Заміщені піридини та їх застосування як антагоністів рецептору метаботропного глутамату
Номер патенту: 74419
Опубліковано: 15.12.2005
Автори: Айзек Метвін Бенджамін, Макліод Дональд А., Моу Скотт Т., ван Вагенен Бредфорд, Шіхен Сузан М., Слессі Абделмалік, Сторманн Томас М., Сміт Деріл Л.



































































































Формула / Реферат
1. Сполука або її фармацевтично прийнятна сіль, вибрана з групи, яка складається зі сполук, представлених у таблиці:
№
сполука
В57
В58
В59
В60
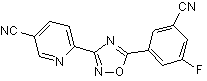
В61
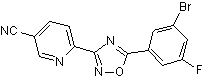
В62
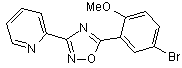
В63
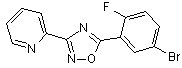
B64
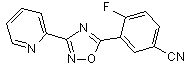
B65
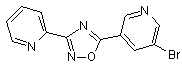
B66
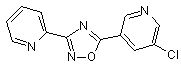
B67
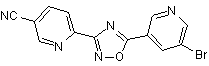
B68
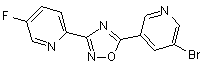
B69
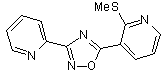
B70
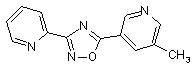
B71
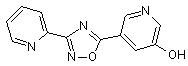
B72
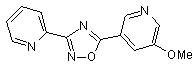
B73
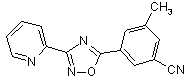
B74
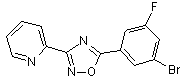
B75
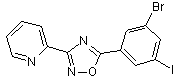
B76
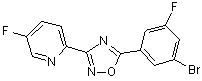
B77
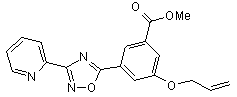
B78
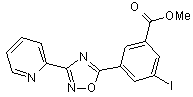
В79
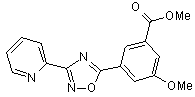
B80
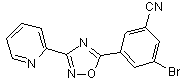
B81
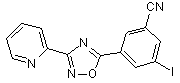
B82
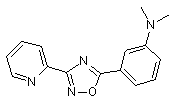
B83
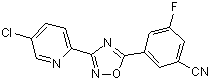
B84
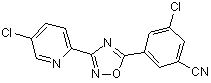
B85
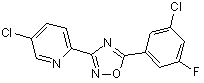
B86
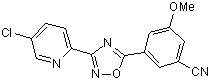
B87
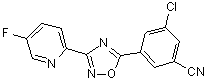
B88
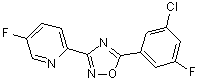
B89
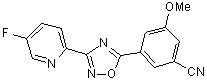
B90
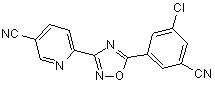
B91
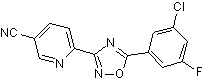
B92
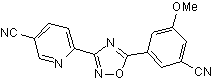
B93
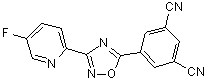
B94
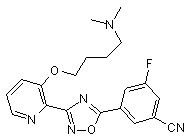
B95
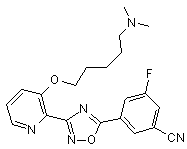
B96
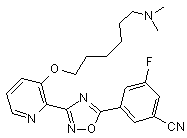
B97
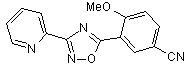
B98
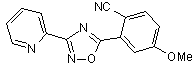
B99
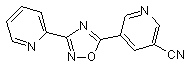
B100
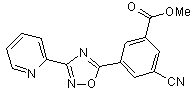
B101
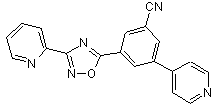
B102
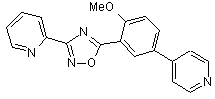
B103
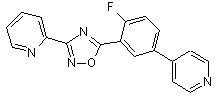
В104
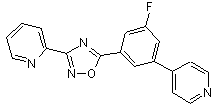
В105
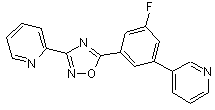
В106
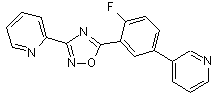
В107
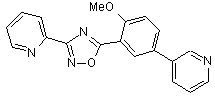
B108
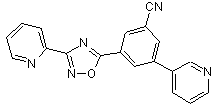
В109
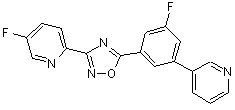
В110
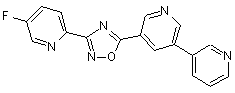
B111
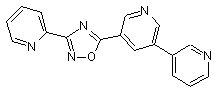
B112
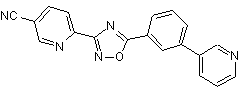
В113
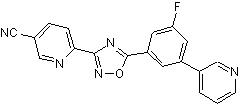
B114
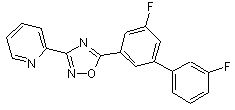
B115
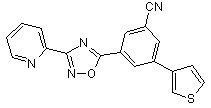
B116
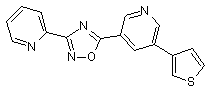
В11 7
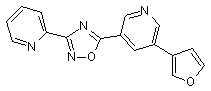
В118
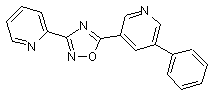
B119
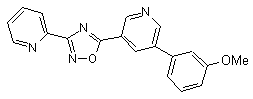
B120
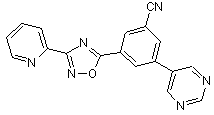
B121
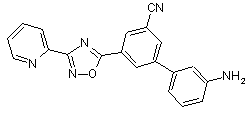
B122
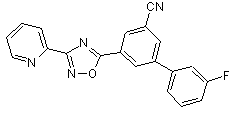
B123
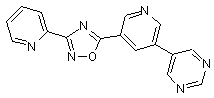
B124
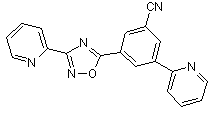
B125
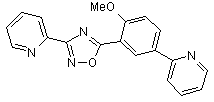
B126
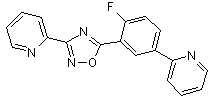
B127
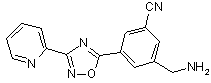
B128
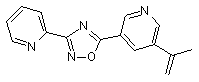
B129
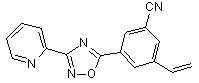
B130
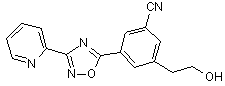
B131
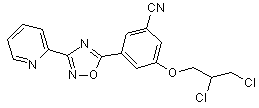
B132
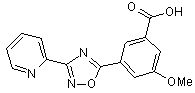
B133
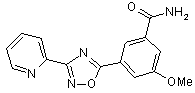
B134
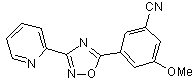
B135
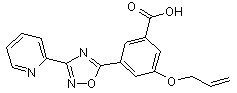
B136
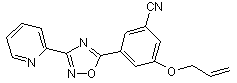
B137
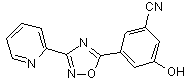
B138
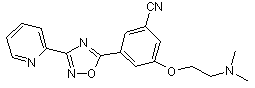
B139
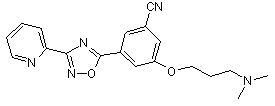
B140
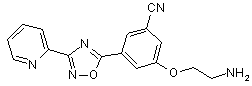
B141
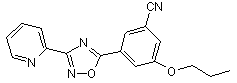
B142
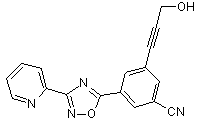
B143
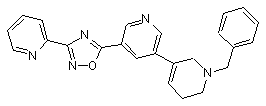
B144
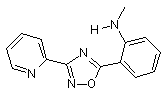
B145
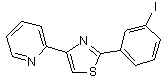
B146
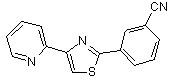
B147
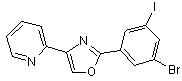
B148
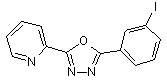
B149
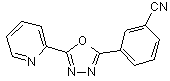
B150
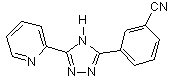
B151
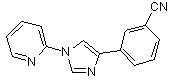
B152
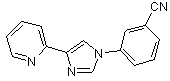
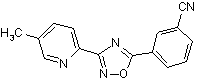
2. Сполука або її фармацевтично прийнятна сіль за п. 1, яка відрізняється тим, що являє собою:
В59
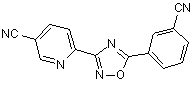
3. Сполука або її фармацевтично прийнятна сіль за п. 1, яка відрізняється тим, що являє собою:
В60
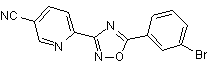
4. Сполука або її фармацевтично прийнятна сіль за п. 1, яка відрізняється тим, що являє собою:
B87
5. Фармацевтична композиція, яка містить терапевтично ефективну нетоксичну кількість сполуки за п. 1 та фармацевтично прийнятний носій.
6. Спосіб лікування хвороби, пов'язаної з активацією mGluR Групи І, при якому здійснюють введення пацієнтові, який потребує такого лікування, фармацевтичної композиції за п. 5.
7. Спосіб за п. 6, який відрізняється тим, що хвороба є хворобою, пов'язаною з активацією mGluR .
8. Спосіб за п. 7, який відрізняється тим, що хвороба є неврологічною хворобою.
9. Спосіб за п. 7, який відрізняється тим, що хвороба є психічною хворобою.
10. Спосіб за п. 7, який відрізняється тим, що хвороба належить до групи, яка складається з інсульту, травми голови, аноксичних уражень, ішемічних уражень, гіпоглікемії, епілепсії, болю, мігреней, хвороби Паркінсона, старечого слабоумства, хореї Гантингтона та хвороби Альцгеймера.
11. Сполука або її фармацевтично прийнятна сіль, вибрана з групи, яка складається зі сполук, представлених у таблиці:
№
сполука
B153
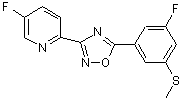
B154
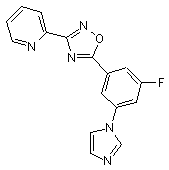
B250
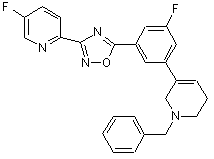
B212
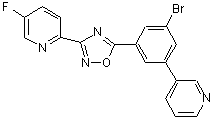
B251
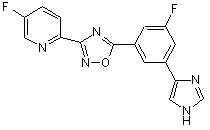
B213
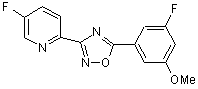
B252
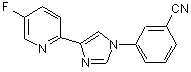
B253
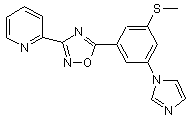
В254
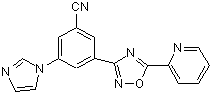
В255
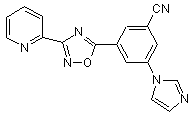
В269
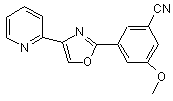
В272
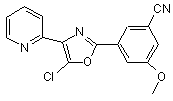
В216
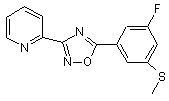
В217
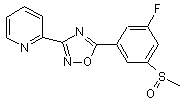
В214
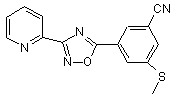
В215
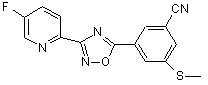
В218
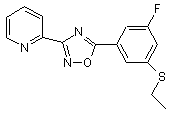
B219
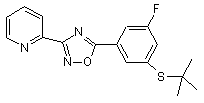
B155
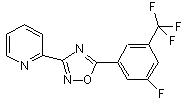
B156
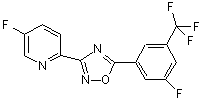
B223
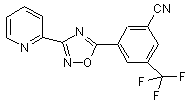
B224
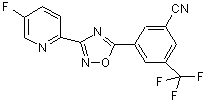
B258
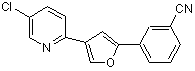
B257
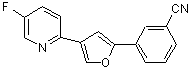
B256
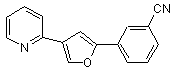
B244
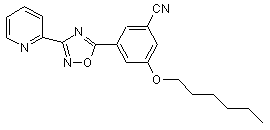
В167
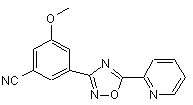
В168
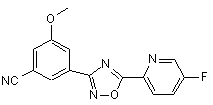
В169
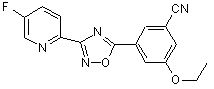
В170
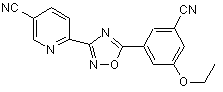
В171
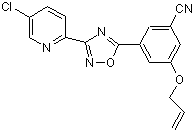
В172
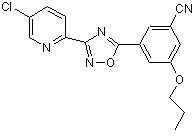
В173
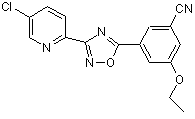
В174
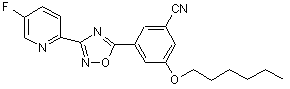
В175
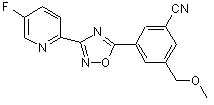
B245
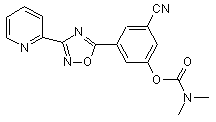
B246
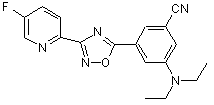
B247
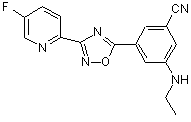
B260
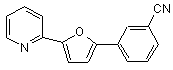
B261
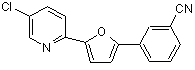
B263
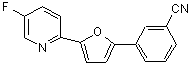
B262
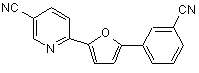
B259
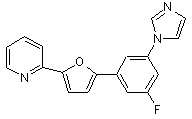
B264
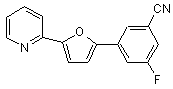
B274
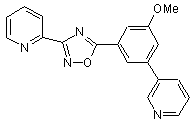
B275
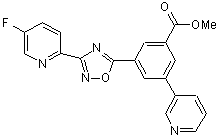
B276
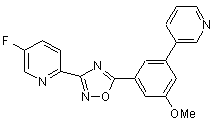
B277
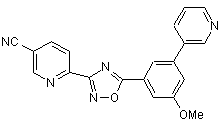
B278
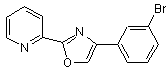
B279
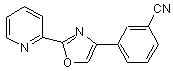
B280
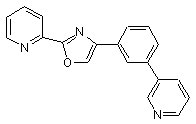
B220
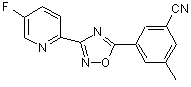
B221
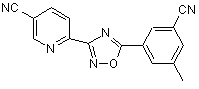
B265
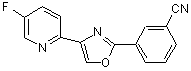
B267
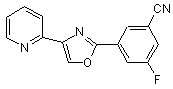
B266
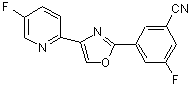
B268
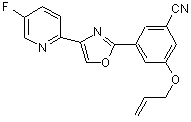
B270
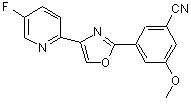
B271
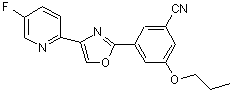
B222
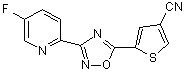
B176
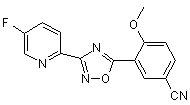
B177
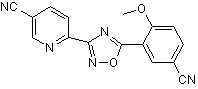
B225
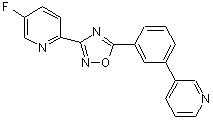
B226
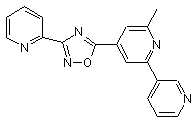
B227
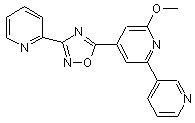
B181
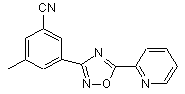
B182
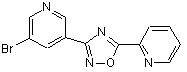
B228
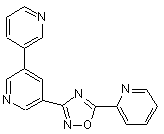
B183
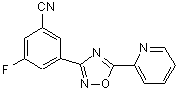
B184
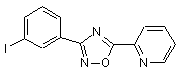
B186
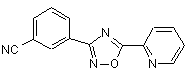
B187
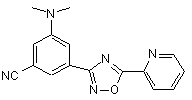
B188
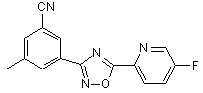
B189
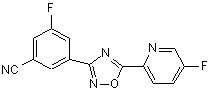
B229
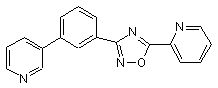
B190
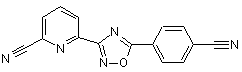
B191
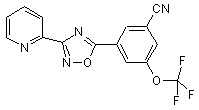
B192
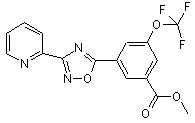
B193
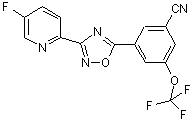
B194
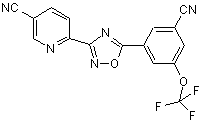
B195
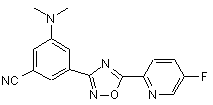
B230
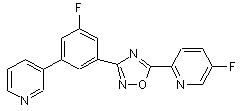
B197
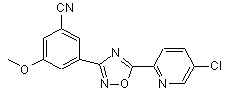
B196
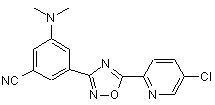
B198
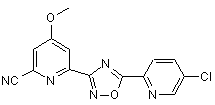
B200
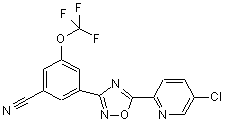
B201
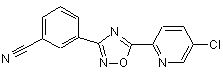
B202
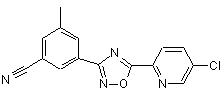
B203
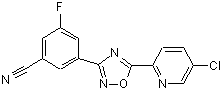
B199
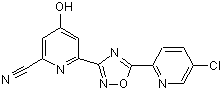
B204
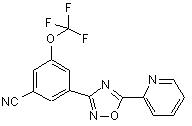
B205
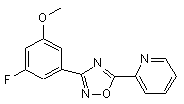
B206
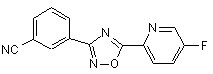
B208
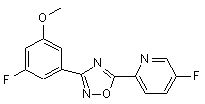
B207
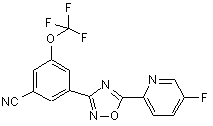
B209
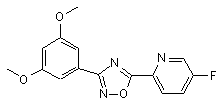
B248
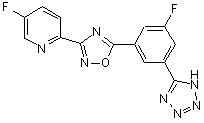
B249
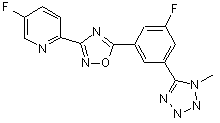
В210
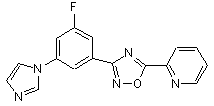
В231
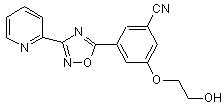
В157
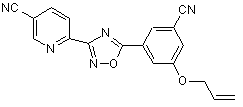
В158
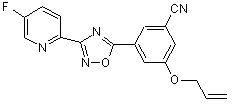
В159
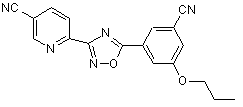
В160
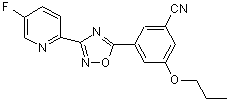
В232
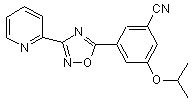
В233
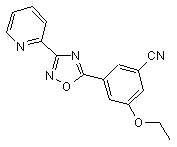
B234
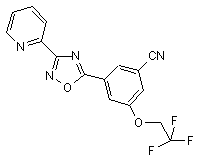
B161
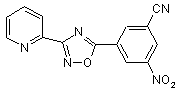
B162
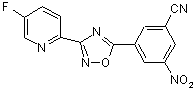
B235
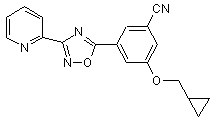
B236
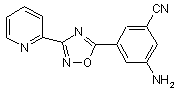
B237
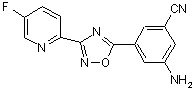
B238
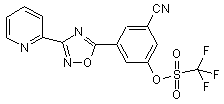
B239
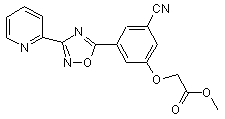
B240
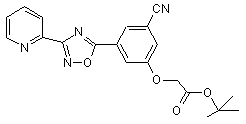
B241
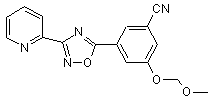
B163
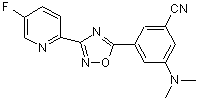
B242
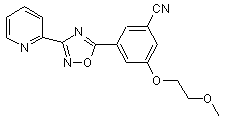
B164
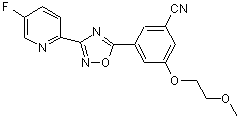
B165
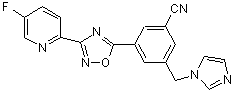
B243
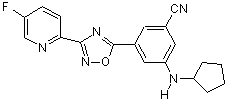
B166
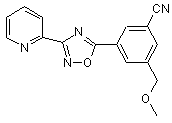
12. Фармацевтична композиція, яка містить терапевтично ефективну нетоксичну кількість сполуки за п. 11 та фармацевтично прийнятний носій.
13. Спосіб лікування хвороби, пов'язаної з активацією mGluR Групи І, при якому здійснюють введення пацієнтові, який потребує такого лікування, фармацевтичної композиції за п. 12.
14. Спосіб за п. 13, який відрізняється тим, що хвороба є хворобою, пов'язаною з активацією mGluR .
15. Спосіб за п. 14, який відрізняється тим, що хвороба є неврологічною хворобою.
16. Спосіб за п. 14, який відрізняється тим, що хвороба є психічною хворобою.
17. Спосіб за п. 14, який відрізняється тим, що хвороба належить до групи, яка складається з інсульту, травми голови, аноксичних уражень, ішемічних уражень, гіпоглікемії, епілепсії, болю, мігреней, хвороби Паркінсона, старечого слабоумства, хореї Гантингтона, страху та хвороби Альцгеймера.
Текст
Даний винахід забезпечує сполуки, які є активними на метаботропних рецепторах глутамату, зокрема, сполуки, які є активними як антагоністи на метаботропних рецепторах глутамату, а саме, на рецептор глутамату mGluR5. Останні успіхи у дослідженні нейрофізіологічної ролі метаботропних рецепторів глутамату роблять ці рецептори перспективними об'єктами медикаментозного впливу в лікуванні гострих та хронічних неврологічних та психіатричних порушень та захворювань. Однак, найскладнішим завданням у реалізації цих перспектив є розробка селективних щодо підтипу метаботропного рецептора глутамату сполук. Глутамат є головним збуджувальним медіатором у центральній нервовій системі (ЦНС) ссавців. Глутамат справляє свій вплив на центральні нейрони, зв'язуючись із рецепторами поверхні клітин і, таким чином, збуджуючи їх. Ці рецептори було поділено на два основні класи, монотропні та метаботропні рецептори глутамату, залежно від особливостей структури рецепторних білків, засобів, за допомогою яких рецептори передають сигнали у клітину, та фармакологічних профілів. Метаботропні рецептори глутамату (mGluR) є зв'язаними з G-білком рецепторами, які активують різні внутрішньоклітинні системиВторинних месенджерів після зв'язування глутамату. Активація mGluR в інтактних нейронах ссавців викликає одну або кілька з таких реакцій: активація фосфоліпази С; збільшення гідролізу фосфоінозитидів (РІ); вивільнення внутрішньоклітинного кальцію; активація фосфоліпази D; активація або інгібування аденілциклази; збільшення або зменшення утворення циклічного аденозинмонофосфату (сАМР); активація гуанілілциклази; збільшення утворення циклічного гуанозинмонофосфату (cGMP); активація фосфоліпази А2; збільшення вивільнення арахідонової кислоти та збільшення або зменшення активності регульованих напругою або лігандами іонних каналів. Schoepp ef al., Trends Pharmacol. Sci. 14:13 (1993); Schoepp, Neurochem. Int. 24:439 (1994); Pin etal., Neuropharmacology 34:1 (1995). Шляхом молекулярного клонування було розпізнано вісім різних підтипів mGluR, від mGluR1 до mGluR8. Див., наприклад, Nakanishi, Neuron 73:1031 (1994); Pin ef al., Neuropharmacology 34:1 (1 995); Knopfel et al., J. Med. Chem. 38:1417 (1995). Подальше розрізнення рецепторів відбувається через експресію переміжно з'єднаних форм деяких підтипів mGluR. Pin et al., PNAS 89:10331 (1992); Minakami et al., BBRC 799:1136 (1994); Joly ef al., J. Neurosci. 15:3970 (1995). Підтипи метаботропного рецептора глутамату можуть бути поділені на три групи: mGluR Групи І, Групи II та Групи III, залежно від гомології амінокислотної послідовності, систем вторинних месенджерів, використовуваних рецепторами, та їх фармакологічних характеристик. Nakanishi, Neuron 73:1031 (1994); Pin ef al., Neuropharmacology 34:1 (1995); Knopfel etal., J. Med. Chem. 38:1417 (1995). До mGluR Групи І належать mGluR1, mGluR5 та їх переміжно з'єднані варіанти. Зв'язування агоністів з цими рецепторами в результаті веде до активації фосфоліпази С та наступної мобілізації внутрішньоклітинного кальцію. Для демонстрації цього впливу, наприклад, у яйцеклітинах Xenopus, які експресують рекомбінантні рецептори mGluR1, застосовували електрофізіологічні вимірювання. Див., наприклад, Masu et al., Nature 349:760 (1991); Pin et al., PNAS 89:10331 (1992). Подібних результатів досягали з яйцеклітинами, що експресують рекомбінантні рецептори mGluR5. Abe et al., J. Вiol. Chem. 267:13361 (1992); Minakami et al., BBRC 799:1136 (1994); Jolyef al., J. Neurosci. 75:3970 (1995). В альтернативному варіанті активація Агоністом рекомбінантних рецепторів mGluR1, експресованих у клітинах яєчника китайського хом'яка (СНО), стимулює гідроліз РІ, утворення сАМР та вивільнення арахідонової кислоти, згідно з вимірюваннями шляхом стандартних біохімічних аналізів. Aramori et al., Neuron 8:757 (1992). Для порівняння, активація рецепторів mGluR5, експресованих у клітинах СНО, стимулює гідроліз РІ та наступні перехідні процеси внутрішньоклітинного кальцію, але не спостерігається стимуляції утворення сАМР або вивільнення арахідонової кислоти. Abe et al., J. Biol. Chem. 267:13361 (1992). Однак активація рецепторів mGluR5, експресованих у клітинах LLC-PK1 в результаті веде до гідролізу РІ і збільшує утворення сАМР. Joly et al., J. Neurosci. 15:3970 (1995). Профіль активності Агоніста для mGluR Групи І є таким: квісквалат>глутамат і= ботенат >(2S,1'S,2'S)-2-карбоксициклопропіл)гліцин (L-CCG-I)>(1S,3R)-1аміноциклопентан-1,3-дикарбонова кислота (ACPD). Квісквалат є відносно селективним для рецепторів Групи І порівняно з mGluR Групи II та Групи" III, але він також є ефективним активатором іонотропних рецепторів АМРА. Pin et al., Neuropharmacology 34:1, Knopfel et al., J. Med. Chem. 33:1417 (1995). Брак специфічних до підтипу Агоністів та антагоністів mGluR перешкоджає з'ясуванню фізіологічної ролі конкретних mGluR, і пов'язані з mGluR патофізіологічні процеси, які впливають на ЦНС, ще мають бути визначені. Однак робота з наявними неспецифічними агоністами та антагоністами дала певне загальне розуміння щодо mGluR Групи І порівняно з mGluR Групи II та Групи III. Спроби пояснення фізіологічної ролі mGluR Групи І показують, що активація цих рецепторів викликає нейрональне збудження. Різні дослідження продемонстрували, що ACPD може забезпечувати постсинаптичне збудження, діючи на нейрони у гіпокампі, корі головного мозку, мозочка та таламуса, а також інші ділянки мозку. Дані свідчать, що це збудження зумовлене прямою активацією постсинаптичних mGluR, але також вказувалося на те, що відбувається активація пресинаптичних mGluR, результатом якої є збільшення вивільнення нейротрансмітера. Baskys, Trends Pharmacol. Sci. 15:92 (1992); Schoepp, Neurochem. Int. 24:439 (1994); Pin etal., Neuropharmacology 34;7(1995). Фармакологічні експерименти охоплюють mGluR Групи І як медіатори цього механізму збудження. Вплив ACPD може бути відтворений низькими концентраціями квісквалату у присутності антагоністів іонотропних GluR. Hu et al.. Brain Res. 568:339 (1991); Greene etal., Eur. J. Pharmacol. 226:279 (1992). Дві фенілгліцинові сполуки, які активують mGluR1, а саме, (S)-3-пдроксифенілгліцин ((S)-3HPG) та (S)-3,5дигідроксифенілгліцин ((S)-DHPG), також викликають збудження. Watkins et ai, Trends Pharmacol. Sci. 75:33 (1994). Крім того, збудження може блокуватися (S)-4-карбоксифенілгліцином ((S)-4CPG), (S)-4-карбокси-3гідроксифенілгліцином ((S)-4C3HPG) та (+)-альфа-метил-4-карбоксифенілгліцином ((+)-MCPG), сполуками, які є антагоністами mGluR1 Eaton et ai, Eur. J. Pharmacol. 244:195 (1993); Watkins et ai, Trends Pharmacol. Sci. 75:333(1994). Метаботропні рецептори глутамату беруть участь у багатьох нормальних процесах у ЦНС ссавців. Було виявлено, що активація mGluR вимагається для викликання гіпокампального довготермінового потенціювання та мозочкової довготермінової депресії. Bashir et al., Nature 363:347 (1993); Bortolotto et al., Nature 368:740 (1994); Aiba et al., Cell 79:365 (1994); Aiba et al., Cell 79:377 (1994). Також було продемонстровано роль активації mGluR у ноцицепції та аналгезії. Meller et al., Neuroreport 4:879 (1993). Крім того, було виявлено, що активація mGluR відіграє модулюючу роль у різних інших нормальних процесах, включаючи синаптичну передачу, нейрональний розвиток, апоптозну нейрональну смерть, синаптичну пластичність, просторове мислення, нюхову пам'ять, центральний контроль серцевої діяльності, неспання, руховий контроль та контроль вестибуло-окулярного рефлексу. Див. Nakanishi, Neuron 13: 1031 (1994); Pin et al., Neuropharmacology 34:1; Knopfel et al.,J. Med. Chem. 38:1417 (1995). Метаботропні рецептори глутамату також відіграють роль у різних патофізіологічних процесах та хворобливих станах, які уражують ЦНС. До них належать інсульт, травма голови, аноксичні та ішемічні ураження, гіпоглікемія, епілепсія та нейродегенеративні хвороби, такі як хвороба Альцгеймера. Schoepp et al., Trends Pharmacol. Sci. 74:13 (1993); Cunningham et al., Life Sci. 54:135 (1994); Hollman et al., Ann. Rev. Neurosci. 17:31 (1994); Pin et al., Neuropharmacology 34:1 (1995); Knopfel et al., J. Med. Chem. 38:1417 (1995). Вважається, що патологія при цих станах зумовлюється, головним чином, надмірним викликаним глутаматом збудженням нейронів ЦНС. Оскільки mGluR Групи І підвищують опосередковане глутаматом нейрональне збудження через постсинаптичні механізми та збільшення пресинаптичного вивільнення глутамату, можливо, що їх активація сприяє патології. Відповідним чином, терапевтично сприятливими можуть бути селективні антагоністи рецепторів mGluR Групи І, зокрема, як нейропротективні засоби, анальгетики або антиконвульсанти. Попередні оцінки терапевтичного потенціалу з наявними Агоністами та антагоністами mGluR дали суперечливі результати. Наприклад, повідомляється, що застосування ACPD на гіпокампальних нейронах веде до судом та нейронального ураження (Sacaan et al., Neurosci. Lett. 139:77 (1992); Lipparti et al., Life Sci. 52:85 (1993). Однак інші дослідження показують, що ACPD інгібує епілептиформну активність, а також може виявляти нейропротективні властивості. Taschenberger et al., Neuroreport 3:629 (1992); Sheardown, Neuroreport 3:916 (1992); Koh et al., Proc. Natl. Acad. Sci. USA 88:9431 (1991); Chiamulera et al., Eur. J. Pharmacol. 216:335 (1992); Siliprandi et al., Eur. J. Pharmacol. 219:173 (1992); Pizzi et al., J. Neurochem. 61:683 (1993). Очевидно, ці суперечливі результати зумовлені браком селективності ACPD, що спричинює активацію кількох різних підтипів mGluR. У дослідженнях з виявлення нейронального ураження було виявлено активацію mGluR Групи І, що підвищує небажану збуджувальну нейротрансмісію. У дослідженнях з виявлення нейропротективного впливу було виявлено активацію mGluR Групи II тNабо Групи III, що інгібує пресинаптичне вивільнення глутамату і зменшує збуджувальну нейротрансмісію. Це пояснення узгоджується зі спостереженням, за яким (S)-4C3HPG, антагоніст mGluR Групи І і агоніст mGluR Групи II, захищає від аудіогенних судом" у мишей DBA/2, тоді як селективні агоністи mGluR Групи II DCG-IV та L-CCG-I захищають нейрони відвикликаної NMDA та КА токсичності. Thomsen et al., J. Neurochem. 62:2492 (1994); Bruno et al., Eur. J. Pharmacol. 256:109 (1994); Pizzi etal., J. Neurochem. 61:683 (1993). На основі викладеного вище стає зрозумілим, що брак активності та селективності обмежує властивості наявних нині агоністів та антагоністів mGluR. Крім того, більшість наявних нині сполук є амінокислотами або похідними амінокислот, які мають обмежену біодоступність, перешкоджаючи, таким чином, in vivo дослідженням для оцінки фізіології, фармакології та терапевтичного потенціалу mGluR. З іншого боку, сполуки, які селективно інгібують активацію підтипів метаботропного рецептора глутамату Групи І, є показаними для лікування неврологічних порушень та хвороб, таких як старече слабоумство, хвороба Паркінсона, хвороба Альцгеймера, хорея Гентингтона, біль, епілепсія, травма голови, аноксичні та ішемічні ураження, та психіатричних порушень, таких як страх, шизофренія та депресія. Відповідно, існує потреба в ефективних агоністах та антагоністах mGluR, які виявляють високу селективність до підтипу mGluR, зокрема, підтипу рецептора Групи І. Даний винахід забезпечує активні щодо метаботропного рецептора глутамату сполуки, які виявляють високий ступінь ефективності і селективності щодо окремих підтипів метаботропного рецептора глутамату, та способи одержання цих сполук. Крім того, цей винахід забезпечує фармацевтичні композиції, що містять сполуки, які виявляють високий ступінь ефективності та селективності щодо окремих підтипів метаботропного рецептора глутамату, та способи одержання цих фармацевтичних композицій. Інший аспект винаходу стосується забезпечення способів інгібування активації рецептора mGluR Групи І, а саме - mGluR5. До медичних станів, пов'язаних із метаботропними рецепторами глутамату, зокрема, належать: інсульт; травма голови; аноксичне ураження; ішемічне ураження; гіпоглікемія; епілепсія; біль; мігрені; хвороба Паркінсона; старече слабоумство; хорея Гентингтона та хвороба Альцгеймера. Винахід забезпечує способи лікування хвороб, пов'язаних зі збуджувальною активацією рецептора mGluR Групи І, та інгібування нейронального ураження, викликаного збуджувальною активацією рецептора mGluR Групи І, зокрема, якщо рецептор mGluR Групи І є рецептором mGluR5. I нарешті, даний винахід забезпечує ефективні антагоністи mGluR Групи І, зокрема, mGluR5. Згідно з першим аспектом винаходу, ці антагоністи можуть бути представлені сполуками загальної формули: де Аr1 є необов'язково заміщеним гетероароматичним компонентом, і Аr2 є необов'язково заміщеним бензольним кільцем. L-компонент є групою, яка не лише ковалентно зв'язується з компонентами Аr1 та Аr2 і сприяє набуттю правильної просторової орієнтації Аr1 та Аr2, але й сама може взаємодіяти з білком для здійснення рецепторного зв'язування. В одному варіанті втілення винаходу L вибирають із групи, яка складається з -NH-, -S-, -О-, -CO-, CONH-, -CONHCH2-, -CH2CONH-, -CNHNH-, -CNHNHCH2-, -С=NO-CH2-, -CH2NHCH2-, -CH2CH2 NH-, NHCH2CO-, -NHCH2CHOH-, -NHCNHNH.-, -NHCONH-, циклопентану, циклопентадієну, фурану, тіофурану, піролідину, піролу, 2-імідазоліну, 3-імідазоліну, 4-імідазоліну, імідазолу, піразоліну, піразолідину, імідазолідину, оксазолу, 2-оксазолу, тіазолу, ізоксазолу, ізотіазолу, 1H-1,2,4-триазолу, 1Н-1,2,3-триазолу, 1,2,4-оксатіазолу, 1,3,4-оксатіазолу, 1,4,2-діоксазолу, 1,4,2-оксатіазолу, 1,2,4-оксадіазолу, 1,2,4-тіадіазолу, 1,2,5-оксадіазолу, 1,2,5-тіадіазолу, 1,3,4-оксадіазолу, 1,3,4-тіадіазолу, 1H-тетразолу, циклогексану, піперидину, тетрагідропіридину, 1,4-дигідропіридину, піридину, бензолу, тетрагідропірану, 3,4-дигідро-2/-/пірану, 2Н-пірану, 4Н-пірану, тетрагідротіопірану, 3,4-дигідро-2H-тіопірану, 2Н-тіїну, 4Н-тіопіран, морфоліну, тіоморфоліну, піперазину, піридазину, піримідину, піразину, 1,2,4-триазину, 1,2,3-триазину, 1,3,5-триазину та 1,2,4,5-тетразину. В іншому варіанті втілення винаходу Аr1 вибирають із групи, яка складається з фенілу, бензилу, нафтилу, флуоренілу, антренілу, інденілу, фенантренілу та бензонафтенілу, і Аr2 вибирають із групи, яка складається з тіазоїлу, фурилу, піранілу, 2Н-піролілу, тієнілу, піроїлу, імідазоїлу, піразоїлу, піридилу, піразинілу, піримідинілу, піридазинілу, бензотіазолу, бензимідазолу, ЗН-індолілу, індолілу, індазоїлу, пуринілу, хінолізинілу, ізохінолілу, хінолілу, фталізинілу, нафтиридинілу, хіназолінілу, цинолінілу, ізотіазолілу, хіноксалінілу, індолізинілу, ізоіндолілу, бензотієнілу, бензофуранілу, ізобензофуранілу та хроменілу. Згідно з другим аспектом винаходу ці антагоністи можуть бути представлені сполуками Формули І: де представляє подвійний або одинарний зв'язок; Χ, Υ та Ζ незалежно вибирають із групи, яка складається з Ν, О, S; і CR1 та принаймні один із Χ, Υ та Ζ є гетероатомом; де R1 вибирають із групи, яка складається з Н, алкілу, -CF3, -OR2, -SR2, -NR2R3, =О, =S, =NR2, та=CR2R3; і де R1 та R3 незалежно можуть бути вибрані з групи, яка складається з Н, алкілу, галоалкілу, алкілокси, алкіламіну, циклоалкілу, гетероциклоалкілу, арилу, гетероарилу, алкіларилу, алкілгетероарилу, галоарилу, алкілоксіарилу, алкеніларилу, алкенілоксіарилу та галогетероарилу; і Аr1 та Аr2 незалежно вибирають із групи, яка складається з арилу та гетероарилу, і принаймні один із Аr1 та Аr2 є заміщеним принаймні одним замісником G; де G вибирають із групи, яка складається з галоалкілу, гетероарилу, циклоалкену, алкенілу, алкінілу, Аалкенілу, А-алкінілу, алкілокси, А-алкілокси, -R2OR3, -R2OC(O)R3, (CH2)m-NR2R3, -OCH2CH(CI)CH2CI та заміщеного арилу, де арильним замісником є R4, і де А є лінкером, вибраним із групи, яка складається з СН2, О, NH, S, SO, SO2, NSO2,- OSO2 та -C(NR2)NR3; m є вибраним 3-поміж 0 та 1; і R4 вибирають із групи, яка складається з гало, -OR2, -SR2, -SOR2, -SO2R2, -SO2NR2R3, -R2OR3, -R2SR3, OCOR2, -OCONR2R3, -NR2COR3, -NR2CO2R3, -CN, -NO2, -C(NR2)NR3, -CO2R2R3, -CONR2R3, -C(O)R2, CH(OR2)R3, -CH2(OR2), -A-(CH2)m-NR2R3, NR2R3, арилу, аралкілу, гетероарилу та гетероаралкілу; і Аr1, Аr2 та замісник G є необов'язково додатково заміщеними одним або кількома замісниками, незалежно вибраними з групи, яка складається з R2 тa R4. В іншому аспекті винаходу забезпечуються антагоністи Формули II де представляє подвійний або одинарний зв'язок; Х2 є вибраним 3-поміж N та С, і Y2 вибирають із групи, яка складається з Ν, О, S та CR5, і принаймні один із Х2 та Υ2 є гетероатомом; де R5 вибирають із групи, яка складається з Н, алкілу, -CF3, -OR6, -SR6, NR6R-, =О, =S, -=NR6 та=CR6R7; і де R6 та R7 незалежно можуть бути вибрані з групи, яка складається з Н, алкілу, галоалкілу, алкілокси, алкіламіну, циклоалкілу, гетероциклоалкілу, арилу, гетероарилу, алкіларилу, алкілгетероарилу, галоарилу, алкілоксіарилу, алкеніларилу, алкенілоксіарилу та галогетероарилу; і Аr3 та Аr4 незалежно вибирають із групи, яка складається з арилу та гетероарилу, і один або обидва з Аr3 та Аr4 є необов'язково заміщеними одним або кількома замісниками G2; де G2 вибирають із групи, яка складається з галоалкілу, гетероарилу, циклоалкену, алкенілу, алкінілу, Аалкенілу, А-алкінілу, алкілокси, А-алкілокси, -R6OR7, -R6OC(O)R7, (CH2)m-NR6R7, -OCH2CH(CI)CH2CI та заміщеного арилу, де арильним замісником є R8; і де А є лінкером, вибраним із групи, яка складається з СН2, О, NH, S, SO, SO2, NSO2, OSO2 та -C(NR6)NR7; та вибраним 3-поміж 0 та 1; і R8 вибирають із групи, яка складається з гало, -OR6, -SR6, -SOR6, -SO2R6, -SO2NR6R7, -R6OR7, R6SR7, OCOR6, -OCONR6R7, -NR6COR7, -NR6CO2R7, -CN, -NО2, -C(NR6)NRr, -CO2R6R7, -CONR6R7, -C(O)R6, CH(OR6)R7, -CH2(OR6), -A-(CH2)m-NR6R7, NR6R7, арилу, аралкілу, гетероарилу та гетероаралкілу; і Аг3, Аr4 та замісник G2 є необов'язково додатково заміщеними одним або кількома замісниками, незалежно вибраними з групи, яка складається з R6 та R8. Інший аспект винаходу стосується забезпечення способів одержання сполук даного винаходу. Ще один аспект винаходу стосується забезпечення способу інгібування активації рецептора mGluR Групи І, зокрема, mGluR5, який включає обробку клітини, що містить вищезгаданий рецептор mGluR Групи І, ефективною кількістю сполуки даного винаходу. Ще один аспект винаходу стосується забезпечення способу інгібування нейронального ураження, викликаного збуджувальною активацією рецептора mGluR Групи І, зокрема, mGluR5, який включає обробку нейронів ефективною кількістю сполуки даного винаходу. Ще один аспект винаходу стосується забезпечення способу лікування хвороби, пов'язаної з активацією mGluR Групи І, або хвороби, яка піддається терапевтичному втручанню з застосуванням антагоніста mGluR Групи І, наприклад, хвороби, пов'язаної з викликаним глутаматом нейрональним ураженням, причому цей спосіб включає етап введення пацієнтові, який потребує такого лікування, наприклад, пацієнтові, який страждає від вищезгаданої хвороби, або якому загрожує вищезгадана хвороба, терапевтично ефективної нетоксичної кількості сполуки даного винаходу. У конкретному аспекті винаходу терапевтично ефективною кількістю згідно з даним винаходом має бути кількість, яка селективно протидіє рецепторові mGluR Групи І, зокрема, рецепторові mGluR5. Інші аспекти, особливості та переваги даного винаходу стануть зрозумілими з представленого нижче детального опису. Однак слід розуміти, що детальний опис та конкретні приклади, хоча вони і вказують на оптимальні варіанти втілення винаходу, представлено лише для пояснення, оскільки з цього детального опису спеціалістам стане зрозумілою можливість різних змін та модифікацій, які відповідають сутності й охоплюються обсягом винаходу. Вжитий авторами термін "алкіл" стосується лінійних або розгалужених алкільних радикалів, що містять від 1, 2, 3, 4, 5 або 6 атомів вуглецю, і включає метил, етил та ін. Вжитий авторами термін "арил" стосується моноциклічної ароматичної групи, такої як феніл та ін., або бензо-злитої ароматичної групи, такої як інданіл, нафтил, флуореніл та ін. Вжитий авторами термін "гетероарил" стосується ароматичних сполук, які містять один або кілька гетероатомів, таких як піридил, фурил, тієніл та ін., або бензо-злитої ароматичної сполуки, що містить один або кілька гетероатомів, такої як індоліл, хінолініл та ін. Вжитий авторами термін "гетероатом" стосується відмінних від вуглецю атомів, таких як N, О, S та ін. Вжитий авторами термін "циклоалкіл" стосується карбоциклічного кільця, що містить 3, 4, 5, 6, 7 або 8 атомів вуглецю, і включає циклопропіл, циклогексил та ін. Вжитий авторами термін "гетероциклоалкіл" стосується 3-, 4-, 5-, 6-, 7-або 8-членних кілець, які містять 1, 2, або 3 гетероатомів, вибраних із групи, яка складається з N, S та О і включає піперидин, піперизин, піран та ін. Вжитий авторами термін "гало" стосується галогенних радикалів фторо, хлоро, бромо та йодо. Вжитий авторами термін "галоалкіл" стосується алкілу, заміщеного одним або кількома галогенами, такого як бромоетил, хлорометил, трихлорометил та ін. Вжитий авторами термін "алкокси" стосується лінійного або розгалуженого алкокси, що містить 1, 2, 3, 4, 5 або 6 атомів вуглецю, і включає метокси, етокси та ін. Вжитий авторами термін "алкілокси" стосується алкілу, заміщеного гідрокси групою, такого як гідроксіетил, гідроксипропіл та ін. Вжитий авторами термін "алкеніл" стосується лінійного або розгалуженого алкілу, який містить один або кілька подвійних зв'язків, такого як пропеніл, вініл та ін. Вжитий авторами термін "аралкіл" стосується алкілу, заміщеного арилом, такого як бензил, фенетил та ін. Вжитий авторами термін "алкіламін" стосується алкілу, заміщеного аміном, такого як амінометил або диметиламіноетил та ін. Вжитий авторами термін "алкіларил" стосується арилу, заміщеного алкільною групою, такого як метилфеніл, ізопропілнафтил та ін. Вжитий авторами термін "алкілгетероарил" стосується гетероарилу, заміщеного алкільною групою. Конкретними прикладами є метилпіридин, етилфуран та ін. Вжитий авторами термін "алкініл" стосується лінійного або розгалуженого алкілу, який містить один або кілька подвійних зв'язків, такого як етиніл, пропініл, вініл та ін. Вжитий авторами термін "галоарил" стосується арилу, заміщеного галогеном, такого як бромофеніл, хлорофеніл та ін. Вжитий авторами термін "алкілоксіарил" стосується арилу, заміщеного алкілоксигрупою, такого як гідроксіетилфеніл та ін. Вжитий авторами термін "алкенілоксіарил" стосується арилу, заміщеного алкенілоксигрупою, такого як пропенілоксифеніл та ін. Вжитий авторами термін "галогетероарил" стосується гетероарилу, заміщеного галогеном. Конкретним прикладом є 4-хлоропіридин. Вжитий авторами термін "циклоалкен" стосується 3-, 4-, 5-, 6-, 7- або 8-членного кільця, яке містить один або кілька подвійних зв'язків і може містити гетероатом. Конкретними прикладами є циклогексен, тетрагідропіридин та ін. Вжитий авторами термін "алкеніларил" стосується арилу, заміщеного алкенільною групою. Конкретним прикладом є вінілбензол. Даний винахід забезпечує сполуки, які є ефективними і селективними антагоністами mGluR5. Сполуки, які охоплюються винаходом, можуть бути представлені загальною формулою: де Аr1 є необов'язково заміщеним гетероциклічним компонентом, і Аr2 є необов'язково заміщеним карбоциклічним компонентом. G-компонент є групою, яка не лише ковалентно зв'язується з компонентами Аr1 та Аr2 і сприяє набуттю правильної просторової орієнтації Аr1 та Аr2, але й сама може взаємодіяти з білком, забезпечуючи рецепторне зв'язування. Компонент Аr1 в загальних рисах визначається як гетероциклічний компонент, а компонент Аr2 в загальних рисах визначається як карбоциклічний компонент. Аr1 та Аr2 можуть бути моноциклічними або злитими біциклічними групами. Аr2 в оптимальному варіанті визначають як арильний або алкарильний компонент. Аr1 в оптимальному варіанті визначають як гетероциклічний, гетероарильний або гетероарилалкільний компонент. Кільцеві системи, які охоплює Аr1, можуть містити до чотирьох гетероатомів, незалежно вибраних із групи, яка складається з N, S та О. Якщо Аr1 є гетероарильним кільцем або кільцевою системою, він в оптимальному варіанті містить один або два гетероатоми. Принаймні один з гетероатомів в оптимальному варіанті є азотом (N). Гетероциклічний або злитий гетероциклічний компонент в оптимальному варіанті вибирають із групи, яка складається з хінолілу, хіназолілу, хіноксалілу, 2-піримідилу, 4-піримідилу, 5-піримідилу, 2-піридилу, 3-піридилу, 4-піридилу та піразилу. До моноциклічних Аr1 груп, крім інших, належать такі компоненти, як тіазоїл, фурил, піраніл, 2Н-піроліл, тієніл, піроїл, імідазоїл, піразоїл, піридил, піразиніл, піримідиніл та піридазиніл. Моноциклічна Аr2 група, крім інших, може означати феніл та бензил. Злитий біциклічний Аr2, крім інших, може означати нафтил, флуореніл, антреніл, інденіл, фенантреніл та бензонафтеніл. До злитих біциклічних Аr1 груп, крім інших, належать такі компоненти, як бензотіазол, бензимідазол, 3Н-індоліл, індоліл, індазоїл, пуриніл, хінолізиніл, ізохіноліл, хіноліл, фталізиніл, нафтиридиніл, хіназолініл, цинолініл, ізотіазоліл, хіноксалініл, індолізиніл, ізоіндоліл, бензотієніл, бензофураніл, ізобензофураніл та хроменіл. Аr1 в оптимальному варіанті є 2піридильним компонентом. Аr2 в оптимальному варіанті є заміщеним фенільним компонентом. Аr1 та Аr2 компоненти необов'язково можуть бути незалежно заміщеними одним або кількома компонентами, вибраними з групи, яка складається з галогену, С1-С3алкілу, С1-С3О-алкілу, -ОН, -OCF3, COOR, -COR, -SOR, -SO2NRR', -NRR', -CN, -CF3, -CO-NRR', -A-(CH2)n-NRR', де А є С, О, Ν, SO, SO2, і R та R' незалежно вибирають із групи, яка складається з С 1-С6 алкілу, Н, циклоалкілу, гетероциклоалкілу, арилу, і n є 1, 2, 3 або 4. L-компонент в цілому складається з 1-14 атомів. L незалежно може бути вибраний з групи атомів: С, Η, Ν, Ο та S. L-компонент, таким чином, може складатися з нециклічного компонента. Прикладами їх можуть бути NH-(aмін), -S-(тioeтep), -О-(етер), -СО-(кетон), -CONH-(aмін), -CONHCH2-, -CH2CONH-, -CNHNH-(aмідин), CNHNHCH2-, -C=NO-СН2-(метоксимін), -CH2 NHCH2-, -CH2CH2 NH-, -NHCH2CO-, -NHCH2CHOH-, -NHCNHNH.(гуанідин) та -NНСОМН-(сечовина). Розташування атомів у L-компоненті також може бути таким, щоб утворювалося п'ятичленне кільце. Прикладами можуть бути циклопентан, циклопентадієн, фуран, тіофуран, піролідин, пірол, 2-імідазолін, 3імідазолін, 4-імідазолін, імідазол, піразолін, піразолідин, імідазолідин, оксазол, 2-оксазол, тіазол, ізоксазол, ізотіазолу, 1H-1,2,4-триазол, 1Н-1,2,3-триазол, 1,2,4-оксатіазол, 1,3,4-оксатіазол, 1,4,2-діоксазол, 1,4,2оксатіазол, 1,2,4-оксадіазол, 1,2,4-тіадіазол, 1,2,5-оксадіазол, 1,2,5-тіадіазол, 1,3,4-оксадіазол, 1,3,4тіадіазол та 1Н-тетразол. Найбільшу перевагу віддають 1,2,4-оксадіазолові. Розташування атомів у L-компоненті також може бути таким, щоб утворювалося шестичленне кільце. Прикладами можуть бути циклогексан, піперидин, тетрагідропіридин, 1,4-дигідропіридин, піридин, бензол, тетрагідропіран, 3,4-дигідро-2Н-піран, 2Н-піран, 4Н-піран, тетрагідротіопіран, 3,4-дигідро-2Н-тіопіран, 2Нтіїн, 4H-тіопіран, морфолін, тіоморфолін, піперазин, піридазин, піримідин, піразин, 1,2,4-триазин, 1,2,3триазин, 1,3,5-триазин та 1,2,4,5-тетразин. Розташування атомів в L-компоненті також може бути таким, щоб утворювалося п'яти- або шестичленне кільце, що містить одну або кілька карбонільних груп. Прикладами можуть бути 2-азетидинон, 1,2-діазетидин-3-он, циклопентанон, 2-циклопентенон, 2-піролідинон, 3-піролін-2-он, сукцинімід, малеімід, 3піразолідинон, 2-імідазолідон, 4-імідазолін-2-он, 2Н-імідазол-2-он, 4-імідазолінон, 3-піразолін-5-он, гідантоїн, 1H-імідазол-2,5-діон, 2-оксазолін-4-он, 2-оксазолідинон, 3-оксазолін-5-он, 3(2Н)-ізоксазолон, 2,4оксазолідиндіон, 1,2,4-триазолін-3,5-діон, 2,4-дигідро-3Н-1,2,4-триазол-3-он, 2Н-піран-2-он, 2(1 Н)-піридон, 2(1Н-піразинон, 4(3Н)-піримідон, 3,4-дигідропіримідин-4-он, глутаримід, 4,6-(1H,5Н)-піримідиндіон, 1,3,5триазин-2(1Н)-он та ціанурова кислота. В оптимальному варіанті втілення L включає гетероциклічну 5-членну кільцеву систему. В оптимальному варіанті L є оксазольним або 1,2,4-оксадіазольним кільцем. L-компонент може мати будь-яку з двох можливих орієнтацій по відношенню до Αη та Аrг груп. Таким чином, наприклад, у винаході перевагу віддають сполукам, які мають конфігурацію 4-(Аr1)-(Аr2)-оксазолу або 3-(Ar1)-5-(Ar2)-1,2,4-оксадіазолу. Згідно з одним аспектом винаходу, забезпечуються сполуки Формули І. Формула І містить п'ятичленне кільце, що містить три змінні Χ, Υ та Ζ. До цього п'ятичленного кільця приєднуються два замісники, Аr1 та Аr2. П'ятичленне кільце може містити 0, 1 або 2 подвійні зв'язки, позначені пунктирними лініями у Формулі 1. В оптимальному варіанті втілення винаходу п'ятичленне кільце має 2 подвійні зв'язки. У варіантах втілення винаходу змінні Χ, Υ та Ζ незалежно вибирають 3-поміж Ν, О, S та заміщеного вуглецю, позначеного CR1, де R1 є таким, як визначено вище. Принаймні один із Χ, Υ або Ζ має бути гетероатомом. В оптимальному варіанті втілення винаходу більш, ніж один із Χ, Υ та Ζ є гетероатомом. В одному аспекті винаходу два з Χ, Υ та Ζ є гетероатомами. Хоча віншому аспекті винаходу всі троє з Χ, Υ та Ζ є гетероатомами. В оптимальному варіанті втілення винаходу принаймні один із Χ, Υ та Ζ є N. У ще кращому варіанті втілення винаходу двоє з Χ, Υ та Ζ є N. У ще одному оптимальному варіанті втілення винаходу X є Ν, Υ є Ν, і Ζ є О. Згідно з іншим аспектом винаходу, групи Аr1 та Аr2 незалежно вибирають 3-поміж арилу та гетероарилу. Зокрема, до варіантів втілення винаходу належать ті, в яких Аr1 та Аr2 незалежно вибирають 3-поміж 5- та 6-членних арильних та гетероарильних кілець. У більш конкретних варіантах втілення винаходу Аr1 та Аr2 вибирають 3-поміж 6-членних арильних та гетероарильних кілець. До ще більш конкретних варіантів втілення винаходу належать ті, в яких Аr1 та Аr2 незалежно вибирають 3-поміж фенілу, піридилу, фуранілу, тієнілу, піразинілу, піримідинілу, піридазинілу, піролілу, піразолілу, імідазолілу, триазолілу, тіазолілу. В оптимальному варіанті втілення винаходу An вибирають 3-поміж фенілу та піридилу. У ще кращому варіанті втілення винаходу Аr1 вибирають 3-поміж піридилу. У ще кращому варіанті втілення Аr1 вибирають 3-поміж 2-піридилу. В іншому оптимальному варіанті втілення Аr2 вибирають 3-поміж фенілу та піридилу. У прийнятному варіанті втілення Аr2 є фенілом. В іншому прийнятному варіанті втілення Аr2 є 3-піридилом. Згідно з іншим аспектом винаходу, принаймні один із Аr1 та Аr2 є заміщеним принаймні одним замісником G. В оптимальних варіантах втілення винаходу Аr2 є заміщеним G. До прийнятних варіантів втілення винаходу належать ті, в яких G вибирають із групи, яка складається з галоалкілу, гетероарилу, циклоалкену, алкенілу, алкінілу, А-алкенілу, А-алкінілу, алкілокси, А-алкілокси, R2OR3, -R2OC(O)R3, (CH2)mNR2R3, -ОСН2СН(СІ)СН2СІ та заміщеного арилу, де R2 та R3 є такими, як визначено вище. В одному варіанті втілення винаходу G є галоалкілом. В іншому варіанті втілення винаходу G є гетероарилом, причому гетероарил вибирають із групи, яка складається з піридилу, фуранілу, тієнілу, піразинілу, піримідинілу, піридазинілу, піролілу, піразолілу, імідазолілу, триазолілу та тіазолілу. В оптимальному варіанті втілення винаходу G вибирають із групи, яка складається з піридину, уранілу, тіонілу та піримідинілу. У ще кращому варіанті втілення винаходу G вибирають із групи, яка складається з 2-піридилу, 3-піридилу, 4-піридилу, 3-тієнілу, 5-піримідинілу та 3фуранілу. У ще одному оптимальному варіанті втілення винаходу G є циклоалкеном. У ще одному оптимальному варіанті втілення винаходу G вибирають 3-поміж 5- та 6-членних карбоцикпічних та гетероциклічних кілець, які містять один або кілька подвійних зв'язків. У ще одному оптимальному варіанті втілення винаходу G є 6членним гетеро циклом, який містить один подвійний зв'язок. У ще одному варіанті втілення G є 3-(1,2,5,6тетрагідропіридилом). У прийнятному варіанті втілення G є N-заміщеним 3-(1,4,5,6-тетрагідропіридилом, наприклад 3-N-бензил-(1,2,5,6-тетрагідропіридилом). Згідно з іншим аспектом винаходу, G є алкенілом. У більш конкретному варіанті втілення винаходу G вибирають із групи, яка складається з вінілу, 2-метилвінілу, пропенілу та бутенілу. Згідно з іншим аспектом винаходу, G є алкінілом. У більш конкретному варіанті втілення винаходу G вибирають 3-поміж пропаргілу та бутинілу. Згідно зі ще одним аспектом винаходу, G вибирають із групи, яка складається з А-алкенілу та Аалкінілу, причому алкеніл або алкініл, відповідно, є зв'язаним з Аr1 або Аr2 через А. У конкретних варіантах втілення винаходу А вибирають із групи, яка складається з СН2, О, NH, S, SO, SO2, NSO2, OSO2 та C(NR2)NR3. У більш конкретному варіанті втілення винаходу А вибирають 3-поміж О та NH. У ще більш конкретному варіанті втілення винаходу G є -ОНСН2СН=СН2. У ще одному варіанті втілення винаходу G вибирають із групи, яка складається з алкілокси та Аалкілокси, причому алкілокси є лінійним або розгалуженим алкільним радикалом, заміщеним гідроксигрупою, і А є лінкером. У більш конкретному варіанті втілення винаходу алкілоксигрупа є зв'язаною з Аr1 або Аr2 через А, і А вибирають із групи, яка складається з СН2, О, NH, S, SO, SO2, NSO2, OSO2 та C(NR2)NR3. У більш конкретному варіанті втілення винаходу А є О, і алкілокси вибирають 3-поміж гідроксиметилу, гідроксіетилу та гідроксипропілу. У більш конкретному варіанті втілення G є ОСН2СН2СН2ОН. У ще одному варіанті втілення винаходу G є R2OCOR3. У конкретному варіанті втілення винаходу G є алкіловим естером, причому естер зв'язується з An або Аr2 через алкільну групу. У більш конкретному варіанті втілення винаходу G є -СН2ОС(О)Н. У ще одному варіанті втілення винаходу G є (CH2)m-NR2R3, де m є 0 або 1. У конкретному варіанті втілення винаходу G є (CH2)m-NR2R3, і R2 та R3 незалежно вибирають 3-поміж Н, алкілу, алкілокси, алкіламіну, циклоалкілу, гетероциклоалкілу, арилу, гетероарилу, алкіларилу, алкілгетероарилу, галоарилу, алкілоксіарилу, алкеніларилу, алкенілоксіарилу, галогетероарилу. У більш конкретному варіанті втілення винаходу R2 та R3 незалежно вибирають 3-поміж Η та алкілу. У ще одному варіанті втілення винаходу R2 та R3 незалежно вибирають 3-поміж Η та метилу. Згідно з іншим аспектом винаходу, G є арилом, заміщеним замісниками R4 Зокрема, арильну групу вибирають із групи, яка складається з фенілу нафтилу антренілу та флуоренілу. Замісник R4 вибирають із групи, яка складається з гало, -OR2, -SR2, -SOR2, -SO2R2, -SO2NR2R3, -R2OR3, R2SR3, -OCOR2, -OCONR2R3, NR2COR3, -NR2CO2R3, -CN, -NO2, OH, -R2OH, -C(NR2)NR3, -CO2R2R3, -CONR2R3, -C(O)R2, -CH(OR2)R3, CH2(OR2), -A-(CH2)m-NR2R3, NR2R3, арилу, аралкілу, гетероарилу та гетероаралкілу. В оптимальному варіанті втілення арил є фенілом. У ще одному оптимальному варіанті втілення R4 вибирають із групи, яка складається з гало, NR2R3, алкокси та CN. У ще одному оптимальному варіанті втілення винаходу R4 вибирають із групи, яка складається з F, NH2, метокси. Згідно з іншим аспектом винаходу, кожен з Аr1, Аr2 та G є необов'язково додатково заміщеним одним або кількома замісниками, вибраними 3-поміж R2 та R4. В оптимальному варіанті втілення винаходу Аr1 є додатково заміщеним замісником, вибраним із групи, яка складається з Н, алкілу, галоалкілу, алкілокси, алкіламіну, гало, -OR2, -SR2, -SOR2, -SO2R2, -SO2NR2R3) -R2OR3, R2SR3, -OCOR2, -OCONR2R3, -NR2COR3, NR2CO2R3, -CN, -NO2, -C(NR2)NR3, -CO2R2R3, -CONR2R3, -C(O)R2, -CH(OR2)R3, -CH2(OR2), -A-(CH2)m-NR2R3, NR2R3, арилу, аралкілу, гетероарилу, гетероаралкілу, циклоалкілу, гетероциклоалкілу, алкіларилу, алкілгетероарилу, галоарилу, алкілоксіарилу, алкеніларилу, алкенілоксіарилу та галогетероарилу. У ще одному оптимальному варіанті втілення винаходу Аr1 є додатково заміщеним замісником, вибраним із групи, яка складається з гало та ціано. В іншому аспекті винаходу, в якому Аr1 є 2-піридилом, ще один замісник є розташованим у 5-й позиції Аr1. У ще одному варіанті втілення винаходу Аr1 є 5-фторо-2-піридилом. У ще одному варіанті втілення винаходу Аr1 є 5-ціано-2-піридилом. Згідно з іншим аспектом винаходу, Аr2 є додатково заміщеним одним або кількома замісниками, вибраними з групи, яка складається з Н, алкілу, галоалкілу, алкілокси, алкіламіну, гало, -OR2, -SR2, -SOR2, SO2R2, -SO2NR2R3, -R2OR3, -R2SR3, -OCOR2, -OCONR2R3, -NR2COR3, -NR2CO2R3, -CN, -NO2, -C(NR2)NR3, CO2R2R3, -CONR2R3, -C(O)R2, -CH(OR2)R3, -CH2(OR2), -A-(CH2)m-NR2R3, NR2R3, арилу, аралкілу, гетероарилу, гетероаралкілу, циклоалкілу, гетероциклоалкілу, алкіларилу, алкілгетероарилу, галоарилу, алкілоксіарилу, алкеніларилу, алкенілоксіарилу та галогетероарилу. В оптимальному варіанті втілення винаходу Аr2 є додатково заміщеним одним або кількома замісниками, вибраними з групи, яка складається з алкілу, алкокси, алкілокси, гідрокси, гало, ціано та нітро. У ще кращому варіанті втілення винаходу Аr2 має додатковий замісник, вибраний із групи, яка складається з ціано, фторо, хлоро, бромо, йодо та метокси. У ще кращому варіанті втілення винаходу Аr2 є фенілом або 3-піридилом і є заміщеним замісником G у мета-позиції і ще одним замісником в іншій мета-позиції. В іншому варіанті втілення винаходу замісник G є необов'язково додатково заміщеними одним або кількома замісниками, вибраними з групи, яка складається з Н, алкілу, галоалкілу, алкілокси, алкіламіну, гало, -OR2, -SR2, -SOR2, -SO2R2, -SO2NR2R3, -R2OR3 R2SR3, -OCOR2, -OCONR2R3, -NR2COR3, -NR2CO2R3, CN, -NO2, -C(NR2)NR3, -CO2R2R3, -CONR2R3, -C(O)R2, -CH(OR2)R3, -CH2(OR2), -A-(CH2)m-NR2R3. NR2R3, арилу, аралкілу, гетероарилу, гетероаралкілу, циклоалкілу, гетероциклоалкілу, алкіларилу, алкілгетероарилу, галоарилу, алкілоксіарилу, алкеніларилу, алкенілоксіарилу та галогетероарилу. У ще кращому варіанті втілення винаходу G є необов'язково додатково заміщеними одним або кількома замісниками, вибраними з групи, яка складається з алкілу, алкокси, алкенілу, гало та ціано. У конкретному варіанті G є -ОСН2СН2СН2ОН і є додатково заміщеним хлоро, що дає сполуку -ОСН2СН(СІ)СН2ОН. В одному аспекті винаходу сполуками Формули І є: 3-(2-піридил)-5-(3-метоксифеніл)-1,2,4-оксадіазол (В1), 3-(2-піридил)-5-(3,5-дихлорофеніл)-1,2,4-оксадіазол (В2), 3-(2-піридил)-5-(3-хлорофеніл)-1,2,4-оксадіазол (В3), 3-(2-піридил)-5-(2-хлорофеніл)-1,2,4-оксадіазол (В5), 3-(2-піридил)-5-[3-(трифторометил)феніл]-1,2,4-оксадіазол (В6), 3-(2-піридил)-5-(3-метилфеніл)-1,2,4-оксадіазол (В9), 3-(2-піридил)-5-(1-нафтил)-1,2,4-оксадіазол (В10), 3-(2-піридил)-5-[3-(трифторометокси)феніл]-1,2,4-оксадіазол (В11), 3-(2-піридил)-5-(2,3-дифторофеніл)-1,2,4-оксадіазол (В16), 3-(2-піридил)-5-(2,5-дифторофеніл)-1,2,4-оксадіазол (В17), 3-(2-піридил)-5-(3,5-дифторофеніл)-1,2,4-оксадіазол (В18), 3-(2-піридил)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В21), 3-(2-піридил)-5-(3,5-диметоксифеніл)-1,2,4-оксадіазол (В23), 3-(2-піридил)-5-(2,3-дихлорофеніл)-1,2,4-оксадіазол (В25), 3-(2-піридил)-5-(3-хлоро-5-ціанофеніл)-1,2,4-оксадіазол (В26), 3-(2-піридил)-5-(3-фторо-5-ціанофеніл)-1,2,4-оксадіазол (В27), 3-(2-піридил)-5-(3-хлоро-5-фторофеніл)-1,2,4-оксадіазол (В28), 3-(5-хлоропірид-2-ил)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В29), 3-(5-фторопірид-2-ил)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В30), 3-(5-фторопірид-2-ил)-5-(3-ціано-5-фторофеніл)-1,2,4-оксадіазол (В31), 3-(3-фторопірид-2-ил)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В32), 3-(5-фторопірид-2-ил)-5-(3,5-диметоксифеніл)-1,2,4-оксадіазол (В33), 3-(5-метоксипірид-2-ил)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В34), 3-(2-хінолініл)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В35), 3-(3-хлоро-5-трифторометилпірид-2-ил)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В36), 3-(2-піридил)-5-(5-хлоро-2-метоксифеніл)-1,2,4-оксадіазол (В37), 3-(2-піридил)-5-(2-хлоро-5-метилтіофеніл)-1,2,4-оксадіазол (В39), 3-(2-піридил)-5-(2-бромо-5-метоксифеніл)-1,2,4-оксадіазол (В42), 3-(2-піридил)-5-(2,5,6-трифторофеніл)-1,2,4-оксадіазол (В45), 3-(2-піридил)-5-(3-нітрофеніл)-1,2,4-оксадіазол (В19), 3-(2-піридил)-5-(3-бромофеніл)-1,2,4-оксадіазол (В22) і їх фармацевтично прийнятні солі. У ще одному аспекті винаходу до конкретних сполук Формули І належать: 2-(3,5-дихлорофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3-хлорофеніл)-4-(2-піридил)-1,3-оксазол (В50), 2-(3-метоксифеніл)-4-(2-піридил)-1,3-оксазол, 2-(2-хлорофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3-трифторофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3-метилфеніл)-4-(2-піридил)-1,3-оксазол, 2-(1-нафтил)-4-(2-піридил)-1,3-оксазол, 2-(3-трифторометоксифеніл)-4-(2-піридил)-1,3-оксазол, 2-(2,3-дифторофеніл)-4-(2-піридил)-1,3-оксазол, 2-(2,5-дифторофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3,5-дифторофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3-ціанофеніл)-4-(2-піридил)-1,3-оксазол (В52), 2-(3,5-диметоксифеніл)-4-(2-піридил)-1,3-оксазол, 2-(2,3-дихлорофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3-хлоро-5-ціанофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3-фторо-5-ціанофеніл)-4-(2-піридил)-1,3-оксазолу, 2-(3-хлоро-5-фторофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3-ціанофеніл)-4-(5-хлоропірид-2-ил)-1,3-оксазол, 2-(3-ціанофеніл)-4-(5-фторопірид-2-ил)-1,3-оксазол, 2-(3-ціано-5-фторофеніл)-4-(5-фторопірид-2-ил)-1,3-оксазол, 2-(3-ціанофеніл)-4-(3-фторопірид-2-ил)-1,3-оксазол, 2-(3,5-диметоксифеніл)-4-(5-фторопірид-2-ил)-1,3-оксазол, 2-(3-ціанофеніл)-4-(5-метоксипірид-2-ил)-1,3-оксазол, 2-(3-ціанофеніл)-4-(2-хінолініл)-1,3-оксазол, 2-(3-ціанофеніл)-4-(3-хлоро-5-трифторометилпірид-2-ил)-1,3-оксазол, 2-(5-хлоро-2-метоксифеніл)-4-(2-піридил)-1,3-оксазол, 2-(2-хлоро-5-метилтіофеніл)-4-(2-піридил)-1,3-оксазол, 2-(2-бромо-5-метоксифеніл)-4-(2-піридил)-1,3-оксазол, 2-(2,5,6-трифторофеніл)-4-(2-піридил)-1,3-оксазол, 2-[3-хлорофеніл]-4-[піридин-2-іл]-1,3-оксазол, 2-(2,5,6-трифторофеніл)-4-(2-піридил}-1,3-оксазол, 2-(3-нітрофеніл)-4-(2-піридил)-1,3-оксазол, 2-(3-бромофеніл)-4-(2-піридил)-1,3-оксазол (В51) та їх фармацевтично прийнятні солі. У ще одному аспекті винаходу сполуками Формули І є: 3-(2-піридил)-5-(3-алілокси-5-(метоксикарбоніл)феніл)-1,2,4-оксадіазол (В77), 3-(2-піридил)-5-{3-N,N-диметиламінофеніл)-1,2,4-оксадіазол (В82), 3-(2-піридил)-5-(3-ціано-5-(4-піридил)феніл)-1,2,4-оксадіазол (В101), 3-(2-піридил)-5-[2-метокси-5-(4-піридил)феніл]-1,2,4-оксадіазол (В102), 3-(2-піридил)-5-[2-фторо-5-(4-піридил)феніл]-1,2,4-оксадіазол (В103), 3-(2-піридил)-5-(3-фторо-5-(4-піридил)феніл)-1,2,4-оксадіазол (В104), 3-(2-піридил)-5-(3-фторо-5-(3-піридил)феніл)-1,2,4-оксадіазол (В105), 3-(2-піридил)-5-[2-фторо-5-(3-піридил)феніл]-1,2,4-оксадіазол (В106), 3-(2-піридил)-5-[2-метокси-5-(3-піридил)феніл]-1,2,4-оксадіазол (В107), 3-(2-піридил)-5-(3-ціано-5-(3-піридил)феніл)-1,2,4-оксадіазол (В108), 3-(5-фторо-2-піридил)-5-(3-фторо-5-(3-піридил)феніл)-1,2,4-оксадіазол (В109), 3-(2-піридил)-5-[5-(3-піридил-пірид-3-ил)]-]-1,2,4-оксадіазол (В111), 3-(5-фторопірид-2-ил)]-5-[5-(3-піридил-пірид-3-ил)]-]-1,2,4-оксадіазол (В110), 3-(5-ціанопірид-2-ил)-5-(3-(пірид-3-ил)феніл)-1,2,4-оксадіазол (В112), 3-(5-ціанопірид-2-ил)-5-(3-фторо-5-(пірид-3-ил)феніл)-1,2,4-оксадіазол (В113), 3-(2-піридил)-5-(3-ціано-5-(2-піридил)феніл)-1,2,4-оксадіазол (В124), 3-(2-піридил)-5-[2-метокси-5-(2-піридил)феніл]-1,2,4-оксадіазол (В125), 3-(2-піридил)-5-[2-фторо-5-(2-піридил)феніл]-1,2,4-оксадіазол (В126), 3-(2-піридил)-5-[(3-(3-фторофеніл)-5-фторофеніл)]-1,2,4-оксадіазол (В114), 3-(2-піридил)-5-(3-ціано-5-(3-тіофен)феніл)-1,2,4-оксадіазол (В115), 3-(2-піридил)-5-[5-(3-тієніл)-пірид-3-ил]-1,2,4-оксадіазол (В116), 3-(2-піридил)-5-[5-(3-фурил)-пірид-3-ил]-1,2,4-оксадіазол (В117), 3-(2-піридил)-5-[5-(3-метоксифеніл)-пірид-3-ил]-1,2,4-оксадіазол (В119), 3-(2-піридил)-5-(3-ціано-5-(5-піримідилу)феніл)-1,2,4-оксадіазол (В120), 3-(2-піридил)-5-(3-ціано-5-(3-амінофеніл)феніл)-1,2,4-оксадіазол (В121), 3-(2-піридил)-5-(3-ціано-5-(3-фторофеніл)феніл)-1,2,4-оксадіазол (В122), 3-(2-піридил)-5-[5-(5-піримідилу)-пірид-3-ил]-1,2,4-оксадіазол (В123), 3-(2-піридил)-5-(3-амінометил-5-ціанофеніл)-1,2,4-оксадіазол (В127), 3-(2-піридил)-5-[5[(2-пропеніл)-пірид-3-ил]-1,2,4-оксадіазол (В128), 3-(2-піридил)-5-(3-ціано-5-вінілфеніл)-1,2,4-оксадіазол (В129), 3-(2-піридил)-5-(3-ціано-5-(2-гідроксіетил)феніл)-1,2,4-оксадіазол (В130), 3-(2-піридил)-5-(3-ціано-5-(2,3-дихлоропропокси)феніл)-1,2,4-оксадіазол (В131), 3-(2-піридил)-5-(3-алілокси-5-карбоксифеніл)-1,2,4-оксадіазол (В135), 3-(2-піридил)-5-(3-алілокси-5-ціанофеніл)-1,2,4-оксадіазол (В136), 3-(2-піридил)-5-(5-ціано-3-[3-гідроксипропін-1-іл]феніл)-1,2,4-оксадіазол (В142), 3-(2-піридил)-5-(2-N-метиламінофеніл)-1,2,4-оксадіазол (В144) і 3-(2-піридил)-5-[5-(3-N-бензил-1,2,5,6,тетрагідропіридин)-пірид-3-ил]-1,2,4-оксадіазол (В фармацевтично прийнятні солі. У ще одному аспекті винаходу забезпечуються такі сполуки Формули І: 3-(5-метил-пірид-2-ил)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В57), 3-(5-ціано-пірид-2-ил)-5-(3-ціанофеніл)-1,2,4-оксадіазол (В58), 3-(2-піридил)-5-(5-бромо-2-метоксифеніл)-1,2,4-оксадіазол (В62), 3-(2-піридил)-5-(5-бромо-2-фторофеніл)-1,2,4-оксадіазол (В63), 3-(2-піридил)-5-(5-ціано-2-фторофеніл)-1,2,4-оксадіазол (В64), 3-(2-піридил)-5-(5-бромопірид-3-ил)-1,2,4-оксадіазол (В65), 3-(2-піридил)-5-(5-хлоро-пірид-3-ил)-1,2,4-оксадіазол (В66), 3-(5-ціанопірид-2-ил)-5-(5-бромо-пірид-3-ил)-1,2,4-оксадіазол (В67), 3-(5-фторопірид-2-ил)-5-(5-бромо-пірид-3-ил-1,2,4-оксадіазол (В68), 3-(2-піридил)-5-(2-тіометокси-пірид-3-ил)]-1,2,4-оксадіазол (В69), 3-(2-піридил)-5-(5-метилпірид-3-ил)-1,2,4-оксадіазол (В70), 3-(2-піридил)-5-(5-метоксипірид-3-ил)-1,2,4-оксадіазол (В72), 3-(2-піридил)-5-(3-ціано-5-метилфеніл)-1,2,4-оксадіазол (В73), 3-{2-піридил)-5-(3-фторо-5-бромофеніл)-1,2,4-оксадіазол (В74), 3-(2-піридил)-5-(3-йодо-5-бромофеніл)-1,2,4-оксадіазол (В75), 3-(5-фторо-2-піридил)-5-(3-фторо-5-бромофеніл)-1,2,4-оксадіазол (В76), 143) та їх 3-(2-піридил)-5-(3-йодо-5-(метилфеніловий естер)-1,2,4-оксадіазол (В78), 3-(2-піридил)-5-(3-метокси-5-(метоксикарбоніл)феніл)-1,2,4-оксадіазол (В79), 3-(2-піридил)-5-(3-бромо-5-ціанофеніл)-1,2,4-оксадіазол (В80), 3-(2-піридил)-5-(5-ціано-3-йодофеніл)-1,2,4-оксадіазол (В81), 3-(5-ціано-2-піридил)-5-(3-бромофеніл)-1,2,4-оксадіазол (В59), 3-(5-ціано-2-піридил)-5-(3-ціано-5-фторофеніл)-1,2,4-оксадіазол (В60), 3-(5-ціано-2-піридил)-5-(3-бромо-5-фторофеніл)-1,2,4-оксадіазол (В61), 3-(2-піридил)-5-(5-ціано-2-метоксифеніл)-1,2,4-оксадіазол (В97), 3-(2-піридил)-5-(2-ціано-5-метоксифеніл)-1,2,4-оксадіазол (В98), 3-(2-піридил)-5-(5-ціано-пірид-3-ил)-1,2,4-оксадіазол (В99), 3-(2-піридил)-5-(3-ціано-5-(метоксикарбоніл)феніл)-1,2,4-оксадіазол (В100), 3-(2-піридил)-5-(5-феніл-пірид-3-ил)-1,2,4-оксадіазол (В118), 3-(2-піридил)-5-(3-ціано-5-метоксифеніл)-1,2,4-оксадіазол (В134), 3-(2-піридил)-5-(3-ціано-5-гідроксифеніл)-1,2,4-оксадіазол (В137), 3-(2-піридил)-5-{3-ціано-5-пропоксифеніл)-1,2,4-оксадіазол (В141), 2-(3-ціанофеніл)-4-(піридин-2-іл)-1,3-тіазол (В146), 2-(3-бромо-5-йодофеніл)-4-піридин-2-іл)-1,3-оксазол (В147), 2-(2-піридил)-5-(3-йодофеніл)-1,3,4-оксадіазол (В148), 2-(2-піридил)-5-(3-ціанофеніл)-1,3,4-оксадіазол (В149), 2-(2-піридил)-5-(3-ціанофеніл)-1,3,4-триазол (В150), 3-(5-хлоропірид-2-ил)-5-(3-ціано-5-фторофеніл)-1,2,4-оксадіазол (В83), 3-(5-хлоропірид-2-ил)-5-(3-ціано-5-хлорофеніл)-1,2,4-оксадіазол (В84), 3-(5-хлоропірид-2-ил)-5-(3-хлоро-5-фторофеніл)-1,2,4-оксадіазол (В85), 3-(5-хлоропірид-2-ил)-5-(3-ціано-5-метоксифеніл)-1,2,4-оксадіазол (В86), 3-(5-фторопірид-2-ил)-5-(3-ціано-5-хлорофеніл)-1,2,4-оксадіазол (В87), 3-(5-фторопірид-2-ил)-5-(3-фторо-5-хлорофеніл)-1,2,4-оксадіазол (В88), 3-(5-фторопірид-2-ил)-5-(3-ціано-5-метоксифеніл)-1,2,4-оксадіазол (В89), 3-(5-ціанопірид-2-ил)-5-(3-ціано-5-хлорофеніл)-1,2,4-оксадіазол (В90), 3-(5-ціанопірид-2-ил)-5-(3-фторо-5-хлорофеніл)-1,2,4-оксадіазол (В91), 3-(5-ціанопірид-2-ил)-5-(3-ціано-5-метоксифеніл)-1,2,4-оксадіазол (В92), 3-(5-фторопірид-2-ил)-5-(3,5-сІі-ціанофеніл)-1,2,4-оксадіазол (В93), 3-(3-(4-диметиламінобутокси)-пірид-2-ил)-5-(3-ціано-5-фторофеніл)-1,2,4-оксадіазол (В94), 3-(3-(5-диметиламінопентилокси)-пірид-2-ил)-5-(3-ціано-5-фторофеніл)-1,2,4-оксадіазол (В95) і [3-(3-(6-диметиламіногексилокси)-пірид-2-ил)-5-(3-ціано-5-фторофеніл)-1,2,4-оксадіазол (В96). Даний винахід також забезпечує сполуки, які є ефективними і селективними антагоністами mGluR5, які можуть бути представлені Формулою II. Згідно з іншим аспектом винаходу, передбачено п'ятичленне кільце, яке містить дві змінні Х2 та Y2. До цього п'ятичленного кільця приєднуються два замісники, Аr3 та Аr4. П'ятичленне кільце може містити 0, 1 або 2 подвійні зв'язки, позначені пунктирними лініями у Формулі II. В оптимальному варіанті втілення винаходу п'ятичленне кільце має два подвійні зв'язки. У варіантах втілення винаходу похідну Х 2 вибирають із групи, яка складається з N та С, і змінну Y2 вибирають із групи, яка складається з N, О, S та CR5, причому принаймні один із Х2 та Y2 має бути гетероатомом. У разі, якщо Y2 є CR5, R5 вибирають із групи, яка складається з Н, алкілу, -CF3, -OR6, -SR6, NR6R7, -C(O), -C(S), -C=NR6 та=CR6R7, причому R6 та R7 незалежно можуть бути вибрані з групи, яка складається з Н, алкілу, галоалкілу, алкілокси, алкіламіну, циклоалкілу, гетероциклоалкілу, арилу, гетероарилу, алкіларилу, алкілгетероарилу, галоарилу, алкілоксіарилу, алкеніларилу, алкенілоксіарилу та галогетероарилу. В оптимальних варіантах втілення винаходу Х 2 та Y2 обидва є гетероатомами. У ще одному оптимальному варіанті втілення винаходу Х 2 є N. У ще кращому варіанті втілення винаходу Υ2 є N. У ще кращому варіанті втілення винаходу Х2 та Υ2 обидва є N. Згідно з іншим аспектом винаходу групу Аr3 та Аr4 незалежно вибирають із групи, яка складається з арилу та гетероарилу. Зокрема, до варіантів втілення винаходу належать ті, в яких Аr3 та Аr4 незалежно вибирають 3-поміж 5- та 6-членних арильних та гетероарильних кілець. У більш конкретних варіантах втілення винаходу Аr3 та Аr4 вибирають 3-поміж 6-членних арильних та гетероарильних кілець. До ще більш конкретних варіантів втілення винаходу належать ті, в яких Аr3 та Аr4 незалежно вибирають 3-поміж фенілу, піридилу, фуранілу, тієнілу, піразинілу, піримідинілу, піридазинілу, піролілу, піразолілу, імідазолілу, триазолілу та тіазолілу. В оптимальному варіанті втілення винаходу Аr3 та Аr4 незалежно вибирають 3поміж фенілу та піридилу. У ще кращому варіанті втілення винаходу один із Аr3 та Аr4 є фенілом, і один є піридилом. Згідно зі ще одним варіантом втілення винаходу, Аr3 та Аr4 є необов'язково заміщеними одним або кількома замісниками G2, причому G2 вибирають із групи, яка складається з галоалкілу, гетероарилу, циклоалкену, алкенілу, алкінілу, А-алкенілу, А-алкінілу, алкілокси, А-алкілокси, -R6OR7, -R6OC(O)R7, (CH2)mNR6R7, -OCH2CH(CI)CH2CI та заміщеного арилу, де арильним замісником є Re. У конкретному варіанті втілення, в якому один або обидва з Аr3 та Аr4 є заміщеними G2, G2 вибирають із групи, яка складається з Аалкенілу, алкінілу та А-алкілокси, і А вибирають із групи, яка складається з СН2, О, NH, S, SO, SO2, OSO2, NSO2 та -C(NR6)NR7. У більш конкретному варіанті втілення G2 є (CH2)m-NR6R7. В одному варіанті втілення винаходу G2 є заміщеним арилом, і замісник R8 вибирають із групи, яка складається з гало, -OR6, -SR6, SOR6, -SO2R6, -SO2NR6R7, -R6OR7 R6SR7, -OCOR6, -OCONR6R7, -NR6COR7, -NR6CO2R7, -CN, -NO2, C(NR6)NR-, -CO2R6R7, -CONR6R7, -C(O)R6, -CH(OR6)R7, -CH2(OR6), -A-(CH2)m-NR6R7, NR6R7, арилу, ар алкілу, гетероарилу та гетероаралкілу. У ще одному варіанті втілення винаходу кожен з Аr3 та Аr4 і G2 є додатково заміщеним одним або кількома замісниками, вибраними 3-поміж R6 та R8. В оптимальному варіанті втілення винаходу кожен з Аr3 та Аr4 є незалежно додатково заміщеним одним або кількома замісниками, вибраними з групи, яка складається з Н, алкілу, галоалкілу, алкілокси, алкіламіну, гало, -OR6, -SR6, -SOR6, -SO2R6, -SO2NR6R6, R6OR7 R6SR7, -OCOR6, -OCONR6R7, -NR6COR7, -NR6CO2R7, -CN, -NO2, -C(NR6)NR7, -CO2R6R7, -CONR6R7, C(O)R6, -CH(OR6)R7, -CH2(OR6), -A-(CH2)m-NR6R7, NR6R7, арилу, аралкілу, гетероарилу, гетероаралкілу, циклоалкілу, гетероциклоалкілу, алкіларилу, алкілгетероарилу, галоарилу, алкілоксіарилу, алкеніларилу, алкенілоксіарилу та галогетероарилу. Згідно з іншим аспектом винаходу, Аr3 та Аr4 є незалежно заміщеними замісником, вибраним із групи, яка складається з гало та ціано. У конкретних варіантах втілення винаходу сполуками Формули II є: 4-(3-ціанофеніл)-1-(2-піридил)-1Н-імідазол (В151) 1-(3-ціанофеніл)-4-(2-піридил)-1Н-імідазол (В152). Випробування сполук на антагоністичну дію щодо mGluR Групи І Фармакологічні властивості сполук винаходу аналізують із застосуванням стандартних аналізів на функціональну активність. Приклади аналізів рецепторів глутамату є добре відомими спеціалістам. Див., наприклад, Аrатогі et al., Neuron 8:757 (1992); Tanabe et al., Neuron 8:169 (1992); Miller et al., J. Neuroscience 15: 6103 (1995); Balazs, et al., J. Neurochemistry 69:151 (1997). Методологію, описану в цих публікаціях, включено авторами шляхом посилання. Зручним є те, що сполуки винаходу можуть бути досліджені шляхом аналізу з вимірюванням мобілізації внутрішньоклітинного кальцію, [Са2+]i, у клітинах, які експресують mGluR5, що може зв'язувати сполуки. Загальновідому лінію клітин, яка є придатною для цієї мети, описано в роботі Miller et al., J. Neuroscience 15: 6103 (1995), зміст якої включено авторами шляхом посилання. Було продемонстровано, що піддавання астроцитів щура дії факторів росту, основного фактора росту фібробластів, FGF, або трансформуючого фактора росту-α, помітно підвищувало експресію білка та функціональну активність ендогенного mGluR5 (Miller et al., J. Neuroscience, 15(9):6103-6109, 1995). Тобто, первинні культури астроцитів одержували з 3-5-денних дитинчат щура Sprague-Dawley, застосовуючи модифікацію, описану у Miller et al., наносили на вкриті полі-L лізином колби у модифіковане Дульбекко середовище Ігла (DMEM), що містило ембріональну сироватку телят (FCS). Для аналізу кювет культури піддавали підвищувальній регуляції факторами росту у колбах протягом 3-5 днів, потім збирали і підготовляли для вимірювання мобілізації [Са2+]і, як було описано вище (Nemeth et al., 1998). Для FLIPR-аналізу клітини висівали на вкриті полі-D-лізином 96-лункові планшети з прозорим дном і чорними краями і аналіз мобілізації [Са2+]i здійснювали через 3 дні після підвищувальної регуляції фактором росту. FLIPR-експерименти здійснювали з застосуванням лазера потужністю 0,800Вт та CCD-камери зі швидкістю затвора 0,4сек. Кожен FLIPR-експеримент розпочинали з 180мкл буфера, присутнього у кожній лунці планшета з клітинами. Після кожного додавання сполуки 50 разів з інтервалами в 1 секунду вимірювали сигнал флуоресценції з наступними 3 вимірюваннями з 5-секундними інтервалами. Реакцію вимірювали як піковий показник реакції за період вимірювання. Визначення ЕС50 та ІС50 здійснювали на основі даних, одержаних за 8 точками кривих реакції на концентрацію (CRC), які виконували по двічі. CRC Агоніста будували шляхом перерахунку всіх реакцій на максимальну реакцію, яку спостерігали для планшета. Блокування антагоністом імунізації Агоністом нормалізували до середнього показника реакції імунізації Агоністом у 14 контрольних лунках на одному планшеті. Детальний протокол випробування сполук винаходу представлено нижче у Прикладі 6. Одержання Фармацевтичних композицій, які містять антагоністи mGluR, та їх застосування в лікуванні неврологічних захворювань Сполуки даного винаходу можуть бути корисними для лікування неврологічних порушень або хвороб. Хоча ці сполуки зазвичай застосовують для терапії людей, вони також можуть застосовуватися у ветеринарній медицині для лікування подібних або ідентичних хвороб. При терапевтичному тNабо діагностичному застосуванні сполуки даного винаходу можуть бути рецептовані для різних режимів введення, включаючи системне та місцеве або локалізоване введення. Загальні відомості про способи та композиції можна знайти у Remington's Pharmaceutical Sciences (18th ed.), Mack Publishing Co. (1990). Сполуки згідно з винаходом є ефективними у широкому діапазоні доз. Наприклад, при лікуванні дорослих людей застосовують дози від приблизно 0,01 до приблизно 1000мг, в оптимальному варіанті — від приблизно 0,5 до приблизно 100мг на день. Найбільш оптимальна доза становить від приблизно 2мг до приблизно 70мг на день. Конкретна доза залежить від шляху введення, форми, в якій вводять сполуку, суб'єкта, якого піддають лікуванню, маси тіла суб'єкта, якого піддають лікуванню, та особистого досвіду лікаря. Фармацевтично прийнятні солі, як правило, є добре відомими спеціалістам, і до них, крім інших, можуть належати, наприклад, ацетат, бензолсульфонат, безилат, бензоат, бікарбонат, бітартрат, бромід, едетат кальцію, камзилат, карбонат, цитрат, едетат, едисилат, естоат, езилат, фумарат, глюцептат, глюконат, глутамат, гліколіларсанілат, гексилрезорцинат, гідрабамін, гідробромід, гідрохлорид, гідроксинафтоат, йодид, ізетіонат, лактат, лактобіонат, малат, малеат, манделат, мезилат, мукат, напсилат, нітрат, памоат (ембонат), пантотенат, фосфат/дифосфат, полігалактуронат, саліцилат, стеарат, субацетат, сукцинат, сульфат, танат, тартрат, або теоклат. Інші фармацевтично прийнятні солі згадуються, наприклад, у Remington's Pharmaceutical Sciences (18th ed.), див. вище. Оптимальними фармацевтично прийнятними солями є, наприклад, ацетат, бензоат, бромід, карбонат, цитрат, глюконат, гідробромід, гідрохлорид, малеат, мезилат, напсилат, памоат (ембонат), фосфат, саліцилат, сукцинат, сульфат або тартрат. Залежно від конкретних станів, які піддають лікуванню, ці агенти рецептують у рідкі або тверді дозовані форми і вводять системно або місцево. Агенти можуть доставлятися, наприклад, у формі затриманого або уповільненого вивільнення, які є відомими спеціалістам. Способи рецептування та введення описано у Remington's Pharmaceutical Sciences (18th ed.), див. вище. Прийнятними шляхами введення можуть бути пероральний, букальний, під'язиковий, ректальний, крізьшкірний, вагінальний, трансмукозальний, назальний або інтестинальний шляхи введення; парентеральне доставления, включаючи внутрішньом'язові, підшкірні, внутрішньомозкові ін'єкції, а також інтратекальні, прямі внутрішньошлуночкові, внутрішньовенні, внутрішньочеревинні, інтраназальні або внутрішньоочні ін'єкції. Для ін'єкцій агенти згідно з винаходом рецептують у водних розчинах, в оптимальному варіанті у фізіологічно сумісних буферах, таких як розчин Хенка, розчин Рингера або фізіологічний соляний буфер. Для такого трансмукозального введення у композиції застосовують придатні для подолання бар'єру проникні рідини. Такі проникні рідини, як правило, є відомими спеціалістам. Застосування фармацевтично прийнятних носіїв для рецептування сполук, описаних авторами з метою практичного втілення винаходу, у дозовані форми, придатні для системного введення, охоплюється обсягом винаходу. Завдяки належному виборові носія та відповідному способові виробництва композиції даного винаходу, зокрема, рецептовані як розчини, можуть вводитись парентерально, наприклад, шляхом внутрішньовенної ін'єкції. Сполуки можуть бути легко рецептовані з застосуванням фармацевтично прийнятних носіїв, добре відомих спеціалістам, у дозовані форми, придатні для перорального введення. Такі носії дозволяють рецептувати сполуки винаходу як таблетки, пілюлі, капсули, рідини, гелі, сиропи, суспензії та ін. для перорального приймання пацієнтом, якого піддають лікуванню. До фармацевтичних композицій, придатних для застосування у даному винаході, належать композиції, в яких активні інгредієнти містяться в ефективній для передбаченого застосування кількості. Визначення ефективної кількості належить до компетенції спеціалістів, особливо, з огляду на представлений авторами детальний опис. Крім активних інгредієнтів, ці фармацевтичні композиції можуть містити відповідні фармацевтично прийнятні носії, включаючи наповнювачі та допоміжні речовини, які сприяють переробці активних сполук на композиції, придатні для фармацевтичного застосування. Композиції, рецептовані для перорального введення, можуть бути у формі таблеток, драже, капсул або розчинів. Фармацевтичні композиції для перорального застосування одержують шляхом комбінування активних сполук з твердими наповнювачами, необов'язково подрібнення одержаної в результаті суміші та обробки суміші гранул після додавання відповідних допоміжних речовин, у разі необхідності, для одержання ядра таблетки або драже. Придатними наповнювачами є, зокрема, такі наповнювачі, як цукри, включаючи лактозу, сахарозу, маніт або сорбіт; целюлозні композиції, наприклад, кукурудзяний крохмаль, пшеничний крохмаль, рисовий крохмаль, картопляний крохмаль, желатин, трагакантову камедь, метилцелюлозу, гідроксипропілметилцелюлозу, натрій-карбоксиметилцелюлозу (CMC) тNабо полівінілпіролідон (PVP: повідон). У разі необхідності додають Агенти розпаду, такі як зшитий полівінілпіролідон, Аrар або альгінова кислота або її сіль, така як альгінат натрію. Ядро драже вкривають відповідним покриттям. Для цього застосовують концентровані розчини цукрів, які необов'язково можуть містити гуміарабік, тальк, полівінілпіролідон, гель карбопол, поліетиленгліколь (PEG) тNабо діоксид титану, розчини глазурі та придатні органічні розчинники або суміші розчинників. До покриття таблеток або драже додають барвники або пігменти для розпізнання або розрізнення різних комбінацій доз активних сполук. Фармацевтичні композиції для перорального застосування включають капсули "push-fit" з желатину, а також м'які, герметично закриті капсули з желатину та пластифікатора, такого як гліцерин або сорбіт. Капсули "push-fit" можуть містити активні інгредієнти у суміші з наповнювачем, таким як лактоза, зв'язувальними речовинами, такими як крохмалі, тNабо мастилами, такими як тальк або стеарат магнію, і, необов'язково, стабілізаторами. У м'яких капсулах активні сполуки можуть бути розчинені або суспендовані у відповідних рідинах, таких як жирні олії, рідкий парафін або рідкі поліетиленгліколі (PEG). Крім того, додають стабілізатори. Даний винахід буде легше зрозуміти шляхом посилання на представлені нижче приклади, які представлено для пояснення і які не обмежують обсягу даного винаходу. У Таблицях 1, 2 та 3 показано конкретні приклади сполук даного винаходу. Одержання антагоністів mGluR Групи І Багато вихідних матеріалів для одержання сполук даного винаходу виробляються серійно такими компаніями, як Aldrich Chemical Company (Milwaukee, Wl). Крім того, сполуки винаходу можуть бути легко одержані з наявних попередників шляхом прямих перетворень, які є добре відомими спеціалістам. Спеціалістові стане зрозуміло, що антагоністи mGluR Групи І згідно з винаходом можуть бути одержані добре відомими способами з застосуванням, загальновизнаних технологій органічної хімії. Придатні реакції описано у стандартних підручниках з органічної хімії. Наприклад, див. March, Advanced Organic Chemistry, 2d ed., McGraw Hill (1977). Тобто, сполуки винаходу, як правило, одержують шляхом утворення G-компонента з двох сполукпопередників, які містять відповідні Аr1 та Аr2 компоненти. Якщо лінкер містить 1,2,4-оксадіазол, гетероцикл утворюють, застосовуючи загальновідомі технології, такі як реакція між амідоксимом та хлорид кислоти, або шляхом реакції амідоксиму та ацилімідазолу. Подібне перетворення пояснюється нижче у Прикладах 4 та 5. Амідоксими одержують, застосовуючи загальновідомі технології шляхом реакції Аr1-заміщеного нітрилу з гідроксиламіном. Подібне перетворення пояснюється нижче у Прикладі 1. У більшості випадків попередники хлориди кислот Аr2 є легко доступними або можуть бути одержані з застосуванням прямих способів органічної хімії. Наприклад, карбонові кислоти можуть бути перетворені на відповідні хлориди кислот шляхом реакції, наприклад, з тіонілхлоридом або оксалілхлоридом. У разі, якщо лінкер містить 1,3-оксазол, сполуки одержували, застосовуючи процедуру, подібну до описаної в роботі Kelly et al., J. Org. Chem. 61, 4623-4633 (1996). Таким чином, 3,5-двозаміщені -1,3оксазоли одержували шляхом змішування галокетону з карбоксамідом у підданому дефлегмації толуолі протягом 3 днів. Одержаній в результаті суміші давали охолонути до кімнатної температури, розчинник видаляли і залишок очищали. На Схемі 1 пояснюється спосіб синтезу сполук даного винаходу. Зокрема, спосіб, який пояснюється на схемі 1, застосовують для одержання таких сполук: В77-В81, В86, В89, В101, В108, В11 5, В120-В122, В124, В129-В141. Схема 2 пояснює інший спосіб синтезу сполук даного винаходу. Зокрема, спосіб, який пояснюється на схемі 2, застосовують для одержання сполуки В144. На Схемі 3 пояснюється ще один спосіб синтезу сполук даного винаходу. Зокрема, спосіб, який пояснюється на схемі 3, застосовують для одержання таких сполук: В57-В76, В82-В85, В87, В88, В90, В91, В93-В100, В102-В107, В109-В114, В116-В119, В123, В125-В128, В142, В143. Інші сполуки даного винаходу можуть бути легко одержані шляхом застосування варіантів реакцій, показаних вище на схемах, які стануть зрозумілими спеціалістам. ПРИКЛАДИ Загальні експериментальні способи Дані капілярної газової хроматографії та мас-спектрометрії одержували, застосовуючи газовий хроматограф Hewlett-Packard (HP) 5890 серії II, сполучений з мас-спектрометричним селективним детектором HP 5971 [високоефективна капілярна колонка Ultra-2 (зшитий 5% РhМе силікон); довжина колонки, 25м; внутр. діаметр колонки 0,20мМ; швидкість потоку гелію 60мNхв; темп, інжектора 250°С; температурна програма 20°С/хв. від 125 до 325°С протягом 10хв з наступним підтриманням постійної температури 325°С протягом 6хв). Тонкошарову хроматографію здійснювали, застосовуючи HF TLC пластинки Analtech Uniplate з 250мкм силікагелю. Іноді застосовували ультрафіолетове світло у поєднанні з нінгідрином та аерозольними реагентами Драгендорфа (Sigma Chemical Co.) для виявлення сполук на TLCпластинках. Більшість реагентів, застосованих у реакціях, отримували від Aldrich Chemical Co. (Milwaukee, Wl), Sigma Chemical Co. (Saint Louis, MO), Fluka Chemical Corp. (Milwaukee, Wl), Fisher Scientific (Pittsburgh, PA), TCI America (Portland, OR) або Lancaster Синтез (Windham, NH). Приклад 1: Синтез проміжних сполук амідоксиму Застосовуючи загальну процедуру згідно з Shine et al., J. Heterocyclic Chem. (1989) 25:125-128, гідроксиламінгідрохлорид (7,65г, 110ммоль) в етанолі (100мл) обробляли 10N розчином гідроксиду натрію (11мл, 110ммоль). Швидко утворювався осад, і реакційну суміш перемішували при кімнатній температурі протягом 30хв. Неорганічний осад фільтрували і промивали етанолом (100мл). Фільтрат та етанолові промиї комбінували і обробляли 2-ціанопіридином (10,4г, 100ммоль). Реакційну суміш після цього нагрівали з дефлегмацією протягом 20 годин. Після охолодження леткі речовини видаляли in vacuo, одержуючи 13,3г (97%) пірид-2-иламідоксиму. Суміш 2-бромо-5-метилпіридину (2,001г, 11,63ммоль), ціаніду цинку (830мг, 7ммоль), цинку (порошок, 35мг, 0,53ммоль), [1,1'-біс(дифенілфосфіно)фероцен] дихлоропаладію(ІІ), комплексу з дихлорометаном (1:1) (192,5мг, 0.24ммоль) у N,N-диметилформаміді (10мл) нагрівали з дефлегмацією протягом 16 годин. Після охолодження реакційну суміш розводили етилацетатом і екстрагували водою та розсолом. Органічний розчин фільтрували крізь шар силікагелю, промивали дихлорометаном. Видалення розчинника in vacuo давало 770мг (56%) 5-метил-2-ціано-піридину. Застосовуючи загальну процедуру синтезу амідоксимів, 5-метил-2-ціано-піридин (770мг, 6,5ммоль), 5М гідроксиламінгідрохлориду (1,5мл, 7,5ммоль) в етанолі (10мл) та 10N гідроксид натрію (0,75мл, 7,5ммоль) нагрівали з дефлегмацією протягом 18 годин. Стандартна обробка давала 594мг (60%) 5-метилпірид-2иламідоксиму. Подібним чином суміш 2,5-диціанопіридину (740мг, 5,74ммоль), 5М гідроксиламінгідрохлориду (1,15мл, 5,75ммоль) та 1М гідроксиду натрію (5,74мл, 5,74ммоль) в етанолі (10мл) нагрівали при 80°С протягом 5 хвилин. Осад збирали шляхом фільтрації, одержуючи 555мг (60%) 5-ціанопірид-2-иламідоксиму. Суміш 2-ціано-5-хлоропіридину (1г, 7,22ммоль) та фториду калію (1,26г, 21,68ммоль) у 1-метил-2піролідиноні (25мл) нагрівали з дефлегмацією протягом 18 годин. Після охолодження реакційну суміш розводили етилацетатом і екстрагували водою та розсолом. Органічні розчинники після цього видаляли in vacuo. Хроматографія залишку на силікагелі давала 425мг (48%) 2-ціано-5-фторопіридину. Застосовуючи загальну процедуру синтезу амідоксимів, 2-ціано-5-фторопіридин (425мг, 3,48ммоль), 5М гідроксиламінгідрохлориду (0,79мл, 3,95ммоль) в етанолі (5мл) та 10N гідроксид натрію (0,398мл, 3,98ммоль) нагрівали з дефлегмацією протягом 24 годин. Стандартна обробка давала 330мг (61%) 5фторопірид-2-иламідоксиму. Суспензію 6-ціанонікотинової кислоти (535мг, 3,6ммоль) у дихлорометані (8мл) при 0°С обробляли 2М оксалілхлориду (3,6мл, 7,2ммоль, дихлорометан) з каталітичною кількістю N,N-диметилформаміду. Реакційну суміш після цього перемішували при кімнатній температурі протягом 2 годин. Розчинник видаляли in vacuo і залишок розчиняли у дихлорометан (10мл). Одержаний в результаті розчин обробляли піридином (2мл) та трет-бутанолом (0,8мл) і реакційну суміш перемішували до наступного дня при кімнатній температурі. Реакційну суміш розводили дихлорометаном (200мл) і послідовно промивали насиченим бікарбонатом натрію (50мл) та розсолом (50мл). Органічний шар висушували над безводним сульфатом натрію, фільтрували і розчинник видаляли in vacuo. Хроматографія на силікагелі з градієнтом від 5% до 10% етилацетату в гексані давала 623мг (84%) 5-трет-бутоксикарбоніл-2-ціано-піридину у вигляді жовтої твердої речовини. Застосовуючи загальну процедуру синтезу амідоксимів, 5-трет-бутоксикарбоніл-2-ціано-піридин (623мг, 3,05ммоль) та 5М гідроксиламінгідрохлориду (0,69мл, 3,4ммоль) в етанолі (7мл) і 10N гідроксид натрію (0,34мл, 3,4ммоль) нагрівали з дефлегмацією протягом 18 годин. Стандартна обробка давала 570мг (79%) 5-трет-бутоксикарбонілпірид-2-иламідоксиму. Одержували розчин диметил-5-гідрокси-ізофталату (6г, 28,6ммоль) та карбонат калію (9г, 65,4ммоль) в ацетоні (120мл). До нього додавали метилйодид (4мл, 63,7ммоль) і реакційну суміш залишали перемішуватися до наступного дня за навколишньої температури. Реакційну суміш фільтрували, а потім концентрували. Залишок розчиняли в етилацетаті і промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. 6,4г (кількісно) диметил-5-метоксиізофталату виділяли у вигляді білуватої твердої речовини. Розчин диметил-5-метокси-ізофталату (2,5г, 11,1ммоль) у тетрагідрофурані/метанолі (56мN20мл) обробляли 2,0N гідроксидом натрію (12мл, 25ммоль). Реакційну суміш залишали перемішуватися протягом 15 годин при кімнатній температурі. Після концентрації розчину тверду речовину розчиняли у воді і підкислювали 2,0N хлористим воднем. Етилацетат застосовували для екстрагування осаду, який після цього промивали розсолом і висушували над безводним сульфатом натрію. Після видалення розчинника in vacuo виділяли загалом 2,5г (кількісно) 5-метокси-ізофталевої кислоти. Розчин 5-метокси-ізофталевої кислоти (2,5г, 13,8ммоль) у дихлорометані (20мл) обробляли 2М оксалілхлориду (38мл, 76,6ммоль) та кількома краплями DMF. Після перемішування протягом 17 годин реакційну суміш концентрували in vacuo, а потім одержану в результаті буру олію переносили у холодний перемішуваний розчин гідроксиду амонію в етилацетаті (70мN200мл) з невеликою кількістю дихлорометану. Реакцію залишали для продовження протягом 1 години, після чого утворювався осад. Воду (100мл) та етилацетат (1500мл) комбінували з реакційною сумішшю у ділильній лійці і органічний шар збирали, промивали розсолом і висушували над сульфатом натрію. Тверду речовину, що залишилася у водному шарі, відокремлювали шляхом фільтрації, промивали водою і висушували. Органічний шар фільтрували і концентрували, а потім розтирали з гексанами. Загалом виділяли 2,5г (кількісно) 5-метокси-ізофталаміду. Суспензію 5-метокси-ізофталаміду (3,1г, 16ммоль) у дихлорометані (27мл) при 0°С обробляли піридином (5,2мл, 65 моль), а потім по краплях додавали трифторооцтовий ангідрид (5,4мл, 39ммоль). Реакційну суміш перемішували при 0°С протягом 20 хвилин, а потім перемішували до наступного дня за навколишньої температури. Реакційну суміш промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Хроматографія на силікагелі з застосуванням гексанів:етилацетату:дихлорометану давала 940мг (46%) 5-метокси-ізофталонітрилу у вигляді білої твердої речовини. Застосовуючи загальну процедуру синтезу амідоксимів, 5-метокси-ізофталонітрил (600мг, 3,8ммоль), 5М гідроксиламінгідрохлориду (0,76мл, 3,8ммоль) в етанолі (6мл) та 1N гідроксид натрію (3,8мл, 3,8ммоль) нагрівали з дефлегмацією протягом 3 годин. Стандартна обробка та колонкова хроматографія з застосуванням 30-40% етилацетату/гексанів давала 329мг (45%) 3-ціано-5-метоксифеніл-амідоксиму. 5-фторо-ізофталонітрил (730мг, 5ммоль) та 5М гідроксиламінгідрохлориду (1мл, 5ммоль) в етанолі (10мл) і N гідроксид натрію (5мл, 5ммоль) нагрівали з дефлегмацією протягом 30 хвилин. Стандартна обробка з наступною колонковою хроматографією давала 473мг (52,8%) 3-ціано-5-фторофеніламідоксиму. 5-бромонікотинонітрил (982мг, 5,4ммоль) та 5М гідроксиламінгідрохлориду (1,098мл, 5,4ммоль) в етанолі (5мл) і 1N гідроксид натрію (5,4мл, 5,4ммоль) нагрівали з дефлегмацією протягом 5 хвилин. Стандартна обробка давала 925мг (79,3%) 5-бромопірид-3-ил-амідоксиму. 3,5-диціанотолуол (1000мг, 7,04ммоль) та 5М гідроксиламінгідрохлориду (1,4мл, 7,04ммоль) в етанолі (5мл) і 1N гідроксид натрію (7,04мл, 7,04ммоль), нагрівали з дефлегмацією протягом 12 хвилин. Стандартна обробка з наступною колонковою хроматографією давала 215мг (17,4%) 3-ціано-5-метилфеніламідоксиму. Ізофталонітрил (640мг, 5ммоль) та 5М гідроксиламінгідрохлориду (1мл, 5ммоль) в етанолі (5мл) і 1N гідроксид натрію (5мл, 5ммоль) нагрівали з дефлегмацією протягом 2,5 години. Стандартна обробка з наступною колонковою хроматографією давала 650мг (80,7%) 3-ціанофеніламідоксиму. 3-йодобензонітрил (1145мг, 5ммоль) та 5М гідроксиламінгідрохлориду (1мл, 5ммоль) в етанолі (5мл) і 1N гідроксид натрію (5мл, 5ммоль) нагрівали з дефлегмацією протягом 2,5 години. Стандартна обробка з наступною колонковою хроматографією давала 920мг (70,2%) 3-йодофеніламідоксиму. 5-диметиламіноізофталонітрил (856мг, 5ммоль) та 5М гідроксиламінгідрохлориду (1мл, 5ммоль) в етанолі (10мл) і 1N гідроксид натрію (5мл, 5ммоль) нагрівали з дефлегмацією протягом 30 хвилин. Стандартна обробка з наступною колонковою хроматографією давала 280мг (27,4%) 3-ціано-5диметиламінофеніламідоксиму. 2,6-диціанопіридин (3,87г, 30ммоль) та 5М гідроксиламінгідрохлориду (6мл, 30ммоль) в етанолі (50мл) і 1N гідроксид натрію (30мл, 30ммоль) нагрівали з дефлегмацією протягом 10 хвилин. Стандартна обробка з наступною колонковою хроматографією давала 2,87г (59%) 6-ціано-пірид-2-ил-амідоксиму. 3-бромо-5-фторобензонітрил (1,9г, 9,5ммоль) та 5М гідроксиламінгідрохлориду (4мл, 20ммоль) в етанолі (20мл) і 1N гідроксид натрію (20мл, 20ммоль) нагрівали з дефлегмацією протягом 1 години. Стандартна обробка з наступною колонковою хроматографією давала 721мг (32,6%) 3-бромо-5фторофеніламідоксиму. 3-фторо-5-метоксибензонітрил (379мг, 2,5ммоль) та 5М кімнатній температурі протягом 16 годин і при 80°С протягом 24 годин. Реакційну суміш виливали у воду (100мл) і екстрагували EtOAc. Органічну фазу промивали розсолом, висушували (MgSO4), фільтрували і концентрували. Проміжну сполуку 3-фторо-5-бромо-(1Н-імідазол-1-іл)бензол використовували безпосередньо на наступному етапі. Застосовуючи стандартну процедуру ціанування, одержували 3-фторо-5-ціано-(1Н-імідазол-1-іл)-бензол у вигляді безбарвної твердої речовини. Неочищений продукт використовували безпосередньо на наступному етапі. Застосовуючи стандартну процедуру омилення, одержували названу сполуку у вигляді безбарвної твердої речовини. Неочищений продукт використовували безпосередньо на наступному етапі. Названий продукт одержували згідно з описаною в літературі процедурою (Fujiki, Kanji; Kashiwagi, Mitsuyoshi; Miyamoto, Hideyuki; Sonoda, Akinari; Ichikawa, Junji; et al., J.Fluorine Chem. 1992, 57, 307-321) із 3,5-біс(трифторометил)бензолу (7,3мл, 47ммоль) та йоду (11,95г, 47ммоль) у 30% олеумі (30мл) при 55 градусах протягом 15год. Після гасіння льодом здійснювали екстрагування неочищеного продукту етером, послідовне промивання водного шару сульфітом натрію (1М) та водою. До неочищеного розчину етеру додавали гідроксид натрію (1N, -150мл) до лужного рівня рН, шари відокремлювали і водний шар після цього підкислювали, застосовуючи НСІ (12N, ~10мл, рН кислий). Після екстрагування етером з наступним висушуванням над сульфатом магнію одержували неочищену кислоту (4,3г, 29%). Матеріал був недостатньо очищеним для використання, тому продукт естерифікували, застосовуючи гідроксиламінгідрохлориду (0,5мл, 2,5ммоль) в етанолі (2,5мл) і 1N гідроксид натрію (2,5мл, 2,5ммоль) нагрівали з дефлегмацією протягом 1 години. Стандартна обробка давала 431мг (93,4%) 3-фторо-5метоксифеніламідоксиму. 1-бромо-3,5-дифторобензол (1,00г, 5,18ммоль) розчиняли у безводному DMF (10мл). Розчин охолоджували у льодяній ванні і додавали NaSMe (0,36г, 5,18ммоль). Через 30 хвилин реакційну суміш виливали у воду (100мл) і екстрагували гексанами. Органічну фазу промивали водою та розсолом, висушували (МgSО4) фільтрували і концентрували для забезпечення названої сполуки у вигляді безбарвної олії. Неочищений продукт використовували безпосередньо на наступному етапі. Застосовуючи стандартну процедуру ціанування, одержували 5-ціано-3-фторо-1-(тіометил)бензол у вигляді жовтої олії. Неочищений продукт використовували безпосередньо на наступному етапі. Застосовуючи стандартну процедуру омилення, одержували названу сполуку у вихідній кількості 0,60г (63% у 3 етапи) у вигляді безбарвної твердої речовини. 1-бромо-3,5-дифторобензол (1,00г, 5,18ммоль) розчиняли у безводному DMF (10мл). Розчин охолоджували у льодяній ванні. Додавали імідазол (0,36г, 5,18ммоль) та К2СО3 (0,72г, 5,18ммоль). Реакційну суміш перемішували при ацетилхлорид у метанолі, очищали, застосовуючи флешхроматографію (силікагель, 20% дихлорометану у гексані) і гідролізували гідроксидом натрію у метанолі, одержуючи на виході 2,95г (69%) чистої 3-йодо-5-трифторометилбензойної кислоти. Суспензію 3-алілокси-5-(метоксикарбоніл)бензойної кислоти (5,5мг, 23ммоль) у тіонілхлориді (30мл) нагрівали з дефлегмацією протягом 2 годин. Надлишок тіонілхлориду після цього видаляли in vacuo і проміжний хлорид кислоти розчиняли у дихлорометані (25мл). Після охолодження до 0°С розчин обробляли 0,5Μ аміаку в 1,4-діоксан (100мл), а потім давали нагрітися до кімнатної температури. Після 2 годин перемішування розчинник видаляли in vacuo і залишок розтирали з водою. Осад збирали, промивали водою і висушували in vacuo, одержуючи 5,0г (92%) метил 3-(алілокси)-(5-амінокарбоніл)бензоату у вигляді білої твердої речовини. Суспензію метил 3-(алілокси)-(5-амінокарбоніл)бензоату (5,0г, 21ммоль) у дихлорометані (70мл) при 0°С обробляли піридином (3,5мл, 43ммоль), а потім по краплях додавали трифторооцтовий ангідрид (3,6мл, 25ммоль). Реакційну суміш перемішували при 0°С протягом 20 хвилин, а потім перемішували до наступного дня за навколишньої температури. Реакційну суміш промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Хроматографія на силікагелі з використанням 10% етилацетату/гексанів давала 3,8г (81%) метил 3-(алілокси)-5-ціанобензоату у вигляді білої твердої речовини. Розчин метил 3-(алілокси)-5-ціанобензоату (1,5г, 6,9ммоль) у метанолі-тетрагідрофурані (1:2, 30мл) обробляли 0,5 N гідроксидом літію (17мл, 8,3ммоль). Реакційну суміш перемішували при 70°С протягом 30 хвилин, а потім розчинник видаляли in vacuo. Залишок розчиняли у невеликій кількості води, а потім підкислювали (рН ~4) шляхом додавання 2N хлористого водню. Осад збирали і висушували, одержуючи 1,0г (74%) 3-алілокси-5-ціанобензойної кислоти у вигляді білої твердої речовини. Суспензію 3-алілокси-5-(метоксикарбоніл)бензойної кислоти (5,5мг, 23ммоль) у тіонілхлориді (30мл) нагрівали з дефлегмацією протягом 2 годин. Надлишок тіонілхлориду після цього видаляли in vacuo і проміжний хлорид кислоти розчиняли у дихлорометані (25мл). Після охолодження до 0°С розчин обробляли 0,5 Μ аміаку у 1,4-діоксані (100мл), а потім давали нагрітися до кімнатної температури. Після 2 годин перемішування розчинник видаляли in vacuo і залишок розтирали з водою. Осад збирали, промивали водою і висушували in vacuo, одержуючи 5,0г (92%) метил 3-(алілокси)-(5-амінокарбоніл) бензоату у вигляді білої твердої речовини. Метанол (20мл) та дихлорометан (20мл) додавали у колбу з круглим дном, яка містила метил 3(алілокси)-(5-амінокарбоніл)бензоат (2,0г, 8,5ммоль) та паладій (10мас.% на активованому вугіллі, 200мг) в атмосфері аргону. Вміст колби відкачували, застосовуючи водний аспіратор, а потім заповнювали її воднем з балона. Балон, наповнений воднем, був під'єднаний до колби під час перемішування реакційної суміші протягом 2 годин. Паладій на вугіллі видаляли шляхом фільтрації крізь целіт. Розчинник видаляли, застосовуючи роторний випарник, а потім зразок висушували у вакуумі, одержуючи 2,0 (97%) метил 3(амінокарбоніл)-5-пропоксибензоату у вигляді білої твердої речовини. Суспензію метил 3-(амінокарбоніл)-5-пропоксибензоату (2,0г, 8,2ммоль) у дихлорометані (25мл) при 0°С обробляли піридином (1,3мл, 17ммоль), а потім по краплях додавали трифторооцтовий ангідрид (1,4мл, 9,9ммоль). Реакційну суміш перемішували при 0°С протягом 20 хвилин, а потім перемішували до наступного дня за навколишньої температури. Реакційну суміш промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Хроматографія на силікагелі з застосуванням 40% дихлорометану/гексанів давала 1,5г (84%) метил 3-ціано-5-пропоксибензоату у вигляді білої твердої речовини. Розчин метил 3-ціано-5-пропоксибензоату (1,5г, 6,8ммоль) у метанолі-тетрагідрофурані (1:2, 30мл) обробляли 0,5 Μ гідроксиду літію (16мл, 8,2ммоль). Реакційну суміш перемішували при 70°С протягом 30 хвилин, а потім розчинник видаляли in vacuo. Залишок розчиняли у невеликій кількості води, а потім підкислювали (рН — 4) шляхом додавання 2N хлористого водню. Осад збирали і висушували, одержуючи 1,2г (86%) 3-ціано-5-пропоксибензойної кислоти. Застосовуючи таку саму процедуру, як і для 3-алілокси-5-ціанобензойної кислоти, одержували 3-ціано5-нітробензойну кислоту (2,0г, 10,7ммоль) з моно-метил 5-нітроізофталату (5,0г, 22ммоль). Метил 3-ціано-2-нітробензоат (6,0г, 29ммоль) та хлорид дигідрат олова (II) (26г, 12ммоль) у метанолі (100мл) нагрівали з дефлегмацією протягом 3 годин. Реакційну суміш переносили в оснащену перемішувальним стержнем 1-літрову колбу Ерленмейєра, яка містила лід. При перемішуванні реакційної суміші додавали 1N гідроксид натрію до рН ~4-5. У цей момент додавали твердий бікарбонат натрію додавали до рН ~8. Суміш переносили до ділильної лійки і екстрагували етилацетатом. Органічний шар промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Хроматографія на силікагелі з застосуванням 30% етилацетату/гексанів давала 2,4г (47%) метил 3аміно-5-ціанобензоату у вигляді світло-бурої твердої речовини. Формальдегід, 37мас.% розчин у воді, (4,3мл, 57ммоль), твердий ціаноборогідрид натрію (752мг, 11ммоль), а потім оцтову кислоту (909мкл, 16ммоль) додавали до метил 3-аміно-5-ціанобензоату (500мг, 2,8ммоль) в ацетонітрилі (10мл). Після перемішування за навколишньої температури протягом 5 годин реакційну суміш переносили до ділильної лійки, а потім додавали етилацетат. Органічний шар промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Хроматографія на силікагелі з застосуванням 15% етилацетату/гексанів давала 390мг (55%) метил-3-ціано-5-диметиламінобензоату. Розчин метил-3-ціано-5-диметиламінобензоату (318мг, 1,6ммоль) у тетрагідрофурані (5мл) обробляли 0,5 N гідроксидом літію (3,7мл, 1,9ммоль). Реакційну суміш перемішували при 70°С протягом 30 хвилин, а потім розчинник видаляли in vacuo. Залишок розчиняли у невеликій кількості води, а потім підкислювали шляхом додавання по краплях 2N хлористого водню до припинення утворення білого осаду. Після екстрагування водного шару діетиловим етером органічний шар промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували, одержуючи 308мг (кількісно) 3-ціано-5-диметиламінобензойної кислоти. Суміш метил 3-алілокси-5-ціанобензоату (1,5г, 6,9ммоль) та йодиду тетрабутиламонію (2,8г, 7,6ммоль) у дихлорометані (38мл) при -78°С в атмосфері аргону обробляли розчином 1М трихлориду бору у дихлорометані (24мл, 24ммоль). Через 5 хвилин при -78°С реакційну суміш перемішували за навколишньої температури протягом 1 години. Реакцію після цього гасили льодяною водою і перемішували протягом додаткових 30 хвилин. Органічний шар промивали насиченим бікарбонатом натрію, висушували над безводним сульфатом натрію, фільтрували і концентрували. Хроматографія на силікагелі з застосуванням градієнта 20-30% етилацетату/гексанів давала 811мг (67%) метил 3-ціано-5-гідроксибензоату у вигляді світло-жовтої твердої речовини. Суміш метил 3-ціано-5-гідроксибензоату (302мг, 1,7ммоль), карбонату калію (471мг, 3,4ммоль) та 2хлороетилметилового етеру (309 (мклмг, 3,4ммоль) у N,N-диметилформаміді (4мл) нагрівали у герметично закритій посудині при 140°С протягом 15 хвилин. Реакційну суміш охолоджували, розводили етилацетатом, промивали водою та насиченим розсолом, фільтрували і концентрували. Фільтрація крізь силікагель з використанням дихлорометану давала 375мг (93%) метил 3-ціано-5-(2-метоксіетокси)бензоату. Розчин метил 3-ціано-5-(2-метоксіетокси)бензоату (375мг, 1,6ммоль) у тетрагідрофуран (4мл) обробляли 0,5N гідроксидом літію (3,8мл, 1,9ммоль). Реакційну суміш перемішували при 70°С протягом 30 хвилин, а потім розчинник видаляли in vacuo. Залишок розчиняли у невеликій кількості води, а потім підкислювали 2N хлористим воднем до рН ~2. Після екстрагування водного шару етилацетатом органічний шар промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували, одержуючи 343мг (97%) 3-ціано-5-(2-метоксіетокси)бензойної кислоти. Суміш метил 3-(бромометил)-5-йодобензоату (500мг, 1,4ммоль), карбонату калію (388мг, 2,8ммоль) та імідазолу (96мг, 1,4ммоль) у Ν,Ν-диметилформамід (4мл) нагрівали при 70°С протягом 3 годин. Після охолодження реакційну суміш розводили водою, а потім екстрагували дихлорометаном. Органічний шар промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Хроматографія на силікагелі з застосуванням 100% етилацетату давала 242мг (51%) метил 3-(імідазол-1-ілметил)-5-йодобензоату у вигляді білої твердої речовини. Після барботування аргоном розчину метил 3-(імідазол-1-ілметил)-5-йодобензоату (242мг, 0,70ммоль) у Ν,Ν-диметилформаміді (2мл) протягом 5 хвилин додавали ціанід цинку (90мг, 0,77ммоль) та тетракіс(трифенілфосфін)паладій(0) (80мг, 0,070ммоль). Реакційну суміш нагрівали при 80°С протягом 30 хвилин в атмосфері аргону. Після охолодження реакційну суміш розводили етилацетатом, а потім утворений осад видаляли шляхом фільтрації. Фільтрат промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Залишок розтирали з 20% діетиловим етером/гексанами, фільтрували і висушували in vacuo, одержуючи 150мг (89%) 3-ціано-5-(1Німідазол-1-іл-метил)бензоату у вигляді білої твердої речовини. Розчин 3-ціано-5-(1Н-імідазол-1-іл-метил)бензоату (150мг, 0,62ммоль) у тетрагідрофурані (2мл) обробляли 0,5 N гідроксидом літію (1,5мл, 0,75ммоль). Реакційну суміш перемішували при 70°С протягом 10 хвилин, а потім розчинник видаляли in vacuo. Залишок розчиняли у невеликій кількості води, а потім підкислювали (рН ~4) шляхом додавання 2N хлористим воднем. Осад збирали і висушували, одержуючи 140мг (кількісно) 3-ціано-5-(1Н-імідазол-1-іл-метил)бензойної кислоти. Суміш метил 3-(бромометил)-5-йодобензоату (400мг, 1,1ммоль) та карбонату калію (311мг, 2,3ммоль) у метанолі/тетрагідрофурані (5мN5мл) нагрівали при 55°С протягом 1 години. Після охолодження реакційну суміш розводили водою, а потім екстрагували етилацетатом. Органічний шар промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Після висушування in vacuo виділяли 325мг (94%) метил 3-(метоксиметил)-5-йодобензоату у вигляді білої твердої речовини. Після барботування аргоном розчину метил 3-(метоксиметил)-5-йодобензоату (316мг, 1,03ммоль) у Ν,Ν-диметилформаміді (3мл) протягом 5 хвилин додавали ціанід цинку (133мг, 1,13ммоль) та тетракіс(трифенілфосфін)паладій(0) (119мг, 0,010ммоль). Реакційну суміш нагрівали при 80°С протягом 15 хвилин в атмосфері аргону. Після охолодження реакційну суміш розводили етилацетатом, а потім утворений осад видаляли шляхом фільтрації. Фільтрат промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували. Хроматографія на силікагелі з застосуванням 10-30% етилацетату/гексанів давала 184мг (86%) метил 3-ціано-5-(метоксиметил)бензоату у вигляді безбарвної олії. Розчин метил 3-ціано-5-(метоксиметил)бензоату (184мг, 0,75ммоль) у тетрагідрофурані (2,1мл) обробляли 0,5N гідроксидом літію (1,8мл, 0,90ммоль). Реакційну суміш перемішували при 70°С протягом 30 хвилин, а потім після охолодження розчинник видаляли in vacuo. Залишок розчиняли у невеликій кількості води, а потім підкислювали 2 N хлористим воднем до рН ~2-3. Після екстрагування водного шару етилацетатом органічний шар промивали водою та насиченим розсолом, висушували над безводним сульфатом натрію, фільтрували і концентрували, одержуючи 145мг (кількісно) 3-ціано-5(метоксиметил)бензойної кислоти. Одержували розчин метил 3-ціано-5-гідроксибензоату (270мг, 1,5ммоль) та карбонату калію (482мг, 3,4ммоль) в ацетоні (5,5мл). До нього додавали етилйодид (272мкл, 3,4ммоль) і реакційну суміш нагрівали при 52°С протягом 2,5 години. Реакційну суміш концентрували, повторно розчиняли в етилацетаті і промивали водою та насиченим розсолом. Шар органічного розчинника збирали, висушували над безводним сульфатом натрію, фільтрували і концентрували. 321мг (99%) метил 3-ціано-5-етоксибензоату виділяли у вигляді світло-бурої твердої речовини. Метил 3-ціано-5-етоксибензоат (312мг, 1,5ммоль) гідролізували, як було описано вище, одержуючи 290мг (кількісно) 3-ціано-5-етоксибензойної кислоти у вигляді білуватої твердої речовини. Одержували розчин метил 3-ціано-5-гідроксибензоату (200мг, 1,3ммоль) та карбонату калію (357мг, 2,6ммоль) в ацетоні (4,0мл). До нього додавали пропілйодид (245мкл, 2,5ммоль) і реакційну суміш нагрівали при 50°С протягом 5 годин. Реакційну суміш концентрували, повторно розчиняли в етилацетаті і промивали водою та насиченим розсолом. Шар органічного розчинника збирали, висушували над безводним сульфатом натрію, фільтрували і концентрували. 222мг (90%) метил 3-ціано-5пропоксибензоату виділяли у вигляді світло-бурої твердої речовини. Метил 3-ціано-5-пропоксибензоат (222мг, 1,0ммоль) гідролізували, як було описано вище, одержуючи 169мг (80%) 3-ціано-5-пропоксибензойної кислоти у вигляді білуватої твердої речовини. Одержували розчин метил 3-ціано-5-гідроксибензоату (170мг, 0,95ммоль) та карбонату калію (304мг, 2,2ммоль) в ацетоні (4,0мл). До нього додавали 1-бромогексан (300мкл, 2,1ммоль) і реакційну суміш нагрівали при 50°С до наступного дня. Реакційну суміш концентрували, повторно розчиняли в етилацетаті і промивали водою та насиченим розсолом. Шар органічного розчинника збирали, висушували над безводним сульфатом натрію, фільтрували і концентрували. Розтирання з гексанами забезпечувало 250мг (кількісно) метил 3-ціано-5-гексилоксибензоату у вигляді світло-бурої твердої речовини. Метил 3-ціано-5-етоксибензоат (250мг, 0,95ммоль) гідролізували, як було описано вище, одержуючи 247мг (кількісно) 3-ціано-5-гексилоксибензойної кислоти у вигляді білуватої твердої речовини. До розчину 4-аміно-3-трифторометоксибензойної кислоти (5г, 22,6ммоль) в оцтовій кислоті (50мл) при кімнатній температурі по краплях додавали бром (3,98г, 24,9ммоль) в оцтовій кислоті (10мл). Після перемішування реакційної суміші протягом однієї години у суміш додавали воду. Тверду фазу фільтрували і промивали водою для одержання 4-аміно-3-бромо-5-трифторометоксибензойної кислоти (3,9г, 54,9%). 4-аміно-3-бромо-5-трифторометоксибензойну кислоту (1,5г, 5ммоль) змішували з етанолом (15мл) при 0°С, а потім додавали концентровану сірчану кислоту (2,26г, 10,2ммоль). По краплях додавали водний розчин (1,2мл) нітриту натрію (0,38г, 5,5ммоль) при 0°С протягом 1 години. Після нагрівання реакційної суміші до кімнатної температури з наступним нагріванням до дефлегмації протягом 45 хвилин додавали воду. Суміш екстрагували дихлорометаном. Шар дихлорометану висушували і концентрували. Залишок розчиняли в 1 Μ гідроксиду натрію і екстрагували етером. Водний розчин підкислювали 2М НСІ до рН=2 для одержання 3-бромо-5-трифторометоксибензойної кислоти (1,08г, 75,5%). До розчину в етері 3-бромо-5-трифторометоксибензойної кислоти (1,08г, 3,79ммоль) додавали триметилсилілметилазид і перемішували при кімнатній температурі протягом 10 хвилин. Реакцію гасили метанолом і пропускали через колонку з 2% етилацетатом у гексанах для одержання безбарвної олії (0,84г). Цю безбарвну олію змішували з ціанідом цинку (0,33г, 2,8ммоль) та тетракіс(трифенілфосфін)паладіем(0) (Pd(PPh3)4, 467мг, 0,404ммоль) у Ν,Ν-диметилформаміді (10мл) в атмосфері аргону при 85°С до наступного дня. Реакційну суміш розводили дихлорометаном і двічі промивали водою. Шар дихлорометану висушували і концентрували. Залишок змішували з 1М гідроксиду натрію (8мл) та метанолом (4мл) і перемішували при кімнатній температурі протягом 2 годин. Суміш підкислювали 1М НСІ до рН=1~2 і екстрагували етилацетатом. Шар етилацетату промивали розсолом і концентрували. Залишок пропускали через колонку з 2% етилацетатом у гексанах для одержання 3-ціано-5трифторометоксибензойної кислоти, яка містила 3-[іміно(метокси)метил]-5-трифторометоксибензойну кислоту (3:1, 145мг, 16,6%) Розчин 3-бромо-5-фторобензойної кислоти (2,00г, 9,13ммоль) у тіонілхлориді (16мл) та диметилформаміді (0,4мл) перемішували при 80°С протягом 1год. Реакційну суміш концентрували in vacuo, залишок розчиняли в метанолі (15мл) і залишали перемішуватися при кімнатній температурі до наступного дня. Реакційну суміш концентрували in vacuo і залишок розчиняли в етилацетаті (50мл). Органічну фазу послідовно промивали водою (50мл), насиченим бікарбонатом натрію (50мл, водний), водою (50мл) та розсолом (50мл), висушували (сульфат натрію), фільтрували і концентрували in vacuo для забезпечення названої сполуки (1,97г, 92%) у вигляді жовтої олії. До розчину метил-3-бромо-5-фторобензоату (1,97г, 8,44ммоль) у толуолі (40мл) послідовно додавали 1,3-пропандіоловий естер піридин-3-борної кислоти (1,79г, 7,63ммоль), карбонат калію (11,66г, 84,4ммоль) та тетракіс(трифенілфосфін) паладій (0) (0,49, 0,42ммоль). Одержану в результаті бурувато-жовту реакційну суміш нагрівали при 120°С в атмосфері аргону до наступного дня. Реакційну суміш охолоджували до кімнатної температури, фільтрували крізь пробку целіту і концентрували in vacuo. Залишок очищали на силікагелі, застосовуючи 1% метанол у дихлорометані для виділення названої сполуки (0,92г, 47%) у вигляді жовтої твердої речовини. У 100мл колбу з круглим дном, оснащену перемішувальним стержнем, додавали метил-3-фторо-5-(3піридил)бензоат (0,92г, 3,93ммоль), метанол (10мл) та гідроксид натрію (5,89мл, 5,89ммоль, 1N водний). Одержану в результаті суміш перемішували при 50°С протягом 2год. Реакційну суміш охолоджували до кімнатної температури, концентрували in vacuo і залишок розчиняли у метанолі (20мл). До цієї суміші по краплях додавали соляну кислоту (1N діетиловий етер) і перемішували при кімнатній температурі протягом 10хв. Реакційну суміш концентрували in vacuo і залишок розтирали з діетиловим етером для забезпечення неочищеного гідрохлориду названої сполуки (1,00г) у вигляді білуватої твердої речовини. Розчин 3-бромо-5-йодобензойної кислоти (5,00г, 15,3ммоль) у метанолі (30мл) та соляну кислоту (15,3мл, 15,3ммоль, 1N діетиловий етер) перемішували при кімнатній температурі протягом 48год. Реакційну суміш концентрували in vacuo і залишок розводили дихлорометаном (100мл). Органічну фазу послідовно промивали гідроксидом натрію (100мл, 1N водний), водою (100мл) та розсолом (100мл), висушували (сульфат натрію), фільтрували і концентрували in vacuo. Неочищений залишок розчиняли в 10% етилацетаті у гексанах (100мл) і фільтрували крізь шар силікагелю. Після концентрування in vacuo виділяли метиловий естер (4,98г, 95%) у вигляді білувато-жовтої твердої речовини. До розчину метил-3-бромо-5-йодобензоату (2,00г, 5,87ммоль) у толуолі (50мл) послідовно додавали 1,3-пропандіоловий естер піридин-3-борної кислоти (1,24г, 7,63ммоль), карбонат калію (8,11г, 58,7ммоль) та тетракіс(трифенілфосфін)паладій (0) (0,34, 0,29ммоль). Одержану в результаті бурувато-жовту реакційну суміш нагрівали при 80°С в атмосфері аргону протягом 10год. Реакційну суміш охолоджували до кімнатної температури, фільтрували крізь пробку целіту і концентрували in vacuo. Залишок очищали на силікагелі, застосовуючи 3% метанол у дихлорометані, для виділення метил-3-бромо-5-(3-піридил)бензоату (1,16г, 67%) у вигляді білої твердої речовини. У 100-мл колбу з круглим дном, оснащену перемішувальним стержнем, додавали метил-3-бромо-5-(3піридил)бензоат (1,16г, 3,96ммоль), метанол (15мл) та гідроксид натрію (5,94мл, 5,94ммоль, 1N водний). Одержану в результаті суміш перемішували при 50°С протягом 2год. Реакційну суміш охолоджували до кімнатної температури, концентрували in vacuo і залишок розчиняли в метанолі (20мл). До цієї суміші по краплях додавали соляну кислоту (1N діетиловий етер) і перемішували при кімнатній температурі протягом 10хв. Реакційну суміш концентрували in vacuo і залишок розтирали з діетиловим етером для забезпечення неочищеного гідрохлориду названої сполуки (1,50г) у вигляді білої твердої речовини. У 250-мл колбу з круглим дном, оснащену перемішувальним стержнем, додавали 3,5дифторобензонітрил (4,2г, 30,4ммоль), метоксид натрію (10,4мл, 45,6ммоль, 25% метанол) та диметилформамід (40мл). Одержану в результаті реакційну суміш перемішували при кімнатній температурі до наступного дня. Реакційну суміш концентрували in vacuo і залишок розчиняли в дихлорометані (200мл). Органічну фазу послідовно промивали водою (150мл) та розсолом (150мл), висушували (сульфат натрію) і концентрували in vacuo. Неочищений залишок очищали на силікагелі, застосовуючи 10% діетиловий етер у гексанах для виділення 3-фторо-5-метоксибензонітрилу (1,63г) у вигляді білої твердої речовини. У 50-мл колбу з круглим дном, оснащену перемішувальним стержнем та дефлегматором, додавали 3фторо-5-метоксибензонітрил (0,62г, 4,10ммоль), метанол (6,2мл) та гідроксид натрію (6,2мл, 6N водний). Одержану в результаті реакційну суміш перемішували при 100°С до наступного дня. Реакційну суміш охолоджували до кімнатної температури і концентрували in vacuo. Залишок розводили дихлорометаном (100мл) в підкислювали, застосовуючи соляну кислоту (1N водна). Органічну фазу відокремлювали, послідовно промивали водою (100мл) та розсолом (100мл), висушували (сульфат натрію) і концентрували in vacuo для одержання названої сполуки (0,64г, 91%) у вигляді білої твердої речовини. 3-бромо-5-тіометилбензойну кислоту (519,6мг, 2,1ммоль) розчиняли в діетиловому етері (20мл). До розчину бензойної кислоти додавали діазометан у діетиловому етері, доки суміш не переставала виділяти газ і не набувала стійкого жовтого кольору. До цього розчину по краплях додавали льодяну оцтову кислоту до зникнення жовтого кольору. Реакційну суміш промивали насиченим бікарбонатом натрію та розсолом, висушували над безводним сульфатом натрію і розчинник видаляли in vacuo для одержання 537мг (98%) 3бромо-5-тіометилового естеру у вигляді безбарвної олії. 3-бромо-5-тіометиловмй естер (536мг, 2,05ммоль) розчиняли в безводному Ν,Ν-диметилформаміді (5мл) в атмосфері аргону. До реакційної суміші додавали ціанід цинку (42мг, 0,36ммоль) та тетракіс (трифенілфосфін) паладій(О) (41мг, 0,356ммоль) і перемішували її при 80°С протягом 10 годин. Після охолодження до кімнатної температури реакційну суміш розводили водою і екстрагували етилацетатом. Органічні екстракти промивали розсолом, висушували над безводним сульфатом натрію і розчинник видаляли in vacuo. Сполуку очищали шляхом колонкової хроматографії на силікагелі для одержання 356мг (85%) 3-ціано-5-тіометилового естеру у вигляді білої твердої речовини. 3-ціано-5-тіометиловий естер (360мг, 1,74 моль) розчиняли в безводному тетрагідрофурані (22мл) і додавали 21,6мл водного розчину гідроксиду літію (0,5М) та 11мл метанолу. Реакційну суміш піддавали дефлегмації протягом 45 хвилин. Реакційну суміш охолоджували і розчинник видаляли in vacuo. Суміш розводили водою і промивали етилацетатом. Водний шар після цього підкислювали до рН 1 за допомогою НСІ (1М) і екстрагували етилацетатом. Органічні екстракти комбінували, висушували над безводним сульфатом натрію і розчинник видаляли in vacuo для одержання 330мг (98%) 3-ціано-5-тіометилбензойної кислоти у вигляді білої твердої речовини. Розчин 3,5-дифторобромобензолу (1,0г, 5,18ммоль) у Ν,Ν-диметилформаміді (15мл) охолоджували до 0°С і додавали тіометоксид натрію (363мг, 5,18ммоль). Реакційну суміш перемішували протягом 30 хвилин, перш, ніж суміш розводили водою і екстрагували гексанами. Органічні екстракти промивали розсолом і висушували над безводним сульфатом натрію. Розчинник видаляли in vacuo і продукт елюювали крізь SPEтрубку (10г) з гексанами для одержання 618мг (54%) безбарвної олії. 5-фторо-3-тіометилбромобензол (618мг, 2,80ммоль) розчиняли в Ν,Ν-диметилформаміді (6мл) і до розчину додавали ціанід цинку (329мг, 2,80ммоль) та тетракіс (трифенілфосфін)паладій (0) (324мг, 0,28ммоль). Реакційну суміш перемішували при 80°С протягом 12 годин. Реакційну суміш охолоджували до кімнатної температури, розводили водою і екстрагували етилацетатом, промивали розсолом, висушували над безводним сульфатом натрію і розчинник видаляли in vacuo. Хроматографія на силікагелі з застосуванням 5% етилацетату у гексанах давала 430мг (92%) білої твердої речовини, 5-фторо-3ціанотіометилбензолу (430мг, 2,57ммоль), який розчиняли у воді (6,0мл) та водному розчині гідроксиду натрію (6М, 6,0мл) і піддавали дефлегмації протягом 12 годин. Реакційну суміш підкислювали до рН 3 і екстрагували етилацетатом. Органічні екстракти промивали розсолом, висушували над безводним сульфатом натрію і розчинник видаляли in vacuo для одержання 470мг (98%) названої сполуки у вигляді білої твердої речовини. Розчин 3,5-дифторобромобензолу (1,0г, 5,18ммоль) у Ν,Ν-диметилформаміді (15мл) охолоджували до 0°С і додавали тіоетоксид натрію (436мг, 5,18ммоль). Реакційну суміш перемішували протягом 30 хвилин, перш, ніж суміш розводили водою і екстрагували гексанами. Органічні екстракти промивали розсолом і висушували над безводним сульфатом натрію. Розчинник видаляли in vacuo і продукт елюювали крізь SPEтрубку (10г) з гексанами для одержання 366мг (30%) безбарвної олії, До розчину додавали 5-фторо-3-тіоетилбромобензол (365мг, 1,55ммоль), який розчиняли в N,Nдиметилформаміді (5мл), та ціанід цинку (182мг, 1,55ммоль) і тетракіс (трифенілфосфін)паладій (0) (179мг, 0,16ммоль). Реакційну суміш перемішували при 80°С протягом 3 годин. Реакційну суміш охолоджували до кімнатної температури, розводили водою і екстрагували етилацетатом, промивали розсолом, висушували над безводним сульфатом натрію і розчинник видаляли in vacuo. Хроматографія на силікагелі з застосуванням 5% етилацетату у гексанах давала 241мг (86%) білої твердої речовини, 5-фторо-3ціанотіоетилбензолу (240мг, 1,32ммоль), який розчиняли у воді (3,0мл) та водному розчині гідроксиду натрію (6М, 3,0мл) і піддавали дефлегмації протягом 12 годин. Реакційну суміш підкислювали до рН 3 і екстрагували етилацетатом. Органічні екстракти промивали розсолом, висушували над безводним сульфатом натрію і розчинник видаляли in vacuo для одержання 274мг (103%) білуватої твердої речовини. 3,5-диметоксибензонітрил (228мг, 1,4ммоль) та 5М гідроксиламінгідрохлорид (0,336мл, 1,68ммоль) в етанолі (2мл) та 1N гідроксиді натрію (1,68мл, 1,68ммоль) нагрівали з дефлегмацією до наступного дня. Стандартна обробка давала 250мг (91%) 3,5-диметоксифеніламідоксиму. 3-фторо-5-(1Н-імідазол-1-іл)бензонітрил (950мг, 5,08ммоль) та 5М гідроксиламінгідрохлорид (1,02мл, 5,08ммоль) в етанолі (5мл) та 1N гідроксиді натрію (5,08мл, 5,08ммоль) нагрівали з дефлегмацією протягом 1 години та 20 хвилин. Стандартна обробка давала 901мг (81,4%) 3-бромо-5-фторофеніламідоксиму. Моногідрат хелідонової кислоти (2,01г, 10ммоль) змішували з 1 Μ НСІ (20мл, 20ммоль, етер) в етанолі (50мл) і нагрівали при 85°С протягом 24 годин. Розчинник видаляли in vacuo і змішували з етилацетатом та водою. Шар етилацетату висушували і концентрували. Залишок розтирали з гексанами та етером для одержання 1,6г (66%) діетил 4-гідрокси-2,6-піридиндикарбоксилату. До суспензії 60% гідриду натрію (0,351г, 8,77ммоль) у диметилформаміді (7,5мл) в атмосфері аргону при кімнатній температурі по краплях додавали розчин діетил 4-гідрокси-2,6-піридиндикарбоксилату (1,4г, 5,85ммоль) у диметилформаміді (9мл) і реакційну суміш перемішували протягом 5 хвилин. Додавали йодометан (1,245г, 8,77ммоль) і реакційну суміш перемішували при кімнатній температурі протягом 20 годин. Суміш виливали у воду і екстрагували дихлорометаном. Шар дихлорометану висушували, концентрували для одержання 1,45г (97,8%) діетил 4-метокси-2,6-піридиндикарбоксилату. Діетил 4-метокси-2,6-піридиндикарбоксилат (1,45г, 5,73ммоль) перемішували з концентрованимамонієм (40мл) при кімнатній температурі протягом 10 хвилин. Осад фільтрували для одержання 0,93г (83%) 4-метоксипіридин-2,6-дикарбоксаміду. 4-метоксипіридин-2,6-дикарбоксамід (900мг, 4,6ммоль) змішували з трифторооцтовим ангідридом (2,32г, 11,1ммоль) та піридином (1,6г, 20,2ммоль) у дихлорометані (20мл) і перемішували до наступного дня при кімнатній температурі. Реакційну суміш розводили дихлорометаном і промивали водою. Стандартна обробка давала 461мг 2,6-диціано-4-метоксипіридину. 2,6-диціано-4-метоксипіридин (460мг, 2,89ммоль) та 5М гідроксиламінгідрохлориду (0,578мл, 2,89ммоль) в етанолі (3мл) і 1N гідроксид натрію (2,89мл, 2,89ммоль) перемішували при кімнатній температурі до наступного дня. Стандартна обробка давала 180мг
ДивитисяДодаткова інформація
Назва патенту англійськоюSubstituted pyridines and use thereof as antagonists at metabotropic glutamate receptors
Автори англійськоюSLASSI ABDELMALIK, Stormann Thomas M., MCLEOD DONALD A
Назва патенту російськоюЗамещенные пиридины и их применение как антагонистов рецептора метаботропного глутамата
Автори російськоюСлесси Абделмалик, Сторманн Томас М., Маклиод Дональд А.
МПК / Мітки
МПК: C07D 417/04, C07D 521/00, C07D 413/04, A61P 25/00, A61K 31/443, A61K 31/444, A61P 25/14, A61P 25/18, A61P 25/08, C07D 213/75, A61P 25/22, A61P 25/16, C07D 213/55, C07D 401/04, C07D 213/78, A61P 25/04, A61P 25/28, C07D 405/04, C07D 213/79, C07D 405/14, A61K 31/4439, A61P 25/06, C07D 413/14
Мітки: антагоністів, заміщені, глутамату, піридини, рецептору, застосування, метаботропного
Код посилання
<a href="https://ua.patents.su/100-74419-zamishheni-piridini-ta-kh-zastosuvannya-yak-antagonistiv-receptoru-metabotropnogo-glutamatu.html" target="_blank" rel="follow" title="База патентів України">Заміщені піридини та їх застосування як антагоністів рецептору метаботропного глутамату</a>
Заміщені піридини як гербіциди і проміжні сполуки

Номер патенту: 74191
Опубліковано: 15.11.2005
Автори: Едмундс Ендрю, де Месмаекер Алайн, Люті, Крістоф, Шатцер Юрген
МПК: C07D 413/06, C07D 401/06, A01P 13/00, C07D 409/06, A01N 43/40, C07D 417/06, C07D 213/50, C07D 405/06
Мітки: проміжні, гербіциди, піридини, сполуки, заміщені
Формула / Реферат:
1. Сполука формули І ,(І)у якійр означає 0 або 1,R1 означає С1-С6алкіленовий, С3-С6алкеніленовий або С3-С6алкініленовий ланцюг, який може бути моно- або полізаміщений галогеном або групою R5, при цьому ненасичені зв'язки ланцюга не приєднані безпосередньо до замісника X1, за умови, що R1 не означає метилен, якщо R2 являє собою незаміщений...
Заміщені піридини, фармацевтична композиція на їх основі та спосіб лікування опосередкованих циклооксигеназою захворювань

Номер патенту: 62935
Опубліковано: 15.01.2004
Автори: Фресен Річард, Фортін Ріджен, Вонг Заойін, Дюб Деніел, Готьє Жак Ів
МПК: A61K 31/00, A61K 31/444, A61P 3/10, C07D 213/82, A61P 17/02, C07D 213/85, A61P 21/00, A61P 19/10, A61P 7/00, A61P 27/02, A61P 1/04, A61P 17/00, A61P 11/06, A61P 19/06, C07D 213/80, C07D 213/64, A61P 29/00, A61P 43/00, A61P 35/04, A61P 25/28, A61K 31/4418, A61P 1/00, C07D 213/89, C07D 213/61, A61P 19/00, C07D 213/34, A61P 27/06, A61P 25/04, A61P 15/06, A61P 13/12, A61P 31/12, A61P 15/00, A61P 19/02, A61K 31/44, C07D 213/65, A61P 15/12, A61P 35/00, C07D 213/75
Мітки: основі, захворювань, фармацевтична, спосіб, композиція, циклооксигеназою, лікування, заміщені, піридини, опосередкованих
Формула / Реферат:
1. Заміщені піридини формули Iабо їх фармацевтично прийнятна сіль, де:R1 вибраний з групи, що включає(a) CH3,(b) NH2,(c) NHC(О)CF3, і(d) NНСН3;Ar являє собою моно-, ди- або три-заміщений феніл або піридиніл (або його N-оксид), де замісники вибрані з групи, що включає(a) водень,(b) галоген,(c) C1-6алкокси,(d) C1-6алкілтіо,(e) CN,(f)...
Спосіб пригнічення росту небажаної рослинності, заміщені гетероциклами піридини, cпособи їх одержання та гербіцидна композиція

Номер патенту: 58478
Опубліковано: 15.08.2003
Автор: Клееман Аксель
МПК: A01N 43/56, C07D 401/12, A01N 43/40, C07D 213/69, A01P 13/00
Мітки: гетероциклами, росту, небажаної, композиція, гербіцидна, cпособи, рослинності, пригнічення, спосіб, одержання, заміщені, піридини
Формула / Реферат:
1. Способ подавления роста нежелательных растений в очаге поражения путем обработки очага поражения эффективным количеством 2,6-замещенного пиридина, отличающийся тем, что в качестве 2,6-замещенного пиридина используют соединение формулы I:, (І)где Х обозначает атом кислорода;А обозначает необязательно замещенную атомом галогена, С1-6-алкилом или...
Застосування 1-аміноалкілциклогексанів як антагоністів 5-нт3 і нейрональних нікотинових рецепторів

Номер патенту: 74194
Опубліковано: 15.11.2005
Автори: Каусс Валєрьянс, Даниш Войцех, Гольд Маркус, Калвіньш Іварс, Парсонс Крістофер Грахам Рафаель, Йіргенсонс Айгарс
МПК: A61P 43/00, A61P 25/28, C07C 211/35, A61P 25/22, A61P 3/00, A61P 1/08, A61P 25/30, A61P 25/24, A61K 31/40, A61P 25/18, C07D 295/02, A61P 25/04, A61P 29/00, A61P 25/32, A61P 25/16, A61K 31/13
Мітки: нейрональних, 5-нт3, застосування, антагоністів, нікотинових, рецепторів, 1-аміноалкілциклогексанів
Формула / Реферат:
1. Спосіб лікування тварин, які потребують інгібування розвитку або полегшення стану за допомогою антагоніста 5-НТ3 або нейрональних нікотинових рецепторів, що включає введення вказаній тварині ефективної кількості сполуки 1-аміноалкілциклогексану формули:,де R* означає -(CH2)n-(CR6R7)m-NR8R9;m+n = 0, 1 або 2;R1-R7 незалежно вибрані з водню і С1-6 алкілу;R8 і R9 незалежно вибрані з водню і С1-6 алкілу або...
Заміщені 2-тіо-3,5-диціано-4-арил-6-амінопіридини та їх застосування

Номер патенту: 73957
Опубліковано: 17.10.2005
Автори: Дембовскі Клаус, Хюбш Вальтер, Кран Томас, Баузер Маркус, Розентретер Ульріх, Ваупель Андреа, Кремер Томас, Шташ Йоханнес-Петер, Хеннінг Рольф, Перцборн Елізабет, Зальхер-Шрауфштеттер Ольга
МПК: A61K 31/4418, A61K 31/4709, A61K 31/4433, A61P 1/16, A61P 25/04, A61K 31/4439, A61P 15/00, A61K 31/4436, A61P 11/00, A61K 31/496, C07D 413/12, C07D 401/12, A61K 31/5377, A61P 29/00, A61K 31/4545, C07D 409/12, A61K 31/444, C07D 405/12, C07D 417/12, C07D 213/85, A61P 9/10, A61P 43/00, A61P 3/10, A61P 35/00, A61P 13/00, A61K 31/443, A61P 25/00
Мітки: 2-тіо-3,5-диціано-4-арил-6-амінопіридини, заміщені, застосування
Формула / Реферат:
1. Похідні 4-арил-6-амінопіридину загальної формули (І): , (I)в якійR1, R2, R3 є однаковими або різними і незалежно вибрані з групи наступних значень:водень;гідрокси;необов'язково, заміщений алкіл з 1-8 атомами вуглецю;необов'язково, заміщений арил з 6-10 атомами вуглецю;необов'язково, заміщений алкокси з 1-8 атомами вуглецю;-O-(СН2)n-СН=СН2, де n = 0, 1 або...
Попередній патент: Фунгіцидна суміш, спосіб боротьби з фітопатогенними грибами та фунгіцидний засіб
Наступний патент: Спосіб дегазації рідини
Випадковий патент: Система розподілу ресурсів при формуванні команд проекту