Заміщені аміносполуки, спосіб одержання та фармацевтична композиція
Номер патенту: 56997
Опубліковано: 16.06.2003
Автори: Бушманн Хельмут Хайнріх, Штрассбургер Вольфганг Вернер Альфред, Енглбергер Вернер, Ціммер Освальд, Фрідеріхс Ельмар Джозеф








Формула / Реферат
1. Заміщені аміносполуки загальної формули (І),
,
де
R1 являє собою Н, ОН, С1-6-алкіл або О-С3-7-циклоалкіл;
(при цьому R1 відрізняється від Н, якщо R2 і R7 = Н, або ОСН3-група заміщена в 7-й позиції);
R2 являє собою Н, ОН, С1-6-алкіл або О-С3-7-циклоалкіл, Сl, F, С1-6-алкокси, арил, гетероцикліл або С1-6-алкілгетероцикліл, С1-6-алкіларил, або 5,6- або 6,7-бензогрупа (незаміщена або моно- або дизаміщена групами Сl, O-арил, С2-6-алкеніларил, F, СF3, С1-6-алкіл, O-С1-6-алкіл або ОН);
при цьому, якщо R1 і R2 або R7 = Н, R2 або R7 не можуть являти собою СF3-групу і R2 або R7 не можуть являти собою ОСН3-групу в 7-й позиції;
або
R2 і R7 разом являють собою -O-(СН2)(1-2)-O- (в 5,6- або 6,7-позиціях);
R3 являє собою Н;
R4 являє собою С1-6-алкіл;
або R3 і R4 разом являють собою -(СН2)(1-4);
R5 являє собою С1-6-алкіл або С3-7-циклоалкіл;
R6 являє собою С1-6-алкіл, С1-6-алкіларил, С1-6-алкілгетероцикліл, -СН2-СН=CH(R8)2, -СН2-( С3-7)-циклоалкіл або С3-7-циклоалкіл;
R7 являє собою Н, ОН, С1-6-алкіл, О-С(3-7)-циклоалкіл, О-арил, С2-6-алкеніларил, С1-6-алкокси, Сl, F, арил, гетероцикліл, С1-6-алкілгетероцикліл, С1-6-алкіларил, або 5,6- або 6,7-бензогрупа (незаміщена або моно- або дизаміщена групами Сl, F, СF3, С1-6-алкіл, O-С1-6-алкіл або ОН);
при цьому, якщо R1 і R2 або R7 = Н, R2 або R7 не можуть представляти СF3-групу і R2 або R7 не можуть представляти ОСН3-групу в 7-й позиції;
і
R8 являє собою Н або СН3,
або їх фармацевтичні солі.
2. Сполуки по п. 1, де
R1 являє собою Н, ОН
(при цьому R1 Н, якщо R2 = Н, або ОСН3- в 7-й позиції);
R2 являє собою Н (якщо R1 Н), ОН, С1-6-алкіл (прямий або розгалужений), О-С1-6алкіл (прямий або розгалужений; без ОСН3- в 7-й позиції, коли R1=H), бензил, О-феніл, феніл, 5,6- або 6,7-бензогрупа [якщо потрібно (ди) заміщена групами Сl, F, СF3, алкіл, O-алкіл], Сl, F, СF3;
R3 являє собою Н;
або R3 + R4 = -( СН2)1-4;
R4 являє собою СН3, С2Р5;
R5 являє собою С1-3-алкіл (прямий або розгалужений), і
R6 являє собою С1-3-алкіл (прямий або розгалужений),
-(СН2)1-2-феніл, -(CH2)1-2-піридил,
-СН2-СН = С (R7)2 (з R7 = Н, СН3)
-СН2 - С3-6-циклоалкіл,
R7 відсутній.
3. Сполуки за п. 1, які відрізняються тим, що R2, R6 і R7 являють собою С1-6-алкілгетероцикліл або С1-6-алкіларил, і що R1, R3 - R5 і R8 мають значення по п. 1.
4. Сполуки за п. 1, які відрізняються тим, що R2 і R7 являють собою арил або гетероцикліл, і що R1, R3 - R6 і R8 мають значення по п. 1.
5. Сполуки за п. 1, які відрізняються тим, що R1 являє собою ОН або -О-С1-6-алкіл, R5 являє собою С1-6-алкіл, R6 являє собою С1-6-алкіл, і що R2 - R4 і R7 мають значення по п. 1.
6. Сполуки за п. 1, які відрізняються тим, що R1 являє собою ОН, R5 являє собою метил, R6 являє собою метил, і що R2 - R4 і R7 мають значення по п. 1.
7. Спосіб одержання сполуки формули (І),
,
де R1 - R7 мають значення по п. 1, які відрізняються тим, що третинний спирт загальної формули (II)
,
де R1 - R7 мають ті ж значення, що і в формулі (І), реагує з напівконцентрованими або концентрованими органічними або неорганічними кислотами в температурному інтервалі від 0°С до 100°С,
причому третинні спирти загальної формули (II) отримують реакцією -амінокетонів загальної формули (III)
,
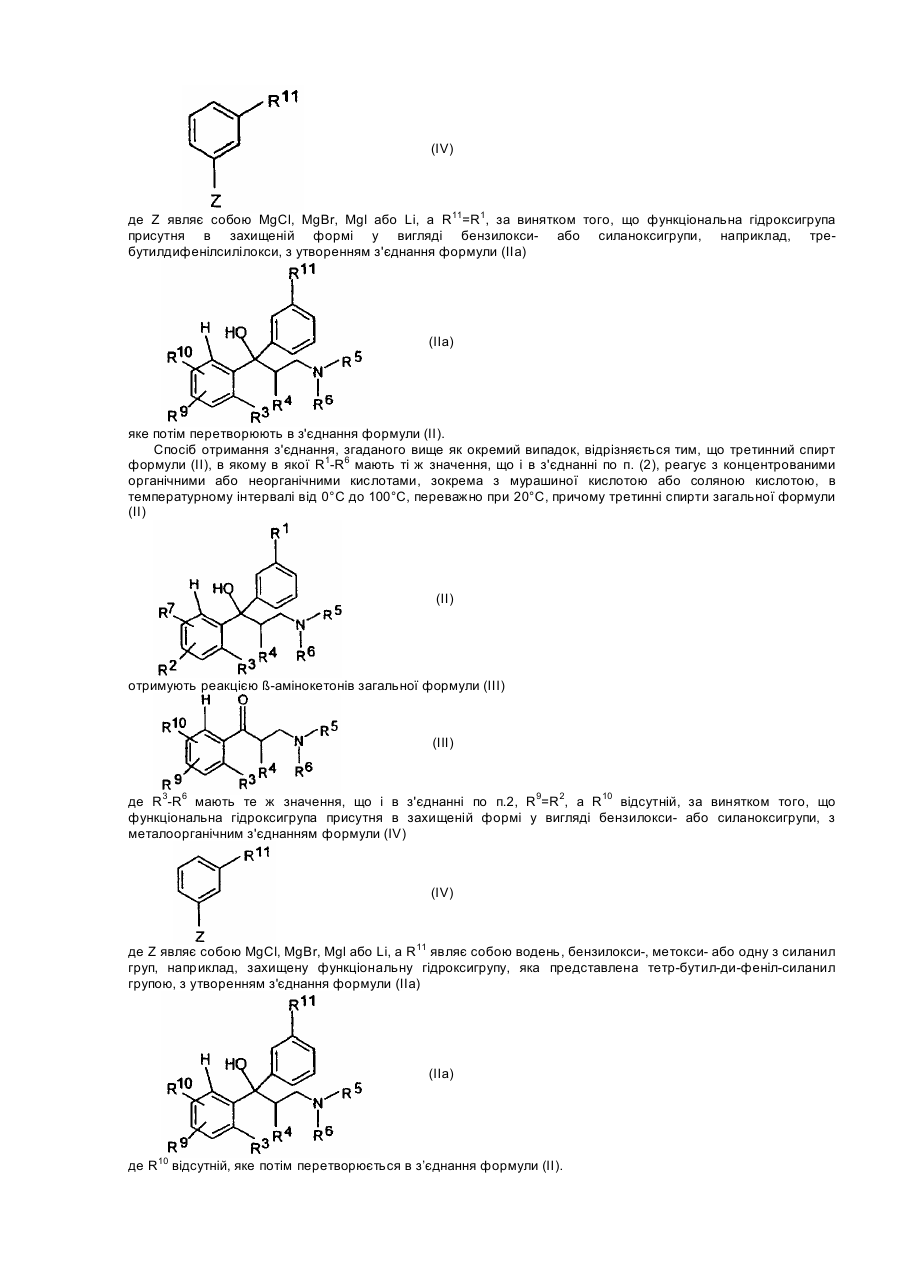
де R3 - R6 мають ті ж значення, що і в формулі (І), R9 = R2, а R10 = R7, за винятком того, що функціональна гідроксигрупа присутня в захищеній формі у вигляді бензилокси- або силаноксигрупи, з металоорганічною сполукою формули (IV)
,
де Z являє собою MgCl, MgBr, MgІ або Li, a R11 являє собою водень, бензилокси-, метокси- або одну з силанілгруп, наприклад захищену функціональну гідроксигрупу, яка представлена тетр-бутилдифеніл-силанілгрупою, з утворенням сполуки формули (ІІа)
,
яку потім перетворюють в сполуку формули (II).
8. Спосіб по п. 7 одержання сполуки по п. 2, який відрізняється тим, що третинний спирт формули (II), в якій R1 - R6 мають ті ж значення, що і в сполуці по п. 2, реагує з концентрованими органічними або неорганічними кислотами, зокрема з мурашиною кислотою або соляною кислотою, в температурному інтервалі від 0°С до 100°С, переважно при 20°С, причому третинні спирти загальної формули (II)
,
отримують реакцією -амінокетонів загальної формули (III)
,
де R3 - R6 мають ті ж значення, що і в сполуці по п. 2, R9 = R2, a R10 відсутній, за винятком того, що функціональна гідроксигрупа присутня в захищеній формі у вигляді бензилокси- або силаноксигрупи, з металоорганічною сполукою формули (IV)
,
де Z являє собою MgCl, MgBr, Mgl або Li, a R11 являє собою водень, бензилокси-, метокси- або одну з силанілгруп, наприклад захищену функціональну гідроксигрупу, яка представлена тетр-бутилдифеніл-силанілгрупою, з утворенням сполуки формули (IIа)
,
де R10 відсутній, яку потім перетворюють в сполуку формули (II).
9.Фармацевтична композиція, що включає активний інгредієнт і фармацевтично прийнятний носій, яка відрізняється тим, що як активний інгредієнт вона містить сполуку формули (І) за п.1.
10. Фармацевтична композиція за п. 9, яка відрізняється тим, що активний інгредієнт є анальгетиком.
Текст
Винахід відноситься до заміщених аміноз'єднань загальної формули (І) (І) або їх фармацевтично сумісних солей, до способу їх отримання і до їх використання в якості ліків. Класичні опіоїди, такі як морфін, дуже ефективні для подолання болів в діапазоні від сильного до дуже сильного. Однак їх застосування обмежене із-за відомих побічних ефектів, таких як придушення дихання, блювота, седативний ефект і стійкий замок, а також із-за розвитку звикання. Крім того, вони менш ефективні для невропатичних і побічних болів, від яких особливо страждають онкохворі. Опіоїди виявляють свій анальгезуючий ефект за рахунок приєднання до мембранних рецепторів, які утворять частину сімейства так званих G-протеїн-зчеплених рецепторів. Біохімічний і фармакологічний опис підтипів цих рецепторів в даний час дає надію на те, що підтип-специфічні опіоїди можуть показувати профіль ефекту/побічного ефекту, відмінний від профілю, наприклад, морфіну. Тоді як морфін виборче зв'язується з так званими m-рецепторами, ендогенні енкефалини охарактеризовані як d виборчі пептиди. У той же час, поглиблені фармакологічні дослідження показали, що, мабуть, існує декілька підтипів цих опіоїдних рецепторів (m 1, m 2, k1, k2, k3 , d 1 i d 2). Уявлення про фізіологічну важливість d-рецептор-виборчих речовин було значно розширене завдяки відкриттю непептидного антагоніста налтриндола. У той же час було встановлено, що d-агоністи виявляють автономний антиноцицептивний потенціал. У доповнення до множини експериментів на тваринах, в цій області також було проведене дослідження по впливу пептидного агоніста DADL на онкопацієнтів, на яких морфін більше не давав анальгезуючої дії. При внутрішньооболонковому вживанні DADL показав анальгезуючу дію, що підтримується протягом тривалого часу. d-агоністи істотно відрізняються від m-агоністів відносно їх взаємодії з "ендогенним опіоїдним антагоністом" холецистокиніном (ХЦК). Крім відмінності профілів ефектів, можливо, що профіль побічних ефектів d-агоністів також відрізняється від профілю m-агоністів, наприклад, зниженим придушенням дихання. Тому основною метою справжнього винаходу було виявлення речовин, що володіють анальгезуючим ефектом, біологічна активність яких частково або в основному здійснюється через d-опіатні рецептори. Було виявлено, що цим вимогам задовольняють аміноз'єднання загальної формули (І). Відповідно, справжній винахід відноситься до заміщених аміноз’єднань загальної формули (І), (І) де R1 являє собою Н, ОН, С1-6-алкіл або О-С3-7-циклоалкіл; (при цьому R1 відрізняється від Н, якщо R2 i R7=H, або ОСН3-група заміщена в 7-й позиції); R2 являє собою Н (якщо R1 відрізняється від Н), ОН, С1-6-алкіл або О-С3-7-циклоалкіл, СІ, О-арил, С2-61алкенил-арил, F, С1-6-алкокси, арил, гетероцикліл, або С1-6-алкіл-гетероцикліл, С1-6-алкіл-арил, або 5,6- або 6,7-бензогрупа (незаміщена або моно- або дизаміщена групами СІ, F, CF3, С1-6-алкіл, О-C1-6-алкіл або ОН); (при цьому, якщо R1 і R2 або R7=Н, R2 або R7 не можуть представляти CF3-групу і R2 або R7 не можуть представляти ОСН3-групу в 7-й позиції); або R2 i R7 разом являють собою -О-(СН2) (1-2)-О-(в 5,6- або 6,7-позиціях); R3 являє собою Н; R4 являє собою С1-6-алкіл; або R3 i R4 разом являють собою -(СН2)(1-4)-; R5 являє собою С1-6-алкіл або С3-7-циклоалкіл; R6 являє собою С1-6-алкіл, С1-6-алкіл-арил, С1-6-алкіл-гетероцикліл, -СН2-CH=CH(R8)2, -CH2-(C3-7)циклоалкіл або С3-7-циклоалкіл; R7 являє собою Н (якщо R1 відрізняється від Н), ОН, С1-6-алкіл, О-(С3-7)-циклоалкіл, О-арил, С2-6-алкениларил, С1-6-алкокси, СІ, F, арил, гетероцикліл, С1-6-алкіл-гетероцикліл, С1-6-алкіл-арил, або 5,6- або 6,7бензогрупа (незаміщена або моно- або дизаміщена групами СІ, F, CF3, С1-6-алкіл, О-С1-6-алкіл або ОН); (при цьому, якщо R1 і R2 або R7=H, R2 або R7 не можуть представляти CF3-групу і R2 або R7 не можуть представляти ОСН3-групу в 7-й позиції); і R8 являє собою Н або СН3, або їх фармацевтичні солі. Як окремий випадок з'єднання по винаходу можуть бути такими, в яких: R1 являє собою Н, ОН (при цьому R1≠Н, якщо R2=Н, або ОСН3-групу в 7-й позиції); R2 являє собою Н (якщо R1≠Н), ОН, С1-С6-алкіл (прямий або розгалужений), О-алкіл (С1-6, прямий або розгалужений; без ОСН3- в 7-й позиції, коли R1=H), бензил, О-феніл, феніл, 5, 6- або 6, 7-бензогрупа [якщо потрібно (ди) заміщена групами СІ, F, CF3, алкіл, О-алкіл], СІ, F, CF3; R3 являє собою Н; або R3+R4=-(CH2)1-4; R4 являє собою СН3, С2Р5; R5 являє собою C1-3-алкіл (прямий або розгалужений), і R6 являє собою С1-3-алкіл (прямий або розгалужений), -(СН2)1-2-феніл, -(СН2 )1-2-піридил, -СН2-СН=С(R7)2(з R7=H, CH3) -СН2-С3-6-циклоалкіл, R7 відсутній. Переважні заміщені аміноз'єднання загальної формули (І) включають такі, в яких: R2, R6 i R7 являють собою С1-6-алкіл-арил або С1-6-алкіл-гетероцикліл, a R1, R3-R5 i R8 мають вищезгадане значення по загальній формулі (І), або R2 i R7 являють собою арил або гетероцикліл, a R1, R3-R6 i R8 мають вищезгадане значення по загальній формулі (І), або R1 являє собою ОН або О-С1-6-алкіл, R5 i R6 являють собою С1-6-алкіл, a R2-R4 і R7 мають значення, детально описане вище. Особливо переважними з'єднаннями є такі, в яких R1 являє собою ОН, R5 i R6 являють собою метил, a R2R4 і R7 мають вищезгадане значення відповідно до загальної формули (І). У цьому винаході вираження "С1-6-алкіл" має на увазі прямі або розгалужені вуглеводні, що містять 1-6 вуглецевих атомів. Прикладами можуть служити метил, етил, пропил, ізопропил, n-бутил, втор-бутил, требутил, n-пентил, неопентил і n-гексил. У контексті цього винаходу вираження "С1-6-алкокси" має на увазі прямі або розгалужені вуглеводні, що містять 1-6 вуглецевих атомів, як зазначено вище, які пов'язані через атом кисню. У контексті цього винаходу вираження "арил" має на увазі фенільні групи, незаміщені або одно- або багатозаміщена групами ОН, F, СІ, CF3, С1-6-алкіл, С1-6-алкокси, С1-7-циклоалкокси, С3-7-циклоалкіл, С2-6алкілен, гетероцикліл або феніл. Радикали гетероцикліла або феніла можливо, як варіант, вбудовані шляхом конденсації. Це вираження може також, як варіант, означати нафтил. У контексті цього винаходу вираження "гетероцикліл" потрібно розуміти як 5- або 6-ланкові, насичені або ненасичені гетероциклічні з'єднання, в які, можливо, конденсується арильна система, і які містять від 1 до 2гетероатомів з групи, що включає азот, кисень і/або сірку. Прикладами насичених гетероциклілів можуть служити 1,4-диоксан, тетрагідрофуран і 1,4-тиоксан. Прикладами груп, що являють собою ненасичені гетероцикліли, можуть служити фуран, тіофен, піридин, піримідин, тіазол, оксазол, ізооксазол, піридазін, пиразін, хінолін, ізохінолін, фталазін і хіназолін. У контексті цього винаходу вираження "С1-6-алкіл-арил" або "С1-6-алкіл-гетероцикліл" означає, що "арили" або "гетероцикліли", визначені вище, пов'язані через С1-6-алкільну групу. У контексті цього винаходу вираження "силанільне з'єднання" потрібно розуміти як триалкіл- або триарилсиліли, диалкіларилсиліли або диарилалкілсиліли, які використовуються як захисні групи для функціональної гідроксигрупи. Приклади включають триетилсиліл, трипропилсиліл, диметил-фенілсиліл, дитрет-бутилфенілсиліл, триізопропилсиліл, диметилізопропилсиліл диетилізопропилсиліл, диметилгексилсиліл, трет-бутилдиметилсиліл, трет-бутилдифенілсиліл, трибензилсиліл, три-р-ксилілсиліл, трифенілсиліл, дифенілметилсиліл або пропіл-дифенілсиліл. Цей винахід також відноситься до способу отримання заміщених аміноз'єднань загальної формули (І), відмінного реакцією третичного спирту загальної формули (II) (ІІ) де R1-R7 мають те ж значення, що і в формулі (І), з напівконцентрованими або концентрованими органічними або неорганічними кислотами, зокрема, мурашиною кислотою або соляною кислотою, в температурному інтервалі від 0°С до 100°С, причому третичні спирти загальної формули (II) отримують реакцією ßамінокетонів загальної формули (III) (ІІІ) де R3-R6 мають те ж значення, що і в формулі (І), R9=R2, a R10=R7, за винятком того, що функціональна гідроксигрупа присутня в захищеній формі у вигляді бензилокси- або силаноксигрупи, з металоорганічним з'єднанням формули (IV) (IV) де Z являє собою MgCl, MgBr, Mgl або Li, a R11=R1, за винятком того, що функціональна гідроксигрупа присутня в захищеній формі у вигляді бензилокси- або силаноксигрупи, наприклад, требутилдифенілсилілокси, з утворенням з'єднання формули (ІІа) (ІІа) яке потім перетворюють в з'єднання формули (II). Спосіб отримання з'єднання, згаданого вище як окремий випадок, відрізняється тим, що третинний спирт формули (II), в якому в якої R1-R6 мають ті ж значення, що і в з'єднанні по п. (2), реагує з концентрованими органічними або неорганічними кислотами, зокрема з мурашиної кислотою або соляною кислотою, в температурному інтервалі від 0°С до 100°С, переважно при 20°С, причому третинні спирти загальної формули (II) (ІІ) отримують реакцією ß-амінокетонів загальної формули (III) (ІІІ) де R3-R6 мають те ж значення, що і в з'єднанні по п.2, R9=R2, a R10 відсутній, за винятком того, що функціональна гідроксигрупа присутня в захищеній формі у вигляді бензилокси- або силаноксигрупи, з металоорганічним з'єднанням формули (IV) (IV) де Z являє собою MgCl, MgBr, Mgl або Li, a R11 являє собою водень, бензилокси-, метокси- або одну з силанил груп, наприклад, захищену функціональну гідроксигрупу, яка представлена тетр-бутил-ди-феніл-силанил групою, з утворенням з'єднання формули (ІІа) (ІІа) де R10 відсутній, яке потім перетворюється в з’єднання формули (ІІ). Реакція з’єднань (ІІІ) і (IV) проводиться в аліфатичному ефірі, наприклад, диетиловому ефірі і/або тетрагідрофурані, при температурах між -70°С і +60°С. З’єднання формули (ІV), в яких Z являє собою атом літію, отримують із з’єднань формули (IV), в яких Z являє собою Br або I, шляхом галоген-літієвого обміну, наприклад, за допомогою розчину n-бутиллітію в n-гексані. Для здійснення реакції з’єднання формули (ІІа) з отриманням з’єднання формули (ІІ), в залежність від R9,R10і R11, відповідно, існують різні способи. Якщо R9,R10 і/або R11 являють собою бензилоксигрупу, ця реакція переважно здійснюється шляхом відновного дебензилировання каталітично активованим воднем, де як каталізатор використовуються платина або паладій, адсорбовані на носії, такому як активоване вугілля. Реакція проводиться в розчиннику, такому як оцтова кислота або С1-4-алкіловий спирт, при тиску 1-100 бар і температурі від +20°С до +100°С, причому з’єднання (ІІа) переважно використовується у вигляді однієї з його солей. Якщо R9,R10і/або R11, являють собою силіл-групу, захисна група відщеплюється шляхом реакції відповідного з’єднання формули (ІІа), при 20°С і в інертному розчиннику, наприклад, тетрагідрофурані, диоксані або диетиловому ефірі, з тетра-n-бутиламонійфторидом або з метанольним розчином хлороводню. Якщо R9,R10і/або R11, в з’єднаннях формули (ІІа) являють собою метоксирадикали, з’єднання формули (ІІа), в якому R1 являє собою гідроксигрупу, може бути отримане шляхом реакції з диізобутилалюмінійгідридом в ароматичному вуглеводні, такому як толуен або ксилен, при температурі між 60°С і 130°С. Аналогічне з'єднання формули (II) може бути також отримане безпосередньо шляхом нагріву (ІІа) при зрошуванні з розчином бромистого водню в крижаній оцтовій кислоті або з концентрованою бромистоводневою кислотою. З'єднання формули (І), в яких R1, R2 і/або R7 являють собою ОН, також можуть бути отримані із з'єднань формули (І), в яких R1, R2 і/або R7 являють собою метоксигрупу, шляхом реакції з диізобутилалюмінійгідридом, як описано вище. Способами, відомими в технології, з'єднання формули (І) можуть бути перетворені в їх солі взаємодією з фізіологічно сумісними кислотами, такою як соляна кислота, бромисто-воднева кислота, сірчана кислота, метансульфонова кислота, мурашина кислота, оцтова кислота, щавлева кислота, сукцинова кислота, винна кислота, мигдалева кислота, фумарова кислота, лимонна кислота, глютамінова кислота і/або аспарагінова кислота. Солеутворення переважно проводиться в розчиннику, такому як диетиловий ефір, диізопропиловий ефір, складний алкільний ефір оцтової кислоти, ацетон і/або 2-бутанон. Для отримання гідрохлоридів особливо переважний триметилхлорсилан в розчині, який містить воду. Цей винахід, крім того, відноситься до використання заміщених аміноз'єднань загальної формули (І) по винаходу в якості ліків. Крім як мінімум одного з'єднання формули (І) по винаходу, препарат, що володіє анальгезуючим ефектом, містить допоміжні речовини, наприклад, носії, розчинники, розрихлювачі, барвники і зв'язуючі. Вибір допоміжних речовин, що використовуються, і їх кількості залежить від того, чи буде препарат застосовуватися орально, внутрішньовенно, внутрішньочеревинно, внутрішньошкірно, внутрішньом'язево, інтраназально, буккально або локально. Для орального застосування зручні препарати у вигляді таблеток, жувальних таблеток, драже, капсул, гранул, капель, соків або сиропів. Для парентерального і місцевого застосування, а також для інгаляцій зручні розчини, суспензії, сухі препарати, що легко приготовляються, і аерозолі. З'єднання по винаходу в розчиненому вигляді, нанесені на плівку-носій або на наклейку, можливо, з доданням агентів, що посилюють проникність шкіри, є прикладами зручних черезшкірних форм застосування. З препаратів, що застосовуються орально або черезшкірно, з'єднання по винаходу можуть вивільнятися із затримкою. Кількість активного інгредієнту, що призначається пацієнту, залежить від ваги пацієнта, типу застосування, призначення і міри тяжкості захворювання. Наступні приклади більш детально пояснюють спосіб по винаходу. Як нерухома фаза в колонкової хроматографії використовувався силікагель 60 (0,40-0,063мм), що випускається Е. Merck, Darmstadt. Тонкоплівкові хроматографічні дослідження проводились з викостінням готових плівок HPTLC силікагелю 60F254 від фірми Е. Merck, Darmstadt. Співвідношення змішаних жвавих фаз у всіх хроматографічних дослідженнях приводиться в об'ємних одиницях (об'єм/об'єм). Приклад 1 3-(2-диметиламінометил-3,4-дигідро-нафт-1–іл)-фенолу гідрохлорид 1-й етап: (RS)-2-диметиламінометил-3,4-дигідро-2Н-нафтален-1 Розчин 21мл 3,4-дигідро-2Н-нафтален-1-она в 200мл крижаної оцтової кислоти обробили по черзі 8,2г диметиламину гідрохлоридом і 3,0г параформальдегідом. Суміш нагрівали до 100°С протягом 2 годин, потім випарили розчинник під вакуумом, а залишок вмістили в 200мл води. Потім його екстрагували 3 рази по 100мл диетилового ефіру. Водну фазу довели до pH10 доданням карбонату калію порціями, при ретельному перемішуванні. Потім продукт екстрагували 3 рази по 150мл етилацетату. Екстракти промили насиченим розчином хлористого натрію і висушили над сульфатом натрію. Після фільтрування і після концентрування фільтрату шляхом випаровування під вакуумом отримали залишок - 15,4г (75,6% теоретично) (RS)-2диметиламінометил-3,4-дигідро-2Н-нафтален-1 у вигляді жовтуватого масла. 2-й етап: (1RS, 2RS)-2-диметиламінометил-1-(3-метокси-феніл)-1,2,3,4-тетрагідро-нафт-1-ол Розчин 7,5г 1-бром-3-метоксибензола в 15мл безводного тетрагідрофурана обробили по краплях, при 50°С, при перемішуванні і в захисній атмосфері азоту, 25мл 1,6-молярного розчину n-бутиллітію в n-гексані. Суміш перемішували при -30°С протягом 30 хвилин і потім по краплях додали розчин 6,1г продукту з етапу 1 в 120мл безводного тетрагідрофурана. Після цього суміш перемішували протягом 3 годин при -50°С і протягом 12 годин при -20°С. Після додання 100мл соляної кислоти (10%) суміш екстрагували двічі по 100мл етилацетату. Хлористоводневу фазу довели до pH10 доданням карбонату калію, а потім тричі екстрагували по 50мл дихлорметану. Екстракти висушили над сульфатом натрію, розчинник випарили під вакуумом, а залишок очистили колоночною хроматографією з використанням в якості елюенту суміш 3/1 етилацетата/метанола. Отримали 5,3г (56,5% теоретично) (1RS, 2RS)-2-диметиламінометил-1-(3-метокси-феніл)-1,2,3,4-тетрагідронафт-1-ола у вигляді в'язкого масла. 3-й етап: 3-(2-диметиламінометил-3,4-дигідро-нафт-1-іл)-фенолу гідрохлорид 5,2г продукту з етапу 2 нагрівали при зрошуванні протягом 6 годин з 160мл розчину бромистого водню в крижаній оцтовій кислоті (33% НВr). Потім цю порцію концентрували шляхом випаровування під вакуумом, а залишок вмістили в 150мл води. Його олужнили карбонатом натрію і тричі екстрагували по 50мл дихлорметану. Після промивання екстрактів насиченим розчином хлористого натрію і сушки над сульфатом натрію їх концентрували шляхом випаровування, а залишок очистили колоночною хроматографією з використанням в якості елюенту суміш 5/1 етилацетата/метанола. Отриману основу названого з'єднання перетворили в гідрохлорид з використанням триметилхлорсилана/вода в 2-бутаноні. Вихід: 2,3г (43,8% теоретично) Температура плавлення: 197-199°С Приклад 2 З використанням індан-1-она, 3,4-дигідро-2Н-фенантрен-1-она, 6,7,8,9-тетрагідробензоциклогептена-5она, 7,8,9,10-тетрагідро-6Н-бензоциклооктена-5-она, 5-феніл-3,4-дигідро-2Н-нафтален-1-она, 6-феніл-3,4дигідро-2Н-нафтален-1-она, 6-(3-хлорофеніл)-3,4-дигідро-2Н-нафтален-1-она, 8,9,10-тетрагідроціклопента[a]нафтален-1-она, 3,4-дигідро-2Н-антрацен-1-она або 6-(4-хлорфенил)-3,4-дигідро-2Н-нафтален-1она замість 3,4-дигідро-2Н-нафтален-1-она, і, можливо, інших амінів, на етапі 1 по методиці, описаній в прикладі 1, отримували наступні з'єднання: 2а: 3-(6-диметиламінометил-8,9-дигідро-7Н-бензо-циклогептен-5-іл)-фенолу гідрохлорид Температура плавлення: 218-220°С 2b: 3-(6-диетиламінометил-8,9-дигідро-7Н-бензоциклогептен-5-іл)-фенолу гідрохлорид температура плавлення: 208-211°С 2с: 3-(6-ди-n-пропиламінометил-8,9-дигідро-7Н-бензоцклогептен-5-іл)-фенолу гідрохлорид Температура плавлення: 199-201°С 2d: 3-{6-[(метил-фенетіл-аміно)-метил]-8,9-дигідро-7Н-бензоциклогептен-5-іл}-фенолу гідрохлорид Температура плавлення: розкладання вище за 117°С 2е: 3-{6-[(бензил-метил-аміно)-метил]-8,9-дигідро-7Н-бензоциклогептен-5-мул}-фенолу гідрохлорид Температура плавлення: розкладання вище за 80°С 2f: 3-(6-диметиламінометил-7,8,9,10-тетрагідро-7Н-бензоциклогептен-5-іл)-фенолу гідрохлорид Температура плавлення: 251-253,5°С 2g: 3-{6-[(циклопропилметил-метил-аміно)-метил]-8,9-дигідро-7Н-бензоциклогептен-5-іл}-фенолу гідрохлорид Температура плавлення: 200-202°С 2h: 3-{6-[(метил-(2-піридин-2-іл-етил)-аміно]-метил)-8,9-дигідро-7Н-бензоциклогептен-5-іл}-фенолу гідрохлорид Температура плавлення: 100-105°С 2і: 3-(2-диметиламінометил-3H-інден-1-іл)-фенолу гідрохлорид Температура плавлення: 210-212°С 2j: 3-(2-диметиламінометил-3,4-дигідро-фенантрен-1-іл)-фенолу гідрохлорид Температура плавлення: 253-254°С 2k: 3-(2-диметиламінометил-5-феніл-3,4-дигідронафт-1-іл)-фенолу гідрохлорид Температура плавлення: 250-253,5°С 2l: 3-(2-диметиламінометил-6-феніл-3,4-дигідронафт-1-іл)-фенолу гідрохлорид Температура плавлення: 242-243°С 2m: 3-[6-(3-хлор-феніл)-2-диметиламінометил-3,4-дигідро-нафт-1-іл]-фенолу гідрохлорид Температура плавлення: розкладання вище за 152°С 2n: 3-(8-диметиламінометил-10,11-дигідро-9Н-циклогепта[a]нафт-7-іл)-фенолу гідрохлорид Температура плавлення: 264-267°С 2о: 3-(2-диметиламінометил-3,4-дигідро-антрацен-1-іл)-фенолу гідрохлорид Температура плавлення: 220-222°С 2р: 3-[6-(4-хлор-феніл)-2-диметиламінометил-3,4-дигідро-нафт-1-іл]-фенолу гідрохлорид Температура плавлення: 245-247°С 2q: 3-{6-[(фуран-3-ілметил-аміно)-метил]-8,9-дигідро-7Н-бензоциклогептен-5-іл}-фенолу гідрохлорид Приклад 3 (6-метокси-1-феніл-3,4-дигідро-нафт-2-ілметил)-диметиламіна гідрохлорид 1-й етап: Розчин 50г 6-метокси-3,4-дигідро-2Н-нафтален-1-она в 500мл ацетонитрила обробили 26,6г N,Nдиметилметилен-імонійхлорида і двома краплями ацетилхлорида, і потім перемішували суміш протягом 30 годин при 20°С. Кристалічний продукт відділили, промили ацетоном і висушили під вакуумом при 40°С. Таким чином отримали 70,9г (92,5%) гідрохлориду названої речовини (температура плавлення: 180-182°С), з якої вивільнили основу за допомогою розбавленого водного розчину гідроксиду натрію і екстрагували його дихлорметаном. Після сушки екстрактів над сульфатом натрію і випаровування розчинника під вакуумом отримали залишок - 56,3г (RS)-2-диметиламіно-метил-6-метокси-3,4-дигідро-2Н у вигляді жовтуватого масла. 2-й етап: (1RS, 2RS)-2-диметиламінометил-6-метокси-1-феніл-1,2,3,4-тетрагідро-нафт-1-ол Розчин 20,5г продукту з етапу 1 в 300мл безводного диетилового ефіру обробили по краплях, при -60°С, при перемішуванні і пропущенні сухого азоту, 50мл 2-молярного розчину феніллітію в суміші 70/30 циклогексана/диетилового ефіру. Суміш перемішували при -60°С протягом 2 годин і потім піддали розщепленню за допомогою 150мл насиченого розчину хлорида амонію. Органічну фазу відділили, а водну фазу двічі екстрагували етилацетатом. Об'єднані органічні фази промили насиченим розчином хлористого натрію і висушили над сульфатом натрію. Масло, що залишилося після випаровування розчинника під вакуумом, очистили колонковою хроматографією з використанням як елюенту диізопропилового ефіру. Таким чином отримали 21,3г (78,0% теоретично) (1RS, 2RS)-2-диметиламінометил-6-метокси-1-феніл-1,2,3,4тетрагідро-нафт-1-ола у вигляді масла. 3-й етап: (6-метокси-1-феніл-3,4-дигідро-нафт-2- ілметил)-диметиламіна гідрохлорид Розчин 6,3г продукту з етапу 2 в 100мл соляної кислоти (10%) перемішували протягом 12 годин при 20°С. Потім його олужнили 1N водним розчином гідроксиду натрію і тричі екстрагували дихлорметаном. Екстракти промили насиченим розчином хлористого натрію і висушили над сульфатом натрію. Неочищений продукт, отриманий після випаровування розчинника під вакуумом, очистили колонковою хроматографією з використанням як елюенту сумішшю 3/1 етилацетата/метанола. Основу перетворили в гідрохлорид за допомогою суміші хлор-триметилсилан/вода в 2-бутаноне. (Вихід: 5,4г=81% теоретично; температура плавлення 189-191 °С). Приклад 4 (5-метокси-1-феніл-3,4-дигідро-нафт-2-ілметил)-диметиламіна гідрохлорид Назване з'єднання отримували, у вигляді кристалів, з використанням методики і послідовності реакцій, описаної в прикладі 3, і з використанням 5-метокси-3,4-дигідро-2Н-нафтален-1-она. Температура плавлення: 205-206°С Приклад 5 5а: 6-диметиламінометил-5-феніл-7,8-дигідро-нафт-2-ол гідрохлорид 3,5г продукту з прикладу 3 піддали реакції з 100мл розчину бромистого водню в крижаній оцтовій кислоті (33% НВr), як описано в прикладі 1, етап 3. Після відповідної обробки, очищення колонковою хроматографією і реакції з триметилхлорсиланом/вода отримали 2,4г (63,7% теоретично) названого з'єднання у вигляді білих кристалів. Температура плавлення: 189-191 °С 5b: 6-диметиламінометил-5-феніл-7,8-дигідро-нафт-1-ол гідрохлорид Назване з'єднання отримували аналогічно продукту з прикладу 4 з використанням методики, описаної в прикладі 5а. Температура плавлення: 245-247°С Приклад 6 6-диметиламінометил-5-(3-гідрокси-феніл)-7,8-дигідро-нафт-2-ола гідрохлорид 1-й етап: (1RS, 2RS)-2-диметиламінометил-6-метокси-1-(3-метокси-феніл)-1,2,3,4-тетрагідро-нафт-1-ол Відповідний реагент Грігнара приготували при слабкому кипінні 7,3г магнієвих стружок і 56,1г 1-бром-3метокси-бензолу в 200мл безводного тетрагідрофурана. Потім по краплях додали розчин 46,7г (RS)-2диметиламінометил-6-метокси-3,4-дигідро-2Н-(продукт з прикладу 3, 1-й етап) в 100мл безводного тетрагідрофурана при температурі від +5°С до +10°С. Суміш перемішували протягом 16 годин при +22°С і після охолоджування приблизно до 10°С обробили 100мл насиченого розчину хлорида амонію. У реакційну суміш розбавили 100мл води і 200мл диетилового ефіру, фази розділили, і водну фазу двічі екстрагували по 100мл диетилового ефіру. Об'єднані органічні фази висушили над сульфатом натрію і звільнили від летючих компонентів під вакуумом. Масляний залишок очистили колонковою хроматографією з використанням в якості елюенту етилацетату, після чого отримали 43,9г (64,3% теоретично) (1RS, 2RS)-2-диметиламінометил-6метокси-1-(3-метокси-феніл)-1,2,3,4-тетрагідро-нафт-1-ола. 2-й етап: 6-диметиламінометил-5-(3-гідрокси-феніл)-7,8-дигідро-нафт-2-ола гідрохлорид 34,2г продукту з етапу 1 перемішували з 350мл розчину бромистого водню в крижаній оцтовій кислоті (33% НВr) протягом 20 годин при температурі від 100°С до 110°С. Реакційну масу концентрували шляхом випаровування під вакуумом, а залишок вмістили в 500мл води. Після обробки, описаної в прикладі 1, етап 2, продукт очистили колонковою хроматографією з використанням як елюенту сумішшю 3/1 етилацетата/метанола. Основа названого з'єднання, отримана таким чином, перетворили в гідрохлорид розчином триметилхлорсилана/вода в 2-бутаноні. Вихід: 12,2г (41,2% теоретично) Температура плавлення: 210-212°С Приклад 7 З'єднання 7а і 7b, що є ізомерами продукту з прикладу 6, отримували по методиці, описаній в прикладі 6, з використанням відповідних початкових речовин: 7а: 6-диметиламінометил-5-(3-гідрокси-феніл)-7,8-дигідронафт-1-ола гідрохлорид Температура плавлення: 260-262°С 7b: 7-диметиламінометил-8-(3-гідрокси-феніл)-5,6-дигідронафт-2-ола гідрохлорид Температура плавлення: 239-242°С Заміна (RS)-2-диметиламінометил-6-метокси-3,4-дигідро-2Н-нафтален-1-она в прикладі 6, етап 1, на (RS)6-диметиламінометил-2-метокси-6,7,8,9-тетрагідро-бензоциклогептен-5-он (7с) або (RS)-2диметиламінометил-6-(3-метокси-феніл)-3,4-дигідро-2Н-нафтален-1-он (7d) і наступна реакція з використанням методики, описаної в прикладі 6, дають: 7с: 6-диметиламінометил-5-(3-гідрокси-феніл)-8,9-дигідро-7Н-бензоциклогептен-2-ола гідрохлорид Температура плавлення: розкладання вище за 110°С 7d: 3-[2-диметиламінометил-6-(3-гідрокси-феніл)-3,4-дигідро-нафт-1-іл]-фенолу гідрохлорид Приклад 8 3-(2-диметиламінометил-7-фенокси-3,4-дигідро-нафт-1-іл)-фенолу гідрохлорид 1-й етап: 7-фенокси-3,4-дигідро-2Н-нафтален-1-он Суспензію 60,8г карбонату калію в 300мл безводного піридину обробили 35,7г 7-гідрокси-3,4-дигідро-2Ннафтален-1-она. Потім при перемішуванні додали 19,9г оксиду міді (II) з наступним доданням по краплях 39,6г бромбензола. Реакційну суміш нагрівали протягом 4 днів при зрошуванні. Після повного випаровування піридину під вакуумом залишок змішали з 200мл етилацетату і профільтрували через силікагель. Фільтрат промили насиченими розчинами хлориду амонію і хлориду натрію, висушили над сульфатом натрію і концентрували шляхом випаровування під вакуумом. Залишок очистили колонковою хроматографією з використанням як елюенту сумішшю n-гексана/етилацетата, після чого отримали 41,2г (78,6% теоретично) 7фенокси-3,4-дигідро-2Н-нафтален-1-она. 2-й етап: 2-диметиламінометил-7-фенокси-3,4-дигідро-2Н-1-он 40,5г продукту з етапу 1 в 500мл ацетонитрила піддали реакції, аналогічно прикладу 3, етап 1, з 16,0г N, Nдиметилметилен-імонійхлорида. Після відповідної обробки отримали 45,8г (91,3% теоретично) 2диметиламінометил-7-фенокси-3,4-дигідро-2Н-нафтален-1-она у вигляді масла. 3-й етап: (1RS,2RS)-1-[3-(тре-бутил-дифеніл-силанокси)-феніл]-диметиламінометил-7-фенокси-1,2,3,4тетрагідронафт-1-ол Розчин 41,2г (3-бром-фенокси)-тре-бутил-дифеніл-силана в 300мл безводного тетрагідрофурана обробили по краплях, при температурі -40°С, при перемішуванні і пропущенні сухого азоту, 62,5мл 1,6M розчину n-бутиллітію в n-гексані. Реакційну масу перемішували ще 30 хвилин і потім по краплях додали розчин 25,1г продукту з етапу 2 в 75мл безводного тетрагідрофурана. Реакційну суміш залишили нагріватися до +20°С більш 12 годин і потім піддали розщепленню шляхом додання 100мл насиченого розчину хлориду амонію. Після розбавлення 200мл води і 200мл етилацетату органічну фазу відділили, а водну фазу двічі екстрагували 100мл етилацетату. Об'єднані органічні фази промили насиченим розчином хлористого натрію, висушили над сульфатом натрію і концентрували шляхом випаровування під вакуумом. Залишок очистили колонковою хроматографією з використанням в якості елюенту етилацетату, в результаті чого отримали 32,9г (61,6% теоретично) (1RS,2RS)-1-[3-(тре-бутил-дифеніл-силанокси)-феніл]-диметиламінометил-7-фенокси1,2,3,4-тетрагідро-нафт-1-ола у вигляді майже безбарвного, в'язкого масла. 4-й етап: (1RS,2RS)-2-диметиламінометил-1-(3-гідрокси-феніл)-7-фенокси-1,2,3,4-тетрагідронафт-1-ол Розчин 31,4г продукту з етапу 3 в 360мл безводного тетрагідрофурана обробили по краплях, при температурі від +5°С до +10°С, при перемішуванні, 57,5мл 1-молярного розчину тетра-n-бутиламонійфториду в тетрагідрофурані. Після завершення додання суміш перемішували протягом 3 годин при 20°С, обробили 150мл насиченого розчину хлорида натрію і тричі екстрагували по 150мл етилацетату. Екстракти висушили над сульфатом натрію і концентрували шляхом випаровування під вакуумом. Залишок очистили колонковою хроматографією з використанням як елюенту сумішшю 5/1 етилацетата/метанола, в результаті чого отримали 17,3г (88,7% теоретично) (1RS,2RS)-2-диметиламінометил-1-(3-гідрокси-феніл)-7-фенокси-1,2,3,4тетрагідронафт-1-ола у вигляді в'язкого масла жовтуватого забарвлення. 5-й етап: 3-(2-диметиламінометил-7-фенокси-3,4-дигідро-нафт-1-іл)-фенолу гідрохлорид 15,6г продукту з етапу 4 піддали реакції з 150мл 6N соляної кислоти, як описано в прикладі 3, етап 3. Після аналогічної обробки і перетворення в гідрохлорид отримали 12,1г (73,8% теоретично) 3-(2диметиламінометил-7-фенокси-3,4-дигідронафт-10іл)-фенолу гідрохлориду у вигляді білих кристалів. Температура плавлення: 210-212°С Приклад 9 У прикладі 8, на етапі 2, 7-фенокси-3,4-дигідро-2Н-нафтален-1-он бy(9g). Змінили відповідним 5-метокси (9а), 7-феніл (9b), 7-фенетил (9d), 5-фенокси (9е), 6-фенокси (9f) і 7-n-бутил-похідними (9с) або 2-фенокси6,7,8,9-тетрагідробензоциклогептен-5-оном (9g). З них, шляхом реакційної послідовності і методики, аналогічної прикладу 8, були отримані наступні продукти: 9а: 3-(2-диметиламінометил-5-метокси-3,4-дигідронафт-1-іл)-фенолу гідрохлорид Температура плавлення: 245-247°С 9b: 3-(2-диметиламінометил-7-феніл-3,4-дигідронафт-1-іл)-фенолу гідрохлорид Температура плавлення: 232-233°С 9с: 3-(7-n-бутил-2-диметиламінометил-3,4-дигідронафт-1-іл)-фенолу гідрохлорид Температура плавлення: 232-233°С 9d: 3-(2-диметиламіномстил-7-фенетил-3,4-дигідронафт-1-іл)-фенолу гідрохлорид Температура плавлення: 233-237°С 9е: 3-(2-диметиламінометил-5-фенокси-3,4-дигідронафт-1-іл)-фенолу гідрохлорид Температура плавлення: 197-198°С 9f: 3-(2-диметиламінометил-6-фенокси-З,4-дигідронафт-1-іл)-фенолу гідрохлорид Температура плавлення: 224,5-226°С 9g: 3-(6-диметиламінометил-2-фенокси-8,9-дигідро-7Н-бензоциклогептен-5-іл)-фенолу гідрохлорид Температура плавлення: 245-247°С Приклад 10 3-{6-[алил-метил-аміно)-метил]-8,9-дигідро-7Н-бензоциклогептен-5-он}-фенолу гідрохлорид 1-й етап: 6-[аллил-метил-аміно)-метил]-6,7,8,9-тетрагідро-бензоциклогептен-5-он 5,2г 6,7,8,9-тетрагідро-бензоциклогептен-5-она, 0,96г параформальдегіду і 10,0г аллил-метиламіна гідрохлориду в 60мл крижаної оцтової кислоти піддали реакції по методиці прикладу 1, етап 1. Після аналогічної обробки отримали 6,7г (84,8% теоретично) 6-[аллил-метил-аміно)-метил]-6,7,8,9-тетрагідробензоциклогептен-5-вонана у вигляді жовтуватого масла. Кінцевий етап: 3-{6-[аллил-метил-аміно)-метил]-8,9-дигідро-7Н-бензоциклогептен-5-іл}-фенолу гідрохлорид Продукт з етапу 1 піддали перетворенню з використанням методики, описаної в прикладі 8, етапи 3-5. Отримали 3-{6-[аллил-метил-аміно)-метил]-8,9-дигідро-7Н-бензоциклогептен-5-іл}-фенолу гідрохлорид у вигляді білих кристалів. Температура плавлення: 156-159°С (розкладання) Приклад 11 а) 3-[3-дитметиламіно-1-(3-гідроксифеніл)-2-метил-пропенил]-фенолу гідрохлорид 1-й етап: (2RS)-3-диметиламіно-1,1-біс-(3-метокси-феніл)-2-метил-пропан-1-ола гідрохлорид 27,0г магнієвих стружок перемісили в 150мл безводного тетрагідрофурана. 207,6г 1-бром-3-метоксибензолу, розчиненого в 400мл безводного тетрагідрофурана, додавали по краплях таким чином, щоб реакційна суміш слабо кипіла. Після завершення додання суміш нагрівали при зрошуванні ще протягом години, потім охолодили до 5-10°С і при цій температурі додали по краплях 166,0г (RS)-3-диметиламіно-1-(3метокси-феніл)-2-метил-пропан-1-она, розчиненого в 400мл безводного тетрагідрофурана. Реакційну суміш залишили на ніч і потім знов охолодили до 5-10°С. Розчин Грігнара піддали розщепленню доданням 300мл 20%-го розчину хлориду амонію. Реакційну суміш розбавили 400мл ефіру, фази розділили, і водну фазу двічі екстрагували 250мл ефіру. Об'єднані органічні фази висушили над сульфатом натрію і звільнили від розчинника. Залишок (342г) вмістили в 4000мл 2-бутанона і обробили 81,5г триметилхлорсилана і 13,5мл води. Отримали 165,0г (60% теоретично) 3,3-біс-(2-гідрокси-феніл-2-метил-проп-2-еніл)-диметиламіна гідрохлориду, що кристалізувався протягом ночі при 4-5°С. Температура плавлення: 158-160°С 2-й етап: 3-[3-дитметиламіно-1-(3-гідроксифеніл)-2-метил-пропенил]-фенолу гідрохлорид 58г (2RS)-3-диметиламіно-1,1-біс-(3-метокси-феніл)-2-метил-пропан-1-ола гідрохлориду з етапу 1 розчинили в 2000мл концентрованої бромистоводневої кислоти (47%) і нагрівали при зрошуванні протягом 7 годин. Після охолоджування до кімнатної температури реакційну суміш обробили 800мл води, 2000мл дихлорметану і 80г гідрокарбонату натрію. Після відділення фази дихлорметану водну фазу двічі екстрагували 1000мл етилацетату. Об'єднані органічні фази висушили над сульфатом натрію і звільнили від розчинника. Залишок (42г) розчинили в суміші, що складається з 100мл етилацетату і 100мл тетрагідрофурана і очистили колонковою хроматографією. Елюірування сумішшю 4/1 етилацетата/метанола дало 22г основи. Його розчинили в 500мл 2-бутанона і обробили 8,5г триметилхлорсилана і 16мл води. Гідрохлорид, який викристалізувався (14,6г), відфільтровувати при відсмоктуванні і утворили суспензію в 1000мл 2-бутанона для очищення. Суспензію перемішували протягом 5 годин при зрошуванні. Після охолоджування до кімнатної температури отримали 13,5г гідрохлориду (26,4% теоретично). Температура плавлення: 222-224°С b) Z-3-(1-біфеніл-4-іл-3-диметиламіно-2-метил-пропеніл)-фенолу гідрохлорид Назване з'єднання отримували аналогічно методиці, описаній в прикладі 11а, з використанням (RS)-1біфеніл-4-іл-3-диметиламіно-2-метил-пропан-1-она. Температура плавлення: 192-194°С Наступні з'єднання отримували шляхом аналогічної реакційної послідовності, з використанням на етапі 1 відповідних заміщених диметиламіно-алкан-1-онів: 11с:Е-3-[1-(3,4-дихлор-феніл)-3-диметиламіно-2-метил-пропенил]-фенолу гідрохлорид Температура плавлення: 176-178°С 11d: Z-3-[1-(4-хлор-феніл)-3-диметиламіно-2-метил-пропенил]-фенолу гідрохлорид Температура плавлення: 144-146°С 11е: Z-3-[3-диметиламіно-2-метил-1-(4-фенокси-феніл)-пропенил]-фенолу гідрохлорид Температура плавлення: 190-192°С 11f: Z-3-[3-диметиламіно-2-метил-1-р-толил-пропенил]-фенолу гідрохлорид Температура плавлення: 200-201 °С 11g: Z-3-(2-диметиламінометил-1-феніл-бут-1-енил)-фенолу гідрохлорид Температура плавлення: 188-190°С 11h: Z-3-[1-(4'-хлор-бифеніл-4-іл)-3-диметиламіно-2-метил-пропенил]-фенолу гідрохлорид Температура плавлення: 156-158°С 11і: Z-3-[3-диметиламіно-2-метил-1-(4-стирил-феніл)-пропенил]-фенолу гідрохлорид Температура плавлення: 236-237° 11j: Z-3-[3-диметиламіно-2-метил-1-(4-фенетил-феніл)-пропенил]-фенолу гідрохлорид Температура плавлення: 183-185°С 11k: Z-3-[3-диметиламіно-1(4-гідрокси-феніл)-2-метил-пропенил]-фенолу гідрохлорид 11l: Z-3-(1-бензо[1,3]диоксол-5-іл-3-диметиламіно-2-метил-пропенил)-фенолу гідрохлорид Температура плавлення: 121-124°С Дослідження здатності скріплення дельта-опіатних рецепторів, описані нижче, показують, що з'єднання формули (І) по винаходу володіють чудовою ефективністю в якості анальгетиків. Дослідження здатності скріплення d-опіатних рецепторів Тести по визначенню спорідненості з'єднань формули (І) по винаходу до 5-опіатних рецепторів виконувалися на гомогенатах мозкової оболонки (гомогенат мозку самців пацюків Wistar, без мозочка, варолієвого моста і довгастого мозку). Мозок пацюка, в кожному випадку свіжопрепарований, гомогенізували для цієї мети, при охолоджуванні у льоду, в 50ммоль/л Tpic-HCl (pH7,4) і центрифугували 10 хвилин при 5000g і 4°С. Після декантування і видалення надосадкової рідини, а потім введення мембранного осаду знов в 50ммоль/л Tpic-HCl (pH7,4) і гомогенізації, гомогенат центрифугували 20 хвилин при 20000g і 4°С. Цей етап промивки повторили ще раз. Після цього декантували надосадкову рідину, мембранний осад гомогенізували в холодній суміші 50ммоль/л Тріс-НСІ, 20% гліцерину (вага/об'єм), 0,01% бацитрацина (вага/об'єм) (pH7,4) і заморозили у вигляді аліквотних частин до виконання тестів. Для тестів по скріпленню рецепторів аліквоти розморожували і розбавляли 1:10 буфером, що застосовується в тесті. У тесті на скріплення в якості буфера використали 50ммоль/л Тріс-НСІ, 5ммоль/л MgCl2 (pH7,4) з 0,1% (вага./про.) альбуміну бичачої сироватки, а також 1ммоль/л 3H-2-D-ала-дельторфина II в якості радіоактивного ліганду. Міру неспецифічного скріплення визначали в присутності 10мкмоль/л налоксону. У інші порції додавали з'єднання по винаходу з різними концентраціями і визначали витіснення радіоактивного ліганду з його специфічного зв'язку. Відповідні потрійні порції термостатували протягом 90 хвилин при 37°С і потім збирали за допомогою фільтрації через скловолокуваті фільтри (GF/B) для визначення радіоактивного ліганду, пов'язаного з мембранним гомогенатом. Радіоактивність дисків скловолокуватого фільтра вимірювали bлічильником після додання сцинтилятору. Спорідненість з'єднань по винаходу до d-опіатним рецепторам розраховували згідно із законом діючої маси, за допомогою нелінійної регресії, як величину ІС 50 Значення Ki розраховували із значень ІС 50 з використанням рівняння Ченга-Пруссова. Значення Ki приводяться у вигляді середніх значень ± стандартне відхилення з≥3 незалежних один від одного тестів. Таблиця 1 Приклад 1 2а 2b 2с 2d 2е 2f 2g 2h 2i 2j 3 4 5 6 7 8 9a 9b 9c 10 11a 11b Скріплення дельта-опіатних рецепторів Кі (нмоль/л) 55.8 ± 4.6 17.2 ± 4.3 728.0 ± 146.0 294.0 ± 106.0 54.3 ± 8.2 936.0 ± 32.0 46.3 ± 10.1 182.0 ± 12.0 223.0 ± 86.0 157.0 ± 16.0 80.8 ± 29.7 456.0 ± 62.0 517.0 ± 45.0 843.0 ± 23.0 40.9 ± 6.7 21.6 ± 6.2 25.7 ± 10.2 23.9 ± 0.9 39.3 ± 2.2 42.2 ± 16.0 679.0 ± 214.0 34.0 ± 5.8 23.7 ± 1.7 Таблиця 1 показує, що аміноз'єднання по винаходу ефективні в якості анальгетиків, причому ця біологічна активність частково або в основному здійснюється через d-опіатні рецептори і виявляється в зв'язку з ними. Як ефективні і виборчі агоністи і антагоністи d-опіоїдів, з'єднання загальної формули (І) по винаходу можуть використовуватися як агенти при лікуванні патологічних станів, які звичайно лікують при допомозі агоністів і антагоністів 5-опіоїдних рецепторів. З'єднання загальної формули (І) переважно можуть використовуватися як анальгетики.
ДивитисяДодаткова інформація
Назва патенту англійськоюSubstituted aminocompounds, a method for preparing thereof and a pharmaceutical composition
Назва патенту російськоюЗамещенные аминосоединения, способ получения и фармацевтическая композиция
Автори російськоюBuschmann, Helmut, Heinrich
МПК / Мітки
МПК: C07C 215/00, A61K 31/135, C07D 213/36, A61K 31/4402, A61P 25/04, A61K 31/44, C07C 211/28, C07C 213/00
Мітки: композиція, одержання, аміносполуки, спосіб, заміщені, фармацевтична
Код посилання
<a href="https://ua.patents.su/9-56997-zamishheni-aminospoluki-sposib-oderzhannya-ta-farmacevtichna-kompoziciya.html" target="_blank" rel="follow" title="База патентів України">Заміщені аміносполуки, спосіб одержання та фармацевтична композиція</a>
Стероїди, заміщені в положенні 11, спосіб їх одержання (варіанти) та фармацевтична композиція

Номер патенту: 54483
Опубліковано: 17.03.2003
Автори: БУАЛІ Іаміна, Ван Де Вельд Патрік, ТЕТШ Жан-Жорж, НІК Франсуа
МПК: C07J 41/00, A61K 31/565, A61P 19/10
Мітки: варіанти, фармацевтична, спосіб, положенні, одержання, заміщені, композиція, стероїди
Формула / Реферат:
1. Сполуки загальної формули (І):,де:n - ціле число, що дорівнює 2 або З,або R1 і R2, ідентичні чи різні, означають атом водню або радикал алкіл, що містить від 1 до 4 атомів вуглецю,або R1 і R2 утворюють разом з атомом азоту, з яким вони зв’язані, моно або поліциклічний гетероцикл, що має від 5 до 15 ланок, ароматичний або не...
6-o-заміщені кетоліди з антибактеріальною активністю, спосіб їх одержання (варіанти), фармацевтична композиція та спосіб регулювання бактеріальної інфекції у ссавців
Номер патенту: 51730
Опубліковано: 16.12.2002
Автори: Платтнер Джекоб Дж., Ма Женкун, Ор Ят Сун, Чу Деніел Т., Кларк Річард Ф.
МПК: C07H 17/08, A61K 31/365, A61K 31/7048
Мітки: 6-o-заміщені, ссавців, варіанти, фармацевтична, бактеріальної, кетоліди, антибактеріальною, регулювання, спосіб, активністю, інфекції, композиція, одержання
Формула / Реферат:
1. Похідна еритроміцину, вибрана з групи, що складається з:, (II), (III) (IV)і, (V)або їх фармацевтично прийнятних солей чи складних ефірів, де Y і Z разом визначають групу X, причому X вибирають з групи, що складається з(1) =O(2) =N-ОН(3) =N-O-R1, де R1 вибраний з групи, що включає(a) незаміщений С1-C12-алкіл,(b) С1-C12-алкіл із замісником у вигляді арильної...
Похідні циклопептидів, спосіб їх одержання, фармацевтична композиція, спосіб її одержання

Номер патенту: 55439
Опубліковано: 15.04.2003
Автори: ФІТТШЕН Клаус, ХЬОЛЬЦЕМАНН Гюнтер, ГОДМАН Сімон
МПК: C07K 7/56
Мітки: одержання, похідні, спосіб, композиція, фармацевтична, циклопептидів
Формула / Реферат:
1. Сполуки формули Іцикло(Arg-X-Asp-R1), (I)в якійХ являє собою Gly, Ala або NH-NH-CO,причому можуть братися до уваги також похідні вказаних амінокислот, і залишки амінокислот з'єднані один з одним через -аміно- і -карбоксигрупи по типу пептидного зв'язку,R1 являє собою залишок формули II(II),R2, R3, R4 кожний незалежно один від одного являє собою Н, А, Аr, R5-Ar, Het абоR5-Het,А...
Заміщені піперидин-2,6,-діони (варіанти), спосіб їх одержання (варіанти), фармацевтична композиція, спосіб моделювання імунного відгуку ссавця, спосіб інгібування антиген-спровокованого синтезу інтерлейкіну-2 у ссавців

Номер патенту: 45408
Опубліковано: 15.04.2002
Автори: Егер Курт, Вінтер Вернер, Внендт Стефан, Ціммер Освальд, Тойберт Уве, Цвінгенбергер Каі
МПК: A61P 37/00, A61K 31/445, A61K 31/4427, A61P 29/00, A61P 43/00, C07D 401/04
Мітки: інгібування, одержання, заміщені, піперидин-2,6,-діони, ссавців, варіанти, антиген-спровокованого, імунного, композиція, моделювання, фармацевтична, синтезу, відгуку, ссавця, інтерлейкіну-2, спосіб
Формула / Реферат:
1. Заміщені піперидин-2,6-діони формули (І)де Z означає –C(R1R2)-CH2- або -C(R1)=CH-,R1 являє собою фталімід, коли Z означає -C(R1R2)-CH2-,або фталімідну групу, яка одно- або двократно заміщена гідрокси-, метокси- або аміногрупами, коли Z означає -C(R1)=CH-,R2 означає водень або С1-С6-алкільну групу,R3 являє собою водень, С1-С6-алкільну групу або ж ароматичну, або гетеро ароматичну кільцеву систему,...
Заміщені похідні 4-бісфеніл-4-гідроксимасляної кислоти як інгібітори матричної металопротеази, спосіб їх одержання, фармацевтична композиція та спосіб лікування на їх основі

Номер патенту: 57047
Опубліковано: 16.06.2003
Автори: Брубейкер Уільям Фредерік, Зад'юра Лайза Марія, Бйорге Сьюзен М., Клуендер Гарольд С.Є.
МПК: A61P 19/02, C07C 231/00, A61P 9/00, A61P 35/04, A61P 13/00, A61P 7/02, C07C 235/42, C07C 323/61, A61P 43/00, C07D 209/48, A61P 19/08, C07C 51/347, C07C 323/56, A61P 27/02, C07C 59/00, C07C 323/62, A61P 1/02, A61K 31/4035, C07C 235/84, A61P 29/00
Мітки: спосіб, основі, одержання, металопротеази, матричної, композиція, фармацевтична, заміщені, лікування, інгібітори, кислоти, 4-бісфеніл-4-гідроксимасляної, похідні
Формула / Реферат:
1. Сполука, яка має інгібуючу активність у відношенні до матричної металопротеази загальної формули,(I)деΤ є фармацевтично придатною замінною групою;х дорівнює 0, 1 або 2;m дорівнює 0 або цілому числу від 1 до 4;n дорівнює 0 або 1; іабоА і G обидва є СН2,абоА є хімічним зв'язком і G є...
Попередній патент: Колошниковий затвор для шахтних печей (варіанти)
Наступний патент: Амортизатор
Випадковий патент: Термопластичний композит