Спосіб одержання похідних хлорамбуціла /його варіанти/
Номер патенту: 6043
Опубліковано: 29.12.1994
Автори: Молотков Володимир Ніканорович, Хуміо Тамура, Сатору Єномото, Кіро Асано





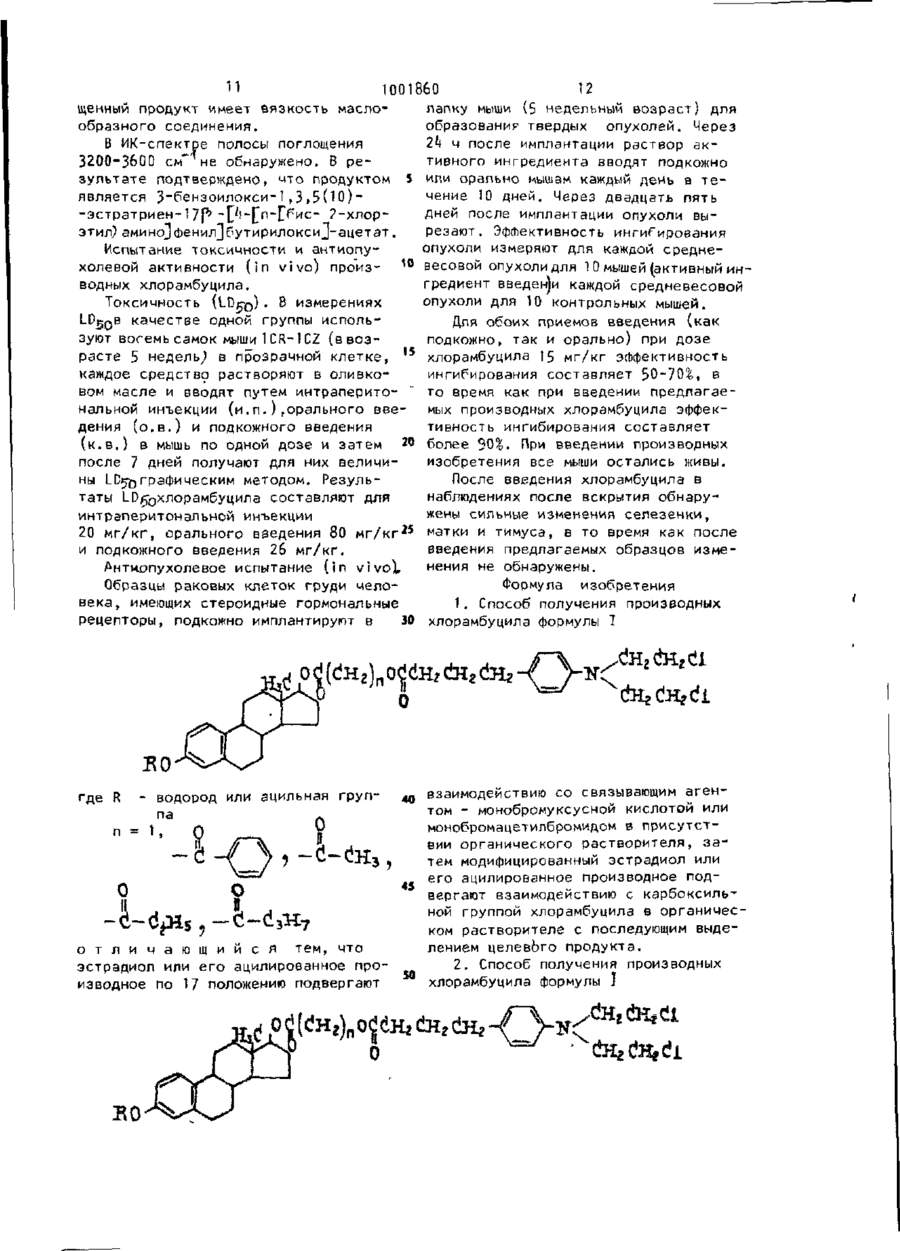


Формула / Реферат
1. Способ получения производных хлорамбуцила формулы І
где R - водород или ацильная группа,
n=1,
отличающийся тем, что эстрадиол или его ацилированное производное по 17 положению подвергают взаимодействию со связывающим агентом - моно-бромуксусной кислотой или монобромацетилбромидом в присутствии органического растворителя, затем модифицированный эстрадиол или его ацилированное производное подвергают взаимодействию с карбоксильной группой хлорамбуцила в органическом растворителе с последующим выделением целевого продукта.
2. Способ получения производных хлорамбуцила формулы 1
где R - водород или ацильная группа
отличающийся тем, что хлорамбуцил подвергают взаимодействию по карбоксиатной группе со связующим агентом - монобромуксусной кислотой или монобромацетилбромидом в присутствии органического растворителя, затем модифицированный хлорамбуцил подвергают взаимодействию с гидроксильной группой в 17 положении эстрадиола или его ацилированного производного в органическом растворителе с последующим выделением целевого продукта.
Текст
Союз Советских Социалистических Республик (И), ТІ И К ПАТЕНТУ (61) Дополнительный к патенту(22)3аявлено 13.08.79 (21) 280^752/23-0^ (23) ПриоритетГосударственный комитет СССР (33) Япония Опубликовано 28.02.83.БюллетеньМ 8 по делам изобретений и открытий (72) Авторы изобретения (71) Заявитель С 07 J 1/00 (32)1^.08.78 (31) 98795/1978 (51)М. Кл. Дата опубликования описания 28.02.83 (53) УДК 5*7.689, .07(088.8) Иностранцы Киро Асэно, Хумио Тамура, Хиромицу Танака и Сатору Еномото (Япония) Иностранная фирма "Куреха Кагаку Когио Кабусики Кайся" (Япония) (5*0 СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХЛОРАМБУЦИЛА (ЕГО ВАРИАНТЫ) 1 Изобретение относится к способу получения новых производных хлорамбуцила общей формулы 1 (I) где R - водород или зцильная группа о о которые обладают противоопухолевой активностью. Известно, что соединений, содержащее бис- (р>-хлорэтил) -аминогруппу являются ал копирующими цитостатическими веществами и применяются в качестве противоопухолевых препаратов. 15 К таким препаратам относятся сарколизин, хлорбутин, хлорамбуцил и другие [ 1 > [ Z ] . Цель изобретения - расширение ассортимента средств, воздействующих 20 на живой организм. Поставленная цель достигается тем, что согласно способу получения производных хлорамбуцила формулы I эстрадиол или~ его ацилированное производное по 17 положению подвергают н и, • ' 1 U 1001860 пряженное аещество хлорамбуцил-эствзаимодействию со связующим агентомрадиол , монобромуксусной кислотой или моно200 мг серебро \_k- І п [бис (2бромацетилбромидом в присутствии ор-хлорэтил) aMM4oJfbeHnnj6yTHpaTaJ (сеганического растворителя, затем модифицированный эстрадиол или его аци5 ребряная соль хлорамРуцила) добавлялированное производное подвергают вза- ют в 1 0 мл ДМСО с образованием белого коллоидного раствора. имодействию с карбоксилатной группой хлорамбуцилэ в органическом раствоЗатем 190,8 мг Зтидрокси-1 ,3,5( 10j рителе с последующим выделением целе-эстратри єн-17$ "монобромацетэта Дового продукта. Ю бавляют и смесь перемешивают при комнатВариант описываемого способа,закной температуре в течение 64 ч в темлючается в том, что хлорамбуцил подноте. Осадок меняет свой цвет от же л• вергают взаимодействию по карбоксилатто-зеленого до темно-зеленого при обной группе со связующим агентом - молучении светом. Фильтрат бесцветный нобромуксусной кислотой или монобром- 15 и прозрачный. ДМСО отгоняют при пониацетилбромидом в присутствии органиженном давлении на водяной бане при ческого растворителя, затем модифици8С С и добавляют 10Q мл для получерованный хлорамбуцил подвергают взания белых кристаллов. Кристаллы от- „ имодействию с гидроксильной группой стаивают в течение } ч для удаления в 17 положении эстрадиола или его аци-то ДМСО и далее отделяют через Фильтр лированного производного в органичесQ-ht тщательно промывают дистиллироком растворителе. Мелевой продукт выванной водой и сушат при пониженном деляют известным способом. давлении в эксикаторе. Выход сыроП р и м е р 1. Зтидрокси-1 ,3,5 (10?- го продукта 330,5 мг. -эстратриен-17 р - Р ' - [ П [ 6 И С (2-хлор- и Очистка продукта. 330,5 мг полуэтил) аминоЗфенилЗбутирилокси3 ацетат ченных кристаллов растворяют в смеПолучение З-гиррокси-1,3»5(Ю)~эстшанном растворителе из 50 об. ч. цикратриен-17 р-монобромацетата. логексана и 10 об. ч. этилацетата. ' 10 г 1,3,5{Ю)-эстратриен-3,17р-ди- Раствор медленно пропускают через коола растворяют в 400 мл безводного J0 лонку, наполненную * 0 г силикагеля, + тетрагидрофурана (ТГф) и затем добави продукт постепенно отделяют с полуляют пиридин (8,8 г ) . чением 188,2 мг чистого продукта (выход 62,86%) , Затем раствор 22,5 г монобромацеРезультаты элементного анализа, тилбромида в 74 г четыреххлористого температура плавления и ИК-спектры углерода добавляют по каплям к полу- 35 продукта следующие. ченному раствору при -5-7 С, Смесь Найдено Д : С 66,0; Н 7,0; N 2,3; поддерживают при этой температуре в С! 11,0 течение одной ночи. После реакции ' Рассчитано,?: С 66,22; Н 6,98; полученный осадок отделяют фильтраN 2,27; С1 И,52 Q цией. Растворитель из фильтрата отТ.пл. (полужидкий) 25 С. гоняют. Осадок растворяют в эфире и П р и м е р 2. 3-бензоилоксиперекристаллизовывают из эфира с по-1,3,5(Ю)эстратриен-17р>-[4 [п-^бис лучением 1,3,5О0)-эстратриен-3,ир> (2-хлорэтил)амино]фемилЗбутирилокси -бис(монобромацетата). 2 г продукта -ацетат. растворяют в 900 мл метанола и раст- 45 Ю г 1,3,5(Ю_)-эстратриен-3,17рвор охлаждают до -> С. К полученному -диола растворяют в 100 мл ТГФ и дораствору добавляют по каплям растбавляют 10 W] водного раствора, совор 0,24 г углекислого калия в 20 мл держащего 1,^7 гидроокиси натрия. воды. После реакции в течение 30 мин Смесь перемешивают при комнатной добавляют 1000 мл воды и получающийтемпературе в течение 30 мин. Затем ся осадок отделяют и высушивают. Элереакционную смесь концентрируют сушментным анализом и ИК-спектрами подкой при пониженном давлении на вотверждено, что продуктом является 3дяной бане при 80°С для удаления во-гидрокси-1,3,5(10) -эстрадиен- 17В ~ ды. Остаток растворяют в безводном -монобромаиетат. тетрагидрофуране, к полученному растПолучение З-гидрокси-t ,3,5(Ю-эствору добавляют по каплям 50 мл этилового эфира, содержащего 5*5 г бенамино]фенилЗбутирилокси]ацетата.(Созоилхлорида, и реакцию проводят при Р [ ^ [ л £ р ) 5 1001 860 6 комнатной температуре в течение 16 ч. *t00 мл воды добавляют к фильтрату. ПоПосле реакции получающийся хлористый лучающийся белый осадок отделяют центнатрий отделяют подходящим методом. рифугированием. Осадок растворяют в Фильтрат выпаривают досуха при пони50 мл ацетона и нерастворенное веженном давлении. Лля того, чтобы уда-5 щество отделяют фильтрацией через лить непрореатировавший бензоилхлорид фильтр G-*i, добавляют 200 мл 0,1н. водного растФильтрат выпаривают досуха при повора гидроокиси натрия и смесь перениженном давлении с получением 165 мг мешивают при комнатной температуре в маслообразного продукта. 10 течение 15 мин. Получающиеся белые Продукт реакции хроматографируют кристаллы отделяют с помощью фильтра на силикагеле со смешанным раствориG-3 и тщательно промывают дистиллителем (этилацетат и циклогексан в рованной водой и высушивают при пообъемном соотношении 10:50) с получениженном давлении в эксикаторе. нием чистого продукта. Очищенный про1 п - - 15 дукт - белое кристаллическое соетиПродукт анализируют тонкослойной нение при 20 С. хроматографией на силикагеле со смеП р и м е р З . 3-пропионилоксишанным проявляющим растворителем . -1,3,5(Ю)-эстратриен-17р-О* -[n{этилацетат и циклогексан в объемном -бис(2-хлорэтил)аминоЗфенил бутирилсоотношении 50=30) с получением осокси) ацетата. новного пятна Rf:0,3^*° 10 г 1,3,5(Ю)-эстратриен-3,17ро Сырые кристаллы перекристаллизовы-диола растворяют в 100 мл тетрагидвают из этилацетата и получают 8,6 г рофурана, добавляют 10 мл водного рабелых кристаллов. Продуктом является створа, содержащего 1,^*7 гидроокиси 17р* -гидрокси-1 , 3 ,5 (Ю)-эстратриеннатрия в 10 мл воды, и смесь переме-З-^ензоат согласно температуре плав* шивают при комнатной температуре в теления, элементному анализу и ИК-спек^ чение 30 мин.Продукт реакции конценттрам. рируют досуха при пониженном давле7,0 г полученного продукта раствонии на водяной бане при 80°С для уда и ряют- в тетрагидрофуране, добавляют ления воды. Остаток растворяют в беач 2,0 г пиридина и смесь охлаждают до М водном тетрагидрофуране, раствор, со-5°С держащий 3,^ г пропионилхлорида в К полученной смеси постепенно по 50 мл безводного тетрагидрофурана, докаплям добавляют раствор, содержащий бавляют по каплям и реакцию проводят 15,5 г 30%-ного раствора монобромацепри комнатной температуре в течение тилбромида в четыреххлористом углеро-as 16 ч. После реакции осадок хлористоде в 50 мл тетрагидрофурана. После дого натрия отделяют, фильтрат выпарибавления смесь перемешивают при -$ С вают досуха при пониженном давлении в течение 2 ч и затем на ледяной бане и остаток перекристаллизовывают из 4 ч и хранят в холодильнике 16 ч. Посэтанола с получением 9 г белых крисле реакции полученный белый осадок от^о таллов. деляют с помощью фильтра Q-k, высушиПродуктом является 17 р-гидроксивают при пониженном давлении на водя-1 ,3,5(Ю)-эстратриен-3'пропионат согной бане при 30°С, добавляют 200 мл ласно элементному анализу и ИК-спектэтилового эфира и смесь перемешивают . , с получением 5,3 г белых кристаллов. 4 5 рам. 7 г продукта растворяют в 70 мл Результаты элементного анализа и безводного тетрагидрофурана и 3,0 г температуры плавления следующие. пиридина добавляют к этому раствору Найдено,%: С 6^,3; Н 5,8; Вг 15,7 и смесь охлаждают до -5°С. К полученРассчитаноД: С 6*1,23; H 5,78: ной смеси по каплям добавляют растВг 15,8. 50 вор, содержащий 17,3 г 30%-ного моноТ.пл. бромацетилбромида в четыреххлористом 182,3 мг З-бензоилокси-1 ^ . углероде в50 млтетрагидрофурана.По^CTpaTpneH-i^ [ -монобромацетата и ^ сле добавления смесь поддерживают при 1А8,5 мг серебро -*4-п-бис(2-хлорэтил} -5 °С в течение 2 ч и затем в холодильамино-Ленилбутирата добавляют в 5 мл SS нике втечение 16 ч для проведения реДМСО и реакцию проводят при комнатакции. После реакции полученный осадок ной температуре в течение 3 дней в темноте. После реакции осадок бромиотделяют фильтрацией. Фильтрат да серебра отделяют фильтрацией и выпаривают досуха при пониженном Г 8 7 1001860 давлении на водяной бане при }0°tt рат выпаривают досуха при пониженном) затем добавляют 200 мл этилового эфидавлении с получением 1 ,2 г маслообразного продукта .Продукт хроматографирура и смесь перемешивают с получениют насиликагеле со смешанным растворием 6,0 г белых кристаллов.Фильтрат телем этмлацетат-циклогексанпри соотдалее концентрируют с получениношении 10:50по объему.Очищенный проем 3,5 г белых кристаллов. Кристаллы дукт имеет вязкость маслообразного перекристаллизовывают из смешанного материала при 20 С. растворителя эфира с этанолом. Результат элементного анализа Результаты элементного анализа (О следующий. следующие. Найдено,%: С 66,0; Н 6,5; W 2,0 Найдено,*: С 61,5; Н 6,5;Вг 17,9 С1 10,9. Рассчитано,%: С 61,43 ; H 6,45; Рассчитано*,?:: С 65,64; Н 6,84; Вг 17,78. N 2jl3; C1 10,79. Продуктом является 3-пропионияок- ( П р и м є р 5. 1 г 3-аиетоксиси-1 ,3,5(Ю)-эстратриен-17р' -монобром - ~1,3,5(12)-эстратриен-17 (Ь -моноаиетат. бромаиетата и 0,8 г натрий 4-[п1 г продукта и 0,91 г ceoeFpo -k[бис (2-хлорэтил) амино] Фенил] бути-£п-[бис (2-хлорэтил) амино3^енил]будобавляют к 50 мл тетрагидротирзта диспергируют и растроряют в проводят реакцию при 60 С 50 мг ДМСО,реакцию выполняют при комв течение 24 ч. натной температуре У течение 3 дней в темноте. После реакиии осадок бромида После реакции осадок отделяют серебра отделяют фильтрацией и добавфильтрацией и Фильтрат концентриляют 4 л воды. Осадок отделяют центруют и высушивают. Продукт отделяют 25 рифугированием и белый осадок раствои очищают с помошью колонки с силикаряют в 50 мл ацетона, а нерастворимь-е гелем со смешанным растворителем материалы отделяют фильтрацией через этилаиетатциклогексан с получением фильтр G-4. Фильтрат выпаривают досу0,9 г очищенного продукта.3-ацетокси. ха при пониженном давлении с получе- ! ,3,5(1 0)-эстратриен-1 7Р> - К~{/ьбис * нием 1,3 г маслообразного продукта.' (2-хлорэтил) -амино] йенил_] бутирилПродукт хроматографируютна силикаокси] аиетат. геле со смешанным растворителем этилП р и м е р 6. З-эиетокси-1,3,5(10) іацетат-циклогексан всоотношении 10:50 -эстратриен-17 Ъ -Г^Тн-Цбис (2-хлорпо объему для его очистки. Очишенный продукт имеет вазкость-маслообразного35 этил)амино] сЬенил^бутирилокси] ацетат, продукта при 20° С. . 200 мг серебро ^-[п-[_бис (2-хлор, этил) амино]Фенил] бутиратэ (серебрян Результаты элементного анализа и ная соль хлорамбуиила) добавляют в ИК-слектры. 10 мл ДМСО с образованием белого коЛНзйдемоД: С 67,1; Н 7,0; Н 2;1; 40 лоидного раствора. Далее 190,8 мг С1 11,0. Рассчитано,^: С 66,0; Н 6,99; З-гидрокси-1 ,3,5(Ю?-эстратриен-17[} К 2,08; С1 10,56. -монобромацетата добавляют к коллоидному раствору и смесь перемешивают Ghbv при комнатной температуре в течение П р и м е р 4.3-ацетокси-1,3,5 (10)-эстратриен-17 ( ~[4(п-бис (2-хлорэтил) 45 64 ч. Осадок меняет цвет до желтова5 то-зеленого. К осадку добавляют неамино фенил бутирилокси ацетат. большое количество ацетона и осадок 1,0 г 3~ацетокси-1,3,5 С10)-эстраотделяют фильтрацией через Фильтр триен-17 { -моноЄромаиетата, полученЬ G-4. фильтрат бесцветный и прозрачного таким же способом, как в примере 2, и 0,9 г серебро 4-|_п-{бис (2-хлор?v 5Q ный, ДМСО отгоняют на водяной бане при 80°С и затем добавляют 100 мл воэтил) амино]фения]бутирата добавляют ды для осаждения белых кристаллов. в 50 мл ДМСО и реакцию проводят при 25 С втечение Здней в темноте.После Смесь отстаивают в течение 1 ч и зареакции осадок бромида серебра отделятем ДМСО отгоняют. Белые кристаллы ют и к фильтрату добавляют Члводы. Поотделяют фильтрацией через фильтр 55 ручающийся белый осадок отделяют цент- G-4, затем промывают дистиллированрифугированием. Осадок растворяют в ной водой и высушивают при пониженном 50 мл ацетона. Нерастворимое вещество давлении в эксикаторе. Выход сырого отделяют с помощью фильтра G-** и филы- продукта 330,5 мг. 9 1001 860 10 П р И М Є р 7 , - 3"ПРОПИОНИЛПКСИ-1 ,3,5 33^,5 мг сырого продукта растворяют а смешанном растворителе цикло(10) -эстратриен-17 p-Di-fn-Сбис (2гексана с этилацетатом а соотноше-хлорэтил) амино j сменил}бутирилоксиЗ нии 50:10 по объему. Раствор медацетата. ленно пропускают через колонку, на5 50 мг 3-гидрокси-1,3,5(10)-эстра" п полненную ^0 г силикагеля для постетриен-17р> -С^"[Г "Сбис- (2-хлорэтил) пенного отделения продукта. Получают аминоЗЛенил^бутирилоксиJ- ацетата раст188,2 мг (выход 62,86 ^чистого проворяют в 1 мл безводного пиридина, дукта. добавляют 1,5 мл пропионового ангидРезультаты элементного анализа и '" рида и смесь держат в холодильнике в течение дня. Реакционную смесь выпаритемпература плавления продукта слевают досуха при пониженном давлении дующие. на водяной бане при 30 С. Остаток смеНайдено,?': С 66,0; Н 7,0; N 2,3; шивают є дистиллированной водой и смесь С1 11 ,0. 5 держат в течение 2 ч с образованием Рассчитано,*: С 66,22; Н 6,98; * коллоидального маслообразного продукМ 2,27; С1 11,52. та. Пиридин и уксусную кислоту удаляют Температура кипения при 25 С в дистиллированной водой и продукт прорасплавленном состоянии. мывают нейтральной водой. Водную фазу Подтверждено, что продуктом являотделяют и масляную фазу высуживают ется З-гигрокси-1 ,3,5О0)-эстратриен 2° при пониженном давлении и эксикаторе с -17р -[*4-[п-Цбис (2-хлорэтил) aMHHoJ получением ^0 мг маслообразного профенчл^-бутирилокси J ацетат. дукта. 50 мг продукта растворяют в 1 мл Продукт хроматографируют на силибезводного пиридина, к этому раствокагеле со смешанным растворителем ру добавляют 1 мл уксусного ангидриэтилацетат-циклогексан в соотношеда и проводят реакцию в холодильнике нии 10:50 по объему для его очистки. в течение 16 ч. Затем реакционную В результате подтверждено, что смесь концентрируют и высушивают при продуктом является 3-пропионилоксипониженном давлении на водяной бане при 30 С.Остаток смешивают сдистил30 -1 ,3,5(1С9 -эстратриен-17 р>-[>-[п£бис- (2-хлорэтил) амино^Фенил^утилированной водой и смесь отстаивают рило кс и]]- ацетат. 1 ч для осаждения маслообразного проП р и м е р 8. 3'бензок,юкси-1,3,5 дукта в виде белой коллоидной пены. (10)-эстратриен-17В-[**-ГгЧГис( 2-хлорПиридин и уксусную кислоту удаляют с дистиллированной водой и продукт 35 этил) аминоЗфенил](*утирилокси^ -ацетат. промывают водой, которая была нейт50 мг Зтидрокси-1 ,3,5(Ю,)'эстраральной. Маслообразный продукт от- -[^:-£п-Пбис- ?-хлоррезают. Эффективность ингиГироеания этил? аминоЗФенил^бутирилоKcnj-ацетат опухоли измеряют для каждой среднеИспытание токсичности и антиопувесовой опухоли для 10 мышей (активный инхолевой активности (in vivo) произгредиент введен)и каждой средневесовой водных хлорамбуцила. опухоли для 10 контрольных мышей. Токсичность (LD^ 0 ). В измерениях качестве одной группы испольДля обоих приемов введения (как зуют вогемь самок мыши 1CR-1CZ (в возподкожно, так и орально) при дозе расте 5 недель,) в прозрачной клетке, IS хлорамбуцила 15 мг/кг эффективность каждое средств9 растворяют в оливкоингибирования составляет 50~70%, в вом масле и вводят путем интраперитото время как при введении предлагаенальной инъекции (и.п.)горального вве- мых производных хлорамбуцила эффекдения (о.в.) и подкожного введения тивность ингибирования составляет (к.в.) в мышь по одной дозе и затем 20 более 90%. При введении производных после 7 дней получают для них величиизобретения все мыши остались живы. ны LD^oграфическим методом. РезульПосле введения хлорзмбуцила в таты 1_Р£0хлорамбуцила составляют для наблюдениях после вскрытия обнаруинтраперитональной инъекции жены сильные изменения селезенки, 20 мг/кг, орального веедения 80 мг/кг*5 матки и тимуса, в то время как после и подкожного введения 26 мг/кг. введения предлагаемых образцов изменения не обнаружены. Антиопухолевое испытание (in vivo). Образцы раковых клеток груди челоФормула изобретения века, имеющих стероидные гормональные 1, Способ получения производных рецепторы, подкожно имплантируют в 30 хлорамбуцила формулы 1 взаимодействию со связывающим агентом - монобромуксусной кислотой или монобромацетилбромидом в присутствии органического растворителя, затем модифицированный эстрадиол или его ацилировэнное производное подвергают взаимодействию с карбоксильной группой хлорамбуцила в органическом растворителе с последующим выделением целевого продукта. о т л и ч а ю щ и й с я тем, что 2. Способ получения производных эстрадиол или его ацилировзнное про50 хлорамбуцила формулы I изводное по 17 положению подвергают где ацильная груп О &ь 40 і 13 001860 I* где Р - родород или ацильная группаприсутствии органического растворителя, затем модифицированный хлорамбуцил подвергают взаимодействию с гидроксипьной группой в 17 положении эстрадиола или его ацилированного производного в органическом растворителе с последующим выделением целеО О вого продукта. Источники информации, 59 ^ V принятые во внимание при экспертизе 1. Патент СССР f 379088, T о т л и ч а ю щ и й с я тем, что і кл. С 07 J 1/00, опублик. 197 * , выхлорамбуцил подвергают взаимодейстданный инофирме "Сандос АГ," Швейцария, вию по карбоксиатнои группе со связу2. Мзшковский М. Д. Лекарственные ющим агентом - монобромуксуснои кис15 средства. Т. 2, 1972 , с. лотой ипи монобромацетилбромидом в Л Редактор А. Фролова Составитель Л. Иоффе Техред Т.Фанта Корректор И. Шулла Заказ ЙбЗ/78 Тираж 385 Подписное ВНИИПИ Государственного комитета СССР по делам изобретений и открытий П3035, Москва, Ж-35, Рауюская наб., д. V 5 Филиал ППП "Патент", г. Ужгород, ул. Проектная, h
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for producing chloroambucyl derivatives (variants)
Назва патенту російськоюСпособ получения производных хлорамбуцила (его варианты)
МПК / Мітки
МПК: A61K 31/565, C07J 41/00, C07J 1/00, A61P 35/00
Мітки: його, похідних, спосіб, хлорамбуціла, одержання
Код посилання
<a href="https://ua.patents.su/8-6043-sposib-oderzhannya-pokhidnikh-khlorambucila-jjogo-varianti.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідних хлорамбуціла /його варіанти/</a>
Спосіб одержання похідних 1-ціклогексіл-4-аріл4-піперідінкарбонових кислот або їх кислотноадитивних солей, або їх стереохімічних ізомерних форм (його варіанти)

Номер патенту: 2700
Опубліковано: 26.12.1994
Автори: Марсель Геребернус Марія Льюікс, Джоан Уільямс, Раймон Стокброекс
Мітки: кислотноадітівних, 1-ціклогексіл-4-аріл4-піперідінкарбонових, кислот, варіанти, солей, ізомерних, стереохімічних, форм, одержання, спосіб, його, похідних
Формула / Реферат:
(57) 1. Способ получения производных 1-циклогексил-4-арил-4-пиперидинкар-боновой кислоты общей формулыгде R1 - водород или низший алкил; R2 - гидроксил, низший алкоксил, низший алкокси - низший алкоксил, фенокси - низший алкил, фенокси - низший алкоксил, в котором фенил может быть замещен низшим алкоксилом или низшим алкилом, амино, ди(низший алкил)амино, ди (низший алкил)амино-низший алкоксил, 4-морфолинил, 1-пирролидинил,...
Спосіб одержання похідних ціс-4-феніл-1,2,3,4-тетрагідра-1-нафтіламіна або його солей
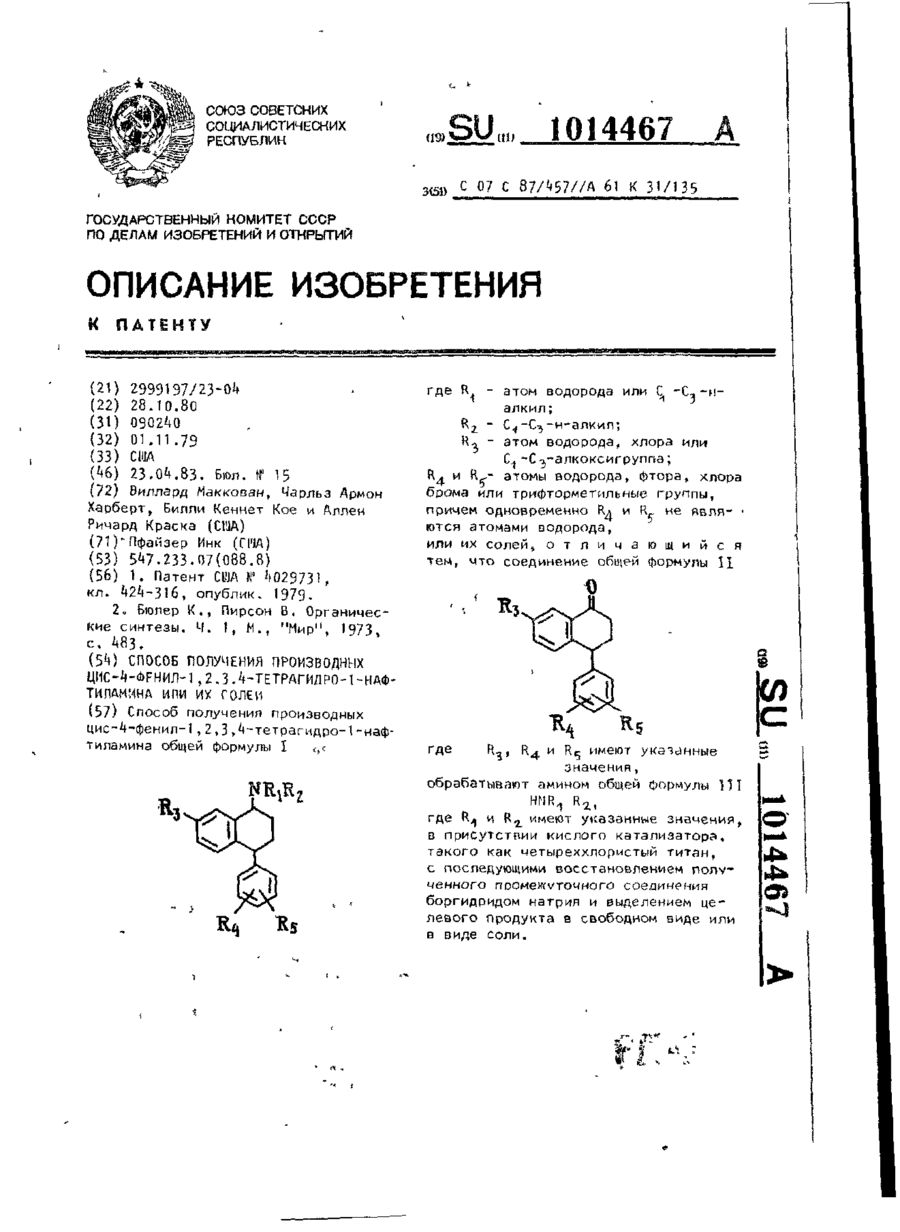
Номер патенту: 6301
Опубліковано: 29.12.1994
Автори: Віллард Маккован, Біллі Кеннет Кое, Чарльз Армон Херберт, Аллен Річард Краска
МПК: A61K 31/137, C07C 213/00, C07C 67/00, C07C 217/74, C07C 209/00, C07C 45/28, A61K 31/135, A61K 31/165, C07C 211/41, A61P 25/26, C07C 255/58, C07C 45/46, C07C 43/21, C07C 49/697, A61K 31/13, C07C 45/00, C07C 57/00, C07C 211/42, A61P 25/24
Мітки: одержання, похідних, ціс-4-феніл-1,2,3,4-тетрагідра-1-нафтіламіна, спосіб, солей
Формула / Реферат:
Способ получения производных цис-4-фенил-1,2,3,4-тетрагидро-1-нафтиламина общей формулы Ігде R1 - атом водорода или С1-С3-н-алкил; R2-С1-С3-н-алкил; R3 - атом водорода, хлора или С1-С3 - алкоксигруппа; R4 и R5 - атомы водорода, фтора, хлора, брома или трифторметильные группы, причем одновременно R4 и R5 не являются атомами водорода, или их солей, отличающийся тем, что соединение общей формулы IIгде R3, R4 и R5...
Спосіб одержання похідних пірідіна (його варіанти)

Номер патенту: 3633
Опубліковано: 27.12.1994
Автори: Педер Бєрнхарт Бернтссон, Стіг Айл Інгемар Карлссон, Бенгт Ріхард Люнг, Ян Ернульф Гаардер
МПК: A61P 9/08, A61K 31/455, C07D 211/90, A61P 9/12
Мітки: його, варіанти, пірідіна, похідних, одержання, спосіб
Формула / Реферат:
Способ получения производных пиридина общей формулы где R1 - метил, этил, метоксиэтил, метоксиэтокси-этил, этоксиэтил, диэтоксиметил; R2-этил, изопропил, трет-бутил, метилпропил, метоксиметилатил, изопропилоксиэтил, метоксидиметилэтил, этоксиэтил, пропаргил, метилаллил, причем R1 и R2 не имеют одинаковое значение; R3 - хлор, метокси; R4 - хлор, метил, метокси, отличающийся тем, что...
Спосіб одержання стимулятору росту рослин на основі похідних пектину

Номер патенту: 3271
Опубліковано: 27.12.1994
Автори: Шепа Василь Васильович, Єськов Вячеслав Іванович, Повстяний Михайло Васильович, Кубрак Микола Леонідович, Шелюк Володимир Павлович, Кухта Євген Петрович, Сабелко Олександр Михайлович, Толкачова Наталія Василівна, Харєбов Валерій Олексійович
МПК: C08B 37/06, A01P 21/00
Мітки: росту, похідних, стимулятору, рослин, одержання, спосіб, пектину, основі
Формула / Реферат:
Способ получения стимулятора роста растений на основе производных пектина, включающий обработку пектина гексаметилендиамином, отличающийся тем, что пектин предварительно растворяют в 0,1 н. водном растворе гидроокиси натрия до концентрации 1,0 %, выдерживают 10 - 12 час, вводят изопропиловый спирт, выделяют осадок, растворяют его в воде до концентрации раствора 1,25 % или в диметилсульфоксиде до концентраций 1,0 %, обрабатывают...
Спосіб одержання похідних пірідіна (його варіанти)

Номер патенту: 3637
Опубліковано: 27.12.1994
Автори: Бенгт Ріхард Люнг, Стіг Айл Інгемар Карлссон, Ян Ернульф Гаардер, Педер Бєрнхарт Бернтссон
Мітки: варіанти, пірідіна, одержання, похідних, спосіб, його
Текст:
...снижение кровяного давления на 20%. Соединения общей формулы ( I ) облаРезультаты сведены в табл.2. дают антигипертенсивным действием. Специфичность релаксации гладкой П р и м е р } . Ufk г 2-3-ДИХлор- 3S мышцы определяется следующим путем. бензальдегида, 3»2 г сложного этилоВ ванну подают изолированную ворот вого эфира З'аминокротоновой кислоную вену крыс и изолированную папилты, **,0 г 2-сложного метоксиэтилолярную сердечную мышцу тех же...
Попередній патент: Спосіб одержання сполучень beta-лактама
Наступний патент: Спосіб одержання макролідних сполучень
Випадковий патент: Зубець для сільськогосподарського пристрою