Спосіб одержання галогенідів n-алкілнальтрексону








Формула / Реферат
1. Двозарядний іон N-метилнальтрексону формули:
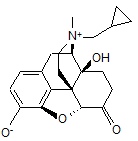 (I)
(I)
у стані безводної форми або гідрату.
2. Двозарядний іон N-метилнальтрексону за п. 1 у конфігурації (R) відносно атома азоту.
3. Двозарядний іон N-метилнальтрексону за п. 1 у конфігурації (S) відносно атома азоту.
4. Двозарядний іон N-метилнальтрексону за будь-яким з пп. 1-3 у стані гідрату, вибраного серед напівгідрату, дигідрату і тригідрату.
5. Двозарядний іон (R)-N-метилнальтрексону, дигідрат.
6. Спосіб одержання сполуки двозарядного іона N-метилнальтрексону, такої, як описано в будь-якому з попередніх пунктів, в якому здійснюють стадію взаємодії метилсульфату N-метилнальтрексону у водному розчині з лужним агентом, вибраним із групи, що включає карбонат натрію, карбонат калію, карбонат кальцію, карбонат магнію, карбонат цезію, карбонат стронцію і їх суміші, так, щоб величина рН водного реакційного середовища знаходилася в інтервалі від 7 до 10.
7. Спосіб одержання броміду N-метилнальтрексону, в якому здійснюють щонайменше стадії:
(і) взаємодії метилсульфату N-метилнальтрексону у водному розчині з лужним агентом, вибраним із групи, що включає карбонат натрію, карбонат калію, карбонат кальцію, карбонат магнію, карбонат цезію, карбонат стронцію і їх суміші, так, щоб величина рН водного реакційного середовища знаходилася в інтервалі від 7 до 10, потім у
(іі) взаємодії продукту, одержаного таким чином, із бромистоводневою кислотою, яку додають для того, щоб величина рН водного реакційного середовища знаходилася в інтервалі від 0,5 до 5, і в такий спосіб одержують бромід N-метилнальтрексону.
8. Спосіб за п. 6 або 7, який відрізняється тим, що лужний агент вибирають із групи, що включає карбонат натрію, карбонат калію і їх суміші.
9. Спосіб за п. 7 або 8, в якому наприкінці стадії (іі) додають метанол, нагрівають реакційне середовище до температури, що знаходиться в інтервалі від 20 до 80 °С, потім фільтруванням відділяють нерозчинний продукт, що залишився, щоб потім остудити фільтрат, з якого кристалізується бажаний бромід N-метилнальтрексону.
10. Спосіб за будь-яким з пп. 7-9, в якому нерозчинний продукт, одержаний наприкінці стадії (і), виділяють після осушування, потім суспендують у суміші метанол/вода, утворюючи в такий спосіб водне реакційне середовище для стадії (іі), на якій при температурі в інтервалі від 20 до 80 °С здійснюють реакцію з бромистоводневою кислотою, потім фільтруванням відділяють нерозчинний продукт, що залишився, щоб остудити потім фільтрат, з якого кристалізується бажаний бромід N-метилнальтрексону.
11. Спосіб за будь-яким з пп. 7-10, в якому одержаний у такий спосіб бромід N-метилнальтрексону піддають стадії очищення розчиненням у суміші ацетон/вода, кип'ятінням зі зворотним холодильником, потім поділом гарячим фільтруванням, осадженням броміду N-метилнальтрексону контактуванням гарячого фільтрату з гарячим ацетоном, охолодженням реакційного середовища до температури нижче 0 °С, при цьому осаджений у такий спосіб бромід N-метилнальтрексону відділяють фільтруванням.
12. Спосіб за будь-яким з пп. 7-11, в якому метилсульфат N-метилнальтрексону одержують, піддаючи метилсульфат О-бензил-N-метилнальтрексону гідруванню.
13. Спосіб за п. 12, в якому метилсульфат О-бензил-N-метилнальтрексону одержують взаємодією О-бензилнальтрексону з диметилсульфатом в ацетоні в присутності гідрокарбонату натрію, при цьому реакційне середовище кип'ятять зі зворотним холодильником протягом часу, достатнього для прийнятного зникнення сполуки О-бензилнальтрексону.
14. Метилсульфат О-бензил-N-метилнальтрексону.
15. Метилсульфат О-бензил-N-метилнальтрексону за п. 14 у конфігурації (R) відносно атома азоту.
16. Метилсульфат О-бензил-N-метилнальтрексону за п. 14 у конфігурації (S) відносно атома азоту.
17. Спосіб за п. 13, в якому О-бензилнальтрексон одержують взаємодією гідрохлориду нальтрексону, або основи нальтрексону, з бензилбромідом в ацетоні в присутності карбонату калію, при цьому реакційне середовище кип'ятять зі зворотним холодильником, потім охолоджують, щоб потім відфільтрувати, і випарюють ацетон з фільтрату з одержанням бажаної сполуки у формі олії.
18. Спосіб за п. 17, в якому гідрохлорид нальтрексону, або основу нальтрексону, одержують взаємодією гідрохлориду нороксиморфону з бромметилциклопропаном у диметилацетаміді в присутності гідрокарбонату натрію, при цьому реакційне середовище нагрівають до температури, що знаходиться в інтервалі від 60 до 75 °С.
Текст
1. Двозарядний іон N-метилнальтрексону формули: 2 (19) 1 3 96957 4 охолодженням реакційного середовища до температури нижче 0 °С, при цьому осаджений у такий спосіб бромід N-метилнальтрексону відділяють фільтруванням. 12. Спосіб за будь-яким з пп. 7-11, в якому метилсульфат N-метилнальтрексону одержують, піддаючи метилсульфат О-бензил-Nметилнальтрексону гідруванню. 13. Спосіб за п. 12, в якому метилсульфат Обензил-N-метилнальтрексону одержують взаємодією О-бензилнальтрексону з диметилсульфатом в ацетоні в присутності гідрокарбонату натрію, при цьому реакційне середовище кип'ятять зі зворотним холодильником протягом часу, достатнього для прийнятного зникнення сполуки Обензилнальтрексону. 14. Метилсульфат О-бензил-Nметилнальтрексону. 15. Метилсульфат О-бензил-N-метилнальтрексону за п. 14 у конфігурації (R) відносно атома азоту. 16. Метилсульфат О-бензил-N-метилнальтрексону за п. 14 у конфігурації (S) відносно атома азоту. 17. Спосіб за п. 13, в якому О-бензилнальтрексон одержують взаємодією гідрохлориду нальтрексону, або основи нальтрексону, з бензилбромідом в ацетоні в присутності карбонату калію, при цьому реакційне середовище кип'ятять зі зворотним холодильником, потім охолоджують, щоб потім відфільтрувати, і випарюють ацетон з фільтрату з одержанням бажаної сполуки у формі олії. 18. Спосіб за п. 17, в якому гідрохлорид нальтрексону, або основу нальтрексону, одержують взаємодією гідрохлориду нороксиморфону з бромметилциклопропаном у диметилацетаміді в присутності гідрокарбонату натрію, при цьому реакційне середовище нагрівають до температури, що знаходиться в інтервалі від 60 до 75 °С. Даний винахід стосується способу одержання галогенідів N-алкілнальтрексону. Четвертинні похідні N-алкілнальтрексону (номенклатурна назва нальтрексону (5)-17(циклопропілметил)-4,5-епокси-3,14дигідроксиморфінан-6-он або Nциклопропілметилнороксиморфон) відомі через їх терапевтичні застосування, зокрема Nметилнальтрексон, застосування якого дозволяє проводити морфінове лікування пацієнта, значно зменшуючи небажані побічні впливи морфіну і його похідних, зокрема на рівні шлунково-кишкового тракту. Під N-метилнальтрексоном мають на увазі, зокрема (R)-N-метилнальтрексон, тобто сполуку конфігурації (R) відносно атома азоту, при цьому фахівцям у даній галузі добре відомо, що (S)-Nметил має активність, протилежну активності, бажаної для проведення морфінового лікування. Конфігурація четвертинної амонієвої групи Nметилнальтрексону наступної формули була визначена в результаті дослідження методом ЯМР 1 H окремих діастереоізомерів (R) і (S): R1 позначає метилциклопропільну групу. 1 Хімічні зрушення в спектрах ЯМР H метальної групи (стандарт TMC (TMS) або тетраметилсилан) становлять 3,62 м.ч. для конфігурації (R) і 3,13 м.ч. для конфігурації (S). Патент US 4176186 (Boehringer Ingelheim GmbH) описує четвертинні похідні нороксиморфону, а також способи їх одержання. Однак, описані способи містять у собі умови, зокрема, тиск, необхідну кількість реагенту, конверсію в аніонообмінній колонці, несумісні із шуканим промисловим застосуванням. Міжнародна заявка на патент WO 2004/043964 описує спосіб при менш високих тисках, що містить в собі використання системи безводного розчинника, зокрема 1-метил-2-піролідону, але який, проте, має ще незручності у відношенні домішок, досить низький необхідний вміст яких неминуче призводить до незадовільного виходу. Таким чином, був безупинно зростаючий інтерес у можливості мати у своєму розпорядженні спосіб, що дозволяє робити такі похідні в промисловому масштабі в найкращих умовах виробництва (безпека, навколишнє середовище) з хорошим виходом. Тепер, несподіваним і дивним чином, знайдений спосіб, що дозволяє дуже вигідним чином одночасно поліпшити умови здійснення (у сенсі безпеки) як для персоналу, так і для навколишнього середовища, і вихід бажаного кінцевого продукту, а саме, галогеніду N-алкілнальтрексону, зокрема броміду N-метилнальтрексону. Відповідно до винаходу, можна здійснити спосіб, що містить в собі стадії відповідно до схеми 1, яка наведена нижче. - конфігурація (S) амонієвої групи з екваторіальною метальною групою: R1 позначає метальну групу, і R2 позначає метилциклопропільну групу, і - конфігурація (R) амонієвої групи з аксіальною метальною групою: R2 позначає метальну групу, і 5 У тому, що наведене нижче, вихідні сполуки і реактиви, зазначені для способу відповідно до винаходу, коли спосіб їх одержання не описаний, є доступними в торгівлі або описаними в літературі, або можуть бути одержані відповідно до методів, що описані або які відомі фахівцям у даній галузі. Таким чином, предметом даного винаходу є, зокрема, нова проміжна сполука, яка, усе-таки без бажання бути пов'язаною з будь-якою теорією, знаходиться у формі двозарядного іона, що має 96957 6 наступну формулу (І) (яка, таким чином, може називатися двозарядним іоном Nметилнальтрексону): 7 Відповідні діастереоізомери конфігурацій (R) і (S) відносно атома азоту двозарядного іона Nметилнальтрексону, а також їхньої суміші, включаючи рацемічні суміші, складають частину винаходу. Крім його безводної форми, двозарядний іон N-метилнальтрексону може також існувати в стані гідрату. Під гідратом, відповідно до винаходу, мають на увазі асоційовану або комбіновану форму сполуки формули (І) з однією або декількома молекулами кристалізаційної води в кристалічній решітці, тобто за винятком води, впровадженої в мікроканали кристалів (або «просочувальної води»), при цьому гідрат спочатку може бути визначений аналізом монокристалу, потім підтверджений рутинним способом за допомогою порівняльного аналізу дифрактограм (або порошкових рентгенограм), як добре відомо фахівцям у даній галузі і проілюстровано в прикладі 1. Такі гідрати теж складають частину винаходу. Наприклад, можна назвати напівгідратну, дигідратну і тригідратну форми. Згідно з окремим способом здійснення винаходу, двозарядний іон формули (І) має конфігурацію (R) відносно атома азоту і знаходиться в стані дигідрату. Дана нова сполука - двозарядний іон Nметилнальтрексону формули (І), переважно, може бути одержаний способом, що містить в собі стадію, яка полягає в здійсненні взаємодії метилсульфату N-метилнальтрексону у водяному розчині з лужним агентом, вибраним в групі, утвореній карбонатом натрію (Na2CO3), карбонатом калію, карбонатом кальцію, карбонатом магнію, карбонатом цезію, карбонатом стронцію і їх сумішей, так, щоб величина рН водяного розчину знаходилася в інтервалі від 7 до 10, переважно, від 9,5 до 9,8, і при температурі, що знаходиться в інтервалі від 15 до 30°C, переважно, близько 20°С. Предметом даного винаходу є також спосіб одержання броміду N-метилнальтрексону, що містить в собі щонайменше стадії, які полягають у: (i) здійсненні взаємодії метилсульфату Nметилнальтрексону у водяному розчині з лужним агентом, вибраним в групі, утвореній карбонатом натрію, карбонатом калію, карбонатом кальцію, карбонатом магнію, карбонатом цезію, карбонатом стронцію і їх сумішей, так, щоб величина рН водяного реакційного середовища знаходилася в інтервалі від 7 до 10, переважно від 9,5 до 9,8, і при температурі, що знаходиться в інтервалі від 15 до 30°С, переважно близько 20°С, потім (ii) у здійсненні взаємодії продукту, одержаного таким чином, із бромисто-водневою кислотою, переважно 48%-ною, яку додають для того, щоб 96957 8 величина рН водяного реакційного середовища знаходилася в інтервалі від 0,5 до 5, переважно була близько 1; переважно, підтримують контакт при перемішуванні протягом ще однієї години, щоб одержати, таким чином, бромід Nметилнальтрексону. Переважно, лужний агент вибирають в групі, утвореній карбонатом натрію, карбонатом калію і їх сумішами. Згідно з окремим способом здійснення, в кінці стадії (іі), описаної вище, можна додати метанол, реакційне середовище нагрівають до температури, що знаходиться в інтервалі від 20 до 80°С, наприклад, від 50 до 70°С, переважно близько 60°С, до практично повного розчинення, потім фільтруванням відокремлюють легкий нерозчинний продукт, що залишився, щоб остудити потім фільтрат метанол/вода, переважно приблизно до 0°С, і змусити кристалізуватися бажаний бромід Nметилнальтрексону. Відповідно до іншого, особливо переважного способу здійснення, нерозчинний продукт, одержаний наприкінці стадії (і), описаної вище, виділяють після осушування, потім суспендують у суміші метанол/вода, переважно, 4/1, утворюючи, таким чином, водяне реакційне середовище для стадії (іі), в якій реакція з бромисто-водневою кислотою, переважно 48%-ною, яку додають, щоб величина рН водяного реакційного середовища знаходилася в інтервалі від 0,5 до 5, переважно була близько 3, здійснюється при температурі в інтервалі від 20 до 80°С, наприклад від 50 до 70°С, переважно 60°C аж до практично повного розчинення, потім фільтруванням відокремлюють легкий нерозчинний продукт, що залишився, щоб остудити потім фільтрат, переважно приблизно до 0°С, і змусити кристалізуватися бажаний бромід Nметилнальтрексону. Перекристалізація із суміші метанол/вода (броміду N-метилнальтрексону) або можливе промивання виділеного продукту (двозарядний іон Nметилнальтрексону) органічним розчинником (наприклад, метанолом) дозволяє видалити ліпофільну домішку броміду O-бензил-Nметилнальтрексону, присутню ще у відомих випадках. Сприятливо, спосіб відповідно до винаходу може містити в собі стадію очищення броміду Nметилнальтрексону, одержаного таким чином, розчиненням у суміші ацетон/вода, переважно 80/20, кип'ятінням зі зворотним холодильником, переважно протягом щонайменше 15 хвилин, потім поділенням фільтруванням у гарячому стані, осадженням броміду N-метилнальтрексону контактуванням гарячого фільтрату з ацетоном, нагрітим, переважно приблизно до 50°С, охолодженням реакційного середовища до температури нижче 0°С, переважно -2°C, при цьому осаджений у такий спосіб бромід N-метилнальтрексону виділяють фільтруванням і сушать. Дана стадія очищення броміду бромід Nметилнальтрексону може бути також здійснена розчиненням у суміші метанол/вода або в одній воді, у такому випадку одержують подібні виходи і кількості тієї ж самої хімічної речовини. 9 У способі, описаному нижче, метилсульфат Nметилнальтрексону може бути вигідно одержаний, піддаючи метилсульфат О-бензил-Nметилнальтрексону стадії гідрування. Згадана стадія гідрування може бути вигідно здійснена як описано в прикладі 1, що йде нижче, і, звичайно, піддаючи метилсульфат О-бензил-Nметилнальтрексону у формі водяного розчину гідруванню на 5%-ному паладованому куті, при цьому реакційне середовище підтримують при температурі, що знаходиться в інтервалі від 30 до 50°С, переважно, 40°C при тиску водню близько 2,5 бар протягом, приблизно, щонайменше 2 годин, для повного O-дебензилування. Реакційне середовище потім прохолоджують і каталітичну систему видаляють фільтруванням. Сприятливо, одержаний продукт можна не виділяти з реакційного середовища, що дозволяє уникнути будь-якого контакту із залишковим диметилсульфатом (дуже токсичний продукт). У способі відповідно до винаходу, метилсульфат О-бензил-N-метилнальтрексону може бути одержаний взаємодією О-бензилнальтрексону з диметилсульфатом в ацетоні в присутності гідрокарбонату натрію, при цьому реакційне середовище кип'ятять зі зворотним холодильником протягом часу, переважно щонайменше близько 72 годин, достатнього для прийнятного зникнення сполуки О-бензилнальтрексон, причому контроль протікання реакції може бути здійснений відомим способом, наприклад, методом ВЕРХ (HPLC). Предметом даного винаходу є також нова проміжна сполука, метилсульфат О-бензил-Nметилнальтрексону, одержана в такий спосіб. Відповідні діастереоізомери конфігурацій (R) і (S) відносно атома азоту метилсульфату Oбензил-N-метилнальтрексону, так само, як їх суміші, включаючи рацемічні суміші, є частиною винаходу. Зокрема захисна бензильна група на фенольному кисні представляє, зовсім конкретно, подвійний інтерес: - використане відщіплення, без введення й утворення іонного продукту, одного водню, і утворений толуол легко видаляється; - гідрування дозволяє зменшити кількість 7,8дидегідро-N-метилнальтрексону (небажаний спряжений кетон) у кінцевому продукті після гідрування подвійного зв'язку. Крім того, спосіб відповідно до винаходу забезпечує відмінну діастереоселективність раніше і при виділенні N-метилнальтрексону у формі двозарядного іона, і при одержанні бажаного кінцевого продукту, а саме, (R)-N-метилнальтрексону. У способі відповідно до винаходу Обензилнальтрексон може бути вигідно одержаний реакцією гідрохлориду нальтрексону, або основи нальтрексону, з бензилбромідом в ацетоні в присутності карбонату калію, при цьому реакційне середовище кип'ятять зі зворотним холодильником при температурі, переважно, близько 60°C на протязі, приблизно, 2 годин, потім прохолоджують до кімнатної температури (близько 20°C), щоб потім відфільтрувати, у відомих випадках, промити ацетоном, і випарюють ацетон з фільтрату з одер 96957 10 жанням бажаної сполуки у формі олії. Переважно, витягають одержану олію, наприклад, дихлорметаном і промивають, наприклад, розведеним їдким натром (3%). Згадана рідинна екстракція лужним середовищем дозволяє повністю видалити залишковий небензильований нальтрексон і уникнути утворення домішки 3-O-метилнальтрексону на стадії алкілування кватернізації. Сприятливо, продукт можна не виділяти, що дозволяє уникнути маніпулювання із середовищем, що містить бензилбромід, сльозоточивий і токсичний продукт. Нарешті, у способі відповідно до винаходу гідрохлорид нальтрексону, або основа нальтрексону, можуть бути вигідно одержані реакцією гідрохлориду нороксиморфону з бромметилциклопропаном у диметилацетаміді в присутності гідрокарбонату натрію, при цьому реакційне середовище нагрівають до температури, що знаходиться в інтервалі від 60 до 75°С, переважно, від 65 до 69°С, як описано, наприклад, на стадії 1 способу за прикладом 1. Фіг. 1 являє собою теоретичну дифрактограму (або порошкову рентгенограму), одержану для монокристалу двозарядного іона, такого, як описаного у прикладі 1 (частина 1.5.2). Фіг. 2 являє собою експериментальну дифрактограму (або порошкову рентгенограму), одержану для двозарядного іона, такого, як описаного у прикладі 1 (частина 1.5.2). Наступні приклади призначені для ілюстрації даного винаходу не обмежувальним чином і, отже, не повинні бути інтерпретовані як здатні обмежити обсяг патентної охорони винаходу. Приклад 1: одержання броміду Nметилнальтрексону 1.1 Одержання неочищеної основи нальтрексону (Стадія 1: N-алкілування) В реактор об'ємом 500 мл, обладнаний холодильником і механічною мішалкою, послідовно вводили 100 г (0,27 моль) гідрохлориду нороксиморфону, 80,8 г (0,96 моль; 3,55 екв.) гідрокарбонату натрію і 300 мл диметилацетаміду. Реакційне середовище нагрівали до температури в інтервалі від 65°C до 69°С. По закінченні спостережуваного виділення газоподібних продуктів (близько 10 хв) протягом 30 хв вводили 35 мл бромметилциклопропану (0,44 моль; 1,6 екв.), підтримуючи температуру 69°С. N-алкілування закінчувалося приблизно за 6 годин, і глибину реакції контролювали методом ВЕРХ (залишковий вміст нороксиморфону менший або дорівнює 0,5%). Реакційне середовище охолоджували до 50°С, потім при перемішуванні протягом 1 години виливали в суміш 1000 мл води і 100 г хлориду натрію, попередньо нагріту до 50°С. Величину рН доводили до 8,6-9 додаванням 8 мл 30%-ного гідроксиду натрію. Одержаний продукт виділяли фільтруванням при 15°С і сушили у вакуумній сушильній шафі при 50°C протягом 14 год. Зрештою одержували 86 г неочищеного нальтрексону (вихід 88,6%) (відповідно до ВЕРХ відносно стандарту й відповідно до структури, визначе 11 1 ної методом ЯМР H, 13 96957 12 C, і маси). 1 ЯМР H : (м. ч. ± 0,01 м. ч.): 0,45 0,65 (2Н, CH2 (20/21), масив); 0,41 і 0,66 (2Н, CH2 (20/21), два мультиплети); 1,11 (1Н, CH (19), мультиплет); 1,47 і 2,72 (2Н, CH2 (15), мультиплет і тд); 1,50 і 2,05 (2Н, CH2 (8), два мультиплети); 2,10 і 3,03 (2Н, CH2 (7), два мультиплети); 2,48 і 3,03 (2Н, CH2 (16), два мультиплети); 2,97 і 3,32 (2Н, CH2 (18), два мультиплети); 4,02 (1Н, CH (9), дублет; J=6,0 H2 ± 0,5 Гц); 5,04 (1Н, CH (5), синглет); 6,71 (1Н, СН(2), дублет; J=8 Гц ± 0,5 Гц); 7,11 (1Н, СОН (14), синглет); 9,05 (1Н, NH, синглет); 9,05 (1Н, СОН (3), синглет). 13 ЯМР C (м. ч. ± 0,1 м. ч.): 2,6 і 5,0 (С20 і С21); 5,6 (С19); 22,8 (С10); 27,1 (С15); 30,6 (С8), 35 (С7); 46 (С16); 48,5 (С13); 56,6 (С18); 60,8 (С9); 69,7 (С14); 88,5 (С5); 118,0 (С2); 119,7 (C1); 120,4 (С11); 127,8 (С12); 140,1 (С4); 143,5 (С6). + Маса (хімічна іонізація MH) = 342,2. 1.2 Одержання О-бензилнальтрексону (Стадія 2: О-бензилування) В реактор об'ємом 50 мл, обладнаний холодильником і механічною мішалкою, послідовно додавали 5,0 г (0,014 моль) гідрохлориду нальтрексону (може бути використана основа), 5,0 г (0,036 моль; 2,58 екв.) карбонату калію і 25 мл ацетону. Потім протягом 10 хв при 20°C при перемішуванні додавали 2,6 г (0,015 моль; 1,08 екв.) бензилброміду. Реакційне середовище кип'ятили зі зворотним холодильником (60°C) протягом 2 годин, потім охолоджували до 20°C і фільтрували. Осад на фільтрі промивали 2 рази 25 мл ацетону. Ацетон випарювали у вакуумі й олію, що залишилася, вилучали в 40 мл дихлорметану, потім промивали 3 рази 25 мл розведеного гідроксиду натрію (3%). Описана рідинна екстракція в лужне середовище дозволяла повністю видалити залишковий небензильований нальтрексон і уникнути утворення домішки 3-О-метил-N-метилнальтрексону на стадії 3 кватернізації. Після операцій декантації й екстракції хлорметиленовий розчин концентрували до відсутності дистиляції, потім направляли на наступну стадію без додаткового очищення. Продукт не виділяли, щоб уникнути маніпулювання із середовищем, що містить бензилбромід, який являє собою сльозоточивий і токсичний продукт. Структурний аналіз: відбирали зразок одержаної олії з виділенням продукту Обензилнальтрексон у формі гідрохлориду (гідрохлорид О-бензилнальтрексону одержували розчиненням основи у формі олії в МТБЕ (MTBE) - або метил-трет-бутиловому ефірі - і додаванням 35%ної соляної кислоти). 1 ЯМР H (м. ч. ± 0,01 м. ч.): 1,2 (2Н, CH2 (20), мультиплет J=6 Гц); 0,46 і 1,20 (2Н, CH2 (20'), мультиплет, J=5 Гц); 1,2 1Н, CH (19), мультиплет, J=7,0 Гц); 3,2 (2Н, CH2, широкі сигнали); 1,67 і 3,2 (2Н, С Гц (15), дд; J=13,8 Гц, J=3,0 Гц, широкі сигнали); 1,64 і 2,51 (2Н, С Гц (8), тд, J=3,2 Гц, широкі сигнали); 2,33 і 3,25 (2Н, CH2 (7), д, J=14,5 Гц, J=5,0, тд J=14,6 Гц, J=2,0 Гц); 2,51 і 3,45 (2Н, CH2 (16), широкий сигнал); 2,94 і 3,45 (2Н, CH2 (18), дд; J=12,5 Гц, J=7,2 Гц, широкий сигнал), 4,51 (1Н, CH (9), широкий синглет); 5,22 і 5,30 (2Н, CH2 (21) і СН2 (21'); J=12,1 Гц); 5,00 (1H, CH (5); широкий синглет); 6,79 (1H, CH (2) і CH (1), система AB; J=8,3 Гц); 6,65 (1Н, CH (1) і CH (2), система AB, J=8,3 Гц); 6,65 (1Н, CH (23) CH (24) бензильна система) 6,65 (1Н, CH (25), бензильна система); 6,65 (1H, CH (24), CH (23), бензильна система). 13 ЯМР C (м. ч. ± 0,1 м. ч.): 3,8 (С20); (С20'); 6,1 (С19); 24,2 (С10); 27,5 (С15); 31,2 (С8); 35,4 (С7); 47,0 (С16); 49,2 (С13); 58,4 (С18); 61,2 (С9); 70,4 (С14); 72,1 (С21 і 21'); 89,8 (С5); 118,9 (С2 і C1); 119,9 (C1 і С2); 121,6 (С22); 127,8 (С23 і С24); 128 (С25); 128,5 (С24 і С23); 137 (С3); 142,8 (С11 і С12); 145,9 (С12 і С11); 207,1 (С6). + Маса (хімічна іонізація MH) = 432,5. 1.3 Одержання метилсульфату N-метил-Обензилнальтрексону (стадія 3: N-метилування, кватернізація) Олію, одержану на попередній стадії, розчиняли в 20 мл ацетону, потім при 20°C при перемішуванні виливали в сухий реактор об'ємом 50 мл, що містить 1,3 г (0,015 моль; 1,08 екв.) гідрокарбонату натрію; потім протягом 10 хв вводили 6,7 г (0,053 моль; 3,53 екв.) диметилсульфату. Реакційне середовище при перемішуванні кип'ятили зі зворотним холодильником мінімум протягом 72 годин до повного зникнення Oбензилнальтрексону (контроль методом ВЕРХ). Реакційне середовище охолоджували до 20°С, потім фільтрували. Осад на фільтрі промивали 2 рази (10 мл) ацетону, потім занурювали в лужний розчин (NaHCO3 або NaOH). Одержаний фільтрат зберігали при 20°C, щоб ввести його на наступну стадію без виділення. Продукт не виділяли, щоб уникнути маніпулювання з продуктом, що містить диметилсульфат. Такий же осад на фільтрі (NaHCO3 + залишок диметилсульфату) розчиняли з використанням лужного середовища на фільтрі без виділення таким чином, щоб зруйнувати диметилсульфат і одержа 13 ти метилсульфат натрію (нетоксичний). Структурний аналіз: відбирали невелику кількість продукту, яку очищували препаративною хроматографією з одержанням зразка, аналізованого в такий спосіб. 1 ЯМР H (м. ч. ± 0,01 м. ч.); 0,41 і 0,88 (2Н, CH2 (20); мультиплет, J=5,0 Гц); 1,2 (1H, CH (19), мультиплет, J=5,0 Гц); 0,55 і 1,06 (2Н CH2 (20'); мультиплет, J=5,0 Гц); 1,75 і 3,0 (2Н, CH2 (15), д; J=12,5 Гц); 3,1 і 3,41 (2Н, CH2 (10), мультиплет д, J=5,5 Гц, J=20,1 Гц); 1,63 і 2,43 (2Н, CH2 (8), тд, дублет мультиплетів, J=13,7 Гц, J=3,2 Гц, J=11,5 Гц); 2,25 і 3,16 (2Н, CH2 (7), дт, масив, J=14,9 Гц; J=2,8 Гц); 3,66 (ЗН, CH3 (17), с); 2,9 і 3,15 (2Н, CH2 (16), мультиплет, J=3 Гц); 5,03 (1Н, CH (9), д, J=4,1 Гц) 5,20 і 5,28 (2Н, CH2 (21) і (21', д, J=12,0 Гц); 2,60 і 3,77 (2Н, CH2 (18); дд, дд, J=13,5 Гц, J=9,4 Гц ; J=13,5 Гц, J=3,6 Гц); 5,05 (1H, CH (5), с); 6,82 (2Н, CH (2) і CH (1), система AB, J=8,3 Гц); 6,68 (2Н, CH (1) і CH (2), система AB, J=8,3 Гц); 7,33 (2Н, CH (23) і CH (24), бензильна система) 7,33 (1Н, CH (25), бензильна система); 7,33 (2Н CH (23) і CH (24), бензильна система). 13 ЯМР C (м. ч. ± 0,1 м. ч,): 3,6 (С20); 4,2 (С19); 7,1 (С20'); 25 (С15); 27,9 (С10); 32,5 (С8); 35,3 (С7); 49,0 (С13); 53,8 (С17); 58 (С16); 71,4 (С9); 72 (С14); 7,21 (С21 і 21'); 73,2 (С18); 89,6 (С5); 119,0 (С2 і C1); 120,3 (С (1) і С (2)); 121,1 (С22); 127,8 (С23 і С24) 128,1 (С25); 128,5 (С24 і С23); 136,8 (С3); 143,3 (С11 і С12); 146,0 (С12 і С11); 206,8 (С6). + Маса (хімічна іонізація M ) = 446. Методом ВЕРХ підтверджували існування відповідних конфігурацій (R) і (S) відносно атома азоту, згідно з співвідношенням конфігурацій RIS 96,6/3,4. 1.4 Одержання метилсульфату Nметилнальтрексону (стадія 4: O-дебензилування) Попередній ацетоновий розчин концентрували втроє перед тим, як додати 100 мл води і здійснити вакуумну дистиляцію до видалення ацетону. Після охолодження до 20°C попередній розчин додавали до (0,3 г) 5%-ного паладію на куті. Тоді реакційне середовище доводили до 40°С. Перед установленням тиску водню 2,5 бар, здійснювали послідовні продувки (N2/H2). О-дебензилування закінчувалося приблизно через 2 години при вмісті метилсульфату N-метилО-бензилнальтрексону менше 0,5% (контроль методом ВЕРХ). Реакційне середовище охолоджували до 20°C і фільтрували, щоб видалити каталізатор. 96957 14 Одержаний у такий спосіб водяний розчин метилсульфату N-метилнальтрексону безпосередньо направляли на наступну стадію. Бензильна захисна група на фенольному кисні становила подвійний інтерес: - використане відщіплення, без введення й утворення іонного продукту, одного водню, і толуол, що утворюється, легко видаляється; - гідрування дозволяє зменшити кількість 7,8дидегідро-N-метилнальтрексону (спряжений кетон, отже, що насторожливої структури) у кінцевому продукті після гідрування подвійного зв'язку. Продукт не виділяли, щоб уникнути контакту із залишковим диметилсульфатом (дуже токсичний продукт). 1.5 Одержання броміду N-метилнальтрексону (стадія 5: обмін метилсульфат/бромід) 1.5.1 Двозарядний іон N-метилнальтрексону (виділення даної сполуки) Водяний розчин, одержаний на стадії 4, концентрували у вакуумі до одержання залишкового об'єму 30 мл, потім вводили 1 г Na2CO3 до одержання рН, приблизно, від 9,5 до 9,8 (природна величина рН карбонату натрію у воді). Реакційне середовище підтримували при 20°C при перемішуванні протягом 1 години. Використання карбонату натрію на даній стадії дозволяло, зокрема зруйнувати диметилсульфат після 1 хвилини контакту. Нерозчинну речовину, що утворилася, зневоднювали й у такий спосіб виявлялося, що двозарядний іон N-метилнальтрексону може існувати в зазначених окремих умовах рН (з використанням карбонату натрію Na2CO3). Структурний аналіз: частину зневодненої нерозчинної речовини, одержаної вище, суспендували у воді при рН близько 9,5 (що дозволяло очистити двозарядний іон перед аналізом «знесоленням»), потім виділяли зневоднюванням і сушінням. 1 ЯМР H (м. ч. ± 0,01 м. ч.): 0,0 і 0,48 (2Н, CH2 (С20); мультиплет, J=5,0 Гц, J=4,5 Гц); 0,88 (1Н, CH (19), мультиплет, J=4,0 Гц); 0,29 і 0,60 (2Н, С Гц (20'), мультиплет, J=4,8 Гц); 1,49 і 2,51 (2Н, CH2 (15), дублет мультиплетів, J=10,4 Гц) 2,79 і 3,29 (2Н, CH2 (10), д, J=19,9 Гц); 1,57 і 1,97 (2Н, CH2 (8) або (7); дд, дублет мультиплетів, J=13,8 Гц, J=3,9 Гц, J=15,2 Гц); 1,77 і 2,71 (2Н, CH2 (7) або (8), дублет мультиплетів, дт, J=13,9 Гц, J=14,9 Гц, J=5,4 Гц); 3,38 (3Н, CH3 (17), с); 2,80 і 3,03 (2Н, CH2 (16); дд ; J=13,0 Гц, J=3,5 Гц); 3,72 (1Н, CH (9), д, J=4,6 Гц); 2,47 і 3,60 (2Н, CH2 (18); т, дд, J=9,8 Гц, J=13,9 Гц, J=3,5 Гц); 4,54 (1H, CH (5), с), 6,35 (2Н, CH (2) і CH (1), система AB, J=8,2 Гц); 6,26 (2Н, CH (1) і CH (2), система AB, J=8,1 Гц). 15 13 ЯМР (м. ч. ± 0,1 м. ч.)=0,0 (С20); 1,3 (С19); 3,7 (С20'); 22,2 (С15); 25,4 (С10); 30,2 (С8 або С7); 30,3 (С7 або С8); 47,0 (С13); 51,0 (С17); 55,5 (С16); 69,8 (С9); 70,3 (С18); 70,5 (С14); 111,9 (С5); 118,9 (С2 і C1); 119,6 (C1 і С2); 124,1 (С3); 143,8 (CH11 i С12): 147,8 (С12 і С11); 211,5 (С6). + Маса (хімічна іонізація MH ) = 356. Елементний аналіз: - розраховані теоретичні величини (C 60,7%; H 7,68%; N 3,37%; O 28,24%), - експериментальні величини (C 61,64%; H 7,6%; N3,19%). Дві приведені величини беруть до уваги вміст води 14,45%, що може бути апріорі інтерпретоване як ступінь гідратації тригідратної форми (3H2O). Однак, були також проведені наступні аналізи. Рентгенодифракційний аналіз порошку РДА(DRX). Аналіз здійснювали на дифрактометрі D5005 фірми Брюкер (Brüker). Діапазон зміни кутів складав від 2,00 до 40,00°28 із кроком 0,02°20 і 2 секундами на крок. Генератор фіксували на 50 кв-40 мА для мідної трубки, довжина хвилі падаючого пучка якої складала 1,54056 Å. Двозарядний іон, очищений «знесоленням», як описано вище, дає експериментальну дифрактограму (дивися фіг. 2), що представляється ідентичною в порівнянні з теоретичною дифрактограмою, що відповідає кристалічній структурі дигідрату (2H2O). Дана теоретична дифрактограма одержана моделюванням (дивися фіг. 1; програмний засіб MERCURY®), виходячи з результатів кристалографічного дослідження, виконаного на монокристалі такого ж двозарядного іона, очищеного «знесоленням». Розходження в ступені дегідратації, одержане на монокристалі (2H2O) і при елементному аналізі (3H2O), пояснюється присутністю двох молекул кристалізаційної води в структурі кристалічної решітки і однієї молекули води, яка походить з води включення в мікроканалах кристалів (просочувальна вода). Методом ВЕРХ підтверджували існування відповідних конфігурацій (R) і (S) відносно атома азоту, згідно з співвідношенням конфігурацій R/S 98/2. 1.5.2 Бромід N-метилнальтрексону Попередню нерозчинну речовину суспендували в 20 мл суміші (4/1) МеОН/вода, додавали бромистоводневу кислоту (кількість, достатня для рН=3), потім реакційне середовище витримували при 60°C до практично повного розчинення. Нерозчинну легку речовину (Nметилнальтрексон не розчиняється) відфільтровували, потім фільтрат охолоджували до 0°C. Неочищений бромід N-метилнальтрексону кристалізувався при охолодженні, потім його зневоднювали. Перекристалізація із суміші метанол/вода (броміду N-метилнальтрексону) або можливе промивання виділеного продукту («двозарядний іон») органічним розчинником (наприклад, метанол) дозволяла видалити ліпофільну домішку броміду O-бензил-N-метилнальтрексону. 1.6 Одержання чистого броміду Nметилнальтрексону (стадія 6: перекристалізація із 96957 16 суміші ацетон/вода) В реактор об'ємом 50 мл, оснащений холодильником, послідовно вводили 5,6 г неочищеного (сухого) броміду N-метилнальтрексону, 7,5 мл води і 22 мл ацетону (або 5 об'ємів суміші 80:20 ацетон/вода). Середовище кип'ятили зі зворотним холодильником протягом 15 хв. Завись (бромід Nметилнальтрексону нерозчинний) фільтрували в гарячому стані (60°C) і гарячий фільтрат вливали в 10 мл ацетону при 50°С. Продукт випадав в осад у розчині, розчин прохолоджували до -2°C і осад відфільтровували. Продукт сушили у вакуумі при 20°C протягом 48 год. Зрештою, одержували 4,3 г чисті броміду Nметилнальтрексону (вихід 76% відносно неочищеного броміду N-метилнальтрексону і 70% відносно вихідного гідрохлориду N-метилнальтрексону). Фізичні властивості: T плавлення: (ДСК) (DSC): 262°C 1 Спектр ЯМР H (м. ч., ±0,01): ідентичний спектру нальтрексону, за винятком 3,7 (3Н, C (22), синглет); 13С (м. ч. +0,01) ідентично нальтрексону, за винятком 58 (C (22)). В цілому, точки узгоджуються з літературними даними. Приклад 2: Одержання броміду Nметилнальтрексону (стадія 5: обмін метилсульфат/бромід, варіант без виділення проміжного продукту) Водяний розчин, одержаний на стадії 4 прикладу 1, концентрували у вакуумі до одержання залишкового об'єму 30 мл, потім вводили 1 г Na2CO3 до одержання рН, приблизно, від 9,5 до 9,8 (природна величина рН карбонату натрію у воді). Реакційне середовище підтримували при 20°C при перемішуванні протягом 1 години, потім за 1 годину додавали 2,1 мл 48%-ної бромистоводневої кислоти до рН близько 1 і зберігали контакт при перемішуванні протягом 1 додаткової години. Нерозчинну речовину з реакційного середовища зневоднювали, і одержаний осад промивали 10 мл ацетону, потім сушили у вакуумній сушильній шафі (10 мм рт. ст.) при 40°C протягом 12 годин. Одержували 9,35 г суміші неочищеного броміду N-метилнальтрексону і неорганічних солей (NaBr і MeSO4Na; вміст неочищеного броміду Nметилнальтрексону 50%). Приклад 3: Одержання броміду Nметилнальтрексону (стадія 5; обмін метилсульфат/бромід, варіант без виділення проміжного продукту, з MeOH) 17 На стадії 5 способу за прикладом 1, після обробки HBr додавали 40 мл метанолу, потім витримували систему при 60°C до практично повного розчинення. Нерозчинну легку речовину (бромід N-метилнальтрексону не розчиняється) відфільтровували. Фільтрат (суміш MeOH/H2O) охолоджували до Комп’ютерна верстка А. Крулевський 96957 18 0°C. Неочищений бромід N-метилнальтрексону кристалізувався при охолодженні, потім його зневоднювали. Найбільша вигода даного варіанта полягає в солюбілізації неорганічних солей (NaBr, CH3SO4Na) у суміші метанол/вода, тоді як NaBr слабко розчинний в суміші етанол/вода. Підписне Тираж 23 прим. Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparing n-methylnaltrexone halogenides
Автори англійськоюDlubala, Alain
Назва патенту російськоюСпособ получения галогенидов n-алкилнальтрексона
Автори російськоюДлюбала Ален
МПК / Мітки
МПК: C07D 498/08
Мітки: галогенідів, спосіб, одержання, n-алкілнальтрексону
Код посилання
<a href="https://ua.patents.su/9-96957-sposib-oderzhannya-galogenidiv-n-alkilnaltreksonu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання галогенідів n-алкілнальтрексону</a>
Спосіб одержання монокристалів пі-комплексів галогенідів міді (1)

Номер патенту: 25459
Опубліковано: 30.10.1998
Автори: Михалічко Борис Миронович, Миськів Мар'ян Григорович
Мітки: галогенідів, монокристалів, міді, пі-комплексів, одержання, спосіб
Формула / Реферат:
Спосіб одержання монокристалів p-комплексів галогенідів міді (I), що включає електрохімічне відновлення солі міді (II) до міді (I) в присутності органічного ліганда, який відрізняється тим, що як органічний ліганд використовують ациклічну алкенінову сполуку.
Спосіб одержання монокристалів галогенідів лужних металів та пристрій для його реалізації

Номер патенту: 28250
Опубліковано: 16.10.2000
Автори: Дідик Роман Іванович, Гарапин Ірина Володимирівна, Антонів Іван Петрович
МПК: C30B 29/12, C30B 11/00
Мітки: пристрій, монокристалів, реалізації, лужних, металів, одержання, спосіб, галогенідів
Формула / Реферат:
1. Спосіб одержання монокристалів галогенідів лужних металів, який включає їх плавлення у вакуумі в присутності вуглецевого сорбенту в реакційній ампулі, розділення продуктів реакції та розплавленої солі в процесі транспортування розплаву з реакційної ампули а ростову та наступну направлену кристалізацію, який відрізняється тим, що розділення продуктів реакції та солі проводять шляхом випаровування солі з реакційної ампули з наступною...
Спосіб електролітичного одержання титану в розплаві галогенідів лужних і лужноземельних металів

Номер патенту: 27190
Опубліковано: 15.08.2000
Автори: Кужель Вячеслав Анатольович, Нерубащенко Володимир Васильович, Петрунько Анатолій Миколаєвич, Проценко Віктор Максимович
Мітки: електролітичного, титану, галогенідів, лужних, металів, спосіб, лужноземельних, одержання, розплаві
Текст:
...аиолнта низшими хлоридами титана, будет равно 365 -24/30 "24 к 12 остановок. Общее количество остановок при данном режиме симально допустимая толщина осадка электролиза составит 4 + 12 - 16 в готитана на диафрагме, т.е. до замыкаду. Производительность процесса электния его с катодом, на котором также ролиза будет равна (365*24 - 16 *24)^ выделяется осадок титана; £ - время хО,55'1ОО = 460,7 кг/год, где электролиза, ч; о ~ толщина осадка...
Спосіб одержання полікристалічних блоків галогенідів рідкісноземельних металів, полікристалічні блоки та монокристали, одержані з полікристалічних блоків

Номер патенту: 87656
Опубліковано: 10.08.2009
Автор: Ільті Ален
МПК: C01F 17/00, C30B 29/12, C09K 11/77
Мітки: рідкісноземельних, полікристалічних, металів, блоки, монокристали, галогенідів, спосіб, одержання, полікристалічні, блоків, одержані
Формула / Реферат:
1. Спосіб одержання полікристалічного блока масою щонайменше 10 г, який є галогенідом загальної формули LnfX3f, в якій Ln означає один або більше рідкісноземельних металів, Х означає один або більше атомів галогену, вибраних з Сl, Вr або І, f більше або дорівнює 1, де згаданий блок містить менше ніж 0,1 мас.% води і менше ніж 0,2 мас.% оксигалогеніду рідкісноземельного металу, який містить стадію нагрівання суміші щонайменше однієї сполуки,...
Спосіб здійснення полум’яного гідролізу галогенідів або органогалогенідів металів

Номер патенту: 11470
Опубліковано: 15.12.2005
Автори: Яремчук Богдан Миколайович, Миронюк Іван Федорович
МПК: C01G 1/02, C01B 33/18
Мітки: органогалогенідів, здійснення, металів, полум'яного, гідролізу, спосіб, галогенідів
Формула / Реферат:
1. Спосіб здійснення полум'яного гідролізу галогенідів або органогалогенідів металів, що включає одержання високодисперсних оксидів металів шляхом спалювання за допомогою пальника в реакторі полум'яного гідролізу приготовленої суміші горючого газу, парів галогенідів чи органогалогенідів і повітря в захисній оболонці з горючого газу, подальше охолодження продуктів гідролізу, коагулювання і відділення частинок оксидів від газового потоку та...
Попередній патент: Газогенератор рідинного ракетного двигуна
Наступний патент: Деталь з твердим покриттям та спосіб її виготовлення
Випадковий патент: Пристрій для черезкісткового остеосинтезу діафізарних переломів кісток передпліччя