Сечовинозаміщені імідазохінолінові етери
Номер патенту: 74852
Опубліковано: 15.02.2006
Автори: Крукс Стефен Л., Хеппнер Філіп Д., Меррілл Брайон А., Грайсграбер Джордж У.








Формула / Реферат
1. Сполука формули (І):
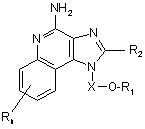 , (I)
, (I)
де X являє собою -CHR5-, -СНR5-алкіл- або -СНR5-алкеніл-;
R1 вибраний з групи, яка складається з:
R4-NR8-CR3-NR5-Z-R6-алкілу;
R4-NR8-CR3-NR5-Z-R6-алкенілу;
R4-NR8-CR3-NR5-Z-R6-арилу;
R4-NR8-CR3-NR5-Z-R6-гетероарилу;
R4-NR8-CR3-NR5-Z-R6-гетероциклілу;
R4-NR8-CR3-NR5R7;
R4-NR8-CR3-NR9-Z-R6-алкілу;
R4-NR8-CR3-NR9-Z-R6-алкенілу;
R4-NR8-CR3-NR9-Z-R6-арилу;
R4-NR8-CR3-NR9-Z-R6-гетероарилу та
R4-NR8-CR3-NR9-Z-R6-гетероциклілу;
R2 вибраний з групи, яка складається з:
водню;
алкілу;
алкенілу;
арилу;
гетероарилу;
гетероциклілу;
алкіл-Y-алкілу;
алкіл-Y-алкенілу;
алкіл-Y-арилу та
алкілу або алкенілу, заміщеного одним або більше замісниками, вибраними з групи, яка складається з:
-ОН;
галогену;
N(R5)2;
CO-N(R5)2;
СО-С1-10алкілу;
СО-O-С1-10алкілу;
N3;
арилу;
гетероарилу;
гетероциклілу;
СО-арилу та
СО-гетероарилу;
кожний R3 являє собою =O або =S;
кожний R4 незалежно являє собою алкіл або алкеніл, які можуть перериватися однією або більше -О-групами;
кожний R5 незалежно являє собою Н або С1-10алкіл;
R6 являє собою зв'язок, алкіл або алкеніл, що можуть перериватися однією або більше -О- групами;
R7 являє собою Н або С1-10алкіл, що можуть перериватися гетероатомом, або R7 може об'єднуватися з R5 з утворенням кільця;
R8 являє собою Н або С1-10алкіл або арилалкіл; або R4 та R8 можуть об'єднуватися разом з утворенням кільця;
R9 являє собою С1-10алкіл, який може об'єднуватися з R8 з утворенням кільця;
кожний Y незалежно являє собою -О- або -S(O)0-2-;
Z являє собою зв'язок, -CO- або -SO2-;
n є числом від 0 до 4; та
кожний присутній R незалежно вибраний з групи, яка складається з С1-10алкілу, С1-10алкокси, гідрокси, галогену та трифторметилу;
або її фармацевтично прийнятна сіль.
2. Сполука або сіль за пунктом 1, де Х є -СН(алкіл)(алкіл)-, де алкільні групи можуть бути однакові або різні.
3. Сполука або сіль за пунктом 1, де Х є -СН2-СН2-.
4. Сполука або сіль за пунктом 1, де Х є -CH(C2H5)(CH2)-.
5. Сполука або сіль за пунктом 1, де R2 є Н.
6. Сполука або сіль за пунктом 1, де R2 є алкілом.
7. Сполука або сіль за пунктом 1, де R2 є алкіл-O-алкілом.
8. Сполука згідно з пунктом 1, вибрана з групи, яка складається з:
N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5-с]хінолін-1-іл]етоксі)етил)-N'-фенілсечовини;
N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5-с]хінолін-1-іл]етоксі}етил)морфолін-4-карбоксаміду;
N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5-с]хінолін-1-іл]етоксі}етил)-N-метилморфолін-4-карбоксаміду та
N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5-с]хінолін-1-іл]етоксі}етил)-N-метил-N'-фенілсечовини;
або її фармацевтично прийнятна сіль.
9. Сполука формули (II)
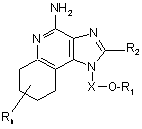 , (II)
, (II)
де X являє собою -CHR5-, -СНR5-алкіл- або -СНR5-алкеніл-;
R1 вибраний з групи, яка складається з:
R4-NR8-CR3-NR5-Z-R6-алкілу;
R4-NR8-CR3-NR5-Z-R6-алкенілу;
R4-NR8-CR3-NR5-Z-R6-арилу;
R4-NR8-CR3-NR5-Z-R6-гетероарилу;
R4-NR8-CR3-NR5-Z-R6-гетероциклілу;
R4-NR8-CR3-NR5R7;
R4-NR8-CR3-NR9-Z-R6-алкілу;
R4-NR8-CR3-NR9-Z-R6-алкенілу;
R4-NR8-CR3-NR9-Z-R6-арилу;
R4-NR8-CR3-NR9-Z-R6-гетероарилу та
R4-NR8-CR3-NR9-Z-R6-гетероциклілу;
R2 вибраний з групи, яка складається з:
водню;
алкілу;
алкенілу;
арилу;
гетероарилу;
гетероциклілу;
алкіл-Y-алкілу;
алкіл-Y-алкенілу;
алкіл-Y-арилу та
алкілу або алкенілу, заміщених одним або більше замісниками, вибраними з групи, яка складається з:
ОН;
галогену;
N(R5)2;
CO-N(R5)2;
СО-С1-10алкілу;
СО-О-С1-10алкілу;
N3;
арилу;
гетероарилу;
гетероциклілу;
СО-арилу та
СО-гетероарилу;
кожний R3 являє собой =O або =S;
кожний R4 незалежно являє собою алкіл або алкеніл, які можуть перериватися однією або більше -О-групами;
кожний R5 незалежно являє собою Н або С1-10алкіл;
R6 являє собою зв'язок, алкіл або алкеніл, що можуть перериватися однією або більше -О- групами;
R7 являє собою Н або С1-10алкіл, що можуть перериватися гетероатомом, або R7 може об'єднуватися з R5 з утворенням кільця;
R8 являє собою Н або С1-10алкіл або арилалкіл; або R4 та R8 можуть об'єднуватися разом з утворенням кільця;
R9 являє собою С1-10алкіл, який може об'єднуватися з R8 з утворенням кільця;
кожний Y незалежно являє собою -О- або -S(O)0-2-;
Z являє собою зв'язок, -СО- або -SO2-;
n є числом від 0 до 4; та
кожний присутній R незалежно вибраний з групи, яка складається з С1-10алкілу, С1-10алкокси, гідрокси, галогену та трифторметилу;
або її фармацевтично прийнятна сіль.
10. Сполука або сіль за пунктом 9, де R2 є Н або алкілом.
11. Сполука або сіль за пунктом 9, де R2 є алкіл-O-алкілом.
12. Сполука згідно з пунктом 9, вибрана з групи, яка складається з:
N-(2-{2-[4-аміно-2-(2-метоксіетил)-6,7,8,9-тетрагідро-1Н-імідазо[4,5-с]хінолін-1-іл]етоксі}етил)-N'-фенілсечовини та
N-(2-{2-[4-аміно-2-(2-метоксіетил)-6,7,8,9-тетрагідро-1Н-імідазо[4,5-с]хінолін-1-іл]етоксі}етил)- N-метил-N'-фенілсечовини;
або її фармацевтично прийнятна сіль.
13. Фармацевтична композиція, що містить терапевтично ефективну кількість сполуки або солі за пунктом 1 або 9 та фармацевтично прийнятний носій.
14. Спосіб індукції біосинтезу цитокінів у тварин, при якому вводять терапевтично ефективну кількість сполуки або солі за пунктом 1 або 9 тварині.
15. Спосіб за пунктом 14, де цитокін являє собою ![]() .
.
16. Спосіб лікування вірусного захворювання у тварини, при якому вводять терапевтично ефективну кількість сполуки або солі за пунктом 1 або 9 тварині.
17. Спосіб лікування неопластичного захворювання у тварини, при якому вводять терапевтично ефективну кількість сполуки або солі за пунктом 1 або 9 тварині.
Текст
1. Сполука формули (І): 2 (19) 1 3 74852 4 2. Сполука або сіль за пунктом 1, де Х є CO-N(R5)2; СН(алкіл)(алкіл)-, де алкільні групи можуть бути СО-С1-10алкілу; однакові або різні. СО-О-С1-10алкілу; 3. Сполука або сіль за пунктом 1, де Х є -СН2-СН2-. N3; 4. Сполука або сіль за пунктом 1, де Х є арилу; CH(C2H5)(CH2)-. гетероарилу; 5. Сполука або сіль за пунктом 1, де R2 є Н. гетероциклілу; 6. Сполука або сіль за пунктом 1, де R2 є алкілом. СО-арилу та 7. Сполука або сіль за пунктом 1, де R2 є алкіл-OСО-гетероарилу; алкілом. кожний R3 являє собой =O або =S; 8. Сполука згідно з пунктом 1, вибрана з групи, яка кожний R4 незалежно являє собою алкіл або алкескладається з: ніл, які можуть перериватися однією або більше N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5О-групами; с]хінолін-1-іл]етоксі)етил)-N'-фенілсечовини; кожний R5 незалежно являє собою Н або С1N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,510алкіл; с]хінолін-1-іл]етоксі}етил)морфолін-4R6 являє собою зв'язок, алкіл або алкеніл, що мокарбоксаміду; жуть перериватися однією або більше -О- групами; N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5R7 являє собою Н або С1-10алкіл, що можуть перес]хінолін-1-іл]етоксі}етил)-N-метилморфолін-4риватися гетероатомом, або R7 може об'єднуватикарбоксаміду та ся з R5 з утворенням кільця; N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5R8 являє собою Н або С1-10алкіл або арилалкіл; с]хінолін-1-іл]етоксі}етил)-N-метил-N'або R4 та R8 можуть об'єднуватися разом з утвофенілсечовини; ренням кільця; або її фармацевтично прийнятна сіль. R9 являє собою С1-10алкіл, який може об'єднувати9. Сполука формули (II) ся з R8 з утворенням кільця; NH2 кожний Y незалежно являє собою -О- або -S(O)0-2-; Z являє собою зв'язок, -СО- або -SO2-; N N n є числом від 0 до 4; та R2 кожний присутній R незалежно вибраний з групи, N яка складається з С1-10алкілу, С1-10алкокси, гідрокX O-R1 си, галогену та трифторметилу; Rn або її фармацевтично прийнятна сіль. , (II) 10. Сполука або сіль за пунктом 9, де R2 є Н або де X являє собою -CHR5-, -СНR5-алкіл- або -СНR5алкілом. алкеніл-; 11. Сполука або сіль за пунктом 9, де R2 є алкіл-OR1 вибраний з групи, яка складається з: алкілом. R4-NR8-CR3-NR5-Z-R6-алкілу; 12. Сполука згідно з пунктом 9, вибрана з групи, R4-NR8-CR3-NR5-Z-R6-алкенілу; яка складається з: R4-NR8-CR3-NR5-Z-R6-арилу; N-(2-{2-[4-аміно-2-(2-метоксіетил)-6,7,8,9R4-NR8-CR3-NR5-Z-R6-гетероарилу; тетрагідро-1Н-імідазо[4,5-с]хінолін-1R4-NR8-CR3-NR5-Z-R6-гетероциклілу; іл]етоксі}етил)-N'-фенілсечовини та R4-NR8-CR3-NR5R7; N-(2-{2-[4-аміно-2-(2-метоксіетил)-6,7,8,9R4-NR8-CR3-NR9-Z-R6-алкілу; тетрагідро-1Н-імідазо[4,5-с]хінолін-1R4-NR8-CR3-NR9-Z-R6-алкенілу; іл]етоксі}етил)- N-метил-N'-фенілсечовини; R4-NR8-CR3-NR9-Z-R6-арилу; або її фармацевтично прийнятна сіль. R4-NR8-CR3-NR9-Z-R6-гетероарилу та 13. Фармацевтична композиція, що містить тераR4-NR8-CR3-NR9-Z-R6-гетероциклілу; певтично ефективну кількість сполуки або солі за R2 вибраний з групи, яка складається з: пунктом 1 або 9 та фармацевтично прийнятний водню; носій. алкілу; 14. Спосіб індукції біосинтезу цитокінів у тварин, алкенілу; при якому вводять терапевтично ефективну кільарилу; кість сполуки або солі за пунктом 1 або 9 тварині. гетероарилу; 15. Спосіб за пунктом 14, де цитокін являє собою гетероциклілу; IFN . алкіл-Y-алкілу; 16. Спосіб лікування вірусного захворювання у алкіл-Y-алкенілу; тварини, при якому вводять терапевтично ефектиалкіл-Y-арилу та вну кількість сполуки або солі за пунктом 1 або 9 алкілу або алкенілу, заміщених одним або більше тварині. замісниками, вибраними з групи, яка складається 17. Спосіб лікування неопластичного захворюванз: ня у тварини, при якому вводять терапевтично ОН; ефективну кількість сполуки або солі за пунктом 1 галогену; або 9 тварині. N(R5)2; 5 Винахід відноситься до імідазохінолінових сполук, що мають етерну групу та залишок сечовини у положенні 1, та до фармацевтичних композицій, які містять такі сполуки. Додатковий аспект даного винаходу пов'язаний з використанням цих сполук як імуномодуляторів для індукції біосинтезу цитокінів у тварин, та при лікуванні захворювань, включаючи вірусні та неопластичні захворювання. Перший достовірний звіт стосовно 1Німідазо[4,5-с]хінолінової кільцевої системи, Backman та ін., [J.Org.Chem. 15, 1278-1284 (1950)] описує синтез 1-(6-метокси-8-хінолініл)-2-метил1Н-імідазо[4,5-с]хіноліну для можливого застосування як антималярійного агента. Згідно з цим були описані синтези різноманітних заміщених 1Німідазо[4,5-с]хінолінів. Наприклад, Jain та ін., [J.Med. Chem. 11, pp.87-92 (1968)], синтезували сполуку 1-[-2-(4-піперидил)етил]-1Н-імідазо[4,5с]хінолін як можливий протисудомний та кардіоваскулярний агент. Крім того, Baranov та ін., [Chem. Abs. 85, 94362 (1976)], повідомили про декілька 2оксоімідазо[4,5-с]хіноліни, а Веrеnуі та ін., [J.Heterocyctic Chem. 18, 1537-1540 (1981)] повідомили про декілька окремих 2-оксоімідазо[4,5с]хінолінів. Окремі 1Н-імідазо[4,5-с]хінолін-4-аміни та їх 1та 2-заміщені похідні були пізніше знайдені корисними як антивірусні агенти, бронходилятори та імуномодулятори. Вони описані, серед інших, [в патентах США №№4,689,338; 4,698,348; 4,929,624; 5,037,986; 5,268,376; 5,346,905; 5,389,640], які усі введені у дану заявку як посилання. Імідазохінолінові системи все ще представляють інтерес. Відомі окремі 1Н-імідазо[4,5-с]нафтиридин-4аміни, 1Н-імідазо[4,5-с]піридин-4-аміни, 1Німідазо[4,5-с]хінолін-4-аміни, що мають етервмісний замісник у положенні 1. Вони описані [у патентах США №№5,268,376; 5,389,640; 5,494,916 та WO 99/29693]. Незважаючи на ці спроби ідентифікувати сполуки, що є корисними як модифікатори імунної відповіді, залишається необхідність у сполуках, що мають здатність модулювати імунну відповідь шляхом індукції біосинтезу цитокінів або інших механізмів. Нами було виявлено новий клас сполук, що є корисними для індукції біосинтезу цитокінів у тварин. Згідно з цим, винахід забезпечує імідазохінолін-4-аміни та тетрагідроімідазохінолін-4-аміни, що мають етерну групу та залишок сечовини як замісники у положенні 1. Сполуки визначаються Формулами (I) та (II), які визначені більш детально нижче. Ці сполуки відповідають загальній структурній формулі: де X, R1, R2 та R є такими, як визначено для кожного класу сполук, що мають Формули (I) та (II). Сполуки Формул (I) та (II) є корисними як модифікатори імунної відповіді завдяки своїй здатно 74852 6 сті індукувати біосинтез цитокінів і, таким чином, модулювати імунну відповідь при введенні тваринам. Це робить ці сполуки корисними при лікуванні різних станів, таких, як вірусні захворювання та пухлини, що є чутливими до таких змін імунної відповіді. Винахід також забезпечує фармацевтичні композиції, що містять сполуки, які модифікують імунну відповідь, та способи індукції біосинтезу цитокінів у тварин, лікування вірусної інфекції у тварини та/або лікування неопластичного захворювання у тварини шляхом введення сполуки Формули (I) або (II) тварині. Крім того, винахід забезпечує способи синтезу сполук згідно з винаходом та нові проміжні сполуки, що є корисними для синтезу цих сполук. Як було зазначено раніше, ми виявили деякі сполуки, що індукують біосинтез цитокінів та модифікують імунну відповідь у тварин. Такі сполуки представлені Формулами (I) та (II), як представлено нижче. Iмідазохінолінові сполуки згідно з винаходом, які мають етерну групу та залишок сечовини у положенні 1, представлені Формулою (I): (I) де: X представляє собою –CHR5-, -СНR5-алкіл, або –CHR5-алкеніл-; R1 вибраний з групи, яка складається з: -R4-NR8-CR3-NR5-Z-R6-алкілу; -R4-NR8-CR3-NR5-Z-R6-алкенілу; -R4-NR8-CR3-NR5-Z-R6-арилу; -R4-NR8-CR3-NR5-Z-R6-гетероарилу; -R4-NR8-CR3-NR5-Z-R6-гетероциклілу; -R4-NR8-CR3-NR5-R7; -R4-NR8-CR3-NR9-Z-R6-алкілу; -R4-NR8-CR3-NR9-Z-R6-алкенілу; -R4-NR8-CR3-NR9-Z-R6-арилу; -R4-NR8-CR3-NR9-Z-R6-гетероарилу; та -R4-NR8-CR3-NR9-Z-R6-гетероциклілу; R2 вибраний з групи, яка складається з: - водню; - алкілу; - алкенілу; - арилу; - гетероарилу; - гетероциклілу; - алкіл-Y-алкілу; - алкіл-Y-алкенілу; - алкіл-Y-арил; та - алкілу або алкенілу, заміщеного одним або більше замісниками, вибраними з групи, яка складається з: -ОН; -галогену; -N(R5)2; -CO-N(R5)2; -СО-С1-10алкілу; -СО-О-С1-10алкілу; -N3; -арилу; 7 74852 8 -гетероарилу; -ОН; -гетероциклілу; -галогену; -СО-арилу; та -N(R5)2; -СО-гетероарилу; -CO-N(R5)2; кожний R3 представляє собой =O або =S; -СО-С1-10алкілу; кожний R4 незалежно представляє собою ал-СО-О-С1-10алкілу; кіл або алкеніл, які можуть перериватися однією -N3; або більше -О- групами; -арилу; кожний R5 незалежно представляє собою Η -гетероарилу; або С1-10алкіл; -гетероциклілу; R6 представляє собою зв'язок, алкіл або алке-СО-арилу; та ніл, що можуть перериватися однією або більше -СО-гетероарилу; О- групами; кожний R3 представляє собой =O або =S; R7 представляє собою Η або С1-10алкіл, що кожний R4 незалежно представляє собою алможуть перериватися гетероатомом, або R7 може кіл або алкеніл, які можуть перериватися однією об'єднуватися з R5 з утворенням кільця; або більше -О- групами; R8 представляє собою Η або С1-10алкіл або кожний R5 незалежно представляє собою Η арилалкіл; або R4 та R8 можуть об'єднуватися раабо С1-10алкіл; зом з утворенням кільця; R6 представляє собою зв'язок, алкіл або алкеR9 представляє собою С1-10алкіл, який може ніл, що можуть перериватися однією або більше об'єднуватися з R8 з утворенням кільця; кожний Υ О- групами; незалежно представляє собою -О- або -S(O)0-2; R7 представляє собою Η або С1-10алкіл, що Ζ представляє собою зв'язок, -CO-, або –SO2-; можуть перериватися гетероатомом, або R7 може n є числом від 0 до 4; та об'єднуватися з R5 з утворенням кільця; кожний присутній R незалежно вибраний з R8 представляє собою Η або С1-10алкіл або групи, яка складається з С1-10алкілу, С1-10алкокси, арилалкіл; або R4 та R8 можуть об'єднуватися рагідрокси, галогену та трифторметилу; або їх фарзом з утворенням кільця; мацевтично прийнятна сіль. R9 представляє собою С1-10алкіл, який може Винахід також включає тетрагідроімідазохінооб'єднуватися з R8 з утворенням кільця; кожний Υ лінові сполуки, що несуть етерну групу та залишок незалежно представляє собою -О- або -S(O)0-2-; сечовини, які містять замісник в положенні 1. Такі Ζ представляє собою зв'язок, -CO-, або –SO2-; тетрагідроімідазохінолінові сполуки представлені n є числом від 0 до 4; та Формулою (ll): кожний присутній R незалежно вибраний з групи, яка складається з С1-10алкілу, С1-10алкокси, гідрокси, галогену та трифторметилу; або їх фармацевтично прийнятна сіль. (II) Одержання сполук Сполуки згідно з винаходом можна одержувати згідно з реакційною схемою l, де R, R2, R3, R4, R5, R8. X та n такі, як визначено вище, ВОС є де: X представляє собою –CHR5-, -CHR5-алкілтрет-бутоксикарбонілом та R11 є Z-R6-алкілом, Z, або –СHR5-алкеніл-; R6-алкенілом, Z-R6-арилом, Z-R6-гетероарилом, ZR1 вибраний з групи, яка складається з: R6-гетероциклілом або R11 є R7, причому R6, R7 та -R4-NR8-CR3-NR5-Z-R6-алкілу; Ζ є такими, як визначено вище. -R4-NR8-CR3-NR5-Z-R6-алкенілу; На етапі (1) реакційної схеми I аміногрупа амі-R4-NR8-CR3-NR5-Z-R6-арилу; носпирту Формули X захищена за допомогою трет-R4-NR8-CR3-NR5-Z-R6-гетероарилу; бутоксикарбонільної групи. Розчин аміноспирту в -R4-NR8-CR3-NR5-Z-R6-гетероциклілу; тетрагідрофурані обробляють за допомогою ди-R4-NR8-CR3-NR5-R7; трет-бутилдикарбонату в присутності основи, такої -R4-NR8-CR3-NR9-Z-R6-алкілу; як гідроксид натрію. Багато аміноспиртів Формули -R4-NR8-CR3-NR9-Z-R6-алкенілу; X є комерційно доступними; інші можуть бути при-R4-NR8-CR3-NR9-Z-R6-арилу; готовлені при використанні відомих способів син-R4-NR8-CR3-NR9-Z-R6-гетероарилу; та тезу. -R4-NR8-CR3-NR9-Z-R6-гетероциклілу; На етапі (2) реакційної схеми і захищений аміR2 вибраний з групи, яка складається з: носпирт Формули XI перетворюють на йодид Фор- водню; мули ХII. Йод додають до розчину трифенілфос- алкілу; фіну та імідазолу в дихлорметані; потім додають - алкенілу; розчин захищеного аміноспирту Формули XI в ди- арилу; хлорметані. Реакцію проводять при кімнатній тем- гетероарилу; пературі. - гетероциклілу; На етапі (3) реакційної схеми I спирт 1Н- алкіл-Y-алкілу; імідазо[4,5-с]хінолін-1-іловий спирт Формули ХIII - алкіл-Y-алкенілу; алкілують за допомогою йодиду Формули ΧII для - алкіл-Y-арил; та одержання 1Н-імідазо[4,5-с]хінолін-1-ілового етеру - алкілу або алкенілу, заміщеного одним або Формули XIV. Спирт Формули ХIII реагує з гідрибільше замісниками, вибраними з групи, яка складом натрію у прийнятному розчиннику, такому, як дається з: 9 74852 10 Ν,Ν'-диметилформамід для утворення алкоксиду. гувати з тіоізоціанатом формули R12-N=C=S, ациЙодид додають до розчину алкоксиду при кімнатлізоціанатом формули R12-C(O)-N=C=O, сульфоніл ній температурі. Після того, як додання закінчують, ізоціанатом формули R12-S(O2)-N=C=O або карбапроводять перемішування реакційної суміші при моїлхлоридом формули R13-N-C(O)Cl, де R13 є R12 підвищеній температурі (~100°С). Відомо багато або R7. Продукт або його фармацевтично прийнясполук Формули ХIII, [див., наприклад, Gerster, тна сіль можуть бути ізольовані при використанні Патент США №4,689,338]; інші можуть бути легко традиційних способів. приготовлені при використанні відомих способів Реакційна схема І синтезу, [див., наприклад, Gerster, Патент США №5,605,899 та Патент США №5,175,296]. На етапі (4) реакційної схеми I 1Н-імідазо[4,5с]хінолін-1-іловий етер Формули XIV окиснюють для забезпечення 1Н-імідазо[4,5-с]хінолін-5Nоксиду Формули XV, використовуючи традиційний окиснювальний агент, здатний до утворення Nоксидів. Бажано, коли сполуку Формули XIV у хлороформі окиснюють, використовуючи 3хлорпероксибензойну кислоту при кімнатній температурі. На етапі (5) реакційної схеми I 1Н-імідазо[4,5с]хінолін-5N-оксид формули XV амінують для одержання 1Н-імідазо[4,5-с]хінолін-4-аміну Формули XVI. Етап (5) включає (і) реакцію сполуки Формули XV з ацилюючим агентом, і потім (іі) реакцію продукту з амінуючим агентом. Частина (і) етапу (5) включає реакцію N-оксиду Формули XV з ацилюючим агентом. Прийнятні ацилюючі агенти включають алкіл- або арилсульфоніл хлориди (наприклад, бензолсульфоніл хлорид, метансульфоніл хлорид, п-толуолсульфоніл хлорид). Арилсульфоніл хлориди є бажаними. Пара-толуолсульфоніл Сполуки згідно з винаходом можуть бути одержані згідно з реакційною схемою ll, де R, R2, R3, хлорид є найбільш бажаним. Частина (іі) етапу (5) включає реакцію продукту частини (і) з надлишком R4, R5, R8, R11, X та n є такими, як визначено вище, а ВОС представляє собою трет-бутоксикарбоніл. амінуюючого агента. Прийнятні амінуючі агенти включають аміак (наприклад, у формі гідроксиду На етапі (1) реакційної схеми ll аміногрупу аміамонію) та солі амонію (наприклад, карбонат амоноспирту Формули XlX захищають за допомогою нію, бікарбонат амонію, фосфат амонію) Гідроксид трет-бутоксикарбонільної групи. Розчин аміноспиамонію є бажаним. Реакцію переважно виконують рту в тетрагідрофурані обробляють дитретшляхом розчинення N-оксиду Формули XV в інертбутилдикарбонатом у присутності основи, такої, як ному розчиннику, такому, як дихлорметан або 1,2гідроксид натрію. дихлоретан при нагріванні у разі необхідності, при Багато спиртів Формули XlX є комерційно досдоданні амінуючого агента до розчину, при повільтупними; інші можуть бути одержані при викорисному доданні ацилюючого агента. Необов'язково танні відомих способів синтезу. реакцію можна бути проводити у герметичній реаНа етапі (2) реакційної схеми ll захищений амікційній колбі для підвищеного тиску при підвищеноспирт Формули XX перетворюють до метансуній температурі (85-100°С). льфонату Формули ΧΧl. Розчин сполуки XX у приНа етапі (6) реакційної схеми ! захисну групу йнятному розчиннику, такому, як дихлорметан, видаляють шляхом гідролізу в кислих умовах для обробляють хлоридом метансульфонілу у присутності основи, такої, як триетиламін. Реакцію можна одержання 1Н-імідазо[4,5-с]хінолін-4-аміну Формули XVll. Бажано, коли сполуку Формули XVl обробпроводити при зниженій температурі (0°С). На етапі (3а) реакційної схеми ll метансульфоляють хлористоводневою кислотою/етанолом при кімнатній температурі або при слабкому нагрінат Формули XXl перетворюють до азиду Формули ΧΧll. Азид натрію додають до розчину сполуки Фованні. На етапі (7) реакційної схеми l 1Н-імідазо[4,5рмули XXl у прийнятному розчиннику, такому, як Ν,Ν-диметилформамід. Реакцію можна проводити с]хінолін-4-амін Формули XVll перетворюють на сечовину або тіосечовин Формули XVlll при викопри підвищеній температурі (80-100°С). На етапі (3b) реакційної схеми ll сполуку Форристанні традиційних способів синтезу. Наприклад, сполука Формули XVll може реагувати з ізомули XXll алкілують за допомогою галогеніду формули Hal-R8 для забезпечення сполуки Формули ціанатом формули R12-N=C=O, де R12 є –R6алкілом, -R6-алкенілом, -R6-арилом, -R6ΧΧlll. Для сполук, в яких R8 є воднем, цей етап опускають. Сполука Формули ХХll реагує з гідригетероарилом або –R6-гетероциклілом. Реакцію можна виконувати шляхом додання розчину ізоцідом натрію у прийнятному розчиннику, такому, як Ν,Ν-диметилформамід або тетрагідрофуран, для анату в прийнятному розчиннику, такому, як дихлорматен або 1-метил-2-піролідинон до розчину утворення аніону і потім поєднують з галогенідом. Реакцію можна проводити при кімнатній темпесполуки Формули XVll при кімнатній температурі. Альтернативно, сполука Формули XVll може реаратурі. 11 74852 12 На етапі (4) реакційної схеми ll азид Формули Реакційна схема ІІ ΧΧll або ΧΧlll відновлюють для одержання аміну Формули XXlV. Бажано, коли відновлення проводять при використанні традиційних гетерогенних каталізаторів гідрування, таких, як паладій на вуглеці. Реакцію можна традиційно проводити в апараті Парра у прийнятному розчиннику, такому, як метанол або ізопропанол. На етапі (5) реакційної схеми ll 4-хлоро-3нітрохінолін Формули XXV реагує з аміном Формули XXlV для одержання 3-нітрохіноліну Формули XXVl. Реакцію можна проводити шляхом додання аміну Формули XXlV до розчину сполуки Формули XXV у прийнятному розчиннику, такому як дихлорметан у присутності основи, такої, як триетиламін. Багато хінолінів Формули XXV є відомими сполуками або можуть бути одержані при використанні відомих способів синтезу, [див., наприклад, Gerster, Патент США №4,689,338] та посилання, які там цитуються. На етапі (6) реакційної схеми ll 3-нітрохінолін Формули XXVl відновлюють для одержання 3амінохіноліну Формули XXVll відновлюють для забезпечення 3-амінохіноліну Формули XXVll. Бажано, коли відновлення проводять при використанні традиційного гетерогенного каталізатора гідрування, такого, як платина на вуглеці. Реакцію можна традиційно проводити в апараті Парра у прийнятному розчиннику, такому, як толуол. На етапі (7) реакційної схеми ll сполука Формули XXVll реагує з карбоновою кислотою або її еквівалентом для одержання 1Н-імідазо[4,5с]хіноліну Формули XlV. Прийнятні еквіваленти карбонової кислоти включають ортоестери та 1,1діалкоксіалкіл алканоати. Карбонову кислоту або еквіваленти вибирають так, щоб вони забезпечуСполуки за винаходом можуть бути одержані вали бажаний R2 замісник у сполуці Формули XlV. згідно з реакційною схемою lll, де R, R2, Я3, R4, R5, Наприклад, триетилортоформат буде забезпечуR8, R11, X та n є такими, як визначено вище. вати сполуку, де R2 представляє собою водень, і На етапі (1) реакційної схеми lll 1Н-імідазо[4,5триетилортовалерат буде забезпечувати сполуку, с]хінолін-4-амін Формули XVll відновлюють для де R2 представляє собою бутил. Реакцію провоодержання 6,7,8,9-тетрагідро-1Н-імідазо[4,5дять при відсутності розчинника або в інертному с]хінолін-4-аміну Формули ΧΧVIII. Бажано, коли розчиннику, такому, як толуол. Реакція проходить відновлення проводять шляхом суспендування при достатньому нагріванні для відгону будь-якого або розчинення сполуки Формули XVll у трифтоспирту або води, що утворюються як побічний роцтовій кислоти при доданні каталітичної кількоспродукт реакції. Необов'язково можна включати ті оксиду платини (lV), та потім гідруванні. Реакцію каталітичну кількість гідрохлориду піридину. можна легко проводити в апараті Парра. Альтернативно, етап (7) можна проводити за Етап (2) виконують так само, як і етап (7) реадопомогою (і) реакції сполуки Формули XXVll з кційної схеми і для одержання 6,7,8,9-тетрагідроацилгалідом формули R2C(O)Cl і потім (іі) цикліза1Н-імідазо[4,5-с]хінолін-4-аміну Формули ХХlХ. ції. У частині (і) додають ацилгалід до розчину Продукт або фармацевтично прийнятна сіль мосполуки Формули XXVll в інертному розчиннику, жуть бути ізольовані при використанні традиційних такому, як ацетонітрил або дихлорметан. Реакцію способів. можна проводити при кімнатній температурі або Реакційна схема ІІl при зниженій температурі. У частині (іі) продукт частини (і) підігрівають в спиртовому розчиннику у присутності основи. Бажано, коли продукт частини (і) кип'ятять зі зворотним холодильником в етанолі у присутності надлишку триетиламіну або підігрівають з метанолом, насиченим аміаком. Етапи (8), (9), (10) та (11) виконують тим самим чином, що й етапи (4), (5). (6) та (7) реакційної схеми l. 13 74852 14 Сполуки згідно з винаходом можна також одеФормули XXXV). ржувати згідно з реакційною схемою lll, де R, R1, На етапі (4) реакційної схеми V 1Н-імідазо[4,5R2, X та n є такими, як визначено вище. с]хінолін-5Н-оксид Формули XXXVl амінують, викоНа етапі (1) реакційної схеми lV 4-хлоро-3ристовуючи спосіб етапу (5) реакційної схеми ί для нітрохінолін Формули XXV реагує з аміном формуодержання 1Н-імідазо[4,5-с]хінолін-4-аміну Формули R1-O-X-NH2 для одержання 3-нітрохінолін-4ли XVlll. Продукт або його фармацевтично прийняаміну Формули XXX. Реакцію проводять при дотна сіль можуть бути ізольовані традиційними споданні аміну до розчину сполуки Формули XXV у собами. прийнятному розчиннику, такому, як хлороформ Реакційна схема V або дихлорметан, та при необов'язковому нагріванні. Багато хінолінів Формули XXV є відомими сполуками, [див., наприклад, Gerster, Патент США 4,689,338] та посилання, які там цитуються. На етапі (2) реакційної схеми lV 3-нітрохінолін4-амін Формули XXX відновлюють при використанні способу етапу (6) реакційної схеми ll для одержання хінолін-3,4-діаміну Формули ХХХН. На етапі (3) реакційної схеми lV хінолін-3,4діамін Формули XXXl циклізують при використанні способу етапу (7) реакційної схеми ll для одержання 1Н-імідазо[4,5-с]хіноліну Формули ΧΧΧll. На етапі (4) реакційної схеми lV 1Н-імідазо[4,5с]хінолін Формули ΧΧΧll окиснюють, використовуючи спосіб етапу (4) реакційної схеми l для одержання 1Н-імідазо[4,5-с]хінолін-5N-оксиду Формули ΧΧΧlll. На етапі (5) реакційної схеми lV 1Н-імідазо[4,5с]хінолін-5N-оксид Формули ΧΧΧlll амінують, використовуючи спосіб етапу (5) реакційної схеми l для одержання 1Н-імідазо[4,5-с]хінолін-4-аміну Формули l. Продукт або його фармацевтично прийнятна Винахід також забезпечує нові сполуки, кориссіль можуть бути ізольовані при використанні трані як проміжні сполуки при синтезі сполук Формул диційних способів. (l) та (ll). Ці проміжні сполуки мають структурні ФоРеакційна схема ІV рмули (lll) та (lV), що описані більш детально нижче. Один клас проміжних сполук має формулу (lll): (III) Сполуки згідно з винаходом можуть бути одержані згідно з реакційною схемою V, де R, R2, R3, R4, R5, R8, R11, X та n є такими, як визначено вище. На етапі (1) реакційної схеми V групу ВОС видаляють із сполуки Формули XlV, використовуючи спосіб етапу (6) реакційної схеми l для одержання 1Н-імідазо[4,5-с]хіноліну Формули XXXlV. На етапі (2) реакційної схеми V 1Н-імідазо[4,5с]хінолін Формули XXXlV перетворюють до сечовини або тіосечовини Формули XXXV, використовуючи спосіб етапу (7) реакційної схеми l. На етапі (3) реакційної схеми V 1Н-імідазо[4,5с]хінолін Формули XXXV окиснюють, використовуючи спосіб етапу (4) реакційної схеми t для одержання 1Н-імідазо[4,5-с]хінолін-5№оксиду де: X представляє собою –CHR5-, -CHR5-алкіл, або –CHR5-алкеніл-; R1 вибраний з групи, яка складається з: -R4-NR8-CR3-NR5-Z-R6-алкілу; -R4-NR8-CR3-NR5-Z-R6-алкенілу; -R4-NR8-CR3-NR5-Z-R6-арилу; -R4-NR8-CR3-NR5-Z-R6-гетероарилу; -R4-NR8-CR3-NR5-Z-R6-гетероциклілу; -R4-NR8-CR3-NR5-R7; -R4-NR8-CR3-NR9-Z-R6-алкілу; -R4-NR8-CR3-NR9-Z-R6-алкенілу; -R4-NR8-CR3-NR9-Z-R6-арилу; -R4-NR8-CR3-NR9-Z-R6-гетероарилу; та -R4-NR8-CR3-NR9-Z-R6-гетероциклілу; R2 вибраний з групи, яка складається з: - водню; - алкілу; - алкенілу; - арилу; - гетероарилу; - гетероциклілу; - алкіл-Y-алкілу; - алкіл-Y-алкенілу; - алкіл-Y-арил; та - алкілу або алкенілу, заміщеного одним або 15 74852 16 більше замісниками, вибраними з групи, яка склаR6 представляє собою зв'язок, алкіл або алкедається з: ніл, що можуть перериватися однією або більше -ОН; О- групами; -галогену; R7 представляє собою Η або С1-10алкіл, що -N(R5)2; можуть перериватися гетероатомом, або R7 може -CO-N(R5)2; об'єднуватися з R5 з утворенням кільця; -СО-С1-10алкілу; R8 представляє собою Η або С1-10алкіл або -СО-О-С1-10алкілу; арилалкіл; або R4 та R8 можуть об'єднуватися ра-N3; зом з утворенням кільця; -арилу; R9 представляє собою С1-10алкіл, який може -гетероарилу; об'єднуватися з R8 з утворенням кільця; -гетероциклілу; n є числом від 0 до 4; та -СО-арилу; та кожний присутній R незалежно вибраний з -СО-гетероарилу; групи, яка складається з С1-10алкілу, С1-10алкокси, кожний R3 представляє собой =O або =S; галогену та трифторметилу; кожний R4 незалежно представляє собою алабо їх фармацевтично прийнятна сіль. кіл або алкеніл, які можуть перериватися однією Як такі, що використовуються у контексті даної або більше -О- групами; заявки, терміни “алкіл”, “алкені” та префікс „алк-” кожний R5 незалежно представляє собою Η включають як нерозгалужений ланцюг, так і розгаабо С1-10алкіл; лужені ланцюгові групи або циклічні групи, наприR6 представляє собою зв'язок, алкіл або алкеклад, циклоалкіл та циклоалкеніл. Якщо не вказаніл, що можуть перериватися однією або більше но інше, ці групи містять від 1 до 20 атомів О- групами; вуглецю, з алкенільними групами, що містять від 2 R7 представляє собою Η або С1-10алкіл, що до 20 атомів вуглецю. Переважні групи мають заможуть перериватися гетероатомом, або R7 може галом до 10 атомів вуглецю. Циклічні групи можуть об'єднуватися з R5 з утворенням кільця; бути моноциклічними або поліциклічними і переR8 представляє собою Η або С1-10алкіл або важно мають від 3 до 10 кільцевих атомів вуглецю. арилалкіл; або R4 та R8 можуть об'єднуватися раТипові циклічні групи включають циклопропіл, цикзом з утворенням кільця; лопропілметил, циклопентил, циклогексил та адаR9 представляє собою С1-10алкіл, який може мантил. об'єднуватися з R8 з утворенням кільця; кожний Υ Крім того, алкільні та алкенільні частини -Хнезалежно представляє собою -О- або -S(O)0-2-; груп можуть бути незаміщеними або заміщеними Ζ представляє собою зв'язок, -CO-, або –SO2-; одним або більше замісниками, при цьому замісn є числом від 0 до 4; та ники вибрані з груп, що містять алікіл, алкеніл, кожний присутній R незалежно вибраний з арил, гетероарил, гетероцикліл, арилалкіл, гетегрупи, яка складається з С1-10алкілу, С1-10алкокси, роарилалкіл та гетероцикіл алкіл. гідрокси, галогену та трифторметилу; Термін “гало алкіл” включає групи, які є заміабо їх фармацевтично прийнятна сіль. щеними одним або більше атомами галогену, Інший клас проміжних сполук представляє совключаючи перфторовані групи. Це також дійсно бою імідазохінолін-N-оксидні сполуки Фордля груп, які включають префікс “гало-”. Прикламули (lV): дами прийнятних галоалкільних груп є хлорметил, трифторметил, тощо. Термін “арил”, як такий, що використовується у даній заявці, включає карбоциклічні ароматичні (IV) кільця або кільцеві системи. Приклади арильних груп включають феніл, нафтил, біфеніл, флуореніл та інденіл. Термін “гетероарил” включає ароде: X представляє собою –CHR5-, -CHR5матичні кільця або кільцеві системи, що містять алкілен-, або -CHR5-алкенілен-; принаймні одне кільце гетероатомів (наприклад, R1 вибраний з групи, яка складається з: О, S, N). Прийнятні гетероарильні групи включа-R4-NR8-CR3-NR5-Z-R6-алкілу; ють фурил, тієніл, піридил, хінолініл, ізохінолініл, -R4-NR8-CR3-NR5-Z-R6-алкенілу; індоліл, ізоіндоліл, триазоліл, піроліл, тетразоліл, -R4-NR8-CR3-NR5-Z-R6-арилу; імідазоліл, піразоліл, оксазоліл, тіазоліл, бензофу-R4-NR8-CR3-NR5-Z-R6-гетероарилу; раніл, бензотіофеніл, карбазоліл, бензоксазоліл, -R4-NR8-CR3-NR5-Z-R6-гетероциклілу; піримідиніл, бензімідазоліл, хіноксалініл, бензотіа-R4-NR8-CR3-NR5-R7; золіл, нафтиридиніл, ізоксазоліл, ізотіазоліл, пури-R4-NR8-CR3-NR9-Z-R6-алкілу; ніл, хіназолініл і т.п. -R4-NR8-CR3-NR9-Z-R6-алкенілу; “Гетероцикліл” включає неароматичні кільця -R4-NR8-CR3-NR9-Z-R6-арилу; або кільцеві системи, що містять, принаймні, один -R4-NR8-CR3-NR9-Z-R6-гетероарилу; та кільцевий гетероатом (наприклад, О, S, N) та -R4-NR8-CR3-NR9-Z-R6-гетероциклілу; включають усі повністю заміщені або частково кожний R4 незалежно представляє собою алзаміщені похідні вказаних вище гетероарильних кіл або алкеніл, які можуть перериватися однією груп. Типові гетероциклічні групи включають піроабо більше -О- групами; лідиніл, тетрагідрофураніл, морфолініл, тіоморкожний R5 незалежно представляє собою Η фолініл, піперидиніл, піперазиніл, тіазолідиніл, або С1-10алкіл; імідазолідиніл, ізотіазолідиніл, тощо. Арильні, ге 17 74852 18 тероарильні та гетероциклільні групи можуть бути рмацевтично прийнятним носієм. незаміщеними або заміщеними одним або більше Термін “терапевтично ефективна кількість” замісниками, незалежно вибраними з групи, яка означає кількість сполуки, достатню для індукції складається з алкілу, алкокси, алкілтіо, галоалкілу, терапевтичного ефекту, такого, як індукція цитокігалоалкокси, галоалкілтіо, галогену, нітро, гідрокнів, протипухлинна активність та/або антивірусна си, меркапто, ціано, карбокси, формілу, арилу, активність. Незважаючи на те, що точна кількість арилокси, арилтіо, арилалкокси, арилалкілтіо, геактивної сполуки, що використовується у фарматероарилу, гетероарилокси, гетероарилтіо, гетецевтичній композиції згідно з винаходом буде вароарилалкокси, гетероарилалкілтіо, аміно, алкіларіювати в залежності від факторів, що відомі спеміно, діалкіламіно, гетероциклілу, ціалісту у даній галузі, такі, як фізична та хімічна гетероциклоалкілу, алкілкарбонілу, алкенілкарбоприрода сполуки, природа носія, прописаний ренілу, алкоксикарбонілу, галоалкілкарбонілу, галоажим дозування, передбачається, що композиції лкоксикарбонілу, алкілтіокарбонілу, арилкарбонізгідно з винаходом будуть містити достатню кільлу, гетероарилкарбонілу, арилоксикарбонілу, кість активного інгредієнту для забезпечення дози гетероарилоксикарбонілу, арилтіокарбонілу, гетевід приблизно 100нг/кг до приблизно 50мг/кг, пероарилтіокарбонілу, алканоїлокси, алканоїлтіо, реважно від приблизно 10мкг/кг до приблизно алканоїламіно, арилкарбонілокси, арилкарбоніл5мг/кг сполуки для пацієнта. Можуть використовутіо, алкіламіносульфонілу, алкілсульфонілу, арилватися будь-які традиційні дозовані форми, такі, як сульфонілу, гетероарилсульфонілу, арилдіазинілу, таблетки, продовгуваті таблетки, парентеральні алкілсульфоніламіно, арилсульфоніламіно, арикомпозиції, сиропи, креми, мазі, аерозольні комполалкілсульфоніламіно, алкілкарбоніламіно, алкезиції, трансдермальні пластирі, трансмукозальні нілкарбоніламіно, арилкарбоніламіно, арилалкілпластирі, тощо. карбоніламіно, гетероарилкарбоніламіно, Сполуки згідно з винаходом можуть вводитися гетероарилкарбоніламіно, алкілсульфоніламіно, як єдиний терапевтичний агент в режимі лікуваналкенілсульфоніламіно, арилсульфоніламіно, ня, або сполуки згідно з винаходом можуть вводиарилалкілсульфоніламіно, гетероарилсульфонітися у комбінації ще з одним іншим або з іншими ламіно, гетероарилалкілсульфоніламіно, алкіламіактивними агентами, включаючи додаткові модинокарбоніламіно, алкеніламінокарбоніламіно, арифікатори імунної відповіді, антивірусні агенти, анламінокарбоніламіно, тибіотики, тощо. арилалкіламінокарбоніламіно, гетероариламінокаБуло показано, що сполуки згідно з винаходом рбоніламіно, гетероарилалкіламінокарбоніламіно індукують продукцію певних цитокінів в експерита, у випадку гетероциклілу, оксо. Якщо будь-які ментах, проведених згідно з набором тестів, що інші групи ідентифіковані, як такі, що “заміщені” представлені нижче. Ці результати показують, що або “необов'язково заміщені”, то ці групи можуть сполуки є корисними як модифікатори імунної відтакож бути заміщені одним або більше вказаними повіді, які модулюють імунну відповідь багатьма вище замісниками. різними шляхами, і це робить їх корисними при Певні замісники є особливо бажаними. Наприлікуванні цілого ряду розладів. клад, бажані R1 групи включають -R4-NR8-CR3-NR5Цитокіни, продукція яких може індукуватися Z-R6-алкіл та -R4-NR8-CR3-NR5-Z-R6-арил, де алкішляхом введення сполук згідно з винаходом, в льна та арильна групи можуть бути незаміщені або загальному випадку включають інтерферон-α (lFNзаміщені; R4 бажано є етиленом або n-бутиленом α) та/або фактор некрозу пухлин α (TNF-α), а таабо R4 та R8 зв'язані, утворюючи кільце. Бажано, кож деякі інтерлейкіни (lL). Цитокіни, біосинтез коли жодний з R замісників не присутній (тобто, n яких може індукуватися сполуками за винаходом, дорівнює 0). Переважні R2 групи включають вовключають lFN-α, TNF-α, lL-1, lL-6, lL-10 та lL-12 та день, алкільні групи, що мають від 1 до 4 атомів різноманітність інших цитокінів. Серед інших впливуглецю (тобто, метил, етил, пропіл, ізопропіл, нвів ці та інші цитокіни можуть інгібувати вірусну бутил, втор-бутил, ізобутил та циклопропілметил), продукцію та ріст пухлинних клітин, що робить ці метоксіетил та етоксиметил. Для заміщених груп, сполуки корисними при лікуванні вірусних захвотаких, як заміщені алкільні або заміщені арильні рювань та пухлин. Згідно з цим винахід забезпечує групи, бажані замісники включають галоген, нітспосіб індукції біосинтезу цитокінів у тварин, що рил, метокси, метилтіо, трифторметил та трифтопередбачає введення ефективної кількості сполурметокси. Один або більше цих бажаних замісники або композиції за винаходом тварині. ків, в разі присутності, можуть бути присутніми у Окремі сполуки за винаходом були виявлені як сполуках згідно з винаходом у будь-якій комбінації. такі, що переважно індукують експресію lFN-α в Винахід включає сполуки, описані у даній популяції гематопоетичних клітин, таких, як РВМС заявці у формі будь-яких прийнятних солей, вклю(мононуклеарні клітини периферійної крові), що чаючи ізомери (наприклад, діастереомери та енамістять pDC2 клітини (попередники дендритних нтіомери), солі, сольвати, поліморфні форми, токлітин типу 2) без супутньої продукції значних рівщо. Зокрема, якщо сполуки є оптично активними, нів запальних цитокінів. винахід конкретно включає кожний енантіомерів У доповнення до здатності індукувати продуксполуки, а також рацемічні суміші енантіомерів. цію цитокінів сполуки згідно з винаходом викликаФармацевтичні композиції та біологічна активють інші аспекти природженої імунної відповіді. ність Наприклад, може бути стимульована активність Фармацевтичні композиції згідно з винаходом природних клітин-кілерів, ефект, який може винимістять терапевтично ефективну кількість сполуки кати завдяки індукції цитокінів. Сполуки можуть за винаходом, як описано вище, у комбінації з фатакож активувати макрофаги, які, у свою чергу, 19 74852 20 стимулюють секрецію оксиду азоту та продукцію ки можуть бути корисними при лікуванні опортунісдодаткових цитокінів. Крім того, сполуки можуть тичних інфекцій та пухлин, що виникають після викликати проліферацію та диференціацію Впригнічення імунітету, опосередкованого клітиналімфоцитів. ми, наприклад, у пацієнтів, яким вживляють трансСполуки за винаходом також можуть мати плантат, ракових хворих та пацієнтів, хворих на вплив на набуту імунну відповідь. Наприклад, неΒІЧ. зважаючи на те, що не передбачається існування Кількістю сполуки, ефективною для індукції бібудь-якого безпосереднього впливу на Т-клітини осинтезу цитокінів, є кількість, що достатня для або безпосередню індукцію цитокінів Т-клітин, того, щоб викликати продукцію певної кількості продукція цитокіну lFN-γ Т-хелперами типу 1 (Th1) одного або більше цитокінів, таких, як lFN-α, TNFіндукується опосередковано і продукція цитокінів α, lL-1, lL-6, lL-10 та lL-12, одним або більше типаlL-4, lL-5 та lL-13 інгібується при введенні сполук ми клітин, таких як моноцити, макрофаги, дендризгідно з винаходом. Ця активність означає, що тні клітини та B-клітини, що збільшується понад сполуки є корисними при лікуванні захворювань, вихідного рівня цих цитокінів. Точна кількість буде при яких підвищення Тh1 відповіді та/або зниженваріювати в залежності від факторів, що відомі у ня Тh2 відповіді є бажаним. З огляду на здатність галузі техніки, але очікується, що це буде доза від сполук за винаходом інгібувати Th2 імунну відпоприблизно 100нг/кг до приблизно 50мг/кг, перевавідь, сполуки, як очікується, будуть корисними при жно від приблизно 10мкг/кг до приблизно 5мг/кг. лікуванні атопічних захворювань, наприклад, атоКількість, ефективна для лікування або інгібування пічного дерматиту, астми, алергії, алергічного ривірусної інфекції, є кількістю, що буде спричинюваніту; системної еритематозної вовчанки; як вакти зменшення одного або більше проявів вірусної цинний ад'ювант для імунітету, опосередкованого інфекції, таких як вірусні пошкодження, вірусне клітинами; та можливі як засіб для лікування ренавантаження, швидкість продукції вірусу та смерцидивуючих грибкових захворювань та хламідій. тність у порівнянні з необробленими контрольними Модифікуючі впливи на імунну відповідь цих тваринами. Точна кількість буде варіювати в засполук роблять їх корисними при лікуванні широкої лежності від факторів, що відомі у галузі техніки, різноманітності станів. Завдяки їх здатності індукуале очікується, що доза складає від приблизно вати продукцію цитокінів таких, як lFN-α та/або 100нг/кг до приблизно 50мг/кг, бажано від приблиTNF-α, сполуки є особливо корисними при лікузно 10мкг/кг до приблизно 5мг/кг. Кількість сполуванні вірусних захворювань та раку. Ця імуномоки, ефективна для лікування неопластичного стадулююча активність передбачає, що сполуки згідну, є кількістю, що буде спричинювати зменшення но з винаходом є корисними при лікуванні розміру пухлини або кількості пухлинних локусів. З захворювань таких, як, але не обмежуються, віруіншого боку точна кількість буде варіювати в залесні захворювання, включаючи генітальні бородавжності від факторів, що відомі у даній галузі, але ки, звичайні бородавки, підошвенні бородавки, очікується, що доза буде складати від приблизно гепатит В, гепатит С, вірус звичайного герпесу 100нг/кг до приблизно 50мг/кг, переважно від притипу 1 та типу 2, контагіозний молюск, віспа, особблизно 10мг/кг до приблизно 5мг/кг. ливо, натуральна віспа, ВІЧ, ЦМВ, вітряна віспа, Винахід також описується наступними приклариновірус, аденовірус, вірус грипу та парагрипу, дами, які призначені тільки для ілюстрації і не приінтраепітеліальні неоплазії, такі, як цервікальна значені для обмеження будь-яким чином. інтраепітеліальна неоплазія, папіломавірус людиУ прикладах, що наведені нижче деякі з сполук ни (HPV) та асоційовані неоплазії, грибкові захвоочищають при використанні напівпрепаративного рювання, наприклад, кандидоз, аспергільоз та ВЕРХ. Використовували автоматичну систему криптококовий менінгіт, неопластичні захворюваночистки Waters Fraction Lynx. Фракції напівпрепаня, наприклад, карцинома базальних клітин, лейративної ВЕРХ аналізували при використанні кемія ворсинчастих клітин, саркома Капоші, карциMicromass LC-TOFMS та прийнятні фракції поєднома ниркових клітин, карцинома лускатих клітин, нували та випарювали за допомогою центрифугумієлогенна лейкемія, множинна мієлома, мелановання для одержання трифторацетатної солі бама, лімфома не Ходжкіна, шкірна Т-клітинна лімжаної сполуки. Структури підтверджували за фома, та інші види раку, паразитарні захворювандопомогою 1Н ядерного магнітного резонансу. ня, наприклад, пневмоцистіс карнії, Колонка: Phenomenex Luna C18(2), 10 50мм, криптоспорідіоз, гістоплазмоз, токсоплазмоз, трирозмір частинок 5 мікрон, розмір пор 100А, швидпаносомна інфекція та лешманіоз, бактеріальні кість витікання: 25мл/хв.; градієнт елюювання від 5 інфекції, наприклад, туберкульоз та мікобактеріум до 65% В протягом 4 хвилин, потім від 65 до 95% авіум. Додаткові захворювання або стани, які моВ протягом 0,1хв., потім витримували при 95% В жуть лікуватися при використанні сполук за винапротягом 0,4хв., коли А=0,05% трифтороцтової ходом, включають актинічний кератоз, екзему, кислоти/вода та В=0,05% трифтороцтової кислоеозинофілію, есенціальну тромбоцитемію, прокати/ацетонітрил, збір фракцій проводять за допомозу, розсіяний склероз, синдром Оммена, дисковигою мас-селективного запуску. дну вовчанку, захворювання Бовена, бовеноїдний Приклад 1 папульоз, гніздну алопецію, інгібування утворення N-(2-{2-[4-aмiнo-2-(2-мeτoкcieтил)-1Hрубців після хірургічного втручання та інших типів імідaзo[4,5-c]xінoлiн-1-iл]eтoкcи}eτил)-N'шрамів після хірургічного втручання. Крім того, ці фенілсечовина сполуки можуть покращувати або стимулювати заживления ран, включаючи хронічні рани. Сполу 21 74852 22 (2-азидоетокси)етилкарбамату як світлокоричневої олії. Частина D Розчин трет-бутил-2-(2азидоетокси)етилкарбамату (47,0г, 0,204моль) у МеОН обробляли 4г 10% Pd на вуглеці та струшували під Н2 (3кг/см2) протягом 24 годин. Розчин потім фільтрували через целітову м'яку прокладку та концентрували для одержання 35,3г сирового трет-бутил-2-(2-аміноетокси)етилкарбамату як безбарвної рідини, що може використовуватися Частина А без подальшої очистки. Розчин 2-(2-аміноетокси)етанолу (29,0г, Частина Ε 0,276моль) в 180мл тетрагідрофурану (ТГФ), під Перемішаний розчин 4-хлоро-3-нітрохінолін N2 охолоджували до температури 0°С та обробля(31,4г, 0,151моль) в 500мл безводного СН2Cl2 під ли за допомогою 140мл 2N розчину NaOH. Розчин N2 обробляли триетиламіном (43мл, 0,308моль) та дитрет-бутил дикарбонату (60,2г, 0276моль) в трет-бутил-2-(2-аміноетокси)етилкарбаматом 180мл ТГФ потім додавали по краплях протягом 1 (0,151моль). Після перемішування протягом ночі години до розчину, який швидко перемішували. реакційну суміш промивали Н2О (2 300мл) та наРеакційну суміш потім нагрівали до кімнатної темсиченим розчином хлориду натрію (300мл). Оргаператури та перемішували протягом додаткових нічну частину висушували над Na2SO4 та концент18 годин. ТГФ потім видаляли під зниженим тисрували для одержання яскраво жовтої твердої ком, а гідророзчин, що залишився, доводили до речовини. Перекристалізація з етилацетаpH3 шляхом додання 150мл 1М розчину H2SO4. ту/гексану давала 43,6г трет-бутил-2-{2-[(3Гідрозчин екстрагували за допомогою етилацетату нітрохінолін-4-іл)аміно]етокси}етилкарбамату у (300мл, 100мл) і поєднані органічні шари промивавигляді яскраво жовтих кристалів. ли за допомогою Н2О (2X) та насиченого розчину Частина F хлориду натрію. Органічну частину висушували Розчин трет-бутил-2-{2-[(3-нітрохінолін-4над Na2SO4 та концентрували для одержання іл)аміно]етокси}етилкарбамату (7,52г, 20,0ммоль) трет-бутил 2-(2-гідроксіетокси)етилсарбамату як в толуолі оброблювали 1,5г 5% Pt на вуглеці та безбарвної олії (47,1г). струшували під Н2 (3кг/см2) протягом 24 годин. Частина В Розчин потім фільтрували через целітову м'яку Розчин трет-бутил-2-(2прокладку та концентрували для одержання 6,92г гідроксіетокси)етилсарбамату, що швидко перемісирового трет-бутил-2-{2-[(3-амінохінолін-4шується (47,1г, 0,230моль), в 1л безводного іл)аміно]етокси}етилкарбамату як жовтого сиропу. СН2Cl2 охолоджували до температури 0°С в атмоЧастина G сфері N2 та обробляли триетиламіном (48,0мл, Розчин трет-бутил-2-{2-[(3-амінохінолін-40,345моль). Метансульфоніл хлорид (19,6мл, іл)аміно]етокси}етилкарбамату (10,2г, 29,5ммоль) 0,253моль) потім додавали по краплях протягом в 250мл безводного СН2Cl2 охолоджували до 0°С 30 хвилин. Реакційну суміш потім нагрівали до та оброблювали триетиламіном (4,18мл, кімнатної температури та перемішували протягом 30,0ммол). Метоксипропіоніл хлорид (3,30мл, додаткових 22 годин. Реакцію зупиняли шляхом 30,3ммоль) потім додавали по краплях протягом 5 додання 500мл насиченого розчину NaНСО3 та хвилин. Реакційну суміш потім підігрівали до кіморганічний шар відокремлювали. Органічну фазу натної температури і перемішування продовжувапотім промивали за допомогою Н2О (3 500мл) та ли протягом 1 години. Реакційну суміш потім коннасиченим водним розчином хлориду натрію. Орцентрували під зниженим тиском для одержання ганічну частину висушували над Na2SO4 та концетвердої речовини оранжевого кольору. Цю речонтрували для одержання 2-{2-[(третвину розчиняли в 250мл EtOH і додавли 12,5мл бутоксикарботриетиламіну. Суміш потім підігрівали до темпераніл)аміно]етокси}етилметансульфонату як коричтури кипіння зі зворотним холодильником та переневої олії (63,5г). мішували під N2 протягом ночі. Реакційну суміш Частина С потім концентрували до осушення під зниженим Перемішаний розчин 2-{2-[(треттиском та обробляли 300мл Et2O. Суміш потім бутоксикарбофільтрували і фільтрат концентрували під зниженіл)аміно]етокси}етилметансульфонату (63,5г, ним тиском для одержання коричневої твердої 0,224моль) у 400мл Ν,Ν-диметилформаміду (ДМФ) речовини. Тверду речовину потім розчиняли у обробляли за допомогою NaN3 (16,1г, 0,247моль) 200мл гарячого МеОН і оброблювали за допомота реакційну суміш підігрівали до 90°С під N2 Чегою активованого деревного вугілля. Гарячий розрез 5 годин суміш охолоджували до кімнатної темчин потім фільтрували та концентрували для одеператури та обробляли 500мл холодної Н2О. Реаржання 11,1г трет-бутил 2-{2-[2-(2-метоксіетил)кційну суміш потім екстрагували за допомогою 1Н-імідазо[4,5-с]хінолін-1-іл]етокси}етилкарбамату Еt2О (3 300мл) Поєднані органічні екстракти прояк жовтого сиропу. мивали за допомогою Н2О (4 100мл) та насичеЧастина Η ним розчином хлориду натрію (2 100мл). ОрганічРозчин 2-{2-[2-(2-метоксіетил)-1Н-імідазо[4,5ну частину висушували над МgSО4 та с]хінолін-1-іл]етокси} етилкарбамату (10,22г, концентрували для одержання 52,0г трет-бутил 224,7ммоль) в 250мл СНCl3 оброблювали за допо 23 74852 24 могою 3-хлоропероксибензойної кислоти (МСРВА, концентрували під зниженим тиском для одержан77%, 9,12г, 40,8ммоль). Після перемішування проня твердої речовини жовтого кольору. Жовту тветягом 30 хвилин реакційну суміш потім промивали рду речовину розчиняли в мінімальній кількості СН2Сl2 та EtOAc додавали доти, поки розчин не за допомогою 1%-ного розчину Na2СО3 (2 75мл) помутнів. Суміш поміщали у морозильну камеру на та насиченим розчином хлориду натрію. Органічніч та отримували білі кристали. Кристали ізолюний шар висушували над Na2SO4 і концентрували вали шляхом фільтрації та висушували під вакуудля одержання 10,6г трет-бутил-2-{2-[2-(2мом для одержання 126мг N-(2-{2-[4-аміно-2-(2метоксіетил)-5-оксидо-1Н-імідазо[4,5-с]хінолін-1метоксіетил)-1Н-імідазо [4,5-с]хінолін-1іл]етокси}етилкарбамату як оранжевої піни, яку іл]етокси}етил)-N'-фенілсечовини. Точка плавленвикористовували без подальшої очистки. ня 171,0-174,0°С; Частина l MS 449 (М+Н)+; Розчин трет-бутил-2-{2-[2-(метоксіетил)-51 H ЯМР (300МГц, ДМСО-d6): δ 8,50 (с, 1Н); оксидо-1Н-імідазо[4,5-с]хінолін-18,05 (l, J=7,7Гц, 1Н); 7,62 (l, J=8,8Гц, 1Н); 7,44-7,18 іл]етокси}етилкарбамату (10,6г, 24, 6ммоль) в (м, 3Н), 7,27-7,18 (м, 3Н); 6,88 (т, J=7,3Гц, 1Н); 6,54 100мл 1,2-дихлоретану підігрівали до 60°С та об(с, 2Н); 6,12 (т, J=5,5Гц, 2Н); 4,76 (т, J=4,8Гц, 2Н); робляли за допомогою 10мл концентрованого роз3,88 (т, J=5,3Гц, 2Н); 3,81 (т, J=6,7Гц, 2Н); 3,40 (т, чину ΝΗ4ΟΗ. До швидко перемішаного розчину J=6,0Гц, 2Н); 3,28 (с, 3Н); 3,25-3,14 (м, 4Н); додавали твердий п-толуолсульфоніл хлорид 13 С (75МГц, ДМСО-d6): δ 155,5, 152,0, 144,9, (7,05г, 37,0ммоль) протягом періоду часу 10 хви140,8, 132,7, 129,0, 126,8, 126,5, 121,5, 121,4, лин. Реакційну суміш потім обробляли за допомо120,5, 117,9, 115,1, 70,5, 69,4, 58,4, 45,5, 27,6; гою додаткового 1мл концентрованого розчину Розраховано для C24H28N6O3·0,21 Н2О: %С ΝΗ4ΟΗ і потім герметично закупорювали у ємнос63,73, %Н, 6,33, %Ν, 18,58. тях високого тиску і нагрівання продовжували 2 Знайдено: %С, 63,33, %Н, 6,28, %Ν, 18,67. години. Реакційну суміш потім охолоджували та Приклад 2 обробляли 100мл СНCl3. Реакційну суміш потім N-(2-{2-[4-аміно-2-(2-метоксіетил)-6,7,8,9промивали за допомогою Н2О, 1%-ного розчину тетрагідро-1Н-імідазо[4,5-с]хінолін-1Na2СО3 (2Х) і насиченого розчину хлориду натрію. іл]етокси}етил)-N'-фенілсечовина Органічну частину висушували над Na2SO4 і концентрували для одержання 10,6г трет-бутил-2-{2[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5-с]хінолін1-іл]етокси}етилкарбамату як коричневої піни. Частина J Трет-бутил 2-{2-[4-аміно-2-(2-метоксіетил)-1Німідазо[4,5-с]хінолін-1-іл]етокси}етилкарбамат (10,6г, 24,6моль) обробляли за допомогою 75мл 2М НСІ в EtOH і суміш підігрівали до температури кипіння зі зворотним холодильником при перемішуванні. Через 1,5 годин реакційну суміш охолоЧастина А джували та фільтрували для одержання липкої 1-[2-(2-аміноетокси)етил]-2-(2-метоксіетил)-1Нречовини. Цю речовину промивали EtOH та Et2O імідазо [4,5-с]хінолін-4-амін (10,0г, 27,3ммоль) рота висушували під вакуумом для одержання гідрозчиняли в 50мл трифтороцтової кислоти та обробхлоридної солі як світло-коричневої твердої речоляли за допомогою PtO2 (1,0г). Реакційну суміш вини. Вільну основу одержували шляхом розчиструшували під Н2 (3кг/см2). Через 4 дні додавали нення гідрохлоридної солі в 50мл Н2О та обробкою додаткові 0,5г PtO2 та гідрування продовжували за допомогою 10%-ного розчину NaOH. Водну судодатково протягом 3 днів. Реакційну суміш потім спензію потім концентрували до осушення та зафільтрували через целіт та концентрували при лишок обробляли СНСl3. Одержані солі потім визниженому тиску для одержання коричневої олії. даляли шляхом фільтрації та фільтрат Олію потім розчиняли у 200мл Н2О, потім підлужуконцентрували для одержання 3,82г 1-[2-(2вали (рН-11) шляхом додання 10% розчину NaOH. аміноетокси)етил]-2-(2метоксіетил)-1Н-імідазо[4,5Сполуку потім екстрагували за допомогою СНСl3 с}хінолін-4-аміну як жовто-коричневого порошку. (5 75мл) і поєднані органічні шари висушували + MS 330(М+Н) ; над Na2SO4 та концентрували для одержання 1 H ЯМР (300МГц, ДМСО-d6): δ 8,10 (д, J=8,1Гц, 5,17г 1-[2-(2-аміноетокси)етил]-2-(2-метоксіетил)1Н); 7,66 (д, J=8,2Гц 1Н); 7,40 (м, 1Н), 7,25 (м, 1Н); 6,7,8,9-тетрагідро-1Н-імідазо [4,5-с]хінолін-4-аміну 6,88 (шс, 2Н); 4,78 (т, J=5,4Гц, 2Н); 3,89 (т, J=4,8Гц, як жовто-коричневої твердої речовини. 2Н); 3,84 (т, J=6,9Гц, 2Н); 3,54 (т, J=5,4Гц, 2Н); 3,31 MS 334 (М+Н)+; (с, 3Н); 3,23 (т, J=6,6Гц, 2Н); 2,88 (т, J=5,3Гц, 2Н). 1 H ЯМР (300МГц, CDCl3): δ 5,19 (с, 2Н); 4,49 (т, Частина K J=5,4Гц, 2H); 3,48 (т, J=6,6Гц, 2Н); 3,71 (т, J=5,4Гц, 1-[2-(2-аміноетокси)етил]-2-(2-метоксіетил)-1Н2H); 3,36 (т, J=5,2Гц, 2H); 3,51 (с, 3Н); 3,15 (т, імідазо[4,5-с}хінолін-4-амін (750мг, 2,28моль) розJ=56,6Гц, 2Н); 2,95 (м, 2Н); 2,82 (м, 2Н); 2,76 (т, чиняли в 30мл безводного СН2Cl2 та охолоджуваJ=5,1Гц, 2Н); 1,84 (м, 4Н); 1,47 (шс, 2Н). ли до 0°С під N2. Реакційну суміш потім оброблюЧастина В вали за допомогою феніл ізоціанату (247мкл, 1-[2-(2-аміноетокси)етил]-2-(2-метоксіетил)2,28ммоль) та Et2N (0,64мл, 4,56ммоль) та нагрі6,7,8,9-тетрагідро-1Н-імідазо [4,5-с]хінолін-4-амін вали повільно до кімнатної температури. Після (919мг, 2,76моля) розчиняли в 30мл безводного перемішування протягом 2 годин реакційну суміш 25 74852 26 СH2Сl2 та охолоджували до 0°С під N2. Реакційну азидоетокси)етил(метил)карбамату як жовтої рісуміш потім обробляли фенілізоціанатом (300мкл, дини. 2,76ммоль) і Et3N (0,77мл, 5,51ммоль) та повільно Частина В нагрівали до кімнатної температури. Після переРозчин трет-бутил-2-(2-азидоетокси) етил (мемішування протягом ночі реакційну суміш потім тил)карбамату (41,9г, 170ммоль) в 600мл МеОН гасили доданням насиченого розчину NaНСО3 обробляли 2,5г 10% Pd на вуглеці та струшували (30мл). Органічний шар відокремлювали та пропід Н2 (3кг/см2) протягом 24 годин. Розчин потім мивали за допомогою Н2О та розчину хлориду фільтрували через колонку з целітовою прокладнатрію, висушували над NagSO4 та концентрували кою та концентрували для одержання 37,2г сиропід зниженим тиском для одержання жовтої твервого трет-бутил-2-(2дої речовини. Тверду речовину обробляли Et2O аміноетокси)етил(метил)карбамату як світло(30мл) та декількома краплями МеОН. Тверду режовтої рідини. човину ізолювали фільтрацією та висушували під Частина С вакуумом для одержання 460мг N-(2-{2-[аміно-2-(2Перемішаний розчин 4-хлоро-3-нітрохіноліну метоксіетил)-6,7,8,9-тетрагідро-1Н-імідазо[4,5(32,3г, 155ммоль) в 400мл безводного СН2О2 під с]хінолін-1-іл]етокси}етил)-N'-фенілсечовини як N2 обробляли триетиламіном (43,1мл, 310ммоль) білого порошку. та трет-бутил-2-(2Температура плавлення 180-182°С; аміноетокси)етил(метил)карбаматом (37,2г, MS 453 (М+Н)+; 171ммоль). Після перемішування протягом ночі 1 Н ЯМР (300Мгц, ДМСО-d6) δ 8,51 (с, 1Н); 7,37 реакційну суміш потім промивали Н2О (2 300мл) (д, J=7,7Гц, 2Н); 7,19 (т, J=8,2Гц, 2Н); 6,86 (т, та насиченим розчином хлориду натрію (300мл). J=7,7Гц, 1Н); 6,11 (т, J=5,5Гц, 2Н); 5,70 (с, 2Н); 4,43 Органічну частину висушували над Na2SO4 та кон(т, J=5,1Гц, 2Н); 3,78-3,69 (м, 4Н); 3,39 (т, J=5,6Гц, центрували для одержання коричневої олії. При 2Н); 3,25 (с, 3Н); 3,19 (м, 2Н); 3,10 (т, J=6,8Гц, 2Н); використанні колонкової хроматографії (SiO2, 33% 2,91 (м, 2Н);1,72(м,4Н); етилацетат/гексан-67% етилацетат/гексан) одер13 С (75МГц), ДМСО-d6): δ 155,5, 151,3, 149,3, жували 46,7г трет-бутил-метил(2-{2-[(3140,8, 138,5, 129,0, 125,0, 121,4, 118,0, 105,6, 70,6, нітрохінолін-4-іл)аміно]етокси}етил)карбамату як 70,5, 70,4, 58,4, 44,6, 39,2, 32,7, 27,6, 23,8, 23,1, твердої речовини жовтого кольору. 23,0; Частина D Розраховане для C24H32N6O3: %С 63,70, %Н, Розчин трет-бутил-метил(2-{2-[(3-нітрохінолін7,13, %Ν, 18,57. 4-іл)аміно]етокси}етил)карбамату (6,56г, Знайдено: %С, 63,33, %Н, 7,16, %Ν, 18,66. 16,8ммоль) в 75мл толуолу оброблювали за доПриклад 3 помогою 0,5г 5% Pt на вуглеці та струшували під N-(2-{2-[4-аміно-2-(2-метоксіетил)-1НН2 (3кг/см2) протягом 24 годин. Розчин потім фільімідазо[4,5-с]хінолін-1-іл]етокси}етил)-N-метил-N'трували через целітову прокладку та концентруфенілсечовина вали для одержання 6,8г сирового трет-бутилметил(2-{2-[(3-амінохінолін-4іл)аміно]етокси}етил(метил))карбамату як оранжевого сиропу, який використовували далі без подальшої очистки. Частина Ε Розчин трет-бутил-метил(2-{2-[(3-амінохінолін4-іл)аміно]етокси}етил(метил))карбамату (6,05г, 16,8ммоль) у 200мл безводного СH2Сl2 охолоджували до 0°С та оброблювали триетиламіном (2,40мл, 17,2ммоль). Метоксипропіоніл хлорид Частина А (1,72мл, 17,2ммоль) потім додавали по краплях Гідрид натрію (60% масляна дисперсія, 9,1г, протягом 5 хвилин. Реакційну суміш потім підігрі228ммоль) поміщали у флакони з круглим дном та вали до кімнатної температури та перемішування промивали гексаном (3X) під N2. Висушений гідрид продовжували протягом 3 годин. Реакційну суміш натрію потім обробляли 800мл безводного ТГФ. потім концентрували під зниженим тиском для Розчин трет-бутил-2-(2одержання твердої речовини оранжевого кольору. азидоетокси)етилкарбамату) (41,9г, 182ммоль) в Цю речовину потім розчиняли у 200мл EtOH та 200мл ТГФ потім додавали до перемішаного роздодавали 7,2мл триетиламіну. Суміш підігрівали чину гідриду натрію протягом 40 хвилин. Після до температури кипіння зі зворотним холодильнитого, як додання було закінчене, реакційну суміш ком та перемішували під N2 протягом ночі. Реакперемішували проятгом додаткових 20 хвилин, ційну суміш потім концентрували до осушення під після чого додавали метилйодид (13,6мл, зниженим тиском та оброблювали 300мл Et2O. 218ммоль). Після перемішування протягом ночі Суміш потім фільтрували та фільтрат концентруреакцію зупиняли за допомогою 300мл насиченого вали під зниженим тиском для одержання твердої розчину NaНСО3. Реакційну суміш потім обробляречовини коричневого кольору. Цю речовину потім ли за допомогою 200мл Н2О та 1л Et2O. Органічну розчиняли у 300мл СН2СІ2 і промивали Н2О та фазу потім відокремлювали та промивали Н2О та насиченим розчином хлориду натрію. Органічну насиченим розчином хлориду натрію. Органічну частину висушували над Na2SO4 та концентрували частину потім висушували над MgSO4 та концентпід зниженим тиском для одержання коричневої рували під зниженим тиском для одержання 41,9г олії. Олію потім розчиняли у 100мл МеОН та обтрет-бутил-2-(2 27 74852 28 роблювали активованим деревним вугіллям. Гаря7,1, 8,2Гц, 1Н); 7,22 (ддд, J=1,1, 7,1, 8,2Гц, 1Н); чий розчин фільтрували та концентрували для 4,75 (т, J=5,1Гц, 2Н); 3,83 (т, J=6,8Гц, 4H); 3,35 (т, одержання 7,20г трет-бутил-2-{2-[2-(2J=5,6Гц, 2H); 3,30 (с, 3H); 3,21 (т, J=6,9Гц, 2Н); 2,45 метоксіетил)-1Н-імідазо[4,5-с]хінолін-1(т, J=5,6Гц, 2Н); 2,12 (с, 3Н). іл]етокси}етил(метил))карбамату у вигляді жовтого Частина l сиропу. 2-(2-метоксіетил)-1-{2-[2Частина F (метиламіно)етокси]етил}-1Н-імідазо[4,5-с]хінолінРозчин трет-бутил-2-{2-[2-(2-метоксіетил)-1Н4-амін (929мг, 2,71ммоль) розчиняли в 30мл безімідазо[4,5-с]хінолін-1водного СН2О2 та обробляли феніл ізоціанатом іл]етокси}етил(метил))карбамату (7,20г, (300мкл, 2,76ммоль). Після перемішування під N2 16,8ммоль) в 200мл СН2О2 оброблювали за допопротягом ночі реакційну суміш концентрували під могою МСРВА (77%, 4,32г, 19,3ммоль). Після пезниженим тиском. При використанні очистки за ремішування протягом 6 годин реакційну суміш допомогою колонкової хроматографії (SiO2, 3% оброблювали насиченим розчином NaНСО3 та МеОН/СНCl3 насичений за допомогою водного розділяли шари. Органічну частину промивали NH4OH) одержували продукт у вигляді білої тверН2О та насиченим розчином хлориду натрію, а дої речовини. Кристалізація з Н2О та МеОН давапотім висушували над Na2SO4 та концентрували ла 610мг N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Ндля одержання трет-бутил-2-{2-[2-(2імідазо[4,5-с]хінолін-1-іл]етокси}етил)-N-метил-N'метоксіетил)5-оксидо-1Н-імідазо[4,5-с]хінолін-1фенілсечовини у вигляді крихких білих кристалів. іл]етокси}етил(метил))карбамату як світлоТочка плавлення 184,8-185,8°С; коричневої твердої речовини. MC 463 (M+H)+; 1 Частина G H ЯМР (300МГц, ДМСО-d6): δ 8,16 (с, 1Н); Розчин трет-бутил-2-{2-[2-(2-метоксіетил)58,06 (д, J=7,7Гц, 1Н); 7,61 (дд, J=1,0, 8,3Гц, 1Н); оксидо-1Н-імідазо[4,5-с]хінолін-17,43-7,38 (м, 3Н); 7,25-7,17 (м, 3Н); 6,91 (т, J=7,3Гц, іл]етокси}етил(метил))карбамату (7,05г, 1Н); 6,47 (с, 2Н); 4,76 (т, J=5,0Гц, 2Н); 3,39 (т, 15,9ммоль) в 100мл 1,2-дихлоретану нагрівали до J=5,1Гц, 2Н); 3,78 (т, J=6,8Гц, 2Н); 3,48 (т, J=5,2Гц, 80°С та оброблювали за допомогою 5мл концент2Н); 3,39 (т, J=5,4Гц, 2Н); 3,27 (с, 3Н); 3,20 (т, рованого розчину NH4OH. До розчину, що швидко J=6,8Гц, 2Н); 2,82 (с, 3Н); 13 перемішується, додавали твердий пС ЯМР (75МГц, ДМСО-d6): δ 155,6, 152,0, толуолсульфоніл хлорид (3,33г, 17,5ммоль) протя151,9, 145,1, 140,9, 132,7, 128,5, 126,7, 126,6, гом 10 хвилин. Реакційну суміш потім оброблюва122,0, 121,4, 120,5, 120,1,115,1, 70,5, 69,6, 69,4, ли за допомогою додаткових 5мл концентрованого 58,4, 47,7, 45,5, 35,4, 27,6. розчину NH4OH та потім запечатували у ємності Розраховане для C25H30N6O3·0,12 Н2О: %С високого тиску та нагрівання продовжували протя64,62, %Н 6,56; %Ν, 18,08. гом 4 годин. Реакційну суміш потім охолоджували Знайдено: %С, 64,69; %Н, 6,65; %Ν, 18,09. та оброблювали за допомогою CH2Cl2. Реакційну Приклад 4 суміш потім промивали за допомогою Н2О, 1% Н-(2-{2-[4-аміно-2-(2-метоксіетил)-6,7,8,9розчину Na2СО3 (3Х) та насиченого розчину хлотетрагідро-1Н-імідазо[4,5-с]хінолін-1риду натрію. Органічну частину потім висушували iл]eτoкcи}eτил)-N-мeтил-N'-фeнілceчoвинa над Na2SO4 та концентрували для одержання 6,50г трет-бутил-2-{2-[4-аміно-2-(2-метоксіетил)1Н-імідазо[4,5-с]хінолін-1іл]етокси}етил(метил))карбамату як коричневої олії. Частина Η Трет-бутил-2-{2-[4-аміно-2-(2-метоксіетил)-1Німідазо[4,5-с]хінолін-1іл]етокси}етил(метил))карбамат (6,50г, 14,7ммоль) розчиняли в 100 EtOH та оброблювали 20мл 2М HCl в EtOH, суміш підігрівали до температури киЧастина А піння зі зворотним холодильником при перемішу2-(2-метоксіетил)-1-{2-[2ванні. Через 6 годин реакційну суміш охолоджува(метиламіно)етокси]етил}-1Н-імідазо[4,5-с]хінолінли та фільтрували для одержання липкої твердої 4-амін (4,22г, 12,3ммоль) розчиняли в 25мл триречовини. Тверду речовину потім промивали за фтороцтової кислоти та обробляли за допомогою допомогою EtOH та Et2O та висушували під вакууPtO2 (0,5г). Реакційну суміш струшували під Н2 мом для одержання гідрохлоридної солі у 50мл (3кг/см2). Через 4 дні додавали додаткові 0,5г PtO2 Н2О та оброблювали 5мл концентрованого розчита гідрування продовжували протягом ще 3 днів. ну ΝΗ4ΟΗ. Водну суспензію екстрагували за допоРеакційну суміш потім фільтрували через целіт та могою СН2Cl2 (5 50мл). Поєднані органічні шари концентрували під зниженим тиском для одержанпотім висушували над Na2SO4 та концентрували ня жовтої олії. Жовту олію розчиняли у 50мл Н2О для одержання 3,93г 2-(2-метоксіетил)-1-{2-[2та екстрагували за допомогою СНCl3. Органічну (метиламіно)етокси]етил}-1Н-імідазо[4,5-с]хінолінчастину потім видаляли та відкидали. Водну час4-аміну як порошку жовто-коричневого кольору. тину потім підлужували (рН-12) шляхом додання + MC 344(М+Н) ; 10%-ного розчину NaOH. Цю речовину потім екст1 H ЯМР (300МГц, ДМСО-d6): δ 8,07 (д, J=7,7Гц, рагували за допомогою СНCl3 (6 50мл) та поєдна1Н); 7,62 (дд, J=1,0, 8,3Гц, 1Н); 7,42 (ддд, J=1,0, 29 74852 30 ні органічні шари потім висушували над Na2SO4 та дин. Реакцію зупиняли доданням насиченого розконцентрували до коричневої олії. Коричневу олію чину бікарбонату натрію (25мл). Фази розділяли та потім розчиняли у 100мл гарячого МеОН та оброорганічний шар промивали водою (3 25мл), насиблювали 1г активованого деревного вугілля. Гаряченим розчином хлориду натрію (25мл), висушучий розчин потім фільтрували через целіт та конвали (Na2SO4), фільтрували та концентрували для центрували до осушення. Одержану липку тверду одержання жовтої піни. Продукт повторно кристаречовину потім концентрували декілька разів за лізували з дихлорметану та етилацетату. Кристадопомогою Et2O для одержання 3,19г 2-(2ли розтирали у порошок за допомогою етеру метоксіетил)-1-{2-[2-(метиламіно)етокси]етил}(2 5мл) для видалення залишку розчинника. Кін6,7,8,9-тетрагідро-1Н-імідазо[4,5-с]хінолін-4-аміну цевий продукт висушували у вакуумній духовій як білого порошку. шафі для забезпечення 200мг N-(2-{2-{4-аміно-2MC 348 (М+Н)+; (2-метоксіетил)-1Н-імідазо[4,5-с]хінолін-1-іл1 H ЯМР (300МГц, CDCl3): δ 4,84 (с, 2Н); 4,48 (т, ]етокси}етил)морфолін-4-карбоксаміду як жовтоJ=5,7Гц, 2Н); 3,84 (τ, J=6,7Гц, 2Н); 3,70 (т, J=5,7Гц, коричневої кристалічної твердої речовини. 2Н); 3,46 (т, J=5,1Гц, 2Н); 3,46 (с, 3Н); 3,14 (т, Точка плавлення 164-166°С. 1 J=6,7Гц, 2Н); 2,96 (м, 2Н); 2,83 (м, 2Н); 2,65 (т, Н ЯМР (300МГц, ДМСО-d6): δ 8,06 (д, J=8,1Гц, J=5,1Гц, 2Н); 2,36 (с, 3Н); 1,85 (м, 4Н). 7,61 (д, J=7,3Гц, 1Н), 7,42 (т, J=7,2Гц, 1H), 7,23 (т, Частина В J=7,8Гц, 1H), 6,51 (с, 2Н), 6,33 (т, J=Гц, 1Н), 4,74 (т, 2-(2-метоксіетил)-1-{2-[2J=4,3Гц, 2Н), 3,85-3,81 (м, 4Н), 3,49 (т, J=4,3Гц, (метиламіно)етокси]етил}-6,7,8,9-тетрагідро-1Н4Н), 3,33 (т, J=5,9Гц, 2Н), 3,30 (с, 3Н), 3,21 (т, імідазо [4,5-с]хінолін-4-амін (750мг, 2,16ммоль) J=6,8Гц, 2Н), 3,14 (т, J=4,5Гц, 4Н), 3,08 (т, J=6,0Гц, розчиняли у 30мл безводного CH2Cl2 та оброблю2Н); 13 вали фенілізоціанатом (239мкл, 2,20ммоль). Після С ЯМР (75МГц, ДМСО-d6): δ 157,8, 151,9, перемішування під N2 протягом ночі реакційну су145,0, 132,7, 126,7, 121,4, 120,5, 115,1, 70,4, 70,2, міш концентрували під зниженим тиском. Криста69,2, 58,4, 45,5, 44,0, 27,6; лізація з EtOAc та СН2Cl2 давала 170мг N-(2-{2-[4Розраховано для C22H30N6O4: %С, 59,71; %Н аміно-2-(2-метоксіетил)-6,7,8,9-тетрагідро-1Н6,83; %Ν. 18,99. імідазо[4,5-с]хінолін-1-іл-]етокси}етил-N-метил-N'Знайдено: %С, 59,71; %Н 6,80; %Ν. 18,78. фенілсечовини у вигляді крихких білих кристалів. MC (хімічна іонізація) m/e 443 (М+Н). Точка плавлення 167,7-170,0°С; Приклад 6 MC 467 (М+Н)+; N-(2-{2-[4-аміно-2-(2-метоксіетил)-1Н1 Н ЯМР (300МГц, ДМСО-d6): δ 8,17 (с, 1Н); імідазо[4,5-с]хінолін-1-іл-]eтoкcи}eтил)-N7,43 (д, J=7,6Гц, 2Н); 7,21 (т, J=7,9Гц, 2Н); 6,91 (т, метилморфолін-4-карбоксамід J=7,3Гц, 1H); 5,65 (с, 2Н); 4,43 (т, J=5,0, 2Н); 3,72 (т, J=7,0Гц, 2Н); 3,70 (т, J=5,2Гц, 2Н); 3,46-3,41 (м, 4Н); 3,24 (с, 3Н); 3,07 (т, J=6,9Гц, 2Н); 2,92 (м, 2Н); 2,85 (с, 3Н); 2,64 (м,2Н);1,72(м,4Н); 13 С ЯМР (75МГц, ДМСО-d6): δ 155,6, 151,2, 149,3, 146,3, 140,9, 138,4, 128,5, 124,9, 122,0, 120,1, 105,5, 70,7, 70,5, 69,5, 58,4, 48,0, 44,6, 35,5, 32,8, 27,6, 23,8, 23,1, 23,0. Розраховано для C25H34N6O3: %C, 64,36; %Н 2-(2-метоксіетил)-1-{2-[27,35; %N. 18,01. (метиламіно)етокси]етил}-1Н-імідазо [4,5-с]хінолінЗнайдено: %С, 64,04; %Н 7,38; %N. 18,02. 4-амін (802мг, 2,34ммоль) розчиняли в 30мл безПриклад 5. водного СН2Cl2 та охолоджували до 0°С під N2. До N-(2-{2-[4-aмінo-2-(2-мeтoкcіeτил)-1Hперемішаного розчину додавали Et3N (0,65мл, імідазо[4,5-c]xінoлін-1-iл-]етокси}етил)морфолін-44,68ммоль) і морфолінкарбонілхлорид (273мкл, карбоксамід 2,34ммоль) та реакційну суміш залишали при кімнатній температурі протягом ночі. Реакцію потім зупиняли шляхом додання насиченого розчину NaНСО3 (30мл) та СН2Cl2 (30мл). Органічний шар відокремлювали та промивали за допомогою Н2О та насиченого розчину хлориду натрію, висушували над Na2SO4 та концентрувлаи під зниженим тиском. Очистка шляхом колонкової хроматографії (SiO2, 2-5% МеОН/СНСl3 насичений за допомогою водного NH4OH) давала продукт у вигляді безбарВ атмосфері азоту, 1-[2-(2-аміноетокси)етил]вної піни. Кристалізація з EtOAc давала 640мг N2-(2-метоксіетил)-1Н-імідазо[4,5-с]хінолін-4-амін (2-{2-[4-аміно-2-(2-метоксіетил)-1Н-імідазо[4,5(0,75г, 2,3ммоль) розчиняли в дихлорметані (30мл) с]хінолін-1-іл-]етокси}етил)-N-метилморфолін-4та триетиламіні (0,64мл, 4,6ммоль), використовукарбоксаміду у вигляді білих кристалів. ючи помірне нагрівання та енергійне перемішуТочка плавлення дорівнює: 121,8-122,3°С. вання. Розчин охолоджували на крижаній бані та MC 457 (М+Н)+; по краплях додавали 4-морфолінкарбонілхлорид 1 Н ЯМР (500 МГц, ДМСО-d6): δ 8,06 (дд, (0,27мл, 2,3ммоль). Видаляли охолоджувальну J=0,9,8,3Гц, 1Н); 7,61 (дд, J=1,1, 8,3Гц, 1Н); 7,41 баню та перемішували протягом додаткових 4 го(ддд, J=1,2, 7,0, 8,3Гц, 1Н); 7,22 (ддд, J=1,3, 7,0, 31 74852 32 8,1Гц, 1Н); 6,44 (с, 2Н); 4,74 (т, J=5,2Гц, 2Н); 3,84 гом 10 хвилин. Реакційну суміш потім запечатува(т, J=5,2Гц, 2Н); 3,84 (т, J=5,2Гц, 2Н); 3,82 (т, ли у ємності високого тиску та нагрівання продовJ=6,9Гц, 2Н); 3,50-3,43 (м, 6Н); 3,30 (с, 3Н); 3,20 (т, жували протягом 2 годин. Реакційну суміш потім J=6,9Гц, 2Н); 3,16 (т, J=5,5Гц, 2Н); 2,88 (т, J=4,7Гц, охолоджували та оброблювали 100мл СН2Cl2. Ре4Н); 2,59 (с, 3Н); акційну суміш потім промивали Н2О, 1%-ним роз13 С ЯМР (75МГц, ДМСО-d6):δ 163,8, 152,0, чином Na2СО3 (3Х) та насиченим розчином хлори151,8, 145,2, 132,7, 126,7, 121,3, 120,6, 115,1, 70,4, ду натрію. Органічну частину висушували над 69,4, 68,9, 66,1, 58,5, 49,1, 47,3, 45,5, 36,9, 27,7. Na2SO4 та концентрували для одержання 3,68г Розраховано для C23H32N6O4: %С, 60,51; %Н трет-бутил-2-[2-(4-аміно-2-бутил-1Н-амідазо[4,57,07; %N. 18,41. с]хінолін-1-іл)етокси]етилкарбамату як світлоЗнайдено: %С, 60,56; %Н 6,85; %N. 18,19. коричневої піни. Приклади 7-21 Частина D Частина А Трет-бутил-2-[2-(4-аміно-2-бутил-1НРозчин трет-бутил-2-{2-[(3-амінохінолін-4амідазо[4,5-с]хінолін-1-іл)етокси]етилкарбамат іл)аміно]етокси}етилкарбамату (3,46, 10,0ммолів) в (3,68г, 8,60ммоль) суспендували в 20мл 2М НСl в 50мл толуолу оброблювали триетилортовалераEtOH та суміш підігрівали до температури кипіння том (2,5мл, 14,5ммолів) та реакційну суміш підігрізі зворотним холодильником при перемішуванні. вали до температури кипіння зі зворотним холоЧерез 3 години реакційну суміш концентрували дильником. Порцію гідрохлориду піридинію вагою для одержання твердої речовини. Тверду речови25мг додавали та кип'ятіння зі зворотним холодину розтирали у порошок за допомогою EtOH льником продовжували протягом 4 годин. Реакцій(50мл) та фільтрували для одержання 2,90г прону суміш потім концентрували до осушення під дукту у вигляді гідрохлоридної солі. Вільну основу зниженим тиском. Залишок розчиняли в 50мл готували шляхом розчинення гідрохлоридної солі СН2СІ2 та промивали насиченим розчином в 50мл Н2О та обробкою за допомогою 5мл концеNaНСО3, Н2О та насиченим розчином хлориду нтрованого розчину NH4OH. Водну суспензію екстнатрію. Органічну частину висушували над Na2SO4 рагували за допомогою СН2Cl2 (3 50мл). Поєднані та концентрували для одержання зеленої олії. органічні шари висушували над Na2SO4 та конценЗелену олію розчиняли в 50мл гарячого МеОН та трували для одержання 1-[2-(2-аміноетокси)етил]оброблювали активованим деревним вугіллям. 2-бутил-1Н-амідазо[4,5-с]хінолін-4-аміну у вигляді Гарячий розчин фільтрували та концентрували жовто-коричневого порошку. для одержання 4,12г трет-бутил-2-[2-(2-бутил-1НMC 328 (М+Н)+; 1 амідазо[4,5-с]хінолін-1-іл)етокси]етилкарбамату як H ЯМР (300МГц, CDCl3): δ 7,95 (д, J=8,3Гц, жовтої олії. 1Н); 7,83 (д, J=8,4Гц, 1Н); 7,50 (м, 1Н); 7,30 (м, 1Н); Частина В 5,41 (с, 2Н); 4,69 (т, J=5,6Гц, 2Н); 3,93 (т, J=5,6Гц, Розчин трет-бутил-2-[2-(2-бутил-1Н2Н); 3,39 (т, J=5,1Гц, 2Н); 2,97 (т, J=7,9Гц, 2Н); 2,76 амідазо[4,5-с]хінолін-1-іл)етокси]етилкарбамату (т, J=5,1Гц, 2Н); 1,89 (м, 2Н); 1,52 (м, 2Н); 1,26 (4,12г, 10,0ммоль) в 50мл СН2Cl2 оброблювали 3(ш.с, 2Н); 1,01 (т, J=7,3, 3Н). хлорпероксибензойною кислотою (МСРВА, 77%, Частина Е 2,5г, 11,2ммоль). Після перемішування протягом 5 Сполуки у таблиці, що представлена нижче, годин реакційну суміш оброблювали насиченим готували згідно з синтетичним способом етапу (7) розчином NaНСО3 та відокремлювали шари. Орреакційної схеми 1, що представлена вище, при ганічну частину промивали Н2О та насиченим розвикористанні наступного загального способу. чином хлориду натрію, а потім висушували над Ізоціанат (84мкмоль) додавали до тестової Na2SO4 та концентрували для одержання 3,68г пробірки, що містить розчин 1-[2-(2трет-бутил-2-[2-(2-бутил-5-оксидо-1Н-амідазо[4,5аміноетокси)етил]-2-бутил-1Н-амідазо[4,5с]хінолін-1-іл)етокси]етилкарбамату у вигляді світс]хінолін-4-аміну (25мг, 76мкмолів) в дихлорметані ло-коричневої піни. (5мл). Тестову пробірку потім запечатували та Частина С поміщали на качалку при кімнатній температурі на Розчин трет-бутил-2-[2-(2-бутил-5-оксидо-1Н20 годин. Розчинник видаляли шляхом вакуумного амідазо[4,5-с]хінолін-1-іл)етокси]етилкарбамату центрифугування. Осад очищали при використанні (3,68г, 8,60ммоль) в 100мл 1,2-дихлоретану підігнапів-препаративної ВЕРХ, використовуючи спорівали до температури 80°С та оброблювали 10мл сіб, описаний вище. Таблиця, представлена нижконцентрованого розчину NH4OH. До енергійно че, показує структуру вільної основи та точну масу перемішаного розчину додавали твердий п(М+Н). толуолсульфонілхлорид (1,87г, 9,81ммоль) протя 33 74852 34 35 Приклади 22-36 Частина А Використовуючи загальний спосіб частини А Прикладів 7-21, 4-піперидинетанол (10г, 77,4ммоль) піддавали реакції з дитретбутилдикарбонатом (17,7г, 81,3ммоль) для одержання 13,1г трет-бутил-4-(2гідроксіетил)піперидин-1-карбоксилату у вигляді прозорої олії. Частина В Йод (7,97г) додавали трьома порціями до розчину імідазолу (3,89г, 57,1ммоль) та трифенілфосфіну (14,98г, 57,1ммоль) в дихлорметані (350мл). Через 5 хвилин додавали розчин сполуки частини А в дихлорметані (70мл). Реакційну суміш перемішували при кімнатній температурі протягом ночі. Додавали ще йод (7,97г) та реакційну суміш перемішували при кімнатній температурі протягом 1 години. Реакційну суміш потім промивали насиченим розчином тіосульфату натрію (2Х) та насиченим розчином хлориду натрію, висушували над сульфатом натрію, фільтрували та потім концентрували під зниженим тиском для одержання маслянистого залишку. Залишок очищали за допомогою колонкової хроматографії (силікагель, елюювання 20% етилацетатом в гексані) для одержання 15,52г трет-бутил-4-(2-йодетил)піперидин1-карбоксилату у вигляді світло-жовтої олії. Частина С В атмосфері азоту 2-(1Н-імідазо[4,5-с]хінолін1-іл)бутан-1-ол (6,5г, 26,9ммоль) додавали трьома порціями до суспензії гідриду натрію (1,4г 60%, 35,0ммоль) в безводному Ν,Ν-диметилформаміді. Реакційну суміш потім перемішували протягом 45 хвилин, протягом яких припинялося виділення газу. Трет-бутил-4-(2-йодетил)піперидин-1карбоксилат (10,5г, 29,6ммоль) додавали по краплях протягом періоду часу 15 хвилин. Реакційну суміш перемішували, потім її нагрівали до 100°С та перемішували протягом ночі. Аналіз за допомогою ВЕРХ показав, що реакцію закінчено приблизно на 35%. Додавали насичений розчин хлориду амонію, одержану суміш потім перемішували протягом 20 хвилин, а потім екстрагували за допомогою етилацетату (2Х). Етилацетатні екстракти промивали водою (2Х), а потім насиченим розчином хлориду натрію, поєднували, висушували на сульфатом натрію, фільтрували, а потім концентрували під зниженим тиском для одержання коричневої олії. Олію очищали за допомогою колонкової хроматографії (силікагель, елюювання послідовно за допомогою 30% етилацетату в гексані, 50% етилацетату в гексані та етилацетату) для одержання 2,2г трет-бутил-4-{2-[2-(1-Німідазо[4,5-с]хінолін-1-іл)бутокси]етил}піперидин-1карбоксилату. Частина D Використовуючи загальний спосіб Прикладів 7-21 частини Η сполуку частини С піддавали окисненню для одержання трет-бутил-4-{2-[2-(5оксидо-1-Н-імідазо[4,5-с]хінолін-1іл)бутокси]етил}піперидин-1-карбоксилату у вигляді олії. Частина Е Розчин гідроксиду амонію (20мл) додавали до 74852 36 розчину сполуку частини D в дихлорметані (20мл). Розчин тозилхлориду (0,99г, 5,2ммоль) у дихлорметані (10мл) додавали протягом 5 хвилин. Одержану двофазну реакційну суміш піддавали перемішуванню протягом ночі. Реакційну суміш розводили хлороформом та насиченим розчином бікарбонату натрію. Шари розділяли. Органічний шар висушували над сульфатом натрію, фільтрували та потім концентрували в умовах зниженого тиску для одержання коричневої склоподібної речовини. Цей матеріал очищали колонковою хроматографією (силікагель, елюювання спочатку за допомогою 50% етилацетату у гексані, а потім за допомогою етилацетату) для одержання 1,0г третбутил-4-{2-[2-(4-аміно-1-Н-імідазо[4,5-с]хінолін-1іл)бутокси]етил}піперидин-1-карбоксилату у вигляді світло-жовтої прозорої піни. Частина F В атмосфері азоту трет-бутил-4-{2-[2-(4-аміно1Н-імідазо[4,5-с]хінолін-1іл)бутокси]етил}піперидин-1-карбоксилат (1,00г, 2,1ммоль) та 2Ν етанольну хлористоводневу кислоту (10мл, 20ммоль) поєднували та розчин перемішували при кімнатній температурі протягом 14 годин. Розчинник видаляли у вакуумі та одержану жовто-коричневу тверду речовину розчиняли у воді. Насичений водний розчин карбонату натрію додавали до тих пір, поки pH не досягало 10. Після екстрагування дихлорметаном (3Х), органічні фракції поєднували, промивали насиченим розчином хлориду натрію, висушували (Na2SO4), фільтрували, і основну частину розчинника видаляли під вакуумом. Гексан додавали для утворення преципітату. За допомогою вакуумної фільтрації одержували 0,5г 1-{1-[2-піперидин-4ілетокси)метил]пропіл}-1Н-імідазо[4,5-с]хінолін-4аміну у вигляді жовто-коричневого порошку. 1 H ЯМР (300МГц, ДМСО-d6): δ 8,34 (шс, 1Н), 8,19 (д, J=8,49Гц, 1Н), 7,61 (дд, J=8,31, 1,13Гц, 1Н), 7,45-7,39 (м, 1Н), 7,25-7,19 (м, 1Н), 6,55 (с, 2Н), 5,25-5,15 (м, 1Н), 4,00-3,80 (м, 2Н), 3,55-3,3 (м, 2Н), 2,8-2,64 (м, 2Н), 2,22-2,11 (м, 2Н), 209-1,99 (м, 2Н), 1,8-1,63 (шс, 1Н), 1,37-1,0 (м, 5Н), 0,95-0,7 (м, 5Н); 13 С-ЯМР (75МГц, ДМСО-d6): δ 152,8, 145,8, 140,6, 133,0, 127,8, 127,0, 126,9, 121,3, 121,0, 115,5, 71,8, 68,1, 58,4, 46,1, 36,3, 33,1, 32,7, 24,5, 9,9; МС (хімічна іонізація) m/e 368,2459 (368,2450 розраховане для C21H30N5O). Частина G Сполуки, представлені у таблиці, що наведена нижче, готували згідно зі способом синтезу етапу (7) реакційної схеми l, що наведена вище, використовуючи наступний загальний спосіб. lзоціанат або ізотіоціанат (75мкмоль) додавали до тестової пробірки, що містить розчин 1-{1[(2-піперидин-4-ілетокси)метил]пропіл}-1Німідазо[4,5-с]хінолін-4-аміну (25мг, 68мкмолів) у дихлорметані (5мл). Тестову пробірку запечатували та потім поміщали на качалку при кімнатній температурі протягом 20 годин. Розчинник видаляли шляхом вакуумного центрифугування. Залишок очищали за допомогою напів-препаративної ВЕРХ, використовуючи спосіб, описаний вище. 37 74852 Таблиця, що представлена нижче, показує структуру вільної основи та виявлені точні маси (М+Н). 38 Приклади 37-44 Частина А Розчин трет-бутил-2-{2-[(3-амінохінолін-4іл)аміно]етокси}етилкарбамату (6,92г, 20,0ммоль) у 100мл толуолу оброблювали триетилортоформатом (4,65мл, 28,0ммоль) та реакційну суміш підігрівали до температури кипіння зі зворотним холодильником. Додавали 100мг гідрохлориду піридинію додавали та кип'ятіння продовжували протягом 2 годин. Реакційну суміш потім концентрували до осушення в умовах зниженого тиску. Залишок розчиняли у 200мл CH2Cl2 та промивали насиченим розчином NaНСО3, Н2О та насиченим розчином хлориду натрію. Органічну частину висушували над Na2SO4 та концентрували для одержання зеленої олії. Зелену олію розчиняли у 200мл гарячого МеОН та обробляли за допомогою 10г активованого деревного вугілля. Гарячий розчин фільтрували та концентрували для одержання 5,25г трет-бутил-2-[2-(1Н-імідазо[4,5-с]хінолін-1іл)етокси]етилкарбамату як світло-жовтого сиропу. Частина В Розчин трет-бутил-2-[2-(1Н-імідазо[4,5с]хінолін-1-іл)3-амінохінолін-1іл)етокси]етилкарбамату (5,25г, 14,7ммолів) в 39 74852 40 CH2Cl2 обробляли за допомогою МСРВА (77%, а потім поміщали на качалку при кімнатній темпе3,63г, 16,3ммоль). Після перемішування протягом ратурі протягом 20 годин. Розчинник потім виданочі реакційну суміш оброблювали насиченим роляли шляхом вакуумного центрифугування. Зализчином NаНСО3 та розділяли шари. Органічну часшок очищали за допомогою напів-препаритивної тину промивали Н2О та насиченим розчином хлоВЕРХ, використовуючи спосіб, описаний вище. риду натрію, а потім висушували над Na2SO4 та Таблиця, представлена нижче, показує структуру концентрували для одержання 4,60г трет-бутил-2вільної основи та виявлені точні маси (М+Н). [2-(5-оксидо-1Н-імідазо[4,5-с]хінолін-1іл)етокси]етилкарбамату у вигляді світлокоричневої піни. Частина С Розчин трет-бутил-2-[2-(5-оксидо-1Німідазо[4,5-с]хінолін-1-іл)етокси]етилкарбамату (4,60г, 12,4ммоль) в 150мл 1,2-дихлоретані підігрівали до температури 80°С та оброблювали 10мл концентрованого розчину NH4OH. Розчин енергійно перемішували і при перемішуванні додавали твердий п-толуолсульфоніл хлорид (2,71г, 14,2ммоль) протягом 10 хвилин. Реакційну суміш потім оброблювали додатковими 2мл концентрованого розчину NH4OH і потім запечатували у ємності високого тиску та нагрівання продовжували протягом 3 годин. Реакційну суміш потім охолоджували та обробляли 100мл СН2СІ2. Реакційну суміш промивали Н2О, 1% розчину Na2СО3 (3Х) та насиченого розчину хлориду натрію. Органічну частину висушували над Na2SO4 та концентрували для одержання 4,56г трет-бутил-2-[2-(4-аміно-1Німідазо[4,5-с]хінолін-1-іл)етокси]етилкарбамату у вигляді світло-коричневої піни. Частина D Трет-бутил-2-[2-(4-аміно-1Н-імідазо[4,5с]хінолін-1-іл)етокси]етилкарбамат (4,56г, 12,3ммоль) розчиняли у 100мл EtOH та оброблювали 30мл 2М НСl в EtOH, суміш підігрівали до температури кипіння зі зворотним холодильником при перемішуванні. Через 3 години реакційну суміш концентрували для одержання твердої речовини. Тверду речовину розтирали у порошок у гарячому EtOH (100мл) та фільтрували для одержання продукту у вигляді гідрохлоридної солі. Вільну основу одержували шляхом розчинення гідрохлоридної солі у 50мл Н2О та обробкою 5мл концентрованого розчину NH4OH. Водну суспензію екстрагували СН2Сl2 (5 50мл). Поєднані органічні шари висушували над Na2SO4 та концентрували для одержання 1,35г 1-[2-(2-аміноетокси)етил]-1Німідазо[4,5-с]хінолін-4-аміну у вигляді жовтокоричневого порошку. MC 272 (М+Н)+; 1 Н ЯМР (300МГц, CDCl3) δ 7,98 (д, J=8,2Гц, 1Н); 7,88 (с, 1Н); 7,84 (д, J=8,4Гц, 1Н); 7,54 (м, 1Н); 7,32 (м, 1Н); 5,43 (с, 2Н); 4,74 (т, J=5,2Гц, 2Н); 3,97 (т, J=5,2Гц, 2Н); 3,42 (т, J=5,1Гц, 2Н); 2,78 (т, J=5,1Гц, 2Н); 1,10 (шс, 2Н). Частина Е Сполуки, що представлені у таблиці нижче, готували згідно зі способом синтезу етапу (7) реакційної схеми l, що приведена вище, використовуючи наступний загальний спосіб. 1-[2-(2-аміноетокси)етил]-1Н-імідазо[4,5с]хінолін-4-амін (20мг, 74мкмоль) та 1-метил-2Індукція цитокінів у клітинах людини піролідинон (5мл) поєднували у тестовій пробірці Систему in vitro клітин крові людини використа потім піддавали обробці ультразвуком при натовували для оцінки індукції цитокінів. Активність гріванні для одержання розчину. Додавали ізоціабазується на вимірюванні інтерферону (α) та факнат (81мкмоль) та тестову пробірку запечатували, 41 74852 42 тора некрозу пухлин (α) (lFN та TNF, відповідно) Таблиця, представлена нижче показує найнисекретуються в культуральне середовище, як опижчу концентрацію, яка індукує інтерферон та найсано Тестерманом та ін. У “Індукція цитокінів імунижчу концентрацію, яка індукує фактор некрозу номодуляторами іміквімодом та S-27609", [Journal пухлин для кожної сполуки. "*" показує, що не споof Leukocyte Biotogy, 58, 365-372 (вересень, 1995)]. стерігали ніякої індукції при будь-якій з тестових Приготування клітин крові для культури концентрацій; взагалі, що найвища з тестованих Цільну кров від здорових донорів збирали концентрацій складала 10 або 30мкМ. шляхом венопунктури у пробірки, що містять ЕДТА. Мононуклеарні клітини периферичної крові Таблиця (РВМС) відокремлювали від цільної крові шляхом центрифугування в градієнті щільності, використоІндукція цитокінів у клітинах людини вуючи Histopaque®-1077. РВМС промивали двічі за допомогою збалансованого сольового розчину Найнижча ефективна концентрація Приклад, Хенкса та потім суспендували у повному розчині (мкМ) номер RPMl в концентрації 3-4 106клітин/мл. Суспензію Інтерферон Фактор некрозу пухлин РВМС вносили до стерильних планшетів для куль3 0,01 0,37 тури тканин на 48 комірок (Costar, Cambridge, MA 7 0,0001 10 або Becton Dickinson Labware, Lincoin Park, NJ), що 8 0,0001 10 містили еквівалентний об'єм повного середовища 9 0,0001 1 RPMl, яке містить тестову сполуку. 10 0,0001 10 Приготування сполуки 11 0,0001 0,1 Сполуки розчиняли у диметилсульфоксиді 12 0,0001 1 (ДМСО). Для додання до комірок планшету для 13 0,0001 1 культури концентрації ДМСО не повинні переви14 0,0001 10 щувати заключну концентрацію 1%. 15 0,0001 0,1 Інкубація 16 0,0001 10 Розчин тестової сполуки додавали до першої 17 * 10 комірки, що містить повне середовище RPMlЮ, та 18 1 * готували серійні розведення у комірках. Суспензію 19 0,1 10 РВМС потім додавали до комірок у рівному об'ємі, 20 0,01 10 доводили концентрації тестової сполуки до бажа21 1 10 ного інтервалу. Заключна концентрація суспензії 6 22 0,1 1 РВМС складала 1,5-2,0 10 клітин/мл. Планшети 23 1 1 покривали стерильними пластиковими кришками, 24 0,1 1 обережно перемішували та потім інкубували про25 0,1 1 тягом 18-24 годин при температурі 37°С в 5% ат26 0,1 1 мосфері діоксиду вуглецю. Відокремлення Після інкубації планшети центрифугували протягом 5-10 27 0,1 1 хвилин при 1000об/хв. (-200 g) при 4°С. Суперна28 0,1 1 танти культури, вільні від клітин, видаляли за до29 0,1 1 помогою стерильної поліпропіленової піпетки та 30 1 1 переносили до стерильних поліпропіленових про31 1 10 бірок. Зразки витримували при –30 - -70°С до ана32 0,1 1 лізу. Зразки аналізували на інтерферон (α) та фак33 1 10 тор некрозу пухлин (α) шляхом ELlSA. 34 1 10 Аналіз на інтерферон (α) та фактор некрозу 35 1 1 пухлин (α) шляхом ELlSA 36 0,1 * Концентрацію інтерферону (α) визначали шля37 10 10 хом ELlSA, використовуючи набір від PBL 38 10 10 Biomedical Laboratiries, New Brunswick, NJ. Резуль39 10 10 тати виражали у пг/мл. 40 10 10 Концентрацію фактора некрозу пухлини (α) ви41 10 10 значали шляхом ELlSA, використовуючи набори 42 10 * від Genzyme, Cambridge, ΜΑ; R&D Systems, 43 10 10 Mineapotis, MN; Pharmingen, San Diego, CA. Ре44 * * зультати виражали у пг/мл. Комп’ютерна верстка Т. Чепелева Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюUrea-substituted imidazoquinoline ethers
Назва патенту російськоюМочевинозамещенные имидазохинолиновые этеры
МПК / Мітки
МПК: A61P 31/16, A61K 31/4745, A61P 31/08, A61P 31/12, A61P 25/00, A61P 33/02, A61P 31/22, A61P 37/02, A61P 17/00, A61P 25/28, A61P 33/08, A61P 37/08, A61P 17/04, A61P 31/10, A61P 43/00, A61K 31/506, A61P 37/04, A61P 33/00, C07D 471/04, A61P 37/06, A61P 31/00, A61P 31/14, A61P 31/20, A61K 31/655, A61P 31/04, A61P 1/16, A61P 17/02, A61K 31/5377, A61P 37/00, A61P 15/00, A61P 29/00, C12P 21/02, A61P 31/18, A61P 35/00, A61P 11/06, A61P 27/02, A61P 17/14, A61P 7/00, A61P 31/06, A61P 35/02, A61K 31/437
Мітки: імідазохінолінові, етери, сечовинозаміщені
Код посилання
<a href="https://ua.patents.su/21-74852-sechovinozamishheni-imidazokhinolinovi-eteri.html" target="_blank" rel="follow" title="База патентів України">Сечовинозаміщені імідазохінолінові етери</a>
Заміщені сульфонамідом та сульфамідом імідазохіноліни

Номер патенту: 73503
Опубліковано: 15.08.2005
Автори: Ліндстрьом Кайл Дж., Крукс Стефен Л., Меррілл Брайон А., Райс Майкл Дж.
МПК: C07D 471/04, A61K 9/00, A61P 31/12, A61P 35/00, A61K 45/06, A61K 31/4745, A61P 37/02
Мітки: імідазохіноліни, заміщені, сульфонамідом, сульфамідом
Формула / Реферат:
1. Сполука формули (І):, (I)в якійR1 є -алкіл-NR3-SO2-X-R4 або -алкеніл-NR3-SO2-X-R4;Х є зв'язком або -NR5-;R4 є арилом, гетероарилом, гетероциклілом, алкілом або алкенілом, кожен з яких може бути незаміщеним або заміщеним одним або більше замісниками, вибраними з групи, яка складається...
Галогенпіримідиніларил(тіо)етери, засіб та спосіб боротьби з шкідниками, проміжні сполуки

Номер патенту: 59380
Опубліковано: 15.09.2003
Автори: Гайєр Герберт, Крюгер Бернд-Віланд, Штенцель Клаус, Гердес Петер, Тіманн Ральф, Дутцманн Штефан, Хенсслер Герд, Хайнеманн Ульріх
МПК: A01N 43/54, C07D 239/56, C07D 239/52
Мітки: галогенпіримідиніларил(тіо)етери, боротьби, проміжні, шкідниками, спосіб, сполуки, засіб
Формула / Реферат:
1. Сполуки загальної формули (І),(I)в якійZ означає заміщений або незаміщений циклоалкіл, арил або гетероцикліл,R означає водень або алкіл,Q означає кисень або сірку,Х означає галоген іL1, L2, L3, L4-однакові або різні і, незалежно один від одного, означають відповідно водень, галоген, ціаногрупу, нітрогрупу, заміщений або незаміщений галогеном алкіл, алкокси, алкілтіо, алкілсульфініл або...
Спосіб одержання хінолінкарбальдегіду

Номер патенту: 64824
Опубліковано: 15.03.2004
Автори: Шімізу Таканорі, Мацумото Хіру, Такада Ясутака
МПК: C07D 215/14
Мітки: спосіб, одержання, хінолінкарбальдегіду
Формула / Реферат:
1. Спосіб одержання 2-циклопропіл-4-(4-фторфеніл)хінолін-3-карбальдегіду формули (III):, [III]при якому здійснюють оксидування 2-циклопропіл-4-(4-фторфеніл)хінолін-3-гідроксиметилхіноліну формули (І): ,[I]сіллю гіпогалогеноводневої кислоти в присутності солі четвертинного амонію формули (II): ,[II]де кожний з R1, R2, R3 і R4, що однакові чи відмінні один від іншого, – алкільна група С1-16 чи бензильна...
Заміщені сечовиною імідазохіноліни

Номер патенту: 73504
Опубліковано: 15.08.2005
Автори: Райс Майкл Дж., Крукс Стефен Л., Меррілл Брайон А.
МПК: A61P 35/00, A61P 43/00, A61K 31/5377, A61P 11/06, A61P 37/02, A61P 1/02, A61K 31/4745, A61P 31/10, A61P 31/12, C07D 471/04, A61P 37/08, A61P 31/00, A61P 27/16
Мітки: заміщені, сечовиною, імідазохіноліни
Формула / Реферат:
1. Сполука формули (І):, (I)в якійR1 є -алкіл-NR3-CY-NR5-X-R4 або -алкеніл-NR3-CY-NR5-X-R4; деY є =O або =S;Х є зв'язком, -CO- або -SO2-;R4 є арилом, гетероарилом, гетероциклілом, алкілом або алкенілом, кожен з яких може бути незаміщеним або заміщеним одним або більше замісниками, вибраними з групи, яка складається...
Заміщені 2-арил-3-(гетероарил)-імідазо[1,2-a]піримідини, спосіб їх одержання (варіанти) та фармацевтична композиція на їх основі

Номер патенту: 70350
Опубліковано: 15.10.2004
Автори: Додд Джон Х., Генрі Джеймс Р., Руперт Кеннет С.
МПК: A61P 1/16, A61P 1/04, A61P 19/02, A61P 25/04, A61P 1/02, C07D 487/04, A61P 29/00, A61P 19/10, A61P 17/14, A61P 31/04, A61P 37/08, A61K 31/519, A61P 17/06, A61P 25/28, A61P 9/10, A61P 11/00, A61P 37/06, A61P 1/18, A61P 31/18, A61P 31/12, A61P 9/00, A61P 3/10
Мітки: спосіб, одержання, варіанти, фармацевтична, 2-арил-3-(гетероарил)-імідазо[1,2-a]піримідини, композиція, заміщені, основі
Формула / Реферат:
1. Заміщений 2-арил-3-(гетероарил)-імідазо[1,2-a]піримідин формули І, (I)або його фармацевтично прийнятна сіль, де(а) R1 вибрано із NH2, C1-5алкіламіно, діC1-5алкіламіно, гідрокси, С1-5алкокси, фенілметиламіно, гетероциклілметил, С1-5алкілкарбоніламіно та заміщеного фенілкарбоніламіно, дезазначені фенілметиламіно та гетероциклілметил можуть бути заміщені по своїй фенольній складовій одним або більшою кількістю...