Препарат інсуліну пролонгованої дії
Номер патенту: 110935
Опубліковано: 10.03.2016
Автори: Бодерке Петер, Фрік Аннке, Шеттлє Ізабелль, Лоос Петра, Фюрст Крістіане, Мюллер Вернер, ВЕРНЕР Ульріх, Беккер Райнхард, Терч Катрін

















































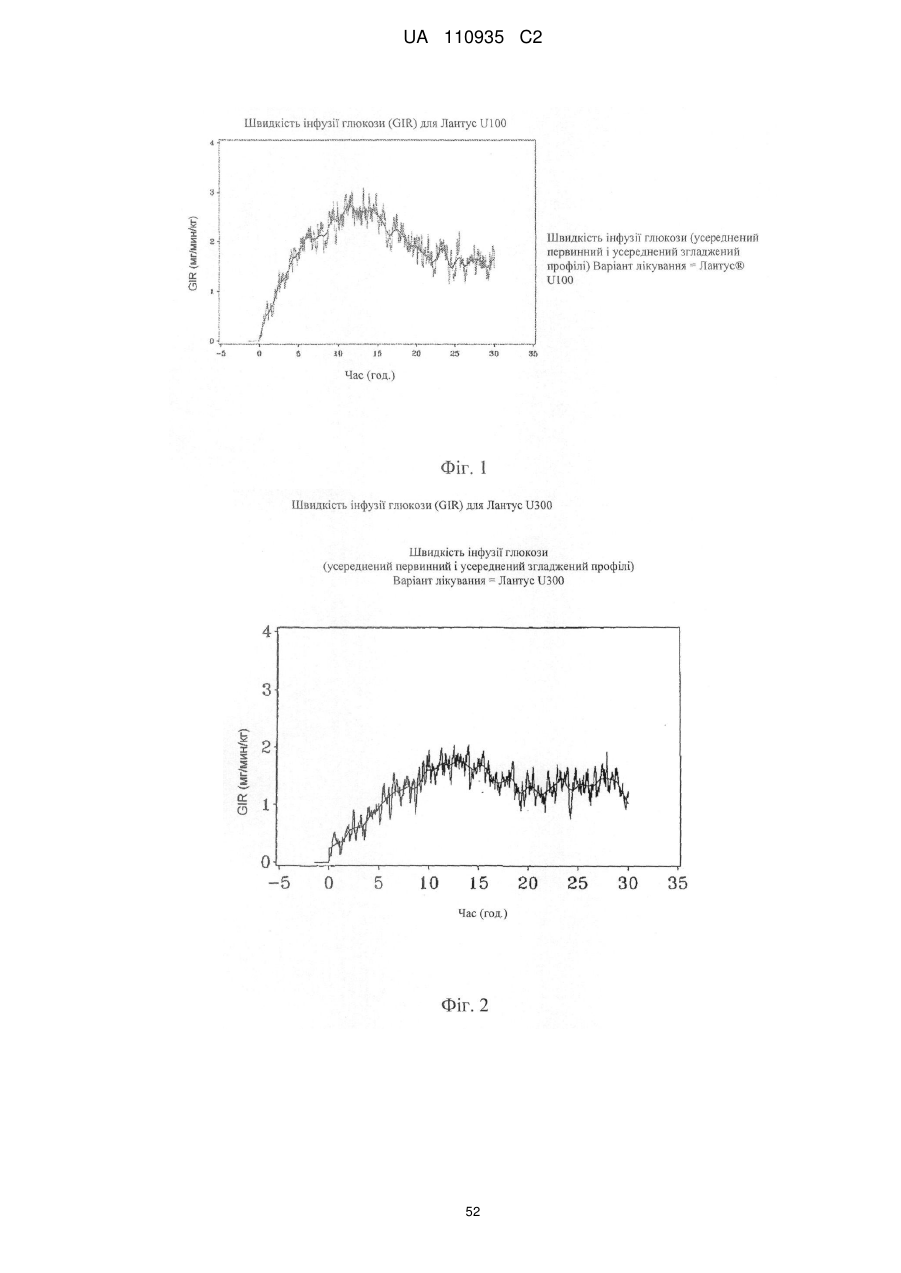
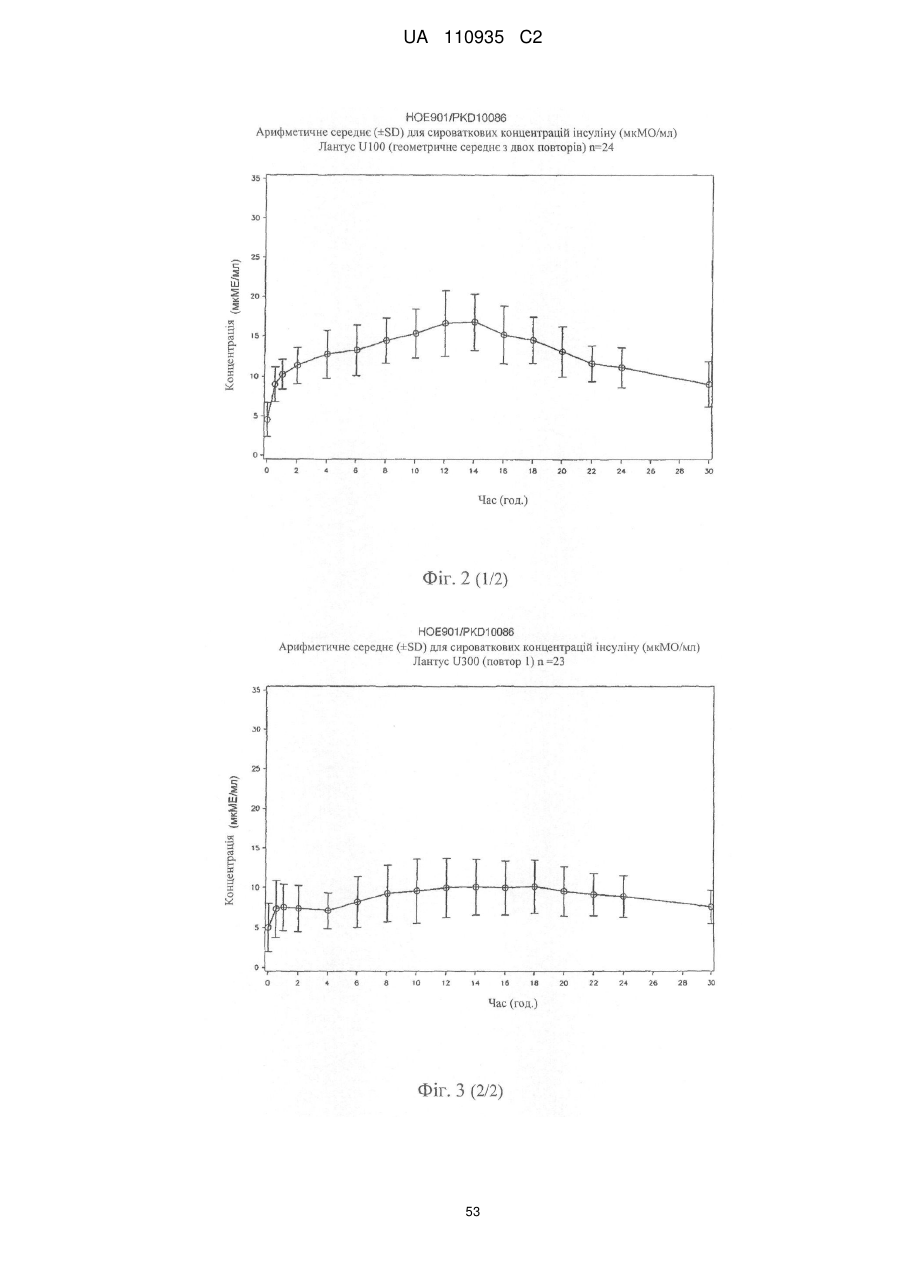
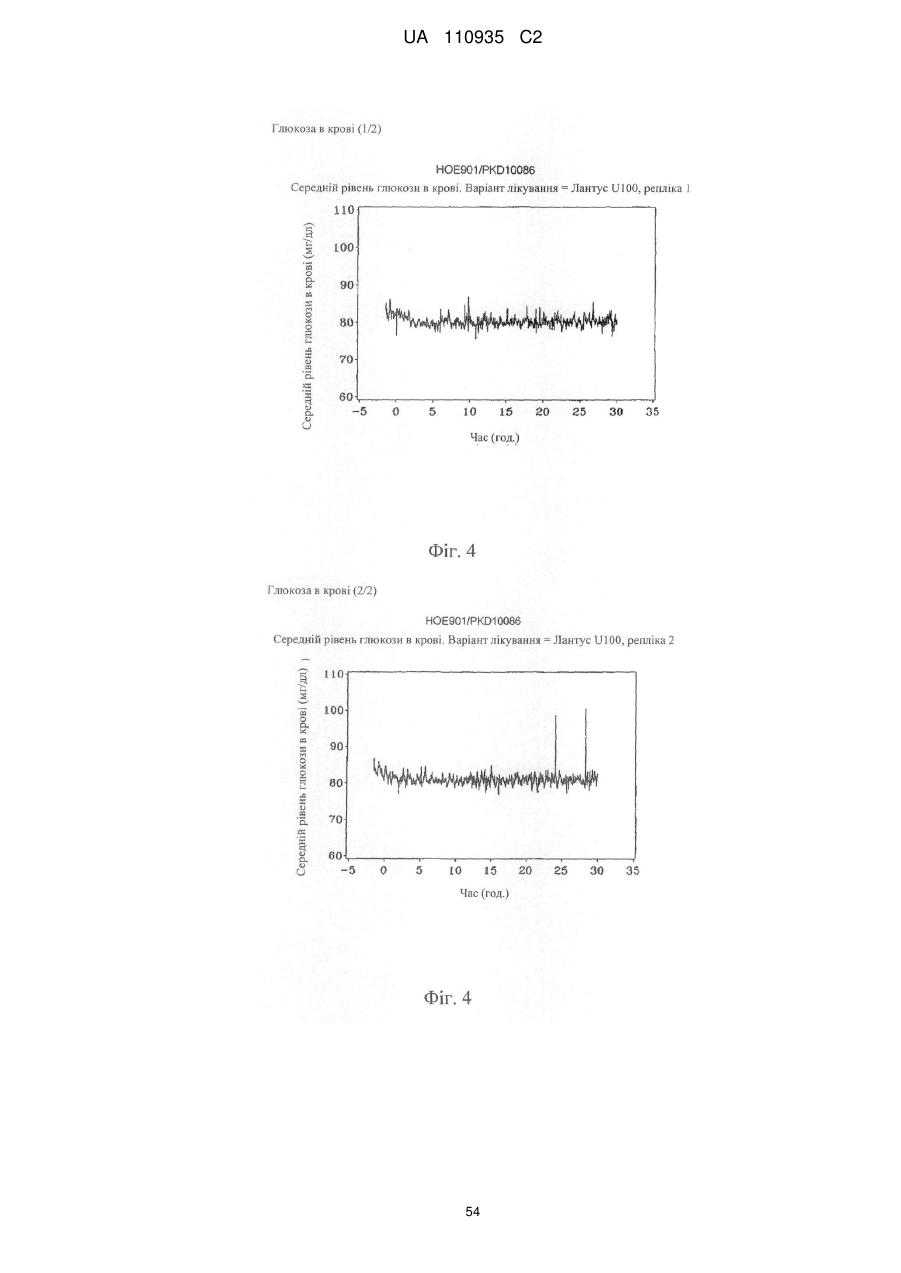
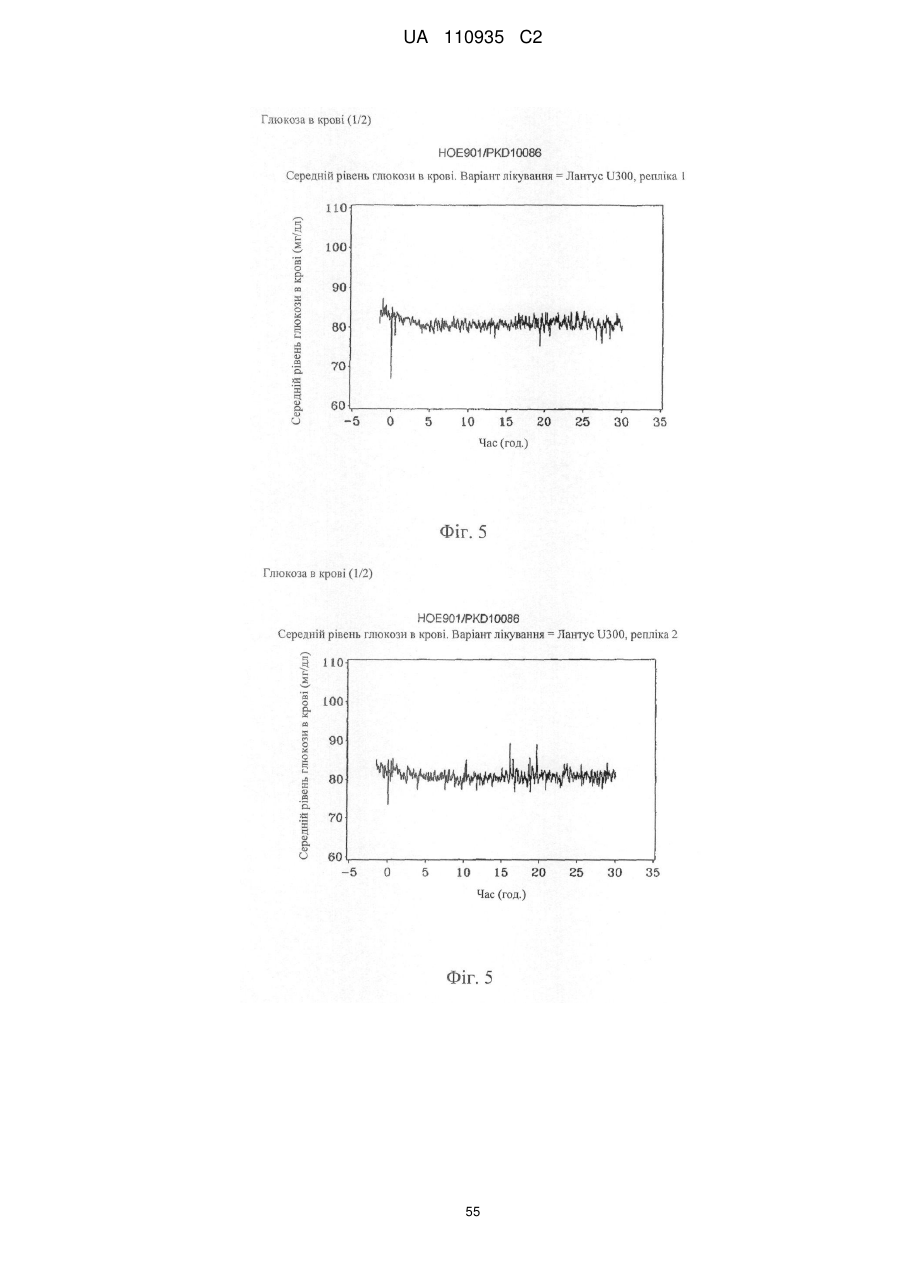


Формула / Реферат
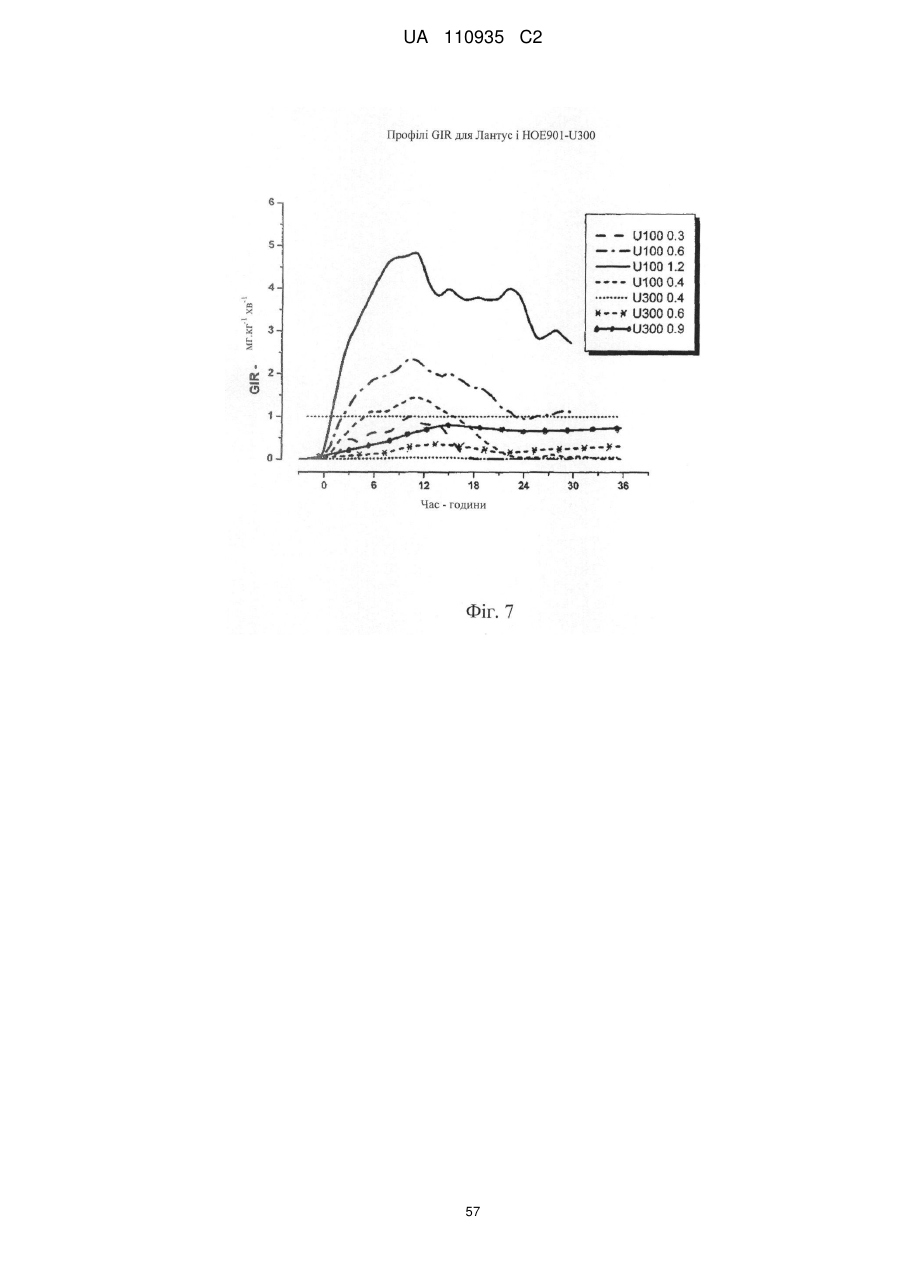
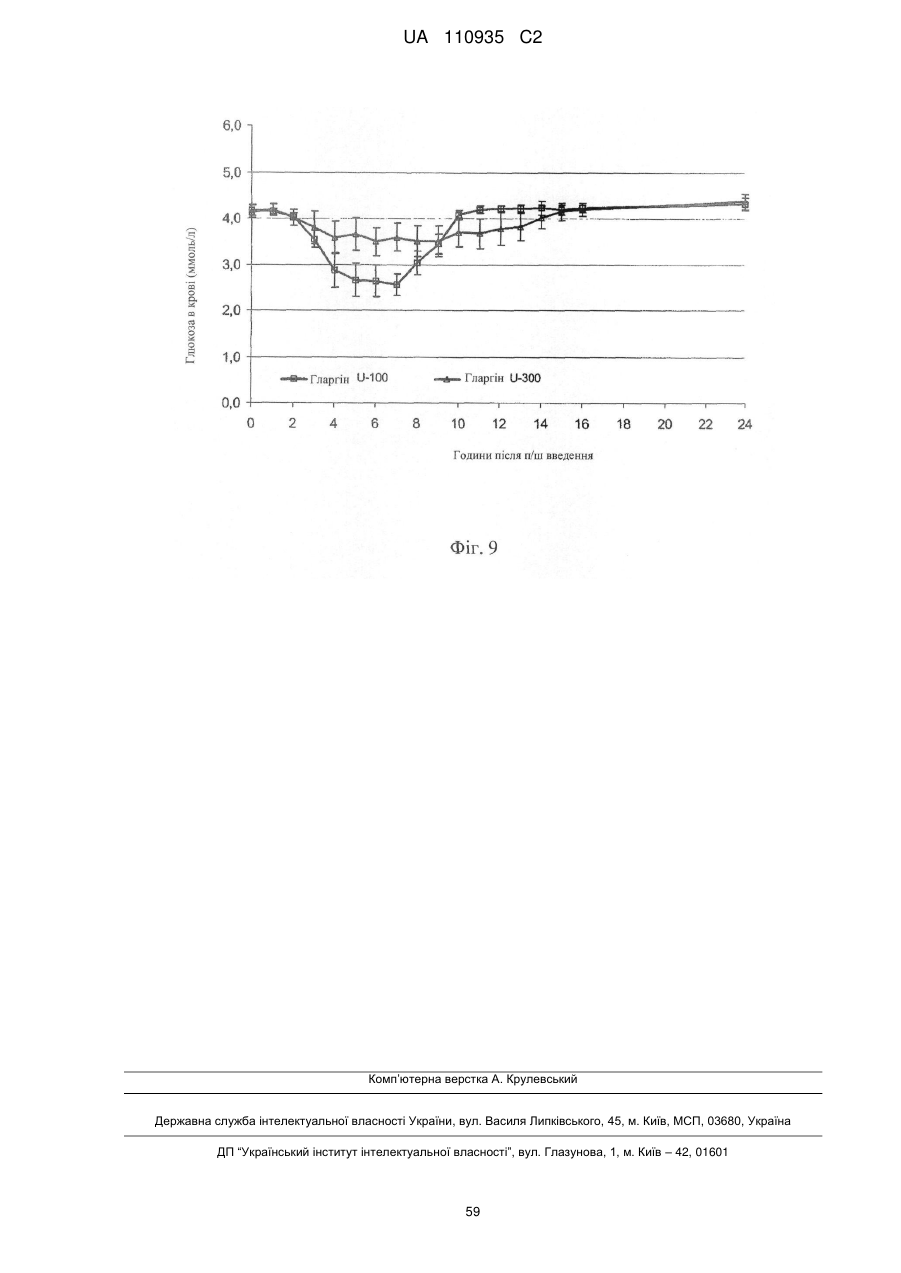
1. Водний фармацевтичний препарат, який містить 270-330 Од/мл інсуліну гларгіну [еквімолярно 270-330 МО людського інсуліну].
2. Водний препарат за п. 1, який містить 300 Од/мл інсуліну гларгіну [еквімолярно 300 МО людського інсуліну].
3. Водний фармацевтичний препарат за будь-яким з попередніх пунктів, що містить аналог ексендину-4.
4. Водний препарат за п. 3, у якому аналог ексендину-4 вибраний із групи, що включає ліксисентатид, ексенатид і ліраглутид.
5. Водний препарат за п. 4, який містить від 0,1 мкг до 10 мкг ліксисентатиду на Од інсуліну гларгіну.
6. Водний препарат за п. 5, який містить від 0,2 мкг до 1 мкг ліксисентатиду на Од інсуліну гларгіну.
7. Водний препарат за п. 6, який містить від 0,25 мкг до 0,7 мкг ліксисентатиду на Од інсуліну гларгіну.
8. Водний препарат за будь-яким з попередніх пунктів, який містить один або більше ексципієнтів, вибраних із групи, що включає цинк, м-крезол, гліцерин, полісорбат 20 і натрій.
9. Водний препарат за п. 8, який містить 90 мкг/мл цинку, 2,7 мг/мл м-крезолу і 20 мг/мл гліцерину 85 %.
10. Водний препарат за п. 8, який містить 90 мкг/мл цинку, 2,7 мг/мл м-крезолу, 20 мкг/мл полісорбату 20 і 20 мг/мл гліцерину 85 %.
11. Водний препарат за будь-яким з попередніх пунктів, у якому рН складає від 3,4 до 4,6.
12. Водний препарат за п. 11, у якому рН складає 4.
13. Водний препарат за п. 11, у якому рН складає 4,5.
14. Спосіб лікування цукрового діабету І типу і II типу у пацієнта, який включає введення зазначеному пацієнту водної фармацевтичної композиції, що містить інсулін гларгін у концентрації 300 Од/мл.
15. Спосіб за п. 14, у якому зазначена фармацевтична композиція додатково містить ексципієнти, вибрані з групи, яка складається з цинку, м-крезолу, гліцерину, полісорбату 20 і натрію.
16. Спосіб за п. 14, у якому зазначена фармацевтична композиція додатково містить від 0,1 мкг до 10 мкг ліксисентатиду на Од інсуліну гларгіну.
17. Спосіб збільшення тривалості експозиції інсуліну пролонгованої дії при лікуванні цукрового діабету І типу і II типу у пацієнта, який включає введення зазначеному пацієнту водної фармацевтичної композиції, що містить інсулін гларгін у концентрації 300 Од/мл.
18. Спосіб за п. 17, у якому зазначена фармацевтична композиція додатково містить ексципієнти, вибрані з групи, яка складається з цинку, м-крезолу, гліцерину, полісорбату 20 і натрію.
19. Спосіб за п. 17, у якому зазначена фармацевтична композиція додатково містить від 0,1 мкг до 10 мкг ліксисентатиду на Од інсуліну гларгіну.
20. Спосіб зниження частоти виникнення гіпоглікемії при лікуванні цукрового діабету І типу і ІІ типу у пацієнта, що застосовує інсулін пролонгованої дії, який включає введення зазначеному пацієнту водної фармацевтичної композиції, що містить інсулін гларгін у концентрації 300 Од/мл.
21. Спосіб за п. 20, у якому зазначена фармацевтична композиція додатково містить ексципієнти, вибрані з групи, яка складається з цинку, м-крезолу, гліцерину, полісорбату 20 і натрію.
22. Спосіб за п. 20, у якому зазначена фармацевтична композиція додатково містить від 0,1 мкг до 10 мкг ліксисентатиду на Од інсуліну гларгіну.
23. Спосіб забезпечення рівномірного вмісту базального інсуліну пролонгованої дії при лікуванні цукрового діабету І типу і II типу у пацієнта, який включає введення зазначеному пацієнту водної фармацевтичної композиції, що містить інсулін гларгін у концентрації 300 Од/мл.
24. Спосіб за п. 23, у якому зазначена фармацевтична композиція додатково містить ексципієнти, вибрані з групи, яка складається з цинку, м-крезолу, гліцерину, полісорбату 20 і натрію.
25. Спосіб за п. 23, у якому зазначена фармацевтична композиція додатково містить від 0,1 мкг до 10 мкг ліксисентатиду на Од інсуліну гларгіну.
26. Застосування водного препарату за будь-яким з попередніх пунктів у лікуванні цукрового діабету І типу і цукрового діабету II типу.
Текст
Реферат: Винахід стосується водного фармацевтичного препарату, що містить 270-330 Од/мл інсуліну гларгіну [еквімолярно 270-330 МО людського інсуліну], а також його застосування. UA 110935 C2 (12) UA 110935 C2 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 Заявка на винахід стосується водного фармацевтичного препарату, який містить 200-1000 Од/мл [еквімолярно 200-1000 МО (IU) людського інсуліну] інсуліну гларгіну, за умови, що концентрація зазначеного препарату не дорівнює 684 Од/мл інсуліну гларгіну, а також його застосування. B B Інсулін гларгін являє собою 31 -32 -Di-Arg людський інсулін, аналог людського інсуліну з додатковою заміною аспарагіну в положенні A21 гліцином. Лантус® є інсуліновим препаратом, який містить інсулін гларгін, що забезпечує 24-годинне постачання базальним інсуліном після підшкірної ін'єкції однієї дози. По глюкодинамічному ефекту Лантус® відрізняється від інших наявних на ринку на даний час інсулінових препаратів через уповільнену і передбачувану абсорбцію інсуліну гларгіну з місця підшкірної ін'єкції, результатом чого є згладжений профіль концентрації й активності залежно від часу протягом 24 годин без чітко вираженого піка. Лантус® був розроблений для задоволення медичної потреби в інсуліновому препараті пролонгованої дії, який можна вводити однократною щоденною ін'єкцією для забезпечення нормального або майже нормального контролю рівня глюкози в крові з профілем базального інсуліну, якомога більш згладженим протягом 24-годинного періоду. Такий препарат забезпечує гарний контроль рівня глюкози в крові протягом цілої доби, при цьому зводячи до мінімуму тенденцію розвитку гіпоглікемії, яка спостерігається у випадку інших препаратів інсуліну з більш чітко вираженим "піковим" ефектом. Значне число пацієнтів, зокрема пацієнти з підвищеною резистентністю до інсуліну внаслідок ожиріння, використовують великі дози для контролю рівня глюкози в крові. Наприклад, для одержання дози 100 Од необхідна ін'єкція 1 мл Лантус® U100, що може викликати деякий дискомфорт; кожен мл Лантус® U100 містить 100 Од (3,6378 мг) інсуліну гларгіну. Для зменшення об’єму ін'єкції був розроблений препарат, що містить 300 Од інсуліну гларгіну в мл. Хоча винахід не обмежений препаратом інсуліну гларгіну U300, клінічні дослідження, описані в даному документі, були проведені з препаратом інсуліну гларгіну U300; кожен мл інсуліну гларгіну U300 містить 300 Од (10,9134 мг) інсуліну гларгіну. Даний препарат дозволяє пацієнтам вводити те ж число одиниць в одній третині об’єму ін'єкції. Очікувалося, що обидва препарати інсуліну гларгіну, U100 і U300, забезпечать однакову експозицію інсуліну й однакову ефективність, тобто профілі залежності від часу. Докладний опис Експозицію й активність інсуліну гларгіну U300, тестованого (T) препарату, перевіряли на здорових суб'єктах без діабету методом фіксації стану еуглікемії з погляду еквівалентності по експозиції й активності препарату Лантус U100, схваленому референсному (R) препарату. З урахуванням тривалості дії інсуліну гларгіну був вибраний час 30 годин після підшкірного введення. Експозицію оцінювали на основі профілів концентрації інсуліну гларгіну залежно від часу після підшкірного введення, при цьому одночасно оцінювали активність як утилізацію глюкози на одиницю інсуліну. Дизайн експерименту з репліками дозволяв скоротити число суб'єктів для оцінки біоеквівалентності і варіабельності відповідно до рекомендацій керівництва FDA "Guidance for Industry, Statistical Approaches to Establishing Bioequivalence". Очікувалося, що відповідне клінічне дослідження встановить еквівалентність по експозиції й активності. Для даного дослідження вибрали дозу 0,4 Од/кг маси тіла; це відповідає середній дозі базального інсуліну у пацієнтів. У здорових суб'єктів без діабету ця доза викликає значне підвищення концентрації інсуліну в плазмі і тривалий ефект зниження рівня глюкози, що можна кількісно визначати в умовах фіксації стану еуглікемії. Дизайн експерименту з репліками, рекомендований керівництвом, передбачає дві репліки однодозових ін'єкцій будь-якого ДП (IP) (R: Лантус® U100, T: інсулін гларгін U300) у попередньо визначених чотириваріантних перехресних послідовностях (RTTR або TRRT) відповідно до плану рандомізації. Це було виконано в періоди (P) 1-4 у чотири різних дні. В результаті кожен суб'єкт одержував по дві репліки введених підшкірно однократних доз 0,4 Од/кг Лантус® U100 (R) і інсуліну гларгіну U300 (T) поперемінно в дві протилежних ділянки зони пупка. Кожний із днів введення доз був відділений періодом вимивання, що складає від 4 до 18 днів. Тривалість періоду вимивання варіювалася індивідуально, що дозволяло пристосовуватися до потреб як учасника, так і дослідника. З досвіду, 4 дні складали мінімальний період відновлення, дозволяючи проводити 1 фіксацію на тиждень для учасника, тоді як 18 днів означали перерву в 3 тижні між днями фіксації, що надавало суб'єктам більше свободи у виконанні своїх зобов'язань, не пов'язаних з дослідженням. Перед відвідуваннями для фіксації стану еуглікемії під час SCR (скринінгового відвідування) кандидати піддавалися скринінгу на придатність, а під час відвідування EOS (після закінчення 1 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 дослідження) суб'єкти приходили для остаточного обстеження з метою підтвердження нормального стану здоров'я. Проміжок часу між скринінгом і P1 складав не більше 21 дня, при тому, що відвідування EOS відбувалися не раніше, ніж у той же день тижня, що і день 1 у P4, на наступному тижні, тобто через додаткові 4 дні, і не пізніше, ніж через два тижні після дня 2 у P4, тобто через додаткових 14 днів. Це було однодозове дослідження з використанням, в цілому, 4 реплікативних введень. Ефект ДП повинен був продовжуватися протягом приблизно 24 годин, тому суб'єкти повинні були залишатися в інституті протягом 2 днів. Суб'єктів піддавали впливу 4 рази. Основна мета дослідження полягала в оцінці середньої біоеквівалентності (ABE) препарату Лантус® U100 (препарат, що є у продажу) і інсуліну гларгіну U300 з погляду біодоступності (експозиції) і біоефективності (активності) за допомогою методу фіксації стану еуглікемії. Друга мета дослідження полягала в оцінці безпеки і переносимості інсуліну гларгіну U300. Як згадувалося вище, обидва препарати інсуліну гларгіну, U100 і U300, повинні були, як очікувалося, забезпечити однакову експозицію інсуліну й однакову ефективність. Однак дивно, але експозиція й ефективність препаратів інсуліну виявилися не однаковими. Інсулін гларгін U100 і інсулін гларгін U300 не еквівалентні по біодоступності (експозиції) і біоефективності (активності). Експозиція й активність після введення інсуліну гларгіну U300 були менше приблизно на 40% у порівнянні з експозицією й активністю після введення тієї ж кількості (0,4 Од/кг) інсуліну гларгіну U100. Однак інсулін гларгін U300 продемонстрував навіть більш плоскі профілі ФК (PK) (експозиція) і ФД (PD) (активність), ніж інсулін гларгін U100, що було б бажано для базального інсуліну. Ці дивні і несподівані відмінності в експозиції й активності між препаратами інсуліну гларгіну U100 і інсуліну гларгіну U300 після однієї і тієї ж підшкірної дози здоровим суб'єктам ефективно показані на фігурах, наведених нижче. Слід зазначити, що в той же час рівень глюкози в крові був постійним. Ефект інсуліну гларгіну на зниження рівня глюкози в крові додатково оцінювали на здорових нормоглікемічних собаках породи бігль. Зі збільшенням концентрації інсуліну гларгіну середній час дії зростав з 6,8 год. (U100) до 7,69 год. (U300), відповідно. У собак, зі збільшенням концентрації інсуліну гларгіну від 100 до 300 Од/мл профіль залежності активності, що знижує рівень глюкози в крові, від часу змінювався у бік більш рівномірної і тривалої активності. Ці дані, одержані на собаках, узгоджуються з даними, одержаними за участі людей, демонструючи, що підвищення лікарської концентрації інсуліну гларгіну позитивно корелює з профілем і більшою тривалістю дії. Крім того, осади препаратів інсуліну гларгіну з концентраціями 100 Од/мл, 300 Од/мл, 500 Од/мл, 700 Од/мл і 1000 Од/мл досліджували за допомогою мікроскопа. Ці дослідження виявили відмінності в характеристиках осадів, що приводять до утворення значно більш великих частинок при збільшенні концентрації. Крім того, досліджували вплив збільшення концентрацій препаратів інсуліну гларгіну з погляду розчинності, використовуючи in vitro тест-системи. Для цього проводили дослідження осадження, використовуючи фосфатний буфер з pН 7,4, що моделює умови in vivo. Супернатант осадженого інсуліну досліджували методом ВЕРХ для визначення вмісту інсуліну гларгіну. У WO 2008/013938 A2 описаний водний фармацевтичний препарат, який містить інсулін гларгін у концентрації 684 Од/мл. Хоча винахід не обмежений препаратом інсуліну гларгіну U300, а охоплює й інші більш концентровані препарати інсуліну гларгіну, як докладно описано в специфікації, клінічні дослідження, описані в даному документі, проводили з препаратом інсуліну гларгіну U300. A B B 1 мл препарату інсуліну гларгіну U300 містить 10,913 мг 21 -Gly-30 a-L-Arg-30 b-L-Arg людського інсуліну [еквімолярно 300 МО людського інсуліну], 90 мкг цинку, 2,7 мг м-крезолу, 20 мг гліцерину 85%, HCl і NaOH до рН 4,0; питома вага 1,006 г/мл. Однак можливі варіації, що стосуються видів ексципієнтів і їх концентрацій. Фармацевтичний препарат містить 200-1000 Од/мл інсуліну гларгіну [еквімолярно 200-1000 МО людського інсуліну], причому концентрація зазначеного препарату не дорівнює 684 Од/мл, переважно 250-500 Од/мл інсуліну гларгіну [еквімолярно 250-500 МО людського інсуліну], більш переважно 270-330 Од/мл інсуліну гларгіну [еквімолярно 270-330 МО людського інсуліну] і навіть більш переважно 300 Од/мл інсуліну гларгіну [еквімолярно 300 МО людського інсуліну]. У фармацевтичний препарат можна додавати сурфактанти, наприклад, у числі інших, неіонні сурфактанти. Зокрема, переважними є загальноприйняті у фармацевтиці сурфактанти, такі як, наприклад: часткові складні ефіри і складні ефіри жирних кислот, а також ефіри багатоатомних спиртів, таких як гліцерин, сорбіт і тому подібні (Span®, Tween®, зокрема Tween® 20 і Tween® 80, Myrj®, 2 UA 110935 C2 ® 5 10 15 20 25 30 35 40 45 50 55 60 Brij®), Cremophor або полоксамери. Сурфактанти присутні у фармацевтичній композиції в концентрації 5-200 мкг/мл, переважно 5-120 мкг/мл і особливо переважно 20-75 мкг/мл. Препарат може додатково містити консерванти (наприклад фенол, м-крезол, п-крезол, парабени), ізотонічні засоби (наприклад маніт, сорбіт, лактозу, декстрозу, трегалозу, хлорид натрію, гліцерин), буферні речовини, солі, кислоти і луги, а також інші ексципієнти. Ці речовини можуть у кожному випадку бути присутніми індивідуально або, альтернативно, у вигляді сумішей. Гліцерин, декстроза, лактоза, сорбіт і маніт можуть бути присутніми у фармацевтичному препараті в концентрації 100-250 мм, NaCl у концентрації до 150 мм. Буферні речовини, такі як, наприклад, фосфатний, ацетатний, цитратний, аргініновий гліцилгліциновий або тріс (тобто 2аміно-2-гідроксиметил-1,3-пропандіол) буфер і відповідні солі, присутні в концентрації 5-250 мм, переважно 10-100 мм. Додатковими ексципієнтами можуть бути, у числі інших, солі або аргінін. Концентрація цинку в препараті знаходиться в діапазоні концентрацій, що досягаються за рахунок присутності 0-1000 мкг/мл, переважно 20-400 мкг/мл цинку, найбільш переважно 90 мкг/мл. Однак цинк може знаходитися у формі хлориду цинку, але сіль може бути не тільки хлоридом цинку. У фармацевтичному препараті гліцерин і/або маніт може бути присутнім у концентрації 100250 ммоль/л і/або NaCl переважно є присутнім у концентрації до 150 ммоль/л. У фармацевтичному препараті буферна речовина може бути присутньою у концентрації 5250 ммоль/л. Наступним об'єктом винаходу є фармацевтичний препарат інсуліну, який містить додаткові добавки, такі як, наприклад, солі, що уповільнюють вивільнення інсуліну. Сюди також включені суміші таких інсулінів уповільненого вивільнення з препаратами, описаними вище. Ще одним об'єктом винаходу є спосіб одержання таких фармацевтичних препаратів. Для одержання препаратів інгредієнти розчиняють у воді і pН доводять, використовуючи HCl і/або NaOH. Крім того, наступним об'єктом винаходу є застосування таких препаратів для лікування цукрового діабету. Ще одним об'єктом винаходу є застосування або додавання сурфактантів як стабілізатора у процесі виробництва інсуліну, аналогів інсуліну або похідних інсуліну, або їх препаратів. Винахід також стосується препарату, описаного вище, який, крім того, додатково містить глюкагоноподібний пептид-1 (GLP-1), або його аналог або похідне, або ексендин-3 або -4, або його або аналог похідне, переважно ексендин-4. Винахід також стосується препарату, описаного вище, у якому аналог ексендину-4 вибраний із групи, що включає 36 H-десPro -ексендин-4-Lys6-NH2, 36,37 H-дес(Pro )-ексендин-4-Lys4-NH2 і 36,37 H-дес(Pro )-ексендин-4-Lys5-NH2, або їх фармакологічно прийнятну сіль. Винахід також стосується препарату, описаного вище, у якому аналог ексендину-4 вибраний із групи, що включає 36 28 десPro [Asp ]ексендин-4(1-39), 36 28 десPro [IsoAsp ]ексендин-4(1-39), 36 14 28 десPro [Met(O) ,Asp ]ексендин-4(1-39), 36 14 28 десPro [Met(O) ,IsoAsp ]ексендин-4(1-39), 36 25 28 десPro [Trp(O2) ,Asp ]ексендин-2(1-39), 36 25 28 десPro [Trp(O2) ,IsoAsp ]ексендин-2(1-39), 36 14 25 28 десPro [Met(O) Trp(O2) ,Asp ]ексендин-4(1-39) і 36 14 25 28 десPro [Met(O) Trp(O2) ,IsoAsp ]ексендин-4(1-39), або їх фармакологічно прийнятну сіль. Винахід також стосується препарату, описаного в попередньому параграфі, у якому пептид Lys6-NH2 приєднаний до C-кінця аналогів ексендину-4. Винахід також стосується препарату, описаного вище, у якому аналог ексендину-4 вибраний із групи, що включає 36 28 H-(Lys)6-десPro [Asp ]ексендин-4(1-39)-Lys6-NH2, 28 36 37 38 десAsp Pro ,Pro ,Pro ексендин-4(1-39)-NH2, 36 37 38 28 H-(Lys)6-десPro ,Pro ,Pro [Asp ]ексендин-4(1-39)-NH2, 36 37 38 28 H-Asn-(Glu)5десPro ,Pro ,Pro [Asp ]ексендин-4(1-39)-NH2, 36 37 38 28 десPro ,Pro ,Pro [Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 37 38 28 H-(Lys)6-десPro ,Pro ,Pro [Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 37 38 28 H-Asn-(Glu)5-десPro ,Pro ,Pro [Asp ]ексендин-4(1-39)-(Lys)6-NH2, 3 UA 110935 C2 36 5 10 15 20 25 30 35 40 45 50 55 25 28 H-(Lys)6-десPro [Trp(O2) ,Asp ]ексендин-4(1-39)-Lys6-NH2, 28 36 37 38 25 H-десAsp Pro ,Pro ,Pro [Trp(O2) ]ексендин-4(1-39)-NH2, 36 37 38 25 28 H-(Lys)6-десPro ,Pro ,Pro [Trp(O2) ,Asp ]ексендин-4(1-39)-NH2, 36 37 38 25 28 H-Asn-(Glu)5-десPro ,Pro ,Pro [Trp(O2) ,Asp ]ексендин-4(1-39)-NH2, 36 37 38 25 28 десPro ,Pro ,Pro [Trp(O2) ,Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 37 38 25 28 H-(Lys)6-десPro ,Pro ,Pro [Trp(O2) ,Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 37 38 25 28 H-Asn-(Glu)5-десPro ,Pro ,Pro [Trp(O2) ,Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 14 28 H-(Lys)6-десPro [Met(O) ,Asp ]ексендин-4(1-39)-Lys6-NH2, 14 28 36 37 38 десMet(O) Asp Pro ,Pro ,Pro ексендин-4(1-39)-NH2, 36 37 38 14 28 H-(Lys)6-десPro ,Pro ,Pro [Met(O) ,Asp ]ексендин-4(1-39)-NH2, 36 37 38 14 28 H-Asn-(Glu)5-десPro ,Pro ,Pro [Met(O) ,Asp ]ексендин-4(1-39)-NH2, 36 37 38 14 28 десPro ,Pro ,Pro [Met(O) ,Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 37 38 14 28 H-(Lys)6-десPro ,Pro ,Pro [Met(O) ,Asp ]ексендин-4(1-39)-Lys6-NH2, 36 37 38 14 28 H-Asn-(Glu)5десPro ,Pro ,Pro [Met(O) ,Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 14 25 28 H-(Lys)6-десPro [Met(O) ,Trp(O2) ,Asp ]ексендин-4(1-39)-Lys6-NH2, 28 36 37 38 14 25 десAsp Pro ,Pro ,Pro [Met(O) ,Trp(O2) ]ексендин-4(1-39)-NH2, 36 37 38 14 25 28 H-(Lys)6-десPro ,Pro ,Pro [Met(O) ,Trp(O2) ,Asp ]ексендин-4(1-39)-NH2, 36 37 38 14 28 H-Asn-(Glu)5-десPro ,Pro ,Pro [Met(O) ,Asp ]ексендин-4(1-39)-NH2, 36 37 38 14 25 28 десPro ,Pro ,Pro [Met(O) ,Trp(O2) ,Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 37 38 14 25 28 H-(Lys)6-десPro ,Pro ,Pro [Met(O) ,Trp(O2) ,Asp ]ексендин-4(1-39)-(Lys)6-NH2, 36 37 38 14 25 28 H-Asn-(Glu)5-десPro ,Pro ,Pro [Met(O) ,Trp(O2) ,Asp ]ексендин-4(1-39)-(Lys)6-NH2, або їх фармакологічно прийнятну сіль. Винахід також стосується препарату, описаного вище, який додатково містить 34 26 ε α Arg ,Lys (N (γ-глютаміл(N -гексадеканоїл)))GLP-1(7-37) [ліраглютид] або його фармакологічно прийнятну сіль. В одному варіанті здійснення даний винахід стосується водного фармацевтичного препарату, що містить інсулін гларгін у діапазоні концентрацій 200-1000 Од/мл [еквімолярно 200-1000 МО людського інсуліну], переважно від 200 Од/мл до 650 Од/мл, ще більш переважно від 700 Од/мл до 1000 Од/мл, більш переважно 270-330 Од/мл і найбільш переважно в концентрації 300 Од/мл, за умови, що концентрація зазначеного препарату не дорівнює 684 Од/мл інсуліну гларгіну. Крім того, препарат також містить аналог ексендину-4, такий як, наприклад, ліксисентатид, ексенатид і ліраглутид. Ці аналоги ексендину-4 присутні в препараті в діапазоні від 0,1 мкг до 10 мкг на Од інсуліну гларгіну, переважно від 0,2 до 1 мкг на Од інсуліну гларгіну і більш переважно від 0,25 мкг до 0,7 мкг на Од інсуліну гларгіну. Ліксисентатид є переважним. Крім того, водний фармацевтичний препарат може містити один або більше ексципієнтів, вибраних із групи, що включає цинк, м-крезол, гліцерин, полісорбат 20 і натрій. Зокрема, водний фармацевтичний препарат може містити 90 мкг/мл цинку, 2,7 мг/мл м-крезолу і 20 мг/мл гліцерину 85%. Необов'язково, водний фармацевтичний препарат може містити 20 мкг/мл полісорбату 20. pН водного фармацевтичного препарату складає від 3,4 до 4,6, переважно 4 або 4,5. Даний винахід стосується способу лікування цукрового діабету I типу і II типу, який включає введення водної фармацевтичної композиції за даним винаходом зазначеному пацієнту з діабетом. Переважною серед різних описаних діапазонів концентрацій є концентрація 300 Од/мл, а переважним аналогом інсуліну є інсулін гларгін. Додатково водний фармацевтичний препарат може також містити цинк, м-крезол, гліцерин, полісорбат 20 і натрій, а також їх суміші в діапазонах, розкритих у даному документі стосовно водного фармацевтичного препарату за даним винаходом. У переважному варіанті здійснення водний фармацевтичний препарат також містить від 0,1 мкг до 10 мкг ліксисенатиду на Од інсуліну гларгіну. Інсулін переважно вводять один раз на добу, але можна вводити два рази на добу, якщо необхідно. Потреби в дозах залежать від потреб окремого пацієнта, що визначаються шляхом досягнення нормальних або прийнятних рівнів глюкози в крові. Даний винахід також стосується способу збільшення тривалості експозиції інсуліну гларгіну при лікуванні цукрового діабету I типу і II типу у пацієнта, який включає введення зазначеному пацієнту водного фармацевтичного препарату за даним винаходом. Переважною серед різних описаних діапазонів концентрацій є концентрація 300 Од/мл. Додатково водний фармацевтичний препарат може також містити цинк, м-крезол, гліцерин, полісорбат 20 і натрій, а також їх суміші в діапазонах, розкритих у даному документі стосовно водного фармацевтичного препарату за даним винаходом. У переважному варіанті здійснення водний 4 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 фармацевтичний препарат також містить від 0,1 мкг до 10 мкг ліксисенатиду на Од інсуліну гларгіну. Даний винахід також стосується способу зниження частоти виникнення гіпоглікемії при лікуванні цукрового діабету I типу і II типу у пацієнта, що застосовує інсулін гларгін, який включає введення зазначеному пацієнту водного фармацевтичного препарату за даним винаходом. Переважною серед різних описаних діапазонів концентрацій є концентрація 300 Од/мл. Додатково водний фармацевтичний препарат може також містити цинк, м-крезол, гліцерин, полісорбат 20 і натрій, а також їх суміші в діапазонах, розкритих у даному документі стосовно водного фармацевтичного препарату за даним винаходом. У переважному варіанті здійснення водний фармацевтичний препарат також містить від 0,1 мкг до 10 мкг ліксисенатиду на Од інсуліну гларгіну. Даний винахід також стосується способу забезпечення рівномірного вмісту базального інсуліну пролонгованої дії при лікуванні цукрового діабету I типу і II типу у пацієнта, що застосовує інсулін гларгін, який включає введення зазначеному пацієнту водного фармацевтичного препарату за даним винаходом. Переважною серед різних описаних діапазонів концентрацій є концентрація 300 Од/мл. Додатково водний фармацевтичний препарат може також містити цинк, м-крезол, гліцерин, полісорбат 20 і натрій, а також їх суміші в діапазонах, розкритих у даному документі стосовно водного фармацевтичного препарату за даним винаходом. У переважному варіанті здійснення водний фармацевтичний препарат також містить від 0,1 мкг до 10 мкг ліксисенатиду на Од інсуліну гларгіну. Застосування водного препарату за будь-яким з вищевказаних пунктів у лікуванні цукрового діабету I типу і цукрового діабету II типу. Далі винахід описується за допомогою деяких прикладів, що ніяким чином не повинні діяти обмежно. Приклад 1: Опис протоколу Дане дослідження було одноцентровим, рандомізованим, контрольованим, сліпим перехресним дослідженням з чотирма періодами, 2 варіантами лікування і 2 послідовностями лікування на здорових суб'єктах, що включає шість відвідувань: Відвідування 1: скринінг (SCR); Відвідування 2-5, періоди (P) 1-4: лікування, період фіксації стану еуглікемії; Відвідування 6: закінчення дослідження (EOS). Суб'єкти одержували підшкірно по одній дозі 0,4 Од/кг інсуліну гларгіну U100 і інсуліну гларгіну U300, що почергово вводиться в двох протилежних ділянках області пупка (ліворуч, праворуч, ліворуч, праворуч) у чотири різних дні. Досліджуваний препарат вводили з реплікою варіанта лікування R і T у 2 послідовностях, RTTR або TRRT, у періоди P1-P4. Кожний із днів введення дози був відділений періодом вимивання, що складає від 4 до 18 днів. R: 0,4 Од/кг маси тіла інсуліну гларгіну U100 (препарат, що є у продажу; референс); T: 0,4 Од/кг маси тіла інсуліну гларгіну U300 (тестований препарат). P1 повинен починатися не більше ніж через 3-21 день після SCR. Відвідування EOS повинно відбутися через 4-14 днів після P4. Протягом P1-P4 суб'єкти були підключені до Biostator для вимірювання рівня глюкози в крові і коректування швидкості інфузії глюкози. Рівні глюкози в крові і швидкість інфузії глюкози (GIR) контролювали протягом 90 хвилин (вихідний період) до підшкірного введення досліджуваного препарату і протягом 30 годин після введення досліджуваного препарату. Починали інфузію 20% розчину глюкози для підтримання рівнів глюкози в крові на 5% нижче індивідуального рівня глюкози в крові натще, що визначається як середнє з 3 значень рівня глюкози в крові натще, вимірюваних за 60, 30 і 5 хвилин до введення досліджуваного препарату. Одержували профілі GIR. Зразки крові були взяті в попередньо визначені моменти часу протягом періоду фіксації стану еуглікемії для визначення сироваткових концентрацій інсуліну гларгіну. За винятком водопровідної води, суб'єкти голодували протягом періоду фіксації рівня глюкози. Очікувалося, що тривалість даного дослідження для суб'єкта буде складати до 13 тижнів між відвідуваннями SCR і EOS. Протокол був переданий на розгляд незалежним комітетам з етики і/або експертним радам організації для розгляду і письмового затвердження. Протокол відповідав рекомендаціям 18-го Всесвітнього конгресу по здоров'ю (Гельсінкі, 1964) і усім відповідним поправкам. Протокол також відповідав законам і нормативним актам, а також будь-яким відповідним керівним принципам Німеччини, де проводилося дослідження. До проведення будь-яких процедур, що стосуються дослідження, була одержана інформована згода учасників. Приклад 2: Відбір суб'єктів 5 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 За планом, у дослідженні повинні були брати участь двадцять чотири (24) здорових суб'єкти, щоб у підсумку 20 з них завершили дослідження. Суб'єкти, що відповідають усім наступним критеріям, були допущені до участі в дослідженні. Демографія суб'єкти обох статей у віці від 18 до 50 років; 2 маса тіла від 50 кг до 110 кг і індекс маси тіла від 18 до 28 кг/м . Стан здоров'я визнані здоровими після всебічного клінічного обстеження (докладний анамнез і повне фізичне обстеження); які не палять протягом щонайменше 3 місяців; електрокардіограма в 12 відведеннях і основні показники стану організму, за винятком тих випадків, коли дослідник вважає аномалію такою, що не має клінічного значення; нормальні основні показники стану організму через 5 хвилин відпочинку в положенні лежачи на спині: 95 мм ртутного стовпчика ≤ систолічний артеріальний тиск ≤ 140 мм ртутного стовпчика; 45 мм ртутного стовпчика ≤ діастолічний артеріальний тиск ≤ 90 мм ртутного стовпчика; 40 ударів на хвилину ≤ частота серцевих скорочень≤100 ударів у хвилину; нормальна ЕКГ у 12 відведеннях; 120 мс < PR < 220 мс, QRS < 120 мс, QTc ≤ 430 мс (для жінок: QTc ≤ 450 мс); лабораторні показники в межах норми, за винятком тих випадків, коли дослідник вважає аномалію такою, що не має клінічного значення для здорових суб'єктів; однак сироватковий креатинін і печінкові ферменти (AST, ALT) повинні бути строго нижче верхньої лабораторної норми; нормальний метаболічний контроль, визначений як рівень глюкози в сироватці натще (≤100 мг/дл) і глікозилованого гемоглобіну (HbА1c ≤ 6,1%); суб'єкти не повинні регулярно застосовувати лікарські препарати, що відпускаються за рецептом, протягом щонайменше чотирьох (4) тижнів до участі в дослідженні. Зобов'язання для жінок Жінки, здатні до дітонародження (визначувані як такі, що знаходяться у віці до менопаузи і не стерилізовані хірургічно або після менопаузи протягом менше 2 років) і сексуально активні, повинні практикувати адекватний контроль над народжуваністю. Адекватний контроль над народжуваністю визначений як високоефективний метод контрацепції (індекс Пірла 4 чашок або склянок/день); протипоказання за результатами (відповідність нормальним діапазонам - якщо значення знаходиться за межами нормального діапазону, кандидат може бути включений у випадку, коли дослідник вважає це аномальне значення клінічно незначимим): - медичної/хірургічної історії і фізичного обстеження, - лабораторних тестів (гематологія, клінічна біохімія, аналіз сечі за допомогою щупа), - стандартної електрокардіограми в 12 відведеннях, 6 UA 110935 C2 5 10 15 20 25 30 35 - артеріального тиску і частоти серцевих скорочень; будь-яке поточне лікування лікарськими засобами, що відпускаються за рецептом, або будьяке регулярне лікування лікарськими засобами, що відпускаються за рецептом, за 4 тижні до участі в дослідженні; симптоми клінічно значимого захворювання за 3 місяці до дослідження або будь-яке серйозне внутрішнє захворювання за 4 тижні до дослідження, що, на думку дослідника, може перешкодити цілям дослідження; наявність або ускладнення захворювання або інші стани, які, як відомо, перешкоджають абсорбції, розподілу, метаболізму або виведенню лікарських засобів; історія зловживання алкоголем або наркотиками; історія гіперчутливості до досліджуваного препарату або лікарських засобів з подібною хімічною структурою; прогресуюче смертельне захворювання; запланована хірургічна операція під час дослідження; донорство крові більше 500 мл протягом попередніх 3 місяців. Нікого не допускали до участі в дослідженні більше одного разу. Загальні положення суб'єкт, що, на думку дослідника, імовірно, не буде виконувати указання у процесі дослідження або не здатний співпрацювати внаслідок мовних проблем або розумової недорозвиненості або внаслідок психічного стану, через який людина не здатна зрозуміти природу, масштаб або можливі наслідки дослідження; суб'єкт, що знаходиться в періоді виведення з попереднього дослідження відповідно до діючого нормами; суб'єкт є дослідником або будь-яким співдослідником, науковим співробітником, фармацевтом, координатором дослідження, іншим дослідницьким персоналом, що безпосередньо бере участь у виконанні протоколу; прийом експериментального лікарського засобу протягом попередніх 30 днів до SCR. Біологічний статус Позитивна реакція в одному з наступних тестів на наявність: HBs антигену, анти-HCV антитіл, анти-ВІЛ1 антитіл, анти-ВІЛ2 антитіл; позитивні результати при перевірці сечі на наркотики під час SCR (амфетаміни/метамфетаміни, барбітурати, бензодіазепіни, канабіноїди, кокаїн, опіати); позитивний тест на алкоголь у видихуваному повітрі. Приклад 3: Варіанти лікування Деталі досліджуваних варіантів лікування Код лікарського засобу: HOE901 (препарат, що (Препарат інсуліну гларгіну U300) є у продажу, Лантус® U100) INN: Інсулін гларгін (рекомбінантний аналог Інсулін гларгін (рекомбінантний аналог людського інсуліну) людського інсуліну) Препарат Картриджі на 3 мл розчину U100 Картриджі на 3 мл розчину U300 A B B A B B (1 мл містить 3,637 мг 21 -Gly-30 a-L-Arg-30 b- (1 мл містить 10,913 мг 21 -Gly-30 a-L-Arg-30 bL-Arg людського інсуліну [еквімолярно 100 МО L-Arg людського інсуліну [еквімолярно 300 МО людського інсуліну], 30 мкг цинку, 2,7 мг млюдського інсуліну], 90 мкг цинку, 2,7 мг мкрезолу, 20 мг гліцерину 85%, HCl і NaOH до крезолу, 20 мг гліцерину 85%, HCl і NaOH до рН pН 4,0; питома вага 1,004 г/мл 4,0; питома вага 1,006 г/мл Доза/спосіб введення 0,4 Од/кг маси тіла; одна п/ш ін'єкція в 0,4 Од/кг маси тіла; одна п/ш ін'єкція в припупкову область живота після голодування припупкову область живота після голодування протягом ночі протягом ночі Виробник: Sanofi-Aventis Deutschland Gmb Виробник: Sanofi-Aventis Deutschland Gmb 40 Розрахунок дози для препарату Лантус®/інсулін гларгін Щоб розрахувати кількість інсуліну гларгіну, що вводиться кожному суб'єкту (0,4 Од/кг), масу тіла (у кг) визначали до одного десяткового знака і розраховану кількість інсуліну округляли у більшу або меншу сторону до цілих чисел, як показано в наступних прикладах: суб'єкт із масою тіла 75,3 кг одержував 30 Од інсуліну (75,3×0,4=30,12, округляли в меншу сторону до 30); суб'єкт із масою тіла 74,4 кг одержував 30 Од інсуліну (74,4×0,4=29,76, округляли у більшу 7 UA 110935 C2 5 10 сторону до 30). Масу тіла, зареєстровану під час дня 1 періоду 1, використовували для розрахунку дози досліджуваного препарату для періодів 2, 3 і 4, якщо маса тіла не змінювалася більше ніж на 2 кг у порівнянні з періодом 1. Кількість в одиницях була однаковою для інсуліну гларгіну U100 і інсуліну гларгіну U300. Ця питома вага однакова для обох лікарських продуктів. Однак, з урахуванням у три рази більш високої концентрації інсуліну гларгіну в інсуліні гларгіні U300 у порівнянні з інсуліном гларгіном U100, об’єм для введення, а отже і вага, складав 1/3 для інсуліну гларгіну U300. Шприци, що забезпечують індивідуальну дозу, готували по вазі. Вага нетто була зареєстрована тільки у вихідних документах дослідника. Розрахунок і підготовка дози для інфузій Таблиця 1 Підготовка інфузії Код лікарського засобу Глюкоза 25 30 35 40 45 Гепарин 0,9% Хлорид натрію 20 Глюкоза Гепарин натрію Intramed 15 INN Хлорид натрію Препарат Виробник Доза/спосіб введення 20% розчин для інфузії Ампула, що містить 5 мл розчину (5000 МО/мл) Сертифіковано, вибрано PROFIL в/в інфузія Сертифіковано, вибрано PROFIL в/в інфузія Сертифіковано, вибрано PROFIL в/в інфузія Розчин Розчин глюкози: 20% розчин глюкози вводили інфузією за допомогою Biostator для підтримання індивідуальних рівнів глюкози в крові суб'єктів на визначеному цільовому рівні. Другий інфузійний насос (частина Biostator) доставляв 0,9% розчин хлориду натрію для підтримання прохідності катетера. У випадку, якщо необхідна кількість 20% розчину глюкози перевищувала інфузійну потужність Biostator, був задіяний другий насос для інфузії глюкози. Гепарин: 10000 МО гепарину в 100 мл 0,9% розчину хлориду натрію вводили інфузією у двопросвітний катетер зі швидкістю приблизно 2 мл/год. для збереження його прохідності з метою вимірювання рівня глюкози за допомогою Biostator. Опис сліпих методів Дане дослідження було сліпим. Різні об’єми ін'єкцій виключають сліпий метод застосовно до препарату. Ін'єкцію виконував уповноважений медичний працівник, що не брав іншої участі в дослідженні. Дослідник мав доступ до коду рандомізації. Метод розподілу суб'єктів по групах лікування Досліджуваний препарат вводили тільки суб'єктам, включеним у дане дослідження відповідно до процедур, викладених в протоколі клінічного дослідження. Була створена схема рандомізації, що прив'язувала номери рандомізації, з розбивкою по статі, до послідовностей лікування двома препаратами Лантус®, які потрібно було вводити в періоди P1-P4. Ранком у день 1 періоду 1, як тільки дослідник підтвердив, що суб'єкти відповідають критеріям, зазначеним у протоколі, придатні суб'єкти були рандомізовані по ділянці. Згодом номер рандомізації був співвіднесений з номером суб'єкта в тому порядку, у якому придатність суб'єкта для дослідження була підтверджена перед P1. Перший суб'єкт, віднесений до гендерної групи після SCR, одержував перший номер рандомізації для відповідної гендерної групи. Наступний суб'єкт, що увійшов у групу, одержував наступний номер рандомізації в групі. Номер рандомізації використовували як номер лікувального набору, щоб співвідносити лікувальний набір із суб'єктом. Кожен суб'єкт одержував досліджуваний препарат під номером лікувального набору, що був призначений для нього. Лікувальний набір, що містить ДП, мав етикетку з загальною інформацією, номером лікувального набору, номером періоду, полем для запису номера суб'єкта на контейнерній коробці, а також додаткові заяви відповідно до вимог місцевого законодавства. Суб'єкти, що остаточно припиняли брати участь у дослідженні, зберігали номер суб'єкта і номер рандомізації, якщо вже встигли їх одержати. Упаковка і маркування 8 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 Досліджуваний препарат був упакований компанією Sanofi-Aventis Deutschland Gmb, Франкфурт-на-Майні, Німеччина, відповідно до плану рандомізації. Картриджі, що містять досліджуваний препарат, і коробки, у які вони були упаковані, мали як маркування номер дослідження, номер рандомізації, номер партії, умови зберігання, найменування спонсора і номер P. Уся партія досліджуваного препарату була одержана одним постачанням. Усі контейнери мали етикетки однакового формату. Крім того, був постачений 1 комплект етикеток для шприців. Досліджуваний препарат і резервний препарат зберігалися в різних холодильниках. Перед введенням досліджуваного препарату фармацевт або особа, призначена ним, готував шприци з відповідним досліджуваним препаратом і маркував шприц номером суб'єкта, номером рандомізації і номером відповідного періоду відповідно до контейнерів, що містять досліджуваний препарат. Зміст маркування відповідав місцевим нормативним правилам і вимогам. Умови зберігання Досліджуваний препарат зберігали в захищеному від світла місці при температурі від +2°C до +8°C. Досліджуваний препарат захищали від замерзання. У процесі підготовки не було необхідності захищати препарат від впливу світла. Резервні зразки (300 картриджів Лантус® U100 і 300 картриджів інсуліну гларгіну U300) зберігали в таких же безпечних умовах на рівні дослідницького центра. Приклад 4: Оцінка досліджуваного препарату Активність або фармакодинаміка Механізмом дії є стимуляція інсулінових рецепторів інсуліном гларгіном. Наступне периферичне споживання глюкози і пригнічення ендогенної продукції глюкози складають глюкодинамічний ефект, що приводить до зниження концентрації глюкози в крові. Утилізація глюкози, що відбувається в результаті, найкраще характеризується шляхом вимірювання кількості глюкози, необхідної для підтримання постійної концентрації глюкози в крові. Був використаний метод фіксації стану еуглікемії для оцінки кількості глюкози, необхідної для підтримання концентрацій глюкози в крові на 5% нижче вихідного рівня після ін'єкції інсуліну гларгіну. Методи клінічної оцінки Он-лайн визначення рівня глюкози в крові виконували за допомогою Biostator (Life Sciences instruments, Elkhart, IN, USA), використовуючи глюкозооксидазний метод. Рівень глюкози в крові за межами дослідницького центру визначали за допомогою аналізатора глюкози Super GL, також за допомогою глюкозооксидазного методу. Фармакодинамічні змінні/Кінцеві критерії оцінки Кількість глюкози, утилізованої на одиницю (дозу) введеного підшкірно інсуліну є мірою глюкодинамічного ефекту. Безупинно реєстрована швидкість інфузії глюкози (GIR) відображає профіль активності введеного інсуліну залежно від часу. Основна змінна/Кінцевий критерій оцінки Основною фармакодинамічною змінною є площа під кривою залежності швидкості інфузії -1 глюкози від часу протягом 24 годин [GIR-AUC(0-24 год.) (мг·кг )]. Вторинна змінна/Кінцевий критерій оцінки Вторинною фармакодинамічною змінною є час до 50% GIR-AUC(0-24 год.) [T50%-GIR-AUC(0-24 год.) (год.)]. Фармакокінетика Час збирання зразків Зразки крові для оцінки сироваткових концентрацій інсуліну гларгіну і С-пептиду брали за 1 годину, 30 хв. і безпосередньо перед підшкірною ін'єкцією досліджуваного препарату, а потім через 30 хв., 1 годину, 2 години і після цього раз на дві години аж до 24 годин, і через 30 годин після ін'єкції. Нумерація зразків інсуліну гларгіну була P00, P01, P02, P03, P04 і так далі, нумерація зразків C-пептиду була C00, C01, C02, C03, C04 і так далі (дивіться також план-графік дослідження). Кількість фармакокінетичних зразків Мінімум по 18 зразків відбирали за одне відвідування з фіксацією (P1-P4). У цілому від кожного суб'єкта було одержано 72 зразка. Виконання ФК-процедур Точний час збирання зразків повинен бути зареєстрований в CRF. Використовували спеціальні процедури для збереження і транспортування фармакокінетичних зразків (інсулін гларгін, С-пептид). 9 UA 110935 C2 5 10 15 Біоаналітичний метод Біоаналіз проводили, узявши за основу правила Належної Лабораторної Практики (GLP), застосовні до даного типу дослідження, викладені в керівництві "OECD Principles of Good Laboratory Practice" (у редакції 1997 р.), ENV/MC/CHEM (98)17, і правила GLP, прийняті в країні проведення дослідження. У зв'язку з відсутністю резервних зразків пріоритет віддавався визначенню інсуліну гларгіну. Інсулін гларгін Сироваткові концентрації інсуліну гларгіну визначали за допомогою радіоімуноаналізу (RIA) для людського інсуліну (Insulin RIA kit, ADALTIS, Italy), відкаліброваного для інсуліну гларгіну. REF набору 10624. Нижня межа кількісного визначення (LLOQ) для даного аналізу складала 4,92 мкОд/мл. C-пептид Сироваткові концентрації C-пептиду визначали за допомогою радіоімуноаналізу (RIA) для Cпептиду (C-peptide RIA kit, ADALTIS, Italy). REF набору для C-пептиду 10282. Нижня межа кількісного визначення (LLOQ) складала 0,090 нмоль/л. Короткий виклад біоаналітичного методу Аналіт Матриця Аналітичний метод Нижня межа кількісного визначення Аналітичний об’єм Посилання на метод 20 25 30 35 інсулін, С-пептид сироватка RIA 4,92 мкОд/мл інсулін; 0,090 нмоль/л C-пептид 100 мкл для інсуліну; 100 мкл для C-пептиду Adaltis S.p.A. Italy; REF набору 10624 інсулін (№ методу 435VAL02) і REF набору C-пептид 10282 (№ методу DMPK/FRA/2003-0002) Фармакокінетичні змінні/Кінцеві критерії оцінки Крива залежності концентрації інсуліну гларгіну від часу була мірою системної експозиції інсуліну з введеного підшкірно ДП. Основна змінна/Кінцевий критерій оцінки Основною фармакокінетичною змінною була площа під кривою залежності сироваткової -1 концентрації інсуліну гларгіну від часу [INS-AUC(0-24 год.) (мкОд·год.·мл )]. Вторинна змінна/Кінцевий критерій оцінки Вторинною фармакокінетичною змінною був час до 50% INS-AUC(0-24 год.) [T50%-INS-AUC(0-24 год.) (год.)]. Об’єм зразків крові Об’єм зразків крові Архівована кров/Генотипування 0 мл Гематологія/Клінічна хімія/Серологія (20+12 мл) 32 мл RBC, Hb, Hct (22 мл) необов'язково 4 мл Глюкоза в крові (2 мл/год.324) 256 мл Глюкоза в крові (0,3 мл434) 41 мл ФК інсуліну гларгіну (3,5 мл184) 252 мл Усього 585 мл Заходи для захисту сліпого характеру дослідження Дане дослідження було сліпим. Біоаналітичні визначення були виконані після завершення клінічної стадії. Лікувальний код був відомий для реєстрації будь-якого серйозного побічного ефекту (SAE), несподіваного й обґрунтовано пов'язаного з використанням ДП відповідно до рішення дослідника і/або спонсора. Приклад 5: Процедури дослідження Програма відвідування Процедури скринінгу Медичні документи кожного потенційного суб'єкта дослідження були перевірені перед початком дослідження для визначення права на участь. Суб'єкти голодували (за винятком води) протягом 10 годин до скринінгового обстеження під час SCR. Оцінювалися наступні параметри/результати обстеження 10 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 Вік і раса; фізичне обстеження (у тому числі серцево-судинної системи, грудної клітки і легень, щитовидної залози, черевної порожнини, нервової системи, шкіри і слизових оболонок, а також опорно-рухового апарата); відповідні медичні і хірургічні історії (повинні бути документовані тільки факти, що стосуються дослідження); -2 антропометричні дані: ріст і вага, розрахунок ІМТ (BMI) [маса в кг·(ріст у м) ]; артеріальний тиск і частота серцевих скорочень (через 5 хвилин у положенні лежачи і 3 хв. у вертикальному положенні); центральна температура тіла (тимпанічна); стандартна ЕКГ у 12 відведеннях; гематологічний статус, клінічна хімія й аналіз сечі (за допомогою щупа); коагуляційний статус (INR, aPPT); аналізсечі на наркотики; перевірка на алкоголь (аналізатор подиху); нормальний метаболічний контроль, що визначається як рівень глюкози в крові натще (≤100 -1 мг·дл ) і глікозилований гемоглобін (HbА1c ≤ 6,1%); Тест на гепатит B/C і ВІЛ. У випадку, якщо суб'єкт не пройшов відбір, усі дані, одержані під час SCR, включаючи лабораторні результати скринінгових тестів, були доступні в медичній карті суб'єкта. Опис по типу відвідування Період(и) Кожен період дослідження (P1-P4) продовжувався 2 дні, день 1 і день 2. День 1 був днем початку фіксації стану еуглікемії і введення досліджуваного препарату. День 2 був днем закінчення фіксації стану еуглікемії, що продовжувалася 30 годин після введення досліджуваного препарату. Між періодами дослідження (P1-P4) був період вимивання, що складає 4-18 днів. Будь-яка напружена діяльність (наприклад, їзда на гірському велосипеді, інтенсивне садівництво і так далі) була під забороною за 2 дні до кожного введення досліджуваного препарату. Не допускалося вживання алкогольних напоїв, грейпфрутового соку і стимулюючих напоїв, що містять похідні ксантину (чаю, шоколаду, кави, напоїв типу кока-коли і так далі) і грейпфрутів, починаючи за 24 години і до завершення фіксації стану еуглікемії. Суб'єкти голодували (за винятком води) протягом 10 годин до дня 1 кожного періоду дослідження (P1-P4) і продовжували голодувати (за винятком води) до завершення фіксації стану еуглікемії. Суб'єкти повинні були залишатися в клініці протягом приблизно 32 годин під час кожного відвідування з фіксацією. Ранком у день 1 періоду 1 суб'єкту був наданий 9-значний номер суб'єкта, починаючи з 276001001. Наступний суб'єкт, допущений до участі при SCR, одержував номер суб'єкта 276001002 і так далі. Перший суб'єкт одержував номер рандомізації 101. Наступний допущений суб'єкт одержував номер рандомізації 102. Суб'єктам пропонували поручитися, що в них не відбувалося клінічно значимих змін фізичного стану і що вони додержувалися загальних і дієтичних обмежень, визначених в протоколі, з часу попередніх періодів. Порушення критеріїв дослідження приводило до відсторонення суб'єктів від участі в дослідженні. Залежно від виду порушення суб'єкт міг бути виключений тільки з конкретного періоду, з можливістю перенесення дня дослідження. Будь-які порушення протоколу в кожному конкретному випадку обговорювалися зі спонсором попередньо. Будь-які зміни в стані здоров'я суб'єктів з моменту останнього періоду були зареєстровані в медичній документації суб'єкта (джерелі) і CRF. Артеріальний тиск, частоту серцевих скорочень і центральну температуру тіла (тимпанічну) реєстрували в положенні лежачи на спині після щонайменше 5-хвилинного відпочинку ранком у день 1, до і після завершення процедур фіксації через 30 годин після кожного введення досліджуваного препарату (день 2). Масу тіла, наявність алкоголю, показники RBC, Hb, Hct (тільки перед періодами фіксації P3 і P4) оцінювали тільки перед початком фіксації ранком у день 1. У день 1 кожного періоду суб'єктів приймали в клініці в 6:30 ранку. Після проведення описаних вище обстежень у суб'єктів підготовляли три венозні лінії. У дорзальну вену руки або в латеральну вену зап'ястя лівої руки вводили канюлю ретроградним чином і з'єднували з Biostator (Life Sciences instruments, Elkhart, IN, USA), щоб постійно забирати артеріалізовану венозну кров для визначення рівня глюкози в крові. Для досягнення артеріалізації ліву руку поміщали в "Hot-Box" при температурі приблизно 55°C. Другу венозну лінію поміщали в ліктьову 11 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 вену на лівій руці і використовували для збирання зразків сироваткового інсуліну гларгіну і визначення референсного рівня глюкози в крові. Третю канюлю поміщали у вену на протилежному передпліччі для проведення інфузії 20% розчину глюкози і 0,9% фізіологічного розчину через Biostator. Biostator визначав рівні глюкози в крові і коректував швидкість інфузії глюкози для підтримання рівнів глюкози в крові на 5% нижче індивідуального рівня глюкози в крові натще, що визначається як середнє з 3 значень рівня глюкози в крові натще, виміряних за 60, 30 і 5 хвилин до введення досліджуваного препарату. Додаткові зразки крові по 0,3 мл для визначення рівня глюкози в крові відбирали за 60, 30 і 5 хвилин до введення досліджуваного препарату для перевірки відносно лабораторного референсу на основі глюкозооксидазного методу. Приблизно в 09:00 ранку або інсулін гларгін U100 (препарат, що є у продажу), або інсулін гларгін U300 вводили ін'єкцією в область пупка на 5 см збоку від пупка (ліворуч, праворуч, ліворуч, праворуч), використовуючи стандартний спосіб шкірної складки. Використовували інсулінові шприци U100 (виробник: Beckton & Dickinson) об’ємом 0,5 мл з голками 0,30×8 мм (30G). Досліджуваний препарат маркували номером відповідного йому лікувального набору, номером суб'єкта (який повинен бути зазначений на контейнерній коробці після рандомізації) і номером періоду (дивіться розділ 8.5. Упаковка і маркування). Після введення досліджуваного препарату починали інфузію 20% розчину глюкози зі змінною швидкістю поки рівень глюкози в крові не падав на 5% від індивідуального рівня глюкози в крові натще, і цей рівень підтримувався. Тривалість періоду фіксації складала 30 годин. Швидкість доставки глюкози коректувалася Biostator у відповідь на зміни рівня глюкози в крові з інтервалами в 1 хвилину за допомогою заданого алгоритму. Значення рівнів глюкози в крові, одержані від Biostator, перевіряли відносно лабораторного референсу на основі глюкозооксидазного методу з 30-хвилинними інтервалами протягом всієї фіксації. За необхідності Biostator заново калібрували відповідно до результатів референсного методу лабораторії. Під час періоду фіксації суб'єкти залишалися в положенні лежачи на спині. Зразки крові для визначення концентрацій сироваткового інсуліну гларгіну і C-пептиду відбирали за 1 годину, 30 хв. і безпосередньо перед введенням препарату, а потім через 30 хв., 1 годину, 2 години і після цього раз на дві години аж до 24 годин, і через 30 годин після введення досліджуваного препарату. У день 2 кожного періоду дослідження (P1-P4) суб'єктів годували після завершення фіксації стану еуглікемії. Реєстрували артеріальний тиск, частоту серцевих скорочень і центральну температуру тіла (тимпанічну) і брали зразок для визначення рівня глюкози в крові. Суб'єктів відпускали з клініки після того, як безпека їх стану була підтверджена дослідником. Місця ін'єкцій оглядали протягом усього періоду фіксації. Будь-які зміни в стані здоров'я суб'єктів реєстрували в медичній документації суб'єкта (джерелі) і CRF. Гематологія для контролю безпеки RBC, Hb і Hct аналізували під час P3 на схильність до анемії під час P4. У випадку позитивного результату інтервал між P3 і P4 збільшували до максимально допустимих 18 днів і проводили додаткову оцінку RBC, Hb і Hct до початку P4. Процедури виведення з дослідження Суб'єкти поверталися для відвідування EOS у термін від 4 до 14 днів після P4. Суб'єкти голодували (за винятком води) протягом 10 годин. Будь-які зміни в стані здоров'я суб'єктів з моменту останнього періоду були зареєстровані в медичній документації суб'єкта (джерелі) і CRF. Оцінювалися наступні параметри/результати обстеження: фізичне обстеження (у тому числі серцево-судинної системи, грудної клітки і легень, щитовидної залози, черевної порожнини, нервової системи, шкіри і слизових оболонок, а також опорно-рухового апарата); маса тіла; артеріальний тиск і частота серцевих скорочень (через 5 хвилин у положенні лежачи на спині); центральна температура тіла (тимпанічна); стандартна ЕКГ у 12 відведеннях; гематологічний статус, клінічна хімія й аналіз сечі (за допомогою щупа); тест на β-HCG у сечі (тільки для жінок). Суб'єктів відпускали в день 2 кожного періоду після повного огляду дослідником наявних даних по безпеці. 12 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 Розклад збирання біологічних зразків Кров SCR (скринінг): Гематологія, клінічна хімія, HbА1c, серологія (тест на гепатит B/C, тест на ВІЛ); збирали приблизно 20 мл крові. P1-P4 (день 1 і 2): Рівень глюкози в крові Biostator автоматично вимірював рівень глюкози в крові з інтервалами в одну хвилину протягом всього періоду фіксації, включаючи період до введення досліджуваного препарату. -1 Об’єм крові, необхідний для Biostator, складав 2 мл·год. . По оцінках, об’єм крові 252 мл був необхідний для визначення рівня глюкози приладом Biostator протягом чотирьох періодів. Зразки крові (0,3 мл) для перевірки значень рівня глюкози в крові, одержаних від Biostator, збирали за 60, 30, 5 і 0 хвилин до введення доз і з 30-хвилинними інтервалами після введення доз до кінця фіксації (30 годин). По оцінках, об’єм крові 41 мл був зібраний за чотири періоди. Сироваткові концентрації інсуліну гларгіну і C-пептиду Зразки венозної крові (3,5 мл) збирали за 1 годину, 30 хв. і безпосередньо перед введенням доз, через 30 хв., 1 годину, 2 години і після цього раз на дві години аж до 24 годин, і через 30 годин після введення доз. По оцінках, об’єм крові 252 мл був зібраний за чотири періоди. Визначення інсуліну гларгіну було пріоритетним. Тільки зайві зразки були використані для визначення концентрації C-пептиду. RBC, Hb, Hct Венозну кров збирали до початку періодів фіксації 3 і 4. Приблизно 4 мл крові було зібрано за два періоди. Відвідування наприкінці дослідження (EOS): гематологія, клінічна хімія; було зібрано приблизно 12 мл крові; тест на β-HCG у сечі (тільки для жінок). Загальний обсяг крові SCR-EOS: В цілому приблизно 585 мл крові було зібрано від кожного суб'єкта протягом усього дослідження. Сеча Якісну перевірку сечі на наркотики проводили під час SCR і EOS. Перевірка сечі на наркотики включала перевірку на амфетаміни/метамфетаміни, барбітурати, бензодіазепіни, канабіноїди, кокаїн, опіати. Якісний аналіз сечі на безпеку за допомогою щупа проводили під час SCR і EOS. Аналіз сечі на безпеку складався з аналізів на: pН, білок, глюкозу, кров, еритроцити, лейкоцити, білірубін, уробіліноген, кетони, питому вагу і нітрити. Розклад вимірювання інших змінних дослідження Фізичне обстеження проводили під час SCR і EOS. Центральну температуру тіла (тимпанічну) вимірювали під час SCR, P1-P4 до і після періоду фіксації, а також під час EOS. Артеріальний тиск і частоту серцевих скорочень вимірювали через приблизно 5 хвилин відпочинку в положенні лежачи на спині, а також після 3 хвилин у вертикальному положенні під час SCR і EOS. У періоди P1-P4 артеріальний тиск і частоту серцевих скорочень реєстрували в положенні лежачи на спині за щонайменше 5 хвилин до початку процедур фіксації ранком у день 1 і після завершення процедур фіксації через 30 годин після кожного введення досліджуваного препарату (день 2). Електрокардіограми (стандартні в 12 відведеннях) реєстрували під час SCR і EOS. Масу тіла і ріст вимірювали під час SCR. Масу тіла реєстрували ранком у день 1 періодів P1-P4 (до введення досліджуваного препарату) і під час EOS. Перевірку на алкоголь (етанол, аналізатор подиху) проводили під час SCR і EOS, а також ранком у день 1 періодів P1-P4 (до введення досліджуваного препарату). Обмеження дослідження З вечора дня -1 (P1-P4) і протягом періодів (дні фіксації) суб'єкти утримувалися від вживання алкоголю, чаю, кави, цитрусових або напоїв типу коли, паління. Їсти цитрусові фрукти також було заборонено протягом усього дослідження. Суб'єктам пропонували вести розміряний спосіб життя протягом всього дослідження до останніх контрольних вимірювань, без інтенсивного фізичного навантаження. Визначення даних джерела Всі оцінки, перераховані нижче, що наведені в CRF, підкріплені відповідним чином підписаними визначеними документальними джерелами, що стосуються: ідентифікації суб'єкта, історії хвороби; клінічного обстеження, основних показників стану організму, маси тіла і росту; 13 UA 110935 C2 5 10 15 20 25 лабораторних результатів, ЕКГ; фармакокінетичних моментів часу; дат і часу відвідувань і обстежень; дат і часу введення, і місця введення; AE; тривалості фіксації (часу початку і закінчення); іншого. Для інших параметрів CRF вважали документальним джерелом. Приклад 6: Статистичні критерії У даному прикладі надана інформація про план статистичного аналізу для дослідження. План статистичного аналізу був складений до включення суб'єктів у дослідження. Визначення розміру вибірки INS-AUC(0-24 год.) був основним параметром, для якого робили розрахунок розміру вибірки. Для розрахунку цього розміру вибірки розглядали декілька внутрішньогрупових SD внутр. перетвореної по натуральному логарифму INS-AUC(0-24 год.) від 0,125 до 0,225. Метод розрахунку розміру вибірки для підходу середньої біоеквівалентності використовували для дизайну з 4 періодами, 2 варіантами лікування, 2 перехресними послідовностями введення. Якщо 90% ДІ (CI) для відношення препаратів повністю знаходилося в межах 0,80-1,25, тоді середню біоеквівалентність приймали за параметр. Дослідження HOE901/1022 стало основою для передбачення про варіабельність. На основі статистичного аналізу дослідження HOE901/1022 можна було передбачати величину 0,175 для внутрішньогрупового стандартного відхилення (SDвнутр.) по шкалі, перетвореній по натуральному логарифму. У таблиці нижче зазначена кількість суб'єктів, необхідна, щоб продемонструвати середню біоеквівалентність відношення скоректованих геометричних середніх (тестований проти референсного препарату) з використанням референсного інтервалу біоеквівалентності: 0,801,25, передбачаюче істинне відношення від 0,85 до 1,15 з 90% статистичною потужністю дослідження. 30 35 40 При даному дизайні дослідження, 20 суб'єктів (10 на послідовність) необхідно для демонстрації еквівалентності двох препаратів інсуліну гларгіну зі статистичною потужністю дослідження 90%, що робить можливим істинне відношення 0,9 у випадку, якщо істинне значення SDвнутр. по шкалі натуральних логарифмів складає 0,175. На випадок потенційного виходу учасників з дослідження участь у ньому приймали 24 рандомізованих суб'єкти. Опис суб'єктів Розподіл суб'єктів Докладні зведені дані про суб'єктів, включаючи число суб'єктів, допущених до дослідження, рандомізованих, підданих впливу (тобто, які одержували будь-яку кількість досліджуваного препарату), які завершили дослідження (тобто суб'єктів, які пройшли всі лікувальні періоди 14 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 дослідження), які перервали дослідження з основних причин для його припинення, були створені для кожної послідовності й в цілому для всіх суб'єктів. Розподіл суб'єктів під час останнього відвідування був представлений у вигляді переліку, що включає групу послідовності, розподільний статус наприкінці дослідження з датою останнього введення досліджуваного препарату, датою останнього відвідування, причиною переривання дослідження. Усі випадки виведення з дослідження, що мають місце під час або після початку першого введення досліджуваного препарату, були повністю документовані в звіті про клінічне дослідження (CSR). Відхилення від протоколу До блокування бази даних була уважно вивчена відповідність протоколу з погляду критеріїв включення в дослідження і виключення з нього, додержання режиму лікування, заборонених препаратів, а також термінів і наявності запланованих оцінок. Відхилення від протоколу були виявлені дослідницькою командою до блокування бази даних і перераховані в звіті про перевірку даних, у тому числі відсутні дані і припинення прийому ДП, і були класифіковані як несуттєві або суттєві відхилення. Були перераховані індивідуальні відхилення від критеріїв включення і виключення, повідомлені дослідником. Інші відхилення були перераховані і/або описані в CSR. Аналізована популяція Популяція, що підлягає аналізу Суб'єкти, виключені з будь-якої аналізованої популяції, були перераховані з указанням послідовності лікування і причини для виключення. Будь-яка інформація, що стосується справи, була повністю документована в CSR. У випадку, якщо суб'єкти одержували лікування, що відрізняється від того, котре було їм запропоноване відповідно до схеми рандомізації, були проведені аналізи відповідно до одержаного лікування, а не відповідно до рандомізованого лікування. Фармакокінетична популяція Усі суб'єкти без яких-небудь суттєвих відхилень, що стосуються введення досліджуваного препарату, і для яких були одержані ФК-параметри, були включені у фармакокінетичну популяцію. Для суб'єктів з незадовільними ФК-профілями в деякі, але не в усі дні дослідження, в аналіз були включені параметри задовільних профілів. Фармакодинамічна популяція Усі суб'єкти без яких-небудь суттєвих відхилень, що стосуються введення досліджуваного препарату, і для яких були одержані ФД-параметри, були включені у фармакодинамічну популяцію. Для суб'єктів з незадовільними GIR-профілями в деякі, але не в усі дні дослідження, в аналіз були включені параметри задовільних профілів. Популяція для оцінки безпеки Оцінка безпеки була основана на показниках суб'єктів, що одержували дозу досліджуваного препарату (експонована популяція), незалежно від кількості введеного препарату, включаючи суб'єктів, передчасно виведених з дослідження. Демографічні і вихідні характеристики Демографічні характеристики суб'єктів, історія хвороби і діагнози Були зібрані наступні дані: стать, вік під час скринінгу, ріст, маса тіла і раса. Для кожного суб'єкта індекс маси тіла (ІМТ) розраховували, виходячи з показників маси тіла і росту: -2 ІМТ=маса тіла [кг]·(ріст [м]) . Усі змінні, що стосуються демографічних і вихідних характеристик, були перераховані індивідуально й узагальнені. Відхилення від критеріїв включення, що стосуються історії хвороби і діагнозів, були перераховані й описані індивідуально. Вихідні фармакодинамічні параметри Вихідні рівні глюкози в крові були узагальнені по послідовностях. Вихідні параметри безпеки Для змінних безпеки, останнє одержане за розкладом значення до введення досліджуваного препарату в рамках періоду або в рамках дослідження, що було застосовно для даної змінної, приймали за вихідне значення. Якщо вихідне значення до введення дози було перевірене ще раз перед введенням дози, перевірене ще раз значення вважали вихідним і використовували в статистичних методах. Тривалість впливу досліджуваного препарату і додержання режиму терапії Подробиці про введення доз досліджуваного препарату і додаткова інформація були наведені індивідуально й узагальнені у відповідних випадках. 15 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 Використовувані раніше/супутні препарати/терапія Використовувані раніше і супутні препарати/терапія (при наявності) були закодовані згідно з довідковим списком лікарських засобів Всесвітньої організації охорони здоров'я (WHO-DRL) і були перераховані індивідуально. Аналіз фармакодинамічних змінних Опис фармакодинамічної змінної(их) Для досягнення порівнянності результатів між суб'єктами при введенні доз інсуліну залежно від маси тіла, для аналізу всі значення для GIR ділили на масу тіла суб'єкта в кг. Таким чином, далі GIR завжди означає стандартизовану по масі тіла швидкість інфузії глюкози. Основною ФД-змінною була площа під кривою залежності стандартизованої по масі тіла швидкості інфузії глюкози від -1 часу [GIR-AUC(0-24 год.) (мг·кг )]. Вторинною ФД-змінною був час (год.) до 50% GIR-AUC(0-24 год.) [T50%-GIR-AUC(0-24 год.) (год.)]. Були одержані наступні додаткові ФД-змінні: площа під кривою залежності стандартизованої по масі тіла швидкості інфузії глюкози від -1 часу аж до кінця фіксації [GIR-AUC(0-кінець) (мг·кг )]; часткові площі під кривою залежності стандартизованої по масі тіла швидкості інфузії -1 глюкози від часу [GIR-AUC(4-20 год.), GIR-AUC(0-12 год.), GIR-AUC(12-24 год.) (мг·кг )]; -1 -1 максимальна стандартизована по масі тіла швидкість інфузії глюкози [GIRmax (мг·кг ·хв. )]; час до GIRmax [GIR-tmax (год.)]. Для одержання значимих і достовірних даних величину GIR max і, відповідно, час до GIRmax одержували зі згладженої кривої GIR для кожного суб'єкта. Основний аналіз -1 Для оцінки відносної біоефективності (активності) для GIR-AUC(0-24 год.) (мг·кг ), неперетворений параметр аналізували за допомогою лінійної моделі зі змішаними ефектами. Змішана модель включає фіксовані умови для послідовності, періоду, препарату і випадкові умови для суб'єкта в межах послідовності, зі специфічними для препарату міжгруповими і внутрішньогруповими дисперсіями, а також дисперсією суб'єктів залежно від препарату. Точкову оцінку і 90% довірчий інтервал для відношення препаратів (T/R) одержували на основі теореми Філлера [Fieller, 1954]. Рішення про еквівалентну біоефективність (активність) приймали, якщо довірчий інтервал для відношення препаратів знаходився в межах 0,80-1,25. Допущення для розподілу змінної були перевірені. Вторинний аналіз/Аналіз вторинних змінних Були побудовані стандартизовані по індивідуальній і середній масі тіла GIR-профілі, а також середні процентні кумулятивні профілі залежно від часу. ФД-параметри були перераховані індивідуально, і була створена описова статистика. Відношення препаратів (T/R) з довірчими границями були одержані для часткових GIR-AUC -1 -1 -1 (мг·кг ) і максимальної стандартизованої швидкості інфузії глюкози [GIR max (мг·кг ·хв. )] за допомогою відповідної лінійної моделі зі змішаними ефектами, як описано для основного аналізу. Час до 50%-GIR-AUC (год.) і час до GIRmax [GIR-tmax (год.)] аналізували непараметрично. Проведення фіксації Були побудовані індивідуальні профілі концентрації глюкози в крові. Аналіз даних про безпеку Всі узагальнені дані про безпеку були одержані на базі популяції для вивчення безпеки. Індивідуальна фаза в період лікування для аналізу даних про безпеку починалася з першим введенням досліджуваного препарату і закінчувалася з відвідуванням EOS. Побічні ефекти (AE) Усі AE кодували за допомогою MedDRA (версії, що знаходиться у використанні). Визначення Виникаючі під час лікування AE Усі AE класифікували наступним чином: виникаючі під час лікування AE (TEAE): AE, що виникали в процесі періоду лікування в перший раз або збільшувалися в процесі періоду лікування, якщо були раніше; виникаючі не під час лікування AE (NTEAE): AE, що виникали поза періодом лікування без погіршення в процесі періоду лікування. Співвіднесення з препаратами 16 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 Для цілей аналізу кожен TEAE був віднесений до останнього препарату, наданого до появи і/або збільшення AE. Якщо TEAE з'являвся у відповідь на один препарат і збільшувався після більш пізнього препарату, його вважали TEAE для обох препаратів. Відсутня інформація У випадку відсутньої або суперечливої інформації AE вважали TEAE, якщо тільки не було повністю очевидно, що це не TEAE (наприклад, за рахунок часткових дат або іншої інформації). Якщо початкова дата AE була неповною або була відсутня, передбачалося, що він виникав після першого введення досліджуваного препарату, за винятком тих випадків, коли неповна дата вказувала на те, що AE виникав до лікування. Виникаючі під час лікування побічні ефекти Усі AE були перераховані індивідуально. Вони були узагальнені по препаратах, включаючи узагальнення по класах систем органів. Смерті, важкі й інші значні побічні ефекти В усіх таких випадках, смерті, важкі AE і інші значні AE були перераховані індивідуально і докладно описані в звіті про дослідження. Побічні ефекти, що призводять до припинення лікування AE, що призводять до припинення лікування, були перераховані індивідуально і докладно описані в звіті про дослідження. Клінічні лабораторні оцінки Потенційно клінічно значимі аномалії (PCSA) і критерії нестандартності були визначені в плані статистичного аналізу дослідження. Визначення потенційно клінічно значимих аномалій (PCSA) і визначення нестандартності були наведені по параметрах. Індивідуальні дані були перераховані по суб'єктах і по візитах, як і додаткова інформація. Суб'єктів з нестандартними показниками і суб'єктів з PCSA аналізували по препаратах, і в цілому для завершальної оцінки. Були перераховані суб'єкти з PCSA, що виникають після вихідного стану. Основні показники стану організму Потенційно клінічно значимі аномалії (PCSA) і критерії нестандартності були визначені в плані статистичного аналізу дослідження. Визначення PCSA і визначення нестандартності були наведені по параметрах. Суб'єктів з PCSA аналізували по препаратах, і в цілому для завершальної оцінки. Були перераховані суб'єкти з PCSA, що виникають після вихідного стану. Первинні значення й одержані параметри узагальнювали по препаратах, і в цілому для завершальної оцінки. Індивідуальні дані були перераховані по суб'єктах і по візитах з позначеними прапорцями аномаліями, як і додаткова інформація. ЕКГ Потенційно клінічно значимі аномалії (PCSA) і критерії нестандартності були визначені в плані статистичного аналізу дослідження. Визначення PCSA і визначення нестандартності були наведені по параметрах. Суб'єкти з PCSA наприкінці дослідження були проаналізовані в цілому. Були перераховані суб'єкти з PCSA, що виникають після вихідного стану. Первинні значення й одержані параметри під час SCR і EOS були узагальнені в цілому. Індивідуальні дані були перераховані по суб'єктах і по візитах з позначеними прапорцями аномаліями, як і додаткова інформація. Аналіз фармакокінетичних даних Фармакокінетичні параметри Фактичні відносні моменти часу були використані для одержання ФК-параметрів. Основною змінною була -1 INS-AUC(0-24 год.) (мкОд·год.·мл ). Вторинною ФК-змінною був час (год.) до 50% INS-AUC(0-24 год.) [T50%-INS-AUC(0-24 год.) (год.)]. Були одержані наступні додаткові ФК-змінні: -1 часткові INS-AUC [INS-AUC(4-20 год.), INS-AUC(0-12 год.), INS-AUC(12-24 год.) (мкОд·год.·мл )], -1 INS-AUC до кінця фіксації [INS-AUC(0-кінець) (мкОд·год.·мл )], -1 максимальна сироваткова концентрація інсуліну [INS-Cmax (мкОд·мл )], час до INS-Cmax [INS-Tmax (год.)]. Статистичний аналіз Описовий аналіз Описова статистика даних по концентрації була представлена по моментах часу в протоколі. 17 UA 110935 C2 5 10 15 20 25 30 35 40 Були побудовані індивідуальні і середні профілі сироваткових концентрацій інсуліну. Сироваткові концентрації інсуліну були перераховані індивідуально, і була створена описова статистика на кожну часову точку. Описову статистику ФК-параметрів створювали по препаратах. Були побудовані й описово охарактеризовані профілі для C-пептиду. Основний аналіз Для оцінки відносної біодоступності для INS-AUC(0-24 год.) логарифмічно перетворений параметр аналізували за допомогою лінійної моделі зі змішаними ефектами. Змішана модель включала фіксовані умови для послідовності, періоду, препарату і випадкові умови для суб'єкта в межах послідовності, зі специфічними для препарату міжгруповими і внутрішньогруповими дисперсіями, а також дисперсією суб'єктів залежно від препарату. Для INS-AUC(0-24 год.) точкову оцінку і 90% довірчі інтервали для відношення препаратів (T/R) одержували шляхом обчислення оцінок і 90% довірчих інтервалів для відмінностей між середніми значеннями препаратів у рамках моделі зі змішаними ефектами і наступного переведення в шкалу відношень шляхом антилогарифмічного перетворення. Рішення про еквівалентну біодоступність приймали, якщо довірчий інтервал для відношення препаратів знаходився в межах 0,80-1,25. Аналіз вторинних і додаткових ФК-параметрів Час до 50%-INS-AUC (год.) і час до максимальної концентрації [INS-Tmax (год.)] аналізували непараметрично. -1 Логарифмічно перетворені часткові INS-AUC і INS-AUC(0-кінець) (мкОд·год.·мл ), а також -1 максимальну сироваткову концентрацію інсуліну гларгіну [INS-Cmax (мкОд·мл )] аналізували з використанням відповідної лінійної моделі зі змішаними ефектами, як описано для основного аналізу. Оцінки точок і довірчих інтервалів наведені. C-пептид У міру можливості були побудовані й описово охарактеризовані профілі для C-пептиду. ФК/ФД-АНАЛІЗ ФК/ФД-аналізи проводили дослідницьким способом, у випадку доцільності. Приклад 6: Результати дослідження Розподіл суб'єктів В цілому 35 суб'єктів, 11 жінок і 24 чоловіки, були піддані скринінгу, з яких 24 здорових придатних суб'єкти були включені в дослідження, рандомізовані й одержали щонайменше одну дозу досліджуваного препарату. З 24 рандомізованих суб'єктів, 1 суб'єкт вибув з дослідження з власного бажання після першого періоду введення доз. Двадцять три (23) суб'єкти завершили дослідження відповідно до протоколу і були включені в аналізи фармакодинаміки (ФД) і фармакокінетики (ФК). Усі 24 суб'єкти, що одержували лікування, були включені в оцінку безпеки. Суттєві відхилення від протоколу були відсутні. Демографічні характеристики Були зібрані наступні дані: стать, вік під час скринінгу, ріст, маса тіла і раса. Для кожного суб'єкта індекс маси тіла (ІМТ) розраховували, виходячи з показників маси тіла і росту: ІМТ = -2 маса тіла [кг]·(ріст [м]) . 18 UA 110935 C2 5 10 15 20 25 30 35 Проведення фіксації Дві групи лікування, Лантус U100 і Лантус U300, були схожі з погляду вихідних концентрацій глюкози в крові у суб'єктів натще, що служили для визначення рівня фіксації глюкози у суб'єктів. Тривалість фіксацій після введення доз складала 30 годин і стільки ж у всіх періодах лікування. Основні кінцеві критерії оцінки Еквівалентність у біодоступності (експозиції) для препаратів Лантус U100 і Лантус U300 не була встановлена. Еквівалентність у біоефективності (активності) для препаратів Лантус U100 і Лантус U300 не була встановлена. Основні змінні Площа під кривою залежності сироваткової концентрації інсуліну гларгіну від часу з 0 до 24 годин (INS-AUC(0-24 год.)) не була еквівалентною для Лантус U100 і Лантус U300. Експозиція була менше приблизно на 40% у випадку U300. Площа під кривою залежності GIR від часу з 0 до 24 годин (GIR-AUC(0-24 год.)) не була еквівалентною для Лантус U100 і Лантус U300. Активність була менше приблизно на 40% у випадку U300. Вторинні змінні Час до 50% INS-AUC(0-24 год.) (год.) був подібним для Лантус U100 і Лантус U300. Час до 50% GIR-AUC(0-24 год.) (год.) був більше на 0,545 (год.) (0,158-1,030) для Лантус U300, що було статистично значимим. Безпека Не були зареєстровані ніякі серйозні побічні ефекти (AE). П'ять (5) суб'єктів за лікування (тестоване і референсне) повідомляли в цілому про 14 TEAE, усі з який були від легкої до помірної інтенсивності і пройшли без ускладнень. Найчастіше повідомляли про головний біль (4 суб'єкти за лікування), супроводжуваний нудотою, блюванням і лихоманкою (1 суб'єкт, що одержував U100), і процедурний біль (1 суб'єкт, що одержував U300). Слід зазначити, що головний біль є звичайним симптомом для досліджень фіксації і пов'язаний з інфузією гіперосмолярних розчинів глюкози. Однак не можна виключати зв'язок з досліджуваними препаратами. Не було зареєстровано реакцій у місцях ін'єкцій. Висновки Інсулін гларгін U100 і інсулін гларгін U300 не еквівалентні по біодоступності (експозиції) і біоефективності (активності). Експозиція й активність після інсуліну гларгіну U300 були менше приблизно на 40% у порівнянні з експозицією й активністю після введення такої ж кількості (0,4 Од/кг) інсуліну гларгіну U100. Однак інсулін гларгін U300 демонструвала навіть більш плоскі ФК (експозиція) і ФД (активність) профілі, ніж інсулін гларгін U100, що було б бажано для базального інсуліну. Ці дивні і несподівані відмінності в експозиції й активності між препаратами інсуліну гларгіну U100 і інсуліну гларгіну U300 після однакових п/ш доз, введених здоровим суб'єктам, ефективно 19 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 продемонстровані на кресленнях нижче. Слід зазначити, що в той же час рівень глюкози в крові був постійним. Введення інсуліну гларгіну U300 пройшло без проблем з безпекою і переносимістю. Приклад 7: Обґрунтування дослідження для вивчення в порівнянні глюкодинамічної активності й експозиції трьох різних підшкірних доз інсуліну гларгіну U300 Результати дослідження на здорових суб'єктах (дивіться приклади 1-6) показали нееквівалентність по експозиції й ефективності між Лантус® U100 і інсуліном гларгіном U300. Суб'єкти одержували одну і ту ж дозу інсуліну гларгіну (0,4 Од/кг) для U100 і U300, однак при доставці однієї і тієї ж кількості в одиницях з U300 спостерігалися приблизно на 40% менші експозиція й ефект, ніж при доставці з U100. Однак інсулін гларгін U300 демонстрував навіть більш плоский фармакодинамічний профіль, ніж Лантус® U100, що було б бажано для базального інсуліну. Тому в новому дослідженні, описаному в наступних прикладах, порівнюється глюкодинамічна активність і експозиція трьох різних підшкірних доз інсуліну гларгіну U300 відносно стандартної дози Лантус® U100 як препарату порівняння в умовах фіксації стану еуглікемії у пацієнтів з діабетом І типу. Дане дослідження спрямоване на зразкове визначення дози U300, яка дорівнює по ефективності 0,4 Од/кг Лантус® U100, що оцінюється по параметрах утилізації глюкози в крові, одержуваних за допомогою методу фіксації. Експозицію інсуліну гларгіну оцінювали на основі профілів залежності концентрації від часу після підшкірного введення, а активність як утилізацію глюкози на одиницю інсуліну. Дане дослідження призначене для оцінки метаболічного ефекту й експозиції різних доз інсуліну гларгіну U300 у порівнянні зі стандартною дозою Лантус® U100 в умовах фіксації стану еуглікемії у пацієнтів з цукровим діабетом І типу. Дослідження включало 4 варіанти лікування (R, T1, T2 і T3), 4 періоди лікування (TP1-4) і 4 послідовності. Було одне скринінгове відвідування (від D-28 до D-3), 4 лікувальні відвідування (від D1 до D2 у TP1-TP4), і одне відвідування наприкінці дослідження (між D5 і D14 після останнього введення доз) із завершальною оцінкою параметрів безпеки. Суб'єктів піддавали кожному з варіантів лікування R, T1, T2 і T3 один раз перехресним, подвійним сліпим і рандомізованим чином за схемою латинського квадрата. Ця схема вважається придатною для оцінки фармакологічного ефекту й експозиції різних доз інсуліну гларгіну U300 у порівнянні з Лантус® U100. Доза Лантус® U100 0,4 Од/кг, вибрана для дослідження, добре вивчена для цілей забезпечення стану еуглікемії у пацієнтів з діабетом І типу і її часто використовують в інших дослідженнях по фіксації у пацієнтів з діабетом І типу. Тестували три різні дози для інсуліну гларгіну U300, 0,4, 0,6 і 0,9 Од/кг. Цей діапазон доз дозволяє інтраполювати зразкову дозу, що дорівнює по ефективності дозі 0,4 Од/кг Лантус® U100. Дозу 0,4 Од/кг інсуліну гларгіну U300 уже тестували на здорових добровольцях (дивіться приклади 1-6) і було встановлено, що вона менш активна, ніж 0,4 Од/кг Лантус® U100 протягом 30 годин, визначеного закінчення періоду спостереження. Біоактивність 0,4 Од/кг інсуліну гларгіну U300, виміряна за рахунок загальної утилізації глюкози, була на 39,4% нижче, ніж така у референсного препарату (0,4 Од/кг Лантус® U100). Відповідно, очікувалося, що більш висока доза інсуліну гларгіну U300, наприклад 0,6 Од/кг інсуліну гларгіну U300, приведе до приблизно еквівалентної глюкодинамічної активності в порівнянні з 0,4 Од/кг Лантус® U100. Крім того, пропорційне збільшення дози дозволяло вивчити профілі експозиції й ефекту для пропорційності доз. Дослідження на пацієнтах з діабетом І типу дозволяло уникнути змішаного впливу ендогенного інсуліну і краще дозволяло оцінити експозицію і тривалість дії. Крім того, відсутність аналізу, специфічного для інсуліну гларгіну, змушувало використовувати аналіз, що визначає весь ендогенний інсулін. Таким чином, будь-яке додаткове джерело інсуліну, крім екзогенного інсуліну гларгіну, було б причиною помилкових занадто завищених концентрацій інсуліну. Дане дослідження мало перехресний дизайн; із практичних і етичних розумінь не більше ніж 3 дози U300 порівнювалися з Лантус® U100. Для оцінки глюкодинамічної активності препаратів інсуліну пролонгованої дії необхідні умови фіксації стану еуглікемії протягом аж до 36 годин унаслідок більшої тривалості дії. Активний фармацевтичний інгредієнт, інсулін гларгін, був однаковим в обох препаратах, U100 і U300. Дози, використовувані в даному дослідженні, знаходилися в звичайно використовуваних межах. Хоча в цілому ризик гіпоглікемії повністю не був виключений, він знаходився під контролем завдяки методу фіксації стану еуглікемії. Фармакодинаміка 20 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 Фармакодинамічну активність інсуліну гларгіну оцінювали методом фіксації стану еуглікемії у пацієнтів з діабетом І типу, що є встановленою стандартною процедурою для оцінки ефекту введених екзогенних препаратів інсуліну на утилізацію глюкози в крові. Параметрами, специфічними для оцінки утилізації глюкози в умовах фіксації стану еуглікемії, були стандартизована по масі тіла швидкість інфузії глюкози (GIR), загальна кількість утилізованої глюкози, GIR-AUC0-36 і часи до визначеної частки у відсотках від GIR-AUC0-36, наприклад час до 50% GIR-AUC0-36. Допоміжними параметрами були максимально згладжені стандартизовані по масі тіла GIR, GIRmax, а також час до GIRmax, GIR-Tmax. Тривалість дії інсуліну гларгіну визначали на основі часу між введенням доз і попередньо обговорених відхилень вище рівня стану еуглікемії (фіксації). Моніторинг рівня глюкози проводили протягом 36 годин у зв'язку з тривалістю дії інсуліну гларгіну після підшкірного введення. Фармакокінетика Внаслідок уповільненого характеру вивільнення інсуліну гларгіну в концентраційному профілі були відсутні виражені піки. Внаслідок цього, час до 50% INS-AUC (T50% INS-AUC0-36) розраховували як міру для положення в часі профілю експозиції інсуліну гларгіну, а INS-Cmax і INS-Tmax служили як додаткові міри. Основні цілі дослідження Основною метою дослідження була оцінка коефіцієнтів метаболічного ефекту трьох різних доз інсуліну гларгіну U300 у порівнянні з 0,4 Од/кг Лантус® U100. Вторинні цілі дослідження Вторинними цілями дослідження були оцінка коефіцієнтів експозиції трьох різних доз інсуліну гларгіну U300 у порівнянні з 0,4 Од/кг Лантус® U100, порівняння тривалості дії різних доз інсуліну гларгіну U300 відносно 0,4 Од/кг Лантус® U100, вивчення залежності ефекту дози й експозиції дози інсуліну гларгіну U300, а також оцінка безпеки і переносимості інсуліну гларгіну U300 для пацієнтів з діабетом І типу. Приклад 8: Дизайн дослідження, опис протоколу Одноцентрове, подвійне сліпе, рандомізоване, перехресне (4 варіанти лікування, 4 періоди лікування і 4 послідовності; латинський квадрат) дослідження фази I з активним контролем, із тривалістю періоду вимивання між періодами лікування (5-18 днів, переважно 7 днів), за участі чоловіків і жінок, що страждають цукровим діабетом І типу, які одержують однократні дози інсуліну гларгіну, що відповідають 0,4 Од/кг Лантус® U100 (=референс R), 0,4 Од/кг інсулін гларгін U300 (=тест T1), 0,6 Од/кг інсулін гларгін U300 (=тест T2), 0,9 Од/кг інсулін гларгін U300 (=тест T3). Чотири варіанти лікування R і T1-3 застосовувалися перехресно під час чотирьох періодів лікування (TP1-TP4) з чотирма послідовностями R-T1-T2-T3, T3-R-T1-T2, T2-T3-R-T1, T1-T2-T3-R, випадковим чином призначеними для суб'єктів (у співвідношенні 1:1:1:1). Тривалість участі в дослідженні Загальна тривалість дослідження для одного суб'єкта: приблизно 4-11 тижнів (мінімальнамаксимальна тривалість, залежно від періоду вимивання, виключаючи скринінг). Тривалість кожної частини дослідження для одного суб'єкта: - скринінг: 3-28 днів (з D-28 по D-3), - періоди лікування 1-4: 2 дні (1 ночівля), - вимивання: 5-18 днів (переважно 7 днів між послідовними введеннями доз), - відвідування наприкінці дослідження: 1 день між D5 і D14 після останнього введення досліджуваного препарату. Приклад 9: Відбір суб'єктів Заплановане число суб'єктів: щонайменше 24 суб'єкти мали бути включені в дослідження, щоб одержати 20 обстежених суб'єктів. Критерії включення: Демографія I 01. Чоловіки або жінки у віці від 18 до 65 років, включно, що страждають цукровим діабетом І типу більше одного року по визначенню Американської діабетичної асоціації (American Diabetic 21 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 Association. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 1998; 21:5-19). I 02. Загальна доза інсуліну 30 МО/л, естрадіол 40 грамів/день). E 11. Паління більше 5 сигарет або їх еквівалента на день, нездатність утримуватися від паління під час дослідження. E 12. Надмірне споживання напоїв, що містять ксантинові основи (>4 чашок або склянок/день). E 13. У жінок вагітність (визначувана по позитивному тесту на β-HCG), грудне вигодовування. Речовини, що заважають E 14. Будь-які лікарські препарати (включаючи звіробій) протягом 14 днів до включення або протягом 5-кратного періоду напіввиведення або фармакодинамічного напіврозпаду даного препарату, залежно від того, що довше, і регулярне використання будь-яких лікарських засобів, крім інсуліну, в останньому місяці перед початком дослідження, за винятком гормонів щитовидної залози, гіполіпідемічних і антигіпертензивних препаратів, і, для жінок, за винятком гормональної контрацепції або замісної гормональної терапії при менопаузі; будь-яка вакцинація протягом останніх 28 днів. Загальні положення E 15. Суб'єкт, який, на думку дослідника, імовірно, не буде виконувати указання у процесі дослідження або не здатний співпрацювати внаслідок мовних проблем або розумової недорозвиненості. E 16. Суб'єкт, що знаходиться в періоді виведення з попереднього дослідження відповідно до діючих норм. E 17. Суб'єкт, з яким неможливо зв'язатися в екстреному випадку. E 18. Суб'єкт є дослідником або будь-яким співдослідником, науковим співробітником, фармацевтом, координатором дослідження, іншим дослідницьким персоналом, що безпосередньо бере участь у виконанні протоколу. Біологічний статус E 19. Позитивна реакція в одному з наступних тестів на наявність: поверхневого антигену вірусу гепатиту B (HBs Аг), антитіл проти ядра вірусу гепатиту B (анти-HBc Ат), якщо сполука має позитивну імунну активність, антитіла проти вірусу гепатиту C (анти-HCV2), антитіла проти вірусу імунодефіциту людини 1 і 2 (анти-ВІЛ1 і анти-ВІЛ2 Ат). E 20. Позитивні результати при перевірці сечі на наркотики (амфетаміни/метамфетаміни, барбітурати, бензодіазепіни, канабіноїди, кокаїн, опіати). E 21. Позитивний тест на алкоголь. Специфічні для дослідження фактори E 22. Відома гіперчутливість до інсуліну гларгіну і ексципієнтів. E 23. Будь-яка історія або наявність тромбозу глибоких вен ніг або часта поява тромбозу глибоких вен ніг у родичів першого ступеня (батьки, брати, сестри і діти). Приклад 10: Варіанти лікування Досліджуваний препарат Інсулін гларгін. Використовували два різних препарати інсуліну гларгіну: - розчин для ін'єкцій Лантус® U100, що містить 100 Од/мл інсуліну гларгіну (препарат, що є у продажу), - розчин для ін'єкцій інсуліну гларгіну U300, що містить 300 Од/мл інсуліну гларгіну. Доза: - Лантус® U100: 0,4 Од/кг (=референс R), - інсулін гларгін U300: 0,4, 0,6 і 0,9 Од/кг (=тест T1-T3). Контейнер: 3-мл скляні картриджі. Спосіб введення: підшкірно горизонтально на відстані 5 см праворуч і ліворуч від пупка. Умови: натще. Тривалість лікування: 1 день у кожному періоді, одна доза. Початок: 09:00 у день 1 (D1) у періоди лікування 1-4 (TP 1-4). Додаткові методи лікування для 100% включених у дослідження суб'єктів надані. 60 23 UA 110935 C2 Таблиця 4 Варіанти лікування Референсне лікування Лантус® U100 Інсулін гларгін (рекомбінантний аналог INN людського інсуліну) Картриджі на 3 мл розчину U100 1 мл містить A B B 3,637 мг 21 -Gly-30 a-L-Arg-30 b-L-Arg людського інсуліну [еквімолярно 100 МО людського інсуліну] Препарат 30 мкг цинку 2,7 мг м-крезолу 20 мг гліцерину 85% HCl і NaOH, pН 4,0 питома вага 1,004 г/мл Доза 0,4 Од/кг Виробник sanofi-aventis Deutschland GmbH Номер партії Препарат, що є у продажу, одержаний через CRO Тестоване лікування Інсулін гларгін U300 Інсулін гларгін (рекомбінантний аналог людського інсуліну) Картриджі на 3 мл розчину U300 1 мл містить A B B 10,913 мг 21 -Gly-30 a-L-Arg-30 b-L-Arg людського інсуліну [еквімолярно 300 МО людського інсуліну] 90 мкг цинку 2,7 мг м-крезолу 20 мг гліцерину 85% HCl і NaOH, pН 4,0 питома вага 1,006 г/мл 0,4 Од/кг 0,6 Од/кг 0,9 Од/кг sanofi-aventis Recherche & Development, Montpelier, France Підлягає визначенню INN = міжнародна непатентована назва. 5 10 15 20 25 30 Введення доз Дослідження було однодозовим, що включає в цілому 4 введення досліджуваного препарату. Суб'єкти були рандомізовані по різних послідовностях референсного і тестованих варіантів лікування так, що кожен суб'єкт одержував референсне лікування (R) і кожне з тестованих варіантів лікування (T1-3) один раз. Ін'єкції робили ліворуч або праворуч від пупка, при цьому обидві ділянки використовували для окремих ін'єкцій. Період вимивання тривалістю 5-18 днів розділяв послідовні дні введення доз, переважно він складав 7 днів (7 днів між послідовним введенням доз). Тривалість періоду вимивання варіювалася індивідуально, що дозволяло пристосовуватися до потреб як учасника, так і дослідника. З досвіду, 5 днів складали мінімальний період відновлення, дозволяючи проводити 1 фіксацію на тиждень для учасника, тоді як 18 днів означали перерву в 3 тижні між днями введення доз, що надавало суб'єктам більше свободи у виконанні своїх зобов'язань, не пов'язаних з дослідженням, якщо це було неминуче. ДП вводили в стані натще; суб'єкт продовжував голодувати протягом усього періоду фіксації. Концентрація глюкози в крові знаходилася в межах 5,5 ммоль/л (100 мг/дл) ± 20% без інфузії глюкози в останню годину перед введенням доз протягом періоду передфіксації. Коли рівень глюкози в крові залишався стабільним протягом щонайменше 1 години без інфузії глюкози, вводили ДП. Введення ДП проводили не раніш 09:00 годин ранку і не пізніше 14:00 у день 1 у періоди лікування 1-4. Якщо рівень глюкози в крові не стабілізувався до 14:00 годин, дозу не вводили. Відвідування закінчувалося і суб'єкту призначали нове відвідування для введення дози через 1-7 днів. Для кожного суб'єкта і кожного введення дози використовували новий картридж. Введення ДП виконував співробітник, який ніяк інакше не залучений у дослідження і не є частиною дослідницького колективу в CRO. Цей співробітник одержував код рандомізації для підготовки введення ДП відповідно до відкритого списку рандомізації і відповідно до нього вводив дози суб'єктам. Підготовка і введення доз супроводжувалися і перевірялися другим незалежним співробітником. Відповідні документи, що стосуються підготовки доз і послідовності лікування, були строго конфіденційними і не розголошувалися ніякій іншій особі. Розрахунок дози ДП (інсулін гларгін) Щоб розрахувати кількість інсуліну гларгіну, що вводиться кожному суб'єкту, масу тіла (у кг) визначали до одного десяткового знака і розраховану кількість інсуліну округляли у більшу або 24 UA 110935 C2 5 10 15 меншу сторону до цілих чисел, як показано в наступних прикладах для дози 0,6 Од/кг інсуліну гларгіну: суб'єкт із масою тіла 75,3 кг одержував 45 Од інсуліну (75,3×0,6=45,18, що округлялося в меншу сторону до 45); суб'єкт із масою тіла 74,4 кг одержував 45 Од інсуліну (74,4×0,6=44,64, що округлялося у більшу сторону до 45). Масу тіла, зареєстровану під час D1 у TP1, використовували для розрахунку дози досліджуваного препарату для всіх періодів лікування. Дозу досліджуваного препарату не змінювали, якщо маса тіла суб'єкта мінялася менше ніж або рівно на 2 кг між TP1 і одним з наступних TP. Якщо маса тіла суб'єкта мінялася більше ніж на 2 кг між TP1 і одним з наступних TP, дозу досліджуваного препарату повторно розраховували, виходячи з маси тіла в D1 відповідного періоду лікування. Шприци і голки Використовували тільки шприци з голками, прикріпленими відповідним чином для точного введення невеликих кількостей ін'єкційного розчину (наприклад, Becton Dickinson, Ref 305502, Розміри: 1-мл 27G 3/8 0,4010). Шприци були надані дослідником. Інші препарати Інші препарати, використовувані в процесі процедури фіксації, описані в таблиці 5. Таблиця 5 Підготовка інфузії Код лікарського засобу Глюкоза Препарат INN Глюкоза Гепарин Натрій Гепарин Intramed 0,9% хлорид Хлорид натрію натрію Інсулін Апідра® Глулізин 20% розчин для інфузії Ампула, що містить 5 мл розчину (5000 МО/мл) Розчин 100 Од/мл для ін'єкцій Виробник Сертифіковано вибрано PROFIL Сертифіковано вибрано PROFIL Сертифіковано вибрано PROFIL sanofi-aventis Доза/спосіб введення в/в інфузія в/в інфузія в/в інфузія в/в інфузія 20 25 30 35 40 45 Розчин глюкози, розчин хлориду натрію, гепарину й інсулін глулізин були надані дослідником. Розчин глюкози: 20% розчин глюкози вводили інфузією за допомогою Biostator™ для підтримання індивідуальних рівнів глюкози в крові суб'єктів на визначеному цільовому рівні. Другий інфузійний насос (частина Biostator™) доставляв 0,9% розчин хлориду натрію для підтримання прохідності катетера. У випадку, якщо необхідна кількість 20% розчину глюкози перевищувала інфузійну потужність Biostator™, був задіяний другий насос для інфузії глюкози. Гепарин: розчин гепарину в низькій дозі (10000 одиниць гепарину/100 мл фізіологічного розчину) вводили інфузією через двопросвітний катетер. Розчин гепарину забирався разом із кров'ю, використовуваною для вимірювання рівня глюкози в крові за допомогою Biostator™, в інший просвіт катетера і призначався для запобігання згортанню крові в системі. Інсулін глулізин: 15 Од Апідра® [100 Од/мл] вносили в 49 мл фізіологічного розчину, до якого додавали 1 мл власної крові суб'єкта для запобігання адгезії, що давало концентрацію 0,3 Од/мл розчину, який вводили інфузією з індивідуальною швидкістю для досягнення еуглікемії. Опис сліпих методів Суб'єкти одержували чотири різних варіанти лікування (R, T1, T2 і T3) у рандомізованому, сліпому і перехресному дизайні дослідження. Для збереження сліпого характеру дослідження, у розподілі і введенні ДП був задіяний співробітник, що володіє інформацією, від третьої сторони. Цей співробітник ніяк інакше не був залучений у дослідження і/або не був частиною дослідницького колективу в CRO, нікому не розголошував інформацію і забезпечував підтримання сліпого характеру дослідження. Він/вона одержував(ла) код рандомізації і відповідно до нього вводив(ла) дози суб'єктам. Готування ДП і введення доз також супроводжувалося і перевірялося другим незалежним співробітником, що також мав доступ до коду рандомізації, але також в однаковій мірі був пов'язаний угодою про конфіденційність. Метод розподілу суб'єктів по групах лікування 25 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 ДП вводили відповідно до протоколу клінічного дослідження тільки суб'єктам, що дали інформовану згоду в письмовому вигляді. Суб'єкти, що відповідають усім критеріям включення/виключення, розподілялися по групах безпосередньо перед введенням досліджуваного препарату в день 1 періоду лікування 1. Порядковий номер суб'єкта - відповідно до хронологічного порядку включення в дослідження ранком у D1 періоду лікування 1. 9-значний номер суб'єкта складався з 3 компонентів (наприклад, 276 001 001, 276 001 002, 276 001 003 і так далі), з яких перші 3 цифри (276) являють собою номер країни, середня 3 цифра являють собою номер центра й останні 3 цифри являють собою порядковий номер суб'єкта в даному центрі. Номер суб'єкта залишався незмінним і дозволяв ідентифікувати суб'єкта протягом всього дослідження. Лікувальний номер - у запланованому порядку відповідно до списку рандомізації, при цьому наступний придатний суб'єкт завжди одержував наступний лікувальний номер відповідно до списку рандомізації. Введення ДП проводили відповідно до рандомізованої послідовності лікування. Суб'єкти, виведені з дослідження, зберігали свій номер суб'єкта і лікувальний номер, якщо вже встигли їх одержати. Узяті на заміну суб'єкти мали інший ідентифікаційний номер (тобто 500 + номер суб'єкта, що припинив участь у дослідженні). Кожен суб'єкт одержував ту ж послідовність лікування, що і суб'єкт, що припинив участь у дослідженні. Суб'єктам, що не пройшли скринінг, надавався інший номер, наприклад 901, 902 (який мав бути записаний у CRF тільки у випадку AE, що має місце в скринінговий період після підписання інформованої згоди). Примітки: рандомізація суб'єктів відбувалася після того, як дослідник підтверджував, що суб'єкт придатний для даного дослідження. Вихідними параметрами були параметри, одержані ближче усього до введення доз. Упаковка і маркування Розчин інсуліну гларгіну U300 був наданий sanofi-aventis у коробках, що перегруповуються, з 3-мл картриджами. Відповідний номер ДП був упакований під відповідальність sanofi-aventis відповідно до вимог належної виробничої практики і місцевих нормативних вимог і був наданий у CRO. Зміст маркування відповідав місцевим нормативним правилам і вимогам. Лантус® U100 є комерційно доступним препаратом і був замовлений CRO. Умови зберігання Усі ДП зберігалися у відповідній замкненій кімнаті під відповідальність дослідника і могли бути доступні тільки для уповноваженого персоналу. ДП повинні були зберігатися при температурі від +2°C до +8°C, захищатися від світла і не заморожуватися. Доступ до коду рандомізації в процесі дослідження Для збереження сліпого характеру дослідження, за розподіл і введення ДП відповідав співробітник, що володіє інформацією, від третьої сторони. Ця людина ніяк інакше не була залучена у дослідження і/або не була частиною дослідницького колективу в CRO, нікому не розголошувала інформацію і забезпечувала підтримання сліпого характеру дослідження. Він/вона одержував(ла) код рандомізації і відповідно до нього вводив(ла) дози суб'єктам. Готування ДП і введення доз також супроводжувалося і перевірялося другим незалежним співробітником, що також мав доступ до коду рандомізації, але також в однаковій мірі був пов'язаний угодою про конфіденційність. У випадку виникнення побічного ефекту, код не мав бути розкритий, за винятком ситуації, коли інформація про досліджуваний препарат важлива для лікування суб'єкта. Для кожного суб'єкта був підготовлений у конверті матеріал, що розкриває код, який містить назву лікувального препарату. Він зберігався в безпечному місці в центрі протягом усього клінічного дослідження. Після завершення клінічного випробування спонсор забирав весь матеріал, що розкриває код (у відкритому або запечатаному вигляді). У випадку порушення сліпого характеру дослідження дослідник документував дату відкриття інформації і причину розкриття коду у вихідних даних. Дослідник, клінічний фармацевт центра або інший персонал, допущений до збереження і розподілу ДП, ніс відповідальність за забезпечення того, щоб ДП, використовуваний у дослідженні, надійно зберігався, відповідно до вказівок спонсора і відповідно до застосовних нормативних вимог. Усі ДП розподілялися відповідно до протоколу клінічного дослідження, і дослідник ніс відповідальність за забезпечення чіткого ведення записів про виданий і повернутий ДП. Супутнє лікування 26 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 Використання супутніх лікувальних препаратів не допускалося протягом дослідження, що зазначено в критерії виключення № E 14, за винятком препаратів, наведених у тому ж пункті, і припинялося у визначені періоди часу (дивіться E14) до включення суб'єкта в день 1 періоду лікування 1. Для запобігання перешкодам від стандартного лікування суб'єкта інсуліном для вимірювань при фіксації, суб'єкти повинні були утримуватися від використання базальних інсулінів і переходити на препарати інсуліну середньої або короткої тривалості дії, починаючи за 48 годин до введення доз у D1 періодів TP1-TP4, якщо звичайно використовують препарати інсуліну пролонгованої дії, тобто Лантус® (інсулін гларгін), Левемір® (детемір) або інсулін ультраленте, препарати інсуліну короткої дії, починаючи за 24 години до введення доз у D1 періодів TP1TP4, якщо звичайно використовують препарати інсуліну середньої тривалості дії, тобто NPHінсулін. Останню підшкірну ін'єкцію інсуліну короткої дії проводили не пізніше ніж за 9 годин до введення досліджуваного препарату. Суб'єкти на помповій терапії припиняли інфузію інсуліну ранком у день 1, щонайменше за 6 годин до кожного введення ДП (приблизно в 03:00 годин, вважаючи початком введення ДП 09:00). Для симптоматичних побічних ефектів, що не ставлять під загрозу безпеку суб'єктів (наприклад, головного болю), супутні препарати були зарезервовані на випадок побічних ефектів сильної інтенсивності або помірної інтенсивності, що продовжуються довго. Зокрема, використання ацетамінофену/парацетамолу було заборонено, якщо відомо про ризик гепатотоксичності або як тільки з'являлися аномалії печінкових ферментів. Однак, якщо специфічне лікування було необхідне з якої-небудь причини, точний запис мав бути зроблений у відповідній формі, включаючи назву препарату (міжнародна непатентована назва), добову дозу і тривалість його використання. Спонсор мав бути проінформований протягом 48 год. по електронній пошті або по факсу, за винятком лікування головного болю. Лікування потенційних алергічних реакцій повинно було здійснюватися відповідно до рекомендацій, опублікованих в іншому місці (Samspon H.A., Munoz-Furlong A., Campbell R.L. et al. Second symposium on the definition and management of anaphylaxis: summary report-Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Journal of Allergy and Clinical Immunology 2006; 117(2): 391-397). Залежно від тяжкості алергічної реакції могло бути розглянуте лікування антигістамінними препаратами, кортикостероїдами й адреналіном. Звіт про лікування і додержання принципів Додержання принципів стосовно ДП: - ДП вводили під безпосереднім спостереженням лікаря, відповідний запис робився співробітником, відповідальним за розподіл і введення ДП, або його/її делегатом; ніяка інформація про послідовність лікування або дозу не розголошувалася і документи знаходилися замкненими без доступу інших осіб, залучених у дослідження, - споживання ДП підтверджувалося вимірними результатами аналізу на препарат. Звітність по ДП: - співробітник, відповідальний за розподіл і введення ДП, або його/її делегат рахував число картриджів, що залишаються в повернутих упакуваннях, потім заповнював форму журналу лікування, - дослідник записував інформацію про день і час введення дози на відповідній сторінці(ках) індивідуальної реєстраційної карти (CRF), - група контролю, відповідальна за дослідження, потім перевіряла дані в CRF, порівнюючи їх з формами для ДП і відповідної звітності після блокування бази даних (для запобігання порушенню сліпого характеру дослідження). Використані картриджі зберігалися дослідником аж до того моменту, коли спонсором було проведено цілком документоване узгодження після завершення дослідження після блокування бази даних. Приклад 11: Оцінка досліджуваного препарату Дане дослідження призначене для оцінки коефіцієнтів метаболічного ефекту й експозиції трьох різних доз інсуліну гларгіну U300 у порівнянні з 0,4 Од/кг Лантус® U100, для порівняння тривалості дії різних доз інсуліну гларгіну U300 відносно 0,4 Од/кг Лантус® U100, для вивчення залежності ефекту дози й експозиції дози інсуліну гларгіну U300, а також для оцінки безпеки і переносимості інсуліну гларгіну в умовах фіксації стану еуглікемії у суб'єктів с цукровим діабетом І типу. Фармакодинаміка 27 UA 110935 C2 5 10 15 20 25 30 35 40 45 50 55 60 Фіксація стану еуглікемії Фармакодинамічний ефект інсуліну гларгіну, головним чином загальну утилізацію глюкози і тривалість дії інсуліну, оцінювали методом фіксації стану еуглікемії. Під час фіксації стану еуглікемії концентрацію глюкози в артеріалізованій венозній крові, що є відображенням джерела для загальної утилізації глюкози всіма тканинами, і швидкість інфузії глюкози (GIR), необхідну для підтримання концентрації глюкози в крові суб'єкта на її цільовому рівні (рівень фіксації), постійно вимірювали і реєстрували за допомогою приладу Biostator™ (система безупинного моніторингу рівня глюкози, Life Sciences Instruments, Elkhart, IN, USA). Необхідна кількість глюкози (GIR-AUC) є мірою засвоєння глюкози в тканинах (утилізація глюкози або знижуюча рівень глюкози активність), опосередковуваного надлишком екзогенного інсуліну. Biostator™ визначає рівні глюкози в крові з інтервалами в 1 хвилину і коректує швидкість інфузії глюкози у відповідь на зміни рівня глюкози в крові, використовуючи заданий алгоритм. Процедура фіксації Для запобігання перешкодам від стандартного лікування суб'єкта інсуліном для вимірювань при фіксації, суб'єкти повинні були утримуватися від використання базальних інсулінів і переходити на препарати інсуліну середньої або короткої тривалості дії, починаючи за 48 годин до введення доз у D1 періодів TP1-TP4, якщо звичайно використовують препарати інсуліну пролонгованої дії, тобто Лантус® (інсулін гларгін), Левемір® (детемір) або інсулін ультраленте, препарати інсуліну короткої дії, починаючи за 24 години до введення доз у D1 періодів TP1TP4, якщо звичайно використовують препарати інсуліну середньої тривалості дії, тобто NPHінсулін. Останню підшкірну ін'єкцію інсуліну короткої дії проводили не пізніше ніж за 9 годин до введення ДП. Суб'єкти на помповій терапії припиняли інфузію інсуліну ранком у день 1, щонайменше за 6 годин до кожного введення ДП (приблизно в 03:00, вважаючи початком введення ДП 09:00). Протягом періодів лікування 1-4 (TP1-TP4), суб'єкти приходили в клініку ранком у день D1 після нічного голодування протягом щонайменше 10 годин. Ранком у день 1 починалася процедура передфіксації і суб'єктів приєднували до Biostator™. Концентрацію глюкози в крові доводили до 4,4-6,6 ммоль/л (80-120 мг/дл) і підтримували в цих межах за допомогою в/в болюсного введення швидкодіючого аналога інсуліну (наприклад, інсуліну глулізину) і наступних індивідуальних інфузій глюкози в міру необхідності. Потім за 60 хв. до введення досліджуваного препарату рівень глюкози в крові доводили до 5,5 ммоль/л (100 мг/дл) ± 20% (рівень фіксації стану еуглікемії) без інфузії глюкози протягом останньої години перед введенням дози. Інфузію інсуліну глулізину припиняли безпосередньо перед введенням досліджуваного препарату. Коли рівень глюкози був стабільним протягом щонайменше 1 години в межах 5,5 ммоль/л (100 мг/дл) ± 20% без інфузії глюкози, вводили ДП (=T0 у D1 під час TP1-TP4, приблизно в 09:00). Суб'єкти одержували референсне або тестовані варіанти лікування (R, T1-3, дивіться таблицю 4) згідно з рандомізацією. Ін'єкції робили ліворуч або праворуч від пупка. Введення ДП проводили не раніш 09:00 годин ранку і не пізніше 14:00 у день 1 у періоди лікування 1-4. Якщо рівень глюкози в крові не стабілізувався під час передфіксації до 14:00 годин, дозу не вводили. Відвідування закінчувалося і суб'єкту призначали нове відвідування для введення дози через 1-7 днів. ДП вводили в стані натще; суб'єкт продовжував голодувати протягом усього періоду фіксації. Рівень глюкози в крові при фіксації стану еуглікемії постійно підтримувався за допомогою в/в інфузії розчину глюкози до закінчення фіксації. Метою будь-якого додаткового введення базального інсуліну є додавання або навіть заміна секреції ендогенного інсуліну між прийомами їжі. У суб'єктів без ендогенної секреції інсуліну, яким було запропоновано взяти участь у цьому дослідженні, екзогенний інсулін повинен був забезпечувати тільки кількість інсуліну, необхідну для утилізації виробництва глюкози в печінці. При правильному підборі не було необхідності в надлишку глюкози для компенсації надлишку інсуліну. В результаті швидкість інфузії глюкози наближалася до нуля. Як тільки припинялася дія інсуліну, концентрація глюкози в крові підвищувалася. Час до початку підйому і до часу, коли концентрації глюкози в крові перевищували визначений поріг, визначався Biostator™. Вибрані дози Лантус® U100 і інсуліну гларгіну U300 перевищували середню базальну потребу, що, у свою чергу, викликало деяку потребу в глюкозі, що знаходить відображення в значній GIR аж до 36 годин. 28
ДивитисяДодаткова інформація
Назва патенту англійськоюLong-acting formulations of insulins
Автори англійськоюBecker, Reinhard, Frick, Annke, Boderke, Peter, Fuerst, Christiane, Mueller, Werner, Tertsch, Katrin, Werner, Ulrich, Loos, Petra, Schoettle, Isabell
Автори російськоюБеккер Райнхард, Фрик Аннке, Бодерке Петер, Фюрст Кристиане, Мюллер Вернер, Терч Катрин, Вернер Ульрих, Лоос Пэтра, Шеттле Изабелль
МПК / Мітки
МПК: A61P 5/50, A61K 9/08, A61K 38/28, A61P 3/10
Мітки: дії, пролонгованої, препарат, інсуліну
Код посилання
<a href="https://ua.patents.su/61-110935-preparat-insulinu-prolongovano-di.html" target="_blank" rel="follow" title="База патентів України">Препарат інсуліну пролонгованої дії</a>
Препарат інсуліну пролонгованої дії у формі супозиторію

Номер патенту: 24529
Опубліковано: 10.07.2007
Автори: Жуков Віктор Іванович, М'ясоедов Валерій Васильович, Каліман Віктор Павлович, Каліман Павло Авксентійович, Клименко Микола Олексійович
МПК: A61K 9/02
Мітки: форми, дії, інсуліну, пролонгованої, супозиторію, препарат
Формула / Реферат:
Гіпоглікемічний засіб тривалої дії, який відрізняється тим, що інсулін застосовують у формі супозиторію, де основна речовина - молекула інсуліну, солюбілізована у інноваційній макромолекулі, а допоміжна речовина являє собою полімер, який дозволяє пролонгувати дію основної речовини.
Препарат інсуліну середньої пролонгованої дії

Номер патенту: 57688
Опубліковано: 15.08.2006
Автори: Стадник Віктор Іванович, Соляник Людмила Павлівна, Сологуб Павло Павлович, Оксамитний Віктор Миколайович, Рибачук Валентина Миколаївна, Мельник Валерій Миколайович, Луців Володимир Романович, Прудієв Дмитро Павлович, Лазарев Олексій Павлович
МПК: A61K 38/28
Мітки: середньої, пролонгованої, дії, препарат, інсуліну
Формула / Реферат:
Антифрикційний мастильний склад містить базове консистентне мастило і присадку у вигляді дрібнодисперсних порошків, причому як присадки використано ультрадисперсні алмази з розміром частинок 3-10 нм.
Спосіб одержання препарату інсуліну пролонгованої дії

Номер патенту: 33799
Опубліковано: 15.02.2001
Автори: Возіанов Сергій Олександрович, Лазарєв Олексій Павлович, Оксамитний Віктор Миколайович, Стефанов Олександр Вікторович, Рибчук Віктор Олександрович, Олейнікова Євгенія Адольфівна
МПК: A61K 38/28
Мітки: пролонгованої, одержання, дії, спосіб, інсуліну, препарату
Текст:
...з використанням компонентів у такому співвідношенні: вміст інсуліну високоочищеного монокомпонентного свиного або телячого або їх суміші- 36-42 ОД, натрію хлориду- 6,5-7,5, натрію оцтовокислого- 1,25-1,45 мг, цинку хлориду- 0,05-0,10 мг або окису цинку, консерванту, наприклад ніпагіну- 0,91,4 мг, води високочищеної для ін'єкцій - до 1мл. Основні переваги запропонованого способу порівняно з прототипом: - зменшено кількість операцій: одна...
Спосіб одержання препарату інсуліну пролонгованої дії

Номер патенту: 57690
Опубліковано: 15.08.2006
Автори: Сологуб Павло Павлович, Рибачук Валентина Миколаївна, Луців Володимир Романович, Соляник Людмила Павлівна, Оксамитний Віктор Миколайович, Стадник Віктор Іванович, Лазарев Олексій Павлович, Мельник Валерій Миколайович, Прудієв Дмитро Павлович
МПК: A61K 38/28
Мітки: препарату, дії, спосіб, одержання, пролонгованої, інсуліну
Формула / Реферат:
Спосіб виробництва комбікормів, що передбачає пров'ялювання сирих кормових трав до вологості 65 - 70%, подрібнення сирих кормових трав до розміру часток 10 - 30мм, змішування сирих кормових трав з підготовленими висівками, зерновими компонентами, шротом соняшниковим, крейдою, сіллю та преміксом та охолоджування суміші компонентів комбікорму, який відрізняється тим, що після подрібнення сирих кормових трав до розміру часток 10 - 30мм їх знову...
Спосіб приготування готової лікарської форми інсуліну пролонгованої дії

Номер патенту: 46669
Опубліковано: 15.12.2005
Автори: Рибачук Валентина Миколаївна, Прудієв Дмитро Павлович, Лесик Ігор Павлович, Сологуб Павло Павлович
МПК: A61K 9/22, A61K 38/28, C07K 14/62, A61P 3/10
Мітки: приготування, дії, форми, лікарської, пролонгованої, інсуліну, готової, спосіб
Формула / Реферат:
Пристрій для виявлення дефектів на працюючому обладнанні, який складається з накладки, що утворює разом з контрольованою поверхнею герметичну порожнину, заповнену рідиною під тиском і з'єднану з датчиком тиску, який відрізняється тим, що на поверхні накладки, яка контактує з контрольованою поверхнею обладнання, виконана сітка з'єднаних між собою каналів.
Попередній патент: Композиції шортенінгів і способи їх отримання та застосування
Наступний патент: Спосіб ущільнення стручка
Випадковий патент: Спосіб діагностики лейкозу великої рогатої худоби