислоты и 0,76г (5,1 ммолей) иодида натрия нагревали до 155° С и при этой температуре смешивали со смесью, полученной в 1 а Во время добавки и затем еще в течение ЗОмин после окончания добавки отгоняли воду Выделенную таким путем воду после охлаждения реакционной смеси до 70°С повторно добавляли в смесь для разбавления последней Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на рН 8,5 - 9 При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое может быть выделено путем декантирования После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 172,5 г 3-метилпиразола (90,55% по отношению к количеству используемого гидрата гидразина) со степенью чистоты 99% (определение согласно ЖХВД) Ткип 88°С/10мбар 2 Получение 4-метилпиразола 2 а Трансформирование метакролеина и гидрата гидразина К 115г (2,3 моля) гидрата гидразина добавляли 177,1г (2,53 моля) метакролеина, причем тем 8 пературу охлаждением поддерживали на уровне 30°С После введения добавки смесь продолжали перемешивать еще в течение ЗОмин при температуре 25 - 30°С 2 6 Трансформирование в 4-метилпиразол Смесь из 720,8г (5,06 молей) 68,8%-ной серной кислоты и 1,00г (6,7 ммолей) иодида натрия нагревали до 155°С и при этой температуре смешивали со смесью, полученной в 1 а Во время добавки и затем еще в течение ЗОмин после окончания добавки отгоняли воду Выделенную таким путем воду после охлаждения реакционной смеси до 50°С повторно добавляли в смесь для разбавления последней Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на рН 8,5 При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое может быть выделено путем декантирования После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 170,5г 4-метилпиразола (89,52% по отношению к количеству используемого гидрата гидразина) со степенью чистоты 99,2% (определение согласно ЖХВД) Ткип 82° С/7 мбар 3 Получение 3,4-диметилпиразола 3а Трансформирование транс-2,3диметилакролеина гидратом гидразина К 15,6г (0,25 моля) 80%-ного гидрата гидразина добавляли 22,1г (0,2625 моля) транс-2,3диметилакролеина, причем температуру охлаждением поддерживали на уровне 30°С После введения добавки смесь продолжали перемешивать еще в течение ЗОмин при температуре 25 - 30°С 3б Трансформирование в 3,4диметилпиразол Смесь из 74,2г (0,52 моля) 68,8%-ной серной кислоты и 0,5г (3,3 ммоля) иодида натрия нагревали до 155°С и при этой температуре смешивали со смесью, полученной в 3 а Во время добавки и в течение последующих ЗОмин после окончания добавки отгоняли воду Выделенную таким путем воду после охлаждения реакционной смеси до 50°С повторно добавляли в смесь для разбавления последней Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на рН 8,5 При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое можно было выделять путем декантирования После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 21,4г 3,4-диметилпиразола (88,4% по отношению к количеству используемого гидрата гидразина) со степенью чистоты 99,2% Ткип 96° С/10 мбар 4 Получение 1,5-диметилпиразола 4 а Трансформирование кротонового альдегида метилгидразином К 92г (2 моля) метилгидразина добавляли 147г (2,1 моля) кротонового альдегида, причем температуру охлаждением поддерживали на уровне 30°С После введения добавки смесь продолжали перемешивать еще в течение 30 мин при температуре 25 - 30° С 4б Трансформирование в 1,5 47392 диметилпиразол Смесь из 626,7г (4,4 моля) 68,8%-ной серной кислоты и 0,66г (4,4 ммоля) иодида натрия нагревали до 155°С и при этой температуре смешивали со смесью, полученной в 4 а Во время добавки и в течение последующих ЗОмин после окончания добавки отгоняли воду Выделенную таким путем воду после охлаждения реакционной смеси до 70°С повторно добавляли в смесь для разбавления последней Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на рН 8,5 - 9 При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое можно было выделять путем декантирования После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 167,8г 1,5-диметилпиразола (86,7% по отношению к количеству используемого метилгидразина) со степенью чистоты 99,2% Ткип 157°С/1013мбар 5 Получение 3-метилпиразола 5 а Трансформирование кротонового альдегида гидратом гидразина К 125г (2,0 моля) 80%-ного гидрата гидразина добавляли 147г (2,1 моля) кротонового альдегида, причем температуру охлаждением поддерживали на уровне 30°С После введения добавки смесь продолжали перемешивать еще в течение ЗОмин при температуре 25 - 30°С 5 6 Трансформирование в 3-метилпиразол Смесь из 449,2г (4,4 моля) 95%-ной серной кислоты и 0,66г (4,4 ммоля) иодида натрия смешивали при 25°С со смесью, полученной в 5 а, причем температуру охлаждением поддерживали на уровне 25°С Затем температуру реакционной смеси доводили в течение 45мин до 125°С и в течение бОмин поддерживали на этом уровне Во время нагрева и перемешивания воду отгоняли Затем выделенную таким путем воду после охлаждения реакционной смеси до 70°С повторно добавляли в смесь для разбавления последней Разбавленную реакционную смесь с помощью 10%-ного едкого натра устанавливали на рН 8,5 9 При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое можно было выделять путем декантирования После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 143,4г 3-метилпиразола (87% по отношению к количеству используемого гидрата гидразина) со степенью чистоты 99,5% Ткип 88°С/1013мбар Сравнение способа по изобретению со способом, известным из европейской патентной заявки 10 ЕР-А-402722 А Получение 3-метилпиразола по способу согласно изобретению АЛ Трансформирование кротонового альдегида и гидрата гидразина К 62,5г (1,0 моль) 80%-ного гидрата гидразина добавляли 73,5г (1,05 моля) кротонового альдегида, причем температуру охлаждением поддерживали на уровне 30°С После введения добавки смесь продолжали перемешивать еще в течение ЗОмин при температуре 25 - 30°С А 2 Трансформирование в 3-метилпиразол Смесь из 313,6г (2,2 моля) 68,8%-ной серной кислоты и 0,33г (2,2 ммоля) иодида натрия нагревали до 155°С и при этой температуре смешивали со смесью, полученной в 1 а Во время добавки и в течение ЗОмин после окончания добавки воду отгоняли Выделенную таким путем воду после охлаждения реакционной смеси повторно добавляли в эту смесь Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на рН 8,5 При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое выделяли путем декантирования После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 73,4г 3-метилпиразола (89% по отношению к количеству используемого гидрата гидразина) со степенью чистоты 99,5% (определение согласно ЖХВД) Ткип 88°С/10мбар Б Получение 3-метилпиразола по способу, известному из европейской патентной заявки ЕРА-402722 Б 1 Смесь из 313,6г (2,2 моля) 68,8%-ной серной кислоты и 0,33г (2,2 ммоля) иодида натрия нагревали до 155°С и при этой температуре одновременно смешивали с 62,5г (1 моль) 80%-ного гидрата гидразина и 73,6г (1,05 моля) кротонового альдегида Во время добавки и в течение ЗОмин после окончания добавки воду отгоняли Выделенную таким путем воду после охлаждения реакционной смеси до 50°С повторно добавляли в смесь для разбавления последней Б 2 Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на рН 8 5 При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое выделяли путем декантирования После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 62,4г 3-метилпиразола (75,7% по отношению к количеству используемого гидрата гидразина) со степенью чистоты 99,5% Ткип 88°С/10мбар 11 47392 ДП «Український інститут промислової власності» (Укрпатент) вул Сім'ї Хохлових, 15, м Київ, 04119, Україна ( 0 4 4 ) 4 5 6 - 2 0 - 90 ТОВ "Міжнародний науковий комітет" вул Артема, 77, м Київ, 04050, Україна (044)216-32-71 12
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for preparing pyrazole and its derivatives
Назва патенту російськоюСпособ получения пиразола и его производных
МПК / Мітки
МПК: C07D 231/12, C07D 521/00
Мітки: одержання, спосіб, піразолу, похідних
Код посилання
<a href="https://ua.patents.su/6-47392-sposib-oderzhannya-pirazolu-i-jjogo-pokhidnikh.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання піразолу і його похідних</a>
Спосіб одержання сульфамоілзаміщених похідних фенетіламіна або його солянокислої солі

Номер патенту: 5990
Опубліковано: 29.12.1994
Автори: Сінаті Хасімото, Такасі Фудзікура, Куніхіро Ніїгата, Казуо Імаї, Тоїті Такєнака
Мітки: сульфамоілзаміщених, солі, солянокислої, спосіб, похідних, одержання, фенетіламіна
Формула / Реферат:
(57) Способ получения сульфамоилзамещенных производных фенэтиламина общей формулыR, - моно- или ди- низшая алкиломинагруппа;R2 – низшая алкоксигруппа;R3 - водород;R4 - низшая алкильная группа;R5, R6, R 7- водород;R8 - низшая алкоксигруппа;Y - кислородили его солянокислая соль, отличающийся тем, что соединение формулы где R1 , R2 , R3 , R4 имеют указанные значения,...
Спосіб виділення похідних піразолу

Номер патенту: 7045
Опубліковано: 31.03.1995
Автори: Фрідріх Лінхарт, Вінфрід Ріхарц, Карл Ейкен, Берн Гіргензон
МПК: C07D 231/20, C07D 231/14, C07D 231/18, C07D 231/12, C07D 521/00
Мітки: виділення, спосіб, похідних, піразолу
Формула / Реферат:
Формула изобретенияСпособ выделения производных пиразола общей формулы Iгде R и R1 - метил или этил; R2 - водород, метил или метоксигруппа; X - хлор или бром, из соответствующего сырья, отличающийся тем, что, с целью упрощения процесса, сырье обрабатывают 36-37%-ной хлористоводородной кислотой или 50-60%-ной серной кислотой, взятой в 2-5-кратном количестве от теоретического, полученный водный раствор...
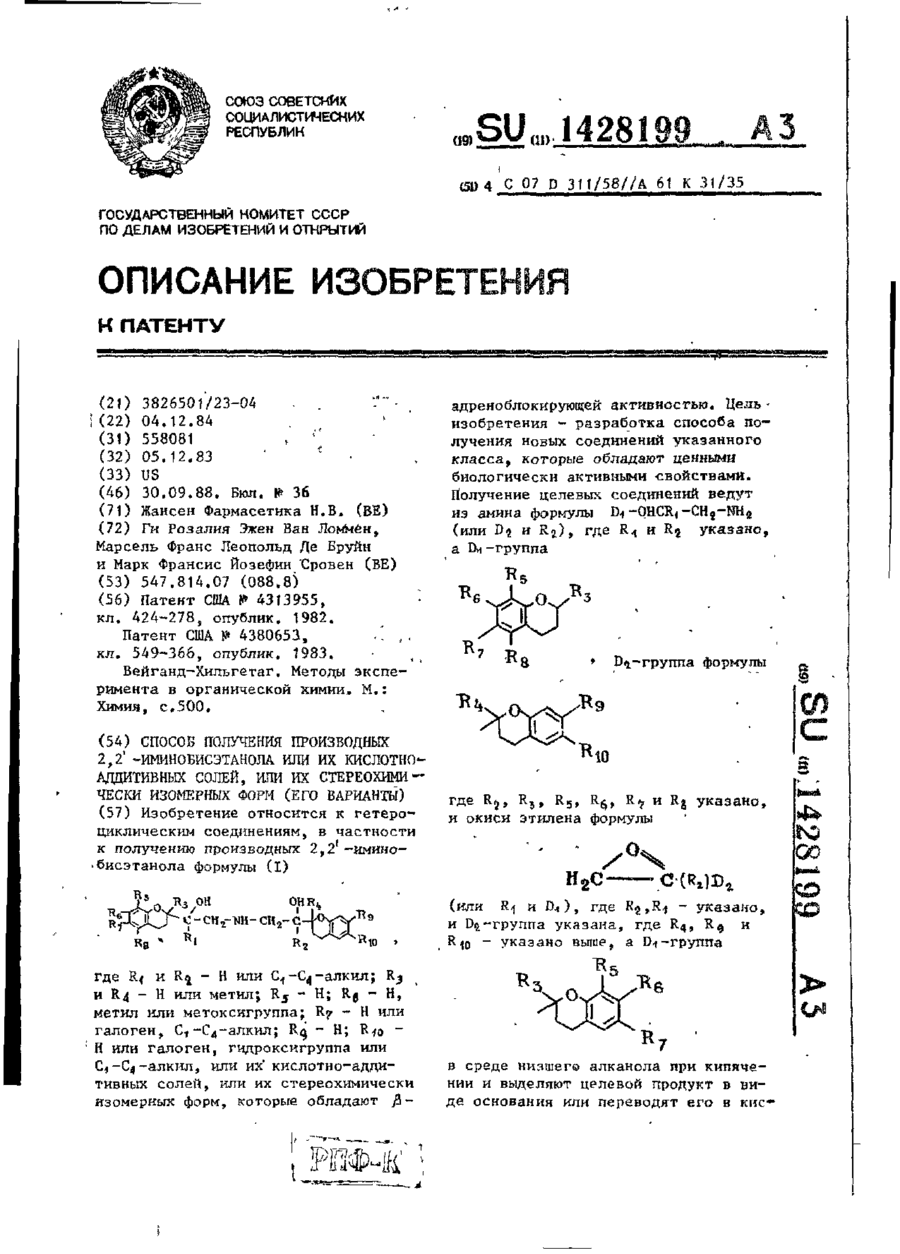
Спосіб одержання похідних 2,2′-імінобіоетанола або їх кислотно-адітівних солей, або їх стереохімічно ізомерних форм (його варіанти)
Номер патенту: 2707
Опубліковано: 26.12.1994
Автори: Гі Розалія Ежен Ван Ломмен, Марсель Франс Леопольд Де Бруйн, Марк Франсіс Йозефін Сровен
МПК: A61K 31/35, C07D 311/70, C07D 407/04, A61K 31/352, A61P 25/02, C07D 311/66, C07D 307/79, C07D 307/81, C07D 311/22, C07D 311/58, C07D 311/92
Мітки: ізомерних, одержання, кислотно-адітивних, похідних, спосіб, форм, варіанти, його, 2,2'-імінобіоетанола, стереохімічно, солей
Формула / Реферат:
1. Способ получения производных 2,2'-иминобисэтанола формулы Ігде R1 и R2— водород или С1 — С2 алкил; R3 и R4— водород или метил; R5 — водород; R6— водород, метил или метоксигруппа; R7 —водород или галоген; С1 — С4-алкид, ацетиламиногруппа, цианогруппа гидроксигруппа или метил-сульфониламиногруппа; R8 — водород или С1 — С4-алкил; R9 — водород; R10 — водород или галоген, гидроксигруппа или С1—С4-алкил, или их кислотно-аддитивных...
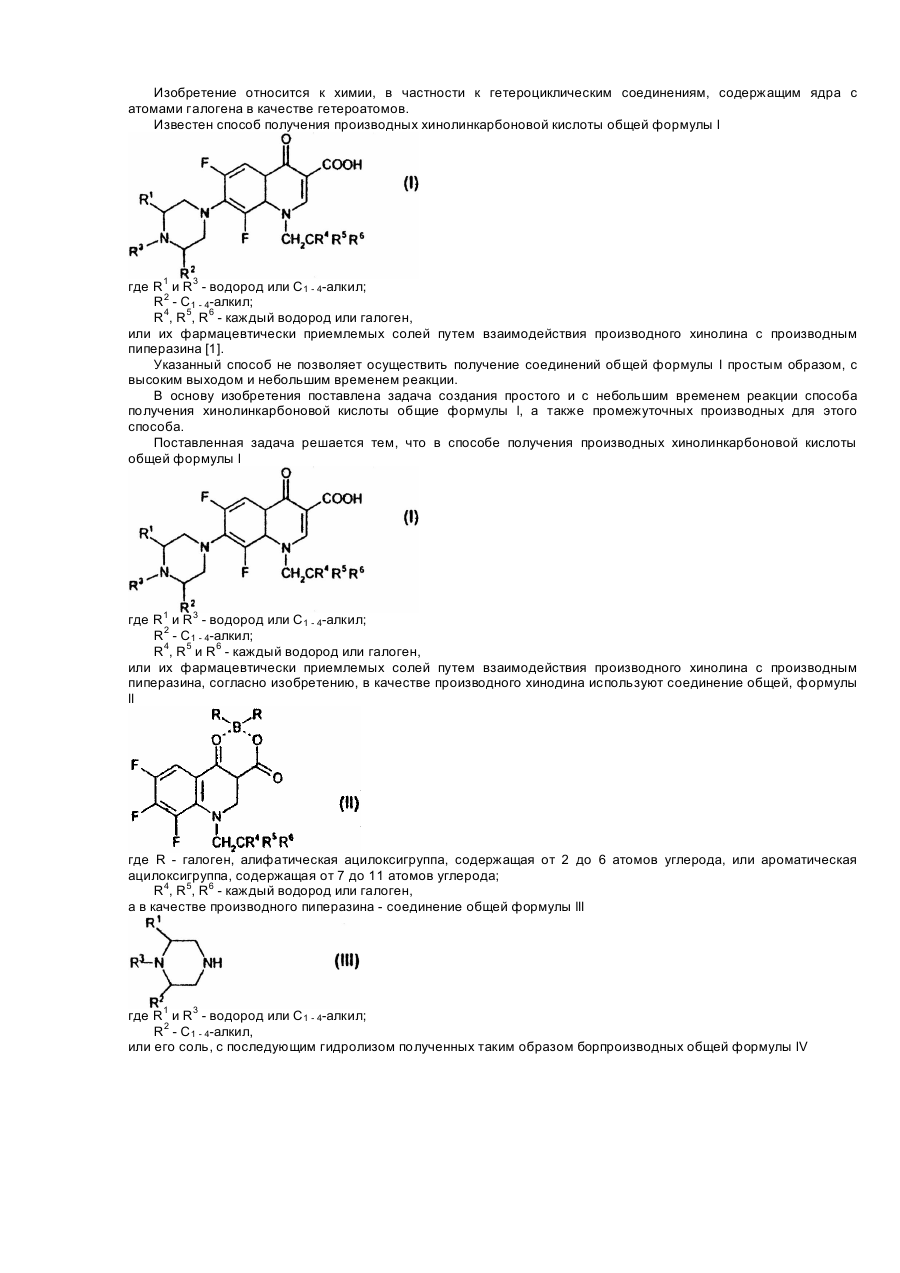
Спосіб одержання похідних хінолінкарбонової кислоти або її фармацевтично придатних солей та проміжні сполуки для їх одержання
Номер патенту: 26568
Опубліковано: 11.10.1999
Автори: Балог Марія, Хорват Агнеш, Рітлі Петер, Керестурі Геза, Хермец Іштван, Вашварі Лелле, Пайор Аніко, Шіпош Юдіт
МПК: A61K 31/495, C07F 5/00, C07D 215/56, C07D 401/04, A61P 31/04
Мітки: придатних, фармацевтично, хінолінкарбонової, спосіб, проміжні, кислоти, одержання, похідних, сполуки, солей
Формула / Реферат:
1. Способ получения производных хинолинкарбоновой кислоты общей формулы lгде R1 и R3 - водород или C1 - 4-алкил;R2 - C1 - 4-алкил;R4, R5 и R6 - каждый - водород или галоген,или ее фармацевтически приемлемых солей, взаимодействием производного хинолина с производным пиперазина, отличающийся тем, что в качестве производного хинолина используют соединение общей формулы llгде R - галоген,...
Спосіб одержання похідних пірімідину

Номер патенту: 3517
Опубліковано: 27.12.1994
Автори: Андраш Ведреш, Габор Блашко, Бела Штефко, Тамаш Мештер, Іштван Хегедюш, Денеш Мате, Андрієн Суховські, Чаба Сантаі, Янош Крайдль, Андраш Немеш, Ерік Богш
МПК: C07D 239/50, C07D 239/48
Мітки: спосіб, одержання, піримідину, похідних
Формула / Реферат:
Способ получения производных пиримидина общей формулыгде Х-Y - группа N(О)=СNНR1, где R1 - водород (Іd), или группа СОR, где R - С1-С6-алкил (Id), а R2 - водород, или Х-Y-группа N(ОСОR)С(=NН)-, где R-С1-С6-алкил, а R2-водород (Id), или группа RСО-, где R-С1-С6-алкил (Id), Z - хлор, бром или замещенная однократно или многократно С1-С3-алкилом, в случае необходимости арилсульфонилоксигруп-па, путем окисления...
Попередній патент: Сходи, що фільтрують, для ванни басейну
Наступний патент: Клейова композиція для зв’язування еластомерів
Випадковий патент: Спосіб прогнозування ризику тромботичних ускладнень у хворих з переломами