Фізична форма (r)-7-ацетил-5-(4-амінофеніл)-8, 9-дигідро-8-метил-7н-1, 3-діоксоло[4, 5-h]-[2, 3]-бензодіазепіну, спосіб її отримання та фармацевтична композиція
Номер патенту: 45309
Опубліковано: 15.04.2002
Автори: Змієвскі Мілтон Джозеф, мол., Андерсон Бенджамін Алан, Віченці Джеффрі Томас, Харкнесс Аллен Роберт, Хансен Марвін Мартін, Вері Девід Лі



Формула / Реферат
1. Физическая форма (R)-7-ацетил-5-(4-аминофенил)-8,9-дигидро-8-метил-7Н-1,3-диоксоло[4,5-h][2,3]бензодиазепина, имеющая порошковую рентгенограмму с d-параметрами 10,61, 8,83, 6,78, 5,83, 4,13 и 3,74 .
2 Физическая форма (R)-7-ацетил-5-(4-аминофенил)-8,9-дигидро-8-метил-7Н-1,3-диоксоло[4,5-h][2,3]бензодиазепина, имеющая порошковую рентгенограмму с dl-параметрами 10,61, 8,83, 6,78, 5,83, 4,13 и 3,74 , которая может быть использована для изготовления медикаментов, применимых в качестве антагониста рецептора АМРА.
3. Способ получения физической формы (R)-7-ацетил-5-(4-аминофенил)-8,9-дигидро-8-метил-7Н-1,3-диоксоло[4,5-h][2,3]бензодиазепина, охарактеризованной в п. 1, который включает
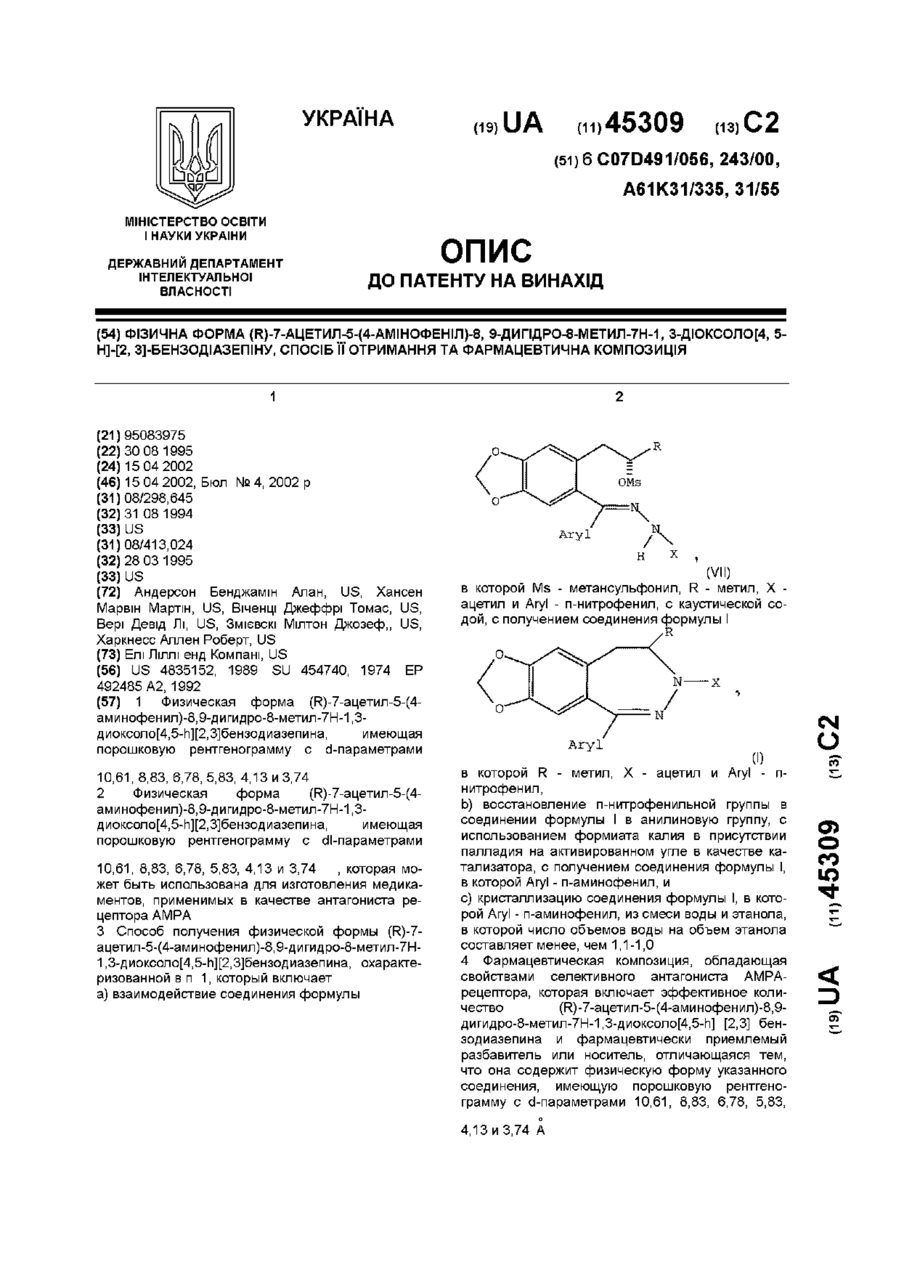
а) взаимодействие соединения формулы
(VII)
в которой Ms - метансульфонил, R - метил, Х - ацетил и Aryl - п-нитрофенил, с каустической содой, с получением соединения формулы I:
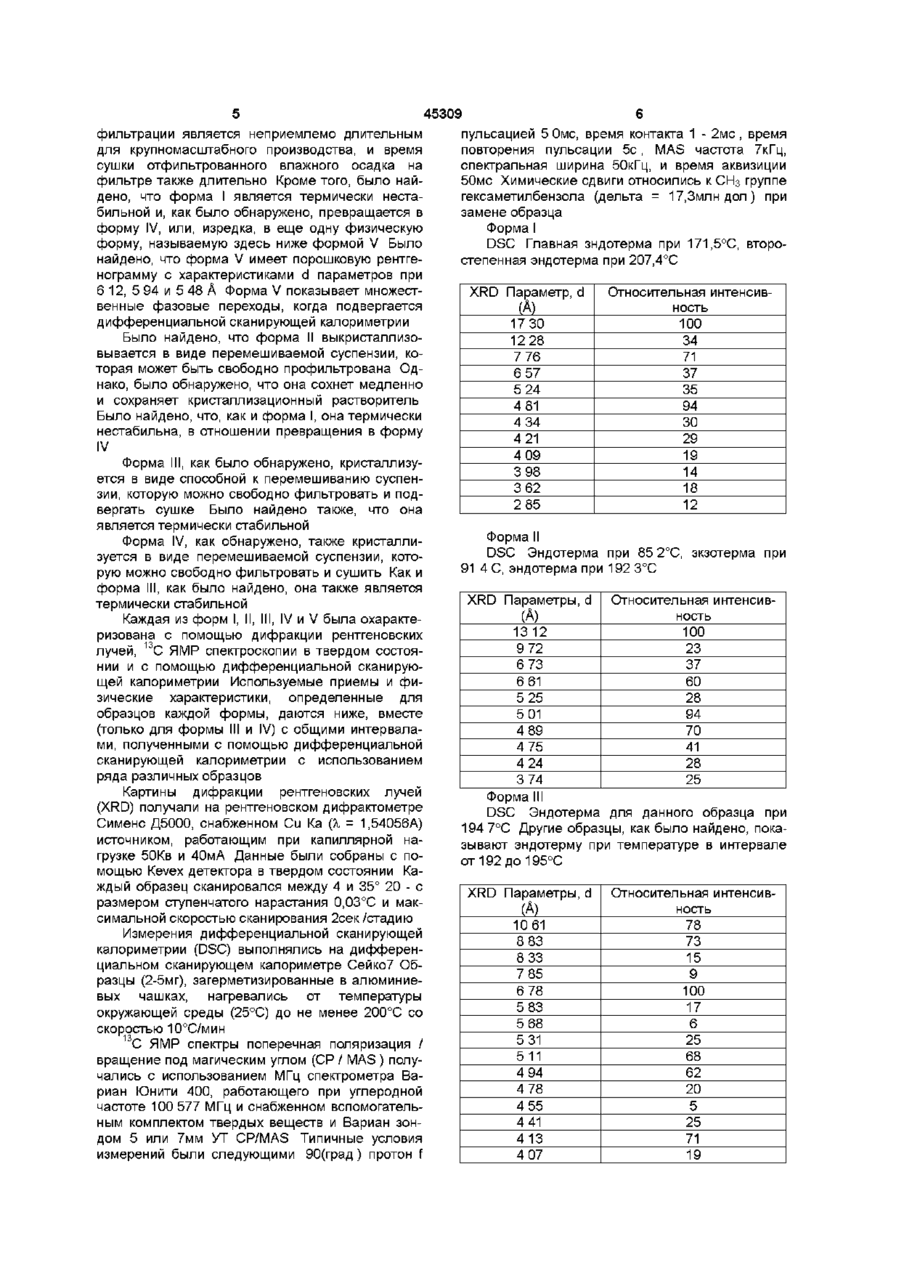
(I)
в которой R - метил, Х - ацетил и Aryl - п-нитрофенил,
b) восстановление п-нитрофенильной группы в соединении формулы I в анилиновую группу, с использованием формиата калия в присутствии палладия на активированном угле в качестве катализатора, с получением соединения формулы І, в которой Aryl - п-аминофенил, и
с) кристаллизацию соединения формулы I, в которой Aryl - п-аминофенил, из смеси воды и этанола, в которой число объемов воды на объем этанола составляет менее, чем 1,1-1,0.
4. Фармацевтическая композиция, обладающая свойствами селективного антагониста АМРА-рецептора, которая включает эффективное количество (R)-7-ацетил-5-(4-аминофенил)-8,9-дигидро-8-метил-7Н-1,3-диоксоло[4,5-h] [2,3] бензодиазепина и фармацевтически приемлемый разбавитель или носитель, отличающаяся тем, что она содержит физическую форму указанного соединения, имеющую порошковую рентгенограмму с d-параметрами 10,61, 8,83, 6,78, 5,83, 4,13 и 3,74 .
Текст
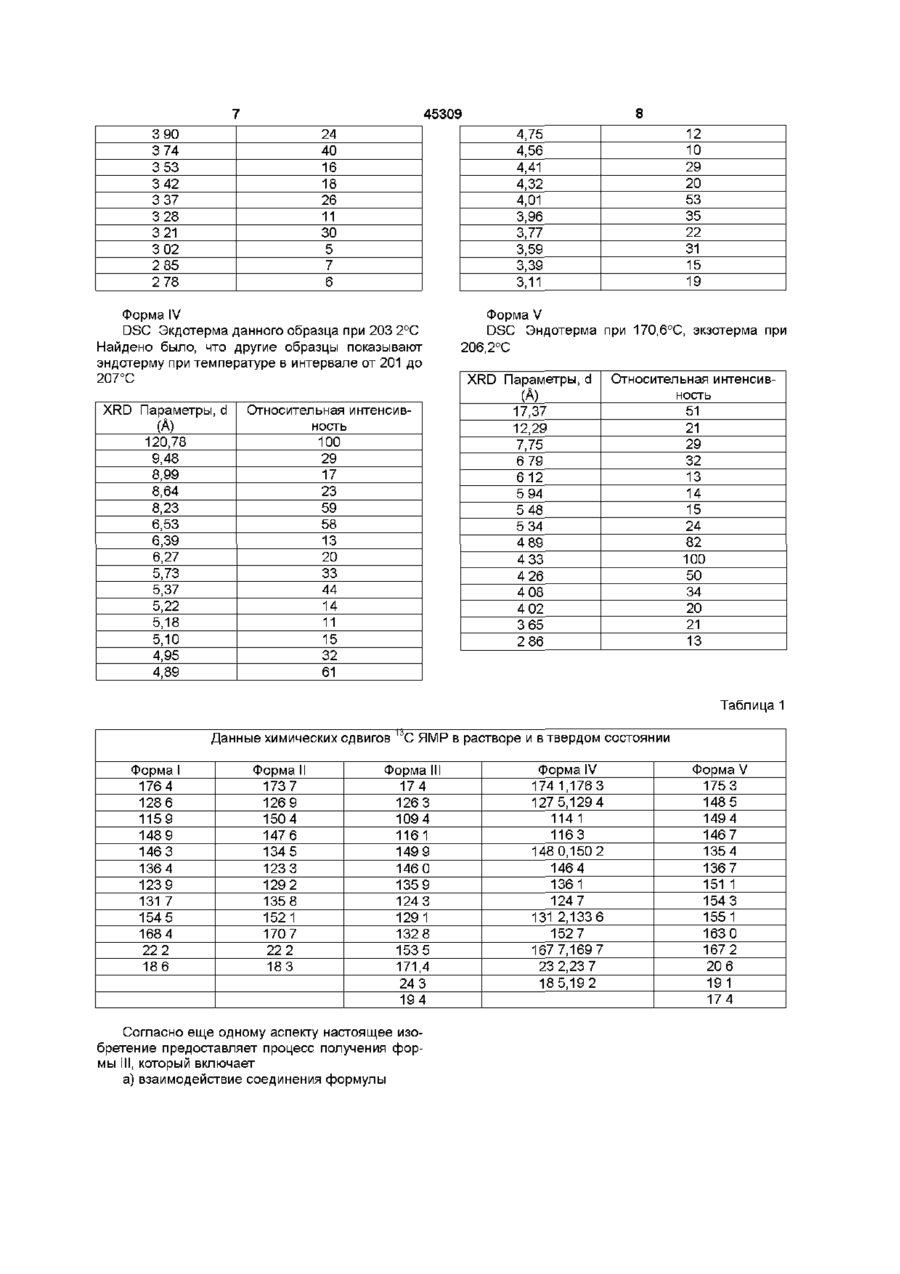
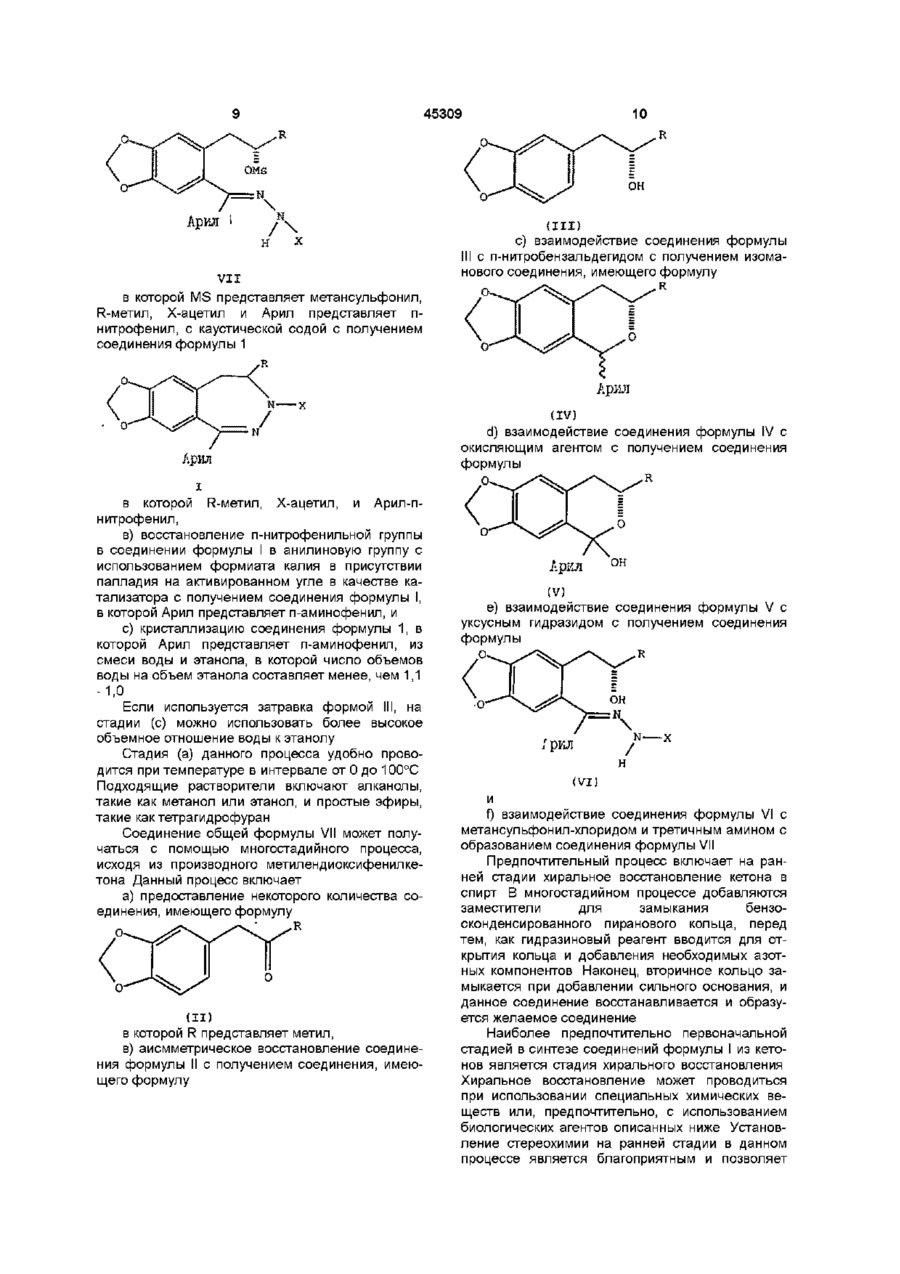
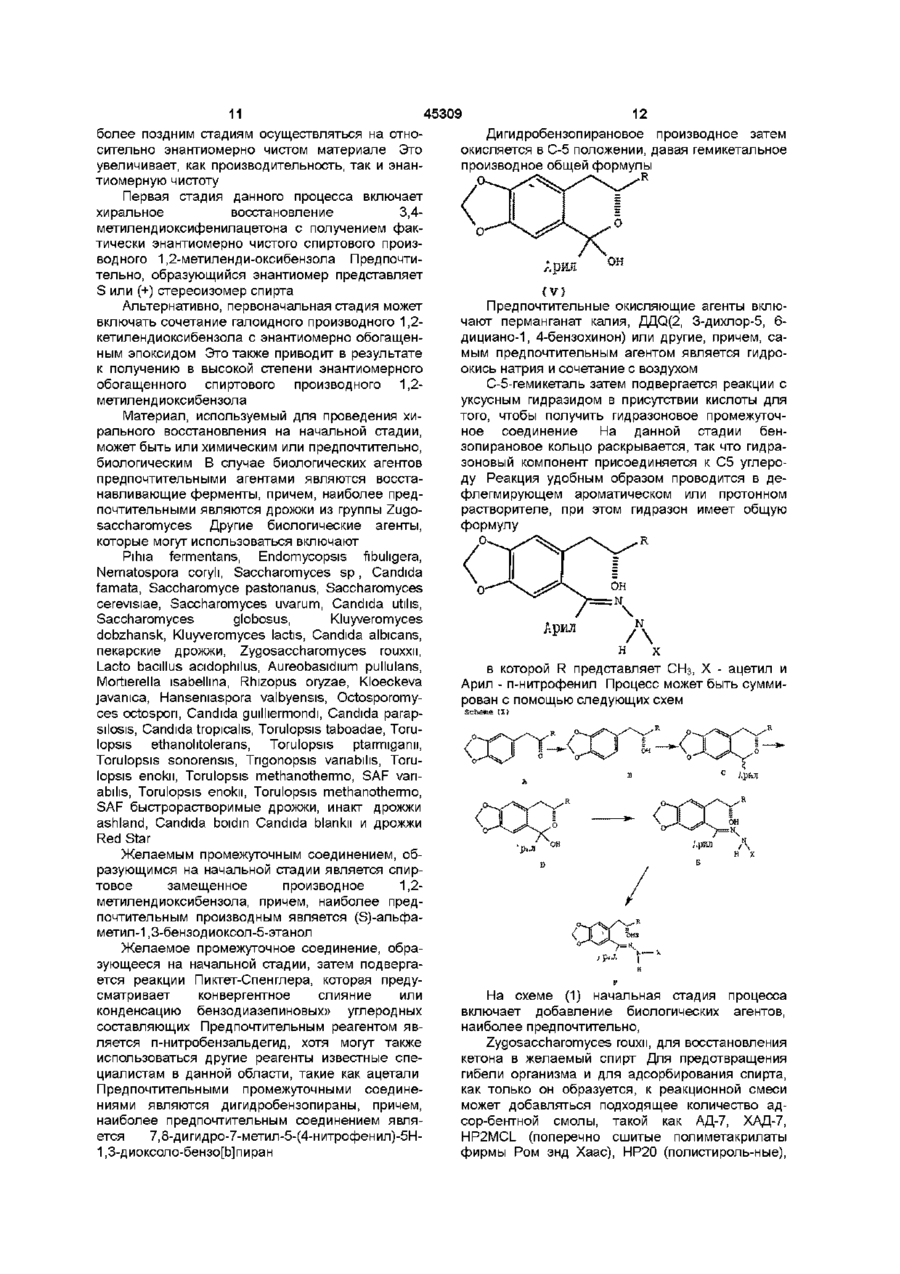
1 Физическая форма (Р)-7-ацетил-5-(4аминофенил)-8,9-дигидро-8-метил-7Н-1,3диоксоло[4,5-п][2,3]бензодиазепина, имеющая порошковую рентгенограмму с d-параметрами 10,61, 8,83, 6,78,5,83, 4,13 и 3,74 2 Физическая форма (Р)-7-ацетил-5-(4аминофенил)-8,9-дигидро-8-метил-7Н-1,3диоксоло[4,5-п][2,3]бензодиазепина, имеющая порошковую рентгенограмму с dl-параметрами 10,61, 8,83, 6,78, 5,83, 4,13 и 3,74 , которая может быть использована для изготовления медикаментов, применимых в качестве антагониста рецептора АМРА 3 Способ получения физической формы (R)-7ацетил-5-(4-аминофенил)-8,9-дигидро-8-метил-7Н1,3-диоксоло[4,5-п][2,3]бензодиазепина, охарактеризованной в п 1, который включает а) взаимодействие соединения формулы N Aryl (I) в которой R - метил, X - ацетил и Aryl - пнитрофенил, b) восстановление п-нитрофенильной группы в соединении формулы І в анилиновую группу, с использованием формиата калия в присутствии палладия на активированном угле в качестве катализатора, с получением соединения формулы I, в которой Aryl - п-аминофенил, и c) кристаллизацию соединения формулы І, в которой Aryl - п-аминофенил, из смеси воды и этанола, в которой число объемов воды на объем этанола составляет менее, чем 1,1-1,0 4 Фармацевтическая композиция, обладающая свойствами селективного антагониста АМРАрецептора, которая включает эффективное количество (Р)-7-ацетил-5-(4-аминофенил)-8,9дигидро-8-метил-7Н-1,3-диоксоло[4,5-п] [2,3] бензодиазепина и фармацевтически приемлемый разбавитель или носитель, отличающаяся тем, что она содержит физическую форму указанного соединения, имеющую порошковую рентгенограмму с d-параметрами 10,61, 8,83, 6,78, 5,83, 4,13 и 3,74 А О о го ю 45309 Данное изобретение относится к новой физической форме производных дигидро-2,3бензодиазепина полезных в качестве фармацевтического средства при лечении расстройства нервной системы В публикации европейской патентной заяви № ЕР-А1 -0492485 раскрывается соединение 1 -(4аминофенил)-3-ацетил-4-метил-7,8метилендиокси-3,4-дигидро-5Н-2,3бензодиазепин Данное соединение является сильным и селективным антагонистом возбудительного аминокислотного АМРА рецептора и считается, что оно обладает способностью лечить разнообразные некрологические нарушения (Р) энантиомер данного соединения, называемый здесь далее (Р)-7-ацетил-5-(4-аминофенил)-8,9дигидро-8-метил-7Н-1,3-диоксоло[4,5 п] [2,3]бензодиазепин, является наиболее сильным энантиомером Настоящее изобретение предоставляет физическую форму (Р)-7-ацетил-5-(4-аминофенил)-8,9дигидро-8-метил-7Н-1,3-диоксоло[4,5 п] [2,3]бензодиазепина, имеющую порошковую рентгенограмму cd параметрами при 10 61, 8 83, 6 78, 5 83, 4 13 и З 74А Оно предоставляет также процесс получения данной формы, фармацевтические композиции содержащие ее и методы ее использования Было найдено, что (Р)-7-ацетил-5-(4аминофенил)-8 9-дигидро-8-метил-7Н-1,3диоксоло[4,5 - п] [2,3]бензодиазепин является полиморфным Первая физическая форма (Р)-7-ацетил-5-(4аминофенил)-8,9-дигидро-8-метил-7Н-1,3диоксоло[4,5 - п] [2,3]бензодиазепина как было найдено, имеет точку плавления примерно 168 172°С и порошковую рентгенограмму с характерными d параметрами при 6 57 и 5 24 А Данная физическая форма называется здесь ниже как форма 1 Она получается с помощью восстановления (Р)-7-ацетил-8,9-дигидро-8-метил-5-(4нитрофенил)-7Н-1,3-диоксоло-[4,5 п] [2,3]бензодиазепина в этаноле с использованием водорода и палладия на угле в качестве катализатора, затем удаления катализатора путем фильтрования, выпаривания этанола, нагревания остатка в 5,7 объемах 1 1 смеси воды и этанола в условиях дефлегмации, и затем оставления полученного в результате раствора охлаждаться Совершенно удивительно, модификация процесса, используемого для получения формы 1, с помощью использования формиата аммония и палладия на угле вместо водорода и палладия на угле дала новую физическую -форму (RJ-7-ацетил5-(4-аминофенил)-8,9-дигидро-8-метил-7Н-1,3диоксоло-[4,5 - п] [2,3] - бензодиазепина, называемую ниже формой II Таким образом, форма II была получена с помощью восстановления (R)-7ацетил-8,9-дигидро-8-метил-5-(4-нитрофенил)-7Н1,3-диоксоло[4,5 - п] [2,3]-бензодиазепина в этаноле с использованием формата аммония и палладия на угле в качестве катализатора, затем уда ления катализатора путем фильтрования, выпаривания этанола, нагревания остатка в 6 объемах смеси 1 1 воды и этанола в условиях дефлегмации и оставления полученного в результате раствора для охлаждения Было найдено, что форма II имеет порошковую рентгенограмму с характерными d параметрами при 13 12 и 5 01 А Модификация процесса, используемого для получения формы II с помощью использования формиата калия и палладия на угле вместо формиата аммония и палладия на угле на удивление дала еще одну физическую форму, называемую здесь ниже формой III Таким образом, форма III была получена с помощью восстановления (R)-7ацетил-8,9-дигидро-8-метил-5-(4-нитрофенил)-7Н1,3-диоксоло[4,5 - п] [2,3]бензодиазепина в этаноле с использованием формиата калия и палладия на угле в качестве катализатора, затем удаления катализатора фильтрованием, выпаривания этанола, нагревания остатка в объемах смеси 1 1 воды и этанола в условиях дефлегмации и оставления полученного в результате раствора для охлаждения Было найдено, что форма III имеет порошковую рентгенограмму с характерными d параметрами при 10 61, 8 83, 6 78, 5 83, 4 13 и 3 84 А Данная физическая форма предоставляется в качестве одного из аспектов настоящего изобретения На удивление была также найдена еще одна физическая форма, называемая ниже как форма IV Первоначально было замечено, что данная форма образуется после того, как форма II нагрета Впоследствии было обнаружено, что форма IV может быть получена непосредственно с помощью модификации процесса, используемого для получения формы III, в частности, путем увеличения объемного соотношения вода / этанол, используемых на стадии кристаллизации Таким образом форма IV получена путем восстановления (Р)-7-ацетил-8,9-дигидро-8-метил-5-(4нитрофенил)-7Н-1,3-диоксоло[4,5 п] [2,3]бензодиазепина в этаноле с использованием формиата калия и палладия на угле в качестве катализатора, затем удаления катализатора с помощью фильтрования, выпаривания этанола, нагревания остатка в 8 объемах смеси 5 3 вода / этанол или в 7 объемах смеси 4 3 вода / этанол в условиях дефлегмации, с необязательным использованием затравки кристаллами формы IV при 70 - 80°С и оставления полученной в результате смеси охлаждаться Было найдено, что форма IV имеет порошковую рентгенограмму с характерными d параметрами при 12 78, 9 48, 8 99, 8 64, 8 23, 6,39, 6,27, 5 73, 4 01 и 3 96 А Данная физическая форма является предметом находящейся одновременно на рассмотрении патентной заявки (ссылочный № Х-9386Д) Было найдено, что форма I обладает несколькими неблагоприятными свойствами В частности, было обнаружено, что она кристаллизуется в виде густой суспензии, которую трудно перемешивать и транспортировать Было найдено, что время 45309 фильтрации является неприемлемо длительным для крупномасштабного производства, и время сушки отфильтрованного влажного осадка на фильтре также длительно Кроме того, было найдено, что форма I является термически нестабильной и, как было обнаружено, превращается в форму IV, или, изредка, в еще одну физическую форму, называемую здесь ниже формой V Было найдено, что форма V имеет порошковую рентгенограмму с характеристиками d параметров при 6 12, 5 94 и 5 48 А Форма V показывает множественные фазовые переходы, когда подвергается дифференциальной сканирующей калориметрии Было найдено, что форма II выкристаллизовывается в виде перемешиваемой суспензии, которая может быть свободно профильтрована Однако, было обнаружено, что она сохнет медленно и сохраняет кристаллизационный растворитель Было найдено, что, как и форма I, она термически нестабильна, в отношении превращения в форму IV Форма III, как было обнаружено, кристаллизуется в виде способной к перемешиванию суспензии, которую можно свободно фильтровать и подвергать сушке Было найдено также, что она является термически стабильной Форма IV, как обнаружено, также кристаллизуется в виде перемешиваемой суспензии, которую можно свободно фильтровать и сушить Как и форма III, как было найдено, она также является термически стабильной Каждая из форм I, II, III, IV и V была охарактеризована с помощью дифракции рентгеновских лучей, 13С ЯМР спектроскопии в твердом состоянии и с помощью дифференциальной сканирующей калориметрии Используемые приемы и физические характеристики, определенные для образцов каждой формы, даются ниже, вместе (только для формы III и IV) с общими интервалами, полученными с помощью дифференциальной сканирующей калориметрии с использованием ряда различных образцов Картины дифракции рентгеновских лучей (XRD) получали на рентгеновском дифрактометре Сименс Д5000, снабженном Си Ка (X = 1.54056А) ИСТОЧНИКОМ, работающим при капиллярной нагрузке 50Кв и 40мА Данные были собраны с помощью Kevex детектора в твердом состоянии Каждый образец сканировался между 4 и 35° 20 - с размером ступенчатого нарастания 0,03°С и максимальной скоростью сканирования 2сек /стадию Измерения дифференциальной сканирующей калориметрии (DSC) выполнялись на дифференциальном сканирующем калориметре Сейко7 Образцы (2-5мг), загерметизированные в алюминиевых чашках, нагревались от температуры окружающей среды (25°С) до не менее 200°С со скоростью 10°С/мин 13 С ЯМР спектры поперечная поляризация / вращение под магическим углом (СР / MAS ) получались с использованием МГц спектрометра Вариан Юнити 400, работающего при углеродной частоте 100 577 МГц и снабженном вспомогательным комплектом твердых веществ и Вариан зондом 5 или 7мм VT CP/MAS Типичные условия измерений были следующими 90(град) протон f пульсацией 5 Оме, время контакта 1 - 2мс , время повторения пульсации 5с, MAS частота 7кГц, спектральная ширина 50кГц, и время аквизиции 50мс Химические сдвиги относились к СНз группе гексаметилбензола (дельта = 17,3млндол) при замене образца Форма I DSC Главная зндотерма при 171,5°С, второстепенная эндотерма при 207,4°С XRD Параметр, d (А) 17 30 12 28 7 76 6 57 5 24 4 81 4 34 4 21 4 09 3 98 3 62 2 85 Относительная интенсивность 100 34 71 37 35 94 30 29 19 14 18 12 Форма II DSC Эндотерма при 85 2°С, зкзотерма при 91 4 С, эндотерма при 192 3°С XRD Параметры, d (А) Относительная интенсивность 100 23 37 60 28 94 70 41 28 25 1312 9 72 6 73 6 61 5 25 5 01 4 89 4 75 4 24 3 74 Форма III DSC Эндотерма для данного образца при 194 7°С Другие образцы, как было найдено, показывают эндотерму при температуре в интервале от192до195°С XRD Параметры, d (А) 10 61 8 83 8 33 7 85 6 78 5 83 5 68 5 31 511 4 94 4 78 4 55 4 41 413 4 07 Относительная интенсивность 78 73 15 9 100 17 6 25 68 62 20 5 25 71 19 45309 3 90 3 74 3 53 3 42 3 37 3 28 3 21 3 02 2 85 2 78 24 40 16 18 26 11 ЗО 5 7 6 Форма IV DSC Экдотерма данного образца при 203 2°С Найдено было, что другие образцы показывают эндотерму при температуре в интервале от 201 до 207°С XRD Параметры, d (А) 120,78 9,48 8,99 8,64 8,23 6,53 6,39 6,27 5,73 5,37 5,22 5,18 5,10 4,95 4,89 12 10 29 20 53 35 22 31 15 19 4,75 4,56 4,41 4,32 4,01 3,96 3,77 3,59 3,39 3,11 Относительная интенсивность 100 29 17 23 59 58 13 20 33 44 14 11 15 32 61 Форма V DSC Эндотерма при 170,6°С, экзотерма при 206,2°С XRD Параметры, d (А) 17,37 12,29 7,75 6 79 612 5 94 5 48 5 34 4 89 4 33 4 26 4 08 4 02 3 65 2 86 Относительная интенсивность 51 21 29 32 13 14 15 24 82 100 50 34 20 21 13 Таблица 1 Данные химических сдвигов '^С ЯМР в растворе и в твердом состоянии Форма I 176 4 128 6 1159 148 9 146 3 136 4 123 9 131 7 154 5 168 4 22 2 186 Форма II 173 7 126 9 150 4 147 6 134 5 123 3 129 2 135 8 152 1 170 7 22 2 183 Форма III 174 126 3 109 4 1161 149 9 146 0 135 9 124 3 129 1 132 8 153 5 171,4 24 3 194 Согласно еще одному аспекту настоящее изобретение предоставляет процесс получения формы III, который включает а) взаимодействие соединения формулы Форма IV 1741,1763 127 5,129 4 1141 1163 148 0,150 2 146 4 136 1 124 7 131 2,133 6 152 7 167 7,169 7 23 2,23 7 185,192 Форма V 175 3 148 5 149 4 146 7 135 4 136 7 151 1 154 3 155 1 163 0 167 2 20 6 191 174 45309 10 он Аркл VII в которой MS представляет метансульфонил, R-метил, Х-ацетил и Арил представляет пнитрофенил, с каустической содой с получением соединения формулы 1 (III) с) взаимодействие соединения формулы III с п-нитробензальдегидом с получением изоманового соединения, имеющего формулу 0~ Арил Арил (IV) d) взаимодействие соединения формулы IV с окисляющим агентом с получением соединения формулы О. в которой R-метил, Х-ацетил, и Арил-пнитрофенил, в) восстановление п-нитрофенильной группы в соединении формулы І в анилиновую группу с использованием формиата калия в присутствии палладия на активированном угле в качестве катализатора с получением соединения формулы I, в которой Арил представляет п-аминофенил, и с) кристаллизацию соединения формулы 1, в которой Арил представляет п-аминофенил, из смеси воды и этанола, в которой число объемов воды на объем этанола составляет менее, чем 1,1 -1,0 Если используется затравка формой III, на стадии (с) можно использовать более высокое объемное отношение воды к этанолу Стадия (а) данного процесса удобно проводится при температуре в интервале от 0 до 100°С Подходящие растворители включают алканолы, такие как метанол или этанол, и простые эфиры, такие кактетрагидрофуран Соединение общей формулы VII может получаться с помощью многостадийного процесса, исходя из производного метилендиоксифенилкетона Данный процесс включает а) предоставление некоторого количества соединения, имеющего формулу (II) в которой R представляет метил, в) аисмметрическое восстановление соединения формулы II с получением соединения, имеющего формулу (V) е) взаимодействие соединения формулы V с уксусным гидразидом с получением соединения формулы ,'ркл (VI) f) взаимодействие соединения формулы VI с метансульфонил-хлоридом и третичным амином с образованием соединения формулы VII Предпочтительный процесс включает на ранней стадии хиральное восстановление кетона в спирт В многостадийном процессе добавляются заместители для замыкания бензосконденсированного пиранового кольца, перед тем, как гидразиновый реагент вводится для открытия кольца и добавления необходимых азотных компонентов Наконец, вторичное кольцо замыкается при добавлении сильного основания, и данное соединение восстанавливается и образуется желаемое соединение Наиболее предпочтительно первоначальной стадией в синтезе соединений формулы I из кетонов является стадия хирального восстановления Хиральное восстановление может проводиться при использовании специальных химических веществ или, предпочтительно, с использованием биологических агентов описанных ниже Установление стереохимии на ранней стадии в данном процессе является благоприятным и позволяет 11 45309 12 более поздним стадиям осуществляться на отноДигидробензопирановое производное затем сительно энантиомерно чистом материале Это окисляется в С-5 положении, давая гемикетальное увеличивает, как производительность, так и энанпроизводное общей формулы тиомерную чистоту Первая стадия данного процесса включает хиральное восстановление 3,4метилендиоксифенилацетона с получением фактически энантиомерно чистого спиртового производного 1,2-метиленди-оксибензола ПредпочтиАрил тельно, образующийся энантиомер представляет S или (+) стереоизомер спирта CV} Альтернативно, первоначальная стадия может Предпочтительные окисляющие агенты вклювключать сочетание галоидного производного 1,2чают перманганат калия, ДДО(2, З-дихлор-5, 6кетилендиоксибензола с энантиомерно обогащендициано-1, 4-бензохинон) или другие, причем, саным эпоксидом Это также приводит в результате мым предпочтительным агентом является гидрок получению в высокой степени энантиомерного окись натрия и сочетание с воздухом обогащенного спиртового производного 1,2С-5-гемикеталь затем подвергается реакции с метилендиоксибензола уксусным гидразидом в присутствии кислоты для того, чтобы получить гидразоновое промежуточМатериал, используемый для проведения хиное соединение На данной стадии бенрального восстановления на начальной стадии, зопирановое кольцо раскрывается, так что гидраможет быть или химическим или предпочтительно, зоновый компонент присоединяется к С5 углеробиологическим В случае биологических агентов ду Реакция удобным образом проводится в депредпочтительными агентами являются восстафлегмирующем ароматическом или протонном навливающие ферменты, причем, наиболее предрастворителе, при этом гидразон имеет общую почтительными являются дрожжи из группы Zugoформулу saccharomyces Другие биологические агенты, которые могут использоваться включают Pihia fermentans, Endomycopsis fibuhgera, Nematospora coryh, Saccharomyces s p , Candida famata, Saccharomyce pastonanus, Saccharomyces cerevisiae, Saccharomyces uvarum, Candida utihs, Saccharomyces globosus, Kluyveromyces Арил dobzhansk, Kluyveromyces lactis, Candida albicans, пекарские дрожжи, Zygosaccharomyces rouxxn, Lacto bacillus acidophilus, Aureobasidium pullulans, в которой R представляет СНз, X - ацетил и Mortierella isabellma, Rhizopus oryzae, Kloeckeva Арил - п-нитрофенил Процесс может быть суммиjavamca, Hanseniaspora valbyensis, Octosporomyрован с помощью следующих схем Scheme ( I ) ces octospon, Candida guilhermondi, Candida parapsilosis, Candida tropicahs, Torulopsis taboadae, Torulopsis ethanohtolerans, Torulopsis ptarmigami, Torulopsis sonorensis, Tngonopsis vanabihs, Torulopsis enokn, Torulopsis methanothermo, SAF vanabihs, Torulopsis enokii, Torulopsis methanothermo, SAF быстрорастворимые дрожжи, инакт дрожжи ashland, Candida boidm Candida blankii и дрожжи Red Star Желаемым промежуточным соединением, образующимся на начальной стадии является спиртовое замещенное производное 1,2метилендиоксибензола, причем, наиболее предпочтительным производным является (Э)-альфаметил-1,3-бензодиоксол-5-этанол Желаемое промежуточное соединение, образующееся на начальной стадии, затем подвергается реакции Пиктет-Спенглера, которая предусматривает конвергентное слияние или конденсацию бензодиазепиновых» углеродных составляющих Предпочтительным реагентом является п-нитробензальдегид, хотя могут также использоваться другие реагенты известные специалистам в данной области, такие как ацетали Предпочтительными промежуточными соединениями являются дигидробензопираны, причем, наиболее предпочтительным соединением является 7,8-дигидро-7-метил-5-(4-нитрофенил)-5Н1,3-диоксоло-бензо[Ь]пиран На схеме (1) начальная стадия процесса включает добавление биологических агентов, наиболее предпочтительно, Zygosaccharomyces rouxii, для восстановления кетона в желаемый спирт Для предотвращения гибели организма и для адсорбирования спирта, как только он образуется, к реакционной смеси может добавляться подходящее количество адсор-бентной смолы, такой как АД-7, ХАД-7, HP2MCL (поперечно сшитые полиметакрилаты фирмы Ром энд Хаас), НР20 (полистироль-ные), 13 45309 или SP 207 (бромированный полистирол фирмы Мицубиси) Могут также использоваться другие аналогичные смолы Схема 11 По схеме 11 начальная стадия процесса включает взаимодействие арилгалогенидного производного, такого как 4-бром-1,2(метилендиокси)бензол, с углеводородным соединением щелочного металла (предпочитается вторбутиллитий) и энантиомерно чистым эпоксидом Альтернативно, арилгалогенид может сначала превращаться в реагент Гриньяра по реакции с магнием, затем подвергаться реакции с энантиомерно чистым эпоксидом в присутствии окиси меди (1) в качестве катализатора Предпочитается (3)-(-)-пропиленоксид И по схеме 1 и по схеме 11 целью является установление стереохимии С8 атома бензодиазепинового кольца как можно раньше По наблюдениям было замечено, что обе схемы достигают данной цели, и образуются энантиомерно обогащенные (ее) спирты с чистотой 98% Известно, что (Р)-7-ацотил-5- (4-амииофеыил) -8,9-дигидро-б-метил-7Н-1,3-диоксоло[4,5-п] [2,3]бензодиазепкн является селективным антагонистом рецептора АМРА Таким образом, согласно еще одному аспекту настоящее изобретение предоставляет использование формы 111 для производства медикамента для блокирования АМРА рецепторов у млекопитающих, которым требуется такое лечение Было показано, что большое разнообразие физиологических функций подвержено влиянию избыточной или несоответствующей стимуляции нейротрансмии возбудительных аминокислот Считается, что (Р)-7-ацетил-5-(4-аминофенил)8,9-дигидро-8-метил-7Н-1,3-диоксоло[4,5-п] [2,3]бензодисізєпин обладает способностью лечить разнообразные нейрологические нарушения у млекопитающих, ассоциированных с данным состоянием, которые включают острые нейрологические нарушения, такие как церебральная недостаточность как следствие хирургического вмешательства с использованием искусственного кровообращения и пересадки органов, удар, церебральная ишемия, травма спинного мозга, травма головы, перинатальная гипоксия, остановка сердца и гипогликемические нейрональные повреждения Считается, что это соединение обладает способностью лечить множество хронических некрологических расстройств, таких как болезнь Альцгеймера, хорея Хантингтона, амиотрофический латеральный склероз, деменция, вызванная СПИДом, окулярные повреждения и ретинопатия, и идиопатическая или вызванная лекарствами болезнь Паркинсона Настоящее изобретение предоставляет также использование формы 11 для производства медикаментов для лечения этих расстройств Считается, что данное соединение обладает 14 способностью лечить множество других нейрологических нарушений млекопитающих , которые связаны с глютаматной дисфункцией, такие как мышечные спазмы, конвульсии, мигрени, мочевое недержание, психоз, толерантность лекарств и отторжение их, беспокойство или страх, рвота, мозговая эдема, хронические боли, и поздняя или запоздалая дискинезия Данное соединение полезно также в качестве анальгетического агента Следовательно, настоящее изобретение предоставляет также использование формы 111 для производства лекарств для лечения этих нарушений Термин "эффективное количество", используемый здесь, относится к количеству формы 111, которое способно блокировать рецептор возбудительной аминокислоты АМРА Конкретная доза соединения, которая назначается для приема согласно данному изобретению, будет конечно определяться в зависимости от конкретных обстоятельств, включающих назначаемое соединение, способ назначения, конкретное состояние, подвергаемое лечению, и аналогичные факторы Данная форма может назначаться с помощью множества способов, включающих оральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный, или интраназальный способы Альтернативно, данная форма может назначаться путем непрерывного вливания Типичная суточная доза содержит примерно от 0 01мг/кг до ЗОмг/кг активного соединения данного изобретения Предпочтительные суточные дозы составляют примерно от 0 05мг/кг до 24мг/кг , более предпочтительно, примерно от 0 1 до 20мг/кг Форма 111 обычно назначается пациентам в виде фармацевтических композиций Согласно еще одному аспекту настоящее изобретение предоставляет фармацевтическую композицию, которая включает форму 111 и фармацевтически приемлемый разбавитель или носитель При изготовлении композиций настоящего изобретения активный ингредиент обычно смешивается с носителем, или разбавляется носителем, или вводится внутрь носителя, который может быть в форме капсулы, саше, бумажного или другого контейнера Когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым, или жидким материалом, который действует как носитель, эксципиент, или среда для активного ингредиента Композиции могут быть в форме, например, таблеток, пилюль, порошков, эликсиров, саше, облаток, суспензий, аэрозолей, мягких и твердых желатиновых капсул и стерильно упакованных порошков Некоторые примеры подходящих носителей, эксципиентов и разбавителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, камедь (акации), фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, метил целлюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло Готовые формы препаратов могут дополнительно включать смазочные агенты, смачивающие агенты, эмульгирующие и суспендирующие агенты, консервирующие или предохра 15 45309 16 няющие агенты, подслащивающие агенты или изопропанол / 7% вода Объем во время данной вкусовые или ароматизирующие агенты Композизамены растворителя варьирует от 11 - 14 объеции изобретения могут формироваться таким обмов, и конечный объем был примерно 11 объемов разом, чтобы обеспечить быстрое, отсроченное Смесь охлаждалась до 0 - 10°С и перемешиваили задержанное или замедленное высвобожделась в течение 1 часа Иглообразные кристаллы ние активного ингредиента после назначения папродукта отфильтровывались и промывались 2 циенту с помощью процедур хорошо известных в раза 1 85 объемами изопропанола и сушились под данной области техники вакуумом при 50 - 60°С Выход целевого соединения ин ситу был 95 + %, в то время как выход выКомпозиции предпочтительно формируются в деленного продукта был 87 - 93% Активность бывиде единичных дозированных форм, причем, ла 99+% и ЕЕ-100% каждая доза содержит примерно от 5 до 5000мг, более предпочтительно примерно от 25 до 3000мг Пример 3 активного ингредиента Наиболее предпочтительАльтернативный синтез (3)-альфа-метил-1,3ные единичные дозированные формы содержат бензодиоксол~5-этанола примерно от 100 до 2000мг активного ингредиен3 47 грамм 4 бром-1,2та Термин "единичная дозированная форма" от(метилендиокси)бензола растворялось в 100мл носится к физически дискретной единице, подхотетрагидрофурана при -78°С, затем добавлялось дящей в виде разовой дозы для людей и других 13 9мл 1 ЗМ втор-бутиллития в циклогексане для млекопитающих, причем, каждая единица содерпотребления арилгалогенида менее, чем через 30 жит заданное количество активного материала, минут С помощью шприца добавлялся 1 ООграмм вычисленное с таким расчетом, чтобы давать же( ) - (-)окиси пропилена в 2мл ТГФ, и раствор пелаемый терапевтический эффект, в сочетании с ремешивался в течение 45 минут Раствор затем подходящим фармацевтическим носителем Слеподогревался до 23°С в течение 16 часов Реакдующие ниже примеры иллюстрируют данное ционная смесь выливалась в З М раствор хлориизобретение стого аммония, и продукт отделялся с помощью экстракции этилацетатом Объединенные экстракПример 1 ты сушились над сульфатом магния, фильтроваСинтез (3)-альфа-метил-1,3-бензодиоксол-5лись через флорисил и кон центри вались с помоэтанола щью роторного испарения Остаточное масло 1 эквив 3,4-метилендиоксифенилацетона, очищалось с помощью хроматографии на силика0 45 эквив динатрийфосфата, 0 03 эквив фосгеле и элюировалось смесью 50 50 гексана и форной кислоты, 12 5 объемов АД-7 смолы и 5 8 диэтилового эфира, давая 1 40г (45%) названного объемов воды смешивалась вместе и перемешив заголовке промежуточного продукта Р спет вались в течение 1 5 - 6 0 минут при 20 - 25°С До/о/зб5 + 117 2° (с 1 0, СНСІз) ТСХ Rf = 0 26 (50 50 бавлялось 2 27 эквив глюкозы, и добавляется гексан эфир), ИК (СНСІз) 3598, 3012, 2973, 28, Z rouxii ATCC 14462 в количестве 1 5 грамма кле1490, 1249, 1041см \ 13С ЯМР (СДСІ3) d 147 75, точной пасты на грамм кетона (то есть 146 19, 132 26, 122 27, 109 68, 108 30, масс спектр, 0 375грамма/грамм на сухой основе) Данная m/z(FD, M+) 180 смесь разбавлялась водой до 25 объемов, а затем осторожно перемешивалась при 33 - 35°С в течеАнализ для СюНі2Оз Вычислено С 66 65, Н 6 71 ние 8-16 часов Смесь фильтровалась через сито Найдено С 66 42, Н 6,66 из нержавеющей стали размером 100 меш (приПример 4 мерно 150 микрон), и смола, которая задерживаАльтернативный синтез (5RS,7S)-7,8-flHrnflpoлась ситом, промывалась 25 объемами воды, раз7-метил-5-(4-нитрофенил)-5Н-1,3-диоксоло-[4,5 деленными на 4 отдельные порции Продукт, G] [2]бензопирана который адсорбировался на смоле, затем десор244 грамма п-нитробензальдегида добавлябировался со смолы 25 объемами ацетона Раслось к раствору ЗООграмм промежуточного соедитвор продукта в ацетоне затем отпаривался досунения, образовавшегося на стадии биокатализиха под вакуумом, давая указанной в заголовке руемого восстановления примера 1 в 4 45л промежуточное соединение в виде желтого масла толуола По каплям на протяжении 1 5 - 2 0 минут средней вязкости Выход на месте был 97 - 100%, добавлялось 166 5мл концентрированной соляной в то время как выход выделенного продукта был кислоты, и получающаяся в результате смесь на85 - 90% Активность составляла 80 - 95% и ЕЕ гревалась до 60°С в течение 2 5 часов Смесь ох100% лаждалась до комнатной температуры и конценПример 2 трировалась с помощью роторного испарения Синтез (5RS, 75)-7,8-дигидро-7-метил-5-(4Добавлялось Зл этанола, и смесь концентрированитрофенил)-5Н-1,4-диоксоло-[4,5-С] лась до твердого вещества Добавлялась еще [2]бензопирана одна 3 л порция этанола, и смесь перемешивалась в течение 1 часа Суспенция охлаждалась на Указанные выше промежуточное соединение протяжении ночи, и кристаллический продукт отрастворялось в 4 64 объемах толуола, фильтроделялся с помощью фильтрования в вакууме валось через гифло и промывалось 1 55 объемаОсадок на фильтре промывался этанолом, а зами толуола Добавлялось 1 05 эквивалента птем сушился в вакуумной печи при 40 - 60°С, данитробензальдегида и 1 05 эквив концентрировая 450г (86%) не совсем белого твердого вещеванной соляной кислоты, и смесь нагревалась до ства, которое по данным определения 55 - 65°С и перемешивалась 1 час Затем произпредставляло изомерную смесь указанного выше водилась замена растворителя при 250мм Но, при в заголовке оптически активного промежуточного этом толуол заменялся 12 4 объемами смеси 93% 17 45309 18 1Н, СН), 2 4 - 22(м , 2Н, СН 2 ), 1 12, 1 10, (д , ЗН, вещества Р chem /o/365 + 55° (с 0 4, СНСІ3) СНз) С ЯМР (СДСІз, 75МГц) d 209 94(G), 173 38, Пример 5 173 43(С), 149 38, 149 62(С), 148 31, 148 58(С), Синтез (5RS, 73)-7,8-дигидро-7-метил-5-(4147 90, 148 18(С), 147 54(С), 142 5, 143 04(С), нитрофенил)-5Н-1,3-диоксоло[4,5 G] 132 64(С), 127 53, 127 61(СН), 123 75, 123 77(СН), [2]бензопиран-5-ола 350 грамм изомерного про122 86, 123 27(С), 112 13(СН), 110 55(СН), 108 03, межуточного соединения из примера 4 добавля10810(СН), 108,10(СН), 101 83(СН2), 6 7 5 1 , лось к раствору 731мл диметилсульфоксида и 68 08(СН), 42 37, 42 97(СН2), 23 48, 23 83(СН3), 2923мл диметилформамида Смесь охлаждалась 23 48, 23 83(СН3), 20 47, 20 55(СН3), /о/589 + ЮЗ 8° до 8 - 12°С и через смесь пропускался сжатый (с 1, СНСІз), масс спектр, m / z (FD, M + )385 воздух Добавлялось 117 5мл 50% водной гидроАнализ дляВычислено С 59 22, Н 4 97, N окиси натрия в виде одной порции, и получающаяся смесь перемешивалась в течение 4 5 чаC19H19N3O6 10 90 сов Реакционная смесь добавлялась с помощью Найдено С 58 99, Н 5 04, N трубочки на протяжении 30 - 60 минут к 8 25л пе10 68 ремешиваемого 1 норм, раствора соляной кислоПример 7 ты при 10 - 15°С Получающийся в результате Синтез (З)-уксусная кислота-[[6-[2осадок отфильтровывался и промывался 3 литра[(метилсульфонил)оксо]пропил]-1,3-бензодиоксолми воды, а затем подвергался воздушной сушке 5-ил](4-нитрофенил)метилен]гидразида до постоянного веса (384г) Влажный осадок пе340 грамм промежуточного соединения приреносился в пример 6 без дальнейшей сушки Р мера 6 растворялось в 2380мл метиленхлорида chem Данные регистрировались по 3 1 изомерРаствор охлаждался до 0 -ь -10°С, и добавлялось ной смеси ТСХ Rf = 0 19 (75 25 гексан этилаце187мл триэтиламина Затем добавлялось 78 2мл тат), ИК (СНСІз) 3605, 3590, 3015, 3000 2960, метансульфонилхлорида, и получающаяся в реj 2910, 1608, 1522, 1484, 1352, 1240, 1042см зультате смесь перемешивалась в течение 1 5 - 3 0 1 минут Добавлялось 510мл воды Отделившаяся Н ЯМР (СДСІз, 300МГц) d (главный изомер) органическая фаза перемывалась 460мл 1 норм, 8 16(д, 2Н, J = 6 9Гц), 7,73(д, 2Н, J = 6,9Гц), 6,55(с, раствора соляной кислоты, а затем 500мл солево1Н), 6,38(с 1Н), 5,86(с, 1Н), 5 83(с, 1Н), 4,38(м , го раствора Метиленхлоридный раствор подогре1Н), 2 70(м , 2Н), 1,39(д , ЗН, J = 6 3Гц), d (второвался до 35 - 45°С, и на протяжении 90 минут достепенный изомер) 8 27(д , 2Н, J = 8 9Гц), 7 90(д , бавлялось 4760мл гексана Смесь медленно 2Н, J = 8 6Гц), 6 87(с , 1Н), 6 73(с , 1Н), 6 03(с , 1Н), охлаждалась до комнатной температуры, а затем 6 02(С, 1Н), 3 95(м, 1Н), 2 7(смутный, м , 2Н), + охлаждалась далее до 0 - 5°С Продукт отделялся 1 24(д , ЗН, J = 6,71 Гц), масс спектр, m / z (FD, M ) с помощью фильтрования и сушился в вакуумной 329 печи при 40 - 50°С, давая 356 2 грамма (87%) изоАнализ дляВычислено С 62 01, Н 4 59, N мерной смеси названного в заголовке соединения C 1 7 H 1 5 NO 6 4 25 в виде желтого твердого вещества Р chem ДанНайдено С 62 22, Н 4 79, ные, зарегистрированные по 3 1 изомерной смеN4 29 си Т п л 150 5 - 152 5°С, ТСХ Rf = 0 80 и 0 73 Пример 6 (этилацетат), ИК (СНСІз) 1696, 1520, 1486, 1346, Синтез (З)-уксусная кислота-[[6-(21175, 1041, 923 с м 1 , 1 Н ЯМР (СДСІз, 300МГц) d гидроксипропил)-1,3-бензодиоксол-5-ил] (48 44(с, 1Н, NH), 8 20 (д , 2Н, J = 8 8Гц, Аг-Н), 7 73 нитрофенил)метилен]гидразида (д , 2Н, J = 8 6Гц), 6 94(д , 1Н, J = 2 7Гц, Аг-Н), 6 57 К 350г влажного осадка на фильтре из приме(д , 1Н, 2 6Гц, Аг-Н) 6,08(д , J = 5,4Гц), 4 77(м , 1Н, ра 5 в 2300мл этанола добавлялось 94 5г уксусноСН), 2 90(с , ЗН, SCH 3 , главный), 2 83(с , ЗН, SCH 3 , го гидразида и 1мл концентрированной соляной второстепенный), 2 66 - 2 57(м , 2Н, СН 2 ), 1 30(д , кислоты Получающийся в результате раствор ЗН, СНз, второстепенный), 1 26(д , ЗН, СНз, главнагревался до температуры дефлегмации в теченый), масс спектр, m/z(FD, M+) 385 ние 2 5 часов Смесь охлаждалась до комнатной Анализ дляВычислено С 51,83, Н 4 57, N температуры и концентрировалась до желтой пены с помощью роторного испарения Концентрат C20H21N3O8S 9 07, S 6 92 растворялся в 4 9л этилацетата и промывался 1 5 Найдено С 52 05, Н 4 53, N литрами насыщенного бикарбоната натрия, а за8 84, S 6 96 тем 1 5л солевого раствора Органическая фаза Пример 8 сушилась над сульфатом натрия, фильтровалась Синтез ^)-7-ацетил-8,9-дигидро-8-метил-5-(4и концентрировалась, давая 373г желтой пены нитрофенил)-7Н-1,3-диоксоло[4,5 - h] [2, 3] бензо(91%) Данный материал идентифицировался как диазепина 325г промежуточного соединения при1 1 неразделимая смесь изомеров названного в мера 7 растворялось в 3174мл метанола К перезаголовке соединения (97% чистый по данным мешиваемому раствору добавлялось 38 1мл 50% ВЭЖХ) Р chem Данные регистрировались по 1 1 раствора каустической соды Получающаяся изомерной смеси смесь перемешивалась в течение 4 часов К смеТпл 167,8 169 7°С, ТСХ Rf = си добавлялось 6348мл воды, и содержимое пе0 55(этилацетат), ИК (СНСІз) 3590 3485, 3310, ремешивалось в течение 3 часов, после чего по1 1 1694, 1673, 1520, 1485, 1346см см Н ЯМР лучающийся в результате осадок отделялся с (СДСІз, 300МГц) d 8 64, 8 50(с , 1Н, NH), 8 18(д , помощью вакуумного фильтрования Материал 2Н, Аг - Н), 7 74, 7 71 (д , 2Н, J = 8, Аг - Н), 6,99, сушился в вакуумной печи при 45 - 55°С, давая 6 95(с , 1Н, АГ - Н), 6 52, 6 50(с , 1Н, Аг - Н), 6 06, 255 грамм (97%) названного в заголовке соедине6 05(д , 2Н, J = 5, О 2 СН 2 ), 2 44(с ,ЗН, СН3), 3 87(м , ния, которое было на 97 6% чистым по % области 19 45309 20 ВЭЖХ 221 грамм высушенного материала дополлась до дефлегмации в течение 3 5 часов с уданительно очищался с помощью повторного суслением воды с помощью ловушки Дина-Старка пендирования в 1105мл этанола, который нагреРеакционная смесь концентрировалась с помовался до температуры дефлегмации щью вакуумной перегонки до 1 объема КонценПолучающаяся в результате смесь охлаждалась трат разбавлялся 105мл метиленхлорида и продо комнатной температуры, и осадок отделялся с мывался 50 - 55мл каждого из насыщенного помощью вакуумного фильтрования Изолят сураствора бикарбоната натрия и солевого раствошился в вакуумной печи при 45 - 55°С, давая 199 ра Организаций раствор сушился над сульфатом грамм (90%) названного в заголовке соединения, магния (0,25 вес %) и фильтровался через осадок которое было 100% чистоты по данным анализа гифло Фильтр прополаскивался 1 объемом метиактивности ВЭЖХ ленхлорида Объединенная органическая фаза, содержащая указанное в заголовке промежуточПример 9 ное соединение, переносилась на следующую Синтез формы IV (Р)-7-ацетил-5-(4стадию аминофенил)-8,9-дигидро-8-метил-7Н-1,3диоксоло[4,5 - п] [2,3]бензодиазепина Пример 12 К 5 граммам промежуточного соединения Синтез (З)-уксусная ки ел ота-[[6-[2примера 8 в 50мл этанола добавлялось 0 5 грам[(метилсульфонил)окси]пропил]-1,3-бензодиоксолма 10% Pd / С, увлажненного водой Перемеши5-ил](4-нитрофенил)метилен]гидразида ваемая суспензия обрабатывалась раствором 4 Метиленхлорид ный раствор, содержащий грамм формиата калия в 4мл воды Получающаяпромежуточное соединение примера 11, охлажся смесь перемешивалась в течение 2 5 часов, а дался до 0 + -5°С, и добавлялось 10мл триэтилазатем фильтровалась над гифло Фильтрат конмина Медленно добавлялось 4,1мл метансульцентрировался до 10 - 20мл с помощью перегонки, фонилхлорида для поддержания температуры и медленно добавлялось 22мл воды для подогререакции = 0°С К получающемуся раствору добаввания (78°) раствора Получающаяся смесь нагрелялось 1,5 объема воды Органическая фаза отвалась до 90°, а затем медленно охлаждалась до делялась и промывалась 2 5 объемами 1 норм, комнатной температуры Продукт отделялся с пораствора соляной кислоты Органическая фаза мощью вакуумного фильтрования и промывался отделялась и концентрировалась до половины 10 - 20мл воды первоначального объема с помощью атмосферной перегонки Продукт осаждался добавлением к Отделенное твердое вещество сушилось под раствору при 45°С по каплям гептана (2 1 объема вакуумом при 50°С, давая 4 17 грамма (93%) укагептана к органическому концентрату) Перемешизанного в заголовке конечного соединения, котоваемая смесь охлаждалась до 20 - 25°С в течение рое имело 100% чистоту по данным анализа 1 часа, затем охлаждалась до 0 -ь -5°С в течение 1 ВЭЖХ - 2 часов Осадок отделялся с помощью вакуумно/о/зб5 - - 303 7 (с = 1, метанол) го фильтрования и промывался 3 объемами 4 1 Позднее было найдено, что продукт, который смеси гептан метиленхлорида Затем сушился в был кристаллическим, оказался формой IV вакуумной печи при 45 - 50°С Поучалось 17 43 Пример 10 грамма названного в заголовке промежуточного Синтез (5RS, 73)-7,8-дигидро-7-метил-5-(4продукта (78%) в виде оптически активной смеси нитрофенил) -5Н-1,3-диоксоло[4,5 G] изомеров гидразона, которая была на 97 7% чис[2]бензопиран-5-ола той по данным анализа активности ВЭЖХ 15 грамм промежуточного соединения примеПример 13 ра 4 (получаемого по реакции кетонного восстановления с использованием Z гоихм) растворялось Синтез (Р)-7-ацетил-8,9-дигидро-8-метил-5-(4в растворе 75мл диметилсульфоксида и 75мл динитрофенил)-7Н-1,3-диоксоло[4,5 п] метилформамида Раствор охлаждался до 7 - 9°С, [2,3]бензодиазепина а затем аэрировался 40% кислородом в азоте 17,5 грамм промежуточного соединения приДобавлялось 7,62 грамм 50% гидроокиси натрия в мера 12 суспендировалось в 175мл этилового воде, и получающаяся в результате смесь переспирта К перемешиваемой смеси добавлялось мешивалась в течение 3 - 4 часов Реакция пре1 7 грамма порошкообразной гидроокиси натрия рывалась, и при поддержании температуры = 12°С Получающаяся смесь перемешивалась в течение добавлялось 120мл толуола с последующим до1 часа К смеси добавлялось 88мл воды, и содербавлением смеси 45мл воды и 10мл соляной кижимое перемешивалось в течение 1 часа, по исслоты Фазы разделялись, и органический слой течение которого получающийся в результате промывался 75 мл-ми 10% водяного раствора осадок отделялся с помощью вакуумного фильттиосульфата натрия Органический слой, содеррования и промывался 175мл воды Вещество жащий указанное в заголовке промежуточное сосушилось в вакуумной печи при 70°С, давая 12,2 единение, переносился на следующую стадию грамм (86%) названного в заголовке соединения, которое имело 99,9% чистоту по данным анализа Пример 11 активности ВЭЖХ Синтез (З)-уксусная ки ел ота-[[6-(2Пример 14 гидроксипропил)-1,3-бензодиоксол-5-ил](4(Р)-7-ацетил-5-(4-аминофенил)-8,9-дигидро-8нитрофенил)метилен]гидразида метил-7Н-1,3-диоксоло[4,5 п] [2,3]К толуольному раствору промежуточного собензодиазепин единения примера 10 добавлялось 4 26 грамма При использовании продукта примера 13 поуксусного гидразида и 0 01 объема соляной кислолучалось целевое соединение с помощью экспеты Получающаяся в результате смесь нагрева 21 45309 22 риментальной процедуры, той же самой, что опидиоксоло[4,5 - п] [2,3]бензодиазепина (R)-7сана в примере 9 ацетил-8-9-дигидро-8-метил-5-(4-нитрофенил)-7Н1,3-диоксоло[4,5 - п] [2,3]бензодиазепин (38,93г) Пример 15 гидрировался в 730мл (19 объемов) 2В-3 этанола (Р)-7-ацетил-8,9-дигидро-8-метил-5-(4с использованием 7,79г 10% палладия на угле и нитрофенил)-7Н-1,3-диоксоло[4,5 п] [2,3]при 1 атмосфере водорода Когда анализ ВЭЖХ бензодиазепин показал, что исходный материал израсходовался, 1 05 грамма (З)-уксусная ки ел ота-[[6-[2катализатор удалялся фильтрованием, и фильт(гидрокси)пропил]-1,3-бензодиоксол-5-ил](4рат выпаривался, давая 38,7г сырого продукта нитрофенил)метилен]гидразида и 0,78 грамма Сырой продукт растворялся в 220мл (5,7 объемов) трифенилфосфина в 70мл тетрагидрофурана ох1 1 смеси вода этанол с помощью нагревания лаждалось до 0°С По каплям на протяжении 15 до кипения Смесь оставлялась охлаждаться, и минут добавлялось 0 57 грамма диэтилазодикарпродукт осаждался при температуре близкой к боксилата в 5мл тетрагидрофурана Получающаякомнатной Получающаяся в результате густая, ся смесь перемешивалась в течение 2 часов, заплохо перемешивающаяся суспензия перемешитем подогревалась до комнатной температуры в валась при комнатной температуре, а затем охлатечение 2 часов Смесь переносилась в делительждалась на водно-ледяной бане Твердое вещестную воронку, и раствор промывался 1 норм НСІ, во отделялось с помощью фильтрования и водой и солевым раствором Органическая фаза сушилось в вакуумной печи при 55°С на протяжесушилась над сульфатом магния, фильтровалась нии ночи, давая 31,6г очищенного продукта Еще и концентрировалась с помощью роторного испаодна перекристаллизация с использованием тех рения Остаток элюировался через силикагельную же условий давала 28,7г (80%) продукта после колонку (1 1 этилацетат гексан) Фракции, соСушки под вакуумом в течение 3 дней при 65°С и держащие требуемое соединение, концентриро3 дней при комнатной температуре Продукт сувались до желтого масла, которое затвердевало шился очень медленно, и в этот момент в образце при стоянии Желтое кристаллическое вещество все еще было 1,6% этанола Анализ с помощью суспендировалось в 30мл метиленхлорида и гекдифракции рентгеновских лучей (XRD), ЯМР в сана (3 7) при 0°С Осадок удалялся фильтроатвердом состоянии (SS ЯМР) и дифференциальнием, и фильтрат концентрировался до желтой ная сканирующая калориметрия (DSC) указывали пены Остаток суспендировался в 10мл этанола, на то, что образовалась полиморфная форма I который подогревался до температуры дефлегмации, затем медленно охлаждался до комнатной Пример 20 температуры Осадок собирался фильтрованием и Синтез формы II (Р)-7-ацетил-5-(4сушилдя в вакуумной печи при 60°С, давая 0,51 аминофенил)-8 9-дигидро-8-метил-7Н-1,3грамма (50%) названного в заголовке продукта диоксоло[4,5 - п] [2,3]бензодиазепина (100% ЕЕ), который имел чистоту 98,3% по дан(Р)-7-ацетил-8,9-дигидро-8-метил-5-(4ным анализа ВЭЖХ активности нитрофенил)-7Н-1,3-диоксоло[4,5 п] Пример 16 -18 [2,3]бензодиазепин (8 63г) гидрировался в 170мл (19 объемов) 2В-3 этанола с использованием 0 5мл суспензии замороженных дрожжей, со0,86г 10% палладия на угле и 4,59г карбоната амдержащей микроорганизм указанный в таблице 1, мония в 5мл воды в качестве источника переноса добавлялось к 50мл солодово - дрожжевой среды водорода Когда анализ ВЭЖХ указывал на то, что в 250мл колбе После встряхивания в течение 48 исходное вещество израсходовалось, катализатор часов 1,0мл культуры добавляется к дополниудалялся с помощью фильтрования, и фильтрат тельным 50мл среды и встряхивается еще в течевыпаривался, давая 8,19г сырого продукта Сырой ние 48 часов Добавляется 3,4продукт растворялся в 50мл (6 0 объемов) 1 1 метилендиоксифенилацетон до тех пор, пока ковода этанол при нагревании до кипения Смесь нечная концентрация не будет Юграмм/литр, наоставлялась охлаждаться до комнатной темпераряду с 1мл 10% глюкозы Культуры инкубируются туры, а затем охлаждалась в ванне из смеси льда и встряхиваются в течение 24 часов, затем аналии воды Твердое вещество отделялось фильтрозируются с помощью ВЭЖХ на присутствие хиванием и сушилось в вакуумной печи при 60°С на рального спиртового промежуточного продукта протяжении ночи, давая 7 41 г (93%) очищенного примера 1 продукта Крупные кристаллы содержали 5,0% этанола (ГХ) и 4 2% воды (КГ) Анализ с помощью Таблица 1 XRD, SS ЯМР и DSC указывал, что образовалась форма II полиморфа Пример Микроорганизм /ІСТОЧНИК % кон- % № версии ЕЕ Пример 21 16 Candida famata (С f ) A T C C 00 — Синтез формы III (Р)-7-ацетил-5-(4аминофенил)-8,9-дигидро-8-метил-7Н-1,326418 диоксоло[4,5 - п] [2,3]бензодиазепина 17 Zygosaccharomyces A T C C 77 8 99 5 (Р)-7-ацетил-8,9-дигидро-8-метил-5-(4rouxn (Z ro) 14462 нитрофенил)-7Н-1,3-диоксоло[4,5 п] 18 Vlortierrela isobellma N R R L 1 7 94 3 [2,3]бензодиазепин (2,04г) гидрировался в 20мл (Mi) 1557 (10 объемов) 2В-3 этанола с использованием 0,20г 10% палладия на угле и 1,47г формиата каПример 19 I I(JHIVIC(J І О лия в 4мл воды в качестве источника переноса Синтез Синтез формы 1 (Р)-7-ацетил-5-(4водорода Когда анализ ВЭЖХ показывал, что аминофенил)-8,9-дигидро-8-метил-7Н-1,3 23 45309 24 исходный материал израсходовался, катализатор казал необнаруживаемый уровень этанола (ГХ) и удалялся фильтрованием, и фильтрат выпари1 0% воды (KF) вался, давая 2 09г сырого продукта Сырой проАнализ XRD, SS ЯМР и DSC указывал на то, дукт растворялся в 12мл (6,0 объемов) 1 1 смеси что образовалась форма IV полиморфа вода этанол при нагревании до кипения Смесь Пример 23 оставлялась охлаждаться и затравливалась криАльтернативный синтез (3)-альфа-метил-1,3сталлами формы II примерно при 40°С После бензодиоксол-5-этанола достижения комнатной температуры смесь охлажК суспензии магниевых стружек (17г) в 50мл далась в ванне из смеси льда и воды Твердое тетрагидрофурана добавлялся по каплям раствор вещество отделялось фильтрованием и сушилось 5-бром-1,3-бензодиоксола (93,6г) После завершев вакуумной печи при 50°С в течение 24 часов, ния добавления смесь разбавлялась 250мл тетрадавая 1,45г (77%) очищенного продукта Анализ гидрофурана и получающаяся смесь перемешипоказал 0,05% этанола (ГХ) и 0 75% воды (KF) валась на протяжении ночи 13мл раствора Несмотря на использование затравки кристалла(0.78М) переносилось в круглодонную колбу, соми формы II полиморфа, аналих XRD, SS ЯМР и держащую иодид меди (1) (0,12г) Получающаяся DSC показал, что образовалась форма III полисмесь охлаждалась до -50°С, и медленно добавморфа лялся раствор (3)-(-)-пропиленоксида в Змл тетрагидрофурана, а затем перемешивался в течение Пример 22 10 минут Синтез формы IV (Р)-7-ацетил-5-(4аминофенил)-8,9-дигидро-8-метил-7Н-1,3Смесь разбавлялась эфиром Отделенная ордиоксоло[4,5 - h] [2,3]бензодиазепина (R)-7ганическая фаза промывалась водой и солевым ацетил-8,9-дигидро-8-метил-5-(4-нитрофенил)-7Нраствором Водные промывки воды экстрагирова1,3-диоксоло[4,5 - h] [2,3]бензодиазепин (25 2) лись эфиром (2 х), а объединенные органические гидрировался в 250мл (10 объемов) 2В-3 этанола растворы сушились над сульфатом магния, с использованием 2 Ог 10% палладия на угле и фильтровались и концентрировались Остаток 18 9г формиата калия в 20мл воды в качестве исочищался с помощью хроматографии на силикаточника переноса водорода Когда анализ ВЭЖХ геле (50% эфир в пентане), давая 1 66г желаемого показывал, что исходный материал израсходовалпродукта (91%) Анализ хиральной ВЭЖХ показал, ся, катализатор удалялся фильтрованием Фильтчто оптическая чистота вещества было 98 3% рат концентрировался с помощью перегонки до Пример 24 тех пор, пока не оставалось примерно 70мл (3 объема) этанола К раствору при дефлегмации Фармацевтическая готовая препаративная форма добавлялась вода, (93мл, 4 объема) Смесь осАктивный ингредиент 1мг 10мг 50мг 100мг тавлялась охлаждаться и затравливалась криКрахмал 444 5мг 435 8мг 396 2мг 346 6мг сталлическим продуктом примера 9 при 80°С ПоСиликоновая жидкость 4 49мг 4 22мг 3 84мг 3 36мг лучающаяся в результате суспензия оставлялась охлаждаться до комнатной температуры, и переИнгредиенты смешивались и заполнялись в мешиваться на протяжении ночи Твердое вещетвердые желатиновые капсулы размером 0 до ство отделялось фильтрованием и сушилось в веса содержимого 450мг вакуумной печи при 50°С в течение 24 часов, давая 19,8г (85%) очищенного продукта Анализ по ДП «Український інститут промислової власності» (Укрпатент) вул Сім'ї Хохлових, 15, м Київ, 04119, Україна (044)456-20- 90 ТОВ "Міжнародний науковий комітет" вул Артема, 77, м Київ, 04050, Україна (044)216-32-71
ДивитисяДодаткова інформація
Автори англійськоюAnderson Benjamin Alan, Hansen Marvin Martin, Vicenzi Jeffry Thomas, Very David Li
Автори російськоюАндерсон Бенджамин Алан, Хансен Марвин Мартин, Виченци Джеффри Томас, Вери Дэвид Ли
МПК / Мітки
МПК: A61P 27/02, A61P 25/14, C07D 317/58, A61K 31/55, A61P 25/18, C07B 63/00, A61P 25/28, A61P 25/06, A61P 25/16, C07D 491/056, A61P 25/00, C07D 491/04, A61P 25/22, A61P 43/00, A61P 25/04, A61P 25/20, A61P 1/08, C07D 491/113, C07D 493/04, A61P 25/08
Мітки: r)-7-ацетил-5-(4-амінофеніл)-8, 3]-бензодіазепіну, фармацевтична, композиція, 3-діоксоло[4, фізична, 5-h]-[2, форма, 9-дигідро-8-метил-7н-1, отримання, спосіб
Код посилання
<a href="https://ua.patents.su/12-45309-fizichna-forma-r-7-acetil-5-4-aminofenil-8-9-digidro-8-metil-7n-1-3-dioksolo4-5-h-2-3-benzodiazepinu-sposib-otrimannya-ta-farmacevtichna-kompoziciya.html" target="_blank" rel="follow" title="База патентів України">Фізична форма (r)-7-ацетил-5-(4-амінофеніл)-8, 9-дигідро-8-метил-7н-1, 3-діоксоло[4, 5-h]-[2, 3]-бензодіазепіну, спосіб її отримання та фармацевтична композиція</a>
Фізична форма (r)-7-ацетил-5-(4-амінофеніл)-8,9-дигідро-8-метил-7н-1,3-діоксоло[4,5-h][2,3]бензодіазепіну, спосіб її одержання, фармацевтична композиція, що її містить

Номер патенту: 44698
Опубліковано: 15.03.2002
Автори: Змієвскі Мілтон Джозеф, мол., Хансен Марвін Мартін, Гролоу Едвард Грант, Вері Девід Лі, Віченці Джеффрі Томас, Андерсон Бенджамін Алан
МПК: C07D 491/056, C07B 63/00, A61P 25/28, A61P 25/16, A61P 25/08, A61P 25/04, A61P 25/18, A61P 1/08, A61K 31/55, A61P 25/22, A61P 27/02, A61P 25/14, C07D 491/04, A61P 25/00, C07D 493/04, A61P 25/06, C07D 317/58, A61P 43/00
Мітки: композиція, містить, форма, фізична, одержання, спосіб, r)-7-ацетил-5-(4-амінофеніл)-8,9-дигідро-8-метил-7н-1,3-діоксоло[4,5-h][2,3]бензодіазепіну, фармацевтична
Формула / Реферат:
1. Физическая форма (R)-7-ацетил-5-(4-аминофенил)-8,9-дигидро-8-метил-7Н-1,3-диоксоло[4,5-h][2,3]бензодиазепина, имеющая порошковую рентгенограмму с d параметрами 12,78, 9,48, 8,99, 8,64, 8,23, 6,39, 6,27, 5,73, 4,01 и 3,96 .2. Физическая форма (R)-7-ацетил-5-(4-аминофенил)-8,9-дигидро-8-метил-7Н-1,3-диоксоло [4,5-h][2,3] бензодиазепина по п. 1 для использования при изготовлении медикаментов, применяемых в качестве антагониста...
Спосіб отримання 1-(гідразинокарбоніл)-метил-1, 2-дигідро-зн-1, 4-бенздиазепін-2-онів

Номер патенту: 9415
Опубліковано: 30.09.1996
Автори: Меринова Серафима Василівна, Гарібова Таісія Леонівна, Андронаті Сергій Андрійович, Козловська Марина Михайлівна, Богатський Олексій Всеволодович, Жиліна Зінаїда Іванівна, Смольникова Ніна Михайлівна, Яворський Олександр Степанович, Якубовська Лідія Миколаївна, Вороніна Тетяна Олександрівна, Кобзарева Ольга Василівна, Вальдман Артур Вікторович
МПК: C07D 243/26
Мітки: спосіб, отримання, 4-бенздиазепін-2-онів, 2-дигідро-зн-1, 1-(гідразинокарбоніл)-метил-1
Формула / Реферат:
Способ получения 1-(гидразинокарбонил)-метил-1,2-дигидро-3Н-1,4-бенздиазепин-2-онов общей формулы Iгде R1-СН3, Н, Сl, Вr, NO2;R2 - H, Cl,путем взаимодействия 1-(метоксикарбонил)-метил-1,2-дигидро-3Н-1,4-бенздиазепин-2-онов с гидразингидратом в среде этанола при температуре от комнатной до температуры кипения реакционной смеси, отличающийся тем, что, с целью повышения выхода целевого продукта и ускорения процесса,...
Кристалічна форма (-)-цис-4-аміно-1-(2-гідроксиметил-1,3-оксатіолан-5-іл)-(1н)-піримідин-2-ону (варіанти), способи її отримання та фармацевтична композиція

Номер патенту: 41265
Опубліковано: 17.09.2001
Автори: РАВЕНСКРОФТ Пол, ЕВАНС Пол, РОБЕРТС Тоні Гордон
МПК: A61K 31/505, C07D 411/00, A61P 31/12
Мітки: кристалічна, способи, отримання, варіанти, цис-4-аміно-1-(2-гідроксиметил-1,3-оксатіолан-5-іл)-(1н)-піримідин-2-ону, фармацевтична, форма, композиція
Формула / Реферат:
1. Кристаллическая форма (-)-цис-4-амино-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-(1Н)-пиримидин-2-она в виде бипирамидальных кристаллов, имеющих точку плавления выше 170 °С и полосы поглощения в ИК-спектре при значениях 920 и 850 волновых чисел.2. Кристаллическая форма по п. 1, имеющая точку плавления 177-178 °С.3. Кристаллическая форма по любому из пп. 1-2, практически свободная от кристаллической формы, имеющей полосу...
N-метил-n-[(1s)-1-феніл-2-((3s)-3-гідроксипіролідин-1-іл)етил]-2,2- дифенілацетамід гідрохлорид, спосіб його отримання та фармацевтична композиція

Номер патенту: 41978
Опубліковано: 15.10.2001
Автори: Барбер Ендрю, Бешманн Клаус, Берес Холгер, Нейенфельд Стеффен, Штеін Інге
МПК: A61K 31/40, C07B 63/00, A61P 29/00, A61P 1/00, A61P 25/04, C07D 207/12
Мітки: отримання, n-метил-n-[(1s)-1-феніл-2-((3s)-3-гідроксипіролідин-1-іл)етил]-2,2, дифенілацетамід, гідрохлорид, фармацевтична, спосіб, композиція
Формула / Реферат:
1. N-метил-N-[(1S)-1-фенил-2-((3S)-3 -гидроксипирролидин-1-ил)этил] -2,2-дифенилацетамид гидрохлорид в термостойкой кристаллической модификации.2 N-метил-N-[(1S)-1-фенил-2-((3S)-3-гидроксипирролидин-1-ил)этил]-2,2-дифенилацетамид гидрохлорид по п. 1 с точкой плавления 221-226°С.3. Лекарственное средство, содержащее активнодействующее вещество и обычные добавки, отличающееся тем, что в качестве активного вещества оно содержит...
Спосіб отримання 4-ацил-2,3-дигідро-1,4-бензоксазинів чи бензтиазинів

Номер патенту: 4761
Опубліковано: 28.12.1994
Автор: Ханс Мозер
МПК: C07D 279/00, C07D 265/28, A01N 25/32
Мітки: отримання, спосіб, 4-ацил-2,3-дигідро-1,4-бензоксазинів, бензтиазинів
Формула / Реферат:
Способ получения 4-ацил-2,3-дигидро-1,4-бензаксазинов или -бензтиазинов формулыR1- С1-С2-алкил, замещенный одним, двумя или тремя атомами галогена, или С2-алкенил, замещенный тремя атомами галогена, R2 и R3 независимо друг от друга - водород или метил;R4 и R5 независимо друг от друга - водород или хлор, или метил, отличающийся тем, что соединение формулыгде Х, R2, R3, R4 и R5 имеют указанные...
Попередній патент: Спосіб заповнення порожнеч за кріпленням підземних виробок
Наступний патент: Інгібітори 5-ліпоксигенази, фармацевтична композиція та спосіб лікування
Випадковий патент: Енергетичний вузол пристрою для ударної обробки