Спосіб отримання похідних 1,3-оксазолідин-2 она чи їх кислотно-адитивних солей
Номер патенту: 4759
Опубліковано: 28.12.1994
Автори: Харухіко Сінозакі, Коіті Хасімото, Міцуо Масакі, Тосіро Кімісіро, Наойя Моріто, Мазару Сато







Формула / Реферат
Способ получения производных 1,3-оксазолидин-2-она общей формулы
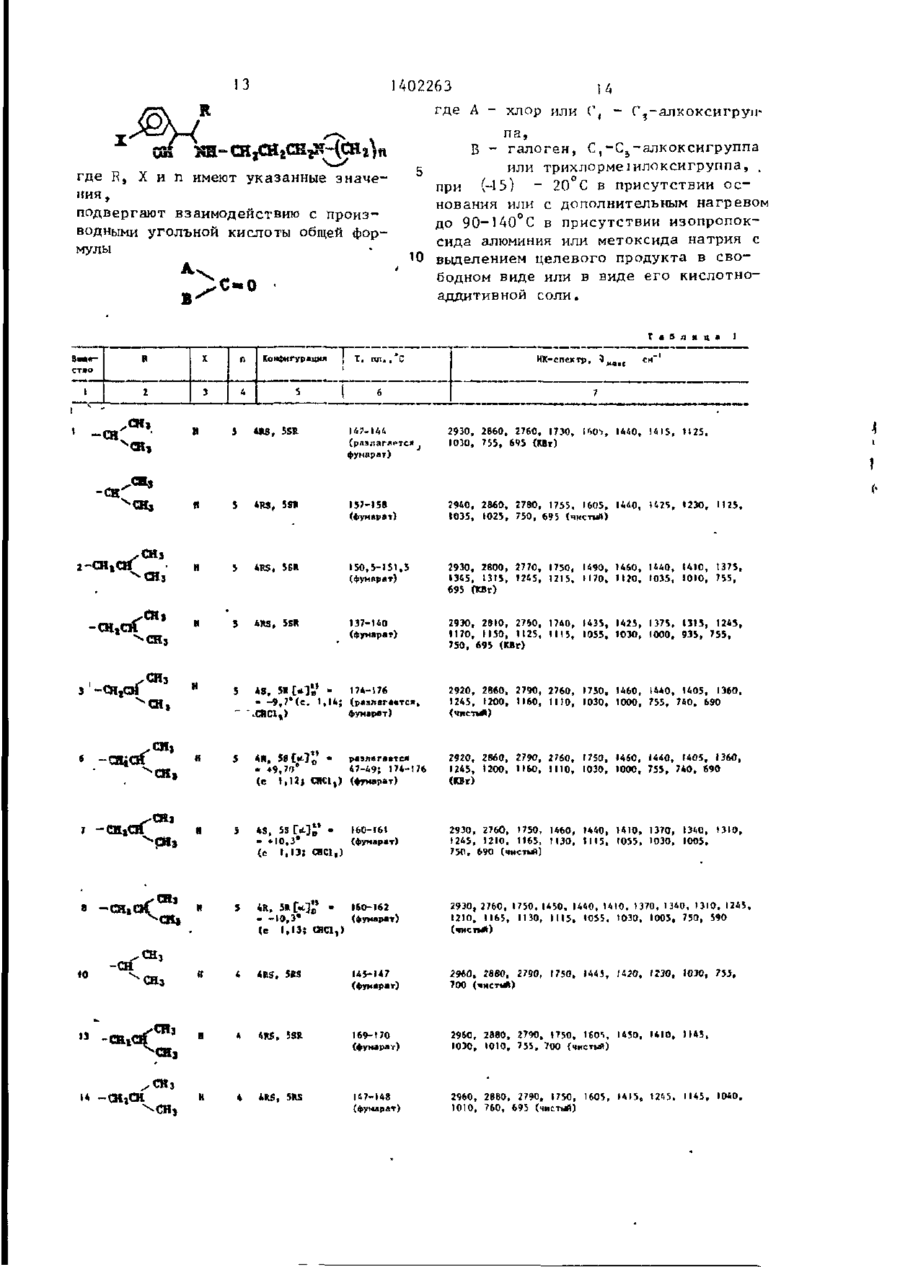
где R - алкильная группа с прямой или разветвленной цепью, имеющая 3-8 атомов углерода; Х- водород, галоген, С1-С3-алкил или С1-С3-алкоксигруппа; n = 4...6, целое число, или их кислотно-аддитивных солей, отличающийся тем, что соединение общей формулы
где R, Х и n имеют указанные значения, подвергают взаимодействию с производными угольной кислоты общей формулы
где А- хлор или С1-С3-алкоксигруппа,
В - галоген, С1-С3-алкоксигруппа или трихлорме-тилоксигруппа, при (-15)-20°С в присутствии основания или с дополнительным нагревом до 90-140°С в присутствии изопропоксида алюминия или метоксида натрия с выделением целевого продукта в свободном виде или в виде его кислотно-аддитивной соли.
Текст
Изобретение к а с а е т с я производных гетероциклических веществ, в ч а с т н о сти 1,3-оксазолидин~2~она общей ф—лы 1; І * Cg-алкил; Х-Н, R - н - или галоген, С 1 -С 5 -алкил или С ( - С 5 алкоксигруппа; п = от 4 до 6, или их кислотно-аддитивных солеи, г проявляющих антиглутаминовую активность. Цель - создание более активных веществ указанного класса. Их синтез ведут из соединений ф-л ( I I ) и ( I I I ) : (АВ)=С=О ( I I ) , Х-С 6 Н 4 -CH(OH)-CRK ( I I I ) , где А - СІ или С,—С^-алкоксил; В - галоген; С н -С 3 ~ -алкокси- или трихлорметилоксигруппа; X, R, п, К указаны выше. Процесс ведут при (-15)-20 С в присутствии основания или с дополнительным нагреванием до 90-140 С в присутствии изопропоксида алюминия или метилата натрия. Целевой продукт выделяют в свободном виде или в виде кислотноаддитивной соли. Новые вещества при концентрации 20 10~5 моль/л обеспечивают степень блокирования глутаминовой кислоты на 53—92% при токсичности LDjo = 18—75,1 мг/кг. 2 и л , , 4 табл. S О го 00 Г РП -л 1402263 1 2 дин-2-она общей формулы Изобретение относится к способу получения производных 1,3-оксазоли (і) или их кислотно-аддитивных солей, обладающих антиглутаминовой а к т и в ностью, которые могут найти применение в медицине и в сельском хозяйстве. Целью изобретения является у с и л е ние антиглутаминовой активности в ряду производных 1,З-оксазолидин-2-она, П р и м е р 1. (4RS, 5 S R ) - 4 - ( l ~Метилэтил)-5-фенил-3-(3-пиперидинопропил)-!,3-оксазолидин-2-он. Дигидрохлорид (IRS, 2SR)-3-MeTmi-1-фенил-2—(3-пиперидинопропиламино)-бутан-1-ола (755 мг, 2,00 ммоль) суспендируют в 12 мл ]0%-ного водного раствора гидроксида натрия, после ч е го добавляют 28 мл диэтилового эфира. Смесь перемешивают, и когда она с т а нет прозрачной, ее охлаждают льдом. В течение 1 ч по каплям добавляют 20%-ный раствор трихлорметилового эфира хлормуравьиной кислоты (ТЭХ) в толуоле ( 4 , 0 м л ) . После добавления ТЭХ смесь перемешивают при комнатной температуре 30 мин. Отделяют органический слой, промывают его один раз солевым раствором, сушат над сульфатом натрия и выпаривают при пониженном давлении. Полученный неочищенный продукт очищают хроматографически на колонке с силикагелем (7 г силикагеля, растворитель — хлороформ: :метанол = 2 0 : 1 ) , получают 613 мг ц е левого продукта в виде светло-желтых кристаллов (выход 93%). 1 ИК-спектр (KBг) } м а к с см" : 2930, 2860, 2760, 1730, 1605, 1440, 1415, 1125, 1030, 755, 695. ЯМР-спектр (CDClj) хим. сдвиг: 0,68 (ЗН, д , J » 7 Гц, СНОТ,); 0,89 (ЗН, д , J = 7 Гц, СНСН3); 1,20-2,01 (9Н, м, С Н ( О Ц ) г 1 2,12-2,25 (6Н, м, & 2 , 9 2 - 3 , 3 0 (1R, м , О ь II о 15 3,54-4,00 (2Н, м, J O ж \ ~~ о 5,60 (1Н, g, J = 8 Гц, (СН-О); 7,1620 7,48 (5Н, м,ароматического кольца протоны), (4RS, 55К)-4-(1-Метилэтил)-5-фенил-З-(З-пиперидинопропил)-!,3-оксазо~с лидин-2*-он, фумарат. Полученное предыдущее соединение (430 мг, 1,30 ммоль) растворяют в 4 мл этанола, добавляют 5 мл горячего этанольного раствора фумаровой кислоты (151 мг, і,30 ммоль). Раствор 30 упаривают при пониженном давлении, остаток растворяют в ацетоне и дают ему отстояться в течение ночи. После охлаждения смеси льдом выпавшие в осадок кристаллы собирают фильтраци35 ей, трижды промывают ацетоном, получают 491 мг целевого вещества в виде белых кристаллов (выход 85%). Т. пл. 142-144°С ( р а з л а г а е т с я ) . ИК-спектр ( К В г ) - 5 м а к с , см"': 3440, 40 2940, 2880, 1740, J700, 1640, J560, 1420, 1250, 1200, 980, 750, 700. ЯМР-спектр (CDClj-CD3OD = 6:1) хим. сдвиг: 0,65 (ЗН, д, J = 7 Гц, СНСН3); 0,86 (ЗН, д , J = 7 Гц, СНСН3); 45 1,35 - 2>,35 (9Н, м, СН(СН,)^, 50 ,70-3,88 (8н, м, СН г СН 2 СН 2 іт' 55 4,04 —* -сн СН 7 — СН о -• - _ 10 (Ш, дублеты, J = 8 Гц, 2 Гц, -к п з 4 402263 ' ' СН-О); 7,10-7,56 (5Н, протоны арома5,75 (1H, д , J = 8 Гц, СН-О); 6,78 тического кольца), (2Н, с , -СН=СН-); 7,16-7,44 (5Н, м, протоны ароматического кольца). (4RS, 53а)-4-(2-Метилпропил)-5П р и м е р 2. (IRS, 2SR)-2-[N-. t -фенил-З-(З-пиперидинопропил)-1,3-ок-Этоксикарбонил-К-(3-пийеридинопро- • сазолидин-2-он. пил)амино] -4-метил-1-фенилпентан-1Толуольный раствор (12 мл) полу-ол. ченного предыдущего вещества (633 мг, Б раствор (1RS, 25И)-4-метил-11,62 ммоль) нагревают на мааляной -фенил-2-(3-пиперидинопропиламино) 10 бане приблизительно до 130 С, чтобы пентан-1-ола (637 мг, 2,00 ммоль) в удалить воду, присутствующую в систе6 мл хлороформа добавляют по каплям, ме. Понижают температуру бани до с охлаждением льдом и перемешиванием 100 С и добавляют мзопропоксид алюполовину раствора (4 мл) этилового миния (16 мг, 0,08 ммоль). Затем эфира хлоругольной кислоты (543 мг, 15 поддерживают температуру бани при 5,0 ммоль) в хлороформе, поддерживая 130-140 С, чтобы посредством дистилтемпературу реакции ниже 15 С. После ляции удалить азеотропную смесь этого оставшуюся половину раствора и (9 мл) этанола и толуола приблизитель8 мл водного раствора гидроксида но в течение 1 ч. После охлаждения натрия (200 мг, 5,0 ммоль) по каплям 20 к остатку добавляют этилацетат. Р а добавляют, поддерживая температуру створ сначала промывают насыщенным - : реакции ниже 15°С т а к , что добавление водным раствором сульфата натрия и обоих растворов завершается одноврезатем раствором соли. Полученный менно. Полученную смесь перемешивают раствор сушат над сульфатом натрия и при 5 С 1 ч , после чего отделяют 25 упаривают при пониженном давлении, хлороформный слой, сушат его над получают 558 мг целевого вещества сульфатом натрия и затем упаривают в виде светло-желтоватых кристаллов при пониженном давлении, получают (выход 10QZ). 781 мг целевого вещества в виде в я з КК-спектр (в чистом виде)^маіхс кого светло—желтоватого масла (выход 30 см' 1 : 2930, 2800, 2770, 1750, 1490, 300%). 1460, 1440, 1410, 1375, 1345, 1315, ИК-спектр (чистого масла) >*ма_кс 1245, 1215, 1170, 1120, 1035, 1010, см"1: 3440, -2940, 2870, 2800, 1680, 755, 6 9 5 . : 1465, 1420, 1310, 1250, 1230, 1100, ЯМР-слектр ) хим сдвиг: 750, 700. 35 0,44-0,68 (ЗН, м СНСНЭ); 0,68-0,92 ЯМР-спектр (CDC13) хим. сдвиг: (ЗН, м, СНСН,); 3 , 0 0 - 1 , 2 6 (ЗН, м 0,67 (ЗН, д . J - . 7 Гц,СНСНэ); 0 , 8 0 С Н „ С Н ( С Н , ) 2 ) ; 1 , 3 0 - 1 , 9 2 (8Н, м (ЗН, д , J * 7 Гц, СНСН3); 0,96-2,72 (18Н, м, СН г СН(СН 3 ) 2 , 2,16-2,52 (бн, 40 «У 2 І сн г сн "у) о 12 ,7 ( Н т, З , J - ' П , . ^ ^ . ; 45 ; о 2,88-3,64 (2Н, м.стСН^ ', 3,90-4,48 о " О Н , м,СКСН * 2,88-3,24 (1Н, м 3 > 50 3,32-3,72 ( Ш , м» " Н 3,84-4,12 (1Н, м, Г ІІ \ ) ; 4 > 1 6 г к в ' ....V-\;:,; * у • J = 55 - 7 Гц, ^ ^ * г / ; 4,76-5,00 ( Ж , м, 3,52 (1Н, д, J = 8 Гц, СН-О); 7,33 (5Н, 'м, ароматические протоны). П р и м е р 3. (4S, 5R)-4-(2-Me'тилпоопилі -5-фенил-3-(3-пиперидино 5 * 1402263 пропил)-!,З-оксазолидин-2-он, фумарат, (1R, 23)-4-Метил-1-фенил-2-(3-пиперидинопропиламино)-пентан-1-ол дигидрохлорид (3,0 г, 2,56 ммоль) растворяют в воде и превращают в основание водным 15%-ным раствором NaOH с последующей экстракцией хлоророфором. Хлороформный экстракт промывают водой и водным насыщенным раствором NaCl. После высушивания хлороформного экстракта при помощи Na,SO4 растворитель о т г о няют при пониженном давлении. Остаток (820 мг) высушивают в вакууме и растворяют в диэтилкарбонате (0,9 мл),К полученному раствору прибавляют NaOMe (4 мг) и смесь перемешивают при 95 С 30 мин. Реакционную смесь выпаривают при пониженном давлении и остаток экстрагируют ЕЦО. Эфирный раствор промывают водой и водным насыщенным раствором NaCl, высушивают над N a 2 S 0 4 . Растворитель отгоняют при пониженном давлении и остаток (820 мг) растворяют в EtOH (2,5 мл) с последующим прибавлением фумаровой кислоты ньй слой экстрагируют бензолом (200 мл). После промывания сначала водой, затем водным раствором бикарбоната натрия и солевим раствором объединенный органический слой сушат над сульфатом натрия и затем выпаривают при пониженном давлении. Остаток обрабатывают этанолом до получе10 ния кристаллов, которые при нагревании растворяют в 80 мл этилового спирта, и добавляют 160 мл гексана. Выпавшие в осадок кристаллы отфильт~ ровывают и промывают смешанным ра15 створом (240 мл) этанола и гексана (1:2) и затем гексаном. Кристаллы 20 25 (277 мг, 2,38 ммоль). Смесь растворяют с нагреванием в фумаровой кислоте 30 и полученному раствору дают выдержку в течение ночи при комнатной температуре. Осажденные кристаллы собирают фильтрованием, промывают дважды EtOH (1,5 мл) и затем высушивают и 35 получают 825 мг целевого соединения в виде белых кристаллов (выход 70%). Получение исходных соединений. П р и м е р А. 2-(1,3-Диоксо-2-азаиндан-2-ііл)— 4-меіил-і-фенилпентан-1-он. Хлористый тионил (3,8 мл, 480 ммоль) добавляют в смесь 2 - ( 1 , 3 -диоксо—2—аэаиндан-2~ил)-4-метилпентановой кислоты (83,61 г, 320 ммоль) и 320 мл бензола. Смесь кипятят с обратным холодильником 2 ч. Растворитель и избыток хлористого тионила удаляют посредством дистилляции при пониженном давлении, добавляют 320мл бензола. Бензол удаляют и добавляют свежую порцию (480 мл) бензола, чтобы образовался раствор. Сразу же в раствор добавляют безводный хлористый алюминии (106,7 г, 800 ммоль) и смесь перемешивают при комнатной температуре 5 ч. Реакционную смесь выливают в ледяную воду (700 м л ) . Вод 40 45 50 55 сушат на воздухе, получают 74,6 г целевого соединения в виде белых кристаллов (выход 7J%). П р и м е р Б. (IRS, 2SR)-2— (1 ,3-Диоксо-2-азаиндан-2-ил}~4-метил-1-фенилпентан-1-ол и (IRS, 2RS)-2-(1,3—диоксо-2-азаиндан-2-ил)-Дметил-1-фенилпентан-1-ол. Цианоборгидрид натрия (177 г, 2,82 ммоль) добавляют в течение 3 ч к раствору вещества (141,4 г, 440 ммоль), полученного в примере 1, в хлороформе (660 мл) и в уксусной кислоте (440 мл) , поддерживая температуру реакционной смеси ниже 30 С, Образовавшуюся смесь перемешивают при комнатной температуре еще 3 ч и добавляют 1 л хлороформа и 1,4 л воды. Отделяют органический слой и промывают его дважды водой, один раз водным раствором бикарбоната натрия и один раз солевым раствором. После высушив ания над сульфатом натрия органический слой упаривают при пониженном давлении, получают142 г белых кристаллов. Эти кристаллы загружают в хроматографическую колонку с силикагелем (2,8 кг силикагеля, растворитель - бензол), получают сначала 94.3 г (IRS, 2SR)-H3OMepa в виде белых кристаллов (выход 66%) и затем 48.4 г (IRS, 2RS)-H3OMepa в виде белых кристаллов (выход 34%), П р и м е р В. (IRS, 2RS)-2-(1,3-Диоксо-2-азаиндан-2-ил) -4 -метил- 1-фениппентан-1-ол. Изопропоксид алюминия (125,6 г, 615 ммоль) добавляют в суспензию 2~0,3-диоксо-2-азаиндан-2-ил)~4-метил-1-фенилпентан-і-она (72,3 г, 225 ммоль) в изопропиловом спирте (1000 мл). Эту смесь кипятят с обрат* ным холодильником 6,5 ч . Изопропило 1402263 8 батывая его гексаном, и полученные выи спирт удаляют посредством дистилкристаллы подвергают перекристаллиляции при пониженном давлении, к остатку добавляют 800 мл этилацетата зации из гексана, получая 12,2 г и водный раствор сульфата натрия. с целевого соединения в виде белых криОбразовавшийся органический слой десталлов (выход 7 3%). кантируют и остаток дважды промывают П р и м е р Е. (IRS, 2RS)-2-AMHHOэтилацетатом (200 мл). Объединенный -1-фенилгептан-1-ол, органический слой промывают солевым Уксусный ангидрид [10 мл) добавляраствором. Органический раствор су10 ют к {1RS, 28Н)-2-амино~1-фенилгептаншат и растворитель удаляют посредст-1-олу (4» 15, 20 ммоль) и смесь навом дистилляции при пониженном давлегревают при 70 С 10 мин. После охнии. Остаток дважды подвергают перелаждения реакционную смесь выливают кристаллизации из бензола, получают в 100 мл воды, в которую добавляют 29,3 г целевого вещества в виде бе15 хлороформ и постепенно водный раствор лых кристаллов (выход 40%). гидроксида натрия. Подщелоченный раствор экстрагируют хлороформом, экстП р и м е р Г. (IRS, 2SR)-2-Амиракт высушивают. Растворитель удаляют но~4-метил-1-фенилпентан-1-ол. дистилляцией при пониженном давлении, При нагревании (50 С) растворяют (IRS, 2SR)-2-(l,3-диоксо -2-азаиндан- 20 получают бесцветное масло. Это масло -2-ил)-4-метил-1~фенилпентан-1-ол охлаждают льдом, добавляют 20 мл (80,0 г, 247 ммоль) в 800 мл этиловохлористого тионила. Смесь перемешиго спирта, после чего добавляют смесь вают при комнатной температуре 20 мин, 85%-ного гидразингидрата (19,0 мл) С предосторожностями, малыми количев 200 мл этилового спирта. Образовав- 25 ствами добавляют воду (30 мл) и смесь нагревают и кипятят с обратным холошуюся смесь кипятят с обратным холо- , дильником 2 ч . Смесь охлаждают, додильником 3 ч и после охлаждения бавляют воду. После промывки см^си льдом добавляют 700 мл соляной кисэфиром полученный водный слой отделоты ( 4 н , ) . Смесь перемешивают при комнатной температуре 30 мин. Нера30 ляют и подщелачивают водным раствостворенное вещество удаляют фильтраром гидроксида натрия. Смесь экстрацией через целлит и затем поомывают гируют три раза хлороформом и высу140 мл соляной кислоты ( 4 н , ) , Промывшивают. Органический раствор упариную жидкость добавляют к фильтрату, вают при пониженном* давлении, полуиз раствора удаляют этиловый спирт 35 чают белые кристаллы. Эти кристаллы посредством конденсации при пониженподвергают перекристаллизации из гек: ном давлении. При охлаждении льдом сана, получают 2,73 г целевого с о е добавляют 6 н.раствор гидроксида натдинения в виде белых кристаллов (вырия (570 мл) и смесь экстрагируют ход 66%). три раза хлороформом. После высушива- 40 П р и м е р Ж. (IRS, 2SR)-2-AWHHOния над сульфатом натрия упаривают -1-(4-метоксифенил)-4-метилпентан-1органический слой, получают 43,2 г -ол. целевого соединения в виде белых Растворяют в 88 мл уксусной кислокристаллов (выход 91%). ты 2-оксиимино—1-(4-метоксифенил)-4П р и м е р Д. 4-Метил-1-(4-ме- 45 -метилпентан-1-он ( 8 , 7 2 г , 36,1 ммоль), тилфенил)-2-(1,3~диоксо-2-азаинданК этому раствору добавляют 0,87 г -2-ил)-пентан-1-он. катализатора - 5%-ного палладия на Тщательно смешивают 2-бром-4-меактивированном у г л е , и реагент к а т а тил—1-(4-метилфенил)-пентан-!-он литически гидрируют при нормальном (13,5 г, 50 ммоль) и фтальимид калия 50 Давлении и 80°С до тех пор, пока не (9,26 г, 50 ммоль) и нагревают при поглотится тройное молярное количест160 С в течение 2 ч . После охлаждево водорода по отношению к реагенту. ния в реакционную смесь добавляют После удаления катальзатора фильтра100 мл этилацетата и 50 мл воды. Орцией при пониженном давлении удаляют ганический слой отделяют и промывают 55 уксусную кислоту. Остаток растворяют раствором соли. Органический раствор в 80 мл соляной кислоты (1 н . ) . Этот сушат над сульфатом натрия и упарираствор дважды промывают 30 мл эфира, вают при пониженном давлении. Остаток водный слой подщелачивают 20%-ным подвергают перекристаллизации, обраводным раствором гидроксида натрии. 1402263 10 Обработанный таким образом водный 5БК)-4-метил-5-фенил-3-(2-пирролидислой экстрагируют три раза хлорофорноэтил)-1,З-оксазолидин-2—он (соедимом, экстракты объединяют и промыванение 5 ) . ют раствором соли. Экстракт сушат Биологический опыт 1. Действие над сульфатом натрия, растворитель на анемичную децеребрационную ригидудаляют посредством дистилляции при ность. пониженном давпении, получают светлоОбразец анемичной децеребрационжелтоватые кристаллы (6,80 г ) . Эти ной ригидности готовят в с о о т в е т с т кристаллы подвергают перекристаллиза- і вии с методикой Фукуды и с о т р . , а ции из бензола н гексана, получают именно: самок крыс Уистара (.вес тела 5,24 г целевого вещества в виде бе270-350 г) держат на спине и под лых кристаллов (выход 63%), эфирным наркозом разрезают им шеи. После тогоу как трахеи и общие сонП р и м е р И. (IRS, 2SR)~4-MeTwi-1-фенил-2-(3-пиперидинопропиламино)~ 15 ные артерии открываются, в трахеи вставляют канюли, а затем на билатеп е н т а н - ї - о л , дигидрохлорид. ральные общие сонные артерии и пищеСмесь (1RS, 25Р)-2-амино-4-метилвод накладывают двойную лигатуру и -1-фенилпентан-1ола ( 4 0 , 6 г, их отрезают. После ^того обнажают 210 ммоль) и 1- (3-хлорпролил) пиперидина (34,0 г, 210 ммоль) сплавляют 20 затылочную кость, сквозь которую просверливают круглое отверстие, чтовместе при 50-70 С в атмосфере а з о т а . бы двукратно наложить лигатуру на Смесь нагревают на масляной бане до простирающуюся центрально основную 110 до 120 С 3 ч , После охлаждения артерию. Когда крыса начинает выхореакционную смесь растворяют в 750мл этанола при нагревании, добавляют 25 дить из анестезированного состояния, ее передние конечности становятся 17 мл концентрированной хлористоводоригидными. Измерения проводят породной кислоты. Смесь охлаждают и высредством записи электромиографичес— павшие в осадок кристаллы отфильтрокого (ЭМГ) отклика от мышцы. Импульвывают» Эти кристаллы снова суспендируют в 1200 мл этанола и кипятят 30 сы ЭМГ преобразуют в интегрированные за каждые 10 с величины и запи— , с обратным холодильником 1 ч . Смесь сывают на самописце в виде гисто-1 охлаждают, выпавшие кристаллы о т граммы. фильтровывают, промывают и сушат, получают 58,8 г целевого соединения в Влияние каждого испытуемого вевиде белых кристаллов (выход 72%), 35 щества на ригидность оценивают как т . пл. 268-270 С ( р а з л а г а е т с я ) . степень подавления. Эту степень вычисляют сначала с помощью определеВ т а б л . 1 представлены физико-хиния площади части с уменьшенным иммические характеристики новых соедипульсом ЭМГ на гистограмме по истенений ( і ) . Эффективность соединений формулы 40 чении 10 мин после введения физиологического солевого раствора каждого ( і ) определяют по блокирующему д е й испытуемого соединения (доза 3 мг/кг) ствию в отношении глутаминовой кисчерез бедренную вену и затем по слелоты, по релаксирующему действию на дующему уравнению: мышцы спинного мозга (понижение ригидности и высвобождающее действие 45 Степень подавления (0) = 100 а/А» на образцах анемичной децеребрационгде а - уменьшенная площадь импульса ной ригидности) и по уровням токсичЭМГ в результате введения ности. Для сравнительных целей и с испытуемого соединения; пользованы следующие соединения: гидА - площадь импульса ЭМГ, когда рохлорид толуперизона (соединение 1 ) ; 50 не вводят никакого испытуемогидрохлорид (4RS, 53К)-4~метил-5-фего соединения (контроль). нил-3-(2-пиперидиноэтил)-1,3-оксазоРезультаты приведены в табл. 2, ЛИДИН-2-ОН (соединение 2 ) ; (4RS, Биологический опыт 2. Блокирующее 5ЕБ)-4-метил-5~фенил-3-(2-пиперидинодействие в отношении глутаминовой этид)-1,3-оксаэолидин-2-он, гидрохло- 55 кислоты в иейромышечных соединениях рид,(соединение 3 ) ; (4RS, 5SR)-4-Meрака. тил~5-фенил-3-(2-пирролидиноэтил)• ,. -І.З-оксазолиднн-2-он, гидрохлорид, Следуют методу, описанному Ишидойи (соединение 4 ) ; гидрохлорид (4RS, с°тР-» т . е. в качестве материалов и 402263 для опыта используют обнаженную мышцу первых ходильных ног рака. Нейромышечный образец выдерживают в 6aHej с помощью которой физиологический раствор состава, ммольі NaCl \95; СаС12 ' l 8 ; KC1 5,4; трис-малеатный буфер (рН 7,5) Ю ; глюкоза 11, для использования в опыте с раком подвергают перфузии при комнатной температур е 10 и при постоянной скорости потока. В центральную часть мышечного волокна вставляют стеклянные микроэлектроды, каждый из которых заполнен ЗМ раство15 ром хлористого калия, для того чтобы записывать внутриклеточно изменения потенциала мембраны мышечной клетки. Блокирующее действие каждого испытуемого вещества в отношении глутами- 20 новой кислоты оценивают как степень подавления деполяризации, которая вызывается применением ванны из L-глу— таминовой кислоты (10" 4 моль/л) при пятиминутной предварительной обработ- 25 ке раствором испытуемого вещества, • Результаты представлены в табл. 3. Биологический опыт 3. Ингибиторное действие в ответ на L-глутамнно— vвую кислоту и ее аналоги в кортикаль- 30 ных нейтронах крыс. !2 ингибирует реакцию на глутаминовую кислоту в прямой зависимости от количества. На фиг. 2 показана регистрация скорости зажигания кортикального нейрона, возбужденного ионофоретическим глутаминовым импульсом (0,2 Гц, продолжительность 2 с ) . Предлагаемое соединение понижает частоту потенциалов действия, вызванных L-глутаминовой кислотой, а также тремя агонистами рецепторов глутамата, т . е . дигеновой кислоты, quisquelic кислотой и К-метил-1>-аспарагиновон кислотой. Из приведенных результатов следует, что исследуемое соединение ингибирует глутаматовую реакцию в кортикальных нейронах крыс. Биологический опыт 4. Острая токсичность. Используя самцов мыши dclN в соответствии с методом "вверх и вниз", определяет уровень острой токсичности каждого испытуемого вещества. Некоторые из испытуемых веществ растворяют в физиологическом солевом растворе и вводят через хвостовую вену. Результаты представлены в табл. 4. Таким образом, соединения ( і ) по своей биологической активности в отношении анемичной децеребрационной ригидности и блокирования глутаминовой кислоты превосходят известные соединения. Предложенные соединения могут быть использованы в медицине. L-Глутаминовую кислоту и соединение (4S, 5В)-4-(2-метилпропил)-3-[3- [(пергидроазепен-1-ил)пропил]-5-фенил-1,3-оксаэолидин-2-он] вводят " ионофореэно в кортикальный нейрон . крысы с использованием многоцитошд— ровой стеклянной микропилетки, наполФ о р м у л а и з о б р е т е н и я ненной L-глутаминовой кислотой (1М) и 40 исследуемым соединением (О,05М).*КонСпособ получения производных 1,3чик этой микропипетки фиксируют с ис-оксазолидин-2-она общей формулы пользованием микроманипулятора в положении, где получается наибольший глутаминовый ответ, который представлен экстрацеллюлярно зарегистрированными потенциалами действия, индуцируемыми ионофоретическим введением L-глутаминовой кислоты. На фиг. 1 показана регистрация скорости зажигания кортикального нейрона во время где R - алкильная группа с прямой введения 1г-глутаминовоЙ кислоты с усили разветвленной цепью, иметойчивыми постоянными потоками в приющая 3-8 атомов углерода; сутствии или отсутствии предлагаемоX - водород, галоген, С - С^-алго соединения в различных количествах кил.или С,-С 3 -алкоксигруппа; (ось ординат - частота потенциалов п * 4 . . . 6 , целое число, = действия, ось абсцисс - время). или их кислотно-аддитивных солей, Приведенные результаты (фиг. I) о т л и ч а ю щ и й ся тем, что означают, что предлагаемое соединение соединение общей формулы 13 402263 |4 где А - хлор или (\ - С,-алкоксигруппа, В - галоген, С,-С 3 ~алкоксигруппа 5 или трихлормеіилоксигруппа, , где R, X и п имеют указанные значепри (-15) - 20°С в присутствии о с ния , нования или с дополнительным нагревом подвергают взаимодействию с произдо 9О-14О°С в присутствии изолропокводными угольной кислоты общей форсида алюминия или метоксида натрия с мулы Ю выделением целевого продукта в свободном виде или в виде его кислотноаддитивной соли. Т а б л и ц а Конфигурация Т. пл, , С ИХ-спектр, ство 1 -си'* 5 « S , 5SR фумарят) -сн; з 157-1S8 (Фумарат) 2940, 2860, 2780, 1755, 1605, І4І0, 1425, 1230, 1035, 1025, 750, 695 (чисть*) 5 4R5, 5SR 150,5-151,5 (фунярат) 2930, 2800, 2770, 1750, 1490, 1460, 1440, 1410, 1375, 1345, 1315, 1265, 1215, 1170, 1120, 1035, 1010, 755, 695 (KBt) 4RS, 5S1 137-1ІО (фумарят) 29ЭО, 2810, 2760, 1740, 1435, 1425, 1375, 1315, 1245, 1170, 1150, 1125, 1115, 1055, 1030, 1000, 9 3 5 , 7 5 5 , 750, 695 ( И г ) 1125, 48, 5R[*lV 174-176 - - 9 , 7 # ( с . 1,14; (разлагается, ~ ".СЙС1%> фумврвт) 2920, 2860, 2790, 2760, 1750, 1460, 1440, 1405, IЭ60, 1245, 1200, 1160, 1110, 1030, 1000, 755, 740, 690 (чисть*) 5 4Н, 5ВС«ОУ * раэплгввтся е - *9,7О 47-49; 174-176 (с І«12і СЯС1,) (фумарат) 2920, 2860, 2790, 2760, 1750, 1460, 1440, 1405, 1360, 1245, 1200, 1160, 1110, 1030, 1000, 755, 740, 690 (Or) Н 5 48, 5S Г * } " - *10,3# (с 1,13; СНЫ.) 160-Г61 (фумарат) 2930, 2760, 1750, 1460, 1440, 1410, 1370, 1340, 1310, 1245, 1210, 1165, И З О , 1115, 1055, 10Э0, 1005, 750, 690 (чист**) Л 5 160-162 (фумарат) 29ЭО, 2760, 1750.1450, 1440, 1410, 1370, 1340, 1310, 1245, 1210, 1165, ИЗО, 1115, 1055. 1030. 1005, 750, 590 (чистій) Ш , 5R3 145-147 (фумарвт) 2960, 28ВО, 2790, 700 ( ч и с т і * ) ; , 5SR 169-170 (фумарат) 2960, 2880, 2790, 1750, 160-і, 1450, 1410, 1030, 1010, 755, 700 (чистій) 147-148 Сфумарат) 2960, 2880, 2790, 1750, 1605, 1415, 12Ь5. 1145, 1040, 1010. 760, 695 (чистьй) СИ, 8 -CHiCH ' СК, 4R, 5К М І - -I0.3(с 14 М25, 4RS, i S * 5 -СЯІСЙ И 2930, 2860, 2760, 1730, 1ft01, 1440, 1*15, 1030, 755, 645 (Иг) 3 Н -сн,сн to а в ( 3 2-СИ 7 } 5и 1 -CH |,1Э| фумарат) щ ^сн, 2920, 2860, 28)0, 171!, 1**5, 760, 695 (чист**) 1*10, 1360, 1025, 1005, 1-СИ, Ш , 5SR 152-153 (розлягя*гся, фумарят) 2950, 2880, 2820, 27В0. 1760, 1445, 1*10, ИЗО, (040, 1010, 755 (чисть*) Э-СЯ, at. 5 3 »RB, SSR 1*2-145 (разлагается^ фумарат) 29ЭО, 2860, 2760, 1750. 1605, U 4 0 , 1*05, И ї О , 1075. 750 Э-СН, 4-СН, 5 4 « , 3RS 5 2920, 2860, 2760, 1750, 1*35, 1030, 775, 750, 690 ( ^ с т м й ) 4RS, SRS 4-Ot, 25 ) 418, 3RS 132-134 (фумярат) Ш 5 , 1210, 1405, 1170, І Ї І 5 , 2940, 28SO, 2780, 1760, 1445, 1413, П 2 3 , 1040, 1015, 815, 760 (чис-пЛІ СИ, 2940, 2870, 27ТО, 1755, 1605, 1440, 1410, 1250, 1120, 10.35, tOlO, &10, 755 (чисти*) 27 -ШгСН 4-ОСН, 5 4RS, 55И 127,5-129,5 2930, 2760, 1750, 1610, 1510, 14Э5, 1250, 1170, 1010, 825, 750 0, 1420, {"исты") 5, 690 Ю25, ( ІИ 41 -«Я,),™, Н 5 4KS, 5SB 92-94 (фумарат) 2920, 2850, 2770. I7S0. 1450, М О , 1010, Ю Н ) , 715, 69S ( ч и с т ь * ) И З О , 1120, 4Ї -(СВ.).СН. » 5 **S, ЬЗЛ 121-123 (фучарет) 2930, (035, 1410. Т а б л и ц а 2 Испытуемое вещество Степень подавления. % Предлагаемое 1 12,0 2 18,0 1605. 1450, 1120, Т а б л и ц а Испытуемое вещество 3 Концентрация испыту^ амого вещества в растворе, 10 моль/л Степень блокирования по отношению к глутаминовой кислоте, 3 20 89 19,8 3 2850, 2770, 1750, 7fcO, 7 0 0 ( ч и с т ы й ) 20 25 21,2 5 37,7 6 14,9 7 64,8 4 20 88 8 12,0 5 20 91 10 18,7 7 20 92 14 10,2 13 20 80 17 93,0 17 20 90 25 10,9 35 2 68 27 8,4 36 2 53 29 9,9 37 2 58 40 2 61 41 2 69 20 4 20 45 Предлагаемое 30 35 40 4Е Сравнительное I 4,8 2 3,6 3 2,2 4 1,0 5 3,0 50 Сравнительное 2 55 402263 19 20 Т а б л и ц а Испытуемое вещество 1 TV LD 50 , мг/кг (в/в) 29,7 18,8 2 3 4 55,5 29,9 4 5 69,4 7 30,4 10 54,6 13 75,1 14 53,2 17 40,1 25 34,9 27 39,8 29 66,1 40 53,6 1 40226 її Время 1 мин Фиг.1 Гц 20 3 І 0 А А 4 1 Риг. 2 Редактор И. Николаичук к к к 20с Составитель X* Жукова Техред А.Кравчук Корректор А. Іяско Заказ 2796/58 Тираж 370 Подписное БНИИПИ Государственного комитета СССР по делам изобретений и открытий 113035, Москва, Ж-35, Раушская наб., д. А/5 Производственно-полиграфическое предприятие, г. Ужгород, ул. Проектная,
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the preparation of 1,3-oxazolidin-2-one derivatives and acid addition salts thereof
Назва патенту російськоюСпособ получения производных 1,3-оксазолидин-2-она или их кислотно-аддитивных солей
МПК / Мітки
МПК: A61K 31/421, C07D 413/06
Мітки: отримання, похідних, солей, спосіб, кислотно-адітивних, 1,3-оксазолідин-2
Код посилання
<a href="https://ua.patents.su/12-4759-sposib-otrimannya-pokhidnikh-13-oksazolidin-2-ona-chi-kh-kislotno-aditivnikh-solejj.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання похідних 1,3-оксазолідин-2 она чи їх кислотно-адитивних солей</a>
Спосіб отримання похідних 1,3-оксазолідин-2 она чи їх кислотно-адитивних солей

Номер патенту: 4758
Опубліковано: 28.12.1994
Автори: Мазару Сато, Тосіро Камісіро, Наойя Моріто, Міцуо Масакі, Харухіко Сінозакі, Коіті Хасімото
Мітки: отримання, солей, спосіб, похідних, кислотно-адітивних, 1,3-оксазолідин-2
Формула / Реферат:
Способ получения производных 1,3 оксазолидин-2-она общей формулы Iгде R-C3-C8-прямой или разветвленный алкил;X - водород, галоид, С1-С3-алкил или С1-С3-алкоксил;n = 4-6, или их кислотно-аддитивных солей, о т л и ч а ю щ и й с я тем, что, соединения общей формулы II
Спосіб отримання похідних бензаміду, або їх кислотно-адитивних солей, або оптичних ізомерів

Номер патенту: 4762
Опубліковано: 28.12.1994
Автори: Свен Ове Егрен, Ян Ола Густав Лундстрем, Стен Інгвар Ремсбі, Геста Леннарт Фрорвалл
МПК: A61K 31/60, C07D 207/09, A61K 31/40, A61P 25/18, A61P 1/08
Мітки: отримання, ізомерів, оптичних, бензаміду, спосіб, кислотно-адітивних, солей, похідних
Формула / Реферат:
Способ получения производных бензамида общей формулы (I)где R1 и R2 одинаковы или различны и означают водород, галоид, низший алкил; R3 - низший алкил или бензил, незамещенный или замещенный фтором;А1 и А2 - оба водород или каждый в отдельности водород или низший алкил,или их кислотно-аддитивных солей, или оптических изомеров, отличающийся тем, что соединение общей формулы (II) где R1, R2 и R3 имеют...
Засіб отримання похідних гуаніну або їх кислотно-адітивних фармацевтично припущених солей

Номер патенту: 2668
Опубліковано: 26.12.1994
Автори: Томас Ентоні Креницкі, Ліліа Мері Бьючемп
Мітки: солей, фармацевтично, засіб, гуаніну, отримання, похідних, припущених, кислотно-адітивних
Формула / Реферат:
1. Способ получения производных гуанина общей формулы Iгде R - группа -СН(СН3)2 или группа-СН(СН3)СН2СН3,или их кислотно-аддитивных фармацевтически приемлемых солей, отличающийся тем, что 9-(2-гид-роксиэтоксиметил)гуанинподвергают взаимодействию с N-защищенным...
Спосіб отримання похідних 3,5-діаміно-1,2,4-триазіну або їх кислотно-адітивних солей

Номер патенту: 5576
Опубліковано: 28.12.1994
Автори: Девід Елан Сойер, Мартін Джордж Бакстер, Алістер Айнслі Міллер, Альберт Реджинальд-Елфік
МПК: C07C 51/347, C07C 51/58, A61K 31/53, C07C 281/00, A61P 25/08, A61P 25/18, C07D 253/00, C07C 51/15
Мітки: солей, отримання, похідних, кислотно-адітивних, 3,5-діаміно-1,2,4-триазіну, спосіб
Формула / Реферат:
1. Способ получения производных 3,5-диамино-1,2,4-триазина общей формулыгде R1 - хлор, бром, иод, метил или трифторметил; R2 - водород или хлор, или R1 и R2 вместе образуют группу -СН=СН-СН=СН-; R3 -водород или бром; R4 - водород, хлор или бром; R5 - водород, метил или фтор; R6 - амино-, ацетиламино- или диметиламинометиленаминогруппа, при условии, что не более чем два заместителя из числа R2 -...
Спосіб отримання похідних пристинамицину пв у формі ізомерів чи їх сумішей, чи адитивних солей з кислотами
Номер патенту: 3573
Опубліковано: 27.12.1994
Автори: Жан-Марк Парі, Клод Котрєль, Жан-Клод Баррьєр
МПК: C07D 498/18, A61P 31/04, C07K 5/078, A61K 31/42
Мітки: форми, солей, отримання, спосіб, пристинамицину, кислотами, сумішей, похідних, адитивних, ізомерів
Формула / Реферат:
Способ получения производных пристинамицина Пв общей формулыгде R - метилпиперидинил, (1-метил-2-пирролидинил) метил, 2-пиперидиноэтил, (4-метил-1-пиперазинил) карбонилоксиэтил или группаа1k - С2-С4-алкил, незамещенный или замещенный метилом, этилом или бензилом, R2 и R3 - оди наковые или разные и означают водород, С1-С10-алкил, циклопентил, циклогексил или R2 и R3 вместе с соседним атомом азота образуют...
Попередній патент: З’єднання сталевих труб
Наступний патент: Спосіб безперервного визначення вологості сипучих харчових продуктів та пристрій для його здійснення
Випадковий патент: Спосіб виробництва натурального десертного вина