Спосіб одержання селективного інгібітора циклооксигенази-2
Номер патенту: 114594
Опубліковано: 10.07.2017
Автори: Містри Ашоккумар Бхікхубхаї, Соланкі Санджай Амратлал, В'яс Ашок Васантрай, Джарівала Вірал Нарендра, Шах Дхармеш Махедра








Формула / Реферат
1. Спосіб одержання етерококсибу, який включає піддавання 5-хлор-3-(4-метилтіо)феніл-2-(2-метил-5-піридиніл)піридину (IV) окисненню за присутності каталізатора окиснення і каталізатора фазового переходу з одержанням еторикоксибу з формулою (І), який відрізняється тим, що каталізатор фазового переходу вибирають із групи, яка складається з метил-три-n-октиламонію хлориду, метил-три-n-бутиламонію хлориду, бензетонію хлориду і метилбензетонію хлориду
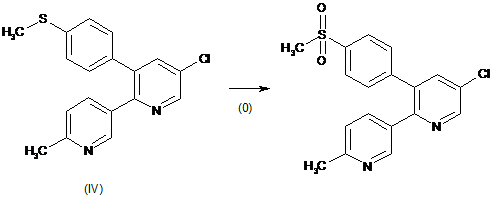 .
.
2. Спосіб за п. 1, який відрізняється тим, що включає:
а) конденсацію 4-метилтіобензилціаніду і метил-6-метилнікотинату за присутності придатної основи і придатного розчинника за температури кипіння з одержанням 1-(6-метил-3-піридиніл)-2-ціано-2-[(4-метилтіо)феніл]етанону (VI);
б) гідроліз сполуки (VI), одержаної на етапі конденсації за присутності кислоти за температури 40-50 °C з наступним декарбоксилюванням in situ за температури кипіння з одержанням 1-(6-метил-3-піридиніл)-2-[4-(метилтіо)феніл]етанону з формулою (V);
в) взаємодію сполуки за формулою (V), одержаної на етапі гідролізу сполуки (VI) з сіллю 2-хлор-N,N-диметиламінотриметиніумгексафторфосфату (III) за присутності основи з наступним додаванням суміші спирту і кислоти, додаванням водного розчину аміаку з наступним додаванням солі аміаку та нагріванням з одержанням 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридину (IV),
г) піддавання одержаної сполуки з формулою (IV) окисненню за присутності каталізатора окиснення і каталізатора фазового переходу з одержанням еторикоксибу з формулою (І).
3. Спосіб за п. 2, який відрізняється тим, що основу для етапу конденсації 4-метилтіобензилціаніду і метил-6-метилнікотинату вибирають із групи, що складається з метоксиду натрію, аміду натрію, гідриду натрію і трет-бутоксиду калію.
4. Спосіб за п. 2, який відрізняється тим, що розчинник для етапу конденсації 4-метилтіобензилціаніду і метил-6-метилнікотинату вибирають із вуглеводнів, таких як гептан, толуол, ксилен або їх суміші.
5. Спосіб за п. 2, який відрізняється тим, що гідроліз сполуки (VI) проводять в суміші органічної кислоти і неорганічної кислоти.
6. Спосіб за п. 5, який відрізняється тим, що органічну кислоту вибирають із групи, що складається з мурашиної кислоти, льодяної оцтової кислоти, пропіонової кислоти, масляної кислоти і пентанової кислоти.
7. Спосіб за п. 6, який відрізняється тим, що неорганічну кислоту вибирають із концентрованої хлористоводневої кислоти або концентрованої сірчаної кислоти.
8. Спосіб за п. 2, який відрізняється тим, що основу для етапу взаємодії сполуки за формулою (V) з сіллю 2-хлор-N,N-диметиламінотриметиніумгексафторфосфату (III) вибирають із групи, яка складається з метоксиду натрію, метоксиду калію, трет-бутоксиду калію, аміду натрію і гідриду натрію.
9. Спосіб за п. 2, який відрізняється тим, що спирт для етапу взаємодії сполуки за формулою (V) з сіллю 2-хлор-N,N-диметиламінотриметиніумгексафторфосфату (ІІІ) вибирають із органічних розчинників, таких як трет-бутанол, ізопропанол, тетрагідрофуран і метил-трет-бутиловий ефір.
10. Спосіб за п. 2, який відрізняється тим, що кислоту для етапу взаємодії сполуки за формулою (V) з сіллю 2-хлор-N,N-диметиламінотриметиніумгексафторфосфату (ІІІ) вибирають із органічних кислот, таких як мурашина кислота, оцтова кислота, n-пропіонова кислота і n-масляна кислота.
11. Спосіб за п. 2, який відрізняється тим, що каталізатор окиснення для етапу піддавання одержаної сполуки з формулою (IV) окисненню вибирають із молібдату натрію, ванадату натрію і вольфрамату натрію.
12. Спосіб за п. 2, який відрізняється тим, що окиснення на етапі піддавання одержаної сполуки з формулою (IV) окисненню проводять за присутності перекису.
13. Спосіб за п. 4, який відрізняється тим, що етап піддавання одержаної сполуки з формулою (IV) окисненню проводять у двофазній системі, що включає галогеновані вуглеводні і воду.
Текст
Реферат: Спосіб одержання етерококсибу, який включає піддавання 5-хлор-3-(4-метитіо)феніл-2-(2метил-5-піридиніл)піридину (IV) окисненню за присутності каталізатора окиснення і каталізатора фазового переходу з одержанням еторикоксибу з формулою (І), де каталізатор фазового переходу вибирають із групи, яка складається з метил-три-n-октиламонію хлориду, метил-триn-бутиламонію хлориду, бензетонію хлориду і метилбензетонію хлориду. UA 114594 C2 (12) UA 114594 C2 UA 114594 C2 ГАЛУЗЬ ТЕХНІКИ: Даний винахід стосується способу синтезу селективного інгібітора циклооксигенази-2. Зокрема, даний винахід відноситься до способу синтезу 5-хлор-3-(4-метилсульфоніл)феніл-2-(2метил-5-піридиніл)піридину з наступною Формулою I із нової сполуки з формулою (IV). 5 10 15 20 Крім того, винахід додатково надає нову сполуку (IV) і спосіб її одержання. Винахід також відноситься до одержання сполуки з Формулою (II), придатної для синтезу зазначеного інгібітора циклооксигенази-2. РІВЕНЬ ТЕХНІКИ: Селективні інгібітори циклооксигенази-2 являють собою важливий клас нестероїдних протизапальних лікарських засобів (НПЗЛЗ), особливо у зв'язку з їх покращеним профілем безпечності. Звичайно використовувані НПЗЛЗ, наприклад арилпропіонова кислота або похідні арилоцтової кислоти при тривалому застосуванні таких препаратів, як відомо, викликають подразнення та виразки шлунка. Інгібітори циклооксигенази-2, які впливають на механізм експресії ізоферменту в тканинах із запаленням, демонструють в цьому відношенні кращу безпечність. Еторикоксиб селективно інгібує ізоформу 2 ферменту циклооксигенази (ЦОГ-2) і на даний час він схвалений у більше ніж 70 країнах для таких терапевтичних показань, як лікування ревматоїдного артриту, псоріатичного артриту, анкілозуючого спондиліту, хронічного болю попереку, гострого болю, остеоартриту і подагри. Сполуки загальної формули (А), включаючи еторикоксиб, розкриті в US5861419, як продемонстровано наступною схемою. 25 Альтернативний спосіб також описаний в US5861419, як продемонстровано наступною схемою. 1 UA 114594 C2 5 10 15 20 25 30 35 Способи, описані в US5861419 включають багатоетапний синтез, що призводить в результаті до зниженого виходу кінцевого продукту. US6040319 описує інший спосіб одержання еторикоксибу відповідно до схеми, наведеної нижче. Він відноситься до конденсації між сполукою з Формулою-II і сполукою з Формулою-IIIa, де Х може бути вибраний з фосфатів, сульфатів, ацетатів, перхлорату, боратів, бензоату, напсилатів, зокрема, гексафторфосфату, сульфату, мезилату, тозилату трифлату, ацетату, трифторацетату, тетрафторборату, тетрафенілборату, гексафторантимонату, хлориду, броміду, фториду, йодиду, бензолату і напсилату. Гідроксиди натрію або калію, карбонат цезію, алкоксиди, аміди і гідриди літію, натрію і калію зображені як придатні основи. WO 99/55830 описує спосіб приготування ЦОГ-2 інгібіторів, включаючи еторикоксиб. Він включає конденсацію між сіллю вінамідініуму і заміщеним похідним бензилпіридилкетону, який, у свою чергу, одержаний з використанням реакції Гріньяра між похідним піридиламіду і тіометилбензилгаліду, з подальшим окисненням. US 6071936 включає заміщені похідні піридину, їх фармацевтичні композицій і спосіб лікування ЦОГ-2-опосередкованих захворювань. Різні сполуки включені до цього документу структурно являють собою похідні 2, 3-діарилпіридину, які мають сульфонові заміщення. Схема синтезу складається із каталізованого паладієм діарилового зв'язування між галогенованим похідним 2-амінопіридину і 4-(метилтіо) фенілборною кислотою з подальшим окисненням і наступним зв'язуванням з іншим арильним фрагментом. US 6001843, US 6596736 В2 і US 6 812346 B2 також розкривають структурно подібні заміщені похідні піридину та застосовувані синтетичні методології. Їх використання для приготування таких фармацевтичних композицій як інгібітори ЦОГ-2, також представлене в цих винаходах. Патентний документ WO2010/097802 A2 описує спосіб приготування лікарської речовини еторикоксибу. Тоді як використання деяких заміщених -хлор вінамідініумних солей повідомлялося раніше, даний винахід відноситься до одержання альтернативних -хлор вінамідініумних солей, що містять циклічну або гетероциклічну групу заміщення. Він також описує спосіб приготування цих альтернативних солей вінамідініуму, а також спосіб очищення. Поліморфні характеристики однієї з таких модифікованих -хлор вінамідініумних солей, а саме 2-хлор-1,3-(біспіперидил) триметиніум гексафторофосфату, який існує у вигляді форми-I і форми-II також задокументовані в патенті. Спосіб приготування еторикоксибу за допомогою таких солей вінамідініуму, що містять циклічні фрагменти також описаний. 2 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 Тим не менше, завжди існує необхідність створювати новий підхід до синтезу, який може привести до більш досконалих і економічно ефективних способів одержання зазначених лікарських речовин. ЦІЛІ ДАНОГО ВИНАХОДУ: Таким чином, метою даного винаходу є забезпечення простого і ефективного способу одержання потужної лікарської речовини, а саме 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил5-піридиніл)піридину з Формулою-I, відомого інгібітора циклооксигенази-2 із встановленою ефективністю. Ще однією метою даного винаходу є забезпечення простого і ефективного способу одержання ключової проміжної сполуки з Формулою-II, а саме 1-(6-метил-3-піридиніл)-2-[4(метилсульфоніл) феніл] етанону, корисного для одержання лікарської речовини 5-хлор-3-(4метилсульфоніл)феніл-2-(2-метил-5-піридиніл) піридину, структура якого представлена Формулою-I. Іншою метою даного винаходу є одержання нової проміжної речовини 5-хлор-3-(4метилтіо)феніл-2-(2-метил-5-піридиніл) піридину, структура якого представлена Формулою-IV, іншої ключової речовини для приготування 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5піридиніл)піридину з Формулою-I. Ще одна мета даного винаходу відноситься до способу одержання сполуки з Формулою-IV, яка є новою проміжною речовиною у приготуванні лікарської речовини з Формулою-І. СУТНІСТЬ ВИНАХОДУ: Згідно до даним винаходом надається спосіб синтезу інгібітора циклооксигенази-2 еторикоксибу з формулою-(I) із сполуки з формулою (IV), який включає піддавання сполуки з формулою (IV) окисненню за присутності каталізатора окиснення і каталізатора фазового переходу. В одному аспекті даний винахід надає нову сполуку з формулою (IV); В іншому аспекті винахід надає спосіб одержання сполуки з формулою (IV), який включає реакцію між похідним бензилпіридилкетону з формулою-(V) і похідним солі вінамідініуму з формулою-(III) за присутності основи. Масу продукту потім піддають серії хімічних та оперативних етапів для одержання проміжної сполуки анельованого піридину, представленої сполукою з формулою-(IV). Інший аспект винаходу полягає у створенні вдосконаленого способу одержання еторикоксибу із 4-метилтіобензилціаніду в якості вихідної сполуки. Зазначену сполуку конденсують з метил 6-метил нікотинатом за присутності основи з одержанням сполуки (VI), яку гідролізують і декарбоксилюють щоб одержати сполуку (V). Сполуку (V) конденсують з похідним солі вінамідініуму з формулою-(III) за присутності основи. Масу продукту потім піддають серії хімічних та оперативних етапів для одержання проміжної сполуки анельованого піридину, представленої сполукою з формулою-(IV), яку потім піддають окисненню, що дає бажану сполуку, еторикоксиб. КОРОТКИЙ ОПИС КРЕСЛЕНЬ: Даний винахід буде описаний докладно з посиланням на супровідні креслення. На супровідних кресленнях: Фігура-1 ілюструє структурний лист Фігура-2 ілюструє інфрачервоний спектр сполуки з формулою-(I) 5-хлор-3-(4метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридину, тобто еторикоксибу. Фігура-3 ілюструє інфрачервоний спектр проміжної сполуки з формулою-(IV), тобто 5-хлор-3(4-метилтіо)феніл-2-(2-метил-5-піридиніл)піридину. Фігура-4 ілюструє ПРД спектр 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5піридиніл)піридину, тобто еторикоксибу. ДЕТАЛЬНИЙ ОПИС ВИНАХОДУ: Тепер винахід буде докладно описаний у зв'язку з певними кращими і необов'язковими варіантами втілення, так що різні його аспекти можуть бути повністю зрозумілі і оцінені. 3 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 Даний винахід у відповідності з об'єктом винаходу, який згадувався раніше, докладно описаний у наступних варіантах втілення. У кращому варіанті втілення, даний винахід надає спосіб одержання еторикоксибу із сполуки 5-хлор-3-(4-метитіо) феніл-2-(2-метил-5-піридиніл) піридину (IV), який включає піддавання зазначеної сполуки з формулою (IV) окисненню за присутності каталізатора окиснення і каталізатора фазового переходу з одержанням еторикоксибу з формулою-(I). Відповідно, сполуку з формулою (IV) 5-хлор-3-(4-метилтіо)феніл-2-(2-метил-5-піридиніл) піридин піддають окисненню за присутності окиснювача, вибраного із перекису водню, перекису натрію, тощо, в умовах каталітичного і двофазного стану за участю органічної і водної фаз. Двофазна маса звичайно містить воду, хлорований вуглеводень, окиснювач, каталізатор окиснення і каталізатор фазового переходу. Як правило, хлорований вуглеводневий розчинник являє собою щонайменше один розчинник, вибраний із групи хлорованих вуглеводневих розчинників, яка складається з таких речовин як хлороформ, дихлоретан, дихлорметан і чотирихлористий вуглець. Звичайно каталізатор окиснення являє собою щонайменше один, вибраний із групи солей, яка складається з молібдату натрію, ванадату натрію і вольфрамату натрію. Як правило, каталізатор фазового переходу являє собою щонайменше один, вибраний із групи таких каталізаторів, яка складається з метил-три-n-октил амонію хлориду, бензетонію хлориду і метилбензетонію хлориду. Реакцію окиснення проводять за участі водного розчину перекису водню в температурному діапазоні 0°-20°C, краще за температури 10°-14°C, з подальшим підвищенням температури до досягнення умов навколишнього середовища, в той самий час контролюючи реакцію шляхом ТШХ до її завершення. Фази розділяють, водну частину екстрагують тим самим органічним розчинником, який використовується в реакції, всі органічні розчини змішують, промивають водним розчином карбонату натрію і водою приблизно до нейтрального рівня рН, висушують над безводним сульфатом натрію, необов’язково очищують активованим вугіллям, фільтрують і концентрують для видалення розчинника у вакуумі. Залишкову масу обробляють спиртовим розчинником, звичайно щонайменше одним, вибраним із групи, яка складається із метанолу, очищеного спирту, ізопропанолу, n-пропанолу і n-бутанолу, що містить 0%-4% води на основі об/об. Масу продукту охолоджують до температури (-)2°-2°C, фільтрують, промивають тим самим холодним розчинником і висушують з одержанням неочищеного продукту еторикоксибу з формулою-(I) Очищення неочищенного еторикоксибу, одержаного в такий спосіб, включає в себе спосіб кристалізації. Неочищений еторикоксиб з формулою-(I) очищують шляхом кристалізації, яка включає розчинення продукту, розчиненого щонайменше в одному спиртовому розчиннику, звичайно вибраному із групи, яка складається із n-пропанолу, ізопропанолу, метанолу, очищеного спирту, ацетону, що містить 0%-6% води на основі об/об. Неочищений продукт розчиняють у розчиннику за підвищеної температури, обробляють активованим вугіллям протягом 15-30 хвилин, фільтрують і охолоджують до температури навколишнього середовища і далі до температури 12°-15°C протягом приблизно однієї години. Затверділий продукт виділяють фільтрацією, потім промивають охолодженим розчинником і висушують. Альтернативно, неочищений еторикоксиб з формулою-(I) може бути очищений шляхом перетворення на його сіль. Таким чином, спосіб включає взаємодію паратолуолсульфонової кислоти з еторикоксибом за присутності органічного розчинника з одержанням солі еторикоксибу і виділенням сольової речовини. Виділену сіль паратолуолсульфонової кислоти еторикоксибу потім розчиняють у воді за умов навколишнього середовища і обробляють водним розчином карбонату натрію, поки вона не стане помірно основною. Масу продукту потім обробляють з толуолом за підвищеної температури, щоб екстрагувати масу продукту в толуолі. Водний шар екстрагують в толуолі і змішують з першим розчином толуолу. Об’єднаний розчин толуолу потім промивають водою, очищують шляхом обробки активованим вугіллям, висушують над безводним сульфатом натрію, фільтрують і концентрують під зниженим тиском з одержанням залишку. Залишок повторно розчиняють щонайменше в одному спиртовому розчиннику, звичайно вибраному із групи, яка складається із метанолу, етанолу, очищеного спирту, ізопропанолу і n-пропанолу, що містить 0%-6% води. Розчин охолоджують до температури 5°-10°C протягом періоду часу, який становить 1-2 год., фільтрують, промивають охолодженим розчинником і висушують з одержанням очищеного еторикоксибу. Даний винахід надає нову проміжну речовину з формулою (IV); 4 UA 114594 C2 5 10 15 20 25 Ще в одному варіанті втілення, даний винахід надає спосіб одержання нової проміжної речовини (IV), що містить взаємодію 1-(6-метил-3-піридиніл)-2[4-(метилтіо)феніл]етанону (V) з 2-хлор-N,N-диметиламіно триметиніум гексафтор фосфатом (III) за присутності основи з наступним додаванням суміші спирту і кислоти за тієї ж температури і додавання водного розчину аміаку з наступним додаванням безводної солі аміаку з одержанням (IV). Ще в одному варіанті втілення, винахід надає простий і ефективний спосіб приготування сполуки з формулою-(I), а саме 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5піридиніл) піридину (еторикоксибу) з високим рівнем чистоти. Зазначений спосіб включає наступні етапи: а. Конденсація 4-метилтіо-бензилціаніду і метил-6-метилнікотинату за присутності придатної основи і придатного розчинника за температури кипіння з одержанням 1-(6-метил-3піридиніл)-2-ціано-2-[(4-метилтіо)феніл]етанону (VI); б. Гідроліз сполуки (VI) етапу (a) за присутності кислоти за температури 40-50°C з наступним декарбоксилюванням in situ за температури кипіння з одержанням 1-(6-метил-3піридиніл)-2[4-(метилтіо)феніл]етанону з формулою (V); в. Взаємодія сполуки з формулою (V) з сіллю 2-хлор-N,N-диметиламіно триметиніум гексафтор фосфату (III) за присутності основи за температури в діапазоні 0°-10°C, з наступним додаванням суміші спирту і кислоти за тієї ж температури, додаванням водного розчину аміаку і солі аміаку, нагріванням, з одержанням проміжної речовини 5-хлор-3-(4-метилсульфоніл)феніл2-(2-метил-5-піридиніл)піридину(IV); і г. Піддавання сполуки з формулою (IV) одержаної на етапі (в)окисненню за присутності каталізатора окиснення і каталізатора фазового переходу у двофазному стані з одержанням еторикоксибу з формулою-(I). Цей спосіб продемонстрований на схемі 1 нижче: Схема 1 5 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 Таким чином, відповідно до схеми 1, етап (a) даного винаходу включає каталізовану основою конденсацію між 4-метилтіо-бензилціанідом і метил-6-метилнікотинатом у вуглеводневому розчиннику. Звичайно, основу вибирають із групи основ, таких як метоксид натрію, амід натрію, гідрид натрію, трет-бутоксид калію, і метоксид калію. Кращою використовуваною основою є метоксид натрію або трет-бутоксид калію. Звичайно використовувані вуглеводні вибирають із гептану, толуолу, ксилену або їх сумішей. Завершення реакції контролюють за допомогою аналізу ТШХ і реакційну масу охолоджують в суміші кригавода. Рівень pH маси коригують розбавленою кислотою, краще розбавленою хлористоводневою кислотою до рівня pH від 5,0 до 6,5, краще від 5,2 до 6,3 і продукт 1-(6метил-3-піридиніл)-2-ціано-2-[(4-метилтіо)феніл]етанон (VI) виділяють шляхом фільтрації. Сполуку з формулою (VI), на етапі (б), гідролізують за присутності кислотного каталізатора з одержанням сполуки карбонової кислоти, яка під час нагрівання декарбоксилюється in situ з виходом 1-(6-метил-3-піридиніл)-2[4-(метилтіо)феніл]етанону з формулою (V). Реакційне середовище складається з суміші органічної карбонової кислоти, вибраної із групи кислот, таких як мурашина кислота, крижана оцтова кислота, пропіонова кислота, масляна кислота, пентанова кислота, і неорганічної кислоти, такої як концентрована хлористоводнева кислота або концентрована сірчана кислота. Звичайно, в способі використовують комбінацію крижаної оцтової кислоти і концентрованої хлористоводневої кислоти. Сполуку з формулою (V) етапу (б) екстрагують вуглеводневим розчинником вибраним із групи розчинників, таких як гексан, гептан, циклогексан, і толуол а потім підлужують до рівня pH від 6,8 до 7,3 водним розчином аміаку. Краще, розчинник, використовуваний у екстрагуванні являє собою гексан, а кращий діапазон рівня pH становить від 6,9 до 7,2. Масу продукту виділяють шляхом фільтрації і висушують за температури 45-60°C. Спосіб екстракції сполуки з формулою (V) (етап б) може необов’язково бути виконаний шляхом екстрагування реакційної маси етапу (б), після завершення реакції і коригування pH до рівня від 6,8 до 7,3 розчинниками вибраними із галогенованих вуглеводнів, таких як хлороформ, дихлорметан, складних ефірів, таких як етилацетат або пропілацетат. Звичайно дихлорметан використовують як кращий розчинник. проміжну сполуку (V) виділяють шляхом концентрації, для видалення розчинника і з наступним додаванням іншого розчинника, вибраного із нижчих спиртів, краще, ізопропанолу, охолодженням маси до температури 10°-12°C, фільтруванням продукту, а потім висушуванням за температури 45°-50°C. Проміжна речовина (V), одержана як описано вище, може необов’язково бути очищеною шляхом її перекристалізації із метанолу. 1-(6-метил-3-піридиніл)-2-[4-(метилсульфоніл)феніл]етанон, сполуку з формулою (V) відповідно до етапу (в)обробляють основним каталізатором в органічному розчиннику за температури в діапазоні 0°-20°C, краще за температури 0°-10°C; більш краще за температури 5°-8°C. Основний каталізатор вибирають із групи, яка складається із метоксиду натрію, метоксиду калію, трет-бутоксиду калію, аміду натрію і гідриду натрію. Звичайно основний використовуваний каталізатор являє собою трет-бутоксид калію. Органічний розчинник вибирають із групи, що складається із трет-бутанолу, ізопропанолу, тетрагідрофурану і метилтрет-бутилового ефіру. Звичайно використовуваний розчинник являє собою ізопропанол. Реакційна маса потім взаємодіє з проміжною сіллю 2-хлор-N,N-диметиламіно триметиніум гексафтор фосфату, сполукою з формулою (III) в діапазоні температур 0°-10°C протягом періоду часу, який становить 2,0-4,0 год., в той час як хід реакції контролюють за допомогою ТШХ доти, поки одна із реакційних проміжних сполук, а саме сполука з формулою (V) практично повністю споживається. Потім реакційну масу обробляють сумішшю спиртових розчинників, вибраних із групи, що складається із метанолу, етанолу, ізопропанолу, трет-бутанолу і nпропанолу; і карбоновою кислотою вибраною із групи кислот, яка складається із мурашиної кислоти, оцтової кислоти, n-пропіонової кислоти і n-масляної кислоти. Звичайно використовуваний спиртовий розчинник являє собою ізопропанол, а використовувана карбонова кислота являє собою оцтову кислоту. Температуру реакційної маси підтримують на рівні 0°-10°C, краще на рівні 5°-10°C протягом періоду часу від 2,5 до 3,0 год. Потім додають водний розчин аміаку з наступним додаванням солі аміаку, вибраної із групи солей, що включає ацетат аміаку, карбонат аміаку, тощо. Звичайно в реакції використовують ацетат аміаку. Масу кип’ятять до завершення реакції, охолоджують до кімнатної температури з наступним додаванням розчину аміаку і розчину формальдегіду. Реакційний розчинник видаляють за допомогою дистиляції в умовах зниженого тиску, додають ще один розчинник толуол, повторно нагрівають реакційну масу до температури 60°-65°C. Органічний і водний шари розділяють. Водний шар промивають толуолом, об’єднані шари толуолу промивають водним розчином карбонату натрію, а потім водою, обробляють активованим вугіллям, фільтрують, висушують 6 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 над безводним сульфатом натрію і концентрують під вакуумом, для видалення розчинника. До залишкової маси додають спиртовий розчинник, вибраний із групи, яка складається із метанолу, ізопропанолу, очищеного спирту, n-пропанолу або їх суміші. Звичайно, для додавання використовують ізопропанол з подальшим охолодженням для затвердіння продукту. Масу продукту фільтрують, промивають ізопропанолом і висушують з одержанням сполуки з формулою (IV). Сполуку з формулою (IV) кристалізують із органічного розчинника, вибраного із групи спиртових розчинників, яка включає етиловий спирт, очищений спирт, n-пропанол, ізопропанол або їх суміш, яка містить 0-6% води на основі об/об. Сполуку з формулою (IV) розчиняють в достатній кількості розчинника, кип’ятять до розчинення, фільтрують, охолоджують для затвердіння продукту і фільтрують для виділення продукту. Звичайно в якості розчинника для очищення використовують ізопропанол. Продукт виділяють і висушують за температури 45°50°C. Сполуку з формулою (IV) 5-хлор-3-(4-метилтіо)феніл-2-(2-метил-5-піридиніл) піридин піддають окисненню за присутності окиснювача, вибраного із перекису водню, перекису натрію, тощо, в умовах каталітичного і двофазного стану за участю органічної і водної фаз. Двофазна маса звичайно містить воду, хлорований вуглеводень, окиснювач, каталізатор окиснення і каталізатор фазового переходу. Як правило, хлорований вуглеводневий розчинник являє собою щонайменше один розчинник, вибраний із групи хлорованих вуглеводневих розчинників, яка складається з таких речовин як хлороформ, дихлоретан, дихлорметан і чотирихлористий вуглець. Звичайно каталізатор окиснення являє собою щонайменше один, вибраний із групи солей, яка складається з молібдату натрію, ванадату натрію і вольфрамату натрію. Як правило, каталізатор фазового переходу являє собою щонайменше один, вибраний із групи таких каталізаторів, яка складається з метил-три-n-октил амонію хлориду, метил-три-nбутил амонію хлориду, метил-три-n-бутил-амонію хлориду, бензетонію хлориду і метилбензетонію хлориду. Реакцію окиснення проводять за участі водного розчину перекису водню в температурному діапазоні 0°-20°C, краще за температури 10°-14°C, з подальшим підвищенням температури до досягнення умов навколишнього середовища, в той самий час контролюючи реакцію шляхом ТШХ до її завершення. Фази розділяють, водну частину екстрагують тим самим органічним розчинником, який використовується в реакції, всі органічні розчини змішують, промивають водним розчином карбонату натрію і водою приблизно до нейтрального рівня рН, висушують над безводним сульфатом натрію, необов’язково очищують активованим вугіллям, фільтрують і концентрують для видалення розчинника у вакуумі. Залишкову масу обробляють спиртовим розчинником, звичайно щонайменше одним, вибраним із групи, яка складається із метанолу, очищеного спирту, ізопропанолу, n-пропанолу і nбутанолу, що містить 0%-4% води на основі об/об. Масу продукту охолоджують до температури (-)2°-2°C, фільтрують, промивають тим самим холодним розчинником і висушують з одержанням неочищеного продукту еторикоксибу з формулою-(I) Очищення неочищеного еторикоксибу, одержаного в такий спосіб, включає в себе спосіб кристалізації. Неочищений еторикоксиб з формулою-(I) очищують шляхом кристалізації, яка включає розчинення продукту, розчиненого щонайменше в одному спиртовому розчиннику, звичайно вибраному із групи, яка складається із n-пропанолу, ізопропанолу, метанолу, очищеного спирту, ацетону, що містить 0%-6% води на основі об/об. Неочищений продукт розчиняють у розчиннику за підвищеної температури, обробляють активованим вугіллям протягом 15-30 хвилин, фільтрують і охолоджують до температури навколишнього середовища і далі до температури 12°-15°C протягом приблизно однієї години. Затверділий продукт виділяють фільтрацією, потім промивають охолодженим розчинником і висушують. Альтернативно, неочищений еторикоксиб з формулою-(I) може бути очищений шляхом перетворення на його сіль. Таким чином, спосіб включає взаємодію паратолуолсульфонової кислоти з еторикоксибом за присутності органічного розчинника з одержанням солі еторикоксибу і виділенням сольової речовини. Виділену сіль паратолуолсульфонової кислоти еторикоксибу потім розчиняють у воді за умов навколишнього середовища і обробляють водним розчином карбонату натрію, поки вона не стане помірно основною. Масу продукту потім обробляють з толуолом за підвищеної температури, щоб екстрагувати масу продукту в толуолі. Водний шар екстрагують в толуолі і змішують з першим розчином толуолу. Об’єднаний розчин толуолу потім промивають водою, очищують шляхом обробки активованим вугіллям, висушують над безводним сульфатом натрію, фільтрують і концентрують під зниженим тиском з одержанням залишку. Залишок повторно розчиняють щонайменше в одному спиртовому розчиннику, звичайно вибраному із групи, яка складається із метанолу, етанолу, очищеного спирту, ізопропанолу і n-пропанолу, що 7 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 містить 0%-6% води. Розчин охолоджують до температури 5°-10°C протягом періоду часу, який становить 1-2 год., фільтрують, промивають охолодженим розчинником і висушують з одержанням очищеного еторикоксибу. Винахід надає спосіб одержання вінамідініумної солі 2-хлор-N, N-диметиламіно триметиніум гексафтор фосфату, представленої формулою (III). Звичайно, N,N-диметилформамід зв’язується з хлорацетил хлоридом за температури приблизно 50°-55°C з наступним додаванням оксихлориду фосфору за підвищеної температури, яка становить 65°-70°C протягом періоду часу, який становить від 5 до 6 год.. Потім сполуку охолоджують до температури навколишнього середовища і обробляють сумішшю води з кригою, що містить гексафторфосфорну кислоту, додають водний розчин гідроксиду натрію, щоб скоригувати pH до рівня приблизно від 2,0 до 2,8, і підтримують перемішуючи протягом періоду часу, що становить приблизно 30 хвилин. Проміжну сполуку з формулою (V) виділяють шляхом фільтрації, промивають з подальшим очищенням у водному ізопропанолі і висушують. Необов’язково може проводитись повторне очищення шляхом розчинення в метанолі за підвищеної температури до кипіння, обробка активованим вугіллям, фільтрування розчину, часткова концентрація шляхом видалення розчинника за допомогою дистиляції, охолодження маси до температури, яка викликає в результаті кристалізацію, фільтрування і висушування. Ще в одному варіанті втілення винахід надає спосіб приготування 1-(6-метил-3-піридиніл)-2[(4-метилсульфоніл) феніл] етанону з формулою (II), який включає; а. Конденсацію 4-метилтіо-бензилціаніду і метил-6-метилнікотинату за присутності придатної основи і придатного розчинника за температури кипіння з одержанням 1-(6-метил-3піридиніл)-2-ціано-2-[(4-метилтіо) феніл] етанону (VI) вологої маси; б. Гідроліз вологої маси сполуки (VI) етапу (a) за присутності кислоти за температури 4050°C з наступним декарбоксилюванням in situ за температури кипіння з одержанням 1-(6-метил3-піридиніл)-2[4-(метилтіо)феніл]етанону з формулою (V); і в. Піддавання сполуки з формулою (V), одержаної на етапі (б) окисненню за присутності каталізатора окиснення і каталізатора фазового переходу з наступним додаванням суміші спирту і кислоти, додаванням водного розчину аміаку з наступним додаванням солі аміаку, нагріванням з одержанням (II). Відповідно до цього способу, 4-метилтіобензил ціанід і метил-6-метил нікотинат піддають каталізованій основою конденсації, як описано вище. Масу продукту у вигляді вологого осаду потім піддають гідролізу, каталізованому кислотою з наступним тепловим декарбоксилюванням з одержанням 1-(6-метил-3-піридиніл)-2-[4-(метилсульфоніл)феніл]етанону, сполуки з формулою-(V), відповідно до параметрів, зазначених раніше. Цю проміжну речовину (V), як масу вологого продукту виділяють із реакційної маси, розчиняють в органічному розчиннику для проведення наступного етапу окиснення. Розчинник вибирають із хлорованих вуглеводнів, таких як хлороформ, дихлоретан, дихлорметан і чотирихлористий вуглець. Каталізатор окиснення вибирають із групи солей, яка складається з молібдату натрію, ванадату натрію і вольфрамату натрію. Каталізатор фазового переходу являє собою щонайменше один, вибраний із групи таких каталізаторів, яка складається з метил-три-n-октил амонію хлориду, бензетонію хлориду і метилбензетонію хлориду. Реакцію окиснення проводять за присутності перекису, такого як перекис водню, перекис натрію тощо, у водному розчині в температурному діапазоні 0°-20°C, краще за температури 10°-14°C, з подальшим підвищенням температури до досягнення умов навколишнього середовища, в той самий час контролюючи реакцію шляхом ТШХ до її завершення. Фази розділяють, водну частину екстрагують тим самим органічним розчинником, який використовується в реакції, всі органічні розчини змішують, промивають водним розчином карбонату натрію і водою приблизно до нейтрального рівня рН, висушують над безводним сульфатом натрію, необов’язково очищують активованим вугіллям, фільтрують і концентрують для видалення розчинника у вакуумі. Залишкову масу обробляють спиртовим розчинником, щонайменше одним, вибраним із групи, яка складається із метанолу, очищеного спирту, ізопропанолу, n-пропанолу і n-бутанолу, що містить 0%-4% води на основі об/об. Масу продукту охолоджують до температури (-)2°-2°C, фільтрують, промивають тим самим холодним розчинником і висушують з одержанням неочищеного продукту 1-(6-метил-3-піридиніл)-2-[(4метилсульфоніл) феніл]етанону (II). Альтернативно, проміжну сполуку з формулою-(II), одержують за допомогою способу, який включає конденсацію між 4-метил сульфоніл феніл оцтовою кислотою і метил-6метилнікотинатом за присутності металоорганічної речовини, такої як трет- бутил магнію хлорид, як описано в даній галузі техніки. Даний винахід тепер буде проілюстрований наступними прикладами, які не призначені для обмеження ефективного об'єму формули винаходу. Як наслідок, будь-які варіації винаходу, 8 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 описані вище, не слід розглядати як відхилення від суті і об'єму заявленого винаходу. Даній винахід був описаний в термінах його конкретних варіантів втілення, і для фахівців в даній галузі техніки різні модифікації, паралелі і еквіваленти будуть очевидними і призначеними для включення в об'єм даного винаходу. Приклад 1 Приготування 1-(6-метил-3-піридиніл)-2-ціано-2-[(4-метилтіо) феніл] етанону (VI) 4-метил тіобензил ціанід (10,0 гр.) розчиняють в (100 мл) толуолу і реакційну масу кип’ятять. Метил-6-метил нікотинат (10,87 гр.) додають повільно за температури кипіння протягом приблизно 30 хвилин. Реакційну масу перемішують протягом 10 хвилин. Розчин метоксиду натрію (30% мас./мас.) 19,96 гр. загружають протягом 30 хвилин. Температуру реакції підтримують за кипіння доти, поки пляма, що відповідає метил-6-метил нікотинату буде практично відсутня згідно з дослідженням за допомогою ТШХ. Реакційну масу охолоджують до температури 25°-30°C під час перемішування. Реакційну масу охолоджують в суміші колотої криги (75 гр.) + води (10 мл). Рівень pH реакційної маси коригують в діапазоні від 5,2 до 6,2 розбавленою хлористоводневою кислотою. Реакційну масу перемішують протягом години, фільтрують, промивають водою і висушують за температури 60-70°C (Вихід 16 гр., Чистота 96,3% визначено за допомогою ВЕРХ). Приклад 2 Приготування 1-(6-метил-3-піридиніл)-2-ціано-2-[(4-метилтіо) феніл] етанону (VI) 4-метил тіобензил ціанід (10,0 гр.) розчиняють в (80 мл) толуолу + 20 мл n-гептану і реакційну масу кип’ятять. Метил-6-метил нікотинат (11,30 гр.) додають повільно за температури кипіння протягом приблизно 30 хвилин. Реакційну масу перемішують протягом 10 хвилин. Розчин метоксиду натрію (30% мас./мас.) 19,96 гр. загружають протягом 30 хвилин. Температуру реакції підтримують за кипіння доти, поки пляма, що відповідає метил-6-метил нікотинату буде практично відсутня згідно з дослідженням за допомогою ТШХ. Реакційну масу охолоджують до температури 25°-30°C під час перемішування. Реакційну масу охолоджують в суміші колотої криги (75 гр.) + води (15 мл). Рівень pH реакційної маси коригують в діапазоні від 5,2 до 6,2 розбавленою хлористоводневою кислотою. Реакційну масу перемішують протягом однієї години, фільтрують, промивають водою і висушують за температури 60-70°C (вихід 15,7 гр., Чистота 95,7 % визначено за допомогою ВЕРХ). Приклад 3 Приготування 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанону (V) 1-(6-метил-3-піридиніл)-2-ціано-2-[(4-метилтіо) феніл] етанон (VI) (10 гр.) додають до суміші концентрованої хлористоводневої кислоти (70 мл) і крижаної оцтової кислоти (25 мл) за температури 40°-50°C, з наступним декарбоксилюванням за температури кипіння. Хід реакції контролюють за допомогою ТШХ до завершення реакції. Реакційну масу охолоджують до кімнатної температури і промивають шляхом екстрагування гексаном. Вищезазначену реакційну масу повільно виливають у суміш (31,75 мл) концентрованого розчину аміаку і (10,00 мл) води. Реакційну масу перемішують протягом 10 хвилин і рівень pH коригують в діапазоні від 6,80 до 7,20 розведеним розчином аміаку за температури 0-5°C. Охолоджену масу перемішують протягом 30-60 хвилин і фільтрують. Одержаний продукт промивають водою, висушують у лотковій сушарці за температури 45-50°C доти, поки вміст вологи буде нижчим за 2% щоб одержати сполуку (V) у вигляді кремового жовтого порошку . (вихід = 8,8 гр., ВЕРХ чистота = 93,89%) Приклад 4 Приготування 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанону (V) 1-(6-метил-3-піридиніл)-2-ціано-2-[(4-метилтіо) феніл] етанон (VI) (10 гр.) додають до суміші концентрованої хлористоводневої кислоти (80 мл) і крижаної оцтової кислоти (30 мл) за температури 40°-50°C, з наступним декарбоксилюванням за температури кипіння. Хід реакції контролюють за допомогою ТШХ до завершення реакції. Реакційну масу охолоджують до кімнатної температури і промивають шляхом екстрагування гексаном. Вищезазначену реакційну масу повільно виливають в суміш (31,75 мл) концентрованого розчину аміаку і (10,00 мл) води. Реакційну масу перемішують протягом 10 хвилин і рівень pH коригують в діапазоні від 7,00 до 7,20 розведеним розчином аміаку за температури 0-5°C. Охолоджену масу перемішують протягом 30-60 хвилин і фільтрують. Одержаний продукт промивають водою, висушують у лотковій сушарці за температури 45-50°C доти, поки вміст вологи буде нижчим за 2%, щоб одержати сполуку (V) у вигляді кремового жовтого порошку . (вихід = 8,70 гр., ВЕРХ чистота = 95,00%) Приклад 5 Приготування 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанону (V) 9 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 Сполуку (VI) (10 гр.) (одержану за допомогою способу, описаного вище в Прикладі 4) додають до суміші концентрованої хлористоводневої кислоти (75 мл) і крижаної оцтової кислоти (25 мл) за температури навколишнього середовища, нагрівають до температури 70-80C. Після завершення реакції її охолоджують до кімнатної температури і екстрагують (12 мл) толуолом. Вищезазначену реакційну масу потім додають у суміш (31,75 мл) розчину аміаку і води 10,00 мл і перемішують. Реакційну масу коригують до рівня pH в діапазоні від 6,8 до 7,2, використовуючи розчин карбонату натрію (5% мас./об.) за температури 0-5°C. Її перемішують протягом 60 хвилин і фільтрують з одержанням вологого осаду. Вологий продукт промивають двічі водою (10 мл). Вологий осад розчиняють в дихлорметані (70 мл) з наступним додаванням (20 мл) води, перемішують протягом 10 хвилин і розділяють шари. Водний шар двічі екстрагують дихлорметаном. Об’єднаний органічний шар, що містить продукт, промивають з (10 мл) водою., висушують над безводним сульфатом натрію і очищують шляхом обробки активованим вугіллям. Шар продукту концентрують шляхом дистилювання дихлорметану в умовах атмосферного тиску з одержанням напівтвердого залишку. Залишкову масу дегазують під вакуумом протягом 30 хвилин. До маси додають ізопропіловий спирт (5 мл) і охолоджують її до температури 1012°C. Після перемішування протягом 1 години за температури 10-12°C, її фільтрують, і промивають охолодженим ізопропіловим спиртом (2,00 мл). Продукт висушують у вакуумній печі за температури 45°-50°C (7 гр., чистота визначена за допомогою ВЕРХ 93,5 %) Одержаний висушений продукт (7 гр.) очищують шляхом обробки метилізобутилкетоном (17,5 мл). Суспензію продукту перемішують протягом 30 хвилин, фільтрують і промивають метилізобутилкетоном (2 мл), і висушують у вакуумній печі за температури 40-45°C. Очищений продукт (5 гр.) демонструє чистоту згідно ВЕРХ 95,00%. Приклад 6 Приготування 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанону (V) До сполуки (VI) (10 гр.) (відповідно до способу, описаному вище в Прикладі 4) додають суміш концентрованої хлористоводневої кислоти (75 мл) і крижаної оцтової кислоти (25 мл). Після завершення реакції її охолоджуютьдо кімнатної температури і екстрагують (12 мл) толуолом. Вищезазначену реакційну масу додають до суміші (31,75 мл) розчину аміаку і води (10,00 мл) і перемішують. Реакційну масу коригують до рівня pH в діапазоні від 6,9 до 7,1, використовуючи розчин карбонату натрію (5% мас./об.) за температури 0-5°C. Перемішують протягом 60 хвилин і фільтрують з одержанням вологого осаду. Вологий продукт промивають двічі водою (10 мл). Продукт у вигляді вологого осаду розчиняють в (70 мл) дихлорметані з наступним додаванням (20 мл) води, перемішують протягом 10 хвилин і розділяють шари. Водний шар двічі екстрагують дихлорметаном. Об’єднаний органічний шар, що містить продукт, промивають водою (10,00 мл), висушують над безводним сульфатом натрію і очищують шляхом обробки активованим вугіллям. Шар продукту концентрують шляхом дистилювання дихлорметану в умовах атмосферного тиску з одержанням напівтвердого залишку. Залишкову масу дегазують під вакуумом протягом 30 хвилин. Ізопропіловий спирт (7,5 мл) додають до маси і охолоджують її до температури 810°C. Після перемішування протягом 1 години за температури 8-10°C її фільтрують і промивають охолодженим ізопропіловим спиртом (2,00 мл). Продукт висушують у вакуумній печі за температури 45°-50°C, щоб одержати висушений продукт (7 гр., чистота визначена за допомогою ВЕРХ 93,24%) Одержаний висушений продукт (7 гр.) очищують шляхом обробки метилізобутилкетоном (17,5 мл). Суспензію продукту перемішують протягом 30 хвилин, фільтрують і промивають метилізобутилкетоном (2 мл), і висушують у вакуумній печі за температури 40-45°C. Очищений продукт (5,2 гр.) демонструє чистоту згідно ВЕРХ 95,40%. Приклад 7 Очищення 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанону (V) Висушений продукт 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанон (V) (10 гр.) і метанол (150 мл) нагрівають до розчинення. Додають активоване вугілля (0,5 гр.), підтримують перемішування реакційної маси протягом 30 хвилин, а потім фільтрують. Фільтрат охолоджують під час перемішування за температури 0°-3°C протягом однієї години, масу продукту фільтрують і промивають охолодженим метанолом (2,5 мл). Одержаний в такий спосіб очищений продукт висушують під вакуумом за температури 45°-50°C. (вихід 4,5 гр., ВЕРХ чистота 98,65%.) Приклад 8 Очищення 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанону (V) 10 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 Висушений продукт 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанон (V) (10 гр.) і метанол (98 мл) + (2 мл) води нагрівають до розчинення. Додають активоване вугілля (0,5 гр.), підтримують перемішування реакційної маси протягом 30 хвилин, а потім фільтрують. Фільтрат охолоджують під час перемішування за температури 0°-3°C протягом однієї години, масу продукту фільтрують і промивають охолодженим метанолом (2,5 мл). Одержаний в такий спосіб очищений продукт висушують під вакуумом за температури 45°-50°C. (вихід 5,5 гр., ВЕРХ чистота 98,22%) Приклад 9 Приготування вінамідініумної солі 2-хлор-N, N-диметиламіно триметиніум гексафтор фосфату (III). N, N-диметил формамід (440 мл) нагрівають до температури 50°-55°C і протягом періоду 3-4 год. повільно додають ацетилхлорид хлору (99 гр.). Реакційну масу далі нагрівають до температури 65°-70°C і поступово додають оксихлорид фосфору (140 гр.), приблизно протягом 5-6 год. Підтримують перемішування маси протягом 5 год. за температури 65-70°C, охолоджують до температури 25°-30°C з наступним додаванням в суміш колотої криги і води, що містить гексафтор фосфорну кислоту (232 гр.) і гідроксид натрію, підтримуючи рівень pH в діапазоні від 2,0 до 2,2. Охолоджену масу перемішують протягом 30 хвилин, фільтрують для виділення твердого продукту і промивають холодною водою (вихід вологого продукту 260-300 гр.) Сіль вінамідіуму хлориду (III) одержану, як описано вище, очищують шляхом обробки водним ізопропіловим спиртом за температури 75°-80°C, з подальшим охолодженням до температури 20°-23°C. Суспензію маси продукту фільтрують, промивають ізопропіловим спиртом і висушують за температури 55°-65°C доти, поки вміст вологи буде нижчим за 0,5% (вихід 174 гр.) т.пл. 124-127°C. Приклад 10 Приготування вінамідініумної солі 2-хлор-N, N-диметиламіно триметиніум гексафтор фосфату (III) N, N-диметил формамід (400 мл) нагрівають до температури 50°-55°C і протягом періоду 3-4 год. повільно додають ацетилхлорид хлору (99 гр.). Реакційну масу далі нагрівають до температури 65°-70°C і поступово додають оксихлорид фосфору (132 гр.), приблизно протягом 5-6 год. Підтримують перемішування маси протягом 5 год. за температури 65-70°C, охолоджують до температури 25°-30°C з наступним повільним додаванням в суміш колотої криги і води, що містить гексафтор фосфорну кислоту (232 гр.) і гідроксид натрію, підтримуючи рівень pH в діапазоні від 2,1 до 2,5. Охолоджену масу перемішують протягом 30 хвилин, фільтрують для виділення твердого продукту і промивають холодною водою (вихід вологого продукту 260-300 гр.) Сіль вінамідіуму хлориду (III) одержану, як описано вище, очищують шляхом обробки водним ізопропіловим спиртом за температури 75°-80°C, з подальшим охолодженням до температури 20°-23°C. Суспензію маси продукту фільтрують, промивають ізопропіловим спиртом і висушують за температури 55°-65°C доти, поки вміст вологи буде нижчим за 0,5% (вихід 165 гр.). т.пл. 124-127°C. Приклад 11 Очищення 2-хлор-N, N-диметиламіно триметиніум гексафтор фосфату (III) Сполуку (III), одержану як в описаному вище прикладі 10, очищують шляхом кристалізації із метанолу. В речовину (10 гр.) додають метанол (150 мл) і нагрівають до температури кипіння. Масу обробляють активованим вугіллям (0,5 гр.) протягом 30 хвилин, фільтрують гарячою, а потім концентрують за допомогою дистиляції 60-70 мл метанолу. Концентровану речовину потім поступово охолоджують до кімнатної температури, а потім до температури 2°-5°C. Очищений продукт фільтрують і промивають холодним метанолом (5 мл). Його висушують за температури 50°-60°C, до досягнення постійної маси (вихід 8,2 гр. ВЕРХ чистота 98,80%) Приклад 12 Очищення 2-хлор-N, N-диметиламіно триметиніум гексафтор фосфату (III) Сполуку (III), одержану як в описаному вище прикладі 10, очищують шляхом кристалізації із метанолу. В речовину (10 гр.) додають метанол (125 мл) і нагрівають до температури кипіння. Масу обробляють активованим вугіллям (0,5 гр.) протягом 30 хвилин, фільтрують гарячою, концентрують за допомогою дистиляції 60-70 мл метанолу. Концентровану речовину поступово охолоджують до кімнатної температури і далі охолоджують до температури 2°-5°C. Очищений продукт фільтрують і промивають холодним метанолом (5 мл). Його висушують за температури 50°-60°C до досягнення постійної маси (вихід 8,5 гр., ВЕРХ чистота 98,95%) Приклад 13 11 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 Очищення 2-хлор-N, N-диметиламіно триметиніум гексафтор фосфату (III) Сполуку (III), одержану як в описаному вище прикладі 10, очищують шляхом кристалізації із метанолу. В речовину (10 гр.) додають метанол (200 мл) і нагрівають до температури кипіння. Масу обробляють активованим вугіллям (0,5 гр.) протягом 30 хвилин, фільтрують гарячою, концентрують за допомогою дистиляції 60-70 мл метанолу. Концентровану речовину поступово охолоджують до кімнатної температури і далі охолоджують до температури 2°-5°C. Очищений продукт фільтрують і промивають холодним метанолом (5 мл). Його висушують у лотковій сушарці за температури 50°-60°C до досягнення постійної маси (вихід 7,2 гр., ВЕРХ чистота 99,14%) Приклад 14 Приготування 5-хлор-3-(4-метилтіо) феніл-2-(2-метил-5-піридиніл) піридину (IV) 1-(6-метил-3-піридиніл)-2-[4-(метилтіо) феніл] етанон, сполуку з формулою (V) (10 гр.) і ізопропанол (150 мл) суспендують і витримують за температури 5°-8°C протягом 20-30 хвилин. Порошок трет-бутоксиду калію (5.01 гр.) поступово додають до реакційної суміші підтримуючи температуру реакційної маси на рівні 5°-8°C протягом півгодини. Підтримують таку ж саму температуру реакційної маси протягом 3 год. під час перемішування. Сіль 2, хлор-N,Nдиметиламіно триметиніум гексафтор фосфату (III,13 гр.) додають до реакційної суміші, підтримуючи температуру реакційної маси на рівні 5°-8°C. Реакція продовжується протягом періоду часу від 2,5 до 4,0 год. до завершення у відповідності з контролем ТШХ щодо відсутності вихідної сполуки з формулою (V). Згадану вище реакційну масу додають приблизно протягом 15-30 хвилин у суміш ізопропанолу (13,0 мл) і крижаної оцтової кислоти (15 мл), підтримують за температури 5°-10°C. Суміш додатково перемішують протягом від 2,5 до 3,0 год. Потім до неї додають розчин аміаку (27 мл) і масу перемішують протягом 10 хвилин. Потім додають безводний ацетат аміаку (2,4 гр.) і реакційну масу повільно нагрівають до температури кипіння протягом періоду часу, який становить 5-6 год. Формування цільового продукту і завершення реакції контролюють за допомогою аналізу ТШХ. Реакційну масу охолоджують до температури навколишнього середовища і знову додають додаткову кількість розчину аміаку (27 мл) з подальшим додаванням розчину формальдегіду (0,85 мл). Ізопропанол видаляють із реакційної маси за температури 45°-55°C під вакуумом. Додають толуол (80 мл), реакційну масу знову нагрівають до температури 60°-65°C протягом 30 хвилин, надають можливість осадження, а потім органічний і водний шари розділяють. Водний шар повторно екстрагують толуолом (40 мл × 2), толуолові екстракти змішують, промивають 10% розчином карбонату натрію (60 мл) і водою (60 мл). Потім суміш обробляють активованим вугіллям (1,0 гр.) висушують над безводним сульфатом натрію, фільтрують і концентрують,для видалення розчинника за температури 55°60°C під вакуумом. Ізопропанол (35 мл) додають до залишкової маси і охолоджують її поступово до температури 0°-3°C протягом 1-2 годин. Масу продукту фільтрують, промивають 3-5 мл холодного ізопропанолу, і висушують за температури 60°-70° з одержанням сполуки (I). (вихід 7,3 гр., т.пл.98°-102°C, чистота визначена за допомогою ВЕРХ 97,60%) Приклад 15 Приготування 5-хлор-3-(4-метилтіо) феніл-2-(2-метил-5-піридиніл) піридину (IV) 1-(6-метил-3-піридиніл)-2-[4-(метилтіо) феніл] етанон, сполуку з формулою (V) (10 гр.) і ізопропанол (150 мл) суспендують і підтримують за температури 5°-8°C протягом 20-30 хвилин. Поступово до реакційної суміші додають порошок метоксиду калію (2,91 гр.), підтримуючи температуру реакційної маси на рівні 5°-8°C протягом півгодини. Підтримують таку ж саму температуру реакційної маси протягом 3 год. під час перемішування. Сіль 2, хлор-N, Nдиметиламіно триметиніум гексафтор фосфату (III,12,6 гр.) додають до реакційної суміші, підтримуючи температуру реакційної маси на рівні 5°-8°C. Реакція продовжується протягом від 2,5 до 4,0 год. до завершення у відповідності з контролем ТШХ щодо відсутності вихідної сполуки з формулою (V). Вищезгадану реакційну масу додають приблизно протягом 15-30 хвилин у суміш ізопропанолу (13,0 мл) і крижаної оцтової кислоти (14,4 мл) і підтримують за температури 5°10°C. Суміш додатково перемішують протягом від 2,5 до 3,0 год. Потім до неї додають розчин аміаку (27 мл) і масу перемішують протягом 10 хвилин. Потім додають безводний ацетат аміаку (2,4 гр.) і реакційну масу повільно нагрівають до температури кипіння протягом періоду часу, який становить 5-6 год. Формування цільового продукту і завершення реакції контролюють за допомогою аналізу ТШХ. Реакційну масу охолоджують до температури навколишнього середовища і знову додають додаткову кількість розчину аміаку (27 мл) з подальшим додаванням розчину формальдегіду (0,9 мл). Ізопропанол видаляють із реакційної маси за температури 45°-55°C під вакуумом. Додають толуол (80 мл), реакційну масу знову нагрівають 12 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 до температури 60°-65°C протягом 30 хвилин, надають можливість осадження, а потім органічний і водний шари розділяють. Водний шар повторно екстрагують толуолом (40 мл × 2), толуолові екстракти змішують, промивають 10% розчином карбонату натрію (60 мл) і водою (60 мл). Потім суміш обробляють активованим вугіллям (1,0 гр.), висушують над безводним сульфатом натрію, фільтрують і концентрують для видалення розчинника за температури 55°60°C під вакуумом. Ізопропанол (35 мл) додають до залишкової маси і поступово охолоджують її до температури 0°-3°C протягом 1-2 годин. Масу продукту фільтрують, промивають 3-5 мл холодного ізопропанолу і висушують за температури 60°-70° з одержанням (I). (вихід 7,0 гр., т.пл. 98°-102°C, чистота визначена за допомогою ВЕРХ 97,34%) Приклад 16 Очищення 5-Хлор-3-(4-метилтіо) феніл-2-(2-метил-5-піридиніл) піридину (IV) Сполуку (IV), одержану як описано вище (18 гр.) розчиняють в ізопропанолі (54 мл) за температури 60°-65°C під час перемішування. Розчин фільтрують гарячим і фільтрат поступово охолоджують спочатку до температури навколишнього середовища, а потім до температури 0°5°C. Суспензію продукту підтримують охолодженою протягом 2,0 год., фільтрують, промивають охолодженим ізопропанолом (5-10 мл), висушують за температури 45°-50° (вихід 15,5 гр., т.пл.103°-106°C, ВЕРХ чистота = 98,6%). Приклад 17 Очищення 5-хлор-3-(4-метилтіо) феніл-2-(2-метил-5-піридиніл) піридину (IV) Сполуку (IV), одержану як описано вище (18 гр.) розчиняють в суміші ізопропанолу (45 мл) + вода (0,90 мл) за температури 60°-65°C під час перемішування. Розчин фільтрують гарячим і фільтрат поступово охолоджують спочатку до температури навколишнього середовища, а потім до температури 0°-5°C. Суспензію продукту підтримують охолодженою протягом 2,0 год., фільтрують, промивають охолодженим ізопропанолом (5-10 мл) і висушують за температури 45°-50°C (вихід 16 гр., т.пл. 103°-107°C, ВЕРХ чистота = 98,8%) Приклад 18 Очищення 5-хлор-3-(4-метитіо) феніл-2-(2-метил-5-піридиніл) піридину (IV) Сполуку (IV), одержану як описано вище (18 гр.) розчиняють в ізопропанолі (90 мл) + вода (3,60 мл) за температури 60°-65°C під час перемішування. Розчин фільтрують гарячим і фільтрат поступово охолоджують спочатку до температури навколишнього середовища, а потім до температури 0°-5°C. Суспензію продукту підтримують охолодженою протягом 2,0 год., фільтрують, промивають охолодженим ізопропанолом (5-10 мл) і висушують за температури 60°-70° (вихід 14 гр., т.пл. 102°-105°C, ВЕРХ чистота = 98,2%) Приклад 19 Приготування 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридил) піридину (I) Сполуку з формулою (IV), а саме 5-хлор-3-(4-метилтіо) феніл-2-(2-метил-5-піридил)-піридин (10 гр.), розчиняють в дихлорметані (100 мл) і перемішують за температури 25°-28°C . Сірчану кислоту (0,94 гр.) розчинену у воді (0,5 мл) повільно додають до зазначеного вище розчину за температури 10°-15°C. Потім до нього поступово додають розчин вольфрамату натрію (0,17 гр.) в воді (1,5 мл), з подальшим додаванням метил-три-n-октиламіноніуму хлориду (0,25 гр.) і дихлорметану (2 мл). Реакційну масу потім піддають окисненню шляхом поступового додавання 50% розчину перекису водню (6,87 гр.) у воді (2 мл), підтримуючи температуру реакції на рівні 810°C протягом періоду часу від приблизно 1,5 до 2,0 год. Температуру реакції поступово підвищують до 28°-30°C і підтримують протягом декількох годин до завершення реакції під контролем ТШХ. В реакційну масу додають воду (50 мл), і 10% розчин бікарбонату натрію, щоб підтримати pH на рівні 6,8-7,0. Дихлорметановий шар, що містить продукт відділяють, а водну масу двічі екстрагують дихлорметаном (25 мл). Дихлорметанові шари змішують, двічі промивають водою (20 мл), висушують над безводним сульфатом натрію і обробляють активованим вугіллям (0,8 гр.). Профільтрований розчин продукту концентрують за допомогою дистиляції з одержанням залишку продукту. Сліди розчинника видаляють застосуванням вакууму і додають ізопропанол (30 мл). Масу охолоджують до температури (-)2°-2°C протягом періоду часу, який становить 2 год. Продукт виділяють шляхом фільтрації, промивають охолодженим ІПС (3 мл) з наступним висушуванням за температури 60°-70°C (вихід 7,2 гр.,т.пл. 129°-131°C ВЕРХ чистота = 97,14%, порошок світло-кремового кольору). Приклад 20 Приготування 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридил) піридину (I) Сполуку з формулою (IV) а саме 5-хлор-3-(4-метилтіо) феніл-2-(2-метил-5-піридил)-піридин (10 гр.) розчиняють в дихлорметані (100 мл) і перемішують за температури 25°-28°C. До зазначеного вище розчину за температури 10°-15°C повільно додають сірчану кислоту (0,94 гр.) розчинену у воді (0,5 мл). Потім до нього поступово додають розчин вольфрамату натрію (0,17 13 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 гр.) у воді (1,5 мл), з подальшим додаванням метил-три-n-октиламіноніуму хлориду (0,25 гр.) і дихлорметану (2 мл). Потім реакційну масу піддають окисненню шляхом поступового додавання 50% розчину перекису водню (5,62 гр.) у воді (2 мл), підтримуючи температуру реакції на рівні 10-12°C протягом періоду часу від приблизно 1,5 до 2,0 год. Температуру реакції поступово підвищують до 28°-30°C і підтримують протягом декількох годин до завершення реакції під контролем ТШХ. В реакційну масу додають воду (50 мл), і 10% розчин бікарбонату натрію, щоб підтримати pH на рівні 6,6-6,8. Дихлорметановий шар, що містить продукт відділяють, а водну масу двічі екстрагують дихлорметаном (25 мл). Дихлорметанові шари змішують, двічі промивають водою (20 мл), висушують над безводним сульфатом натрію і обробляють активованим вугіллям (0,8 гр.). Профільтрований розчин продукту концентрують за допомогою дистиляції з одержанням залишку продукту. Сліди розчинника видаляють застосуванням вакууму і додають ізопропанол (30 мл). Масу охолоджують до температури (-)2°-2°C протягом періоду часу, який становить 2 год. Продукт виділяють шляхом фільтрації, промивають охолодженим ІПС (3 мл) з наступним висушуванням за температури 60°-70°C (вихід 7,1 гр., т.пл. 129°-132°C ВЕРХ чистота = 97,0%, порошок світло-кремового кольору). Приклад 21 Приготування 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридил) піридину (I) Сполуку з формулою (IV), а саме 5-хлор-3-(4-метилтіо) феніл-2-(2-метил-5-піридил)-піридин (10 гр.) розчиняють в дихлорметані (100 мл) і перемішують за температури 25°-28°C. Сірчану кислоту (0,94 гр.) розчинену у воді (0,5 мл) повільно додають до зазначеного вище розчину за температури 10°-15°C. Потім до нього поступово додають розчин вольфрамату натрію (0,17 гр.) в воді (1,5 мл), з подальшим додаванням метил-три-n-октиламіноніуму хлориду (0,25 гр.) і дихлорметану (2 мл). Потім реакційну масу піддають окисненню шляхом поступового додавання 50% розчину перекису водню (8,12 гр.) в воді (2 мл), підтримуючи температуру реакції на рівні 12-14°C протягом періоду часу від приблизно 1,5 до 2,0 год. Температуру реакції поступово підвищують до 28°-30°C і підтримують протягом декількох годин до завершення реакції під контролем ТШХ. В реакційну масу додають воду (50 мл) і 10% розчин бікарбонату натрію, щоб підтримати pH на рівні 6,95-7,15. Дихлорметановий шар, що містить продукт відділяють, а водну масу двічі екстрагують дихлорметаном (25 мл). Дихлорметанові шари змішують, двічі промивають водою (20 мл), висушують над безводним сульфатом натрію і обробляють активованим вугіллям (0,8 гр.). Профільтрований розчин продукту концентрують за допомогою дистиляції з одержанням залишку продукту. Сліди розчинника видаляють застосуванням вакууму і додають ізопропанол (30 мл). Масу охолоджують до температури (-)2°-2°C протягом періоду часу, який становить 2 год. Продукт виділяють шляхом фільтрації, промивають охолодженим ІПС (3 мл) з наступним висушуванням за температури 60°-70°C (вихід 7,5 гр., т.пл. 131°-134°C ВЕРХ чистота = 96,89%, порошок світло-кремового кольору). Приклад 22 Очищення 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридиніл) піридину, еторикоксибу (I) Еторикоксиб (I), одержаний як в прикладі-19, очищують шляхом кристалізації із водного ізопропанолу. Сполуку (20 гр.) розчиняють ізопропанолом (70 мл), що містить 4% мас./мас. води за температури кипіння під час перемішування. Додають активоване вугілля (1,9 гр.) і через 1520 хвилин кипіння розчин продукту фільтрують. Фільтрат повільно охолоджують до температури навколишнього середовища, а потім до температури 12°-15°C протягом приблизно 1 години. Кристалізований продукт збирають шляхом фільтрації з наступним промиванням охолодженим ізопропанолом (5-10 мл). Масу висушують за температури 60°-70°C з одержанням світло-кремового продукту (вихід 14,4 гр., т.пл. 133°-137°,ВЕРХ чистота 99,10%). Приклад 23 Очищення 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридиніл) піридину, еторикоксибу (I) Еторикоксиб (I), одержаний як в прикладі 19, очищують шляхом кристалізації із водного ізопропанолу. Сполуку (20 гр.) розчиняють ізопропанолом (90 мл), що містить 4%мас./мас. води за температури кипіння під час перемішування. Додають активоване вугілля (1,9 гр.) і через 1520 хвилин кипіння розчин продукту фільтрують. Фільтрат повільно охолоджують до температури навколишнього середовища, а потім до температури 12°-15°C протягом приблизно 1 години. Кристалізований продукт збирають шляхом фільтрації з наступним промиванням охолодженим ізопропанолом (5-10 мл). Масу висушують за температури 60°-70°C з одержанням світлокремового продукту (вихід 14,0 гр., т.пл. 134°-137°, ВЕРХ чистота 99,40%). Приклад 24 Очищення 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридиніл) піридину, 14 UA 114594 C2 5 10 15 20 25 30 35 40 45 50 55 60 еторикоксибу (I) Еторикоксиб (I), одержаний як в прикладі 19, очищують шляхом кристалізації із водного ізопропанолу. Сполуку (20 гр.) розчиняють ізопропанолом (70 мл), що містить 3% мас./мас. води за температури кипіння під час перемішування. Додають активоване вугілля (1,9 гр.) і через 1520 хвилин кипіння розчин продукту фільтрують. Фільтрат охолоджують повільно до температури навколишнього середовища, а потім до температури 12°-15°C протягом приблизно 1 години. Кристалізований продукт збирають шляхом фільтрації з наступним промиванням охолодженим ізопропанолом (5-10 мл). Масу висушують за температури 60°-70°C з одержанням світлокремового продукту (вихід 15,3 гр., т.пл. 135°-137°C ВЕРХ чистота 99,18%). Приклад 25 Очищення 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридиніл) піридину, еторикоксибу (I) Еторикоксиб (I), одержаний як в прикладі 19, очищують шляхом кристалізації із водного ізопропанолу. Сполуку (20 гр.) розчиняють в суміші ізопропанолу (60 мл) + ацетону (15 мл), що містить 6% мас./мас. води за температури кипіння під час перемішування. Додають активоване вугілля (1,9 гр.) і через 15-20 хвилин кипіння розчин продукту фільтрують. Фільтрат охолоджують повільно до температури навколишнього середовища, а потім до температури 12°-15°C протягом приблизно 1 години. Кристалізований продукт збирають шляхом фільтрації з наступним промиванням охолодженим ізопропанолом (5-10 мл). Масу висушують за температури 60°-70°C з одержанням світло-кремового продукту (вихід 15 гр., т.пл. 135°-137°C ВЕРХ чистота 99,35%). Приклад 26 Очищення 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридиніл) піридину, еторикоксибу (I) Еторикоксиб (I), одержаний як в прикладі 19, (20 гр.) розчиняють в етилацетаті (110 мл) в умовах навколишнього середовища, під час перемішування. До нього повільно невеликими партіями додають паратолуолсульфонову кислоту (16 гр.). Масу нагрівають до температури 55°-60°C з одержанням прозорого розчину, а потім обробляють 0,5 гр. активованого вугілля протягом 15-20 хвилин. Розчин фільтрують гарячим, і перемішуючи охолоджують до температури 10°-12°C протягом періоду часу, який становить приблизно одну годину. Сіль паратолуолсульфонової кислоти еторикоксибу виділяють шляхом фільтрації, промивають охолодженим етилацетатом (10 мл - 15 мл), і висушують за температури 50°-60° до досягнення постійної маси (вихід 28 гр.) До одержаної вище солі еторикоксиб-ПТСК (28 гр.) додають воду (250 мл) і перемішують за температури навколишнього середовища протягом 15-20 хвилин. Поступово додають 10% розчин бікарбонату натрію до досягнення рівня pH близько 7,90 до 8,10, з наступним додаванням толуолу (50 мл). Реакційну суміш нагрівають до температури 50°-55°C, перемішують протягом 10 хвилин і витримують. Толуоловий шар що містить продукт відділяють, а водний шар двічі екстрагують толуолом (80 мл) аналогічним чином об’єднаний толуоловий шар промивають водою (100 мл), обробляють активованим вугіллям (2,5 гр.), висушують над безводним сульфатом натрію, фільтрують і концентрують за температури 60-65°C під зниженим тиском з одержанням залишку. Ізопропанол (94 мл) що містить 4% води додають до залишку і нагрівають його до температури 55°-65°C для розчинення. Розчин повільно охолоджують до температури навколишнього середовища, а потім до температури 5°-10° протягом 1,5 год. Масу продукту фільтрують, промивають холодним ізопропанолом (10 мл) і висушують за температури 60°-65°C до досягнення постійної маси (вихід 11 гр.,т.пл. 135°-137°C, ВЕРХ чистота = 99,23%) Приклад 27 Приготування 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридиніл) піридину, еторикоксибу (I) Еторикоксиб (I), одержаний як в прикладі 19, (20 гр.) розчиняють в ізопропіловому спирті (70 мл) + вода (4,20 мл) за температури кипіння під час перемішування. Додають активоване вугілля (1,9 гр.) і через 15-20 хвилин розчин продукту фільтрують. Фільтрат повільно охолоджують до температури навколишнього середовища, а потім до температури 12°-15°C протягом приблизно 1 години. Кристалізований продукт збирають шляхом фільтрації з наступним промиванням охолодженим ізопропанолом (5-10 мл). Масу висушують за температури 60°-70°C з одержанням світло-кремового продукту (вихід 15,3 гр., т.пл. 135°-137°C, ВЕРХ чистота 99,10%). Приклад 28 Приготування 5-хлор-3-(4-метилсульфоніл) феніл-2-(2-метил-5-піридиніл) піридину, еторикоксибу (I) 15 UA 114594 C2 5 10 15 20 25 30 35 40 45 Еторикоксиб (I), одержаний як в прикладі 21, (20 гр.) розчиняють в чистому етанолі (80 мл) за температури кипіння під час перемішування. Додають активоване вугілля (1,9 гр.) і через 1520 хвилин розчин продукту фільтрують. Фільтрат повільно охолоджують до температури навколишнього середовища, а потім до температури 12°-15°C протягом приблизно 1 години. Кристалізований продукт збирають шляхом фільтрації з наступним промиванням охолодженим чистим етанолом (5-10 мл). Масу висушують за температури 60°-70°C з одержанням світлокремового продукту (вихід 15,1гр., т.пл. 136°-138°C, ВЕРХ чистота 99,28%) Приклад 29 Приготування 1-(6-метил-3-піридиніл)-2-[(4-метилсульфоніл) феніл] етанону (II) 4-метил тіобензил ціанід (20,0 гр.) розчиняють в (100 мл) толуолі і кип’ятять реакційну масу. Повільно, протягом 45 хвилин додають метил-6-метил нікотинат (21,74 гр.). Порошок метоксиду натрію 12,0 гр. вводять протягом 30 хвилин. Температуру реакції підтримують за кипіння протягом приблизно 4-6 год., доти, поки пляма, що відповідає метил-6-метил нікотинату буде практично відсутня на ТШХ. Реакційну масу охолоджують перемішуючи до температури 25°30°C з наступним повільним додаванням в суміш колотої криги (190 гр.) + води (45 мл). Рівень pH реакційної маси коригують в діапазоні від 5,2 до 6,2 розбавленою хлористоводневою кислотою і перемішують протягом однієї години, фільтрують, промивають водою і висушують відсмоктуванням на фільтрувальній бюхнеровській воронці. Цю вологу масу продукту1-(6метил-3-піридиніл)-2-ціано-2-[(4-метилтіо) феніл] етанону (VI) додають до суміші концентрованої хлористоводневої кислоти (140 мл) і крижаної оцтової кислоти (60 мл) за температури 40°-50°C з наступним декарбоксилюванням за температури кипіння. Реакційну масу охолоджують до кімнатної температури як тільки реакція завершується, про що свідчить ТШХ і промивають шляхом екстрагування гексаном. Вищезазначену реакційну масу повільно виливають у суміш (70 мл) концентрованого розчину аміаку і (25,00 мл) води. Реакційну масу перемішують протягом 10 хвилин і рівень pH коригують в діапазоні від 6,80 до 7,20 розведеним розчином аміаку за температури 0-5°C. Масу продукту витримують протягом 30 хвилин і фільтрують. Одержаний продукт промивають водою і висушують центрифугуванням. Масу продукту (вологу) 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанону (V) розчиняють в дихлорметані (185 мл), перемішують, надають можливість осадження і відділяють водний шар. Дихлорметановий розчин продукту безпосередньо переносять на наступний синтетичний етап окиснення з сумішшю концентрованої сірчаної кислоти (4,10 гр.) + води (4,50 мл) і перемішують протягом 15-20 хвилин. Також загружають розчин вольфрамату натрію (0,74 гр.) в воді (14,0 мл) і реакційну масу перемішують протягом 10 хвилин з наступним додаванням суміші метил-три-n-октил хлориду (0,80 гр.) в дихлорметані (8,0 мл). Реакційну суміш перемішують протягом 10 хвилин і охолоджують до температури 18°-20°C. Протягом 45 хвилин поступово додають суміш 50% перекису водню (19,50 гр.) і води (12,50 мл) за температури 18°20°C. Реакційну масу тримають декілька годин і контролюють за допомогою аналізу ТШХ, доти, поки пляма, що відповідає 1-(6-метил-3-піридиніл)-2-[(4-метилтіо) феніл] етанону (V) буде практично відсутня згідно ТШХ. Рівень pH реакційної маси встановлюють в діапазоні від 6,95 до 7,10 використовуючи суміш розведеного розчину аміаку, перемішуючи протягом 30 хвилин за температури 25°-30°C. ЇЇ перемішують протягом 10 хвилин, водний шар відділяють і екстрагують дихлорметаном (130 мл). Об’єднаний дихлорметановий шар, що містить продукт промивають водою (110 мл), висушують над безводним сульфатом натрію і фільтрують. Дихлорметан в умовах атмосферного тиску з одержанням. Наприкінці застосовують вакуум і масу дегазують Потім додають ізопропіловий спирт (96 мл), перемішують масу і охолоджують до температури 25°-30°C з наступним охолодженням до температури 0°-5°C, підтримують протягом однієї години, фільтрують, промивають охолодженим ізопропіловим спиртом і висушують за температури 50°-60°C (вихід 21 гр., т.пл. 176°-180°C, чистота визначена за допомогою ВЕРХ92,50%). 50 ФОРМУЛА ВИНАХОДУ 55 1. Спосіб одержання етерококсибу, який включає піддавання 5-хлор-3-(4-метилтіо)феніл-2-(2метил-5-піридиніл)піридину (IV) окисненню за присутності каталізатора окиснення і каталізатора фазового переходу з одержанням еторикоксибу з формулою (І), який відрізняється тим, що каталізатор фазового переходу вибирають із групи, яка складається з метил-три-n-октиламонію хлориду, метил-три-n-бутиламонію хлориду, бензетонію хлориду і метилбензетонію хлориду 16 UA 114594 C2 H3C O S Cl N S H3C O Cl (0) N H3C N H3C (IV) 5 10 15 20 25 30 35 40 45 N . 2. Спосіб за п. 1, який відрізняється тим, що включає: а) конденсацію 4-метилтіобензилціаніду і метил-6-метилнікотинату за присутності придатної основи і придатного розчинника за температури кипіння з одержанням 1-(6-метил-3-піридиніл)2-ціано-2-[(4-метилтіо)феніл]етанону (VI); б) гідроліз сполуки (VI), одержаної на етапі конденсації за присутності кислоти за температури 40-50 °C з наступним декарбоксилюванням in situ за температури кипіння з одержанням 1-(6метил-3-піридиніл)-2-[4-(метилтіо)феніл]етанону з формулою (V); в) взаємодію сполуки за формулою (V), одержаної на етапі гідролізу сполуки (VI) з сіллю 2-хлорN,N-диметиламінотриметиніумгексафторфосфату (III) за присутності основи з наступним додаванням суміші спирту і кислоти, додаванням водного розчину аміаку з наступним додаванням солі аміаку та нагріванням з одержанням 5-хлор-3-(4-метилсульфоніл)феніл-2-(2метил-5-піридиніл)піридину (IV), г) піддавання одержаної сполуки з формулою (IV) окисненню за присутності каталізатора окиснення і каталізатора фазового переходу з одержанням еторикоксибу з формулою (І). 3. Спосіб за п. 2, який відрізняється тим, що основу для етапу конденсації 4метилтіобензилціаніду і метил-6-метилнікотинату вибирають із групи, що складається з метоксиду натрію, аміду натрію, гідриду натрію і трет-бутоксиду калію. 4. Спосіб за п. 2, який відрізняється тим, що розчинник для етапу конденсації 4метилтіобензилціаніду і метил-6-метилнікотинату вибирають із вуглеводнів, таких як гептан, толуол, ксилен або їх суміші. 5. Спосіб за п. 2, який відрізняється тим, що гідроліз сполуки (VI) проводять в суміші органічної кислоти і неорганічної кислоти. 6. Спосіб за п. 5, який відрізняється тим, що органічну кислоту вибирають із групи, що складається з мурашиної кислоти, льодяної оцтової кислоти, пропіонової кислоти, масляної кислоти і пентанової кислоти. 7. Спосіб за п. 6, який відрізняється тим, що неорганічну кислоту вибирають із концентрованої хлористоводневої кислоти або концентрованої сірчаної кислоти. 8. Спосіб за п. 2, який відрізняється тим, що основу для етапу взаємодії сполуки за формулою (V) з сіллю 2-хлор-N,N-диметиламінотриметиніумгексафторфосфату (III) вибирають із групи, яка складається з метоксиду натрію, метоксиду калію, трет-бутоксиду калію, аміду натрію і гідриду натрію. 9. Спосіб за п. 2, який відрізняється тим, що спирт для етапу взаємодії сполуки за формулою (V) з сіллю 2-хлор-N,N-диметиламінотриметиніумгексафторфосфату (ІІІ) вибирають із органічних розчинників, таких як трет-бутанол, ізопропанол, тетрагідрофуран і метил-третбутиловий ефір. 10. Спосіб за п. 2, який відрізняється тим, що кислоту для етапу взаємодії сполуки за формулою (V) з сіллю 2-хлор-N,N-диметиламінотриметиніумгексафторфосфату (ІІІ) вибирають із органічних кислот, таких як мурашина кислота, оцтова кислота, n-пропіонова кислота і nмасляна кислота. 11. Спосіб за п. 2, який відрізняється тим, що каталізатор окиснення для етапу піддавання одержаної сполуки з формулою (IV) окисненню вибирають із молібдату натрію, ванадату натрію і вольфрамату натрію. 12. Спосіб за п. 2, який відрізняється тим, що окиснення на етапі піддавання одержаної сполуки з формулою (IV) окисненню проводять за присутності перекису. 13. Спосіб за п. 4, який відрізняється тим, що етап піддавання одержаної сполуки з формулою (IV) окисненню проводять у двофазній системі, що включає галогеновані вуглеводні і воду. 17 UA 114594 C2 18 UA 114594 C2 19 UA 114594 C2 Комп’ютерна верстка А. Крулевський Міністерство економічного розвитку і торгівлі України, вул. М. Грушевського, 12/2, м. Київ, 01008, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 20
ДивитисяДодаткова інформація
Автори англійськоюShah, Dharmesh Mahendra, Solanki, Sanjay Amratlal, Jariwala, Viral Narendra, Vyas, Ashok Vasantray, Mistry, Ashokkumar Bhikhubhai
Автори російськоюШах Дхармеш Махедра, Соланки Санджай Амратлал, Джаривала Вирал Нарендра, Вьяс Ашок Васантрай, Мистри Ашоккумар Бхикхубхайи
МПК / Мітки
МПК: C07D 213/61
Мітки: селективного, спосіб, циклооксигенази-2, інгібітора, одержання
Код посилання
<a href="https://ua.patents.su/22-114594-sposib-oderzhannya-selektivnogo-ingibitora-ciklooksigenazi-2.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання селективного інгібітора циклооксигенази-2</a>
Композиція солей похідних амідину та інгібітора циклооксигенази, спосіб її одержання, застосування та фармацевтична композиція

Номер патенту: 53624
Опубліковано: 17.02.2003
Автори: Шабрієр де Лассон'єр, Броке Колетт
МПК: A61K 31/155, A61K 31/19, A61K 31/195, A61K 31/405, A61K 31/60
Мітки: спосіб, інгібітора, застосування, амідину, солей, циклооксигенази-2, одержання, фармацевтична, похідних, композиція
Формула / Реферат:
1. Композиція солей похідних амідину та інгібітора циклооксигенази, яка складається із сполук A і B,деА є інгібітором циклооксигенази, що представляє карбокси групу,В є сполука загальної формули (Iв), (Iв)деR1 є Н, нітро або феніловий радикал, феніловий радикал, який при необхідності заміщується одним або кількома замісниками,...
Фармацевтична одинична дозована форма опіоїдного анальгетика та інгібітора циклооксигенази-2

Номер патенту: 72193
Опубліковано: 15.02.2005
Автори: Берч Рональд М., Голденхейм Пол Д., Саклер Річард С.
МПК: A61K 9/48, A61F 2/02, A61K 9/28, A61K 9/20
Мітки: одинична, фармацевтична, опіоїдного, анальгетика, форма, циклооксигенази-2, інгібітора, дозована
Формула / Реферат:
1. Фармацевтична одинична дозована форма, що містить аналгезивну комбінацію, яка містить (а) інгібітор СОХ-2 та/або щонайменше одну його фармацевтично прийнятну сіль і (б) оксикодон та/або щонайменше одну його фармацевтично прийнятну сіль, причому зазначений інгібітор СОХ-2 має щонайменше в 9 разів більшу специфічність по відношенню до СОХ-2, ніж до СОХ-1, як in vivo (що визначається шляхом вимірювання ED50), так і/або in vitro (що...
Спосіб синтезу інгібіторів сох-2 (циклооксигенази-2) та проміжні сполуки для їх одержання
Номер патенту: 57143
Опубліковано: 16.06.2003
Автори: Корлі Едвард Г., Девіс Ян В., Ларсен Роберт Д., Пай Філіп Дж., Россен Кай
МПК: A61K 31/4418, C07C 251/30, A61P 29/00, A61K 31/444, A61P 43/00, A61P 31/16, A61P 35/00, C07D 213/34, A61K 31/44, C07D 213/61, A61P 35/04, A61P 31/12, C07C 251/12
Мітки: спосіб, інгібіторів, проміжні, сполуки, сох-2, циклооксигенази-2, синтезу, одержання
Формула / Реферат:
1. Спосіб синтезу сполуки формули (І), (І)деприсутні 0-2 групи R;кожний з R, R' і R", незалежно, являє собою С1-10-алкіл, С-6-10-арил, аралкіл, галоген, -S(O)mH, -S(O)mС1-6-алкіл, -S(О)m-арил, нітро, аміно, С1-6-алкіламіно, ді-С1-6-алкіламіно, -S(O)mNH2, -S(О)mNH-С1-6-алкіл, -S(O)mNНС(O)СF3 і ціано, причому алкільна і арильна групи і алкільна і арильна частини аралкілу, -S(O)m-С1-6-алкілу, -S(O)m-арилу,...
Лікарський засіб для перорального введення, який містить інгібітор циклооксигенази-2, та спосіб його одержання

Номер патенту: 93868
Опубліковано: 25.03.2011
Автори: Мореа Маріне, Ості Нікола
МПК: A61K 9/50
Мітки: циклооксигенази-2, одержання, інгібітор, лікарський, введення, засіб, перорального, містить, спосіб
Формула / Реферат:
1. Лікарський засіб, призначений для перорального введення, який має покращену біодоступність, який містить агломерат на базі інертних твердих частинок на базі як мінімум однієї допоміжної речовини, причому агломерат містить інгібітор циклооксигенази-2 і як мінімум один гідрофільний полімер,який відрізняється тим, що агломерат містить продукт розпилення частинок з розчином або суспензією мікронізованих гранул інгібітора у полімері(ах)...
Поєднання антагоніста nmda-рецептора і селективного інгібітора зворотного захоплення серотоніну для лікування депресії і інших психічних розладів

Номер патенту: 80055
Опубліковано: 10.08.2007
Автори: Саморіскі Гарі, Гупта Сандіп
МПК: A61K 31/13, A61P 25/24, A61K 31/343
Мітки: поєднання, захоплення, психічних, nmda-рецептора, розладів, лікування, селективного, серотоніну, інших, депресії, антагоніста, інгібітора, зворотного
Формула / Реферат:
1. Спосіб лікування пацієнта з психічним порушенням, вибраним з групи, що складається з депресії, дистимії, сезонного афективного розладу, біполярного психозу і післяпологової депресії, який включає в себе введення пацієнту, потребуючому цього, поєднання, що містить терапевтично ефективну(і) першу кількість сполуки, що має властивості функціонального антагоніста рецепторного комплексу N-метил-D-аспартату (NMDA); і(іі) другу...
Попередній патент: Спосіб одержання метаксалону
Наступний патент: Гербіцидні суспензії капсул ацетохлору, що містять знижені кількості антидоту
Випадковий патент: Пристрій для регулювання температури