Засіб отримання похідних гуаніну або їх кислотно-адітивних фармацевтично припущених солей






Формула / Реферат
1. Способ получения производных гуанина общей формулы I
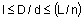
где R - группа -СН(СН3)2 или группа
-СН(СН3)СН2СН3,
или их кислотно-аддитивных фармацевтически приемлемых солей, отличающийся тем, что 9-(2-гид-роксиэтоксиметил)гуанин
подвергают взаимодействию с N-защищенным валином или изолейцином в присутствии дициклогексилкарбодиимида: и, возможно, основания в органическом растворителе с последующим удалением защитной группы и выделением целевого продукта в виде основания или переведением его в соль действием кислоты.
2. Способ по п. 1, отличающийся тем, что в качестве кислоты используют соляную кислоту.
Приоритет по признакам:
15.08.87 при R - группа -СН(СН3)2.
05.11.87 при R - группа -СН(СН3)СН2СН3.
Текст
Изобретение к а с а е т с я производных гуанина, в частности получения соединений обрдей ф-лы вого продукта в виде основания или переводом его в соль действием кислоты (лучше НС1) 0 Новые соединения проявляют антибактериальную активность в отношении вируса пузырькового лишая: концентрация, вызывающая 50%— ное торможение развития вируса» составляет 0,84 и 3,8 мкМ против 0,080,1 мкМ для ацикловира, имеют высокую биоусвояемость [^количество введенной дозы (в моче) в виде ацикловира составляет 63% и 50% против 15% для ацикловира]. 1 з е п л ф-лы, 1 табл0 О5 00 со 00 Изобретение относится к способу' получения новых производных гуанина, обладающих ценными противовирусными свойствами, которые могут найти при" менение в медицине,, Цель изобретения - создание новых производных гуанина, обладающих более высокой антивирусной активно стью за счет более высокой биоусвояемости и меньшей токсичности» П р и м е р К 2-[~(2-Амино-1,6дигидро-6^оксо-9Н-пурин-9-ил)метокси]этил-Ъ-валинат о а) 2-£(2-Амино-1,Ь-дигидро-6-оксо9Н-пурин-9-ил)метокси]-этил-К-^(()ензилокси)карбонил]-Ь-в алинат 0 1634138 Суспензию ацикловира (2,0 г) в су2,0 г суспензии ацикловира в сухом диметилформамиде (ДМ*) 150 мл нахом Д Ф (150 мл) нагревают до 60°С, М гревают до 60°С, в результате получаполучая в результате-бесцветный растют бесцветный раствор» К теплому вор о К теплому раствору добавляют раствору добавляют 3,012 г карбокси— 3,012 г CBZ-L-валинаг, 154 мг ДМАЛ, бензил-Ь-валина (CBZ-L-валин) 154 мг и 2,998 г ДГК. Желтоватому раствору • 4-диметиламинопиридигіа (ДМАП), 2,9981 дают остыть до комнатной температудициклогексилкарбодиимида (ДПК)оБледры и перемешивают в течение ночи. Чено—желтый раствор оставляют охлаж— JQ рез 30 мин наблюдают выпадение белодаться до комнатной температуры и пего осадка с К реакционной смеси снова ремешивают в течение ночИо Через добавляют указанные количества CBZ-*L30 мин наблюдают выпадение белого валина, Д А и ДГК и мутную суспенМП осадка е К реакционной смеси снова дозию перемешивают при комнатной тембавляют указанные количества CBZ-L- щ пературе в течение двух дней, после валина, Д А и ДГК и мутную суспенМП чего фильтруют, получая 1,418 г безию перемешивают в течение двух дней лого твердого вещества0 Бесцветный при комнатной температуре*. фильтрат концентрируют, получая желПосле этого суспензию отфильтротоватую маслянистую жидкость, котовывают 0 Получают 1,418 г твердого 20 рую очищают с,помощью хроматографии вещества белого цвета* Бесцветный на силикагеле, используя в качестве фильтрат концентрируют, получая в р е элюента смеси метанола и дихлормезультате маслянистую жидкость желтотана различного состава (0-15%)о В ватого цвета, которую очищают с порезультате получают 3,751 г целевомощью хроматографии на силикагеле.Элкг- ^ го соединения (92,1%) в виде белого ирование осуществляют смесями метанотвердого вещества» ла и дихлорметана разного состава ' в) 2-£(2-Амино-1,6-дигидро-6-оксо(0-15%)о В результате получают 3,751 г ' 9Н-пурин-9-ил)метокси1-этил-Ь-валицелевого соединения (выход 91,1%) в нат гидрохлорид моногидрато виде белого твердого вещества,, ,« в) 2-£(2-Амино-1,6-дигидро-б-оксоСмесь 3,730 г 2-[.(2-амин о-1,6-ди9Н-пурин-9-ил)метокси1-этил-Ь-валинато гидро-6-оксо-9Н-пурин~9-ил)метокси]~ Смесь 5,0 г 2~[(2-лмино-1,6-диэтил-N-j бензилокси)карбонил^~Ь-вали— гидро-6~оксо-9Н-пурин-9-ил)метокси~[- ' ната, 3/7 мг 5%-ного палладиевого каэтил—N- £(бензилокси)карбоНилТ-С-вали— тализатора на угле, 100 мл метанола, ната, 2 г.5%-ного палладиевого катали-, 100 мл ТГФ и 18 мл 0,5 М соляной кисзатора на угле (50% воды) и 50 мл Д Ф М лоты встряхивают в аппарате Парра встряхивают в аппарате Парра при давпри давлении водорода 50 фунтов на лении водорода 40 фунтов на квоДюйм кводюйм в течение дня. После этого в течение 3 ч„ Реакционную смесь филь-Q реакционную смесь фильтруют через * труют через слой целита и упаривают в слой целита и концентрируютs полувакууме, получая маслянистую жидчая белое твердое вещество, которое кость о Кристаллизацию осуществляют перекристаллизовывают из смеси воды из смеси воды и этанола в объемном со-, и этанола в отношении 1:3в После перекристаллиза- де ции получают 1,5 г целевого соедине— , В результате получают 1,762 г ния. (60,0%) целевого соединения в виде 'белого порошка, т о пл о 150°С (усадка 'твердого вещества) с постепенным пеВычислено,%: С 48,14; Н 6,22; реходом в маслянистую жидкость и N 25,91. 50 разложением со вспениванием при 195 С. Найдено,%: С 47,84; Н 6,26; Вычислено^: С 41,22; Н 6,15; N 25,75 О N 22,19; С1 9,36О П р и м е р 2. 2-Ц2-Амино— 1 ,6— Найдено,%: С 41,09; Н 6,10; дигидро-6-оксо-9Н-пурин-9-ил)метокN 22,12; С1 9,28 О снДэтил-Ь-валинат гидрохлорид моно55 П р и м е р Зо 2-Г(2-Амино-116гидрат с дигидро-6-оксо-9Н-пурин-9~ил)метока) 2- [(2-Амино-і ,6-дигидро-1,6-ок*си |этил-р-валинат гидрохлорид моносо-9Н-пурин-9-ил)метокси]-этил~Ы| гидрат. Цбензилокси)карбонил]~ Ь-валинатв 5 1634138 Суспензию 2,000 г ацикловира в a) 2- р(2-Амино~1,6-дигидро-ь-ок150 мл сухого ДМФ нагревают до 60°С, со-9Н-пурин-9-ил)метокси]этил-Ы- [/бен получая бесцветный раствор. К теплозилоксн)карбонил^]--валинато му раствору добавляют 3,012 г CBZ-DLСуспензию 1,00 г ацикловира в 80 мл сухого Д Ф нагревают до 6О°С, ' 5 валина, 154 мг ДМАП и 2,998 г ДГК в М Раствору дают остыть до комнатной получая бесцветный растворе К теплотемпературы и перемешивают его в те-* му раствору добавляют 1,70 г CBZ-Dчение ночи, После этого к реакционвалина, 74 мг Д А и 1,Ь0 г ДГКО ПроМЛ зрачному раствору дают остыть до ком- 0 ной смеси снова добавляют указанные количества CBZ-DL-валина, ДМАП и ДГК натной температуры и затем перемешии суспензию перемешивают при комнатвают его в атмосфере азота в течение ной температуре в течение 2 дней а За2 дней. После этого к реакционной смеси снова добавляют указанные количества CBZ^D-валина, ДМАП и ДГК и мутную 15 суспензию перемешивают при комнатной температуре в течение 2 дней о После этого суспензию фильтруют, отделяя белый твердый осадок. Бесцветный филь тем ее фильтруют, отделяя твердое белое вещество, а фильтрат концентрируют и очищают с помощью хроматографии на силикагеле, используя в качестве элюента смеси метанола и дихлорметана в различных соотношениях трат концентрируют, остаток раствори- /0 (0-15%). В результате получают целевое соединение,, в) 2- [(2-Амино-1,6-дигидро-6-оксо9Н-пурин-9-ил)метокси]-этилНЭ1.-валиэлюента 10%-ный метанол в дихлорметан а т г И Д р О Х Л О р и д моногидрат„ не 0 В результате получают 1,10 г (52%) 25 С м е с ь 3> 73О г 2-[(2-амино-1,6-дицелевого соединения в виде белого гидро-6-оксо-9Н-пурин-9-ил)метокси]^ твердого вещества, т 0 пл о 155-158 Со этил-Ы-[Хбензилокси)карбонил]-1)Ъ-ваВычислено,%: С 52 0 05; Н 6,01; лината, 377 мг 5%-ного палладиевого N !7,34 О катализатора на угле, 100 мл метано30 ла, 100 мл ТГФ и 18 мл 0,5 М соляной Найдено,%: С 52,02; Н 5,98; N 17,32о кислоты встряхивают в аппарате Парра в) 2- [_(2-Амино-1,6-дигидро~6-оксопри давлении водорода 50 фунтов ні 9Н-пурин-9-ил) меток сиП-этил-О-валинат кв.дюйм в течение дня. После этого гидрохлорид моногидраТо реакционную смесь фильтруют через Смесь 0,94 г 2-[(2-Амино-1,6-дигид-^ 5 С Л ой целита и концентрируют, получая ро-6-оксо-9Н-пурин-9-ил)метокси]этилтвердое вещество, которое затем пеN- Цбензилокси)карбонил~]-В-валината, рекристаллизовывают из смеси воды и 200 мг 5%-ного палладиевого каталиэтанола о Б результате получают целезатора на угле, 25 мг метанола, 25 мл вое соединение. ТГФ и 4,5 мл 0,5 М соляной кислоты 40 Г р и м е р 5 о 2-Г(2~Амино-1,6-днГ встряхивают в аппарате Парра при давгидро-6~оксо-9Н-пурин-9~ил)метокси~1 лении водорода 44 фунтов на кводюйм этил-Ь-изолейцинат гидрохлорид„ в течение 4 ч 0 После этого реакциона) 2- £(2-Амино-1,С-дигидро-6-оксоную смесь фильтруют через микропо,с 9Н-пурин-9-ил)метокси~]этил-Н-Г(бенристую мембрану и фильтрат концентзилокси)карбонилЗ-Ъ-изолейцинат. рируют. Б результате получают целеСмесь 1,0 г (4,4 ммоль) ацикловивое соединение в виде вещества грязра, 74 мг (0,6 ммоль) 4-диметиламиноно-белого цвета, т 0 п л о 185-188 С пиридина, 1,6 г (8,0 ммоль) 1,3-диВычислено,%: С 40,47; Н 6,18; 50 циклогексилкарбодиимида, 1,8 г N 21,37; С1 10,82. (6,6 ммоль) К-карбобензокси-Ь-изолеЙНайдено,%: С 40,62; Н 5,98; цина и 0,3 г молекулярного сита DauN 21,20; С1 10,91* son тип ЗА в 80 мл сухого диметилформП р и м е р 4. 2~£(2-Амино-1,6амида перемешивают при комнатной темдигидро-6-оксо-9Н-пурин-9-ил)метокпе си] этил-БЬ-валинат гидрохлорид моно- 5 5 Р а т У Р е в атмосфере азота а Через гидрате 4 дня к ней добавляют еще 1,Ь г а) 2- [(2-Амино-1,6-дигидро-6-оксо(8,0 ммоль) 1,3-дициклогексилкарбоди* 9Н-пурин-9-ил)метокси]этил-К-[(бенимида и 1,8 г (6,6 ммоль) N-карбозилокси)карбонил]-ОЬ-валинаТо бензокси-Ь-нзолейцина и продолжают ют в смеси метанола и дихлорметана и полученный раствор хроматографируют на силикагеле, используя в качестве a 1634138 перемешивание при комнатной температуре в течение еще 7 дней* Смесь з а тем фильтруют и прозрачный фильтрат концентрируют в вакууме до получения полутвердого остаткао При элюнровании из остатка на силикагеле 60 (ЕМ, 230-400 меш. 8,5*14 см) смесью 2 , 5 57, метанола и метиленхлорида получают 0,8 г (45%) 2-[(2-амино-1,6-дигид-* 0 ро-6-оксо-9Н-пурин-9-ил)метокси]этил^Г(бензилокси)карбонил]-Ъ-изолейцината .в виде белого твердого вещества, т 0 п л о 155-157°Со У (МеОН), н м : ! А м а к с 2 5 5 (£17700) Ф ,5
ДивитисяДодаткова інформація
МПК / Мітки
Мітки: кислотно-адітивних, солей, отримання, похідних, фармацевтично, припущених, гуаніну, засіб
Код посилання
<a href="https://ua.patents.su/6-2668-zasib-otrimannya-pokhidnikh-guaninu-abo-kh-kislotno-aditivnikh-farmacevtichno-pripushhenikh-solejj.html" target="_blank" rel="follow" title="База патентів України">Засіб отримання похідних гуаніну або їх кислотно-адітивних фармацевтично припущених солей</a>
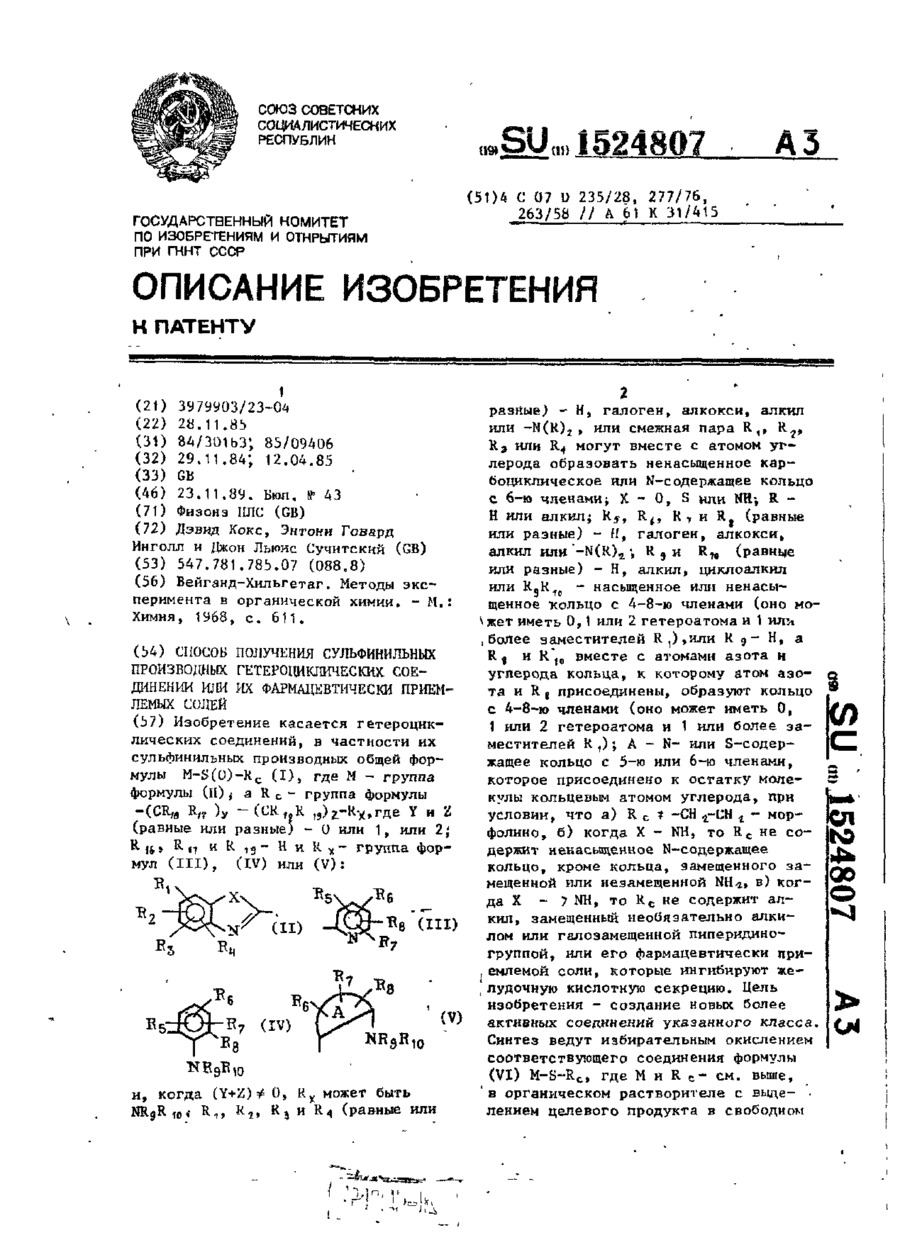
Спосіб одержання сульфінільних похідних гетероциклічних сполук або їх фармацевтично прийнятих солей
Номер патенту: 1944
Опубліковано: 20.12.1994
Автори: Джон Льюіс Сучитский, Ентоні Говард Інголл, Девід Кокс
Мітки: прийнятих, гетероциклічних, похідних, фармацевтично, сульфінільних, спосіб, солей, сполук, одержання
Формула / Реферат:
Формула изобретенияСпособ получения сульфинильных производных гетероциклических соединений общей формулыRC - группа формулы (CR16, R17)y -( CR18R19)z- Rх, где Y и Z - 0 или 1, или 2 каждый, одинаковые или разные;R16, R17, R18 и R19 – каждый водород;Rх – кольцо формули, когда (Y+Z)≠0, Rх может быть NR9R10;R1-R4 - одинаковые или разные, каждый - водород, галоген, алкокси, алкил или...
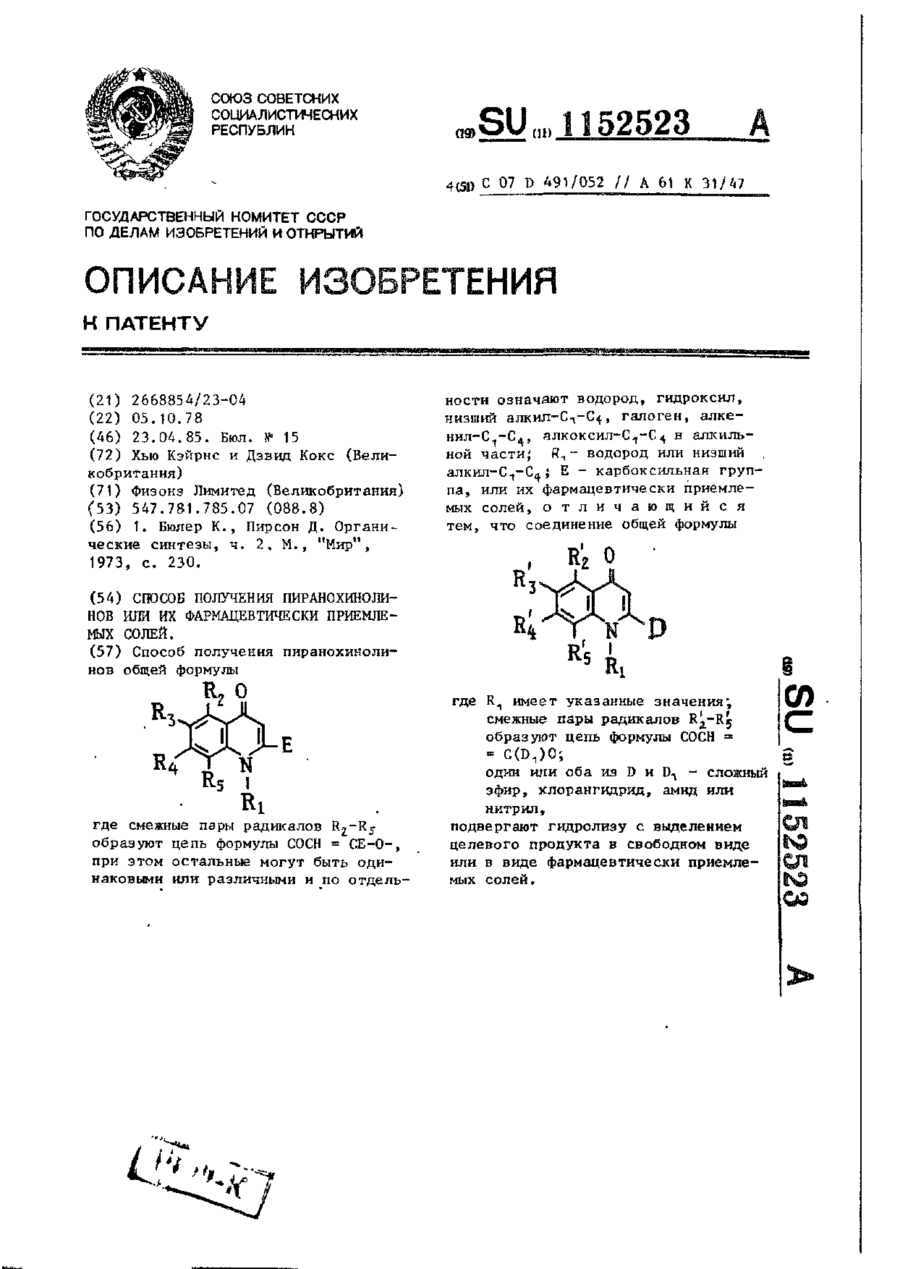
Спосіб одержання піранохінолінів або їх фармацевтично прийнятних солей
Номер патенту: 1784
Опубліковано: 25.10.1994
Автори: Хью Кейрнс, Девід Кокс
МПК: C07D 491/052
Мітки: спосіб, прийнятних, фармацевтично, одержання, солей, піранохінолінів
Формула / Реферат:
Способ получения пиранохинолинов общей формулыгде смежные пары радикалов R2—R5 образуют цепь формулы СОСН == СЕ-О-, при этом остальные могут быть одинаковыми или различными и по отдельности означают водород, гидроксил, низший алкил - С1-С4, галоген, алкенил - С1-С4, алкоксил - С1-С4 в алкильной части; r1 — водород или низший алкил - С1-С4; Е — карбоксильная группа, или их фармацевтически приемлемых солей, отличающийся...
Спосіб одержання похідних 7-[2-/2-амінотіазоліл/2-оксиіміноацетамідо]-3-цефєм-4-карбонових кислот або їх складних ефірів, або їх солей з лужними металами

Номер патенту: 1749
Опубліковано: 25.10.1994
Автори: Такао Такая, Хісасі Такасугі, Кіесі Цудзі, Тосіюкі Тіба
Мітки: металами, одержання, ефірів, солей, кислот, лужними, складних, похідних, спосіб
Формула / Реферат:
Формула изобретенияСпособ получения производных 7-[2-(2-аминотиазолил)-2-оксиимино- ацетамидо ]-3-цефем-4-карбоновых кислот формулыв виде син-изомеров, где R1 - свободная аминогруппа или защищенная формильной или трифторацетильной группой аминогруппа;R2 - водород, C7-C8 -алкил, аллил, пропаргил, С5-С6-циклоалкил, карбоксиметил, этоксикарбонилметил или моно- или тризамещенный хлор или фтор этил;R3 -...
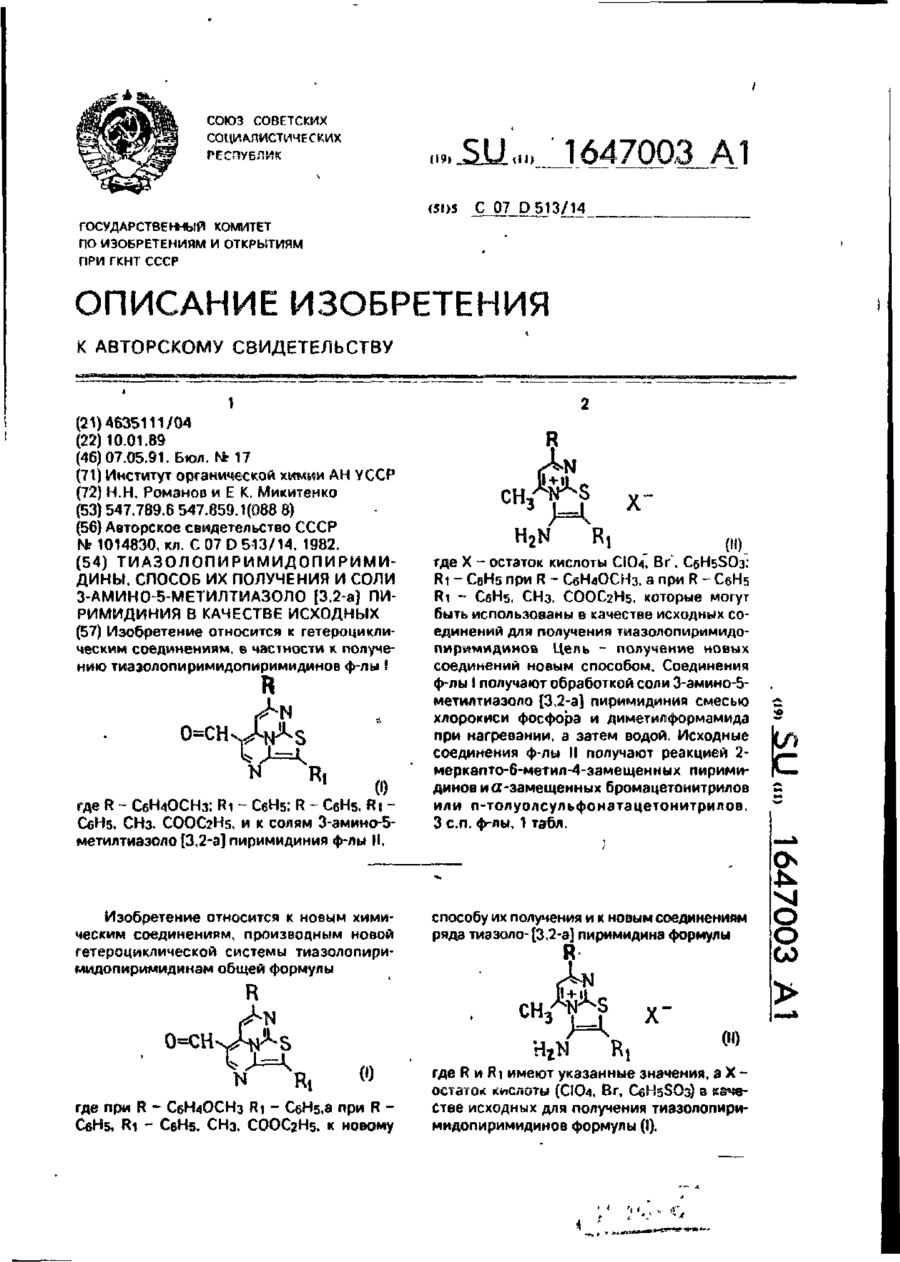
Тіазолопіримідопіримідини, засіб їх отримання та солі 3-аміно-5-метилтіазоло [3, 2 -а] піримідинія як вихідного
Номер патенту: 99
Опубліковано: 30.04.1993
Автори: Романов Микола Миколайович, Микитенко Олена Костянтинівна
МПК: C07D 513/14
Мітки: солі, засіб, 3-аміно-5-метилтіазоло, вихідного, отримання, тіазолопіримідопіримідини, піримідинія
Формула / Реферат:
Формула изобретения1. Тиазолопиримидопиримидины общей формулы (I) где при R–С6Н4ОСНз R1=C6H5. а при R-С6Н5 R1-С6Н5, СН3, СООС2Н5.2. Способ получения тиазолопиримидопиримидинов общей формулы (І), заключающийся в том, что соли 3-змино-5-метилтиазоло [3,2-а] пиримидикия общей формулыгде X - остаток кислоты CIO4, Br, C6H5SO3-.R1-С6Н3 при R–С6Н4ОСН3, а при R-С6Н5...
Спосіб одержання кристалічних солей ацетоацетамід-n-сульфофториду

Номер патенту: 1782
Опубліковано: 25.10.1994
Автори: Дітер Ройшлинг, Адольф Линкис
МПК: C07C 307/00, A23L 1/236, C07B 61/00, C07C 311/00, B01J 27/00, C07C 301/00, C07C 67/00, B01J 27/20, C07D 291/00, C07C 303/00
Мітки: одержання, спосіб, кристалічних, ацетоацетамід-n-сульфофториду, солей
Формула / Реферат:
Способ получения кристаллических солей ацетоацетамид-N-сульфофторида общей формулыгде М — литий, натрий или калий, отличающийся тем, что дикетен формулыподвергают взаимодействию с аминосульфоф-торидом формулы H2NSO2F и с карбонатом или бикарбонатом лития или натрия, или калия в среде ацетона, или ацетонитрила, или диметилформа-мида при температуре от—10 до 0 °С в течение 15—90 мин, а затем при 30—50 °C в течение...
Попередній патент: Гідропривод одноковшового екскаваторанавантажувача
Наступний патент: Спосіб отримання похідних імідазола або його фармацевтично прийнятих солей
Випадковий патент: Контейнер для зберігання та транспортування предметних скелець