Спосіб очищення метил{4,6-діаміно-2-[1-(2-фторбензил)-1н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату та метил{4,6-діаміно-2-[1-(2-фторбензил)-1н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату сульфінілдиметан
Номер патенту: 110779
Опубліковано: 25.02.2016
Автори: Резе Йоахім, Зігель Конрад, Йонтген Вінфрід, Маіс Франц-Йозеф













Формула / Реферат
1. Спосіб очищення метил{4,6-діаміно-2-[1-(2-фторбензил)-1Н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату формули (І)
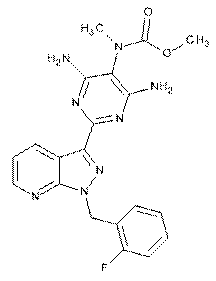 , (І)
, (І)
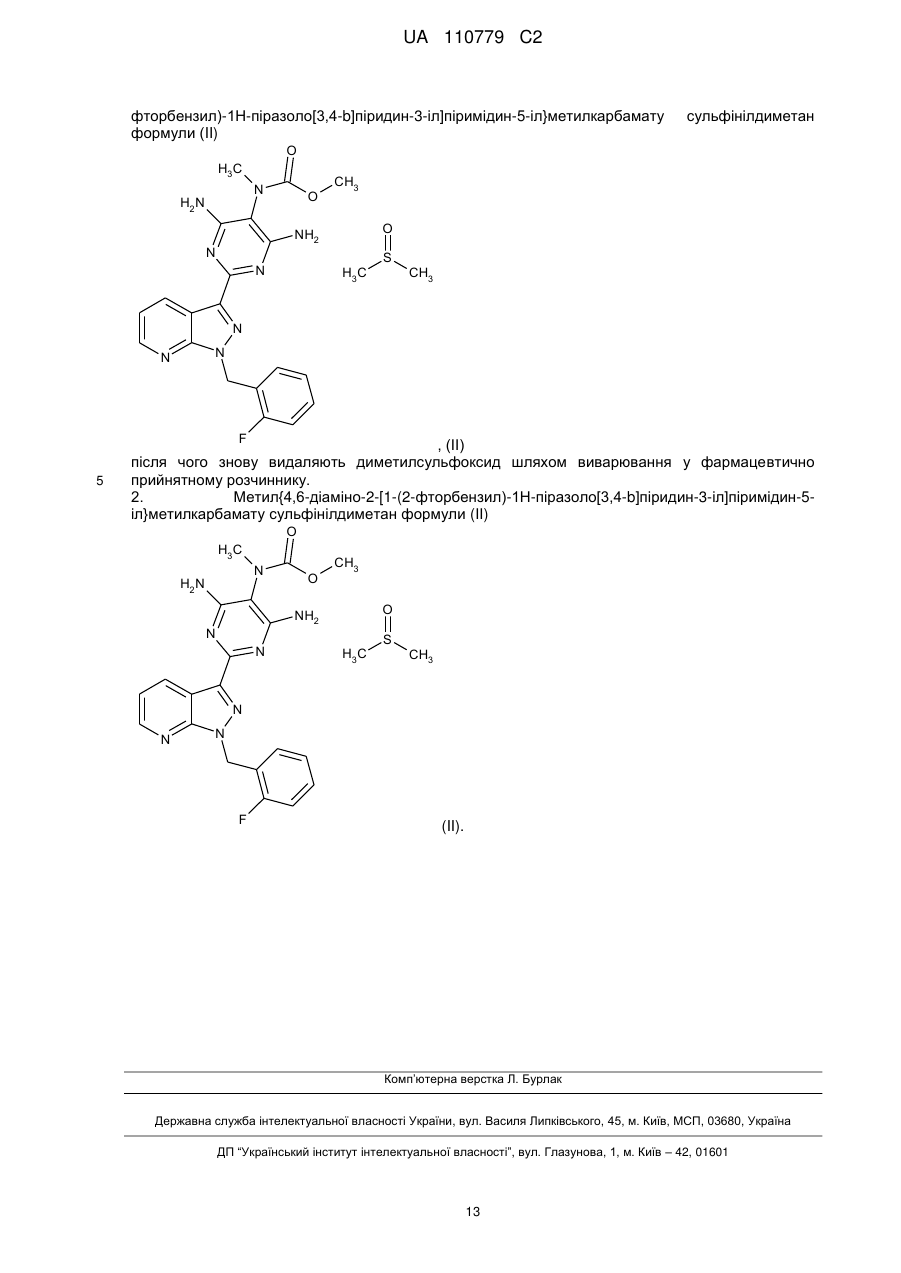
який відрізняється тим, що вихідний продукт сполуки формули (І) розчиняють у диметилсульфоксиді і відокремлюють одержаний при цьому метил{4,6-діаміно-2-[1-(2-фторбензил)-1Н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату сульфінілдиметан формули (II)
 , (ІІ)
, (ІІ)
після чого знову видаляють диметилсульфоксид шляхом виварювання у фармацевтично прийнятному розчиннику.
2. Метил{4,6-діаміно-2-[1-(2-фторбензил)-1Н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату сульфінілдиметан формули (ІІ)
(II).
Текст
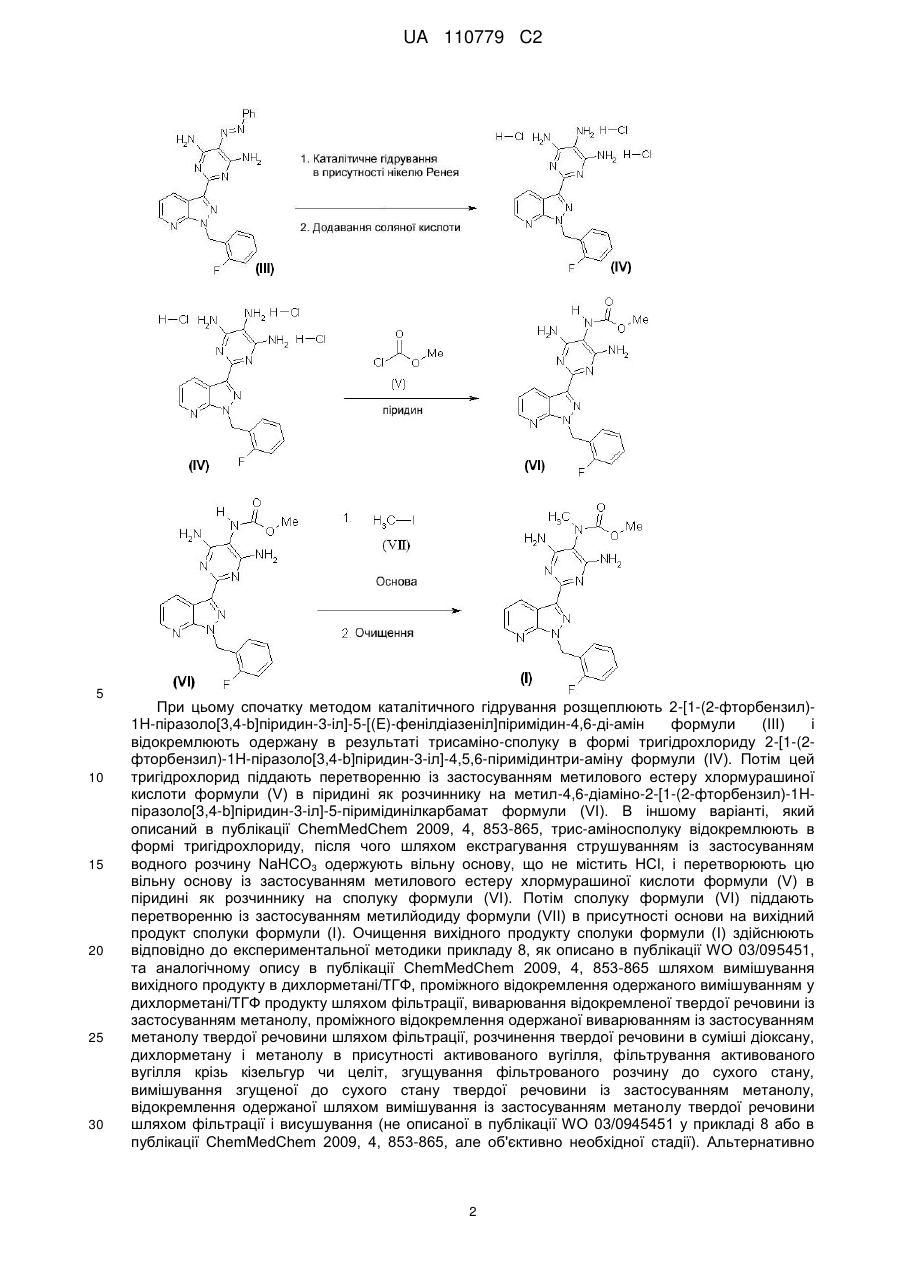
Реферат: UA 110779 C2 (12) UA 110779 C2 Винахід стосується способу очищення метил{4,6-діаміно-2-[1-(2-фторбензил)-1Н-піразоло[3,4b]піридин-3-іл]піримідин-5-іл}метилкарбамату формули (І) O H3 C N H2 N O CH3 NH2 N N N N N F , (I) причому для очищення метил{4,6-діаміно-2-[1-(2-фторбензил)-1Н-піразоло[3,4-b]піридин-3іл]піримідин-5-іл}метилкарбамату сульфінілдиметан (1:1), тобто сполуку формули (II), відокремлюють як проміжний продукт або одержують її як проміжний продукт у цьому способі очищення, залежно від конкретних обставин у формі суміші. UA 110779 C2 Винахід стосується способу одержання метил-{4,6-діаміно-2-[1-(2-фторбензил)-1Hпіразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату формули (I) H3 C H2 N O N O CH3 NH2 N N N N N F 5 (I). Винахід стосується також способу очищення вихідного продукту сполуки формули (I) для використання як фармацевтично активної речовини, причому для очищення метил-{4,6-діаміно2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]піримідин-5іл}метилкарбаматсульфінілдиметан (1:1), тобто сполуку формули (II) O H3 C CH3 N O H2 N O NH2 N S N H3 C CH3 N N N F 10 15 20 (II). відокремлюють як проміжний продукт або одержують як проміжний продукт у цьому способі очищення, залежно від конкретних обставин у формі суміші. Сполука формули (I) діє як стимулятор розчинної гуанілатциклази і може бути застосована як засіб для профілактики та/або лікування серцево-судинних захворювань, наприклад артеріальної гіпертонії та серцевої недостатності, стабільної та нестабільної стенокардії, периферичної та кардіальної ангіопатії, аритмії, для лікування тромбоемболічних захворювань та ішемічних хвороб, наприклад інфаркту міокарда, апоплексії, транзиторних ішемічних атак, порушення периферійного кровообігу, запобігання рестенозам, наприклад після тромболітичної терапії, крізьшкірної транслюмінальної ангіопластики (PTA), крізьшкірної транслюмінальної коронарної ангіопластики (PTCA), шунтування, а також для лікування артеріосклерозу, астматичних захворювань і хвороб урогенітальної системи, наприклад гіпертрофії простати, еректильної дисфункції, жіночої сексуальної дисфункції, остеопорозу, глаукоми, легеневої гіпертонії, гастропарезу та нетримання сечі. Одержання сполуки формули (I) та її очищення в принципі відомі. В публікації WO 03/095451 описане одержання сполуки формули (I) у наведеній далі послідовності: 1 UA 110779 C2 5 10 15 20 25 30 При цьому спочатку методом каталітичного гідрування розщеплюють 2-[1-(2-фторбензил)1H-піразоло[3,4-b]піридин-3-іл]-5-[(E)-фенілдіазеніл]піримідин-4,6-ді-амін формули (III) і відокремлюють одержану в результаті трисаміно-сполуку в формі тригідрохлориду 2-[1-(2фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]-4,5,6-піримідинтри-аміну формули (IV). Потім цей тригідрохлорид піддають перетворенню із застосуванням метилового естеру хлормурашиної кислоти формули (V) в піридині як розчиннику на метил-4,6-діаміно-2-[1-(2-фторбензил)-1Hпіразоло[3,4-b]піридин-3-іл]-5-піримідинілкарбамат формули (VI). В іншому варіанті, який описаний в публікації ChemMedChem 2009, 4, 853-865, трис-аміносполуку відокремлюють в формі тригідрохлориду, після чого шляхом екстрагування струшуванням із застосуванням водного розчину NaHCO3 одержують вільну основу, що не містить HCl, і перетворюють цю вільну основу із застосуванням метилового естеру хлормурашиної кислоти формули (V) в піридині як розчиннику на сполуку формули (VI). Потім сполуку формули (VI) піддають перетворенню із застосуванням метилйодиду формули (VII) в присутності основи на вихідний продукт сполуки формули (I). Очищення вихідного продукту сполуки формули (I) здійснюють відповідно до експериментальної методики прикладу 8, як описано в публікації WO 03/095451, та аналогічному опису в публікації ChemMedChem 2009, 4, 853-865 шляхом вимішування вихідного продукту в дихлорметані/ТГФ, проміжного відокремлення одержаного вимішуванням у дихлорметані/ТГФ продукту шляхом фільтрації, виварювання відокремленої твердої речовини із застосуванням метанолу, проміжного відокремлення одержаної виварюванням із застосуванням метанолу твердої речовини шляхом фільтрації, розчинення твердої речовини в суміші діоксану, дихлорметану і метанолу в присутності активованого вугілля, фільтрування активованого вугілля крізь кізельгур чи целіт, згущування фільтрованого розчину до сухого стану, вимішування згущеної до сухого стану твердої речовини із застосуванням метанолу, відокремлення одержаної шляхом вимішування із застосуванням метанолу твердої речовини шляхом фільтрації і висушування (не описаної в публікації WO 03/0945451 у прикладі 8 або в публікації ChemMedChem 2009, 4, 853-865, але об'єктивно необхідної стадії). Альтернативно 2 UA 110779 C2 5 10 15 20 25 30 35 очищення згущеного до сухого стану вихідного продукту сполуки формули (I) може бути здійснене методом препаративної хроматографії (RP-HPLC) із незадовільним виходом. Ці методи синтезу та очищення мають різні недоліки, які суттєво перешкоджають їх технічній реалізації у великому масштабі. Це стосується насамперед відокремлення трис-аміносполуки в формі тригідрохлориду формули (IV). Додавання соляної кислоти вимагає застосування кислотостійкого технічного устаткування, а вихід стадії складає незадовільні 59,3 % від теоретичного значення (див. зокрема, приклад 8A з публікації WO 03/095451). Здійснення перетворення трис-аміносполуки формули (IV) або відповідної не вміщуючої HCl основи в піридині як розчиннику є недоліком. Сполука формули (VI) може бути відокремлена лише шляхом технічно невигідного повного випарювання реакційної суміші (див., зокрема, приклад 5 в публікації WO 03/095451). Такі стадії у великому масштабі призводять, як правило, до значних проблем, наприклад припікання або термічного розкладення внаслідок суттєвого подовження тривалості дії термічного навантаження при здійснюванні способу в більшому масштабі. Значним недоліком є також очищення продукту формули (VI) згідно з експериментальною методикою прикладу 5 із публікації WO 03/095451 шляхом виварювання в діетиловому етері. Ця стадія внаслідок легкої займистості діетилового етеру може бути здійснена лише за рахунок збільшення технічних витрат. Проте, особливим недоліком є метод очищення вихідного продукту формули (I). Ефективне очищення є обов'язково необхідним для забезпечення можливості застосування як фармацевтично активної речовини. Описане очищення методом високоефективної рідинної хроматографії з оберненою фазою, тобто хроматографічне очищення є лабораторним методом, який в технічному масштабі може бути реалізований лише за рахунок дуже великих витрат. Окрім цього, наведений вихід сягав лише 29 % для стадії синтезу з одержанням вихідного продукту формули (I) і його очищення, тобто був дуже низьким. Альтернативний метод одержання та очищення є дуже складним. Він включає в цілому 5 операцій відокремлення твердої речовини (дві операції згущення до сухого стану і три операції фільтрування), причому, як вже було сказано вище, згущення до сухого стану в технічному масштабі є дуже несприятливим. У цілому 5 операцій відокремлення твердої речовини при здійсненні однієї хімічної стадії для одержання та очищення фармацевтичної активної речовини в технічному масштабі є дуже невигідним. Тому існувала задача розроблення спрощеного способу, який можна було б здійснювати надійно і ефективно також у промисловому масштабі, і який дозволяв би одержувати великий вихід високочистої активної речовини фармацевтично прийнятної якості. Неочікувано було винайдено спосіб одержання метил{4,6-діаміно-2-[1-(2-фторбензил)-1Hпіразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату формули (I) O H3 C Me N O H2 N NH2 N N N N N F 40 (I) та його очищення для застосування як фармацевтично активної речовини. Цей новий спосіб одержання та очищення вихідного продукту сполуки формули (I) відрізняється від донині відомих способів у таких пунктах: - після каталітичного гідрування сполуки формули (III) відокремлюють трис-аміносполуку як вільну основу формули (VIII) без проміжного утворення солей 3 UA 110779 C2 NH2 H2 N NH2 N N N N N F (VIII). - Сполуку формули (VI) одержують із застосуванням метилового естеру хлормурашиної кислоти або диметилдикарбонату як реагенту способом без застосування піридину O H Me N O H2 N NH2 N N N N N F 5 10 (VI). - Сполуку формули (VI) відомим чином із застосуванням засобу для метилювання перетворюють на вихідний продукт формули (I), очищення вихідного продукту формули (I) для застосування як фармацевтичної активної речовини здійснюють із застосуванням сполуки метил-{4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]піримідин-5іл}метилкарбаматсульфінілдиметану (1:1), тобто сполуку формули (II) одержують як відокремлений проміжний продукт або у вигляді суміші H3 C H2 N O N O CH3 O NH2 N N H3 C S CH3 N N 15 N F (II). Ці відмінності дозволяють подолати недоліки донині відомих способів і одержати великий вихід високочистої активної речовини фармацевтично прийнятної якості. Далі наведений докладний опис відповідного винаходові способу одержання сполуки формули (I), а також очищення через одержання проміжного продукту формули (II). Каталітичне гідрування сполуки формули (III) Першу стадію відповідного винаходові способу починають каталітичним гідруванням сполуки формули (III). 4 UA 110779 C2 5 10 15 20 25 30 35 (вільна основа формули (IV)) Ця стадія може бути здійснена в присутності нікелю Ренея або традиційно застосовуваних у техніці платинових чи паладієво-вугільних каталізаторів. Переважними є платинові та паладієво-вугільні каталізатори. Як розчинник застосовують N, N-диметилформамід (ДМФ), N, N-диметилацетамід (ДМА) або N-метил-2-піролідон (N-MП), переважно ДМФ. Гідрування здійснюють в таких умовах: температура 40-80 °C, переважно 50-70 °C, тиск 2-90 бар, переважно 5-70 бар водню, тривалість гідрування 1-72 години, переважно 3-36 годин. Після фільтрування каталізатора здійснюють осадження зі спирту C 1-C4, переважно метанолу або етанолу та/або води. Переважною є суміш із метанолу, ізопропанолу або етанолу і води. Спирт C1-C4 в рамках винаходу означає нерозгалужений або розгалужений спирт, що містить від 1 до 4 атомів вуглецю. Переважними прикладами є метанол, етанол, н-пропанол, ізопропанол, н-бутанол і трет-бутанол. Це визначення стосується також застосовуваних далі спиртів C1-C4. Можливим є також видалення частини розчинника відгонкою перед осадженням, згідно з винаходом здійснюють часткову відгонку 0-80 %, переважно 40-70 % розчинника. Одержаний таким чином вологий продукт висушують у вакуумі. Одержують продукт формули (VIII) (відповідає вільній основі формули (IV)). Перетворення сполуки формули (VIII) із застосуванням метилового естеру хлормурашиної кислоти (V) Продукт формули (VIII) перетворюють із застосуванням, наприклад, метилового естеру хлормурашиної кислоти формули (V) новим способом без застосування піридину на продукт формули (VI). Як розчинник для перетворення використовують спирт C 1-C4, переважно етанол, метанол, ізопропанол, особливо переважно ізопропанол. Кількість метилового естеру хлормурашиної кислоти становить від 1,0 до 3,0 еквівалентів, переважно від 1,0 до 2,0 еквівалентів відносно вихідної речовини формули (VIII). Можлива температура для здійснення реакції становить 0-75 °C, переважно 15-50 °C. У процесі реакції утворюється хлороводень, який у реакційній суміші утворює сполуку формули (IX), тобто гідрохлорид продукту формули (VI). Цей продукт формули (IX) може бути або відокремлений в формі вміщуючого HCl продукту і відщеплений шляхом додавання основи до продукту формули (VI), або він може бути відщеплений шляхом додавання основи ще перед відокремленням, причому відокремлюється безпосередньо продукт формули (VI). 5 UA 110779 C2 5 10 15 20 25 30 35 40 Згідно з винаходом переважним є варіант відщеплення продукту формули (IX) шляхом додавання основи перед відокремленням і безпосереднього відокремлення вільної основи формули (VI). Як основи згідно з винаходом придатними до застосування є всі основи, показник константи основної дисоціації pKB яких є більшим, аніж у сполуки формули (I). Прикладами таких основ є: гідроксиди, карбонати і фосфати лужних і лужноземельних металів, азотовмісні органічні основи, такі як триалкіламін, гуанідини або амідини. Прикладами є гідроксиди або карбонати літію, натрію, калію, рубідію, цезію, магнію, кальцію, стронцію і барію, фосфати натрію і калію, триалкіламіни з нерозгалуженими, циклічними або розгалуженими C 1-C20-алкільними залишками та циклічні або з незамкненими ланцюгами гуанідини або амідини. Переважними згідно з винаходом є триетиламін, трипропіламін, діізопропілетиламін, трибутиламін, дициклогексилетиламін, циклогексилметиламін, циклогексилдіетиламін, триізооктиламін, тридециламін, тридодециламін, тригексадециламін, N-метилморфолін, діазабіциклоундецен (ДБУ), діазабіциклононен (ДБН), тетраметилгуанідин тощо. Особливо переважними є триетиламін, трибутиламін, діізопропілетиламін, N-метилморфолін, ДБУ, ДБН. Кількість основи становить від 1,0 до 2,0 еквівалентів, переважно від 1,0 до 1,5 еквівалентів відносно вихідної речовини – метилового естеру хлормурашиної кислоти формули (V). Можлива температура для здійснення перетворення із застосуванням основи становить 0100 °C, переважно 15-70 °C. Продукт формули (VI) перебуває у вигляді суспензії; його відокремлюють шляхом фільтрації. Його промивають, як зазвичай, спиртом C1-C4 і висушують у вакуумі. Перетворення сполуки формули (VIII) із застосуванням диметилдикарбонату (X) В іншому відповідному винаходові способі продукт формули (VIII) піддають перетворенню із застосуванням диметилдикарбонату формули (X) на продукт формули (VI). Для здійснення цього перетворення немає потреби в застосуванні основи, наприклад піридину. Як розчинник для цього перетворення застосовують спирти C 1-C4, переважно етанол, метанол, ізопропанол, особливо переважно ізопропанол. Кількість диметилдикарбонату становить від 1,0 до 3,0 еквівалентів, переважно від 1,0 до 2,0 еквівалентів відносно вихідної речовини формули (VIII). Можлива температура реакції становить 0-65 °C, переважно 15-40 °C. Продукт формули (VI) випадає в осад; його відокремлюють шляхом фільтрування. Як зазвичай, його промивають спиртом C1-C4 і висушують у вакуумі. При перетворенні із застосуванням диметилдикарбонату одержують безпосередньо продукт формули (VI). Отже, потреби в додаванні основи немає. Обидва способи, тобто перетворення сполуки формули (VIII) із застосуванням метилового естеру хлормурашиної кислоти і наступне відщеплення гідрохлориду формули (IX) із застосуванням основи або перетворення сполуки формули (VIII) із застосуванням диметилдикарбонату, дозволяють одержати продукти формули (VI) порівнянної якості, тобто продукт формули (VI), одержаний будь-яким із цих способів, рівною мірою може бути застосований для подальшого перетворення на продукт формули (I). Обидва способи є переважними згідно з винаходом. 6 UA 110779 C2 5 10 15 20 25 30 35 40 Сполука формули (VI) може утворювати сольвати чи вміщуючі розчинник форми твердої речовини, наприклад вміщуючі метанол, етанол або ізопропанол форми твердої речовини. Тому можливою є ситуація, в якій при розщепленні гідрохлориду формули (IX) на продукт формули (VI) або при безпосередньому синтезі продукту формули (VI) із застосуванням диметилдикарбонату одержують сольват спирту C 1-C4 як розчинника. Цей сольват може бути настільки стабільним, що при висушуванні продукту формули (VI) він не розкладається повністю, внаслідок чого легко виявити залишки розчинника, тобто, наприклад, відповідного спирту C1-C4, у продукті формули (VI). З іншої сторони, температура висушування продукту формули (VI) не може бути довільно високою, оскільки при надто високій температурі він може розкладатися з утворенням побічних продуктів. Тому згідно з винаходом переважним є варіант, в якому продукт формули (VI), одержаний шляхом розщеплення гідрохлориду формули (IX) із застосуванням основи або шляхом прямого синтезу із застосуванням диметилдикарбонату висушують при температурі продукту не вище 110°, особливо переважно не вище 100°. При цьому особливо переважним є варіант, в якому залишки спирту C1-C4 в формі сольвату присутні в продукті формули (VI), і в цій формі його використовують для одержання проміжного продукту формули (II), чи застосовують продукт формули (I)?. Згідно з винаходом цілком переважним є варіант, в якому продукт формули (VI) ще містить ізопропанол як залишок розчинника в кількості від 0 до 13 %. Метилювання сполуки формули (VI) Одержаний таким чином продукт формули (VI) відомим чином, наприклад, згідно з одним із описів, наведених у публікаціях WO 03/0945451 або ChemMedChem 2009, 4, 853-865, із застосуванням засобу для метилування Me-X перетворюють на вихідний продукт, що містить великий відсоток сполуки формули (I). Як засіб для метилювання Me-X згідно з винаходом застосовують метилйодид, диметилсульфат, метиловий естер толуолсульфонової кислоти тощо; переважно застосовують метил йодид або диметилсульфат. Очищення вихідного продукту сполуки формули (I) Вихідний продукт формули (I) згідно з винаходом очищають для застосування як фармацевтичної активної речовини. Для цього спочатку виготовляють суміш, що містить як проміжний продукт великий відсоток сполуки формули (II). Для цього вихідний продукт формули (I) розчиняють в ДМСО, в разі необхідності в присутності фармацевтично прийнятного простого розчинника, вибраного з класу, що включає кетони, етери, естери або спирти. Прикладами таких розчинників є метанол, етанол, ізопропанол, 1-бутанол, 2-бутанол, етилацетат, ізопропіл- або пропілацетат, бутилацетат, третбутил-метиловий етер, діізопропіловий етер, ацетон, метилетилкетон, метилізобутилкетон тощо. Переважно застосовують етанол, ізопропанол, етилацетат, ізопропілацетат, бутилацетат, метилетилкетон, метилізобутилкетон, особливо переважно етилацетат. Можуть бути застосовані також суміші цих розчинників. 7 UA 110779 C2 5 10 15 20 25 30 35 40 45 50 ДМСО додають у кількості від 100 до 750 мас. % відносно застосованої кількості вихідного продукту формули (I), переважно від 150 до 500 мас. %. У разі необхідності до цієї суміші може бути додане активоване вугілля в кількості від 0,25 до 35 мас. % відносно застосованої кількості вихідного продукту формули (I), переважно від 0,5 до 20 % мас. %. Для одержання розчину суміш нагрівають до 40-120 °C, переважно до 50-100 °C. Для одержання фармацевтично прийнятного продукту формули (I) розчин необхідно фільтрувати. Фільтрування слід здійснювати незалежно від того, було чи не було додане активоване вугілля. Кількість фармацевтично прийнятного розчинника, який окрім ДМСО додають до розчину вихідного продукту формули (I), тобто перед фільтруванням, становить від 25 до 200 мас. % відносно загальної кількості ДМСО, переважно від 40 до 100 мас. %. Фільтрування здійснюють при високій температурі, що становить 40-120 °C, переважно 50100 °C. Після фільтрування при високій температурі додають фармацевтично прийнятний розчинник, переважно той самий, що додавали перед цим. Завдяки цьому відбувається кристалізація продукту формули (II). Загальна кількість розчинника, доданого до і після фільтрування, становить від 200 до 1500 мас. % відносно загальної кількості ДМСО, переважно 400-1200 мас. %. Температура, при якій здійснюють додавання, становить 30-110 °C, переважно 35-90 °C. Перед відокремленням твердої речовини, яка містить високий відсоток сполуки формули (II), здійснюють охолодження для остаточного осадження в діапазоні температур 0-35 °C, переважно до нормальної температури, наприклад 20-30 °C. Відокремлення здійснюють за допомогою традиційних пристроїв, наприклад нутч-фільтра або центрифуги. Для видалення маточного розчину відокремлений матеріал при відокремленні промивають фармацевтично прийнятним розчинником, переважно таким самим, який застосовували перед цим. Відокремлений матеріал після повторного розчинення ДМСО містить великий відсоток продукту формули (II). Крім цього, невеликий відсоток продукту формули (I) в звичайному випадку може випадати в осад без утворення сольвату з ДМСО. Можливим є також утворення сольватів із іншою стехіометрією або утворення адуктів розчинника без точно визначеної стехіометрії. Крім цього, матеріал може містити також ДМСО у незв'язаній формі в формі прилиплих залишків розчинника. Вміст ДМСО у відокремленому матеріалі становить в звичайному випадку від 10 до 25 мас. %, переважно 12-17 %. Згідно з винаходом особливо переважним є варіант, в якому продукт формули (II) одержують у формі цієї суміші та використовують для одержання очищеного продукту формули (I). Одержаний таким чином продукт формули (II) може бути підданий висушуванню або у вологій формі, вміщуючій залишки розчинника, тобто прилиплого ДМСО і одного чи кількох розчинників для осадження, також застосований у перетворенні на очищений продукт формули (I). Сполука формули (II) є новою. Вона може бути одержана, як описано у наведених далі прикладах виконання винаходу, в чистій формі; її параметри можуть бути визначені аналітичним шляхом. Для фармацевтичного застосування з продукту формули (II) або суміші, яка містить великий відсоток сполуки формули (II), необхідно видалити ДМСО. Для цього продукт формули (II) або відокремлену суміш, яка містить великий відсоток продукту формули (II), виварюють у фармацевтично прийнятному розчиннику, вибраному з групи, що включає кетони, етери, естери або спирти. Прикладами таких розчинників є метанол, етанол, ізопропанол, 1-бутанол, 2-бутанол, етилацетат, ізопропіл- або пропілацетат, бутилацетат, трет-бутил-метиловий етер, діізопропіловий етер, ацетон, метилетилкетони, 8 UA 110779 C2 5 10 15 20 25 30 35 метилізобутилкетон тощо. Переважними є етанол, ізопропанол, етилацетат, ізопропілацетат, бутилацетат, метилетилкетон, метилізобутилкетон. Можуть бути застосовані також суміші цих розчинників. Особливо переважно застосовують етилацетат або суміш етилацетату з етанолом. Виварювання здійснюють зі зворотним потоком відповідного розчинника або в разі необхідності під дещо підвищеним тиском. Температура становить 50-150 °C, переважно 80120 °C. Відповідний винаходові спосіб пропонує явні переваги порівняно з рівнем техніки. Насамперед неочікуваним є те, що пряме відокремлення сполуки формули (VIII) (вільна основа) без проміжного утворення сполуки формули (IV) (тригідрохлорид) дозволяє значно збільшити вихід при одночасно значному технічному спрощенні здійснення способу (немає потреби в застосуванні кислотостійких компонентів устаткування). Сполука формули (VIII) може бути тепер перетворена новими способами без піридину із застосуванням метилового естеру хлормурашиної кислоти або диметилкарбонату на сполуку формули (VI). Ці нові способи є дуже простими і можуть бути здійснені з мінімальними технічними витратами. Продукт формули (VI) після реакції перебуває в формі суспендованої твердої речовини і може бути відокремлений шляхом фільтрації без здійснення стадії випарювання. При цьому одержуваний вихід є дуже високим. Неочікуваним є також те, що очищення вихідного продукту формули (I) для фармацевтичного застосування здійснюють зокрема шляхом повторного розчинення із застосуванням вміщуючої ДМСО суміші розчинників, а також те, що при цьому одержують нову сполуку формули (II) як проміжний продукт, залежно від конкретних обставин як компонент суміші, вміст якого в суміші є великим. На цій стадії забезпечується відокремлення всіх сторонніх домішок, тому їх залишковий вміст є низьким, завдяки чому після видалення ДМСО шляхом звичайного виварювання одержують високочисту тверду речовину формули (I). Ця тверда речовина, як правило, є безбарвною або злегка жовтуватою, а її аналітична чистота (ВЕРХ) явно перевищує 98 мас. %, що є дуже переважним для фармацевтичного застосування. Технічне здійснення способу є надійним і забезпечує реалізацію виробництва в промисловому масштабі. Він легко може бути узгоджений з конкретним виробничим оснащенням. В особливо переважній формі виконання очищення вихідного продукту формули (I) проміжне відокремлення продукту формули (II) або суміші, що містить великий відсоток сполуки формули (II), здійснюють в нутч-сушарці. Наступне видалення ДМСО з відокремленого на проміжній стадії продукту формули (II) здійснюють шляхом безпосереднього додавання розчинника в нутч-сушарку з проміжним висушуванням або без проміжного висушування продукту формули (II). Це дозволяє уникнути відкритої обробки твердої речовини продукту формули (II), яка пов'язана з ризиком забруднення. Експериментальна частина Скорочення та акроніми: абс. абсолютний кат. каталітичний CI хімічна іонізація (при МС) d доба ТХ тонкошарова хроматографія ДМФ диметилформамід ДМСО диметилсульфоксид теор. теоретичного значення (стосовно виходу) знач. ee надлишок енантіомерів EI іонізація електронним ударом (при МС) ent енантіомер / енантіомерно чистий екв. еквівалент(и) ESI іонізація електроспреєм (при МС) ГХ-МС газова хроматографія/мас-спектрометрія мас. % масовий відсоток год. година (години) ВЕРХ рідинна хроматографія високого тиску, високоефективна рідинна хроматографія конц. концентрований РХ-МС рідинна хроматографія/мас-спектрометрія хв. хвилина (хвилини) МС мас-спектрометрія 9 UA 110779 C2 ЯМР Ph Rf Rt RT v/v водн. 5 10 15 20 25 30 35 40 45 50 спектрометрія ядерного магнітного резонансу феніл індекс утримання (при ТХ) час утримання (при ВЕРХ) кімнатна температура співвідношення об'єм/об'єм (розчину) водний, водний розчин Наведені далі приклади пояснюють, але не обмежують винахід. Приклад 1 Одержання 2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]-4,5,6-піримідинтриаміну (VIII) У автоклаві-конвертері суспендували 1100 г сполуки формули (III) в 5,4 л ДМФ. Додавали 44 г комерційно доступного зволоженого водою (вологість близько 50 %) 5 %-го паладієвовугільного каталізатора і після інертизації азотом і нагнітання водню здійснювали гідрування в закритому автоклаві протягом близько 18 год. під тиском 65 бар водню при внутрішній температурі близько 60 °C. Після охолодження до температури близько 25 °C, зменшення тиску та інертизації виймали одержаний продукт із автоклава, причому додатково промивали його із застосуванням 650 мл ДМФ. Три таких одержаних аналогічним способом порції продукту очищали, видаляли відпрацьований каталізатор фільтруванням, промивали із застосуванням 1,1 л ДМФ і згущували фільтрат у вакуумі приблизно до третини його початкової маси. В залишок масою близько 6,5 кг послідовно вводили 8,25 л метанолу і 8,25 л води, охолоджували суспензію для остаточного завершення кристалізації до температури близько 5 °C, відфільтровували тверду речовину і промивали сумішшю метанол/вода (співвідношення об'ємів 1:1). Продукт висушували у вакуумі при 50 °C. Зважена кількість одержаного продукту становила 2415 г, що відповідало 91,8 % теоретичного значення. Вміст цільового продукту формули (VIII) (вільна основа) становив понад 98 відсотків площі під піком чи понад 97 мас. %. Сторонніми домішками, вміст яких виявився найбільшим, були ДМФ (близько 0,8 мас. %) і вода (близько 0,5 мас. %). Приклад 2 Одержання метил-4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]-5піримідинілкарбамату (VI) У реактор закладали 3063 г сполуки формули (VIII) та 30,7 л технічного ізопропанолу. Перемішуючи, додавали 1641 г диметилдикарбонату при 20-25 °C і продовжували перемішування протягом 22 год. при цій температурі. Відсмоктували продукт, який випав в осад, промивали технічним ізопропанолом і висушували у вакуумі при 50 °C. Зважена кількість одержаного продукту становила 3748 г чи 105,9 % теоретичного значення. Продукт формули (I) містив, зокрема, близько 4,7 % ізопропанолу, який практично не піддавався видаленню висушуванням (ізопропанол перебував частково у формі сольвату), його вміст за результатами аналізу становив 89,5 мас. % (ВЕРХ). Виходячи з цього вмісту, вихід складав 94,8 % теоретичного значення. Приклад 3 Одержання 2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]-4,5,6-піримідинтриаміну (VIII) В автоклав-конвертер закладали 300 г сполуки формули (III), 1600 мл ДМФ та 60 г зволоженого водою нікелю Ренея і після інертизації при внутрішній температурі 60 °C, під тиском водню 65 бар піддавали суміш гідруванню протягом близько 18 год. Після охолодження і зниження тиску відфільтровували відпрацьований каталізатор і промивали 100 мл ДМФ. Фільтрат згущували у вакуумі до 534,5 г і при 35-40 °C додавали до залишку 750 мл метанолу, а потім після охолодження до температури 0-5 °C вливали 750 мл води. Фільтрували тверду речовину і висушували у вакуумі при 50 °C. Зважена кількість одержаного продукту становила 219,7 г чи 91,8 % теоретичного значення. Приклад 4 Одержання метил-4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]-5піримідинілкарбамату (VI) У реактор закладали 1,50 кг сполуки формули (VIII) в 14,25 л ізопропанолу і нагрівали, перемішуючи, до температури 35 °C. До цієї суміші рівномірно протягом 30 хв. швидко додавали 531 г метилового естеру хлормурашиної кислоти, промивали із застосуванням 750 мл ізопропанолу і продовжували перемішування протягом 16 год. при 35 °C. Потім нагрівали суміш до 50 °C, додавали, перемішуючи, при 50 °C 3,85 л метанолу та 606 г триетиламіну і додатково промивали із застосуванням 450 мл метанолу. Потім продовжували перемішування протягом 1 год. при 50 °C, охолоджували до кімнатної температури і продовжували перемішування при 10 UA 110779 C2 5 10 15 20 25 30 35 40 45 50 55 60 кімнатній температурі протягом 1 год. Відсмоктували суспендовану тверду речовину, двічі промивали із застосуванням кожного разу 3,0 л суміші ізопропанол/метанол (4:1) та один раз – із застосуванням 3,0 л ізопропанолу і піддавали відсмоктуванню. Вологий продукт висушували спочатку при 50 °C протягом 1 год., а потім при 100 °C протягом 22 год. у вакуумній сушильній шафі. Зважена кількість одержаного продукту становила 1,793 кг чи 103,3 % теоретичного значення. Продукт формули (VI) містив 6,45 % ізопропанолу, який практично не піддавався видаленню висушуванням (ізопропанол перебував частково у формі сольвату), його вміст за результатами аналізу становив 87,9 мас. % (ВЕРХ). Виходячи з цього вмісту, вихід становив 90,8 % теоретичного значення. Порівняльний приклад 5 Одержання метил-4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]-5піримідиніл(метил)карбамату (I) (метилювання відомим методом згідно з публікацією WO 03/095451, приклад 8, друга методика) 1630 г сполуки формули (VI) суспендували при 20-25 °C у 16,3 л ТГФ. Охолоджували до температури від -6 до -4 °C і додавали 3480 г 1M розчину біс(триметилсиліл)аміду натрію. Продовжували перемішування і додавали 596 г метилйодиду, недовго перемішували і повільно нагрівали до температури близько 5 °C. Перемішували при цій температурі до остаточного завершення перетворення (протягом близько 4 год.). Реакційну суміш промивали 4 рази із застосуванням 4,1 л розчину хлориду амонію концентрацією 15 %. Випарювали органічну фазу до одержання залишку масою близько 6,4 кг і нагрівали до температури близько 25 °C. Тверду речовину, що випала в осад, відфільтровували, промивали із застосуванням у цілому 3 л ТГФ і висушували у вакуумі при 50 °C. Одержали 1112 г вихідного продукту формули (I). Це відповідало виходу 75,2 % теоретичного значення. Приклад 6 Одержання суміші з метил-4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]-5піримідиніл(метил)карбамату (I) і метил-{4,6-діаміно-2-[1-(2-фтор-бензил)-1H-піразоло[3,4b]піридин-3-іл]піримідин-5-іл}метилкарбаматсульфінілдиметану (II) із високим вмістом продукту формули (II) 9,0 г вихідного продукту формули (I), одержаного аналогічно порівняльному прикладу 5, розчиняли в 16 мл ДМСО при 100 °C (просвітлювальне фільтрування, яке є необхідним для одержання продукту фармацевтично прийнятної якості, в цьому лабораторному дослідженні не здійснювали). Потім витримували для охолодження до 75 °C, додавали 110 мл етилацетату і повільно охолоджували до температури близько 25 °C. Тверду речовину, що випала в осад, відфільтровували, промивали із застосуванням у цілому 28 мл етилацетату і висушували у вакуумі при 50 °C. Зважена кількість одержаного продукту становила 9,6 г чи 90,0 % теоретичного значення. Приклад 7 Одержання очищеного метил-4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3іл]-5-піримідиніл(метил)карбамату (I) Всю кількість одержаного у вищенаведеному прикладі 6 продукту формули (II) перемішували в 135 мл етилацетату протягом 1 год. за зворотним холодильником (близько 78 °C) та охолоджували до температури близько 25 °C. Відсмоктували тверду речовину, промивали із застосуванням у цілому 36 мл етилацетату і висушували у вакуумі. Зважена кількість одержаного продукту становила 7,6 г чи 93,8 % теоретичного значення. Вміст продукту явно перевищувала 98 мас. % (ВЕРХ). Вміст застосованого як розчинник етилацетату становив близько 0,2 %. Вміст ДМСО був менше 0,1 %. Приклад 8 Одержання очищеного метил-4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3іл]-5-піримідиніл(метил)карбамату (I) із проміжним відокремленням суміші з великим вмістом метил-{4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3-іл]піримідин-5іл}метилкарбаматсульфінілдиметану (II) в формі вологого продукту 193,5 г вихідного продукту формули (I), одержаного аналогічно порівняльному прикладу 5, розчиняли в 344 мл ДМСО та 172 мл етилацетату при температурі близько 96 °C. Потім додавали 19,4 г активованого вугілля та 172 мл етилацетату і перемішували при високій температурі. Після цього фільтрували активоване вугілля при високій температурі і додатково промивали із застосуванням 172 мл етилацетату. Фільтрат нагрівали до 78 °C і повільно змішували з 1850 мл етилацетату. Протягом близько 2-3 год. охолоджували суміш до температури близько 25 °C, відфільтровували тверду речовину і промивали із застосуванням у цілому 772 мл етилацетату. Вологий продукт, який містив великий відсоток сполуки формули (II) 11 UA 110779 C2 5 10 15 20 25 в суміші, суспендували в 2900 мл етилацетату, протягом 1 год. нагрівали зі зворотним холодильником та охолоджували до температури близько 25 °C.? Відсмоктували тверду речовину, промивали із застосуванням у цілому 774 мл етилацетату і висушували у вакуумі при 50 °C. Зважена кількість одержаного продукту становила 155,1 г чи 80,2 % вихідного продукту. Вміст продукту явно перевищував 98 мас. % (ВЕРХ). Як розчинники продукт містив практично лише етилацетат і ДМСО у невеликій кількості. Приклад 9 Одержання і аналіз метил-{4,6-діаміно-2-[1-(2-фторбензил)-1H-піразоло[3,4-b]піридин-3іл]піримідин-5-іл}метилкарбаматсульфінілдиметану (II) 14,8 г вихідного продукту формули (I), одержаного аналогічно порівняльному прикладу 5, розчиняли в 28,9 г ДМСО та 11,85 г етилацетату при температурі близько 94 °C. Потім додавали 1,5 г активованого вугілля марки Norit A-Supra і ще 11,85 г етилацетату, продовжували перемішування протягом 1 год. зі зворотним холодильником (88-90 °C) і відфільтровували активоване вугілля в гарячому стані. Тверду речовину, яка частково вже випала в осад, знову переводили в розчин, нагріваючи до температури близько 78 °C, після чого залишали розчин для повільного охолодження. Тверду речовину, що випала в осад, відсмоктували при кімнатній температурі, тричі промивали із застосування кожного разу по 50 мл етилацетату і протягом 18 год. при 30 °C висушували в сушильній шафі. Одержали 9,2 г чи 52,5 % теоретичного значення злегка жовтуватого кристалічного порошку сполуки формули (II). ВЕРХ: 99,90 Fl.-% (без урахування ДМСО) ДМСО (ГХ): 14,7 мас. % 1 H-ЯМР (400 МГц в ДМФ-d7): d=2,59 (с, близько 6H, 2 CH3 у ДМСО), 3,13 (с, 3H, N-CH3), 3,58+3,67 (два с, 3H, утруднене обертання O-CH3), 5,91 (с, 2H, -CH2-), 6,53 (с, 4H, 2-NH2), 7,05-7,40 (м, 5H, 4 ароматичних H oфторбензилу та 1H піридинового кільця в мета-положенні відносно атома азоту піридинового кільця), 8,60 (дд, 1H, в орто-положенні відносно атома азоту піридинового кільця), 9,12 (дд, 1H, в пара-положенні відносно атома азоту піридинового кільця). Елементарний аналіз: за даними аналізу C: 52,2 % H: 4,9 % N: 22,7 % 30 за даними розрахунку C: 52,79 % H: 5,03 % N: 22,39 % ФОРМУЛА ВИНАХОДУ 1. Спосіб очищення метил{4,6-діаміно-2-[1-(2-фторбензил)-1Н-піразоло[3,4-b]піридин-3іл]піримідин-5-іл}метилкарбамату формули (І) 35 , (І) який відрізняється тим, що вихідний продукт сполуки формули (І) розчиняють у диметилсульфоксиді і відокремлюють одержаний при цьому метил{4,6-діаміно-2-[1-(2 12 UA 110779 C2 фторбензил)-1Н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату формули (II) сульфінілдиметан O H3 C N H2 N O CH3 O NH2 N N H3 C S CH3 N N N F 5 , (ІІ) після чого знову видаляють диметилсульфоксид шляхом виварювання у фармацевтично прийнятному розчиннику. 2. Метил{4,6-діаміно-2-[1-(2-фторбензил)-1Н-піразоло[3,4-b]піридин-3-іл]піримідин-5іл}метилкарбамату сульфінілдиметан формули (ІІ) O H3 C N H2 N O CH3 O NH2 N N H3 C S CH3 N N N F (II). Комп’ютерна верстка Л. Бурлак Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 13
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for producing methyl-{4,6-diamino-2-[1-(2-fluorobenzyl)-1h-pyrazolo[3,4-b]pyridino-3-yl]pyrimidino-5-yl}methyl carbamate and its purification for use thereof as pharmaceutical substance
Автори англійськоюMais, Franz-Josef, Rehse, Joachim, Joentgen, Winfried, Siegel, Konrad
Автори російськоюМаис Франц-Йозеф, Резе Йоахим, Йонтген Винфрид, Зигель Конрад
МПК / Мітки
МПК: C07D 471/04
Мітки: очищення, метил{4,6-діаміно-2-[1-(2-фторбензил)-1н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату, сульфінілдиметан, спосіб
Код посилання
<a href="https://ua.patents.su/15-110779-sposib-ochishhennya-metil46-diamino-2-1-2-ftorbenzil-1n-pirazolo34-bpiridin-3-ilpirimidin-5-ilmetilkarbamatu-ta-metil46-diamino-2-1-2-ftorbenzil-1n-pirazolo34-bpiridin-3-ilpirimidi.html" target="_blank" rel="follow" title="База патентів України">Спосіб очищення метил{4,6-діаміно-2-[1-(2-фторбензил)-1н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату та метил{4,6-діаміно-2-[1-(2-фторбензил)-1н-піразоло[3,4-b]піридин-3-іл]піримідин-5-іл}метилкарбамату сульфінілдиметан</a>
N-(3,4-диметоксифеніл)-2-{[2-метил-6-(піридин-2-іл)піримідин-4-іл]тіо}ацетамід, що проявляє протисудомну активність

Номер патенту: 76869
Опубліковано: 25.01.2013
Автори: Георгіянц Вікторія Акопівна, Северіна Ганна Іванівна, Волощук Наталія Іванівна, Скупа Ольга Олегівна
МПК: C07D 239/38, A61P 21/02, C07D 239/22, A61K 31/505
Мітки: активність, протисудомну, n-(3,4-диметоксифеніл)-2-{[2-метил-6-(піридин-2-іл)піримідин-4-іл]тіо}ацетамід, проявляє
Формула / Реферат:
N-(3,4-диметоксифеніл)-2-{[2-метил-6-(піридин-2-іл)піримідин-4-іл]тіо}ацетамід формули:,що проявляє протисудомну активність.
Похідні 3-метил-1,4-діарил-1н-піразоло-[4`,3`:5,6]піридо[2,3-d]піримідин-5-онів і спосіб їх одержання

Номер патенту: 89804
Опубліковано: 10.03.2010
Автори: Руденко Роман Володимирович, Муравйова Олена Олександрівна, Чебанов Валентин Анатолійович, Десенко Сергій Михайлович, Афанасіаді Людмила Михайлівна
МПК: C07D 471/12, C07D 487/12
Мітки: 3-метил-1,4-діарил-1н-піразоло-[4`,3`:5,6]піридо[2,3-d]піримідин-5-онів, похідні, одержання, спосіб
Формула / Реферат:
1. Похідні 3-метил-1,4-діарил-1Н-піразоло[4',3':5,6]піридо[2,3-d]піримідин-5-онів загальної формули І де: R=H, СН3; Х=О, S; Ar=C6H5, 4-CH3-C6H4, 4-NO2-C6H4; Аr1=С6Н5, 4-Сl-С6Н4, 4-Br-C6H4, 4F-C6H4, 4-СН3-С6Н4, 4-СН3О-С6Н4, 4-С2Н5-С6Н4, 3-Сl-С6Н4, 3,4-диCH3O-C6H3, 3,5-диCH3O-4-OH-C6H2, ...
N-(3,4-диметоксифеніл)-2-{[2-метил-6-(піридин-2-іл)піримідин-4-іл]тіо}ацетамід, що проявляє протисудомну активність

Номер патенту: 105242
Опубліковано: 25.04.2014
Автори: Скупа Ольга Олегівна, Волощук Наталія Іванівна, Георгіянц Вікторія Акопівна, Северіна Ганна Іванівна
МПК: A61P 21/02, C07D 239/38, A61K 31/505
Мітки: n-(3,4-диметоксифеніл)-2-{[2-метил-6-(піридин-2-іл)піримідин-4-іл]тіо}ацетамід, протисудомну, активність, проявляє
Формула / Реферат:
N-(3,4-диметоксифеніл)-2-{[2-метил-6-(піридин-2-іл)піримідин-4-іл]тіо}ацетамід формули:,що проявляє протисудомну активність.
Спосіб одержання 1-[[3-(6,7-дигідро-1-метил-7-оксо-3-пропіл-1н-піразоло[4,3-d]піримідин-5-іл)-4-етоксифеніл]сульфоніл]-4-метилпіперазину та проміжні сполуки

Номер патенту: 66787
Опубліковано: 15.06.2004
Автори: Леветт Філіп Чарльз, Данн Пітер Джеймс
МПК: C07D 487/04, C07D 231/40, C07D 295/26, C07D 401/06, A61K 31/519, A61P 15/10, C07D 295/22
Мітки: одержання, 1-[[3-(6,7-дигідро-1-метил-7-оксо-3-пропіл-1н-піразоло[4,3-d]піримідин-5-іл)-4-етоксифеніл]сульфоніл]-4-метилпіперазину, спосіб, сполуки, проміжні
Формула / Реферат:
1. Спосіб одержання 1-[[3-(6,7-дигідро-1-метил-7-оксо-3-пропіл-1Н-піразоло[3,4-d]піримідин-5-іл)-4-етоксифеніл]сульфоніл]-4-метилпіперазину формули (ІА), (IA)який включає взаємодію сполуки формули (IIА) (IIA)з -OR, де R означає СН2СН3 і Х означає відщеплювану...
1-[[3-(4,7-дигідро-1-метил-7-оксо-3-пропіл-1н-піразоло[4,3-d]піримідин-5-іл)-4-етоксифеніл]сульфоніл]-4-метилпіперазин та його фармацевтично прийнятні солі

Номер патенту: 80534
Опубліковано: 10.06.2013
Автор: Черкашина Юлія Олександрівна
МПК: C07D 487/00
Мітки: прийнятні, фармацевтично, солі, 1-[[3-(4,7-дигідро-1-метил-7-оксо-3-пропіл-1н-піразоло[4,3-d]піримідин-5-іл)-4-етоксифеніл]сульфоніл]-4-метилпіперазин
Формула / Реферат:
1-[[3-(4,7-Дигідро-1-метил-7-оксо-3-пропіл-1Н-піразоло[4,3-d]піримідин-5-іл)-4-етоксифеніл]сульфоніл]-4-метилпіперазин структурної формули (І) (І)та його фармацевтично прийнятні солі.
Попередній патент: Друкарський станок глибокого друку та рухома каретка такого станка
Наступний патент: Форсунка з суцільним конусом розпилу
Випадковий патент: Спосіб лікування респіраторних захворювань у телят