Спосіб одержання 2-галоацетамидів







Формула / Реферат
Способ получения 2-галоацетамидов общей формулы І
где R - 2,6-диметил-1-циклогексен-1-ил или замещенный фенил формулы
где R1 и R2- С1-С4-алкил; R3 - С1-С4-алкил, хлорметил, С1-С4-алкокси-метил, циклогексил или циклопропилметил; R4 - бромметил, моно- или дихлорметил, взаимодействием соединения общей формулы II
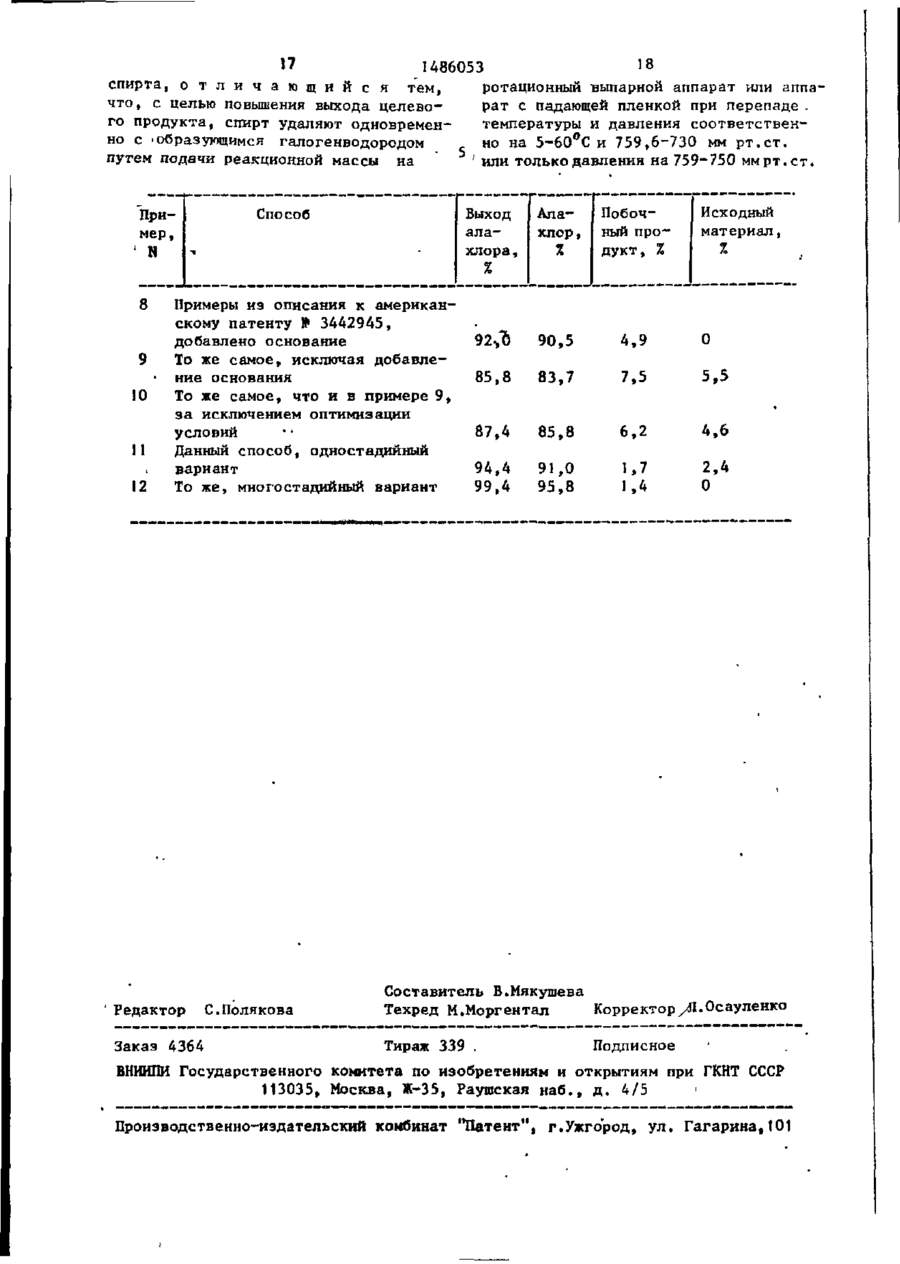
где Х - хлор или бром; R и R4- имеют указанные значения, со спиртом формулы III R3ОН, где R3 - имеет указанное значение при температуре (-20)-(160) °С и молярном соотношении соединений общей формулы II и III, равном 1 : 1-100 : 1 с удалением спирта, отличающийся тем, что, с целью повышения выхода целевого продукта, спирт удаляют одновременно с образующимся галогенводородом путем подачи реакционной массы на ротационный выпарной аппарат или аппарат с падающей пленкой при перепаде температуры и давления соответственно на 5-60 °С и 759,6-730 мм рт. ст. или только давлення на 759-750 мм рт. ст.
Текст
Изобретение относится к амидам в частности к получению 2-галоацета* 4 мидов ф-лы R-N(COR^) |(CH^-ORj) , г д е R - 2,6-диметил-1-циклогексен-1-ил 'или 1 ,3-диалил (С-4- С + )-фенил, которые могут быть использованы в качестве пестицидов и регуляторов роста растений. Цель - повышение выхода целевого продукта. Получение ведут реакцией соединения ф-лы \ R-N(CO-R4)(CH2X), где Х-С1 или Bt; R и R 4 - указано выше со спиртом ф-лы R^ОН,где R 5 - указано выше при молярном соотношении 1:1-100 при (-20)-(160)°С. Процесс проводят с удалением спирта одновременно с обра1зующимся галогенводоро дом путем подачи реакционной массы на ротационный выпарной аппарат или аппарат с падающей пленкой при перепаде температуры и давления соответственно на 5-60°С и 759,6-730 мм р т . с т . или только давления на 759-750 мм р т . с т . Выход 94,4-99,4%. I табл, сл Изобретение относится к области органической химии, а именно к способу получения 2-галоацетамидов общей формулы о R-N C-R. CH2-OR3 где R - 2,6-диметил-1-циклогексен1-ил или замещенный фенил формулы где R и Ry£ - С 4- С-алкил; R. - С ( - С^-алкил, хлорметил, "С|- С 4 -алкокси-метил, циклогексил или циклопропилметил; R^ - бромметил, моно- или дихлорметил, которые могут быть использова! ны в качестве пестицидов и регуляторов роста растений. «РПФ-К 486053 ляюг комплекс 'метанола и практически все оставшееся количество хлористого водорода. Комплекс метанол (хлористый водород из такого выпарного аппарата второй стадии смешивают с 'комплексом метанол), хлористый водород из выпарного аппарата стадии 1 и направляют в систему рекуперации метанола» из которой безводный мета10 нол рекуперируют и возвращают на стаСтадия 1. Расплавленный (при темдию I. пературе 45-55° С) 2\б*-диэтил-(ЛПоток конечных продуктов из выпар-хлорметил)-2-хлорацетанилид подают ного аппарата стадии 2 включает алав смонтированный на линии смеситель хлор в количестве, которое соответс расходом потока 46,67 кг/ч, и смествует практически количественному шивают с практически безводным метавыходу продукта, более 95%-ной степенолом, который направляют в указанный ни чистоты совместно с незначительнысмеситель при расходе потока ми количествами примесей. Этот ала27,24 кг/ч. Эту смесь прокачивают чехлор можно эффективно использовать в рез термостатируемый трубчатый "реак20 качестве гербицида непосредственно в тор, в котором поддерживают температом виде, в котором он получен. туру 40-45°С и длина которого достаточна для достижения продолжительносТехнологическая последовательность ти пребывания по меньшей мере 30 мин. операций реакция (разделение на стаВ результате такой реакции получают 25 дии I) позволяет при ее осуществлес достижением приблизительно 92%-ного нии получать алахлор с достижением выхода 2 ,6 -дизтил-ЇІ-(метоксиметил)высокого выхода продукта. Таким обра-2-хлорацетанилнд(алахлор) и хлорисзом, в оптимальных условиях степени тый водород в пересчете на N-хлорчистоты исходных реагентов и их кон~ метильный промежуточный продукт. Обцентрации» температуры, продолжительразовавшийся хлористый водород раство-30 ности пребывания в реакционном аппараряется в избытке метанола. Отводимый те и зонах разделения и тому подобноиз реактора поток конечных продуктов го осуществление по меньшей мере однаправляют в выпарной аппарат с падаюной технологической последовательносщей пленкой, в котором поддерживают ти операций реакция/разделение, что температуру 100°С и остаточное давле- 35 соответствует упомянутой стадии 1, ние 30 мм рт.ст. Комплекс удаляют и оказалось бы вполне достаточным для направляют в систему рекуперации меполучения алахлора или других соедитанола. нений технического сорта, отвечающих вышеприведенной общей формуле 1. Стадия 2. Поток продуктов из вы40 П р и м е р 2. Получение 2-хлорпарного аппарата "стадии 1, которые -2і ,6 } -диэтнп~1$-(этоксиметил)-ацетанивключают в основном алахлор и непролида. реагировавший 2' ,б'-диэтил-^(хлорме-; Приблизительно 5,5 г (0,02 мол.) тил)-2-хлорацетанилид, направляют 2-ХЛОР-21,6*-диэтил-N-(хлорметил)во второй установленный'на линии сме-1 45 -ацетанилида растворяют в 25 мл этаноситель, в который также вводят дола и раствор выдерживают в бане при полнительное количество метанола с 45°С в течение 30 мин. Избыток этанорасходом потока 27,24 кг/ч. Затем тала быстро удаляют в роторном вакуумкую смесь направляют во вторую ре, ном испарителе при 50°С и остаточном акционную зону, также снабженную тер50 давлении 10 мм рт.ст. В остаточную мостатируемым трубчатым реактором, маслоподобиую массу добавляют 25 мл в котором поддерживают температуру свежего этанола и смесь выдерживают 60-65°С, с достижением продолжительПри 65°С в течение 30 мин. Далее изности пребывания в нем 30 мин. Кобыток этанола вновь удаляют с применечный поток продуктов из этого реактора направляют в выпарной аппарат 55 нением роторного испарителя. При этом получают примерно 5,80 г бледно-янтарс падающей пленкой, в котором поддерного маслоподобного продукта, который живают температуру Ю 0 ° С и остаточсодержит, как это установили газохроное давление 30 мм рт.ст., откуда удаЦелью изобретения является повышение целевого продукта. П р и м е р 1. В данном примере описано осуществление способа в процессе получения алахлора. Этот способ эффективно осуществляют в последовательности операций реакции разделения в две стадии нижеследующим образом. 17 18 1486053 спирта, о т л и ч а ю щ и й с я тем, ротационный выпарной аппарат или аппачто, с целью повышения выхода целеворат с падающей пленкой при перепаде . го продукта, спирт удаляют одноврементемпературы и давления соответственно с ^образующимся галогенводородом но на 5-60°С и 759,6-730 мм рт.ст. путем подачи реакционной массы на или только давления на 759-750 мм рт.ст. Пример, Исходный материал, Выход алахлора, Способ N Примеры из описания к американскому патенту № 3442945, добавлено основание 9 То же самое, исключая добавле• ние основания 10 То же самое, что и в примере 9, эа исключением оптимизации условий •• 11 Данный способ, одностадийный вариант 12 То же, многостадийный вариант Алахлор, Побочный продукт, % 92>0 90,5 4,9 85,8 83,7 7,5 5,5 87,4 85,8 6,2 4,6 94,4 99,4 91,0 95,8 1,7 1,4 2,4 0 8 Редактор Заказ 4364 С.Полякова 0 Составитель В.Мякушева Техред М.Моргентал Корректор уЛ.Осауленко Тираж 339 . Подписное ВНИИПИ Государственного комитета по изобретениям и открытиям при ГКНТ СССР 113035, Москва, Ж-35, Раушская наб., д. 4/5 Производственно-издательский комбинат "Патент", г.Ужгород, ул. Гагарина,101 Взамен ранее изданного СОЮЗ СОВЕТСКИХ СОЦИАЛИСТИЧЕСКИХ РЕСПУБЛИК SU,.,, 1486053 А (SD5 С 07 С 2'33/18 ГОСУДАРСТВЕННЫЙ НОМИТЕТ ПО ИЗОБРЕТЕНИЯМ И ОТНРЫТИЯМ ПРИ ГННТ СССР ОПИСАНИЕ ИЗОБРЕТЕНИЯ И ПАТЕНТУ • ! (21) (22) (31) (32) (33) us . 25&640I/23-04 06,01.78 . 844542 . 26.10.77 ' • • • . , •" • • ...... •• • ' (46) 1 5 . 1 2 . 9 0 . Бнш. № 4 6 ! (71) Монсанто Компани (US) (72) Доналд Едгар Балду с и Эдвард Ервин Дебус;(US) (53) 5 4 7 . 2 9 8 . 1 . 0 7 ( 0 8 8 , 8 ) Г . ; . * . " (56) Патент С А № 344.2945,^ Ш ют. 260^562, опублик. 1964. Патент С А * 3875228* Ш : кл. 260-562, опублик. 1975. Патент СЙА № 3547620, •кл., 71-118, опублик, 1970, (54) СПОСОБ ПОЛУЧЕНИЯ 2-ГАЛОАЦЕТА• (57) Изобретение относится к амидам і в частности к получению 2-галоацетамидов ф^-лы ^ - N ( 0 0 ^ ) j(C%-OR£) , где і R - 2 >ё.-диметил-1 -циклогексенг 1 -ил І или 1 »3-диалил (С « П р и м е р 2. Получение 2-хлорпарного аппарата "стадии I, которые -2* ,6і -диэтнл-Ы-(этаксиметил)-ацетанивключают в основном алахлор и иепролнда. реагировавший 2',6'-диэтнл-Н-(хлорметил)-2-хлорацетанилид, направляют Приблизительно 5,5 г (0,02 мол.) во второй установленный'на линии сме-} 2~хлор-2*,6і -диэтил-Н-(хлорметил).- -ацетанилида растворяют в 25 мл этаноснтель, в который также вводят дополнительное количество'метанола с ла и раствор выдерживают в бане при расходом потока 27,24 кг/ч. Затем 45°С в течение 30 мин» Избыток этанокую смесь направляют во втору» рела быстро удаляют в роторном вакуумакционную зону, также снабженную ном испарителе при 50°С и остаточном мостатируемым трубчатым реактором, давлении 10 мм рт.ст, В остаточную в котором поддерживают температуру йаслоподобную массу добавляют 25 мл 60-б'56С, с достижением продолжитель- і свежего этанола и смесь выдерживают кости пребывания в нем 30 мин. Ко- ' при 65°С в течение 30 мин. Далее изнечный поток продуктов из этого реак-j быток этанола вновь удаляют с приметора направляют в выпарной аппарат 55 нением роторного испарителя. При -этом с падающей пленкой, в котором поддер-* получают примерно 5,80 г бледно-янтарживают температуру 100°С и остаточного маслоподобного продукта, который ное давление 30 мм рт.ст., откуда уда* содержит, как это установили газохро 1486053 матическим анализом, 92,8% целевого торый содержит 98,8% (выход продукта продукта и 1,7% 2~хлор-2',6^-диэтил99%) 2 ,б'-диэтил-М-(н-бутоксимеацетанилида (побочного продукта). Вытил)-2-хлорацетанилида (то есть буход целевого продукта 94,5%. тахлора) и 1 % соответствующего вторичного аминового побочного продукта. П р и м е р 3. В ходе проведения В вышеприведенных примерах анаэксперимента в технологических условилиз с помощью ЯМР-спектрограммы покаях и с использованием исходных реагензал, что соответствующие продукты тов в количествах,"которые аналогичны эксперименту, условиям его проведения 10 были совместимы с их химическими структурными формулами. и количествам исходных реагентов приВ примерах 8-12 целевой продукт мера 2, но с использованием в данном получают по известному способу. случае изопропанола получают 5,92 г П р н м е р в , В данном примере продукта в виде светло-янтарной маслоописан процесс получения 2~хлорподобной массы, который содержит по )) ;> -2 ,6 -диэтил-^(этоксиметил)-ацетаниданным анализа 90,2% 2 .6*-диэтил-Nлида(алахлора). -(изопропоксиметил)-2-хлорацетанилида 100 г 2-хлор-2' ,б'-дизтил-ї$-(хлор(выход продукта 89,4%) и 1,8% вторичметил)-ацетзнилида, который содержит ного амидного побочного продукта, 2 ,6*-диэтил-2-хлорацетанилида. * 20 96,0% (0,350 мол.) основного продукта, растворяют приблизительно в 70 г П р и м е р 4 . В ходе проведения бензола и добавляют раствор в 65,8 г эксперимента по аналогии с изложен(2,054 мол.) метанола, При таком доным в примерах 2 и 3, но с использовабавлении протекает экзотермическая нием в данном случае 1-пропанола в качестве исходного спиртового реаген- 25 реакция. Эту реакционную смесь кипятят с обратным холодильником (при та получают лимонно-желтую маслопотемпературе 63 С) и в течение 1,5 ч добную массу (5,66 г ) , которая по по каплям добавляют в нее избыток данным анализа содержит 92,8% (примерно'63,3 г ) триэтиламина, В про(выход продукта - 87,2%), 2',б'-ди30 цессе этого добавления температура этил-М-(н-пропоксиметил)-2-хлорацеповышается приблизительно до 70 С и танилида и 1,2% соответствующего втоее поддерживают в течение еще примерричного амидного побочного продукта. но 10 мин после завершения операции П р и м е р 5 . В ходе проведения добавления триэтиламина. После охлажэксперимента по аналогии с изложендения до 30°С реакционную смесь проным в примерах 2-4, но с использова35 мывают двумя порциями по I70 мд воды. нием в данном случае изобутанола в Из продукта, тяжелого маслоподобного качестве исходного спиртового реагенслоя, отгоняют растворитель, после та получают 6,20 г маслоподобного чего его обезвоживают вакуумной перепродукта, который содержит 96,4% гонкой до конечной температуры в ки(выход продукта 97%) 2Р ,6' -диэтил40 пятильнике приблизительно 70 С при -Л-(иэобутоксиметил)-2-хлорацетанилиостаточном давлении 1 мм рт.ст. Остада и 3% соответствующего вторичного точная янтарная маслоподобная масса амидного побочного продукта. весом 96,15 г содержит 90,4% целевого П р и м е р 6. Повторяя эксперипродукта и 4,9% 2-хлор-2 > ,6 > -диэтилмент, который описан в примерах 2-5, 45 ацетанилида (побочного продукта) но с использованием в данном случае газохроматографический анализ). Конеч2-хлорэтанола, в качестве исходного ный продукт не содержит непрореагиспиртового реагента получают 6,96 г ровавшего исходного реагента. Выход светло-янтарного маслоподобного процелевого продукта 92,0%. дукта, который содержит 86,0% (вы50 9 у ход продукта 94,0%) 2 ,6 -диэтил-NП р и м е р 9 . В данном примере -(хлорэтоксиметил)-2-хлорацетанилида. описан процесс получения anamtopat но без использования связывающего кислоП р и м е р 7 , В ходе проведения ту агента. эксперимента- по аналогии с изложенным в примерах 2-6, но с использова100 г 2-ХЛОР-2*,6*-диэтил-N-(хлорнием в данном случае н-бутанола в ка- 55 метил) ацетанилида, который содержит честве исходного спиртового реаген96,0% (0,350 мол.) основного продукта, та получают 6,18 г бледно-лимоннорастворенного приблизительно в 70 г желтого маслоподобного продукта, кобензола . добавляют в 66.0 г метанола j ' 1486053 8 (2 s O59 мол.). В результате такого ческого анализа содержит 85,В% целедобавления идет экзотермическая реаквого продукта, 6,2% побочного проция „ после чего реакционную смесь дукта, 2-ХЛОР-2',6*-диэтилацетанилида подвергают кипячению с обратным холои примерно 4,6% непрореагировавшего дильннком в течение 1 ч при темпераисходного реагента. Выход целевого туре 63 С. При этом не добавляют нипродукта 87,4%, какого связывающего кислоту агента. Путем оптимизации реакционных усПосле кипячения с обратным холодильниловий в отсутствии связывающего киском избыток метанола и растворителя 10 лоту агента были достигнуты повыше- ; удаляют вакуумной перегонкой до коние выхода (1,6%) и его качества І нечной температуры в кипятильнике 70 С (2,7%), однако реакция прошла непол-. при остаточном давлении Ї мм рт.ст. ностью. Таким образом получают приблизительно П р и м е р 11. В примере описано 96,83 гбледно-лимоино-желтого маслопо-jc 'проведение; процесса в одностадийном добного продукта, который, по данным реакторе. В этом случае используют те газохроматографического анализа содерже исходные реагенты, что и в примежит 83,7% целевого продукта, 7,5% рах 8-10. > побочного продукта, 2-хлор-2 ,6 -ди/10 г 2~хлор-2 ,6 ~диэтил-Ы-(хлор-'• этил-ацетанилида и 5>5£ непрореаги20 метил)-ацетанилида_, который содержит ровавшего исходного реагента. Выход 96,0% (0,035 мол) основного продукта^ продукта 85,8%, добавляют приблизительно 6,0 г Устранение связывающего кислоту, (0,1873 мол) метанола. При этом идет агента в эксперименте данного примера экзотермическая реакция» в результате привело к снижению выхода целевого 25 которой температура возрастает приб- , продукта из 6,2%. В ходе проведения лизительно до 45°С, после чего ее і этого процесса реакция не сдвинулась поддерживают в течение 30 мин. полностью вправо. В результате хлорисИзбыток метанола быстро удаляют в ва-1товодородный побочный продукт снизил куумном роторном испарителе до конем-*степень конверсии, а в конечном про- 30 ной температуры кипения в кипятильни4дукте в качестве загрязняющей примеке 70 С при остаточном давлении } 1 си содержался непрореагировавший ис1 мм рт.ст. В результате получают ходный реагент. приблизительно 9,80 г бледно-лимонножелтой маслоподобной массы, которая П р и м е р 10. В данном примере по данным газохроматографического описан процесс получения алахлора по 35 анализа содержит 91,0% целевого про-' аналогии с изложенным в примерах опидукта, 1,7% побочного продукта, саний к американскому патенту '2-хлор-2' ^-диэтилацетанилида и № 3442945, но без использования свя2,4% иепрореагировавшего исходного зывающего кислоту агента и в оптимиматериала. Выход продукта 94,4%. зированных температурных условиях. 40 Таким образом, осуществление толь100 г 2-хлор-2*,6 -диэтил-Nко одной стадии предлагаемого спосо-(хлорметил)-ацетанилида, который соба позволило улучшить качество и выдержит 96,0% (0,350 мол.) основного вещества, растворенного приблизитель- / ход целевого продукта в сравнении с известным, несмотря на то, что реакно в 70 г бензола, добавляют в 66 г t ция протекает не до конца (продукт (2,059 мол.) метанола. При этом протесодержит 2,4% непрореагировавшего кает экзотермическая реакция, в реисходного материала). Сопоставление зультате которой температура реакс результатами экпериментов примеционной смеси повышается до 45 С, заров 8-10 показывает очевидное улучтем ее поддерживают в течение еще к шение, хотя в данном случае не испольI ч. В смесь не добавляют никакого зовали никакого связывающего агента. связывающего кислоту агента. Избыток П р и м е р 12. В данном примере метанола и растворитель отгоняют вапроцесс получения алахлора ведут без куумной дистилляцией до конечной темдобавления связывающего кислоту агенпературы кипения в кипятильнике прита с применением многостадийного реакмерно 80° С при остаточном давлении. тора. 1 мм рт.ст. При этом получают прибли10 г 2-хлор-2 ,6 -дизтия-її-(хлорзительно 96,2 г маслоподобной массы, летил)-ацетанилида,который по данным , которая по данным газохроматографи 1486053 0 После отгонки избытка спирта получают 'анализа содержит 96,0% (0^0350 мол) 13,27 г чистого бледно-лимонно-жел* основного вещества, растворяют в того масла (nv 1 ,5245) с содержа6,0 г (0,1873 мол) метанола. Идет экнием 93,9% целевого продукта по данзотермическая реакция, при этом темным гельхроматографического анализа. пература повышается до 45°С, после Выход составляет 92,5%. ЯМР-спектр чего ее поддерживают еще в течение соответствует структуре и является 0,5 ч. Избыток метанола быстро удаляидентичным продукту, полученному разют в вакуумном роторном испарителе до конечной температуры кипения в кипя- j 0 личными методам». тильннке 45Т при остаточном давлении П р и м е р 15. Получение 2-хлорі -2* ,6 -днэтил-№-(2-метоксиэтокси)-ме1 мм рт.ст. Затем добавляют повторную тилаце танилида. порцию метанола, 6,0 г (0,1873 мол), К 13,71 г (0,050 моль) 2*,б'-диреакционную смесь нагревают дл 65 С и выдерживают 0,5 ч. Избыток метанола .5 эт*ш-(К-хлорметил)-2-хлорацетанилида добавляют 38,0 г метилгликоля и дают удаляют аналогично вышеизложенному, в стоять при комнатной температуре результате получают приблизительно 30 мин. Избыток спирта удаляют в ро'9,80 г бледно-лимонно-желтой массы, торном испарителе при65°С/0,5 мм рт.ст, которая по данным анализа содержит 95,8% целевого продукта, 1,4% 2-хлор- 20 В остающееся масло добавляют 38,0 г свежего метилгликоля и выдерживают ~2',6' -диэтилацетанилида, но не содерпри 45°С в течение 30 мин. Избыток жит никакого непрореагировавшего исспирта удаляют с получением 15,46 г ходного материала. Выход продукта бледно-лимонно-желтого масла, Выход 99,4%. составляет 98,5%. Масло растворяют в П р и м е р 13. Получение 2 -метил-25 н-гексане и перекристаллизовывают с -6 -Ь-бутил-(К-метоксиметил)-2-бромполучением белого кристаллического ацетанилида. ; твердого вещества, т.пл.ЗІ,5~32,5 С. } К 15,08 г (О,о4о" моль) 2 -метилП р и м е р 16. Получение 2'-этил' -6* ,1:-бутил-(Н-бромметил)-2-бромацетанилида добавляют 25,0 г безводно-зд -6 -метил-(її-зтоксиметил)-2-хлорацетанилида. го метанола. Смесь нагревают до 45 С и дают стоять 30 мин. Избыток спирта К 10,4 г (0,04 моль) 2-этил-б*-меи НВг удаляют в роторном испарителе тил- (Ы-хлорметил)-2-хлорацетанилида при 45*С/10 мм рт.ст. Маслянистый добавляют 30,0 г этанола и нагревают осадок обрабатывают еще два раза с до 45 С в течение 15-20 мин. Избыток 25,0 г безводного метанола подобным " спирта удаляют под вакуумом в роторобразом. После конечной отгонки полуном испарителе. После третьей обработчают 13,0 г чистого янтарного масла ки вес остающегося масла составляет (n^J 1,5470), которое по даннымгель10,73 г и содержание целевого продукхроматографического анализа содержит та 96,4% по данным гельхроматографи97 s 0% 2 -метил-6*-й-бутил-(И-метоксического анализа. Выход 96,0%. Показаметил)-2~бромацетанилида. Выход тель преломления - п£> 1,5236, ЯМР96>0%. ЯМР-спектр соответствует струкспектр соответствует структуре и является идентичным продукту, получен- • , туре и является идентичным продукту, . ному при использовании вещества для полученному при использовании три, . удаления кислоты (+0стающееся масло этиламина для удаления НВг» обрабатывают 30,0 г свежего этанола П р и м е р 14. Получение 2,6 -дипри 45 С в течение 15 мин и избыток метил-(ї$-изопропоксиметил)—2-хлорэтанола удаляют. ацетанилида. П р и м е р 17. Получение 2* »б' — Приблизительно 12,3 г (0,050 моль) „ ~диэтил-(N-2-хлорэтоксиметил)-(tf-хлор2 \Ь* -диметил-(№-хлорметил)-2-хлорацетанилида. ацетанилида растворяют в 30,0 г безводного изопропанола. Реакционную смесь К 5,5 г (0,020 моль) 2*,6'-диэтилнагревают до 45-50 С в течение 30 мин -(Ы-хлорметил)-2-хлорацетанилида дои избыток спирта отгоняют с примененибавляют 25 мл 2-хлорэтанола и нагреем роторного испарителя при вают до 45 С в течение 30 мин. Избы60°С/10 мм рт.ст. Остаток обрабатываток спирта отгоняют в роторном испают во второй раз с 30,0 г, свежего рителе при 80 С/1 мм рт.ст. Добавляизоцропанола при 45°С в течение 30 мин, ют вторую порцию 25 мл свежего 2-хлор 11 12 1486053 Вторую порцию 40 г свежего спирта этанола, нагревают до 65°С в течение добавляют в остаток и нагревают до 30 мин и избыток спирта удаляют под 45^С в течение 30 мин. Избыток спирвакуумом. Остаток представляет собой та удаляют как показано выше. Остаток светло-янтарное прозрачное масло с представляет собой светло-желтое весом 6,96 г и содержанием 86£ масло, п " 1,5327 (13,0выход 99,7%). 2 ,6' ~диэтил-(Ц-2-хлорэтоксиметил)ЯМР-спектр соответствует структуре. -хлорацетанилида. Выход составляет 94,0% (от теорет.). ЯМР-спектр соотП р и м е р 21. Получение об-хлорветствует структуре. -N-(2,6-диметил-1-циклогексен-1-ил)10 -N-(метоксиметил)-ацетамида. П р и м е р 18» Получение 2 ,6 Приблизительно 5,90 г (23,5 моль. -диэтил-(Ц-метоксиметил)-2,2-дихлор2',6'-диметилциклогексен-1-ил-N"(хлорацетанилида, метил) -2 -хлорацетанилида растворяют К 15,43 г (0,050 моль) І .б^див 28,3 г безводного метанола и дают этил-(М-хлорметил)-2,2дихлорацетани15 стоять 30 мин при комнатной темпералида добавляют 32,0 г безводного метуре. Избыток метанола удаляют под танола. Реакционной смеси дают стовакуумом в роторном испарителе. Вышеять при 45°С в течение 30 мин» Затем названную последовательность повто-" спирт - НС1 удаляют под вакуумом в роторном испарителе. Маслянистый оста- 20 ряют еще два раза и после конечного удаления метанола получают 5,50 г ток обрабатывают еще два раза вышеопи(94,9%) бледно-лимонно-желтого массанным образом и после отгонки избытла (п^1,5050).ЯМР-спектр соответстка спирта получают 15,15 г прозрачновует структуре. го, бледно-лимонно-желтого масла ,26 1 ,5330) с содержанием по данным гельхроматографического анализа 98,3% целевого продукта. Выход составляет 97,9%. ЯМР-спектр соответствует структуре и является идентичным продукту, полученному при использовании 30 основания ддя удаления НС1. П р и м е р 19. Получение 2У-метилJ -6 -Ь-бутил-(М-аллипоксиметил)-2-хлорацетанилида. К 14,5 г (0,05 моль) 2'-метип-б'35 -1:-бутил-(!Я-хлорметил)-2~хлорацетанилида добавляют 29,0 г аллилового спирта и нагревают до 45 С в течение 15 мин* Избыток спирта удаляют под вакуумом в роторном испарителе и заменяют 29,0 г свежего аллилового спирта. Смесь вновь выдерживают при 45 С в течение 15 мин* После отгонки избытка спирта весь процесс повторяют в третий раз. После третьей отгонки получают 14945 г (93,4% выхода) светлоянтарного масла» п^ 1,5338. ЯМР-спектр соответствует структуре. П р и м е р 20, Получение 2 Г -этил~6 -метил-(N-тетрагидрофурфурилоксиметил)-2-хлорацетанилнда. Приблизительно 10,4 г (0,040 моль) 2 -этил-б -метил-(Ы-хлорметил)-2-хлорацетанилида растворяют в 40,8 г (0,40 моль) тетрагидрофурфурилового спирта и дают стоять в течение ночи при комнатной температуре. Избыток спирта удаляют в роторном испяюителе при 65-70°С/О,4 мм рт.ст. П р и м е р 22. Получение 2П,4*,6 танилида. К 22,5 г СО,074 моль) 2-хлор-2'; ,4' ,6' -триэтил-N-(хлорметил)-аце-1 танилида в 30 мл хлорбензола добавля* ют 25 мл (20 г) безводного метанола и дают стоять 30 мин при комнатной температуре. Избыток метанола и небольшое количество хлорбензола удаляют под вакуумом в роторном испарителе и затем к остающему маслу добавт ляют вторую порцию 20 г свежего метанола. Смеси еще раз дают стоять 30 мин при комнатной температуре. Ме-1танол еще раз отгоняют в роторном испарителе и всю процедуру повторяют еще раз. После третьей отгонки по* лучают 21,0 г лимонно-желтого масла ( п ^ 1,5243), которое содержит по данным гельхроматографического анализа 97,7% 2',4\б'-триэтил-(№-метоксиметил)-2-хлорацетанилида, 1,0% 2і ,4*,б'итриэтил-2-хлорацетанилида (побочный продукт) и 0,7% 2',4',6*-триэтил-2,2-дихлорацетанилида (побочный продукт). ЯМР-спектр соответствует структуре. Выход состав ляет 93,1%. П р и м е р 23, Получение 2 * ,6' — -диметил-(N-циклогексилоксиметил)-2-хлорацетанилида. К 12,3 г (0,050 моль) 2',б'-диметил-(1$-хлорметил) -2-хлорацетанилида 13 14 1486053 данным анализа содержит 96,0% основнодобавляют 50,0 г безводного циклого продукта, с перемешиванием при темгексанола и дают стоять в течение D пературе от -20 до -25 C, По истеченочи при комнатной температуре. Избынии примерно 30 мин весь ацетанилид ток спирта отгоняют в роторном испарастворен. По истечении 1 ч при темрителе при 65°С/1 мм рт.ст. Свежий пературе от -20 до -25 С избыток метациклогексанол (50,0 г) добавляют в нола быстро извлекают однократной остаток и раствор нагревают при 45 С перегонкой. ЯМР-спектр кубовых остатв течение 30 мин. Избыток спирта ков показывает превращение 60,8% в отгоняют при 65°С /0,5 мм рт.ст. с получением 15,45 г (99,7% выхода)блед- 10 алахлор. Остаток второй раз обрабатывают 25 г свежего метанола в течено-лимонно-желтого масла. Выкристалние 1 ч при температуре от -20 до лизовавшееся из холодного гексана -25 С, После быстрой однократной пемасло дает полностью кристаллическое, регонки избыточного метанола ЯМРтвердое вещество с точкой плавления 15 спектр кубовых остатков показывает 46-47°С. ЯМР-спектр соответствует превращение 91,6% в алахлор. Остаток структуре. третий раз обрабатывают 25 г свежего ? П р и м е р 24. Получение 2 -метилметанола 2 ч при температуре от ' -6' -t-6yTnn-(N -циклопропилметокси-20 до. -25 С. Однократной перегонкой метил)-2-хлорацетанилида. 20 избыточного метанола получают 2,і 1 г Приблизительно 4,77 г 2 -метилчистого масла бледно-лимонкого цвета. -6*-t-бутил-(N-хлорметил)-2-хлорацеЯМР-спектр показывает полное превратанилида (0,016 моль) растворяют в щение в алахлор. Газохроматографичес9,60 г (0,132 моль) циклопропилкарбикий анализ масла показывает 94,7% нола. Раствору дают стоять 1 ч при 25 2'' ,6 -дизтші~(М-метоксиметил)-2-хлоркомнатной температуре, а затем избыацетанилида (алахлор) и 2,1% 2* ,6 ток спирта удаляют в роторном испари-диэтил-2-хлорацетанилида (побочный телем 55°С/1 мм рт,ст. Приблизительно продукт) . Выход 98,7%. 9,6 г свежего циклопропилкарбинола добавляют в оставшееся масло и раствор П р и м е р 27. В 11,70 г нагревают до 45 С в течение 20 мин. (0,041 моль) 2 ,6 -диэтил-(N-хлорИзбыток спирта еще раз удаляют под метил) -2-хлорацетанилида, который совакуумом (как упомянуто выше) с подержит 96,0% основного продукта, долучением легко-янтарного чистого масбавляют 25 мл безводного метанола и ла, п ^ 1,5280, с весом 5,28 г (98,9%) нагревают до 65 С в течение 5 мин. 35 Избыточный спирт с НС1 быстро удаляют П р и м е р 25. Получение 2'),6|-диметил-К-(2-метокси-1-метилэтоксив роторном испарителе при 65°С при осметил)-2-хлорацетанилида. таточном давлении 10 мм рт.ст.Еще порцию 15 мл свежего метанола добавляют Приблизительно 12,3 г (0,050 моль) в кубовые остатки, нагревают до 85 С 21 ,б'-диметил-К-(хлорметил)~2-хлор40 в течение 2 мин и избыточный спирт ацетанилида, растворенного в 20 мл удаляют под вакуумом. После третьей хлористого этилена, добавляют в 22,5 г обработки с помощью 15 мл свежего (0,25 моль) 2-метокси-і-метил-этанола. метанола при 100°С в течение 2 мин ч Раствору дают стоять 1 ч при комнатизбыточный спирт удаляют под вакууной температуре. Избыток спирта удамом с получением 11,33 г желтого мас-+ ляют под вакуумом в роторном испарила, которое содержит 99,0% 2*,6 'теле. Остающееся масло дополнительно -диэтил-(N-метоксиметил)-2-хлоробрабатывают 22,5 г свежего спирта ацетанилида и 0,9% 2',6*-диэтил-230 мин при 60°С. Избыток спирта уда-хлорацетанилида (побочный продукт). ляют, как показано выше, при 50 Выход 95,4% (от теоретич.). ЯМР, 65°С/1 мм рт.ст. Вес легко-желтого спектр соответствует структуре и являмаслянистого остатка составляет ется идентичным алахлору, полученно14 S 86 г (99,1% выхода). Показатель му по другим методам. преломления масла n*J1,5263. ЯМРспектр соответствует структуре, И р и м е р 28. В 11,17 г П р и м е р 26. В 25,0 г (0,78 моль)55 (o,Q39l м о л ь ) беэводного метанола добавляют 2,15 г метил)-2-хлорацетанилида, который (0,0078 моль) 2*,6 -диэтил-(Н-хлорсодержит 96,0% основного продукта, метмл)-2-хлорацетанилида,который по добавляют 25 мл безводного н-бутано 15 16 486053 9 добавить экономические и экологические преимущества. Преимущество способа в соответствии с настоящим изобретением состоит в том, что исходный- реагент формулы III можно легко выделить из его комплекса с галоидводородным побочным продуктом, очистить и направить на рециркуляцию с возвратом на одну или несколько реакционных стадий процесса. Подобным образом сам галоид водорода можно легко рекуперировать для ис- •• • пользования в ходе проведения многих ценных технологических операций, например для протравливания металлов,оксихлорирования, электролиз а элементарного хлора и водорода и тому подобного» то есть, другими словами , от неСравнительные результаты известного можно избавиться без ущерба го способа (примеры 8-12) сведены в 20 для окружающей среды. таблицу; В таблице под исходным материалом подразумевается непрореагироФ о р м у л а и з о б р е т е н и я вавший 2',6 -диэтил-(И-хлорметил).-2-хлорацетанилид, а термином побочный продукт обозначен 2 ,6 -диэтил-2- 25 Способ получения 2-галоацетамидов -хлорацетанилид, который является особщей формулы' I . • . -....'.. новным ацетанилидным побочным продуктом, образующимся в ходе проведения экспериментов каждого из таких приме-, ров. Совершенно очевидно, что помимо 30 больших количеств выделяемого галоге• S C-R 4 нида водорода образуется небольшое количество ацетанилида и других побочных продуктов, а в ходе проведения эксперимента примера 8 в результате где R - 2 ( 6-диметил-1-циклогексєннейтрализации образуется триэтиламин- 35 -1-ил или замещенный фенші формулы . гидрохлоридный побочный продукт. В таблице выход целевого продукта пригде R, и R a - С^-С^-алкил; веден в процентах в пересчете на коли-, ЇЦ - С.-С^элкил, хлорметил, чество 2 ,6 -диэтил-Н-(хлорметил)-2- < С (-С 4,-алкокси-метил, цикло-хлорацетанилидного исходного матери- : гексил или циклопропилметил; ала. . • і К, - бромметил, моно- или дихлорла. Раствор сразу нагревают до 120 С в масляной бане в течение 2,0 мин, а затем избыточный спирт удаляют под вакуумом в роторном испарителе при 60 С при остаточном давлении 10 ммрт.ст. Остаточное масло третий раз обрабатывают 25 мл н-бутанола при 160°С в течение 5 мин, а затем избыточный спирт удаляют в роторном испарителе. 10 Вес остаточного прозрачного желтогомасла составляет 12,45 г и оно соf держит 94,9% 2 ,6"-диэтил-(1Я-бутоксиметил)-2-хлорацетанилида (бутахлор) с 0,9% 2*(6 -диэтил-2-хлорацетанилида (побочный продукт). Выход 96,9%. ЯМР-спектр является идентичным аутен-. тичному бутахлору. R з метил, Анализ данных, которые указаны в •таблице, показывают преимущества пред-", латаемого способа. Существенно увеличивается выход алахлора, повышается степень чистоты его, заметно уменьша-' ется выход побочного продукта* повышена степень конверсии исходного мате-, риала в ходе проведения процесса без добавления основаниями отсутствует твердый продукт нейтрализации, кото- І рый в больших количествах образуется при осуществлении способа с добавле- 't ниєм основания (пример 8) и являющего-" ск лучшим способом в ранее известной технологии получения алахлора. К этим технологическим преимуществам следует __ взаимодействием соединения, общей формулы II л I I сн«х где X - хлор или бром; R HR,] - имеют указанные значения, со спиртом формулы III где ІЦ- имеет указанное значение, при температуре (-20)-(160 )°С и молярном соотношении соединений общей формулы II и III, равном 1 :1-100 с удалением 17 18 1486053 тем,. ротационный выпарной аппарат или аппаспирта, о т л и ч а ю щ и й с я рат с падающей пленкой при перепаде'; что, с целью повышения выхода целевотемпературы и давления соответственго продукта, спирт удаляют одновременно с * образующимся галогенводородом но на 5-60°Си 759,6-730 мм рт.ст. путем подачи реакционной массы на или только давления на 759-750 мм рт.ст І Способ Пример, АлаВыход хлор, алаX хлора, Примеры из описания к американскому патенту Р 3442945, добавлено основание 92V& 9 То же самое, исключая добавле• нне основания 85,8 10 То же самоё, что и в примере 9, за исключением оптимизации 87,4 условий 11 Данный способ, одностадийный вариант 94 ,4 12 То же, многостадийный вариант 99 ,4 Побоч* ный продукт, % Исходный материал, 8 Редактор С.Полякова Заказ 4364 90,5 4,9 0 83,7 7,5 5,5 85,8 6,2 4,6 91 ,0 95 ,8 1,7 1,4. 2 ,4 0 Составитель В.Мякушева , . Техред, М.Моргеятал КорректоруЛ.Осауленко Тираж 339 . Подписное ВНИИГШ Государственного комитета по изобретениям и открытиям при ГКНТ СССР 113035, Москва, Ж-35, Раушская наб., д. 4/5 Производственно-издательский комбинат "Патент", г.Ужгород, ул. Гагарина,101
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the production of haloacylamides
Автори англійськоюDonald Edgar Baldus, Edward Ervin Debus
Назва патенту російськоюСпособ получения 2-галоацетамидов
Автори російськоюДоналд Эдгар Балдус, Эдвард Эрвин Дебус
МПК / Мітки
МПК: C07C 233/18
Мітки: 2-галоацетамидів, одержання, спосіб
Код посилання
<a href="https://ua.patents.su/14-5921-sposib-oderzhannya-2-galoacetamidiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання 2-галоацетамидів</a>
Спосіб одержання сфінгомієліну

Номер патенту: 1220
Опубліковано: 30.12.1993
Автори: Пінчук Анатолій Миколайович, Краснопольський Юрій Михайлович, Сенніков Георгій Антонович, Швец Віталій Івановіч
МПК: A23J 7/00, A61K 35/30, C11B 1/10
Мітки: сфінгомієліну, спосіб, одержання
Формула / Реферат:
Способ получения сфингомиелина из мозга крупного рогатого скота, включающий очистку сырья, его измельчение, гомогенизацию в ацетоне, центрифугирование, экстракцию осадка этиловым спиртом и петролейным эфиром, гидролиз экстракта, хроматографию, на силикагеле с последующим элюированием целевого продукта смесью хлороформа и метанола и сушкой в вакууме, отличающийся тем, что, с целью повышения выхода целевого продукта, экстракцию проводят...
Спосіб одержання гангліозидів

Номер патенту: 1217
Опубліковано: 30.12.1993
Автори: Мензелеєв Раміль Ферідович, Краснопольський Юрій Михайлович, Швец Віталій Івановіч, Теміров Юрій Павлович, Мезін Ігор Олександрович
МПК: A61K 31/7012, A61K 35/30
Мітки: гангліозидів, спосіб, одержання
Формула / Реферат:
Способ получения ганглиозидов путем гомогенизации мозговой ткани животных, двукратной экстракции гомогената смесью хлороформа и метанола, фильтрации, инкубации, упаривания, хроматографии с последующей элюцией целевого продукта, отличающийся тем, что, с целью повышения выхода целевого продукта и интенсификации способа, к осадку ткани после фильтрации добавляют смесь 70% -ного изопропилового спирта и гексана в объемных соотношениях...
Спосіб отримання 2,6-діалкін-n-(алкоксіметил)-2хлорацетанілідов

Номер патенту: 5577
Опубліковано: 28.12.1994
Автори: Янош Доміан, Ендре Шюмегі, Міклош Надаші, Дьердь Хусак, Ервін Вертеші, Єнє Пеліва, Елемер Тельерді, Ласло Й.Сабо, Андраш Вашш, Ласло Лєндваі, Золтан Колоніг, Балінт Надь, Ласло Кульчар, Бєла Дьєрфі, Міклош Ковач, Андраш Хааш
МПК: C07C 67/00, C07C 231/00, A01N 37/26, A01N 37/22, A01N 37/18, C07C 233/25, C07C 233/88
Мітки: спосіб, 2,6-діалкін-n-(алкоксіметил)-2хлорацетанілідов, отримання
Формула / Реферат:
Способ получения 2',6'-диалкил-N-(алкоксиметил)-2-хлорацетанилидов общей формулы:где R1, R2, R3 - С1-С4 - алкил прямолинейный или разветвленный, с использованием ацилирования аминосоединения хлорацетилхлоридом в среде растворителя с последующей обработкой избытком спирта при 20-40°С и выделением целевого продукта, отличающийся тем, что, с целью увеличения выхода целевого продукта, ксилольный раствор 2,6-диалкиланилина...
Спосіб одержання хлоргідрату 2-(амінометил)-n,nдіетил-1-фенілциклопропанаміду (z)

Номер патенту: 2328
Опубліковано: 26.12.1994
Автори: Жільбер Музен, Бернар Бонно, Анрі Кусс, Жан-Франсуа Патуазо
МПК: C07C 233/12, C07D 307/77, C07C 231/00, C07C 67/00, C07C 237/24, C07D 209/48
Мітки: хлоргідрату, 2-(амінометил)-n,nдіетил-1-фенілциклопропанаміду, спосіб, одержання
Формула / Реферат:
Способ получения хлоргидрата 2-(аминометил) -N, N-диэтил-1 -фенилциклопропанамида (Z) на основе 1-фенил-2-оксо-3-оксабицикло (3, 1, 3) гексана и тионилхлорида формулына основе 1- фенил-2-оксо-З-оксабицикло (3, 1, 3) гексана формулыи тионилхлорида, отличающийся тем, что, с целью повышения выхода целевого продукта, 1-фенил-2-оксо-З-оксабицикло (3, 1, 3) гексан подвергают взаимодействию с фталимидом калия в...
Спосіб одержання фосфатиділсеріну

Номер патенту: 1218
Опубліковано: 30.12.1993
Автори: Теміров Юрій Павлович, Краснопольський Юрій Михайлович, Мензелеєв Раміль Ферідович, Швец Віталій Івановіч, Мензелеєва Гульжан Казбеківна
МПК: G07F 9/10
Мітки: фосфатиділсеріну, одержання, спосіб
Формула / Реферат:
Способ получения фосфатидилсерина путем гомогенизации тканей головного мозга крупного рогатого скота, экстракции, концентрирования экстракта, растворения его в хлороформе, хроматографии, элюции, осаждения целевого продукта с последующим его высушиванием, отличающийся тем, что с целью повышения выхода целевого продукта, перед хроматографией к эстракту добавляют ацетон до объемного соотношения к хлороформу (0,9-1,1):(19-21), отделяют осадок,...
Попередній патент: Пристрій для розтягу та плоскісної сушки плоских шкір
Наступний патент: Полупіковая електростанція
Випадковий патент: Йогурт