Спосіб одержання 2-оксі-4-(метилтіо)-масляної кислоти
Номер патенту: 4916
Опубліковано: 28.12.1994
Автори: Масахару Такано, Лоренс Рассель Вольф, Денніс Артур Руст









Формула / Реферат
Способ получения 2-окси-4-(метилтио)-масляной кислоты гидролизом 2-окси-4-(метилтио)-бутиронитрила водным раствором серной кислоты с использованием нагревания с последующей экстракцией несмешивающимся с водой органическим растворителем и реэкстракцией целевого продукта, отличающийся тем, что, с целью повышения качества целевого продукта, гидролиз 2-окси-4- (метилтио) -бутиронитрила сначала проводят 50-70 % -ной серной кислотой при 25-65°С с последующей обработкой полученного при этом 2-окси-4-(метилтио)-бутирамида 30-50%-ной серной кислотой при 89-120°С, а реэкстракцию проводят в присутствии воды в количестве 5-12,2мас.% в пересчете на экстракт.
Текст
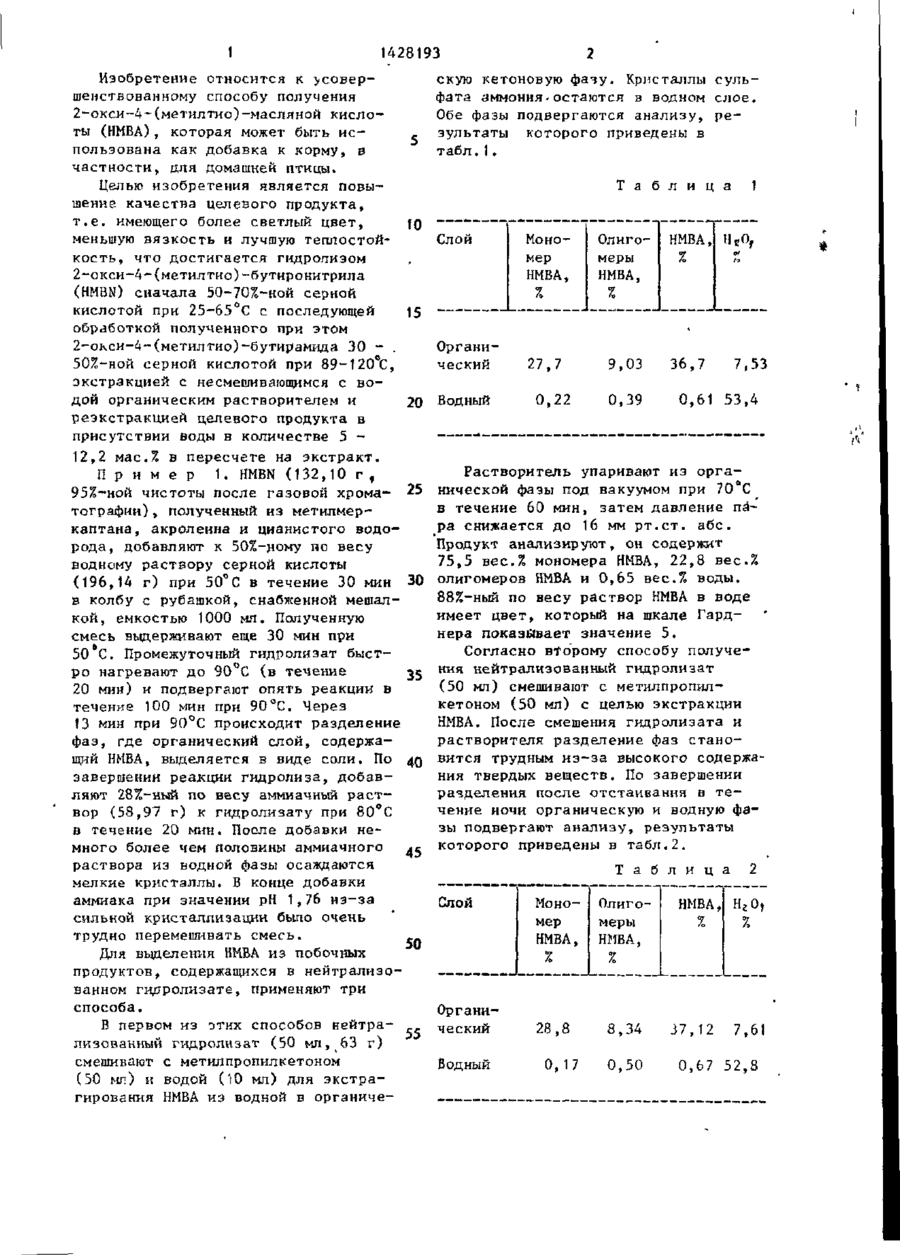
Изобретение касается сероорганических веществ, в частности способа получения 2-окси-4-(метилтио)-масляной кислоты, которая может быть использована как добавка к кор мам в сельском х о з я й с т в е . Цель и з о бретения - повышение качества целевого продукта. Процесс ведут а) гидролизом 2-ОКСН-4-(метилтио)-бутиронитрила 50-;70%-ной H 2 SO 4 при 2 5 - 6 5 ^ ; б) обработкой полученного*2-окси-4-(метилтно)~бутирамида 30-50^-ной H2S0ii при 89-120°С; в) экстракцией с несмешивающнмся с водой органическим растворителем (метилпропил к е тон, 2-пентанол, н-бутилацетат и др^); г) реэкстракцией целевого продукта в присутствии НгО, взятой в количестве 5-12,2 мас.% в пересчете на э к с т р а к т . Способ позволяет получать композиции с отношением мономера и олн' ч омера > 2 , 8 ; цветом (индекс Гарднера) г. 10, вязкостью 90 сСт (против 3,38; 18; 109). 7 т а б л . 8 СО С ГО с» СО 1 1428193 Изобретение относится к с о в е р скую кетоновую фазу. Кристаллы сульшенствованному способу получения фата аммония.остаются в водном слое. 2-ОКСИ-4-Сметилтио)-масляной кислоОбе фазы подвергаются анализу, р е ты (НМВА), которая может быть и с зультаты которого приведены в пользована как добавка к корму, в табл.1. частности, для домашней птицы. Целью изобретения является повыТ а б л и ц а 1 шение качества целевого продукта, т . е . имеющего более светлый цвет, 10 меньшую вязкость и лучшую теплостойСлой МоноОлиго- НМВА, н«о, о* кость, что достигается гидролизом мер меры It X 2-окси-4-(метилтио)-бутиронитрила НМВА, НМВА, (НМВЮ сначала 50-70%-ной серной % % кислотой при 25-65°С с последующей 15 обработкой полученного при этом 2-окси-4~(метилтио)-бутирамида 30 - . Органи50%-ной серной кислотой при 89-120°С, ческий 27,7 9,03 36,7 7,53 экстракцией с несмешивающимся с в о дой органическим растворителем и 0,22 0,61 53,4 0,39 20 Водный реэкстракцией целевого продукта в присутствии воды в количестве 5 12,2 мас.% в пересчете на э к с т р а к т . П р и м е р 1. H B (132,10 г , MN 95%-ной чистоты после газовой хроматографии) , полученный из метилмеркаптана, акролеина и цианистого водорода, добавляют к 50%-ному по весу водному растаору серной кислоты (196,14 г) при 50°С в течение 30 мин в колбу с рубашкой, снабженной мешалкой, емкостью 1000 мл. Полученную смесь выдерживают еще 30 мин при 50 С. Промежуточный гидролизат быстро нагревают до 90 С (в течение 20 мин) и подвергают опять реакции в течение 100 мин при 90°С. Через 13 мин при 90°С происходит разделение фаз, где органический слой, содержащий НИВА, выделяется в виде соли. По завершении реакции гидролиза, д о б а в ляют 28%-ный по весу аммиачный р а с т вор (58,97 г) к гидролизату при 80°С в течение 20 мин. После добавки немного более чем половины аммиачного раствора из водной фазы осаждаются мелкие кристаллы. В конце добавки аммиака при значении рН 1,76 и з - з а сильной кристаллизации было очень трудно перемешивать смесь. Растворитель упаривают из о р г а нической фазы под вакуумом при 70°С в течение 60 мин, затем давление пара снижается до 16 мм р т . с т . а б с . Продукт анализируют, он содержит 75,5 вес.% мономера НМВА, 22,8 вес.% олигомеров Н В и 0,65 вес.% воды. МА 30 88%-ный по весу раствор Н В в воде МА имеет цвет, который на шкале Гарднера показывает значение 5. Согласно второму способу получения нейтрализованный гидролизат 35 (50 мл) смешивают с метилпропилкетоном (50 мл) с целью экстракции НМВА. После смешения гидролизата и растворителя разделение фаз с т а н о 40 вится трудным и з - з а высокого содержания твердых веществ. По завершении разделения после отстаивания в т е чение ночи органическую и водную фазы подвергают анализу, результаты которого приведены в т а б л . 2 . 45 Т а б л и ц а 2 25 Слой 50 Для выделения Н В из побочных МА продуктов, содержащихся в нейтрализованном гидролизате, применяют три способа. В первом из этих способов нейтра55 лизованный гидролизат (50 мл, 63 г) смешивают с метилпропилкетоном (50 мл) и водой (10 мл) для э к с т р а гирования Н В из водной в органичеМА Мономер НМВА, X Органический Водный 28,8 0, 17 Олигомеры НМВА, НМВА, % н г о, % 8, 34 0, 50 37 ,12 7,61 0 ,67 52,8 3 14281 93 Растворитель упаривают из органического слоя под вакуумом при 70°С в течение 60 мин, причем понижается давление пара до 16 мм р т . с т . , про5 дукт Н В на дне анализируют, он с о МА держит 74,9 вес.% мономера НМВА, 23 ,7 вес.% олигомеров Н В и 0,60 вес.% МА воды. Значение цвета 88%-ного р а с т вора продукта Н В в воде на шкале МА 10 Гарднера составляет от 4 до 5. В третьем способе разделения нейтрализованный гидролизат дистиллируют от летучих под вакуумом при 70°С в течение 60 мин, причем давление па-15 ра снижается до 15 мм р т . с т . а б с . В перегонном сосуде образуется очень густой шлам. Отфильтровав твердые вещества, фильтрат анализируют. Он содержит 75,2 вес.% мономера НМВА, 20 20,2 вес.% олигомеров Н В и 3,28 вес.% МА воды. Значение цвета 88%-ного по s e cy раствора продукта Н В в воде МА на шкале Гарднера составляет от 4 до 5. 25 щ снабженный пропеллерной мешалкой. Полученный раствор выдерживают еще в течение 30 мин, затем температуру реакции повышают до 90°С в течение 26-30 мин и выдерживают при 90°С в течение 120 мин. По окончании р е акции порцию гидролизата (1604,4 г) смешивают с метилпропилкетоном (1283,5 г ) при 5О-6О°С в разделительном сосуде емкостью 5 л в т е ч е ние ок. 10 мин, чтобы экстрагировать продукт Н В из гидролизата. МА Затем водную фазу удаляют из сосуда и слой экстракта (2073,2 г) промывают "водой (207_,5 г) при 50°С. Водный слой (48,8 г ; 6,0% НМВА) выливают из сосуда. Раствор упаривают из экстракта под вакуумом при 50°С, продолжая перегонку, пока давление пара не снизилось до 30 мм р т . с т . Затем к остатку под поверхностью добавляют воду (20 мл) и температуру в перегонном сосуде повышают до 70°С, ч т о П р и м е р 2. H B ( 2 0 0 ) , полуMN бы отгонять остаточный растворитель ченный по способу примера 1, медлен-] с водяным паром. Когда давление но добавляют к 50%-ному по весу р а с т пара снижается до 20 мм р т . с т . а б с » при вору серной кислоты (299 г) при 50°С 7б°С дистилляция с водяным паром в течение 30 мин в колбу с рубашкой 30 закончена. Чистый продукт, о с т а в емкостью 1000 мл. Полученную смесь шийся в перегонном сосуде, после выдерживают еще в течение 30мин. Подистилляции с водяным паром подверлученный промежуточный гидролизат гают анализу. Он содержит 74,0 вес.% быстро нагревают до 90 е С (в течение моьомера НМВА, 24,4 вес.% олигомеров 20 мин) и выдерживают еще в течение -зс НМЗА, 1,8 вес.% воды и 0,45 вес.% WQ мин. После 60 мин при 90°С гидсульфатных ионов. Этот продукт р а з ролизат приобретает коричневатый бавляют водой, получая 88%-ный по цвет. Конечный гидролизат состоит из весу продукт НМВА, цвет которого на двух ф а з . шкале Гарднера имеет значения от 5 Не подвергая гидролизат нейтра40 ДО 6 . лизации, его смешивают с одинаковым количеством метилпропилкетона и посП р и м е р 4 . H B (263,16 г ) , MN ле разделения фаз растворитель отполученный способом примера 1, медгоняют в вакууме от экстракта при 70 С в течение 120 мин. Полученный про45 ленно добавляют к 65%-ному по весу раствору серной кислоты (301,45 г) дукт содержит 63,6 вес.% мономера при 50 °С в течение 60 мин в колбу НМВА, 35,2 вес.% олигомеров НМВА, с рубашкой емкостью 1000 мл, снаб0,11 вес.% HMBN, 0,61 вес.% промеженную мешалкой. Полученную смесь жуточного амида, 2,11 вес.% воды выдерживают в течение 30 мин при и 0,27 вес.% сульфатных ионов. Цвет д« 50°С. Затем к промежуточному гидро88%-ного водного раствора продукта лизату добавляют воду (188,91 г ) , на шкале Гарднера имеет значение чтобы снизить концентрацию гидроот 5 до 6. лизующей кислоты до 40% по массе на П р и м е р 3. H B (656 г ) , MN основе отсутствия нитрила. Темпераполученный по способу примера 1, при 55 туру содержимого реактора затем перемешивании медленно добавляют повышают с 50 до 90°С (в течение к 50%-ному водному раствору серной 25 мин) и выдерживают при 90^0 в т е кислоты (981 г) при 50°С в течение чение 115 мин. 60 мин, в реактор вместимостью 2 л , 1428193 На первой стадии гидролиза ( т . е . Продолжение табл.3 во время реакции) в 65%-ном растворе серной кислоты (начальная концентрация при 50°С) вязкость реакМетилизобутилкетон ционной смеси сильно повышается и 2,6 А,7 (116,9 С С) реакционная система начинает образовать две отдельные фазы, одна из 15,4 н-Бутанол (117,3 Q C) 24.0 них содержит промежуточный 2-окси-4-метилтиобутирамид, а другая 10 иэо-Бутанол (107,9°С) 11,2 9.7 только что добавленный к смеси HMBN. Во время второй фазы гидролиза, втор.-Бутанол (99,5°С) 9,6 11,9 т . е . в ходе превращения промежуточного амида в кислотный продукт при трет.Бутанол (82,8°С) Нет 20,5 температуре 90°С, сохраняется одна разде15 фаза без всякого разделения фаз. ления По окончании гидролиза гидролизат фаз подвергают анализу, он содержит 35,21 вес.£ мономера НМВА,0,31 вес.% 2-Пентанол (118,9°С)' 5,2 15,3 димера НМВА, 0.01 вес.% H B и MN 20 0,01 вес.% промежуточного амида. н-Амиловый спирт Другую порцию гидролизата Н В МА (137,5~C) 12,3 15 , 3 данного примера подвергают экстракции, применяя различные растворители, н-Бутиральдегид (75,tTt) 1 2,6 1,4 В каждом примере 100 вес.ч. гид- 25 ролизата смешивают с 60 вес.ч. растНет Этилацетат (77,1 °С) 6,3 ворителя в разделительном сосуде. раздеПосле перемешивания и разделения фаз ления 100 вес,ч. органического слоя профаз мывают 12,5 вес.ч. воды и водный А,9 30 н-Бутилацетат (126,'5°С) 1,9 рафинат (100 в е с . ч . ) , промывают 60 вес.ч. растворителя. Все экстракн-Пропилацетат ( 1 0 1 , 6°С) 2 , 4 7,5 ции проводят при комнатной температуре, т . е . при 25°С.Для каждого-растизо-Пропилацетат (90°С) 2,3 5,4 ворителя определяют коэффициенты распределения при равновесии между ор- 35 Диэтиловый эфир ганической и водной фазами. Коэффи(34,6°С) 2,6 4,5 циент распределения определяют по концентрации Н В в органической фаМА •Диизопропиловый эфир зе в соотношении к концентрации Н В МА (68°С) 2,1 в водной фазе. Результаты экстракций 40 с различными растворителями в данном Хлористый метилен примере приведены в табл.3. ZIIIF" (40°С) Т а б л и ц а З Растворитель (точка кипения) Экстракт в отношении промывочной воды 1 г Метилэтилкетон ( 7 9 , 6 ° С ) 5,4 Метил-н-пропилкетон (102°С) 4,3 Растворитель в отношении рафината 3 14,6 6,2 / і 45 Дихлорэтан (83,5°С) Трихлорэтилен (86,7°С) 50 6 ,7 о;,ь 10 ,4 ,8 9 ,2 и8 Пример 5. Получают Н В по МА следующей схеме. В этой системе гидролизат Н В получают в периодической МА системе реакции, состоящей из одиночного реактора, снабженного мешалкой, 55 однако на двух стадиях реакции. На первой стадии H B медленно добавляMN ют к серной кислоте, причем получают промежуточный гидролизат, содержащий 8 8 1428193 2-окси-4-метилтиобутирамид. Промежуточный гидролизат разбавляют водой и температуру повышают, чтобы превратить промежуточный амид в Н В , Окон- 5 МА чательный гидролизат подают в промежуточный сосуд. Оттуда его непрерывно подают приблизительно в середину экстракционной колонны Карра с ситчатыми тарелками, в которую раство- tO ритель подают возле дна, а промывную воду возле верха. Верхний погон э к стракта предварительно нагревают в теплообменнике и подают в колонну для дистилляции с водяным паром. Н ни- -15 а эу колонны находится жидкий продукт, содержащий Н В и воду. Пары из верхМА ней части колонны конденсируются в конденсаторе и направляются в сепаратор, из которого растворитель р е - 20 циркулируется в нижнюю часть экстракционной колонны, а вода рециркулируется в верхнюю часть экстракционной колонны для промывки. ционной колонны в количестве 100 г/мин. Промывную воду вводят в верхнюю часть колонны. В колонне проводят непрерывную противоточную экстракцию примерно при 59°С при возвратно-поступательном движении тарелок с 140-228 ударами в минуту, получая экстракт, который удаляют из верха колонны, и водный рафинат, который удаляют из низа колонны. Предварительно нагретый в теплообменнике экстракт подают в колонну для перегонки с водяным паром, где дистиллируют растворитель при давлении 2~35 мм р т . с т , в верху, при 82 С в верхней части колонны и 88 С в нижней части колонны, получая низовой продукт в количестве 78 г/мин, к о торый состоит из водного раствора Н В . Пары из верхней части, содерМА жащие 100 г/мин МІВК н 50 г/мин в о ды, конденсируются в конденсаторе и поступают в сепаратор. Выходящий из низа экстракционной 25 колонны рафннат подвергают дистилляции с водяным паром в колонне, чтобы выделить остаточный растворитель из паров в верхней части колонны, которые также направляются к конден- 30 сатору, где они конденсируются и подаются в сепаратор. На дне колонны находятся водные отходы, которые удаляются. Рафинат из низа колонны подвергают перегонке с водяным паром в колонне при давлении в верхней части ко-г лонны 760 мм р т » с т . , температуре вер*ха 97°С и температуре в сосуде 107 в С, получая пар в верхней части, содержащий 0,9 г/мин МІВК и 5 г/мин воды, которые смешивают с парами из верхней части колонны, конденсируют в конденсаторе и направляют в сепаратор. При дистилляции рафината В типичном периодическом гидроли35 в колонке в нижней части колонны зе данного примера 65,1 мас.Х серной получают продукт 144 г/мин, который кислоты (142,3 кг) подают в реактор направляют к отходам. на стадии 1 и в реактор медленно д о бавляют H B (120,1 кг) в течение MN Экстракционная колонна представ61 мин при температуре от 50 до 54 С. ляет собой колонну Карра с ситчатой 40 На стадии ЇА промежуточный гдироли- , тарелкой диаметром 2,54 см и высотой эат разбавляют до концентрации кисло2,1 м. ты 40,1% (на основе отсутствия нитПосле достижения стабилизированнорила) , добавляя воду и нагревая до го состояния гидролизат, поступающий 89 С в течение 30 мин. Затем гидиз сосуда» и водный продукт из низа 45 ролизат еще выдерживают при 90 °С в перегонной колонны периодически о т течение 75 мин. Затем удаляют лету-т бирают для анализа. Результаты аналичиє компоненты, постепенно снижая зов приведены в т а б л . 4 . давление примерно до 110 мм р т . с т . • абс. в течение ок. 45 мин, снижая Т а б л и ц а 4 50 температуру примерно до 65°С. Приблизительно 11 кг вещества улетучиваются . Затем гидролизат вливают в Гидролизат, Продукт, Показатель промежуточный сосуд. % Конечный гидролизат из промежу55 точного сосуда непрерывно подают в колонну в количестве 181 г/мин и метилизобутипкетоновый растворитель (МІВК) подают в нижнюю часть экстрак і г % 3 НМВА 38,2-^42,3 89,2~91,8 Вода 25,1 -28,4 8,20-10,8 9 1428193 Продолжение табл.А 10 и анализируют. В табл.5 приведены результаты этих анализов. 1 і Т а б л и ц а Сульфатный ион 25,6 - 2 8 , 0 0,45^1,3 Мономер НМВА 33,9-35,1 72,8—80,2 5 Олигомеры НМВА Цвет (по Гарднеру) 4, 3~-7,2 Продукт, Показатель 1 1Я4 ^16,9 10 Н В МА 3,5-5,5 41,2 ^ 4 1 , 6 87, W 91 ,9 25,5-26,6 11,8-12,2 Сульфатный ион 2 7 , 1 - 2 7 , 9 0,52-0,62 Мономер Н В МА 74,9~75,4 Олигомеры Н В МА 13,8 ~ 15,0 Цвет -(по Гарднеру) Вода 15 П р и м е р 6. Гидролизат получа,1 ют по способу примера 5. В экстракци онную колонну Карра подают гидролиг" эат в количестве 204 г/мин. Колонна 20 работает при 60°С, вводят растворитель MIBK в количестве 112 г/мин и промывную воду в количестве 23 г/мин, ситчатая тарелка сотрясается с 170 ударами в минуту, получают экстракт, 25 который предварительно нагревают до 99,5°С при абсолютном давлении 451 мм р т . с т , и направляют в перегонную колонну для экстракта. В верхней части колонны давление составля- 30 ет 451 мм р т . с т . и температура 99,5°С, в сосуде - температура 102°С, в нижней части колонны получают концентрированный водный продукт Н В в к о МА личестве 94»0 г/мин, В верхней ч а с 35 ти перегонной колоны для экстракта образуются пары в количестве 112 г/мин MIBK и 42,5 г/мин воды. Эти пары смешивают с верхними парами от перегонки рафината, конденсируют и подают в сепаратор. Рафинат, образующийся на дне экстракционной колонны, подают в колонну для перегонки р а фината, удаляя растворитель перегонкой при давлении 451 мм р т . с т . и температуре 93°С в верхней части колон- 45 ны и 94°С в нижней части, колонны. В верхней части колонны образуются пары в количестве 0,7 г/мин MIBK и ' 12,5 г/мин воды. Эти пары смешивают , с верхними парами от перегонной к о - 50 лонны для э к с т р а к т а , конденсируют и подают в сепаратор. На дне перегонной колонны рафината образуются водные отходы в количестве 129,0 г/мин, которые удаляют.' 55 После достижения стабилизированного состояния в данном примере периодически отбирают пробы продукта ' 3 П р и м е р 7. 63,1 вес.% р а с т в о ра серной кислоты (1555 г , содержащий 980 г, т . е . 10 моль серной кислоты) подают в р е а к т о р , снабженный мешалкой емкостью 5 л . В течение часа H B (1310 г ; 10 моль) добавляMN ют к серной кислоте в реакторе при 50°С, в то время как реактор охлаждают ледяной ванной. По завершении добавки нитрила полученную смесь выдерживают при 50°С в течение получ а с а , затем добавляют воду (900 г) 1 полученную разбавленную смесь (40%4 ная по массе серная кислота на основе отсутствия нитрила) нагревают до 90°С*в течение часа и еще час выдерживают с целью превращения амида в кислотный продукт. Конечный гидролизат упаривают под вакуумом при 7О-9О°С, пока не будет достигнуто конечное давление 100 мм р т . с т . , причем было удалено 37 г л е тучих. Во время отгона летучих из гидроли зата осадилось небольшое количество твердых веществ, добавляют 2,2 г воды, чтобы растворить твердые вещества. Выделяют Н В из порции гидролиМА з а т а , работая с про'тивоточной э к с - ; тракционной системой на четырех ступенях. Согласно этому примеру гидролизат (200 г) и MIBK (40 г) смешивают на первой ступени, получая э к с 11 1428193 12 тракт и рафинат. Порцию (100 г) рафината подают совместно с MIBK (20 г) на вторую ступень. После выделения экстракта на второй стадии 85 г рафината второй ступени направляют на третью ступень, где перемешивают с дополнительной порцией MIBK (17 г ) . После выделения экстракта на третьей .стадии 70 г рафината третьей стадии 10 смешивают с MIBK (14 г) на четвертой ступени. Все экстракции проводят )при комнатной температуре. После разделения фаз на каждой ступени экстракции фазы экстракта и рафината 15 анализируют относительно НМВА, результаты приведены в табл.6. Т а б л и ц а б Ступень Анализ НМВА, вес Л Экстракт Рафинат 57,6 5,67 16,4 t ,82 5,5 0,70 2,0 По завершении гидролиза содержи-, мое реактора обрабатывают под вакуумом, 'упаривая 21 фунтов ( 9 , 5 кг) воды и летучих. После отгонки летучих гидролизат в количестве 204 г/мин вводят в э к стракционную колонну с ситчатыми тарелками диаметром 2,54 см, на месте 61 см ниже верха набора тарелок 244 см. В верх колонны подают воду (23,5 г/мин) и MIBK (112 г/мин) подают в н и з . MIBK составлял непрерывную фазу в экстракционной зоне. Экстракционная колонна работает при температуре примерно 60°С. Экстракт от верхней части колонны проходит через предварительный нагреватель, где он нагревается до 115°С при а т мосферном давлении. При подобных 20 условиях значительное количество MIBK упаривается. Оставшуюся жидкую органическую фазу подают в верх перегонной колонны диаметром 7,6 см и 25 высотой 229 см с выступающей на 0,64 см металлической насадкой Каннона. 0,28 30 П р и м е р 8. H B (107,6 кг) MN добавляют к 64,9%-ному по весу р а с т вору серной кислоты (123,9 кг) в эмалированный реактор вместимостью 38 л , снабженный внешним теплообменником, циркуляционным насосом и трубопроводами для циркуляции и охлаждения содержимого реактора. Нитрил добавляют в течение 59 мин. В т е ч е ние первых девяти минут смесь н а г р е вается с 30 до 60 С и в течение п о следних 50 мин температура поддерживается при 60 Q C. После завершения добавки нитрила смесь перемешивают еще 15 мин при 60°С, получая промежуточный гидролизат. Затем к р е акционной смеси, содержащей 40%-ной по весу серной кислоты на основе отсутствия нитрила, добавляют воду (77,2 кг) и полученную смесь нагревают с 60 до 89° С в течение 30 мин.' Затем смесь выдерживают еще 88 мин при 89°С, получая конечный гидролиз а т , содержащий НМВА. 35 40 45 50 55 В нижнюю часть колонны подают пар в количестве 19 г/мин. Давление на верху колонны атмосферное и тем-\ пература на низу колонны с о с т а в л я ет 116°С. Продукт из низа колонны анализируют, он содержит 88,9% НМВА, 0,56% сульфатных ионов и воду. Цвет , продукта по шкале Гарднера с о с т а в л я ет 4. П р и м е р 9* Гидролизат Н В МА получают пр способу примера 8. Гидролизат экстрагируют, подавая его в количестве 201 г/мин в экстракционную колонну диаметром 2,54 см с сигчатыми тарелками, на месте 61 см ниже верха набора тарелок 244 см. В верх колонны подают воду в количестве 22,5 г/мин и MIBK вводят в низ колонны в количестве 111 г/мин. Фаза растворителя представляла собой непрерывную фазу в экстракционной з о н е . Колонна работала при температуре около 60°С. Экстракт верха экстракционной колонны подают в теплообменник, где он нагревается до 71°С при 147 мм р т „ с т . Значительное количество MIBK упаривается при этих условиях и о с таточную жидкую фазу подают в верхнюю часть перегонной колонны типа, описанного в примере 8. В низ колон- , ны подают пар в количестве 28,5 г/мин. 14 13 H28193 Давление в верху колонны составляет ления воды, затем быстро нагрева1А7 мм р т . с т . ют до температуры примерно 120°С В нижней части получают продукт, и подвергают реакции в течение пекоторый согласно анализу содержит риода, достаточного для превращения 89,0 вес.% НМВА, 0,54 вес.% сульфатобщего количества промежуточного ных ионов и воду. амида в НМВА. Полученный продукт НМВА затем выделяют из конечного П р и м е р JO. H B (ок. 200 г ) , MN гидролизата согласно способу, опиполученный по способу, описанному в примере 1, медленно добавляют к 50%- 10 санному в примере 5. ному по массе раствору серной кислоАнализ композиции НМВА, полученты (299 г) при 65°С. Полученную ной согласно известному способу,осусмесь подвергают реакции в течение ществлялся таким же образом, как и периода, достаточного для превращеанализ композиции, полученной согласния общего количества H B в 2-окси- 15 но предлагаемому способу. Известную MN -4-(метилтно)бутирамид. Промежуточкомпозицию разбавили водой до полуный гидролизат быстро нагревают до чения водного раствора, содержащего температуры примерно 120 °С и под89 вес.% 2-окси-4-(метилтио)маслявергают реакции в течение периода, ной кислоты, что определялось стандостаточного для превращения обще20 дартным титрованием Вг~/ВгОз. Кинего количества промежуточного амида матическая вязкость определялась при в НМВА. Полученный продукт Н В з а МА 25°С,' а цвет оценивался по индексу тем выделяют из конечного гидролицвета Гарднера (диапазон значений зата согласно способу, описанному от 1 до 18). Соотношение мономера в примере 5. 25 с олигомерами НМВА определялось гаП р и м е р 11. H B ( о к . 200 г ) , MN зовой хроматографией. Относительполученный по способу, описанному в ная стабильность определялась темпримере 1 описания, медленно д о пературой, при которой началось выбавляют к 70%-ному по массе р а с т деление газа, когда образец подвору серной кислоты (214 г) при 30 вергли калориметрии с возрастающим 25°С. Полученную смесь подвергают ускорением. Результаты вышеописанреакции в течение периода, д о с т а ного анализа приведены в табл.7. точного для превращения общего к о Сравнение запаха двух образцов в личества H B в 2-окси-4-(метилтио) MN качественном отношении осуществлялось бутирамид. Промежуточный гидролитаким образом, что равновесный пар зат разбавляют до концентрации с е р - 35 ввели в контакт с органами обоняния. ной кислоты 30% по массе (на осноНе было установлено существенного ве отсутствия нитрила) путем добавотличия в запахе. Т а б л и ц а Образец согласно способу * Вязкость (сантистокс при 25°С) Извест ный 109 Предлагаемый 2,8 Цвет 7, Выде (ин- ление декс газа Гард- эндонера) терм., тем., °С 18+ 165 >150 изобретению, по сравнению с композицией известного способа. В то время как данные не подтверждают существен IS 16 1428193 щей экстракцией несмешиваюшимся водой органическим растворителем и реэкстракцией целевого продукта, _ о т л и ч а ю щ и й с я тем, что, с целью повышения качества целевого продукта, гидролиз 2-окси-4-(метилтио)-бутиронитрила сначала проводят 50-70%-ной серной кислотой при Ю 25-65 С с последующей обработкой "Формула и з о б р е т е н и я полученного при этом 2-окси-4-(метилтио)-бутирамида 30-50%-ной с е р Способ получения 2-окси~4-(метилной кислотой при 89-120°С, а р е э к тио)-масляной кислоты гидролизом стракцию проводят в присутствии во 2-окси-4-(метилтио)-бутиронитрила -15 ды в количестве 5-12,2 мас.% в п е водным раствором серной кислоты с ресчете на э к с т р а к т . . .' использованием.нагревания с последуюного улучшения запаха, известная ком позиция уступила предлагаемой в в я з кости и значении цвета. Эти два свойства имеют наибольшее значение, а улучшение этих свойств, наряду с повышением соотношения мономера с олигомерами, приводит к улучшенной композиции. Редактор М.Бандура Составитель Т.Власова Техред А.Кравчук Корректор Э.Лончакова Заказ 4869/58 Тираж 370 . . Подписное ВНИИПИ Государственного комитета СССР по делам изобретений и открытий 113035, Москва, Ж-35, Раушская н а б , , д , 4/5 . Производственно-полиграфическое предприятие, г . Ужгород, ул. Проектная, 4
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation 2-oxy-4-(methylthio)-butyric acid
Автори англійськоюLaurence Russell Wolf, Dennis Arthur Ruest, Masaharu Takano
Назва патенту російськоюСпособ получения 2-окси-4-(метилтио) -масляной кислоты
Автори російськоюЛоренс Рассель Вольф, Деннис Артур Руст, Масахару Такано
МПК / Мітки
МПК: C12P 11/00, A01N 37/02, C07C 327/00, C07C 67/00, C07C 325/00, A23K 1/22, A23K 1/16, C07C 323/52
Мітки: кислоти, одержання, спосіб, 2-оксі-4-(метилтіо)-масляної
Код посилання
<a href="https://ua.patents.su/10-4916-sposib-oderzhannya-2-oksi-4-metiltio-maslyano-kisloti.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання 2-оксі-4-(метилтіо)-масляної кислоти</a>
Спосіб одержання alрнa, alрнa-діфеніл – 4 – аріл – 4 -оксі- 1 – піперідін – бутанамід – n -оксидів або їх стереоізомерів

Номер патенту: 2708
Опубліковано: 26.12.1994
Автори: Людвіг Пауль Кауманс, Лауренс Валс
МПК: A61P 1/12, C07D 211/52, C07D 413/06, A61K 31/445, A61K 31/451, C07D 211/94
Мітки: піперідін, одержання, alрнa-діфеніл, спосіб, оксі, стереоізомерів, бутанамід, alрнa, арил, оксидів
Формула / Реферат:
Способ получения a, a-дифенил-4-арил-4-окси-1 -пиперидинбутанамид-N -оксидов общей формулиили их стереоизомеров, где АІk — группа—СН2— СH2— или СН2—СН(СН3)—; R1, R2 - (C1-4)-алкил; Аr — фенил, возможно имеющий до 2 заместителей, выбранных из галоида и трифтор-метила, отличающийся тем, что производное пиперидина общей формулыподвергают N-окислению 10—50 %-ным раствором Н2О2 или З-хлорбензойной кислотой в...
Спосіб одержання фосфорної кислоти

Номер патенту: 1358
Опубліковано: 25.03.1994
Автори: Арман Лоран Давистер, Фраксіс Артур Тирсон
Мітки: фосфорної, кислоти, спосіб, одержання
Формула / Реферат:
Формула изобретения1. Способ получения фосфорной кислоты, включающий разложение измельченного фосфатного сырья обработкой оборотной фосфорной и серной кислотами в нескольких зонах с образованием реакционной пульпы, состоящей из фосфорной кислоты и осадка сульфата кальция, вакуум-охлаждение пульпы и ее циркуляцию через реакционные зоны в количестве 300-4000% от основного потока, через вакуум-охлаждение в количестве 2000-4000% от...
Спосіб одержання толуолсульфонату 5-метокси-6-метилтіо-2-[5, 5-диметил-3-[5-метокси-6-метилтіо-3етил-2-(3н)-бензотіазоліліден] метил-2-циклогексеніліден] метил-3-етилбензотіазолію

Номер патенту: 2559
Опубліковано: 26.12.1994
Автори: Вальшин Галі Карімович, Романов Микола Миколайович, Сломінський Юрій Леонідович, Смірнова Ганна Леонідівна
МПК: C09B 23/00
Мітки: 5-диметил-3-[5-метокси-6-метилтіо-3етил-2-(3н)-бензотіазоліліден, спосіб, метил-2-циклогексеніліден, метил-3-етилбензотіазолію, одержання, толуолсульфонату, 5-метокси-6-метилтіо-2-[5
Формула / Реферат:
Способ получения толуолсульфоната 5-ме-токси-б-метилтио-2- [5,5-диметил-З- [5-метокси -6-метилтио-3-этил-2-(ЗН) -бензотиазолилиден ]метил-2-циклогексенилиден ]метил-3-этил-бензотиа-золия формулы Івзаимодействием толуолсульфоната бензотиазолия формулы 2с производным циклогексана при повышенной температуре с последующей обработкой полученного плава толуолсульфонатом бензотиазолия в среде смеси пиридина с...
Спосіб одержання 6-аміно-1, 2-дігідро-1-оксі-2іміно-4-піперідінопірімідину

Номер патенту: 3148
Опубліковано: 26.12.1994
Автори: Бела Штефко, Іштван Хегедюш, Чаба Сантаі, Габор Блашко, Денеш Мате, Андраш Ведреш, Тамаш Мештер, Янош Крайдль, Андраш Немеш, Андрієнн Суховскі, Ерік Богш
МПК: C07D 239/50
Мітки: спосіб, 6-аміно-1, 2-дігідро-1-оксі-2іміно-4-піперідінопірімідину, одержання
Формула / Реферат:
1. Способ получения 6-амино-1,2-дигидро-1-окси-2-имино-4 -пиперидинопиримидина формулыпутем взаимодействия производного 6-амино-1, 2-дигидро-1-окси-2-иминопиримидина и пиперидина в органическом растворителе при нагревании, отличающийся тем, что, с целью упрощения процесса и повышения выходного целевого продукта, в качестве производного пиримидина используют пиримидин общей формулыгде К — водород или группа общей...
Спосіб одержання 95%-ої мурашиної кислоти

Номер патенту: 2329
Опубліковано: 26.12.1994
Автори: Хейнц Хохеншуц, Ханс Кіфер, Йоханнес Е.Шмідт
МПК: C07C 53/02, B01J 31/00, C07C 67/00, B01J 31/02, C07C 51/00, C07C 27/00, C07C 51/09, C07B 61/00
Мітки: мурашиної, 95%-ої, спосіб, кислоти, одержання
Формула / Реферат:
Способ получения 95 %-ной муравьиной кислоты путем гидролиза метилформиата при 90— 140 °С и при соответствующем этим температурам давлении в присутствии 1—10 моль воды на 1 моль метилформиата с использованием формамида с последующей перегонкой гидролизной смеси, отличающийся тем, что, с целью упрощения и интенсификации процесса, в качестве формамида используют водорастворимый ди-н-пропилформамид или N-формилморфолин, взятый в эквимолярном...
Попередній патент: Композиція для отримання фарбованих виробів
Наступний патент: Стрілочний перевід з сердечником хрестовини з пересувними основним та допоміжним гостряками
Випадковий патент: Спосіб лікування структур внутрішньо-печінкових жовчних проток