Спосіб одержання рекомбінантного інсуліну людини
Номер патенту: 91281
Опубліковано: 12.07.2010
Автори: Костецький Ігор Євгенович, Луців Володимир Романович, Лесик Ігор Павлович, Лісовський Ігор Леонідович, Лазарєв Олексій Павлович











Формула / Реферат
1. Спосіб одержання рекомбінантного інсуліну людини шляхом конструювання рекомбінантної плазмідної ДНК, що кодує проінсулін, зв'язаний з лідерною послідовністю, одержання і культивування штаму-продуцента гібридного білка Escherichia coli, виділення і дезінтеграції клітин, виділення гібридного білка, його ферментативного розщеплення з наступним очищенням і одержанням цільового продукту, який відрізняється тим, що ділянка рекомбінантної плазмідної ДНК, яка кодує гібридний білок з амінокислотною послідовністю проінсуліну людини (Послідовність №4 або №6, або №8, або №10), є в складі експресуючих векторів, вибраних з групи: pMUT12, pISYN2, pCIM61, pDIM07, або являє собою послідовність ДНК, що гібридизується в жорстких умовах з однією з вищевказаних ділянок рекомбінантної плазмідної ДНК, та які кодують експресію попередника проінсуліну людини, a ділянка рекомбінантної плазмідної ДНК, яка кодує вказаний гібридний білок проінсуліну, вибрана з групи:
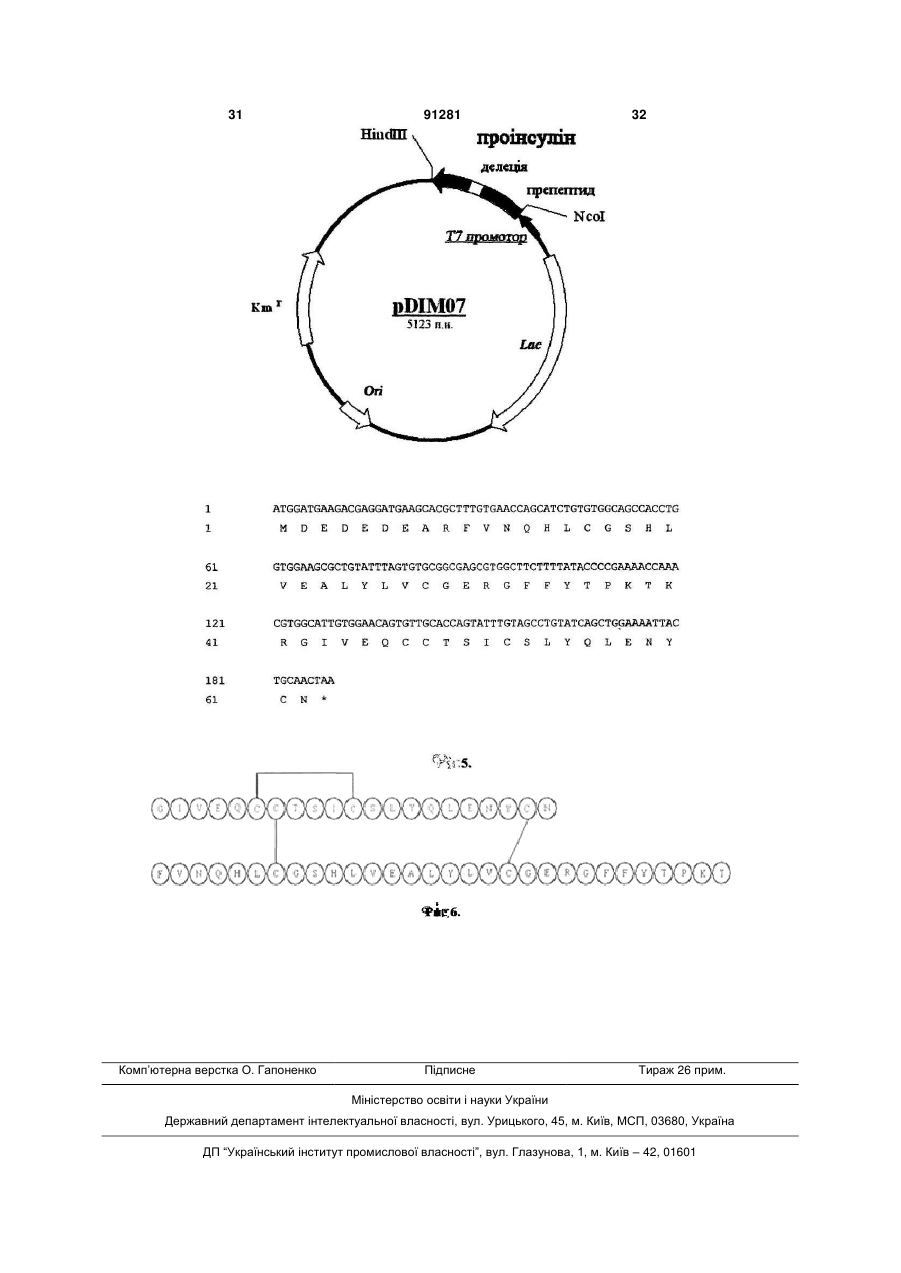
1 ATGGGCAGCAGCCATCATCATCATCATCACAGCAGCGGCCTGGTGCCGCGCGGCAGCCAT
1 M G S S H H H H H H S S G L V P R G S H
61 ATGCGCTTTGTGAACCAACACCTGTGCGGCTCACACCTGGTGGAAGCTCTCTACCTAGTG
21 M R F V N Q H L C G S H L V E A L Y L V
121 TGCGGGGAACGAGGCTTCTTCTACACACCCAAGACCAAGCGTGGCATTGTGGAACAATGC
41 C G E R G F F Y T P K T K R G I V E Q C
181 TGTACCAGCATCTGCTCCCTCTACCAGCTGGAGAACTACTGCAACTAG (Послідовність №3)
61 C T S I C S L Y Q L E N Y C N (Послідовність №4),
1 ATGGGCAGCAGCCATCATCATCATCATCACAGCAGCGGCCTGGTGCCGCGCGGCAGCCAT
1 M G S S H H H H H H S S G L V P R G S H
61 ATGCGCTTTGTGAACCAGCATCTGTGTGGCAGCCACCTGGTGGAAGCGCTGTATTTAGTG
21 M R F V N Q H L C G S H L V E A L Y L V
121 TGCGGCGAGCGTGGCTTCTTTTATACCCCGAAAACCAAACGTGGCATTGTGGAACAGTGT
41 C G E R G F F Y T P K T K R G I V E Q C
181 TGCACCAGTATTTGTAGCCTGTATCAGCTGGAAAATTACTGCAACTAA (Послідовність №5)
61 C T S I C S L Y Q L E N Y C N (Послідовність №6),
1 ATGCGAAAGAAGCGGAAGAAGAAGCGTTTTGTGAACCAGCATCTGTGTGGCAGCCACCTG
1 M R K K R K K K R F V N Q H L C G S H L
61 GTGGAAGCGCTGTATTTAGTGTGCGGCGAGCGTGGCTTCTTTTATACCCCGAAAACCAAA
21 V E A L Y L V C G E R G F F Y T P K T K
121 CGTGGCATTGTGGAACAGTGTTGCACCAGTATTTGTAGCCTGTATCAGCTGGAAAATTAC
41 R G I V E Q C C T S I C S L Y Q L E N Y
181 TGCAACTAA (Послідовність №7)
61 C N (Послідовність №8),
1 ATGGATGAAGACGAGGATGAAGCACGCTTTGTGAACCAGCATCTGTGTGGCAGCCACCTG
1 M D E D E D E A R F V N Q H L C G S H L
61 GTGGAAGCGCTGTATTTAGTGTGCGGCGAGCGTGGCTTCTTTTATACCCCGAAAACCAAA
21 V E A L Y L V C G E R G F F Y T P K T K
121 CGTGGCATTGTGGAACAGTGTTGCACCAGTATTTGTAGCCTGTATCAGCTGGAAAATTAC
41 R G I V E Q C C T S I C S L Y Q L E N Y
181 TGCAACTAA (Послідовність №9)
61 C N (Послідовність №10).
2. Спосіб по п. 1, який відрізняється тим, що стадію створення штамів Escherichia coli здійснюють з використанням плазмідних векторів, що містять нуклеотидну послідовність, яка кодує один чи декілька рекомбінантних поліпептидів, при цьому транскрипція вказаної нуклеотидної послідовності, яка кодує один чи декілька поліпептидів, знаходиться під контролем індукованої системи експресії, потім здійснюють культивування трансформованих штамів Escherichia coli та індукування системи експресії для синтезу рекомбінатного поліпептиду чи поліпептидів в клітинах Escherichia coli, та виділення рекомбінантного поліпептиду чи продукованих поліпептидів.
3. Спосіб по п. 1, який відрізняється тим, що виділення і дезінтеграцію клітин, виділення гібридного поліпептиду здійснюють шляхом руйнування бактеріальної клітини чи фрагментів клітин, сепарування з одночасним відмиванням осаду неіонним детергентом, а виділення гібридного поліпептиду включає розчинення з денатуруючим реагентом, обробку гібридного поліпептиду відновлюючим агентом, рефолдинг гібридного поліпептиду та його концентрування методом абсорбційної хроматографії або ультрафільтрації.
4. Спосіб по п. 1, який відрізняється тим, що ферментативне розщеплення гібридного поліпептиду здійснюють трипсином та карбоксипептидазою одночасно або послідовно з використанням переважно очистки продуктів ферментативної конверсії за допомогою іонообмінної хроматографії.
5. Спосіб по п. 1, який відрізняється тим, що очистку інсуліну після ферментативного розщеплення здійснюють за допомогою іонообмінної хроматографії та/або високоефективної рідинної хроматографії.
Текст
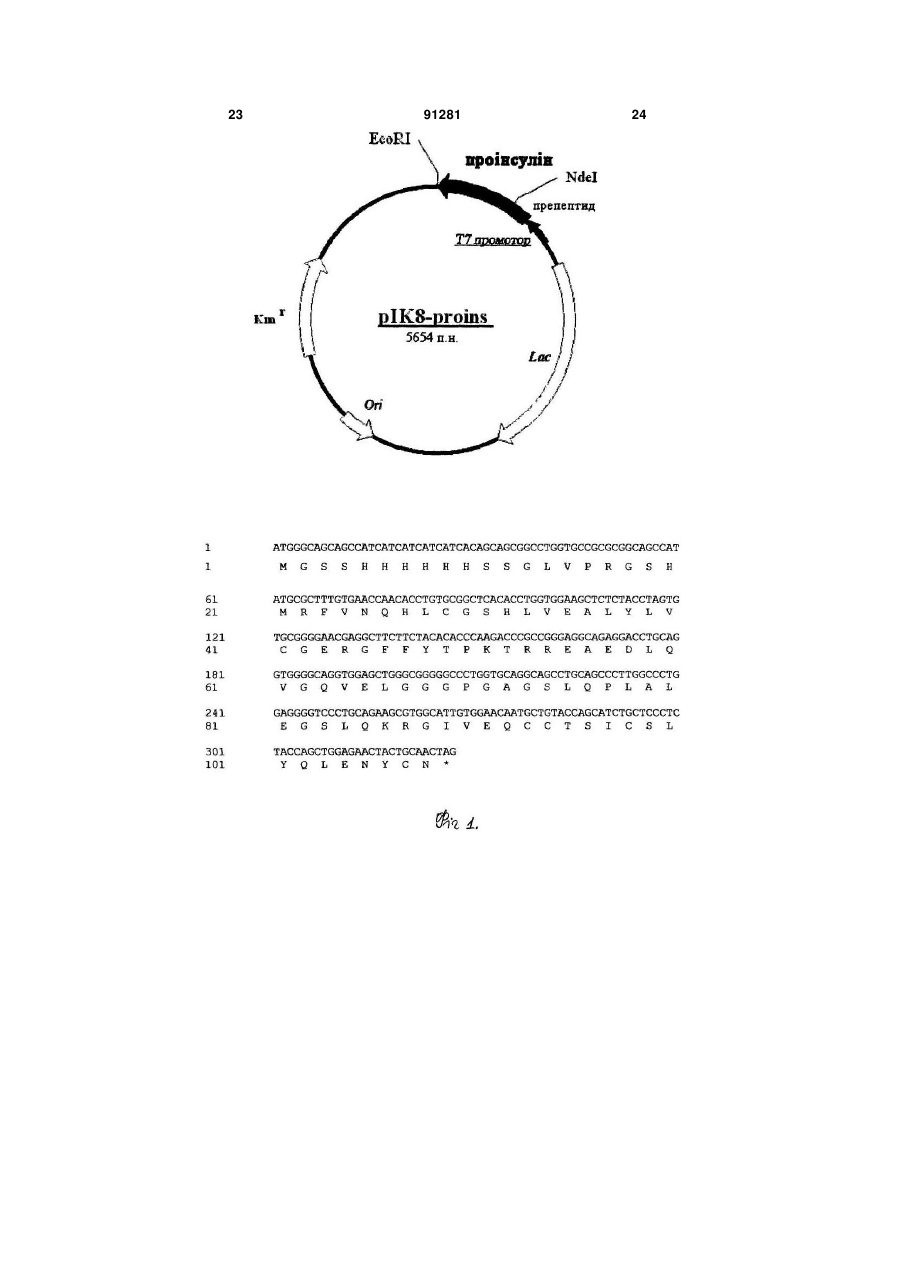
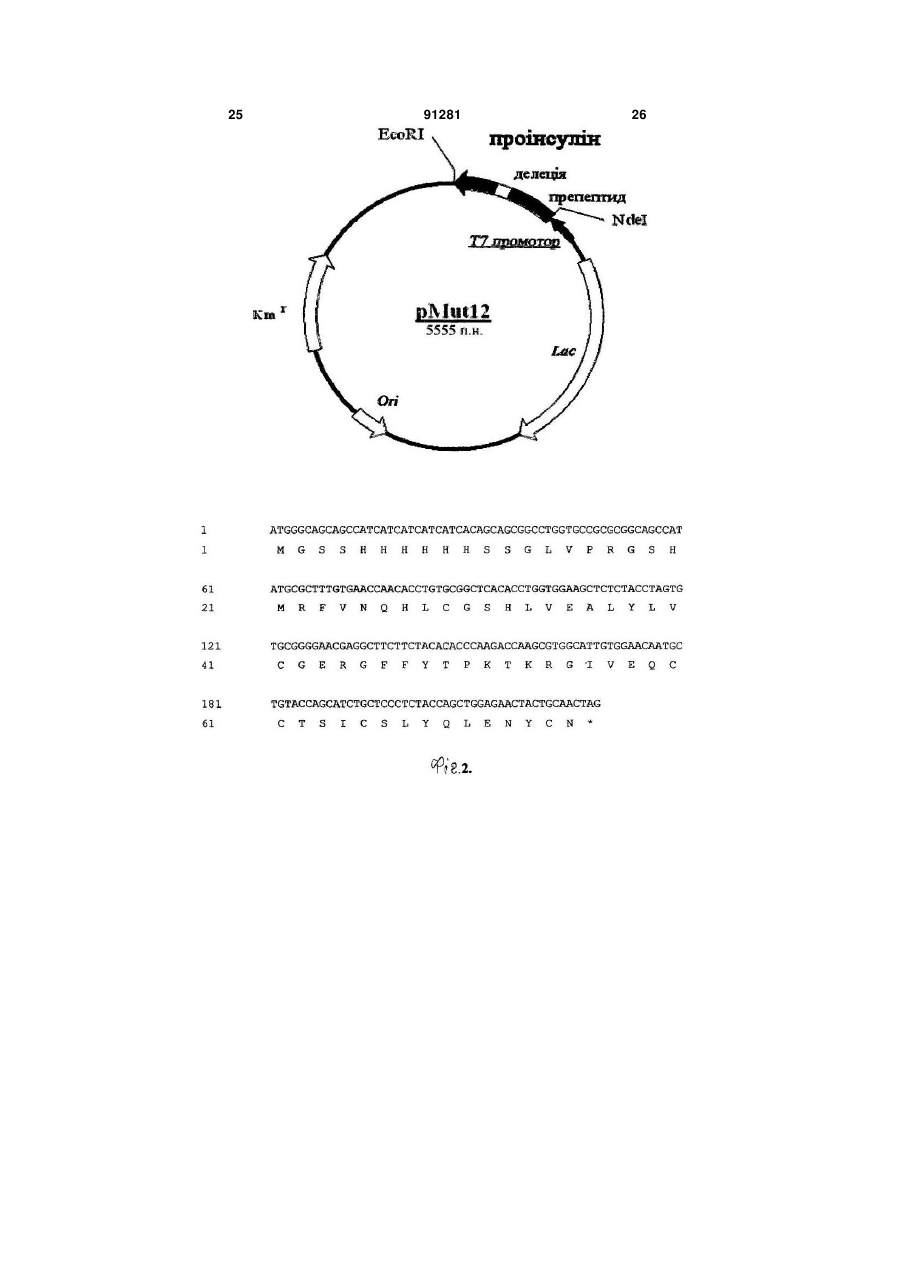
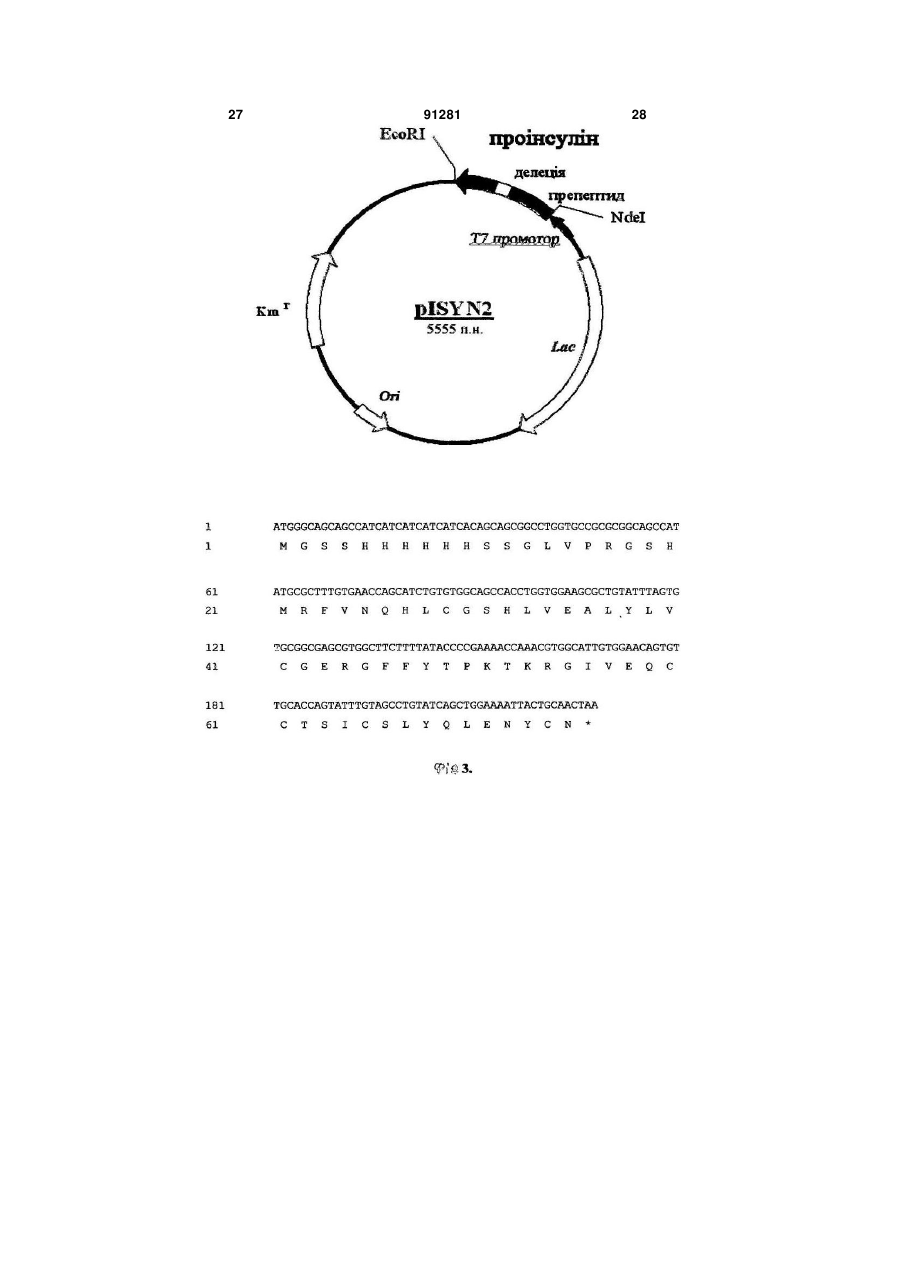
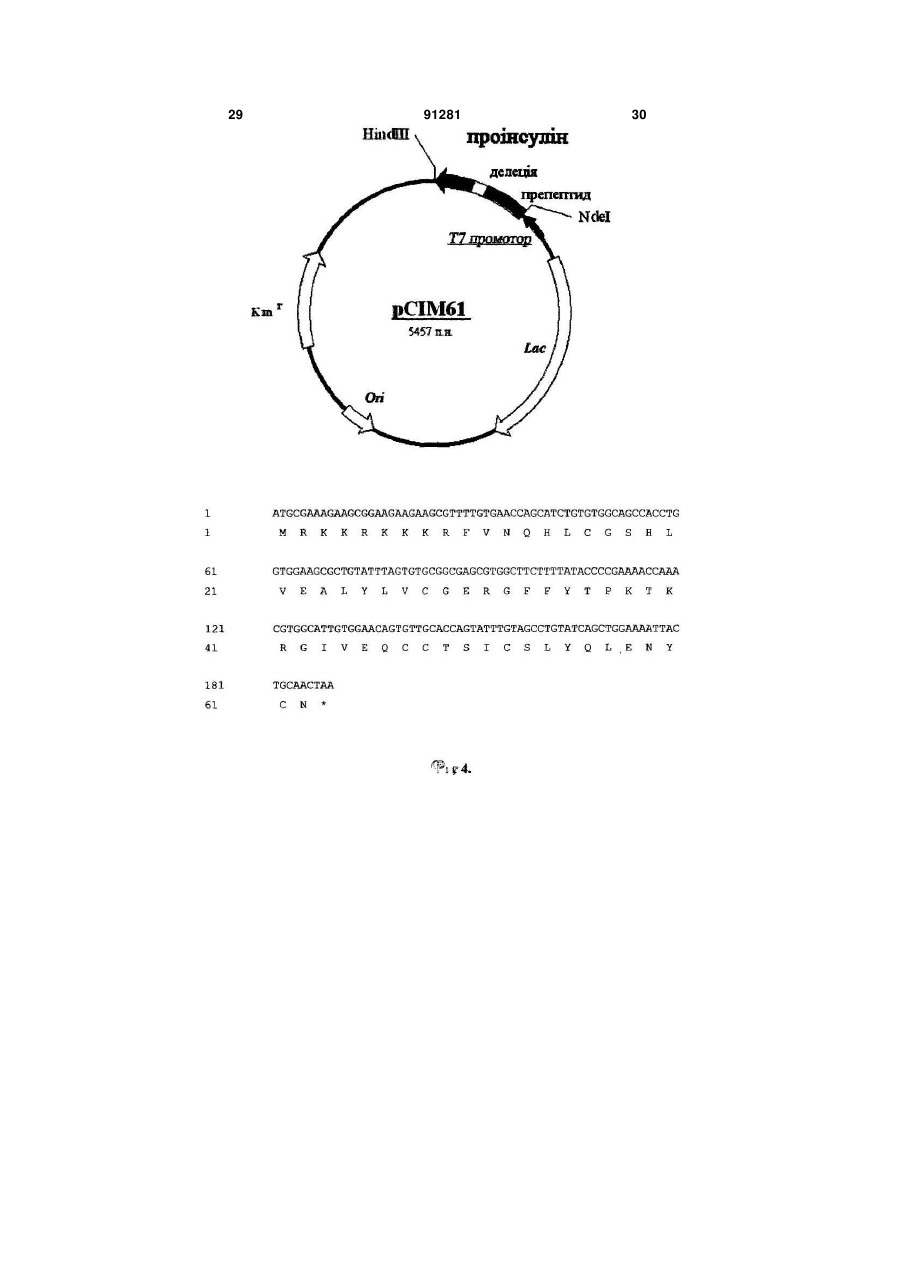
1. Спосіб одержання рекомбінантного інсуліну людини шляхом конструювання рекомбінантної плазмідної ДНК, що кодує проінсулін, зв'язаний з лідерною послідовністю, одержання і культивування штаму-продуцента гібридного білка Escherichia coli, виділення і дезінтеграції клітин, виділення гібридного білка, його ферментативного розщеплення з наступним очищенням і одержанням цільового продукту, який відрізняється тим, що ділянка рекомбінантної плазмідної ДНК, яка кодує гібридний білок з амінокислотною послідовністю проінсуліну людини (Послідовність №4 або №6, або №8, або №10), є в складі експресуючих векторів, вибраних з групи: pMUT12, pISYN2, pCIM61, pDIM07, або являє собою послідовність ДНК, що гібридизується в жорстких умовах з однією з вищевказаних ділянок рекомбінантної плазмідної ДНК, та які кодують експресію попередника проінсуліну людини, a ділянка рекомбінантної плазмідної ДНК, яка кодує вказаний гібридний білок проінсуліну, вибрана з групи: 2 3 91281 4 1 ATGGATGAAGACGAGGATGAAGCACGCTT ваних штамів Escherichia coli та індукування сисTGTGAACCAGCATCTGTGTGGCAGCCACCTG теми експресії для синтезу рекомбінатного поліпе1 M D E D E D E A R F V N Q H L C птиду чи поліпептидів в клітинах Escherichia coli, та G S H L виділення рекомбінантного поліпептиду чи продукованих поліпептидів. 3. Спосіб по п. 1, який відрізняється тим, що ви61 GTGGAAGCGCTGTATTTAGTGTGCGGCGA GCGTGGCTTCTTTTATACCCCGAAAACCAAA ділення і дезінтеграцію клітин, виділення гібридно21 V E A L Y L V C G E R G F F Y T P го поліпептиду здійснюють шляхом руйнування K T K бактеріальної клітини чи фрагментів клітин, сепарування з одночасним відмиванням осаду неіон121 CGTGGCATTGTGGAACAGTGTTGCACCA ним детергентом, а виділення гібридного поліпепGTATTTGTAGCCTGTATCAGCTGGAAAATTAC тиду включає розчинення з денатуруючим 41 R G I V E Q C C T S I C S L Y Q L реагентом, обробку гібридного поліпептиду відноE N Y влюючим агентом, рефолдинг гібридного поліпептиду та його концентрування методом абсорбцій181 TGCAACTAA (Послідовність №9) ної хроматографії або ультрафільтрації. 4. Спосіб по п. 1, який відрізняється тим, що фе61 C N (Послідовність №10). рментативне розщеплення гібридного поліпептиду 2. Спосіб по п. 1, який відрізняється тим, що стаздійснюють трипсином та карбоксипептидазою дію створення штамів Escherichia coli здійснюють з одночасно або послідовно з використанням перевикористанням плазмідних векторів, що містять важно очистки продуктів ферментативної конверсії нуклеотидну послідовність, яка кодує один чи деза допомогою іонообмінної хроматографії. 5. Спосіб по п. 1, який відрізняється тим, що очикілька рекомбінантних поліпептидів, при цьому транскрипція вказаної нуклеотидної послідовності, стку інсуліну після ферментативного розщеплення яка кодує один чи декілька поліпептидів, знахоздійснюють за допомогою іонообмінної хроматогдиться під контролем індукованої системи експрерафії та/або високоефективної рідинної хроматогсії, потім здійснюють культивування трансформорафії. Винахід відноситься до біотехнології, генної інженерії та медицини і може бути використаним у виробництві лікарських препаратів, зокрема препаратів для лікування цукрового діабету. Інсулін - гормональний білок, що складається з кислого А-ланцюга, який містить 21 амінокислотний залишок, і лужного В-ланцюга з 30 амінокислот [1], ці ланцюги зв'язані трьома дисульфідами: один зв'язок знаходиться всередині А-ланцюга (А6А11) і два міжланцюгових зв'язки (А7-В7 та А20-В19). У той час як окремі А- і В-ланцюги інсуліну можуть бути успішно з'єднані in vitro [2], в природних умовах звичайно синтезується єдиний поліпептидний ланцюг (препроінсулін). Він містить сигнальний пептид в аміно термінальній ділянці В-ланцюга і Спептид між А- і В- ланцюгами. Після відщеплення N-кінцевої сигнальної послідовності на ендоплазматичному ретикулумі утворений поліпептид упаковується в секреторні гранули у вигляді проінсуліну [3]. Оброблений далі певним набором протеаз, проінсулін потім перетворюється в інсулін і секретується в потік крові для регуляції рівня цукру. У промисловому масштабі, інсулін людини може бути вироблений методом транспептидації, тобто заміни аланінового залишку в 30-му положенні В-ланцюга інсуліну свині на треонін [4]. Оскільки виробництво інсуліну людини із інсуліну свині є обмеженим через високу собівартість, наукові дослідження були націлені головним чином на розробку та впровадження методів генної інженерії для виробництва інсуліну людини. На сьогодні використовуються три основні методи для виробництва інсуліну із застосуванням мікроорганізмів. Два з них включають використання бактерії Escherichia coli, при цьому попередник інсуліну синтезується як частина великого поліпептиду у вигляді тілець включення або секретується у периплазматичний простір [5]. Третій метод оснований на використанні дріжджів, де попередник інсуліну також може виділятися у навколишнє середовище [6] або формує нерозчинні тільця включення [7]. Процес отримання інсуліну людини за допомогою методів генної інженерії включає в себе наступні стадії: синтез проінсуліну в Escherichia coli у формі злитого поліпептиду, рефолдинг злитого поліпептиду з метою формування правильних дисульфідних зв'язків; протеолітичне розщеплення рефолдованого поліпептиду трипсином і карбоксипептидазою В (carboxypeptidase В); очистка отриманого продукту і одержання інсуліну. Різновид даного підходу використовує також сульфонування злитого поліпептиду і його розщеплення ціанбромідом з метою одержання проінсуліну як проміжного продукту виробництва інсуліну [8]. Також, генетичні маніпуляції дають можливість виробництва злитого поліпептиду з різними амінокислотними послідовностями в проінсуліні та/або Спептиді рекомбінантного поліпептиду. Ці маніпуляції спрямовані на збільшення виходу правильно рефолдованого проінсуліну (препроінсуліну). Однак, вихід рефолдованого проінсуліну, що має правильні дисульфідні зв'язки, зменшується при зростанні концентрації проінсуліну в буфері. Це відбувається через неправильний фолдинг та зростання процесу полімеризації. Таким чином, рефолдинг є критичною стадією в процесі виробництва інсуліну. Також, очистка інсуліну на наступних стадіях виробництва часто є досить трудомісткою і дорогою. З огляду на все це, розробка нових підходів виробництва інсуліну людини направлена не 5 91281 6 тільки на покращення якості готової продукції, але Крім того, в способі відсутня підготовка політакож і на заощадження ресурсів та зменшення пептиду до ферментативної конверсії (цитраконуціни продукції для споживачів. вання). Запропонована ефективна схема трипсиВідомий спосіб одержання рекомбінантного іннолізу при підвищених значеннях рН, при яких суліну людини включає в себе конструювання ремінімізовано утворення дез-треоніну, дозволяє комбінантної плазмідної ДНК, що кодує проінсулін кількісно збільшити вихід цільового продукту та [9], одержання і культивування штаму Escherichia виключити додаткову стадію очистки від цитракоcoli - продуцента гібридного поліпептиду, виділеннового ангідриду. ня і дезінтеграцію клітин, виділення гібридного Оптимізована схема очистки інсуліну з викориполіпептиду, його ферментативне розщеплення з станням іонообмінної хроматографії та високоенаступним очищенням і одержанням цільового фективної рідинної хроматографії дозволяє одерпродукту. жати високоочищений препарат інсуліну з Однак, у цьому способі використовується тільчистотою 98-99%. ки штам Escherichia coli BL21/pIK8-proins, який Таким чином, поставлена задача вирішується характеризується наявністю плазмідної ДНК pIK8за рахунок створення нових конструкцій, удоскоproins, що обмежує коло комбінацій хазяїн/вектор налення технології руйнування клітин і відмивки експресії. тілець включення, оптимізації рефолдингу та конКрім того, у цьому способі перед ферментатицентрування поліпептиду після рефолдингу з вивним розщепленням гібридного поліпептиду прокористанням поверхнево-адсорбційної хроматогводять його обробку цитраконовим ангідридом, що рафії, а також за рахунок відсутності потребує додаткової стадії очистки і кількісно обцитраконування і здійснення трипсинолізу при підмежує вихід цільового продукту. вищених значеннях рН з метою зменшення дезВ основу винаходу поставлено задачу ствотреоніну. рення способу одержання рекомбінантного інсуліТак, на додаток до раніше заявленої плазмідну людини, який відрізняється збільшенням виходу ної ДНК (pIK8-proins) в даному способі заявляютьгібридного поліпептиду та одержанням кінцевого ся інші плазміди, що мають певні переваги: продукту високої якості. - скорочений С-пептид (плазміди pMUT12, Важливий результат способу - підвищення pISYN2, рСІМ61, pDIM07). В результаті цієї модиефективності одержання цільового продукту, в фікації збільшується вихід цільового продукту. При певній мірі скорочення технологічного процесу, використанні плазміди pIK8-proins інсулін складає кількісне підвищення виходу цільового продукту, 47% від гібридного поліпептиду; при використанні підвищення якості за рахунок розширення кола pMUT12, pISYN2 інсулін складає 68% від гібридновекторів експресії, оптимізації амінокислотного го поліпептиду; при використанні рСІМ61, pDIM07 складу препептиду, удосконалення технології руй82%. нування бактеріальних клітин і відмивки тілець - нові плазміди кодують гібридні поліпептиди, включення, а також оптимізації процесу рефолдиякі відрізняються послідовністю препептида. Амінгу та удосконалення технології концентрування і нокислотний склад препептиду підібрано таким очистки. чином, що він змінює заряд (і відповідно ізоелектІснує тісний причинно-наслідковий зв'язок між ричну точку) гібридного поліпептиду, що дозволяє усією сукупністю суттєвих ознак та технічним реваріювати підходи до очистки білка. зультатом, що досягається. ДНК, яка кодує гібридний поліпептид (плазміда Даний винахід стосується покращеного спосоpISYN2), отримана шляхом хімічного синтезу з бу одержання рекомбінатного інсуліну людини урахуванням розподілу кодонів у Escherichia coli, шляхом культивації прокаріотичних клітин, трансщо підвищує ефективність синтезу гібридного поформованих послідовностями ДНК, що кодує ці ліпептиду приблизно на 40% у порівнянні з аналополіпептиди. Рекомбінатний гібридний поліпептид, гічною кДНК з оригінальними кодонами. який включає в себе лідерну послідовність приєдВ способі запропонована високоефективна тенану до проінсуліну, синтезується в клітинах хнологія прес-руйнування бактеріальних клітин Escherichia coli. Інсулін людини утворюється за (ефективність руйнування складає понад 99%), в допомогою обробки правильно рефолдованого той час як ефективність руйнування за допомогою гібридного поліпептиду трипсином та карбоксипепультразвука складає приблизно 40-50%. Крім того, тидазою В з наступною очисткою. Згідно з цим спосіб значно дешевший, має більш високу продувинаходом рекомбінатний інсулін людини може ктивність і дозволяє отримати на виході більш чисбути одержаний з високим відтворенням, в той час тий препарат (тільця включення). як стадії виділення і очистки значно удосконалені. Підготовка гібридного поліпептиду для рефолРозробка нових конструкцій плазмідних ДНК дингу не містить стадії сульфітолізу (ця стадія дозволяє розширити спектр використовуваних зменшує вихід цільового продукту і підвищує його штамів мікроорганізмів і оптимізувати амінокислособівартість). Заявлена схема прямого рефолдинтний склад препептиду. гу відновленого поліпептиду проста у виконанні та Запропонована в способі технологія концентдозволяє проводити рефолдинг при концентраціях рування поліпептиду після рефолдингу шляхом поліпептиду 0,5-1,0 мг/мл, в той час, як відновленповерхнево-адсорбційної хроматографії з викориня сульфітованого поліпептиду проводять при станням сорбенту, який має високі динамічні і ємконцентрації поліпептиду менше 0,1 мг/мл. Таким нісні характеристики, дозволяє значно покращити чином, суттєво зменшуються обсяги реакційної результативність цієї стадії. суміші, необхідні для ренатурації гібридного поліпептиду. 7 91281 8 Суть винаходу пояснюють графічні зображенділянками розпізнавання трипсину в послідовності ня: рекомбінантного поліпептиду; Фіг.1 - фізична карта для експресуючого векб) культивування трансформованих клітинтора pIK8-proins, а також послідовності проінсуліхазяїнів в умовах, які призводять до експресії рену: нуклеотидна (Послідовність №1) та амінокискомбінантної ДНК і продукції тілець включення; лотна (Послідовність №2). в) руйнування клітин і ізоляція продуктів ексФіг.2 - фізична карта для експресуючого векпресії; тора pMUT12, а також послідовності проінсуліну: г) розчинення рекомбінантного поліпептиду в нуклеотидна (Послідовність №3) та амінокислотна присутності денатуратів та відновлюючого агента з (Послідовність №4). наступним рефолдингом; Фіг.3 - фізична карта для експресуючого векд) розщеплення гібридного поліпептиду триптора pISYN2, а також послідовності проінсуліну: сином та карбоксипептидазою В для одержання нуклеотидна (Послідовність №5) та амінокислотна інсуліну; (Послідовність №6). e) виділення і очистка інсуліну. Фіг.4 - фізична карта для експресуючого векСуть винаходу полягає в тому, що в способі тора рСІМ61, а також послідовності проінсуліну: одержання рекомбінантного інсуліну людини шлянуклеотидна (Послідовність №7) та амінокислотна хом конструювання рекомбінантної плазмідної (Послідовність №8). ДНК, що кодує проінсулін, зв'язаний з лідерною Фіг.5 - фізична карта для експресуючого векпослідовністю, одержання і культивування штамутора pDIM07, а також послідовності проінсуліну: продуцента гібридного білка Escherichia coli, видінуклеотидна (Послідовність №9) та амінокислотна лення і дезінтеграції клітин, виділення гібридного (Послідовність №10). білка, його ферментативного розщеплення з наФіг.6 відображає первинну структуру інсуліну ступним очищенням і одержанням цільового пролюдини. дукту, відповідно до винаходу ділянка рекомбінанСпосіб полягає в тому, що одержують рекомтної плазмідної ДНК, яка кодує гібридний білок з бінантну ДНК, що кодує попередник інсуліну люамінокислотною послідовністю проінсуліну людини дини, який експресується в клітинах-хазяїнах (Послідовність №2, або №4, або №6, або №8, або Escherichia соіі у вигляді тілець включення з на№10), є в складі експресуючих векторів, вибраних ступним очищенням рекомбінантного поліпептиду і з групи: pIK8-proins, pMUT12, pISYN2, pCIM61, ренатурацією в умовах, що призводять до правиpDIM07, або являє собою послідовність ДНК, що льного формування дисульфідних зв'язків між загібридизується в жорстких умовах з однією з вилишками цистеїну. Далі рекомбінантний поліпепщевказаних ділянок рекомбінантної плазмідної тид протеолітично розщеплюють та очищають. ДНК, та які кодують експресію попередника проінСпосіб одержання рекомбінантного інсуліну суліну людини, а ділянка рекомбінантної плазмідвключає наступні стадії: ної ДНК, яка кодує вказаний гібридний білок проіна) трансформація клітин Escherichia coli рекосуліну, вибрана з групи: мбінантною ДНК, що кодує попередник інсуліну з 9 91281 10 11 Створення штамів Escherichia coli здійснюють з використанням плазмідних векторів, що містять нуклеотидну послідовність, яка кодує один чи декілька рекомбінантних поліпептидів, при цьому транскрипція вказаної нуклеотидної послідовності, яка кодує один чи декілька поліпептидів, знаходиться під контролем індукованої системи експресії, потім здійснюють культивування трансформованих штамів Escherichia coli та індукування системи експресії для синтезу рекомбінатного поліпептиду чи поліпептидів в клітинах Escherichia coli, та виділення рекомбінантного поліпептиду чи продукованих поліпептидів. Отримання синтезованого продукту здійснюють шляхом руйнування бактеріальної клітини чи фрагментів клітин, сепарування з одночасним відмиванням осаду неіонним детергентом, а подальша його конверсія включає розчинення в буферному розчині з денатуруючим реагентом, обробку гібридного поліпептиду відновлюючим агентом, рефолдинг гібридного поліпептиду та його концентрування методом абсорбційної хроматографії або ультрафільтрації. Ферментативне розщеплення гібридного поліпептиду здійснюють трипсином та карбоксипептидазою В одночасно або послідовно з використанням переважно очистки продуктів ферментативної конверсії за допомогою іонообмінної хроматографії та/або високоефективної рідинної хроматографії. Спосіб здійснюють як описано далі. В заявленій системі експресії рекомбінантної ДНК може використовуватись цілий ряд комбінацій хазяїн/вектор експресії, що забезпечує оптимальну продукцію інсуліну людини. Наприклад, вектори експресії можуть містити промоторну ділянку, сигнал початку трансляції, кДНК проінсуліну людини з/або без модифікацій, введених у послідовність С-пептиду, і оптимізованого препептиду, відділеного від проінсуліну сайтом розпізнавання трипсину. Оптимальною для даного винаходу є рекомбінантна плазмідна ДНК, яка містить інтегровану ДНК, яка описано вище. Вибрані плазміди даного винаходу включають рІK8-proins, pMUT12, pISYN2 і їх похідні. Також у даному винаході описаний процес одержання рекомбінантної молекули ДНК. Цей процес включає в себе клонування у відповідний вектор ДНК-вставки, що кодує повнорозмірну форму проінсуліну, а також аналогів, гомологів і попередників поліпептиду, що перетворюються на інсулін. Переважно вказана послідовність ДНК клонована у правильному положенні та в потрібній рамці зчитування з послідовністю контролю експресії. У подальшому, згідно з даним винаходом, клітини-продуценти трансформуються однією з рекомбінантних молекул ДНК, що здатні до експресії повнорозмірної, модифікованої або редукованої форми попередників інсуліну людини або поліпептиду, що має подібну імунологічну або біологічну активність до інсуліну. В якості експресуючих клітин-хазяїнів використовують відомі штами прокаріот, які включають штами Escherichia coli, такі як Escherichia coli 91281 12 HB101, Escherichia coli X1776, Escherichia coli X2282, Escherichia coli DH5 , Escherichia coli JM103, Escherichia coli BL21, проте не лише їх. Для виробництва поліпептиду попередника інсуліну бактеріальні клітини-продуценти культивуються в рідкому поживному середовищі в умовах, оптимальних для клітин-хазяїнів і вектора експресії. Переважно, клітини-продуценти культивуються в бактеріальному ферментері для широкомасштабного виробництва, проте допускаються інші зручні методи культивування (наприклад, в колбі на шейкері, коли необхідне культивування в об'ємах менших ніж 1 літр). Точні умови росту, вибір часу та добавки середовища, рН культурального середовища, температура й рівень розчиненого кисню, додавання індукуючого агента (де доречно) добираються згідно кожного штаму-продуцента і експресуючого конструкту. Після того, як бактеріальні клітини-продуценти були вирощені до бажаної щільності та була проведена необхідна індукція експресії, клітини збирають. Збирання звичайно виконується центрифугуванням середовища культивування, хоча може бути використана будь-яка інша зручна техніка типу ультрафільтрації. На цьому етапі зібрані бактеріальні клітини-продуценти можуть бути одразу використані відповідно до винаходу, або можуть бути заморожені для переробки пізніше. У такому випадку бактеріальні клітини руйнують шляхом лізису - щоб вивільнити тільця включення, які містять поліпептид-попередник інсуліну. Для цього бактеріальні клітини ресуспендують в буферному розчині при рН від 6,0 до 9,0 та іонній силі в діапазоні від 0,01M до 2M. Будь-яка підходяща сіль, включаючи NaCl, може використовуватися для підтримання відповідного рівня іонної сили. Переважно, лізис клітин проводять в умовах, при яких клітинні фрагменти досить зруйновані і не накопичуються в осаді при низько швидкісному центрифугуванні. Лізис клітин проводять загальновживаними методами, такими як механічні методи (зокрема, циклічне заморожування/танення; використання приладу Microfludizer або French press; чи ультразвукового генератора) або ензиматичні методи (такі як обробка лізоцимом). В цілому, рекомендується проводити лізис клітин при пониженій температурі (тобто, менше ніж 20 градусів С). Тільця включення відділяють від фрагментів клітин, використовуючи любий зручний метод (наприклад, центрифугування), потім відмивають, якщо необхідно. Тільця включення звичайно промивають ресуспендуючи їх в буфері з додаванням детергенту (наприклад, Тритон Х-100), потім знову збирають тільця включення. Відмиті тільця включення потім розчиняють в солюбілізаційному буфері, який містить високу концентрацію хаотропного агента (наприклад, сечовина або гуанідин гідрохлорид) і відновлюючий реагент при оптимальному значенню рН. Концентрація сечовини в солюбілізаційному буфері знаходиться в межах між 6М і 8М, переважно 7М, і концентрація гуанідина гідрохлориду від 5 до 7М, переважно 6М. 13 91281 14 рН солюбілізаційного буфера варіює від 7.5 до тами, наприклад, трипсином і карбоксипептидазою 11.0, переважно від 8.5 до 9.0. Можна використоВ для виробництва готового інсуліну. Концентрувувати будь-який рН буферний агент (такий як вання рефолдованого поліпептиду може проводиTris, HEPES, MOPS, tricine і т.п.). рН буферний тися з використанні будь-якої зручної методики, агент додається до солюбілізаційного буферу при типу ультрафільтрації або хроматографії (наприконцентрації, яка забезпечує ефективну буферну клад, іонообмінної, гідрофобної або афінної хроємність, наприклад, від 10 до приблизно 100 мМ, матографії) і т.п. Процес концентрування, при бапереважно 20 мМ. жанні, може також включати процес заміни Відновлюючі реагенти є складовою частиною буферу. Переважно концентрування проводиться солюбілізаційного буферу - для відновлення дисупри пониженій температурі (наприклад, приблизно льфідних зв'язків і для підтримання залишків цис4-10 градусів С). теїну у відновленій формі. На практиці використоПротеолітичне розщеплення попередника інвуються такі відновлюючі реагенти: бетасуліну проводиться або одночасною обробкою меркаптоетанол, дитіотреітол і т.п. Оптимальна трипсином і карбоксипептидазою В або окремою концентрація бета-меркаптоетанолу - між 20 і 200 обробкою цими двома ферментами. Трипсин вимМ, переважно між 100 і 150 мМ. користовується в концентрації від приблизно 1:200 Оптимально солюбілізаційний буфер містить до 1:20,000 (фермент:cубстрат, вагове співвіднододаткові компоненти типу катіонного комплексону шення), у той час як карбоксипептидаза В викориподібно EDTA. EDTA додається в солюбілізаційстовується в концентрації від приблизно 1:100 до ний буфер до концентрації приблизно 0.5 до 5 мМ, 1:5,000. Протеолітичне розщеплення проводиться переважно - 1 мМ до 2 мМ. Крім того, гліцин (або в буфері з рН приблизно 7,5 до 11,0, переважно інші амінокислоти) додаються у буфер для запобірН 10,5; додавання іонів Са2+, Ζn2+, Μn2+, Mg2+ могання руйнування поліпептиду вільними радикаже поліпшити ефективність реакції. Температура лами. Концентрація гліцину варіює від 5 до 100 інкубації варіює від 0 градусів С до приблизно 30 мМ, переважно від 10 до 20 мМ. градусів С, переважно - близько 6 градусів С, час Розчинення тілець включення в основному інкубації - від 1 до 24 годин, переважно приблизно проводиться за період від шести годин до прибли14 годин. зно 24 годин, переважно протягом 8 годин. РозчиПісля протеолітичного розщеплення біологічно нення може бути проведене при температурі наактивний інсулін очищають. вколишнього середовища або пониженій Нижченаведені приклади ілюструють винахід. температурі, звичайно від 4 градусів до 24 градуСлід розуміти, що ці приклади - тільки для ілюстсів С, переважно при 20 градусах С. Після того, як ративних цілей і не повинні розглядатися в жодрозчинення тілець включення завершено, розчин ному разі як обмеження цього винаходу. відновленого поліпептиду освітлюється для видаПриклад 1 лення нерозчинного залишку. Освітлення може Конструювання плазміни pIK8-proins бути виконане шляхом використання високошвидЗа допомогою полімеразної ланцюгової реакції кісного центрифугування. (ПЛР) кДНК інсуліну людини було ампліфіковано Відновлений попередник проінсуліну потім рона матриці однониткової кДНК підшлункової залозбавляється буфером для рефолдингу. Розведензи людини використовуючи праймери Р1 і Р2: ня проводиться шляхом додавання розчину тілець праймер Р1 (прямий праймер) включення в буфер для рефолдингу або шляхом одночасної подачі в ємність для рефолдингу бупраймер Р2 (зворотній праймер) фера й розчину тілець включення. Розчин тілець включення може бути розведений приблизно від Праймер Р1 співпадає з послідовностю В10 до 200-кратно, переважно 100-кратно буфером ланцюга гену інсуліну людини, починаючи з Arg22 для рефолдингу. Кінцева концентрація поліпептипрепроінсуліну людини, та містить ділянку розпіду після розчинення може складати приблизно від знавання ендонуклеази Ndel (виділена рамкою), 0.01 мг/мл до 5 мг/мл, переважно 1 мг/мл. необхідну для клонування. Зворотній праймер (Р2) Буфер для рефолдингу як правило містить рН є комплементарним аміно-термінальному фрагмебуферний агент, окислювально-відновну пару, нту Α-ланцюга інсуліну та містить EcoRI ділянку катіонний хелатор, реагенти-запобіжники вільно(виділена рамкою) для цілей клонування. ПЛР рерадикального руйнування поліпептиду й деякі інші акцію проводили в розчині, який містив 25 пмоль низькомолекулярні добавки. Процес рефолдингу кожного з вказаних праймерів, 1 ПЛР буфер, 200 проводиться при температурі приблизно від 4 до мкМ кожного з чотирьох нуклеотидів (dA, dC, dG и 24 градусів С, переважніше при 10 градусах С. Час dT), 2 нанограми однониткової кДНК, 5.0 одиниць інкубації в загальному складає приблизно від 2 до Taq полімерази. Умови ПЛР реакції були такими: 20 годин, переважно близько 6 годин. Головні ком95 градусів С протягом 3 хвилин, потім 35 циклів поненти буфера для рефолдингу включають глі(95 градусів С, 1 хвилина; 65 градусів С протягом цин в концентрації приблизно від 1 мМ до 100 мМ, 30 секунд; 72 градусів С протягом 30 секунд), після переважно 10 мМ; цистин в концентрації приблизцього - 5 хвилин при 72 градусах С. ПЛР фрагмент но від 0.5 мМ до 20 мМ; EDTA приблизно від 0.1 довжиною близько 280 п.н. було виділено з агаромМ до 5 мМ; гліцерин - приблизно від 1% до 20%. зного гелю та клоновано в рТА клонувальний векПісля закінчення процесу рефолдиніу поперетор. Лігазну суміш було введено в штам дник інсуліну, що правильно згорнувся, може бути Escherichia coli DH5alpha, трасформанти відібрані сконцентрований, потім очищений, і поліпептид на чашках з агаризованим LB середовищем, яке може бути оброблений протеолітичними фермен 15 91281 16 містить ампіцилін. Декілька окремих колоній були S2: 5'вирощені в LB середовищі протягом ночі, плазмідAAGCCACGCTCGCCGCACACTAAATACAGCGCTT на ДНК була виділена з використанням набору CCA Wizard minipreps DNA isolation kit. Клон IK8 був CCAGGTGGCTGCCACACAGATGCTGGTTCACAAA вибраний для подальшої роботи, оскільки він мав GCGCATATG-3' правильну послідовність ДНК. S3: 5'Для експресії в Escherichia coli плазмідна ДНК, CGGCGAGCGTGGCTTCTTTTATACCCCGAAAACC виділена із клону IK8, була розщеплена ендонукAAA леазами рестрикції Ndel і EcoRI; ДНК фрагмент CGTGGCATTGTGGAACAGTGTTGCACCAGTATTT довжиною близько 270 п.н. був виділений із агароGTAGCCTGT-3' зного гелю та клонований в плазміду рЕТ28а, яка S4: 5'була розщеплена тими ж самими ферментами та CAGGCGTGAATTCTTAGTTGCAGTAATTTTCCAG оброблена лужною фосфатазою із шлунку теляти. CTG ATACAGGCTACAAATACTGGTGC-3' Лігазною сумішшю було трансформовано штам S5: 5'- CAGGCGTGAATTCTTAGTTGC-3' Escherichia coli DH5alpha та трансформанти відібСуміш праймерів SI, S2, S3, S4 (кінцева конрані на LB канаміцинових чашках. Плазмідна ДНК центрація 2 мкМ кожного) в ПЛР буферному розбула ізольована із деяких трансформантів та прочині з 5U rTth ДНК полімерази нагрівали до 94 грааналізована рестрикцією Ndel та EcoRI. Правильдусів С протягом 1 хвилини, охолоджували до 62 ний клон був ідентифікований та названий pIK8градусів С та проводили реакцію при 72 градусах proins. Ця плазміда була в подальшому введена в С протягом 2 хвилин для заповнення розривів. Escherichia coli, наприклад, штам JM109 чи BL21. Наступні 20 циклів ампліфікації були виконані при Приклад 2 наступних умовах: 94 градуси С, 15 сек; 62 градуси Конструювання плазміди pMUT12 С, 30 сек; 72 градуси С, 30 сек. 2 мкл вищезгаданої Плазмідна ДНК pIK8-proins використовувалась суміші використовувалося як матриця для ампліяк матриця для направленого мутагенезу з викофікації кДНК проінсуліну людини з використанням ристанням ПЛР та наступних праймерів: праймерів Sin S5. ПЛР фрагмент довжиною прибМ1: 5'лизно 180 п.н. був очищений із гелю та клонований CTTCTACACACCCAAGACCAAGCGTGGCATTGTG в рТА-клонуючий вектор. Клон IS2 був вибраний GAACAATGCTG-3' для подальшої роботи, оскільки він мав правильну М2: 5'послідовність ДНК. CAGCATTGTTCCACAATGCCACGCTTGGTCTTGG Для експресії в Escherichia coli плазмідна ДНК, GTGTGTAGAAG-3' виділена із клону IS2, була розщеплена ендонукПісля денатурації ДНК при 96 градусах С пролеазами рестрикції Ndel і EcoRI; ДНК фрагмент тягом 3 хвилин було проведено 30 циклів ПЛР з довжиною близько 170 п.н. був виділений із агаровикористанням 5 одиниць rTth ДНК полімерази. зного гелю та клонований в плазмiду рЕТ28а, яка Умови ПЛР реакції були такими: 94 градуси С, 30 була розщеплена тими ж самими ферментами та сек; 59 градусів С, 30 сек; 72 градуси С, 6 хвилин оброблена лужною фосфатазою із шлунку теляти. та завершальний синтез при 72 градусах С протяЛігазною сумішшю було трансформовано штам гом 10 хвилин. Продукт ПЛР реакції було очищено Escherichia coli DH5alpha та трансформанти відібнабором Zymoclean та оброблено 10U рестриктази рані на LB канаміцинових чашках. Плазмідна ДНК Dpnl - для видалення оригінальної матриці. Після була ізольована із деяких трансформантів та проще одного циклу очистки на колонці 2 мкл аліквота аналізована рестрикцією з Ndel та EcoRI. Правибула введена в Escherichia coli штам DH5alpha та льний клон був ідентифікований та названий трансформанти відібрані на LB чашках, які місять pISYN2. Ця плазміда була в подальшому введена канаміцин. Декілька окремих колоній були виров Escherichia coli, наприклад штам JM109 чи BL21. щені в LB середовищі протягом ночі, плазмідна Приклад 4 ДНК була виділена з використанням набору Wizard Створення плазміди pCIM61 minipreps. Клон MUT12 був вибраний для подальПлазмідна ДНК pMUT12 використовувалася як шої роботи, оскільки він мав правильну послідовматриця для ампліфікації кДНЮminі проінсуліну ність ДНК. Для експресії в Escherichia coli ця плазлюдини з модифікованим препептидом. ПЛР реакміда була в подальшому введена в Escherichia цію проводили в умовах описаних в прикладі 1 з coli, наприклад штам JM109 чи BL21. використанням наступних праймерів: Приклад 3 Праймер С (прямий): Конструкція плазміди pISYN2 5'Для створення pCSYN61 ми спочатку синтезуGCCATATGCGAAAGAAGCGGAAGAAGAAGCGTTT вали 5 олігонуклеотидних затравок, опираючись TGTGAACACCTGTG-3' на опубліковану амінокислотну послідовність інсуПраймер ІL4(зворотній): ліну людини [10]. При цьому кодони нуклеотидних 5'затравок відповідали притаманним для Escherichia CCGCAAGCTTTTAGTTGCAGTAGTTCTCCAGCTG coli генам з високим рівнем експресії [11] та рівням G-3' тРНК у Escherichia coli [12]. Ці затравки також місПЛР продукт довжиною приблизно 210 п.н. бутили ділянки розпізнавання ендонуклеаз в різноло виділено з агарозного гелю та клоновано в рТА манітних положеннях - для цілей клонування. вектор. Лігазна суміш була трансформована в Послідовності праймерів: Escherichia coli штам DH5alpha та трансформанти S1: 5'-CATATGCGCTTTGTGAACCAG-3' відібрані на LB чашках, які містять ампіцилін. Клон 17 91281 18 СІМ6 був відібраний як такий, що має правильну 7568 та ССМ 7567 згідно з умовами Будапештської ДНК послідовність. Угоди стосовно Міжнародного договору по розміДля експресії в Escherichia coli, плазмідну ДНК щенню мікроорганізмів з метою патентування. виділену з клону СІМ6, було оброблено ендонукКолонії, відібрані як описано вище, були інокулеазами рестрикції Ndel та Hindlll. Фрагмент ДНК льовані в 1 мл LB середовища (10 г бакто триптодовжиною приблизно 200 п.н. було виділено з гена, 5 г дріжджового екстракту та 10 г NaCl на Л) та лю та клоновано в плазміду рЕТ39а що була обпотім культивувалися при 37 градусів С протягом роблена тими же ферментами. Лігазна суміш була не менше 12 год. Культура була перенесена в котрансформована в Escherichia coli DH5alpha та лбу з 50 мл LB середовища, яке містить 30 мкг/мл трансформанти відібрані на LB чашках з канаміциканаміцину та після досягнення культурою оптичном. Плазмідну ДНК було виділено із декількох ної щільності в межах від 0,5 до 0,8 було добавлетрансформантів та аналізовано розщепленням з но IPTG (ізопропил- -D-тіогалактопіранозід) в куNdel та Hindlll. Правильний клон був ідентифіковальтуру до кінцевої концентрації 1 мМ. ний та названий pCIM61. Ця плазміда була трансВирощування продовжувалося при температурі 37 формована в Escherichia coli, наприклад штам градусів С протягом 4 год при інтенсивному переJM109 чи ВL21. мішуванні (200 об/хв.). Після індукції бактеріальну Приклад 5 культуру центрифугували при 4000 об/хв протягом Створення плазміди pDIM07 15 хв для того, щоб одержати осад клітин Е. coli. Плазмідна ДНК pMUT12 використовувалася як Клітини було лізовано нагріванням до 95 градусів матриця для ампліфікації кДНКmіnі проінсуліну С протягом 5 хв в буфері для нанесення зразків, людини з модифікованим препептидом. ПЛР реаклізати були проаналізовані за допомогою ПААГ цію проводили в умовах описаних в прикладі 1 з електрофорезу в денатуруючих умовах згідно ставикористанням наступних праймерів: ндартної методики [13]. Праймер D (прямий): Приклад 7 5'Ферментація та умови вирощування TACCATGGATGAAGACGAGGATGAAGCACGCTTT 1. Робочий банк GTGAA CCAACACCTGTG-3' Одинична колонія Escherichia coli штаму BL21, Праймер IL4 (зворотній): що містить одну з вище вказаних плазмід, виро5'щувалась в середовищі LB (10 г/л бакто пептон, 5 CCGCAAGCTTTTAGTTGCAGTAGTTCTCCAGCTG г/л дріжджовий екстракт та 5 г/л NaCl) або його G-3' аналогу з додаванням відповідного антибіотику. ПЛР продукт довжиною приблизно 210 п.н. буКультури були потім розбавлені середовищем для ло виділено з агарозного гелю та клоновано в рТА кріоконсервації та зберігалися при температурі -80 вектор. Лігазна суміш була трансформована в градусів С або -170 градусів С. Escherichia coli штам DH5alpha та трансформанти 2. Посів відібрані на LB чашках, які містять ампіцилін. Клон Колба із стерильним середовищем LB з додаDIM1 був відібраний як такий, що має правильну ванням 30 мкг/мл канаміцину чи 50 мкг/мл ампіциДНК послідовність. ліну була засіяна культурою із робочого банку та Для експресії в Escherichia coli плазмідну ДНК, інкубована протягом 15 год на шейкері при 37 гравиділену з клону DIM1, було оброблено ендонукдусах С та приблизно 250 об/хв. При необхідності леазами рестрикції Ncol та Hindlll. Фрагмент ДНК наступні етапи вирощування проводили в аеровадовжиною приблизно 200 п.н. було виділено з гених ферментерах з перемішуванням. Стерильне лю та клоновано в плазміду рЕТ28а, що була обсередовище у ферментері було засіяне бактеріароблена тими же ферментами. Лігазна суміш була льною культурою із колби (об'єм інокулята складав трансформована в Escherichia coli DH5alpha та 5-10% від об'єму ферментера) та інкубувалося трансформанти відібрані на LB чашках з канаміципротягом 5-8 годин при 37 градусах С, рН 6,9±0,5 з ном. Плазмідну ДНК було виділено із декількох перемішуванням та аерацією для того щоб підттрансформантів та аналізовано розщепленням з римувати рівень розчиненого кисню вище 20% Ncol та Hindlll. Правильний клон був ідентифікованасичення. ний та названий рDIМ07. Ця плазміда була транс3. Вирощування формована в Escherichia coli, наприклад штам Виробниче середовище було засіяне посівною JM109 чи BL21. культурою (1-10% від об'єму робочого ферментеПриклад 6 ра) та інкубувалось при 37 градусах С. ПеремішуЕкспресія поліпептидів - попередників інсуліну вання-аерація були виставлені на рівні достатньоПлазміди, в яких підтверджено наявність ДНК му, щоб підтримувати ступінь розчиненого кисню фрагменту що кодує послідовність попередника вище 20% насичення. Показник рН підтримувався інсуліну (pIK8-proins, pMUT12, pCIM61, pISYN2, на рівні 6,9±0,5 за допомогою NEUOH. Пропінол pDIM07 та ін.), використовувалися для трансфорБ400 використовувався як піногасник. мації клітин Е. coli BL21(DE3) у відповідності з процедурами, що описані вище, та клітини колоній Виробниче середовище трансформовані плазмідними ДНК pIK8-proins, pISYN2 та pCIM61 були поміщені в Український бакто пептон 3.5 г/л Центр Мікроорганізмів з номерами доступу відподріжджовий екстракт 3.5 г/л відно IMB В-7169, IMB В-7230 та IMB В-7251 та глюкоза 2.0 г/л депоновані у Чеській Колекції Мікроорганізмів з NaCl 1.0 г/л номерами доступу відповідно ССМ 7511, ССМ (NH4)Cl 3.0 г/л 19 K2НРО4 (ΝΗ4)Η2ΡΟ4 (NH4)2SO4 K2SO4 MgSO4 тіамін мікроелементи канаміцин 91281 4.0 г/л 2.0 г/л 2.0 г/л 3.0 г/л 1.0 г/л 5 мг/л 2 мл/л 30 мг/мл Розчин мікроелементів FeSO4*7H2O ZnSO4*7H2O CuSO4*7H2O MnCl2 Na2B4O7*10H2O CaCl2*2H2O (NH4)6Mo7O24 0.75M EDTA лимонна кислота 10г/л 2,25 г/л 1,0 г/л 0,25 г/л 0,25 г/л 2,0 г/л 0,1 г/л 100 мл/л 60 г/л Стерильний розчин 50% глюкози використовували як джерело вуглецю. Як тільки концентрація клітин досягла (OD600 близько 30, стерильний розчин індуктора, наприклад IPTG (кінцева концентрація 1 мМ) було інфузовано та культивування продовжували протягом 6 год, концентрація клітин досягала приблизно 50 при OD600. Культуральне середовище після цього охолоджували та клітини збирали центрифугуванням. Приклад 8 Очистка тілець включення Бактеріальну масу ресуспендували (у співвідношенні до буферного розчину 1:10) в холодному буферному розчині, що містить 20 мМ Tris, рН 7,5; 1 мМ ЕДТА та 100 мМ NaCl. Клітинна суспензія була пропущена через руйнівник клітин при 17000 psi та тільця включення збирали центрифугуванням. Осад, що містить тільця включення та фрагменти бактерій, був відмитий 20-кратним об'ємом холодного (+10 градусів С) буферу для відмивання який містить 1.0% Triton XI00, 20 мМ Tris-HCl, рН 8.0, 2 мМ ЕДТА, 100 мМ NaCl. Осад був зібраний центрифугуванням приблизно при 14000 об/хв. Стадія відмивання була повторена двічі. 1,5 кг вологих тілець включення, які містять приблизно 1,0 кг гібридного поліпептиду (визначено за методом Бредфорда) були розчинені в 15 л буферного розчину, який містить 8M сечовини, 50 мМ гліцину, рН 8,0; 2 мМ ЕДТА, 150 мМ меркаптоетанолу. Інкубація продовжувалася протягом 6 год при кімнатній температурі. Розчин був освітлений високошвидкісним центрифугуванням та відновлений попередник інсуліну був використаний для рефолдингу. Приклад 9 Очистка тілець включення Водна суспензію тілець включення об'ємом 60 л з загальним вмістом білку 12 кг була оброблена лізоцимом (0,1 мг/мл), РНКазою (0,5 од.акт./мл) та ДНКазою (0,5 од.актУмл) протягом 3 год при 25°С в буфері, що містить 20 мМ Tris-HCl, pH 7,5; 10 мМ MgSO4. 20 Після обробки було визначено вміст нуклеїнових кислот, що становив198 мг/л (656 мг/л - до обробки). Тільця включення були відмиті від надлишку ферментів центрифугуванням і розчинені у 100 л буферного розчину, який містить 8М сечовини, 20 мМ гліцину, рН 8,5; 1 мМ ЕДТА, 150 мМ меркаптоетанолу. Інкубація продовжувалася протягом 4 год при кімнатній температурі. Розчин був освітлений високошвидкісним центрифугуванням та відновлений попередник інсуліну був використаний для рефолдингу. Приклад 10 Рефолдування гібридного поліпептиду Ємність для рефолдингу була наповнена буфером (50 мМ гліцин, 2 мМ ЕДТА, 5 мМ цистин, 10% гліцерол, рН 11,2) та охолоджена до 5 градусів С. Потім освітлений поліпептидний розчин та буфер для рефолдингу швидко змішують в пропорції 1:70 (об/об) шляхом подачі цих розчинів у камеру змішування. Реакційну суміш переносять в ємність для рефолдингу на 10 год при 5 градусах С та при повільному перемішуванні. Концентрація поліпептиду в ємності для рефолдингу складає приблизно 1.0 г/л, як визначено за методом Бредфорда. Концентрування Концентрування рефолдованого поліпептиду зазвичай виконується шляхом ультрафільтрації чи з використанням абсорбційної хроматографії. Хроматографічна колонка розміром 450 500 мм була заповнена 30 л полімерного сорбенту Amberchrom CG-300M. Рефолдована реакційна суміш подавалась на колонку при швидкості потоку 4 л/хв, колонка була відмита трьома об'ємами буферу для врівноваження. Поліпептид елюювали 20-40% розчином 2-пропанолу. Потім елюент був проаналізований методом високоефективної рідинної хроматографії, що показало чистоту отриманого препарату більше 45%, а вихід складає не менше 96%. Приклад 11 Рефолдування гібридного поліпептиду Ємність для рефолдингу була наповнена буфером (10 мМ гліцин, 1 мМ ЕДТА, 2мМ цистин, 1 г/л ПЕГ 1500, рН 11.2) та охолоджена до 10°С. 50 л відновленого гібридного білку змішали з 5000 л буферу для рефолдингу, концентрація білку в ємності для рефолдингу складає 0,5 г/л. Через 6 год в рефолдинг буфер було внесено Zn2+ до концентрації 3 мМ та доведено рН до 6,2. Суспензію висадженого білка було подано на самовигружний сепаратор, та отримано приблизно 150 л водної суспензій аморфного осаду білка, який було розчинено шляхом додавання сечовини до 6М. Приклад 12 Ферментативна конверсія Попередник інсуліну в розчині 50 мМ трис-НСІ, рН 8.0 був розщеплений до інсуліну людини шляхом інкубації з трипсином (1:8,000; вагове співвідношення) та карбоксипептидазою В (1:2000; вагове співвідношення) в присутності 1,5 мМ Zn2+. Реакцію вели на протязі 16 год при 8 градусах С та зупиняли шляхом корекції рН до 2,5 за допомогою 6М НСl. Високоефективний рідинний хроматогра 21 91281 22 фічний аналіз показав, що чистота отриманого deo P1P2 promoters of Escherichia coli. Appl препарату складала 69% чи більше. Інсулін може Microbiol Biotechnol. 1990, 33:424-8. бути висаджений з реакційної суміші сульфатом 3. Davidson HW, Rhodes CJ, Hutton JC. амонію чи хлоридом натрію (кінцева концентрація Intraorganellar calcium and pH control proinsulin 20%). cleavage in the pancreatic beta cell via two distinct Приклад 13 site-specific endopeptidases. Nature, 1988, 333:93Іонообмінна хроматографія 96. 300 г інсуліну було розчинено в 19 л буферно4. Markussen J. Human insulin by tryptic го розчину, що містить 50 мМ Tris-HCl, 1мМ ЕДТА, transpeptidations of porcine insulin and biosynthetic рН 8,3; 32% 2-пропанолу. Розчин інсуліну фільтруprecursors. Boston : MTP Press, 1987. вали через 0.5 мкм фільтр та подавали на колонку 5. Chance RE, Kroeff EP, Hoffmann JA, Frank BH. для іонообмінної хроматографії при швидкості Chemical, physical, and biologic properties of потоку 320 мл/хв. Колонка була заповнена 19 л biosynthetic human insulin. Diabetes Care, 1981, сорбента Matrex PEI-300. Після адсорбції інсуліну 2:147-54. колонка була відмита двома об'ємами буферу для 6. Thim L, Hansen MT, Norris K, Hoegh I, Boel E, врівноваження. Поліпептид був елюйований градіForstrom J, Ammerer G, and Fiil NP. Secretion and єнтом (0-0,3M) концентрації NaCl. Потім фракції Processing of Insulin Precursors in Yeast. PNAS, елюенту, які зібрані в межах 0,1-0,2M NaCl були 1986, 83:6766-6770. проаналізовані за допомогою високоефективної 7. Cousens LS, Shuster JR, Gallegos C, Ku LL, рідинної хроматографії. Аналіз показав, що чистоStempien MM, Urdea MS, Sanchez-Pescador R, та препарату складала не менше 85%, а вихід Taylor A, Tekamp-Olson P. High level expression of продукту - не менше 91%. proinsulin in the yeast, Saccharomyces cerevisiae. Приклад 14 Gene, 1987, 61:265-275. Високоефективна рідинна хроматографічна 8. Cowley DJ, Mackin RB. Expression, purification очистка інсуліну and characterization of recombinant human Кристали інсуліну після іонообмінної хроматоproinsulin. FEBS Letters, 1997, 402:124-130. графії розчиняли при рН 3,0 в розчині, що містив 9. Лазарев ОП, Лесик ІП, Костецький ІЄ, Лісовсь0,05M ацетата натрію та 10% 2-пропанолу і фільткий ІЛ, Рибачук ВМ, Стадник ВІ. Спосіб одержання рували через 0,2 мкм фільтр. Отриманий розчин зі рекомбінантного інсуліну людини Патент України швидкістю 0,8 л/хв подавали на колонку розміром №76661, C12N15/17, C12N1/21, публ. 2006. 10. Le Lay J, Matsuoka ТА, Henderson and Stein R. 200 600мм заповнену 19 л сорбента, наприклад Identification of a novel PDX-1 binding site in the Diasogel SP-120-20-ODS-AP. Попередньо колонку human insulin gene enhancer J. Biol. Chem., 2004, врівноважували 40 л буфера для нанесення 279:22228-22235. (0,05M натрія ацетата, 10% 2-пропанолу, рН 3,0); 11. Grantham R, Gautier C, Gouy M. Codon цим же буферним розчином промивали колонку frequencies in 119 individual genes confirm після нанесення інсуліну. Елюцію інсуліну провоconsistent choices of degenerate bases according to дили лінійним градієнтом 2-пропанолу (10-28%). genome type. Nucleic Acids Res.,1980, 8:1893-1912. Для регенерації сорбента колонку промивали 50 л 12. Ikemura TT. Correlation between the abundance розчину, що містить 0,05M ацетата калію, 50% 2of Escherichia coli transfer RNAs and the occurrence пропанолу, рН 3,0. Аналіз показав, що чистота of the respective codons in its protein genes: a інсуліну складала не менше 98%. proposal for a synonymous codon choice that is Джерела інформації optimal for the E. coli translational system. J. Моl. 1. Brandenburg D, Wollmer A. Insulin: chemistry, Biol., 1981, 151:389-409. structure, and function of insulin and related 13. Sambrook J, Fritsch EF and Maniatis T. Molecular hormones: proceedings of the Second International Cloning: A Laboratory Manual, 2nd edition, Cold Insulin Symposium, Aachen, Germany, September 4Spring Harbor Laboratory Press, 1989. 7, 1979. Publisher: New York: W. de Gruyter, 1980. 2. Fischer M, Fytlovitch S, Amit B, Wortzel A, Beck Y. A constitutive expression vector system driven by the 23 91281 24 25 91281 26 27 91281 28 29 91281 30 31 Комп’ютерна верстка О. Гапоненко 91281 Підписне 32 Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for producing of recombinant human insulin
Автори англійськоюLazariev Oleksii Pavlovych, Lutsiv Volodymyr Romanovych, Kostetskyi Ihor Yevhenovych, Lisovskyi Ihor Leonidovych, Lesyk Ihor Pavlovych
Назва патенту російськоюСпособ получения рекомбинантного инсулина человека
Автори російськоюЛазарев Алексей Павлович, Луцив Владимир Романович, Костецкий Игор Евгеньевич, Лисовский Игорь Леонидович, Лесик Игорь Павлович
МПК / Мітки
МПК: C12N 1/21, C12N 15/17
Мітки: людини, інсуліну, рекомбінантного, одержання, спосіб
Код посилання
<a href="https://ua.patents.su/16-91281-sposib-oderzhannya-rekombinantnogo-insulinu-lyudini.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання рекомбінантного інсуліну людини</a>
Спосіб одержання рекомбінантного інсуліну людини

Номер патенту: 76661
Опубліковано: 15.08.2006
Автори: Лісовський Ігор Леонідович, Стадник Віктор Іванович
МПК: C12N 1/21, C12R 1/19, C12N 15/70, C07K 14/62, C12N 15/17
Мітки: спосіб, людини, інсуліну, рекомбінантного, одержання
Формула / Реферат:
Спосіб одержання рекомбінантного інсуліну людини шляхом конструювання рекомбінантної плазмідної ДНК, яка кодує проінсулін, зв'язаний з лідерною послідовністю через аргінін, одержання і культивування штаму-продуцента гібридного білка ESCHERICHIA COLI, виділення і дезінтеграції клітин, виділення гібридного білка, його ферментативного розщеплення з наступним очищенням і одержанням цільового продукту, який відрізняється тим, що ділянка...
Спосіб одержання напівсинтетичного інсуліну людини

Номер патенту: 46667
Опубліковано: 16.01.2006
Автори: Рибачук Валентина Миколаївна, Сологуб Павло Павлович, Лесик Ігор Павлович, Лазарєв Олексій Павлович, Прудієв Дмитро Павлович, Соляник Людмила Павлівна, Власенко Микола Миколайович, Оксамитний Віктор Миколайович, Луців Володимир Романович, Гавриш Олег Геннадійович, Мельник Валерій Миколайович, Поліванов Анатолій Валентинович, Стадник Віктор Іванович, Коноваленко Вадим Анатолійович, Пояркова Світлана Олексіївна
МПК: A61K 38/28, C12P 21/06, C07K 14/62
Мітки: спосіб, інсуліну, людини, одержання, напівсинтетичного
Формула / Реферат:
П'єзоелектричний перетворювач механічних величин, що містить п'єзоелемент з електродами та два узгоджувальних підсилювачі, який відрізняється тим, що п'єзоелемент виконаний у вигляді прямокутного паралелепіпеда з двома парами електродів, узгоджува-льні підсилювачі виконані у вигляді підсилювачів заряду, вхід та вихід основного підсилювача під'єднані до електродів, які розташовані на різних гранях п'єзоелемента, що паралельні до вектору...
Спосіб одержання комбінованого препарату інсуліну людини

Номер патенту: 46668
Опубліковано: 16.01.2006
Автори: Лесик Ігор Павлович, Лазарєв Олексій Павлович, Власенко Микола Миколайович, Пояркова Світлана Олексіївна, Прудієв Дмитро Павлович, Рибачук Валентина Миколаївна, Соляник Людмила Павлівна, Сологуб Павло Павлович
МПК: C07K 14/62, A61K 38/28
Мітки: препарату, одержання, людини, спосіб, комбінованого, інсуліну
Формула / Реферат:
Даний винахід відноситься до похідних дигідроімідазо[5,1-а]--карболіну загальної формули (І), в якій, зокрема: R1, R2, R3, R4, R6 і R7 однакові або різні, незалежно один від одного, означають атом водню, атом галогену, алкіл, гідроксил, алкоксил, тригалогеналкіл, алкіламіногрупу, діалкіламіногрупу, арил, арилалкіл, карбоксил, алкілкарбонілоксигрупу, ацил, арилоксигрупу або...
Рекомбінантна плазмідна днк psx70, що кодує синтез рекомбінантного людського гранулоцит-колонійстимулюючого фактора (г-ксф), штам escherichia coli sx70, спосіб одержання г-ксф людини та препарат на основі високоочищеного г-ксф

Номер патенту: 87054
Опубліковано: 10.06.2009
Автори: Скрипін Васілій Івановіч, Могутов Міхаіл Алєксандровіч, Чувпіло Сєргєй Альбєртовіч, Яковєнко Андрєй Романовіч, ЯРОЦКІЙ Сєргєй Вікторовіч
МПК: A61K 38/19, C07K 14/53, C12N 15/72, C12N 15/27, C12P 21/02
Мітки: кодує, днк, гранулоцит-колонійстимулюючого, людського, фактора, плазмідна, г-ксф, людини, спосіб, препарат, psx70, основі, sx70, штам, escherichia, одержання, рекомбінантна, високоочищеного, синтез, рекомбінантного
Формула / Реферат:
1. Рекомбінантна плазмідна ДНК pSX70, що кодує синтез рекомбінантного людського гранулоцит-колонійстимулюючого фактора (Г-КСФ), має 4305 пар основ (п.о.) і характеризується наявністю наступних фрагментів: послідовність з 1 н. по 528 н. включає фрагмент ДНК розміром 529 п.о., що містить ген Г-КСФ з наступними нуклеотидними замінами: послідовність з 540 н. по 550 н., яка...
Спосіб одержання кристалічного ліз в28 про в29 інсуліну людини (варіанти)

Номер патенту: 26638
Опубліковано: 11.10.1999
Автори: Френк Брюс Хілл, Картер Ненсі Делорес, Бейкер Джеффрі Клейтон
МПК: A61K 38/28, A61P 5/48, A61P 5/00, C07B 63/00, C07K 1/14, C07K 14/62, A61K 38/00, C07K 14/575
Мітки: варіанти, одержання, кристалічного, людини, спосіб, ліз, інсуліну
Формула / Реферат:
1. Способ получения кристаллического ЛизВ28ПроВ29-инсулина человека, отличающийся тем, что включает: кристаллизацию ЛизВ28ПроВ29-инсулина человека из раствора, содержащего ЛизВ28ПроВ29-инсулин человека, цинк, по меньшей мере 0,3N органическую кислоту, выбранную из группы: уксусная кислота, лимонная кислота или глицин, и фенольное соединение при pH от 5,5 до 6,5.2. Способ по п.1, отличающийся тем, что концентрация ЛизВ28ПроВ29-инсулина...
Попередній патент: Очисник головок коренеплодів
Наступний патент: Спосіб одержання виливків литтям за одноразовими моделями
Випадковий патент: Спосіб обробки зливків, що відливають зі сталі