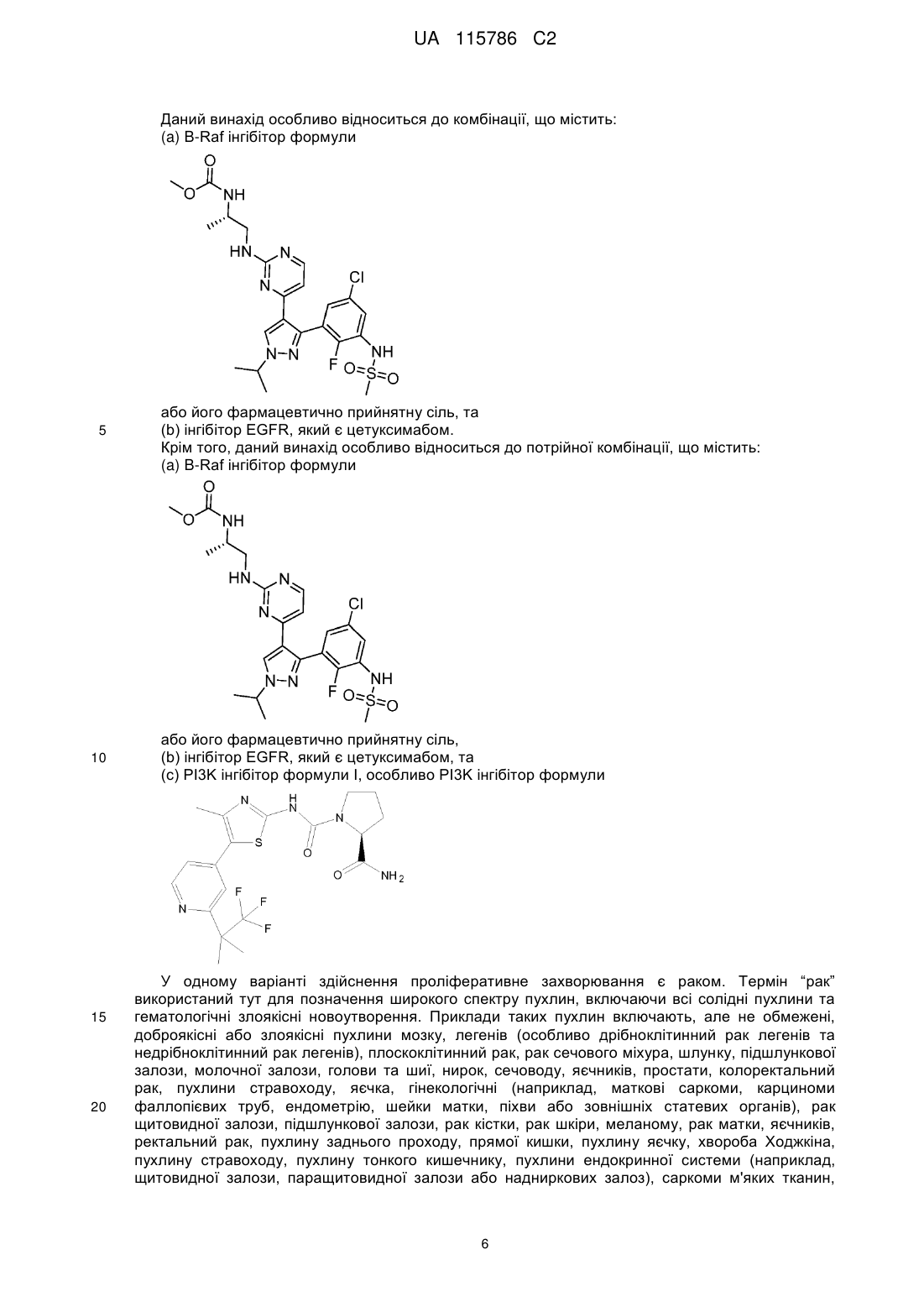
Фармацевтична комбінація, що містить інгібітор b-raf, інгібітор egfr та, необов’язково, інгібітор рі3k-a
Номер патенту: 115786
Опубліковано: 26.12.2017
Автори: Муту-Де Парсеваль Лора, Капонігро Джордано, Стюарт Даррін






Формула / Реферат
1. Фармацевтична комбінація, що містить:
(а) інгібітор B-Raf формули
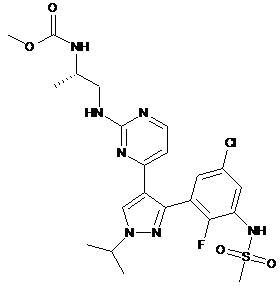
або його фармацевтично прийнятну сіль,
(b) інгібітор EGFR, де інгібітор EGFR являє собою ерлотиніб або цетуксимаб та, необов'язково,
(c) інгібітор РІ3K-a, де інгібітор РІ3К-a являє собою Сполуку В
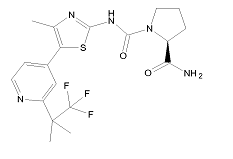 , (В)
, (В)
для одночасного, окремого або послідовного введення.
2. Комбінація за п. 1, у якій інгібітор EGFR являє собою ерлотиніб.
3. Комбінація за п. 1, у якій інгібітор EGFR являє собою цетуксимаб.
4. Застосування комбінації за п. 1 для виробництва лікарського препарату для лікування колоректального раку.
5. Спосіб лікування колоректального раку у пацієнтів-людей, де колоректальний рак характеризується мутацією B-Raf, що включає одночасне, окреме або послідовне введення терапевтично ефективної кількості:
(а) інгібітору B-Raf формули
або його фармацевтично прийнятної солі,
(b) інгібітору EGFR, де інгібітор EGFR являє собою ерлотиніб або цетуксимаб та, необов'язково,
(c) інгібітору РІ3K-a, де інгібітор РІ3К-a являє собою Сполуку В
. (В)
6. Спосіб за п. 5, у якому мутація B-Raf є V600 мутацією.
Текст
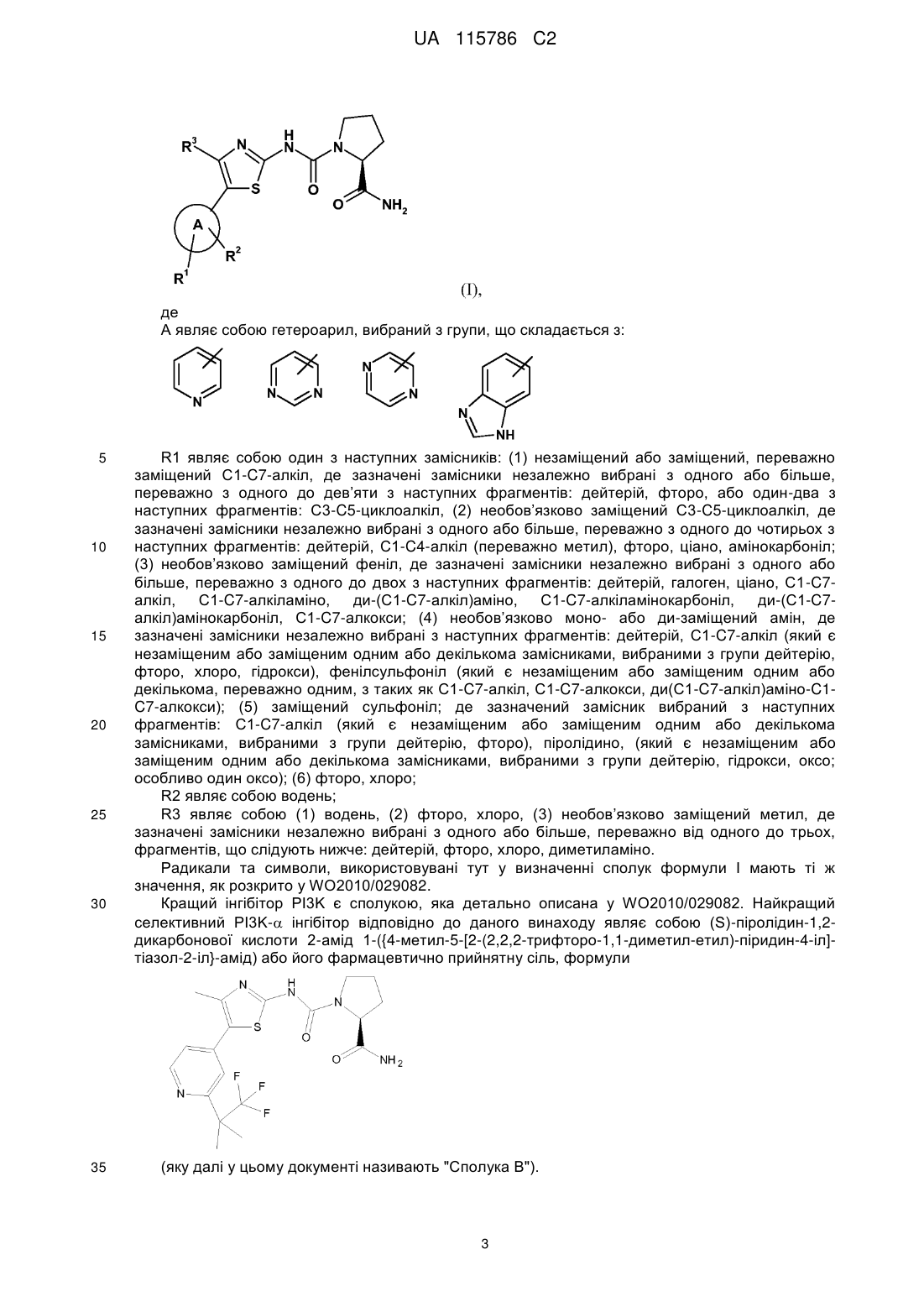
Реферат: Винахід стосується фармацевтичної комбінації, що містить (а) інгібітор B-Raf, (b) інгібітор EGFR та, необов'язково, (с) інгібітор РІ3K, застосування такої комбінації у лікуванні колоректального раку та способу лікування суб'єкта, що страждає на колоректальний рак, що включає введення терапевтично ефективної кількості такої комбінації. UA 115786 C2 (12) UA 115786 C2 UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 Комбінація інгібітору кінази B-Raf та інгібітору рецептору епідермального фактору росту (EGFR, також відомого як ErbB-1 або HER-1) та, необов’язково, інгібітору фосфатидилінозитол 3-кінази (PI 3-кінази або PI3K), який застосовується для лікування проліферативних захворювань. Даний винахід також відноситься до застосування таких комбінацій у лікуванні проліферативних захворювань; до фармацевтичних композицій комбінацій засобів та способів лікування суб’єктів, що страждають на проліферативні захворювання, включаючи введення терапевтично ефективної кількості такої комбінації суб’єкту. РІВЕНЬ ТЕХНІКИ Протеїнкінази являють собою велике сімейство білків, які відіграють центральну роль у регулюванні широкого спектру клітинних процесів та підтримці контролю над клітинними функціями. Аберантна кіназна активність спостерігається на багатьох стадіях захворювання, включаючи доброякісні та злоякісні проліферативні захворювання, також як і захворювання, обумовлені неприпустимою активацією імунної та нервової систем. Сімейство Raf серін/треонінових кіназ включає три члени: C-Raf (або Raf-1), B-Raf та A-Raf. Активуючий алель B-Raf був ідентифікований у 70% меланом, 40% папілярного раку щитовидної залози, 30% низькодиференційованого раку яєчників та 10% колоректального раку. Більшість мутацій B-Raf знайдені всередині кіназного домену, з однією заміною (V600E) відповідальною за 80%. Мутовані B-Raf білки активують Raf-MEK-ERK шлях або за допомогою підвищеної кіназної активності стосовно MEK, або шляхом активування С-Raf. Інгібітор B-Raf у існуючій комбінованій терапії інгібує клітинні процеси, що зачіпають B-Raf кіназу, блокуючи сигнальний каскад у цих ракових клітинах та, у остаточному підсумку, викликаючи зупинку росту та/або смерть клітин. Інгібітори B-Raf, застосовувані у існуючих комбінаціях, загалом та конкретно описані у опублікованій PCT патентній заявці WO2011/025927, яка включена тут за допомогою посилання. Існують три класи PI3-кіназ (PI3K). Ферменти класу I складаються з гетеродимерів, що мають регуляторний (p85) домен та каталітичну (p110) субодиницю, для якої існують чотири ізоформи: p110, p110, p110 та p110. Ізоформи та експресуються убіквітарно; зв'язана вище по сигнальних шляхах, головним чином, з рецепторними тирозинкіназами, тоді як може бути посередником сигналів та від рецепторів, сполучених з G-білком, та від рецепторних тирозинкіназ. Ізоформи та екпресуються насамперед у лімфоцитах та відіграють важливі ролі у регуляції імунних відповідей. Посилення PI3K сигналінгу поширене у багатьох типах людського раку, та включає дезактивацію гену-супресору пухлинного росту PTEN, ампліфікацію/надекспресію або активуючу мутацію деяких рецепторних тирозинкіназ (наприклад, erbB3, erbB2, EGFR), ампліфікацію геномних областей, що містять AKT, ампліфікацію PIK3CA (гену, що кодує p110) та мутації у p110. Як нещодавно було знайдено, більше ніж 30% різних типів солідних пухлин містять мутації PIK3CA. При такій частоті мутацій, PIK3CA є одним з найбільш часто мутованих генів, ідентифікованих при людських ракових утвореннях. Інгібітори PI3K, застосовувані у даному способі, що мають інгібуючу активність для -ізоформи PI3-кіназ, описані у WO2010/029082, який включений тут за допомогою посилання. EGFR є трансмембранними рецепторами, представленими на клітинній мембрані. У них є позаклітинна зв’язуюча частина, трансмембранна частина та внутрішньоклітинна тирозинкіназна частина. EGFR відіграють важливу роль у керуванні нормальним клітинним ростом, апоптозом та іншими клітинними функціями. Порушення активності EGFR може привести до безперервної або аномальної активації рецепторів, що викликає нерегульоване клітинне ділення. У галузі техніки відомі інгібітори рецептору епідермального фактору росту. Як правило, вони є або низькомолекулярними інгібіторами тирозинкіназ, такими як ерлотиніб та гефітиніб, або моноклональними антитілами. Aнти-EGFR моноклональні антитіла, такі як цетуксимаб та панітумумаб, є особливо використовуваними інгібіторами EGFR для застосування відповідно до даного винаходу. Цетуксимаб, його препарат та застосування для лікування проліферативних захворювань розкриті у патенті США № 6217866, який тут включений за допомогою посилання. Панітумумаб, його препарат та застосування для лікування проліферативних захворювань розкриті у патенті США № 6235883, який тут включений за допомогою посилання. СУТЬ ВИНАХОДУ Даний винахід відноситься до терапевтичної комбінації, що містить: (a) інгібітор B-Raf, (b) інгібітор EGFR та, необов’язково, (c) інгібітор PI3K, застосовуваний для окремого, одночасного або послідовного введення суб’єкту, який цього потребує, для лікування або профілактики проліферативного захворювання. Даний винахід особливо відноситься до терапевтичної комбінації, яка містить: 1 UA 115786 C2 (a) B-Raf інгібітор формули 5 10 або його фармацевтично прийнятну сіль (що далі у цьому документі називають Сполука A). (b) інгібітор EGFR та, необов’язково, (c) PI3K інгібітор. ДОКЛАДНИЙ ОПИС ВИНАХОДУ Даний винахід відноситься до терапевтичної комбінації, що містить: (a) інгібітор B-Raf, (b) інгібітор EGFR та, необов'язково, (c) інгібітор PI3K, застосовуваний для роздільного, одночасного або послідовного введення суб'єктові, який цього потребує, для лікування або профілактики проліферативного захворювання. Даний винахід особливо відноситься до терапевтичної комбінації, що містить: (a) B-Raf інгібітор формули або його фармацевтично прийнятну сіль (що далі у цьому документі називають "Сполука 15 A"). 20 (b) інгібітор EGFR та, необов’язково, (c) PI3K інгібітор, особливо селективний PI3K- інгібітор. Даний винахід особливий відноситься до терапевтичної комбінації, у якій інгібітор EGFR є інгібітором тирозинкінази, таким як ерлотиніб або гефітиніб, особливо ерлотиніб, та особливо, у якій інгібітор EGFR є моноклональним антитілом, наприклад, цетуксимабом або панітумумабом, особливо цетуксимабом. У галузі техніки відомі PI3K інгібітори. Додатковий інгібітор PI3K особливо є селективним інгібітором PI3K-, який являє собою похідну 2-карбоксамід циклоаміно сечовини, описану у WO2010/029082, зокрема сполуку формули (I) 2 UA 115786 C2 де А являє собою гетероарил, вибраний з групи, що складається з: 5 10 15 20 25 30 35 R1 являє собою один з наступних замісників: (1) незаміщений або заміщений, переважно заміщений C1-C7-алкіл, де зазначені замісники незалежно вибрані з одного або більше, переважно з одного до дев’яти з наступних фрагментів: дейтерій, фторо, або один-два з наступних фрагментів: C3-C5-циклоалкіл, (2) необов’язково заміщений C3-C5-циклоалкіл, де зазначені замісники незалежно вибрані з одного або більше, переважно з одного до чотирьох з наступних фрагментів: дейтерій, C1-C4-алкіл (переважно метил), фторо, ціано, амінокарбоніл; (3) необов’язково заміщений феніл, де зазначені замісники незалежно вибрані з одного або більше, переважно з одного до двох з наступних фрагментів: дейтерій, галоген, ціано, C1-C7алкіл, C1-C7-алкіламіно, ди-(С1-C7-алкіл)аміно, C1-C7-алкіламінокарбоніл, ди-(С1-C7алкіл)амінокарбоніл, C1-C7-алкокси; (4) необов’язково моно- або ди-заміщений амін, де зазначені замісники незалежно вибрані з наступних фрагментів: дейтерій, C1-C7-алкіл (який є незаміщеним або заміщеним одним або декількома замісниками, вибраними з групи дейтерію, фторо, хлоро, гідрокси), фенілсульфоніл (який є незаміщеним або заміщеним одним або декількома, переважно одним, з таких як C1-C7-алкіл, C1-C7-алкокси, ди(С1-C7-алкіл)аміно-C1C7-алкокси); (5) заміщений сульфоніл; де зазначений замісник вибраний з наступних фрагментів: C1-C7-алкіл (який є незаміщеним або заміщеним одним або декількома замісниками, вибраними з групи дейтерію, фторо), піролідино, (який є незаміщеним або заміщеним одним або декількома замісниками, вибраними з групи дейтерію, гідрокси, оксо; особливо один оксо); (6) фторо, хлоро; R2 являє собою водень; R3 являє собою (1) водень, (2) фторо, хлоро, (3) необов’язково заміщений метил, де зазначені замісники незалежно вибрані з одного або більше, переважно від одного до трьох, фрагментів, що слідують нижче: дейтерій, фторо, хлоро, диметиламіно. Радикали та символи, використовувані тут у визначенні сполук формули I мають ті ж значення, як розкрито у WO2010/029082. Кращий інгібітор PI3K є сполукою, яка детально описана у WO2010/029082. Найкращий селективний PI3K- інгібітор відповідно до даного винаходу являє собою (S)-піролідин-1,2дикарбонової кислоти 2-амід 1-({4-метил-5-[2-(2,2,2-трифторо-1,1-диметил-етил)-піридин-4-іл]тіазол-2-іл}-амід) або його фармацевтично прийнятну сіль, формули (яку далі у цьому документі називають "Сполука В"). 3 UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 Далі тут подвійні комбінації Сполуки A та EGFR інгібітору, потрійні комбінації Сполуки A, EGFR інгібітору, та інгібітор PI3K формули I та, більш конкретно, подвійні комбінації Сполуки A та цетуксимабу та потрійні комбінації Сполуки A та цетуксимабу та Сполуки B будуть згадуватися як КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ. Даний винахід зокрема має відношення до КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ, застосовуваній для окремого, одночасного або послідовного введення суб'єктові, який цього потребує, для лікування або профілактики проліферативного захворювання. Даний винахід також має відношення до КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ для застосування у приготуванні фармацевтичної композиції для лікування або профілактики проліферативного захворювання. Даний винахід додатково має відношення до КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ для приготування фармацевтичної композиції або лікарського препарату для лікування або профілактики проліферативного захворювання. Даний винахід відноситься до способу лікування суб'єкта, що страждає на проліферативне захворювання, що включає введення зазначеному суб'єктові КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ у кількості, яка є сумарно терапевтично ефективною у відношенні проліферативного захворювання. Даний винахід надає комерційне упакування, що включає як терапевтичний засіб КОМБІНАЦІЮ ВІДПОВІДНО ДО ВИНАХОДУ, разом з рекомендаціями з одночасного, окремого або послідовного введення при застосуванні для уповільнення прогресування або для лікування проліферативного захворювання. Загальні терміни, використані тут, мають наступне значення, якщо явно не заявлене інакше: Терміни “що включає” та “що має у своєму складі”, використані тут у їхньому відкритому та необмежуючому змісті, якщо не зазначене інше. Терміни у однині та подібні посилання у контексті опису винаходу (особливо у контексті нижченаведеної формули винаходи) повинні бути витлумачені як покриваючі як однину, так і множину, якщо інакше не позначене або явне не суперечить контексту. При використанні множини для сполук, солей та т.п. мається на увазі, що також може бути використана однина для сполуки, солі тощо. Терміни “комбінація”, “терапевтична комбінація” або “фармацевтична комбінація” при використанні тут означають або фіксовану комбінацію у формі однієї одиниці дозування або набір складових частин для об'єднаного введення, де Сполука A та Сполука B можуть бути введені незалежно у той самий час або окремо у межах часових інтервалів, які дозволяють, щоб партнери по комбінації мали кооперативний, наприклад, синергетичний, ефект. Термін “фармацевтична композиція” визначений тут, як такий, що означає суміш або розчин, що містить щонайменше один терапевтичний засіб, для введення суб'єктові, наприклад, ссавцеві або людині, для профілактики або лікування конкретного захворювання або стану, що стосується ссавця. Термін “фармацевтично прийнятний” визначений тут, як такий, що означає ті сполуки, матеріали, композиції та/або дозовані форми, які є, у межах ретельної медичної оцінки, підходящими для контакту із тканинами суб'єкта, наприклад, ссавця або людини, без надмірної токсичності, подразнюючої алергійної відповіді та інших проблемних ускладнень, співвимірних з розумним співвідношенням користь/ризик. Термін "комбінований лікарський засіб", використовуваний у описі заявки, означає, насамперед, "набір зі складових частин" у тому розумінні, що комбінацію компонентів (а) та (b), зазначену вище, можна вводити незалежно або у вигляді різних фіксованих комбінацій, що містять відомі кількості кожного з компонентів (а) та (b), тобто одночасно або у різний час. У такому випадку компоненти набору зі складових частин можна вводити, наприклад, одночасно або у хронологічному порядку, тобто у різний час та з однаковими або різними інтервалами для будь-якої складової частини набору. Співвідношення загальних кількостей компоненту (а) та компоненту (b), які вводять у вигляді комбінованого препарату, може змінюватися, наприклад, залежно від типу субпопуляції пацієнтів, що підлягають лікуванню, або від стану конкретного пацієнта. Термін “спів-введення” або “спільне введення”, використовуваний у описі заявки, мається на увазі, як охоплюючий введення обраних терапевтичних засобів тому самому пацієнтові й також включає режими лікування, у яких засоби не обов'язково вводяться тим же самим шляхом введення або у той же самий час. Термін “лікувати” або “лікування”, використовуваний у описі заявки, включає лікування, що звільняє від, що скорочує або полегшуюче щонайменше один симптом у пацієнта або, що впливає на затримку прогресування хвороби. Наприклад, лікування може приводити до 4 UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 зменшення одного або декількох симптомів захворювання або до повної ліквідації захворювання, такого як рак. У межах значення даного винаходу термін “лікування” також позначає зупинку, затримку прояву (тобто період до клінічного прояву хвороби) та/або зниження ризику розвитку або погіршення захворювання. Термін "захистити" при використанні тут означає запобігання, затримку або лікування, або всі разом, якщо прийнятно, розвитку або продовження або погіршення захворювання у суб'єкта. Термін "сумарно терапевтично активні" або "сумарний терапевтичний ефект" позначає, що терапевтичні засоби можуть бути дані теплокровній тварині, особливо людині, що підлягає лікуванню, окремо (хронологічно рознесеним чином, особливо послідовним чином) через переважні інтервали часу, за умови демонстрації (переважно синергічної) взаємодії (сумарний терапевтичний ефект). Чи так це насправді, серед іншого, може бути визначене нижченаведеним чином: за рівнями у крові, показавши, що обидві сполуки присутні у крові людини, що одержує лікування, щонайменше, під час певних часових інтервалів. Термін "фармацевтично ефективна кількість" або "клінічно ефективна кількість" або "терапевтично ефективна кількість" комбінації терапевтичних засобів є кількість, достатня для забезпечення помітного поліпшення у порівнянні з вихідним рівнем клінічно спостережуваних ознак та симптомів захворювання, що підлягає лікуванню комбінацією. Термін "суб'єкт" або "пацієнт" при використанні тут включає тварин, які можуть страждати від або має ризик занедужати на рак або інше захворювання, припускаючи, прямо або побічно, рак. Приклади суб'єктів включають ссавців, наприклад, людей, собак, корів, коней, свиней, овець, кіз, кішок, мишей, кроликів, щурів та трансгенних тварин, що не відносяться до людині. У кращому варіанті здійснення суб'єкт є людиною, наприклад, людиною, що страждає від, що має ризик занедужати або що є потенційно здатною занедужати на рак. Термін "близько" або "приблизно" повинен мати значення у межах 10%, більш переважно у межах 5%, даного значення або інтервалу значень. Сполука А та/або Сполука В можуть бути введені у вільній формі або у формі фармацевтично прийнятної солі. Як використовується тут "фармацевтично прийнятна сіль", якщо не позначене інакше, включає солі кислотних та лужних груп, які можуть бути представлені у сполуках відповідно до даному винаходу. Сполуки відповідно до даному винаходу, які мають лужну природу, здатні до формування великої різноманітності солей з різними неорганічними та органічними кислотами. Кислоти, які можуть використовуватися для приготування фармацевтично прийнятних солей приєднання кислоти для таких лужних сполук відповідно до даного винаходу, є такими, які формують нетоксичні солі приєднання кислоти, тобто солі, що містять фармацевтично прийнятні аніони, такі як ацетат, бензоат, бромід, хлорид, цитрат, фумарат, гідробромід, гідрохлорид, йодид, лактат, малеат, манделат, нітрат, оксалат, саліцилат, сукцинат та тартрат. Якщо інакше не визначено, або ясно позначено у тексті, або незастосовно, посилання на терапевтичні засоби, застосовувані КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ, включає як вільну основу сполук, так і всі фармацевтично прийнятні солі цих сполук. Даний винахід особливо має відношення до КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ, застосовуваної для лікування або профілактики проліферативного захворювання у суб'єкта, який цього потребує. У кращому варіанті здійснення даного винаходу КОМБІНАЦІЯ ВІДПОВІДНО ДО ВИНАХОДУ застосовується для лікування або профілактики проліферативного захворювання, включаючи введення суб'єктові комбінованої терапії, що містить ефективну кількість Сполуки A та ефективну кількість інгібітору EGFR, такого як моноклональне антитіло-інгібітор EGFR, конкретно цетуксимаб або панітумумаб, особливо цетуксимаб. Переважно, ці речовини вводяться у терапевтично ефективних дозуваннях, які, у сумі, забезпечують сприятливий ефект. Введення може бути роздільним, одночасним або послідовним. Даний винахід особливо має відношення до КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ, застосовуваної для лікування або профілактики проліферативного захворювання у суб'єкта, який цього потребує. У кращому варіанті здійснення даного винаходу КОМБІНАЦІЯ ВІДПОВІДНО ДО ВИНАХОДУ застосовується для лікування або профілактики проліферативного захворювання, включаючи введення суб'єктові потрійної комбінованої терапії, що містить ефективну кількість Сполуки A, ефективну кількість інгібітору EGFR, такого як моноклональне антитіло-інгібітор EGFR, конкретно цетуксимаб або панітумумаб, особливо цетуксимаб, та ефективну кількість селективного інгібітору PI3K-, конкретно сполуки відповідно до Формули I, переважно Сполуки В. Переважно, ці засоби вводяться у терапевтично ефективних дозуваннях, які, при комбінуванні, забезпечують сприятливий ефект. Введення може бути окремим, одночасним або послідовним. 5 UA 115786 C2 Даний винахід особливо відноситься до комбінації, що містить: (a) B-Raf інгібітор формули 5 10 15 20 або його фармацевтично прийнятну сіль, та (b) інгібітор EGFR, який є цетуксимабом. Крім того, даний винахід особливо відноситься до потрійної комбінації, що містить: (a) B-Raf інгібітор формули або його фармацевтично прийнятну сіль, (b) інгібітор EGFR, який є цетуксимабом, та (c) PI3K інгібітор формули I, особливо PI3K інгібітор формули У одному варіанті здійснення проліферативне захворювання є раком. Термін “рак” використаний тут для позначення широкого спектру пухлин, включаючи всі солідні пухлини та гематологічні злоякісні новоутворення. Приклади таких пухлин включають, але не обмежені, доброякісні або злоякісні пухлини мозку, легенів (особливо дрібноклітинний рак легенів та недрібноклітинний рак легенів), плоскоклітинний рак, рак сечового міхура, шлунку, підшлункової залози, молочної залози, голови та шиї, нирок, сечоводу, яєчників, простати, колоректальний рак, пухлини стравоходу, яєчка, гінекологічні (наприклад, маткові саркоми, карциноми фаллопієвих труб, ендометрію, шейки матки, піхви або зовнішніх статевих органів), рак щитовидної залози, підшлункової залози, рак кістки, рак шкіри, меланому, рак матки, яєчників, ректальний рак, пухлину заднього проходу, прямої кишки, пухлину яєчку, хвороба Ходжкіна, пухлину стравоходу, пухлину тонкого кишечнику, пухлини ендокринної системи (наприклад, щитовидної залози, паращитовидної залози або надниркових залоз), саркоми м'яких тканин, 6 UA 115786 C2 5 10 15 20 25 30 35 40 уретри, пенісу, лейкемії, лімфоми, новоутворення центральної нервової системи, саркоми, мієломи, рак жовчного міхура, рак печінки, нейрофіброматоз, гострий мієлобластний лейкоз (AML), мієлодиспластичні синдроми (MDS) та саркому Капоши. У подальшому варіанті здійснення даного винаходу проліферативне захворювання є меланомою, раком легенів (включаючи недрібноклітинний рак легенів (NSCLC), колоректальним раком (CRC), раком молочної залози, раком нирок, таким як, наприклад, карцинома клітин ниркового епітелію (RCC), раком печінки, ендометріальним раком, гострим мієлобластним лейкозом (AML), мієлодиспластичним синдромом (MDS), раком щитовидної залози, зокрема папілярним раком щитовидної залози, раком підшлункової залози, нейрофіброматозом або гепатоклітинною карциномою. У подальшому варіанті здійснення даного винаходу проліферативне захворювання є солідною пухлиною. Термін "солідна пухлина" особливо означає меланому, рак молочної залози, рак яєчника, рак товстої кишки та у загальному випадку рак шлунково-кишкового тракту, рак шийки матки, рак легенів (включаючи дрібноклітинний рак легенів та недрібноклітинний рак легенів), рак голови та шиї, рак сечового міхура, рак передміхурової залози або саркому Капоши. Дана комбінація пригнічує ріст солідних пухлин і також гемобластозів. Далі, залежно від типу пухлини та конкретної використовуваної комбінації може досягатися зменшення розміру пухлини. Розкриті тут КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ також придатні для запобігання метастазування пухлин та росту або розвитку мікрометастаз. Розкриті тут КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ придатні для лікування пацієнтів з несприятливим прогнозом, особливо тих пацієнтів з несприятливим прогнозом, які мають метастатичну меланому, колоректальний рак або рак підшлункової залози. У наступному варіанті здійснення проліферативне захворювання є меланомою або колоректальним раком, зокрема колоректальним раком. КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ особливо застосовуються для лікування злоякісних пухлин, що мають генетичну зміну у RAS/RAF/MEK шляху сигнальної трансдукції, таку як, наприклад, мутація B-Raf або ампліфікація гену. У важливому варіанті здійснення рак, що підлягає лікуванню, характеризується мутацією BRaf, наприклад, B-Raf-мутований колоректальний рак. Зокрема, мутація B-Raf є мутацією V600, наприклад, V600E, V600K або V600G мутацією. Так, даний винахід особливо відноситься до способу лікування колоректального раку, що характеризується мутацією B-Raf, який включає введення терапевтично ефективної кількості КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ пацієнтові, який цього потребує. Конкретніше, даний винахід відноситься до способу лікування колоректального раку, що характеризується мутацією B-Raf, який включає введення терапевтично ефективної кількості комбінації, що включає (a) B-Raf інгібітор формули або його фармацевтично прийнятну сіль, та (b) інгібітор EGFR, який є цетуксимабом. Додатково, даний винахід особливо відноситься до способу лікування колоректального раку, що характеризується мутацією B-Raf, який включає введення терапевтично ефективної кількості потрійної комбінації, що включає (a) B-Raf інгібітор формули 7 UA 115786 C2 або його фармацевтично прийнятну сіль. (b) інгібітор EGFR, який є цетуксимабом, та (c) PI3K інгібітор формули I, особливо PI3K інгібітор формули 5 10 15 20 25 30 35 У важливому варіанті здійснення кожного з цих способів мутація B-Raf є мутацією V600, наприклад, V600E, V600K або V600G мутацією. Природа проліферативних захворювань залежить від безлічі факторів. За певних умов можна комбінувати лікарські засоби, що діють за різними механізмами. Однак, усього лише розгляд будь-якої комбінації терапевтичних засобів з різними механізмами дії не обов'язково приводить до одержання комбінацій із кращим ефектом. Введення фармацевтичної комбінації відповідно до винаходу може виявляти не тільки сприятливу дію, наприклад синергетичний терапевтичний ефект, наприклад, щодо пом'якшення, уповільнення прогресування або пригнічення симптомів, але, крім того, ще більш несподівані сприятливі дії, наприклад, зниження побічної дії, більш довготривала відповідь, поліпшена якість життя та знижена смертність у порівнянні з монотерапією одним з фармацевтичних терапевтичних засобів, використовуваних у комбінації відповідно до винаходу. Іншою перевагою є той факт, що у КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ можуть бути використані більш низькі дози терапевтичних засобів, наприклад, часто не тільки дозування повинні бути нижче, але й вони повинні вводитися рідше, низькі дозування можуть бути використані з метою зменшити побічні ефекти, спостережувані у випадку введення комбінацій або одного препарату. Це відповідає побажанням та вимогам пацієнта, який повинен проходити лікування. Може бути показане на встановлених експериментальних моделях, що КОМБІНАЦІЯ ВІДПОВІДНО ДО ВИНАХОДУ приводить до сприятливих ефектів, описаних тут колись. Фахівець у даній галузі техніки може вибрати відповідну модель із метою підтвердити такі сприятливі ефекти. Фармакологічну активність КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ можна, наприклад, продемонструвати при клінічному випробуванні або на експериментальній моделі на тварині, як по суті описано тут нижче. Визначення синергетичної взаємодії між одним або більше компонентами, оптимального діапазону для ефекту та абсолютного діапазону дозування кожного компоненту для досягнення ефекту може бути остаточно виміряне шляхом введення компонентів у різних діапазонах співвідношення маса/маса та дозах пацієнтам, що потребують лікування. Для людей складність та вартість проведення клінічних досліджень на пацієнтах можуть зробити непрактичним застосування цієї форми тестування як основної моделі для спільної дії. Однак, спостереження синергізму у одному виді може прогнозувати ефект у інших видах та існуючих тваринних моделях, як описано тут, щоб виміряти синергетичний ефект, та результати таких досліджень із застосуванням фармакокінетичних/фармакодинамічних методів також можуть 8 UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 використовуватися для прогнозування ефективних діапазонів співвідношення доз та абсолютних доз, та плазмових концентрацій, які повинні бути досягнуті для інших видів. Встановлені кореляції між пухлинними моделями та ефектами, поміченими у людини, свідчать, що синергізм у тварин може бути продемонстрований, наприклад, на моделях ксенотрансплантату або у відповідних клітинних лініях. Сполуку A звичайно вводять перорально у дозі у межах від 10 мг до 1000 мг на день, наприклад, 50 мг до 450 мг на день або 100 мг до 400 мг на день. Денна доза може вводитися за розкладом один раз на день (qd) або два рази на день (bid). Вказівки щодо застосування препарату для бренду цетуксимабу ERBUTUX® рекомендує 2 початкове введення у дозі 400 мг/м у вигляді 120-хвилинної внутрішньовенної інфузії з 2 наступними дозами 250 мг/м , що вводяться протягом 60 хвилин. Цетуксимаб вводиться відповідно до вказівок щодо застосування препарату при використанні у даних комбінаціях. Однак, є можливим зниження дозування. Таким чином, відповідно до даного винаходу, 2 цетуксимаб вводиться спочатку у дозі від 200 до 400 мг/м , з наступними щотижневими дозами 2 від 125 до 250 мг/м . Сполуку В звичайно вводять перорально у дозі у межах від 30 мг до 450 мг на день, наприклад, 100 мг до 400 мг на день. Денна доза може вводитися за розкладом один раз на день (qd) або два рази на день (bid). Метою даного винаходу є надання фармацевтичної композиції, що включає КОМБІНАЦІЮ ВІДПОВІДНО ДО ВИНАХОДУ, яка є сумарно терапевтично ефективною у відношенні проліферативного захворювання. У цій композиції компоненти комбінації Сполука A та/або Сполука В можуть бути введені у вигляді одного препарату або дозованої лікарської форми, що вводиться паралельно, але окремо, або можуть бути введені послідовно будь-яким підходящим шляхом. Переважно, пероральні лікарські форми Сполуку А та Сполуку В вводять паралельно, але окремо. Моноклональне антитіло інгібітор EGFR звичайно вводять окремо у вигляді внутрішньовенної інфузії, переважно за розкладом один раз на тиждень, де інгібітор EGFR є цетуксимабом. У одному варіанті здійснення даний винахід також має відношення до КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ для застосування у приготуванні фармацевтичної композиції або лікарського препарату для лікування або профілактики проліферативного захворювання у суб'єкта, який цього потребує. Індивідуальні компоненти КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ можуть вводитися окремо у різний час під час курсу терапії або одночасно у розділених або об'єднаних формах комбінації. Таким чином, мається на увазі, що винахід включає всі такі схеми з одночасним або почерговим введенням та термін "введення" слід інтерпретувати відповідно до вищевикладеного. Ефективна доза кожного з компонентів комбінації, застосовувана у КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ, може змінюватися залежно від застосовуваної конкретної сполуки або композиції, способу введення, стану, що підлягає лікуванню, та важкості стану, що підлягає лікуванню. Таким чином, схему КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ вибирають залежно від безлічі факторів, що включають спосіб введення та стан нирок та печінки пацієнта. Середній клініцист або лікар може легко визначити та прописати ефективну кількість одного з терапевтичних засобів, необхідну для полегшення, протидії або блокування прогресії патологічного стану. Оптимальні співвідношення, індивідуальні та комбіновані дозування та концентрації компонентів комбінації (a) та (b) КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ, які приводять до ефективності без токсичності, основані на кінетиці доступності терапевтичних засобів у цільових сайтах, та визначаються із застосуванням способів, відомих фахівцям у галузі техніки. Ефективне дозування кожного з компонентів комбінації може потребувати більш частого введення однієї зі сполук у порівнянні з іншою сполукою у комбінації. Тому, щоб дозволити відповідне дозування, упаковані фармацевтичні продукти можуть містити одну або більше дозованих форм, які містять комбінацію компонентів, та одну або більше дозованих форм, які містять один з компонентів комбінації, але не містять іншого компоненту комбінації. Якщо комбінація компонентів, які використовуються у КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ, застосовується у формі, що випускається як індивідуальні лікарські засоби, тоді їх доза та спосіб введення можуть відповідати інформації, наведеній на листівці-вкладиші відповідного лікарського засобу. Оптимальне дозування кожного компоненту комбінації для лікування проліферативного захворювання може бути визначене дослідним шляхом для кожної людини з використанням 9 UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 відомих методів, та буде залежати від безлічі факторів, включаючи, хоча й не обмежуючись, ступінь розвитку захворювання; вік, масу тіла, загальний стан здоров'я, стать та дієту людини; час та спосіб введення; та інші лікарські препарати, що приймаються людиною. Оптимальні дозування можуть бути основані на результатах типових досліджень та процедур, відомих у галузі техніки. Оптимальне дозування кожного компоненту комбінації, яка може комбінуватися з речовинами-носіями для створення єдиної дозованої форми, буде варіюватися залежно від індивідуума, що одержує лікування, та від конкретного способу введення. У деяких варіантах здійснення дозовані лікарські форми, що містять описану тут комбінацію засобів, будуть містити кількість кожного компоненту комбінації, яку, як правило, вводять коли засіб вводиться сам по собі. Частота дозування може змінюватися залежно від застосовуваної сполуки та від конкретного стану, що підлягає лікуванню або профілактиці. Пацієнти можуть звичайно спостерігатися для визначення терапевтичної ефективності, використовуючи підходи, що підходять для стану, що підлягає лікуванню або профілактиці, які будуть добре відомі фахівцеві у галузі техніки. Даний винахід відноситься до способу лікування суб'єкта, що страждає від проліферативного захворювання, що включає введення зазначеному суб'єктові, КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ у кількості, яка є сумарно терапевтично ефективною у відношенні проліферативного захворювання. Особливо, проліферативне захворювання, що підлягає лікуванню за допомогою КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ, є колоректальним раком, зокрема В-Raf-мутованим колоректальним раком, наприклад, V600 B-Raf-мутованим колоректальним раком. Додатково, лікування може включати хірургію або радіотерапію. Даний винахід додатково відноситься до КОМБІНАЦІЇ ВІДПОВІДНО ДО ВИНАХОДУ для застосування у лікуванні проліферативного захворювання, зокрема раку, зокрема В-Rafмутованого колоректального раку, такого як V600 B-Raf-мутований колоректальний рак. Даний винахід надає комерційне упакування, що включає як терапевтичний засіб КОМБІНАЦІЮ ВІДПОВІДНО ДО ВИНАХОДУ, разом з рекомендаціями з одночасного, окремого або послідовного введення при застосуванні для уповільнення прогресування або для лікування проліферативного захворювання у суб'єкта, який цього потребує. Нижченаведені Приклади ілюструють винахід, описаний вище; однак, вони не призначені для обмеження обсягу винаходу будь-яким шляхом. Сприятлива дія фармацевтичної комбінації відповідно до даного винаходу може також бути визначена на інших тестових моделях, які відомі фахівцеві у даній галузі техніки. Приклад 1 Це - багатоцентрове відкрите дослідження з фазою Ib з підвищенням дози та рандомізованою фазою II, яке буде охоплювати приблизно 124 пацієнти з B-Raf-мутованим метастатичним колоректальним раком (mCRC). Метою фази Ib (n~24) було визначення максимальної стерпної дози (MTD) та/або рекомендованої для фази два дози (RP2D) та MTD та/або RP2D Сполуки А у комбінації зі Сполукою В та цетуксимабом. У першій стадії підвищення дози когорти пацієнтів будуть одержувати лікування подвійною комбінацією доти, поки MTD/RP2D подвійної комбінації не буде визначений. Потім когорти пацієнтів будуть одержувати лікування потрійною комбінацією протягом другої стадії підвищення дози доти, поки MTD/RP2D потрійної комбінації не буде визначений. Фаза II (n~100) оцінює клінічну ефективність подвійної комбінації та потрійної комбінації та далі охарактеризує безпеку комбінацій лікарських препаратів. Лікування буде проводитися у 28денних циклах до прогресування хвороби, неприпустимої токсичності, скасування інформованої згоди або смерті. Відповідь пухлини буде оцінена на місці дослідником згідно з методичними рекомендаціями, основаними на RECIST версії 1.1. Кожний пацієнт буде оцінений на всі потенційні місця пухлинних осередків при скринінгу/початку дослідження та кожні 6 тижнів після початку досліджуваного лікування до прогресування хвороби. Діагностична візуалізація при скринінгу/початку дослідження може бути проведена протягом 21 дня від початку лікування. Оцінки пухлини протягом дослідження мають вікно ± 7 днів, за винятком першої оцінки пухлини після початку дослідження. Перша оцінка пухлини після початку дослідження може бути проведена протягом 6 тижнів (дозволене вікно +7 днів) від початку лікування. Оцінка пухлини буде проводитися наприкінці лікування (± 3 дні), якщо пацієнт припиняє лікування з якої-небудь причини, відмінної від прогресування захворювання та якщо остання оцінка пухлини проводилася 21 дня до цього дня. Пацієнти, включені у частину дослідження фази II, які 10 UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 припинили лікування з якої-небудь причини, відмінної від прогресування захворювання, повинні одержувати щомісячні телефонні дзвінки та проходити оцінку пухлини кожні 6 тижнів (± 7 днів) до прогресування захворювання або початку наступної протипухлинної терапії, або смерті, що б не сталося раніше. Молекулярний пре-скринінг Щоб увійти у скринінгову фазу дослідження, пацієнти повинні мати письмовий документ про статус KRAS дикого типу та мутації BRAF V600, який повинен бути отриманий на місці на новій біопсії пухлини (переважно) або на доступному найбільш недавньому архівному зразку пухлини. Інформована згода на молекулярний пре-скринінг повинна бути підписана перед будь-якою пов'язаною з дослідженням процедурою молекулярного пре-скринінгу (неприпустимо, якщо мутаційний статус був уже оцінений за межами дослідження). Період лікування Цикл 1 День 1 буде початком періоду лікування. Протягом фази II досліджуване лікування повинне бути почате 1-ого тижня після рандомізації. Досліджуваний вид лікування буде проводитися у 28-денних циклах до прогресування хвороби, неприпустимої токсичності, скасування інформованої згоди або смерті. Закінчення лікування (EOT) Відвідування EOT відбувається протягом 14 днів після останнього проведення досліджуваного лікування (Секція 7.1.5). Усі пацієнти, що беруть участь, повинні зробити це відвідування, навіть якщо вони повинні були передчасно припинити участь. Період наступного спостереження Період наступного спостереження починається після відвідування Закінчення Лікування, та триває до завершення всіх наступних оцінок, включаючи спостереження за виживаністю. Популяція а) Популяція пацієнтів Обидві фази дослідження, фаза Ib та фаза II, будуть проводитися на дорослих пацієнтах з метастатичним колоректальним раком (mCRC), що несе KRAS дикого типу та мутацію BRAF V600, чиє захворювання прогресувало, незважаючи на попередню протипухлинну терапію або для кого ніяка подальша ефективна стандартна терапія не доступна. Пацієнтам, залученим у це дослідження, не дозволено брати участь у паралельних дослідженнях препаратів або обладнань. Крім того, пацієнти, які закінчили дослідження, не повинні бути повторно залучені на другий курс лікування. b) Критерії включення Пацієнти, що мають право на включення у це дослідження, повинні відповідати всім нижченаведеним критеріям: 1. Вік (18 років на початку дозування (фаза Ib) або під час рандомізації (фаза II) 2. Гістологічний або цитологічний доказ метастатичного колоректального раку (mCRC) 3. Прогресія щонайменше після одного попереднього стандартного режиму лікування або не переносять схеми на основі іринотекану. 4. Письмовий документ про статус KRAS дикого типу та мутації BRAF V600 або іншої BRAF V600 мутації 5. Фаза II тільки: нова біопсія пухлини як вихідний рівень 6. Свідчення вимірних проявів хвороби, як визначено у RECIST v1.1 Примітка: Ушкодження у областях попередньої радіотерапії або інших місцево-регіонарних методів лікування (наприклад, підшкірна абляція) не повинні вважатися вимірними, якщо тільки прогрес ушкодження не був зареєстрований після терапії. 7. Тривалість життя 3 місяців 8. Загальний стан за ECOG 2 9. Негативний сироватковий тест на вагітність протягом 72 годин до першої дози досліджуваного лікування для всіх жінок з потенціалом народження дитини 10. Здатний зрозуміти та добровільно підписати форму інформованої згоди та здатний додержуватися графіку відвідувань при дослідженні та інших вимог протоколу. Письмова інформована згода повинна бути отримана до скринінгових процедур. с) Критерії виключення Пацієнти, що мають право на це дослідження, не повинні відповідати жодному з наступних критеріїв: 1. Фаза II тільки: попереднє лікування цетуксимабом, панітумумабом та/або іншими інгібіторами EGFR 2. Фаза II тільки: попереднє лікування Raf-інгібіторами, PI3K-інгібіторами та/або іншими інгібіторами MEK 11 UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 3. Симптоматичне або невилікуване лептоменінгіальне захворювання 4. Симптоматичний метастаз у головний мозок. Можуть брати участь пацієнти, раніше ліковані або неліковані від тих станів, які є безсимптомними під час відсутності терапії кортикостероїдами. Метастаз у головний мозок повинен бути стабільним з підтвердженням діагностичною візуалізацією (наприклад, мозковий MRI або CT, виконаний при скринінгу, що демонструє відсутність поточних свідчень прогресуючих мозкових метастазів). Пацієнтам не дозволяється одержувати фермент-індукуючі протиепілептичні засоби. 5. Пацієнти із цукровим діабетом, що потребують лікування інсуліном, та/або із клінічними ознаками або з рівнем глюкози натще 140 мг/дл/ 7,8 ммоль/л, історією клінічно значних гестаційних цукрових діабетів або задокументованого стероїдного цукрового діабету 6. Відомий гострий або хронічний панкреатит 7. Клінічно значне захворювання серця, включаючи будь-яке з наступних: - Застійна серцева недостатність, що вимагає лікування (NYHA grade 2), LVEF 45%, як визначено MUGA-скануванням або ECHO, або неконтрольована гіпертонія (відповідно до рекомендацій WHO-ISH), - Історія або присутність клінічно значних шлуночкових екстрасистолій або миготливої аритмії - Клінічно значна брадикардія у спокої - Нестабільна стенокардія 3 місяці до початку дослідження препарату - Гострий інфаркт міокарда (AMI) 3 місяці до початку дослідження препарату - QTcF 480 мс 8. Пацієнти з будь-яким наступним лабораторних показників на момент скринінгу/початку дослідження: 3 9 - Абсолютне число нейтрофілів (ANC) 1500/мм [1,510 /л] 3 9 - Тромбоцити 100000/мм [10010 /л] - Гемоглобін 9,0 г/дл - Креатинін сироватки крові 1,5 ULN або обчислений або безпосередньо виміряний CrCl 50% LLN (нижня межа норми) - Загальний білірубін сироватки крові 1,5 ULN - AST/SGOT та/або ALT/SGPT 2,5 ULN, або 5 ULN, якщо є метастази у печінку 9. Недостатність шлунково-кишкової (GI) функції або захворювання ШКТ, яке може значно змінювати адсорбцію перорального співвідношення Сполука A/ Сполука B (наприклад, виразкова хвороба, неконтрольована нудота, блювота, діарея, синдром мальабсорбції, резекція тонкої кишки). 10. Попереднє або супутнє злоякісне новоутворення. Виключення: задовільно пролікований базальноклітинний або плоскоклітинний рак шкіри; in situ карцинома шийки матки, вилікувана радикально та без свідчень повторення протягом щонайменше 3 років до початку участі у дослідженні; або інша солідна пухлина, вилікувана радикально, та без свідчень повторення протягом щонайменше 3 років до початку участі у дослідженні. 11. Вагітні або жінки, що годують грудьми, де вагітність визначається як стан жінки після зачаття та до завершення вагітності, підтверджене позитивним hCG лабораторним тестом ( 5 мМО/мл). Жінки з потенціалом народження дитини, що визначаються як усі жінки, фізіологічно здатні до того, щоб завагітніти, не допускаються до участі у цьому дослідженні, ЯКЩО вони не використовують високоефективні методи контрацепції протягом всього дослідження та протягом 3 місяців після припинення приймання досліджуваного препарату. Високоефективні методи контрацепції включають: - Повне утримання - Чоловіча або жіноча стерилізація - Комбінація будь-яких двох з наступних (a+b або a+c або b+c) а. Використання пероральних, ін’єктуємих або імплантованих гормональних методів контрацепції b. Розміщення внутрішньоматкового обладнання (IUD) або внутрішньоматкової системи (IUS) c. Методи бар'єрної контрацепції: презерватив або перешкоджаючий ковпачок (діафрагма або цервікальні ковпачки) зі сперміцидною піною/гелем/плівкою/кремом/вагінальним супозиторієм Жінкам після менопаузи дозволяють брати участь у цьому дослідженні. Жінок вважають такими, що відносяться до періоду після менопаузи та що не мають потенціалу народження дитини, якщо вони мали 12 місяців природньої (спонтанної) аменореї з відповідним клінічним 12 UA 115786 C2 5 10 15 20 25 30 35 профілем (наприклад, відповідний вік, історія вазомоторних ознак) або шість місяців спонтанної аменореї із сироватковим рівнем фолікуло-стимулюючого гормону (FSH) (40 мМО/мл або мають хірургічну двосторонню оваріектомію (з або без гістеректомії) або трубну лігатуру щонайменше за шість тижнів до скринінгу. У випадку однієї тільки оваріектомії, жінка може бути оцінена як така, що не має потенціалу народження дитини, тільки тоді, коли її репродуктивний статус був підтверджений наступною оцінкою гормонального рівня. 12. Сексуально активні чоловіки повинні використовувати презервативи під час статевого акту, при прийманні лікарського препарату та протягом 3 місяців після припинення лікування та не повинні ставати батьком дитини у цей період. Презерватив повинен використовуватися також чоловіками після вазектомії, для запобігання передачі препарату через насінну рідину. 13. Історія тромбоемболічних або цереброваскулярних подій протягом минулих 6 місяців, включаючи минущу ішемічну атаку, інсульт, тромбоз глибоких вен або легеневу емболію. 14. Пацієнти, які одержували радіаційну терапію (яка включає 30% запасу кісткового мозку), хіміотерапію, біологічну терапію (наприклад, антитіла) у межах 4 тижня (6 тижнів для нітрозосечовини, мітоміцину-C), або ті, які одержували безперервне або переривчасте лікування невеликими молекулами або дослідницькими засобами у межах 5 періодів напіввиведення засобу (або 4 тижнів, коли період напіввиведення невідомий) до початку дослідження препарату, або хто не відновився від побічних ефектів такої терапії (крім облисіння). 15. Пацієнти, які перенесли будь-яке велике оперативне втручання протягом попередніх 2 тижнів до початку дослідження препарату, або хто не повністю відновився від попередньої хірургії 16. Відоме інфікування вірусом імунодефіциту людини (HIV) 17. Інше важке, гостре або хронічне захворювання або розлад психіки або лабораторна анормальність, які можуть збільшити ризик, пов'язаний з участю у дослідженні або введенням досліджуваного препарату, або які можуть впливати на інтерпретацію результатів дослідження та за рішенням дослідника може зробити пацієнта непідходящим для дослідження. 2) Лікування а) Досліджуване лікування Досліджувані лікарські препарати, які будуть використовуватися у цьому дослідженні, є Сполукою A та Сполукою B. Інший препарат, який буде використовуватися у цьому дослідженні, є цетуксимабом. Досліджувані види лікування: - Подвійна комбінація: Сполука A та цетуксимаб - Потрійна комбінація: Сполука A, Сполука В та цетуксимаб i) Режим дозування Пацієнтам призначається (фаза Ib) або рандомізується (фаза II) один з наступних режимів. - Подвійна комбінація: Сполука A (QD або BID) та цетуксимаб (QW) - Потрійна комбінація: Сполука A (QD або BID), Сполука В (QD або BID) та цетуксимаб (QW) 40 Досліджувані види лікування Сполука A цетуксимаб Сполука A Сполука В цетуксимаб Доза та схема лікування Фармацевтична форма та Доза спосіб введення Подвійна комбінація капсула для перорального як призначено прийому 2 400 мг/м початкова інфузія внутрішньовенна інфузія 2 250 мг/м наступна інфузія Потрійна комбінація капсула для перорального як призначено прийому таблетка для як призначено перорального прийому 2 400 мг/м початкова інфузія внутрішньовенна інфузія 2 250 мг/м наступна інфузія 13 Частота один або два рази на день один раз на тиждень один або два рази на день один або два рази на день один раз на тиждень UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 Інструкції щодо введення Сполуки A або Сполука A + Сполука В Сполука A та Сполука В будуть вводиться перорально у щоденному режимі (QD) у вигляді фіксованої дози, поза залежності від маси тіла або площі поверхні тіла. Якщо нові свідчення досліджень, які продовжуються, вказують, що режим(и) два рази на день (BID) може бути кращим, режим BID як для Сполуки A, так і/або для Сполуки B у поєднанні з цетуксимабом може бути досліджений шляхом відкриття нових когорт у частині дослідження фази Ib. Одиничний RP2D та схема будуть вибрані для частини фази II для кожного препарату. - QD Дозування: Пацієнти повинні отримати інструкцію приймати капсули Сполуки A (та таблетки Сполуки B, якщо застосовне) щоденно зранку з великою склянкою води (~250 мл), через приблизно 1 годину після завершення легкого сніданку (наприклад, не грейпфрутовий сік, тост та джем), у приблизно один і той же час кожен день. Пацієнти не повинні їсти впродовж ще 1 години після прийому. Якщо пацієнт забув прийняти дозу зранку, тоді він або вона повинен(повинна) прийняти дозу впродовж 6 годин після пропущеної дози. Якщо пройшло більше, ніж 6 годин, тоді дозу не потрібно приймати у той день, та пацієнт повинен продовжити лікування з наступною запланованою дозою. Якщо з якої-небудь причини сніданок не споживався, тоді пацієнт все ж повинен прийняти заплановану ранкову дозу зі склянкою води. Якщо це відбувається у дні забору зразків для повного фармакокінетичного дослідження (full PK), це повинно бути задокументовано у eCRF. - BID Дозування: Дози Сполуки A (та Сполуки D, якщо застосовне) повинні бути прийняті з інтервалом 12±2 годин. Пацієнти повинні отримати інструкцію приймати дози щоденно з великою склянкою води (~250 мл) зранку, через приблизно 1 годину після завершення легкого сніданку, та ввечері, через приблизно 1 годину після завершення легкого прийому їжі або полуденка, у приблизно один і той же час дня кожен день. Пацієнти не повинні мати впродовж ще 1 години після прийому. Якщо з якої-небудь причини вечірнє приймання їжі не споживалося, тоді пацієнт все-таки повинен прийняти заплановану ранкову дозу зі склянкою води. Якщо тільки один із двох пероральних лікарських засобів (Сполука A, Сполука B) приймається BID, обидва лікарські засоби повинні бути прийняті разом ранком, і тільки BID лікарський засіб повинен бути прийнятий ввечері. - Дози повинні прийматися у приблизно той самий час щодня, за винятком днів, коли намічений збір крові у клініці, у цьому випадку пацієнти повинні прийняти свої ранкові дози у клініці. - Сполука A та Сполука В будуть дозовані у той самий час для пацієнтів, які призначені/рандомізовані для потрійної комбінації. - У дні, коли буде намічений збір крові у клініці, пацієнти приймуть пероральні досліджувані лікарські засоби у клініці під спостереженням дослідника або уповноваженої особи. У всі інші дні пацієнти будуть приймати пероральні досліджувані лікарські засоби вдома. - Контроль рівня глюкози у плазмі натще: У дні контролю рівня глюкози у плазмі натще пацієнти повинні утримуватися від їжі протягом ночі, мінімум 8 годин перед забором крові. Зразок крові на рівень глюкози у плазмі натще повинен бути вибраний перед введенням якихнебудь стероїдів, якщо вони повинні бути введені у той же день для премедикації цетуксимабу. Легкий сніданок може бути вжитий після забору крові на рівень глюкози у плазмі натще. Сполука A (та Сполука В, якщо застосовне) можуть бути прийняті через 1 годину після сніданку. Пацієнти повинні продовжити утримуватися від їжі протягом ще 1 години після приймання Сполуки A (та Сполуки В, якщо застосовне). - Забір зразків на PK: У дні забору зразків на PK пацієнти повинні утримуватися від їжі протягом ночі, мінімум 8 годин перед легким прийманням їжі для досягнення стану легкої ситості. Зразки PK до приймання препарату повинні бути забрані безпосередньо перед прийманням Сполуки A (та Сполуки В, якщо застосовне). - При кожному відвідуванні відповідальний персонал буде забезпечувати приймання відповідної дози кожного досліджуваного препарату та надавати пацієнтові правильну кількість досліджуваного препарату(ів) для наступного дозування. Пацієнти повинні одержати інструкцію повертати невикористані досліджувані препарати на місце дослідження при кожному відвідуванні. - Пацієнти повинні одержати інструкцію ковтати цілі капсули/таблетки, а не жувати або не подрібнювати їх. Будь-які дози, які були пропущені, повинні бути пропущені зовсім та не повинні бути замінені або прийняті під час наступного запланованого дозування або на наступний день, що б не застосовувалося. - Пацієнти повинні уникати вживання грейпфрутів, гранатів, плодів карамболю, помаранчів або продуктів, що містять сік кожного з них під час всього дослідження та переважно за 7 днів 14 UA 115786 C2 5 10 15 20 25 30 35 40 45 50 55 60 до першої дози досліджуваного лікарського препарату, через потенційну взаємодію CYP3A4 з досліджуваними лікарськими препаратами. Апельсиновий сік дозволений. - У випадку виникнення блювоти під час курсу лікування, ніяке повторне дозування пацієнта не дозволене перед наступною запланованою дозою. Виникнення та частота будь-якої блювоти та/або діареї (або збільшена частота випорожнення) повинні бути відзначені у AEs розділі eCRF. Крім того, у дні забору зразків для повного фармакокінетичного дослідження (full PK), час початку будь-яких епізодів блювоти протягом перших 4 годин після дозування у цей день повинен бути відзначений у відповідному Записі про Введення Дози PK eCRF. - Дослідник або відповідальний персонал місця дослідження повинні проінструктувати пацієнта приймати досліджувані лікарські препарати згідно із протоколом (сприяти виконанню умов). Усі дозування, запропоновані та видані пацієнтові, та усі зміни дози та усі пропущені під час дослідження дози, повинні бути зареєстровані у Записі про Введення Дози eCRF. Облік препарату повинен виконуватися на регулярній основі. Пацієнти повинні одержати інструкцію повертати невикористані досліджувані препарати на місце дослідження наприкінці кожного циклу. Відповідальний персонал буде забезпечувати приймання відповідної дози кожного досліджуваного препарату при кожному візиті та надавати пацієнтові правильну кількість досліджуваного препарату(ів) для наступного дозування. Введення цетуксимабу Цетуксимаб буде вводитися внутрішньовенно щотижня у Дні 1, 8, 15 та 22 (± 3 дня) кожного циклу у дослідному центрі згідно із встановленими стандартами. Премедикація повинна вводитися як описано, слідуючи формально встановленим стандартам, 30 хвилин до інфузії 2 цетуксимабу. Доза цетуксимабу, що вводиться спочатку, (Цикл 1 День 1) становить 400 мг/м у / 2 вигляді 120-хвилинної внутрішньовенної інфузії з наступними щотижневими дозами 250 мг м , що вводяться протягом 60 хвилин. Швидкість інфузії не повинна перевищувати 10 мг/хвил. Ретельне спостереження є необхідним протягом інфузії та щонайменше 1 години після закінчення інфузії. При виникненні реакції на інфузію протягом введення цетуксимабу інфузія повинна бути перервана негайно, та пацієнт повинен бути під наглядом та одержувати лікування відповідно до формально встановлених стандартів. Послідовність введення лікарських засобів Премедикація з потенціалом зміни pН верхньої частини шлунково-кишкового тракту (GI), може змінити розчинність Сполуки A та/або Сполуки B та, отже, її біодоступність. Ці засоби включають, але не обмежені, інгібітори протонного насосу (наприклад, омепразол), H2антагоністи (наприклад, ранітидин) та нейтралізуючі кислоту засоби. Таким чином, пероральна доза Сполуки A (та Сполуки B, якщо застосовне) повинна вводитися до цетуксимабу та його премедикації, яка переважно повинна бути основана на комбінації H1-антагоністу (наприклад, дифенгідрамін) та дексаметазону (10 мг IV). Мінімум 1 година повинна пройти від часу введення Сполуки A (та Сполуки В, якщо застосовне) до введення премедикації для цетуксимабу. Рекомендується, щоб інфузія цетуксимабу відбувалася через 0,5 години після пре-медикації (тобто 1,5 години після приймання Сполуки A /Сполуки B). Тривалість лікування Пацієнти можуть продовжувати лікування досліджуваним препаратом до випадку неприпустимої токсичності, прогресування захворювання та/або випадку, коли лікування припинене на розсуд дослідника, або випадку скасування інформованої згоди. Посібник з підвищення дози Обґрунтування стартової дози (1) Подвійна комбінація Стартова доза для подвійної комбінації досліджуваних лікарських препаратів становить 100 2 2 мг QD для Сполуки A, та 400 мг/м початкова доза (Цикл 1 День 1) та 250 мг/м наступні щотижневі дози у вигляді внутрішньовенної інфузії для цетуксимабу. Ці стартові дози основані на доступних даних триваючого першого дослідження на людях Сполуки A та рекомендованій дозі цетуксимабу для метастатичного колоректального раку, згідно з етикеткою цетуксимабу. Беручи до уваги усю інформацію, у цей час доступну про співвідношення доза-DLT Сполуки A та цетуксимабу як окремих засобів, та непевність із приводу токсичності комбінації, попередній розподіл норм DLT вказує на те, що запропонована стартова доза комбінації відповідає критеріям підвищення з контролем над передозуванням (EWOC). (2) Потрійна комбінація Стартові дози Сполуки A, Сполуки В та цетуксимабу у потрійній комбінації основані на доступних даний для всіх трьох лікарських засобів. Сполука A та цетуксимаб будуть вводитися при 50% та 100% від певного MTD/RP2D подвійної комбінації, відповідно. Стартовою дозою 15 UA 115786 C2 5 10 15 20 25 30 35 40 45 Сполуки B, як очікується, буде 100 мг QD, які складуть 25% MTD окремого засобу, визначеною під час фази I клінічного дослідження Сполуки B при введенні пацієнтам із солідними пухлинами. Не очікується DDI на рівні PK між Сполукою B та цетуксимабом. Так як Сполука A є інгібітором BCRP, а Сполука В - субстрат BCRP, тобто потенціал для підвищення впливу Сполуки В, у випадку спільного введення із Сполукою А. Враховуючи сприятливу біодоступність, спостережувану преклінічним чином (58% ADME у щурів) та клінічно, максимальне можливе збільшення впливу Сполуки B, як очікують, складе менше, ніж 60%. Таким чином, стартовою дозою Сполуки B є 100 мг QD для забезпечення достатнього запасу безпеки. Крім того, Сполука B є інгібітором CYP3A4 з часовою залежністю. Сполука A переважно метаболізується CYP3A4. Відповідно до рекомендованої настанови щодо контролю над продуктами та ліками (FDA) механістичною статичною моделлю, доза 100 мг QD Сполуки B, при введенні разом із Сполукою A може збільшити плазмове AUC Сполуки A максимум у 3 рази. Щоб пом'якшити потенційне збільшення вмісту Сполуки A при додаванні Сполуки B, початкова доза Сполуки A у потрійній комбінації (Сполука A, Сполука B, цетуксимаб) буде знижена на 50% його MTD/RP2D, визначеного під час застосування подвійної комбінації, як викладено вище. Крім того, швидка оцінка PK за допомогою in-life PK аналізу буде здійснюватися у фазі підвищення дози для потрійної комбінації для спостереження за День 1 та День 8 PK Сполуки А та Сполуки В для ухвалення інформованого рішення щодо підвищення дози. У випадку спостереження DDI ефекту та передбачуваного передозування Сполуки А, може бути здійснене зниження дози Сполуки А. Перш ніж перший пацієнт буде дозований потрійною комбінацією, байесовська модель буде оновлена новими даними, отриманими у фазі підвищення дози для подвійної комбінації, для підтвердження, що запропоновані стартові дози для Сполуки A та Сполуки B усе ще прийнятні (тобто відповідають критерію EWOC), при введенні з певною дозою цетуксимабу з подвійної комбінації. Якщо передбачувана стартова доза не відповідає критерію, тоді більш низька доза комбінації, яка задовольняє критерію EWOC, буде застосована. Попередні рівні доз Таблиці нижче описують стартові дози, та попередні рівні доз досліджуваних лікарських препаратів для подвійної (Сполука A, цетуксимаб) та потрійної (Сполука A, Сполука B, цетуксимаб) комбінації, які можуть бути визначені під час цього випробування. Доза цетуксимабу не може бути підвищена, але може бути знижена. Додаткові рівні доз, у цей час не визначені, можуть бути зареєстровані, та додаткові пацієнти можуть бути включені у дослідження вже протестованого рівня дози, якщо такі зміни будуть розцінені як необхідні для забезпечення оптимальних даних щодо безпеки та переносимості, фармакокінетики та фармакодинаміки. Якщо коли-небудь під час фази Ib досліджень, дані, що з'являються від інших клінічних випробувань із Сполукою A та/або Сполукою B, вкажуть, що BID режим дозування Сполуки A та/або Сполуки B повинен бути кращим, тоді когорти, що одержують BID режим(и) дозування, можуть бути досліджені у фазі Ib дослідження. Якщо буде ухвалене рішення про перехід на BID, тоді початкова повна щоденна доза (яка буде вводитися як дві розділені дози для BID) буде дозою, яка, як раніше було встановлено, є добре стерпною в якості єдиної щоденної дози, нижче MTD та дозволена BLRM. Рівні дози вище MTDs/RP2Ds, визначених під час попередніх досліджень окремого засобу для Сполуки A та Сполуки B, не будуть оцінюватися у цьому дослідженні. 16 UA 115786 C2 Рівні доз -1** 1 (стартова доза)* 2 3 4 5 Попередні рівні доз (подвійна комбінація) Сполука A QD Цетуксимаб щотижнево 50 мг Знижена доза*** 2 2 100 мг 400 мг/м у цикл 1 день 1 та 250 мг/м щотижнево 2 2 150 мг 400 мг/м у цикл 1 день 1 та 250 мг/м щотижнево 2 2 200 мг 400 мг/м у цикл 1 день 1 та 250 мг/м щотижнево 2 2 300 мг 400 мг/м у цикл 1 день 1 та 250 мг/м щотижнево 2 2 400 мг 400 мг/м ув цикл 1 день 1 та 250 мг/м щотижнево * Можливо, що протягом дослідження будуть додані додаткові та/або середні та більш високі рівні доз. Когорти можуть бути додані на будь-якому рівні доз нижче MTD/RP2D у метою більш точного розуміння безпеки, PK або PD. ** Рівень доз -1 представляє лікувальну дозу для пацієнтів, що потребують зниження дози від початкового рівня. 2 2 2 *** 320 мг/м у Цикл 1 День 1 та 200 мг/м щотижнево; або 240 мг/м у Цикл 1 День 1 та 150 2 мг/м щотижнево Попередні рівні доз (потрійна комбінація) Рівні доз -1** 1 (стартова доза)* 2a 2b 3 4 5 Сполука В Сполука A QD QD 50 мг 50% MTD/RP2D подвійної комбінації Цетуксимаб щотижнево Знижена доза*** 100 мг 50% MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації 100 мг 200 мг 200 мг 300 мг 400 мг MTD/RP2D подвійної комбінації 50% MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації MTD/RP2D подвійної комбінації * Можливо, що протягом дослідження будуть додані додаткові та/або середні та більш високі рівні доз. Когорти можуть бути додані на будь-якому рівні доз нижче MTD/RP2D у метою більш точного розуміння безпеки, PK або PD. ** Рівень доз -1 представляє лікувальну дозу для пацієнтів, що потребують зниження дози від початкового рівня. Доза, нижче ніж зазначена доза, може бути вивчена. 2 2 2 *** 320 мг/м у Цикл 1 День 1 та 200 мг/м щотижнево; або 240 мг/м у Цикл 1 День 1 і 150 2 мг/м щотижнево 5 10 15 Приклад 2 Ефект комбінування Сполуки A або з PI3K-специфічним інгібітором Сполукою B або з інгібітором EGFR ерлотинібом, був перевірений на CRC-похідних клітинних лініях, що швидкопроліферуються, що несуть BRAF мутацію. Обидві комбінації синергетично інгібують проліферацію у більшості клітин, протестованих попарно Сполука A/ Сполука B та Сполука A/ ерлотиніб, активними для 7/8 та 6/9 клітинних ліній, відповідно. Комбінації були активні у клітинах, що несуть як мутантний так і алель дикого типу гену PI3K. У всіх протестованих клітинних лініях тільки Сполука А показала значну активність як окремий засіб, хоча клітинні лінії з мутаціями активації у PI3K або із втратою PTEN були, в основному, несприйнятливі до всіх трьох компонентів. Нарешті, синергізм між Сполукою A та Сполукою B зберігалися, але загальна сила антипроліферативного ефекту збільшувалася у випадку додавання EGFRінгібуючого антитіла цетуксимаб у якості третього засобу. У сукупності, ці дані підтверджують ефект комбінації Сполуки A як з інгібітором EGFR, так і з інгібітором PI3K. Більше того, ці результати свідчать на користь того, що додатковий позитивний результат може бути отриманий при одночасному введенні всіх трьох типів інгібіторів. 17 UA 115786 C2 Результат синергізму CpdA + CpdB + CpdA + CpdA + CpdB 50 нМ ерлотиніб цетуксимаб CpdA Клітинна лінія SW1417 COLO 205 LS411N CL-34 MDST8 HT-29* RKO* SNU-C5* OUMS-23# 5 10 15 20 25 30 35 40 CRC CRC CRC CRC CRC CRC CRC CRC CRC CpdA Ерлотиніб IC50 [нM] 235 5 18 30 319 49 1965 2700 2700 IC50 [нM] 2700 2700 2700 2700 н/в 2700 2700 2700 2700 IC50 [нM] Середнє 2700 2700 2700 2700 2700 2700 2700 2700 2700 2,89 3,80 2,76 4,48 н/в 4,31 5,24 2,44 0,64 SD Середнє SD Середнє SD 0,06 0,06 0,07 0,01 н/в 0,06 0,19 0,10 0,06 4,55 4,02 2,00 4,92 1,40 3,99 0,83 3,51 0,77 0,07 0,06 0,07 0,10 0,14 0,06 0,05 0,07 0,09 3,90 3,68 3,78 3,11 н/в 3,85 4,54 3,23 н/в 0,05 0,05 0,07 0,06 н/в 0,06 0,05 0,08 н/в Дія окремої сполуки та комбінована дія інгібіторів RAF (Сполука A), PI3K (Сполука B) та EGFR (ерлотиніб, цетуксимаб) на проліферацію дев'яти BRAF-мутованих CRC-похідних клітинних ліній. Усі клітинні лінії експресують білок BRAFV600E, за винятком MDST8, яка експресує варіант BRAFV600K. Клітини, що несуть відомі або передбачувані активуючі мутації у гені PI3K, відзначені (*), та клітини із втратою PTEN відзначені (#). Проліферація клітин була виміряна у 72-годинному аналізі клітинного титру glo™, та всі показані результати є результатами щонайменше потрійних вимірювань. Показані значення IC 50 для кожного компоненту як окремої сполуки та вимірювання рівня синергізму для кожної комбінації (описане у Lehar J, Krueger AS, Avery W, et al (2009). Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol 27, 659-666.). Взаємодію вважали синергетичною, коли спостерігався результат 2,0. Для вимірювань синергізму у потрійній комбінації, синергізм Сполуки A та Сполуки B був виміряний у форматі стандартної дозової матриці у присутності фіксованої концентрації цетуксимабу (50 нм). Приклад 3 Оцінка терапевтичних взаємодій між інгібітором B-RAF (Сполука A), інгібітором PI3K- (Сполука B), та цетуксимабом була проведена на моделі підшкірного ксенотрансплантанту аденокарциноми людини HT-29. Клітини, як було повідомлено, були гетерозиготні по V600E мутанту B-RAF (1799TA у B-RAF), P449T мутанту PI3K- (1345CA у PIK3CA) та точковій мутації та вставці у APC; та були гомозиготні по точкових мутаціях у SMAD4 та TP53. У інших 59 генах, зміни в яких часто асоційовані з неоплазією, не було знайдено додаткових, можливо онкогенних мутацій. Сполуку А зберігали при кімнатній температурі та суспендували 2,0 мг/мл у розчині 0,5% карбоксиметилцелюлози (CMC) та 0,5% Tween® 80 у деіонізованій воді (Носій 1). Свіжу суспензію готували кожні два тижні та зберігали при кімнатній температурі. Сполуку B зберігали при 4C та суспендували 2,5 мг/мл у розчині 0,5% метилцелюлози у деіонізованій воді (Носій 2). Свіжу суспензію готували щотижня та зберігали при 4C. Цетуксимаб (ERBITUX®, ImClone/Bristol Myers Squibb, 2 мг/мл, партія # 10COO39SA) був розділений на аліквоти на початку дослідження та зберігався при 4C, свіжа аліквота використовувалася на кожен день дозування. Дозований розчин паклітакселу 3 мг/мл був приготовлений свіжий на кожен лікування шляхом розведення аліквот приготовленого на місці стокового розчину паклітакселу (30 мг/мл паклітакселу у 50% етанол: 50% Cremophor® EL) у десять разів 5% декстрозою у воді. Дозовані розчини були свіжо приготовлені для однієї групи за один раз. Кожний засіб був введений у вигляді одиничної дози, індивідуально та у подвійний та потрійний комбінаціях, які були визначено у День 1 (D1) для самок голих мишей із встановленою підшкірною пухлиною. Протягом дослідження велися спостереження за вагою тіла (BW) та станом здоров'я мишей, а також обсяг пухлини вимірювався двічі на тиждень. Тварин піддали евтаназії на D29 після 4 та 24 годин після останньої дози Сполуки A, та зібрали пухлини від трьох тварин на групу на кожну часову точку. Ефективність була визначена залежно від середніх змін обсягу пухлини між D1 та D29. Prism підсумовує результати тестів як недостовірний (ns) при P 0,05, достовірний (позначуваний “*”) при 0,01 P 0,05, дуже 18 UA 115786 C2 достовірний (“**”) при 0,001P0,01, та надзвичайно достовірний (“***”) при P0,001. Результати представлені у нижченаведеній таблиці. Середній об’єм, T/C мм3 або День День мг/кг шлях режим Зміна T/T0 1 29 -po bid 28 122 1065 943 --po qd 28 Режим лікування Гр. n засіб Статистична Середнє регресія Смерть достовірність BW Nadir vs vs vs vs vs PR CR TR NTR G1 G2 G3 G4 G8 -0,6% ---- -- -- 0 0 0 0 день 8 -0,1% ns --- -- -- 0 0 0 0 день 8 -2,5% ns --- -- -- 0 0 0 0 день 8 -1,2% ns --- -- -- 0 0 0 0 день 8 -0,7% ns ns ns -- *** 0 0 0 0 день 4 1 10 Носій 1 Носій 2 2 10 Спол. А 20 po bid 28 122 1016 894 95% 3 10 Спол. В 25 po qd 28 122 660 538 57% 125 955 830 88% 125 491 366 39% 125 239 114 12% ** ** - ns 0 0 - 0 0 125 584 459 49% ns - ns ns *** 0 0 -1,4% день 4 0 0 123 120 -3 -2% *** *** ** *** - 0 0 - 0 0 qod 5 122 229 107 11% ** - - - 0 0 -10,1% день 11 0 0 4 10 Цетуксимаб 30 35 20 20 po ip Спол. В Цетуксимаб 25 20 po ip Спол. А Спол. В Цетуксимаб 20 25 20 po po ip 9 10 Паклітаксел 25 Спол. А Цетуксимаб 8 10 20 po po 7 10 15 20 25 6 10 10 ip 5 10 5 20 Спол. А Спол. В 30 iv biwk 4 bid 28 qd 28 bid 28 biwk 4 qd 28 biwk 4 bid 28 qd 28 biwk 4 ** - Ефективність лікування була визначена на D29, день, коли дозування Сполуки A було завершено. У цілях статистичного аналізу TV, відмінність у об’ємі пухлини між D1 (початок дозування) та днем очікуваного результату, була визначена для кожної тварини. Для кожної групи, що отримувала лікування, відповідь на день очікуваного результату була підрахована за одним з наступних виразів: T/C (%) = 100 T/C, для T 0 T/T0 (%) = 100 T/T0, для T 0, де T = (середній об’єм пухлини для групи, що отримувала лікування, на день очікуваного результату) – (середній об’єм пухлини для групи, що отримувала лікування на D1) C = (середній об’єм пухлини для контрольної групи на день очікуваного результату) – (середній об’єм пухлини для контрольної групи на D1), та T0 = середній об’єм пухлини для групи, що отримувала лікування, на D1. Негативні значення Т/T0 представляють чисте скорочення пухлини для групи. Значення Т/С рівне 40% або менше свідчить про потенційну терапевтичну активність. Відповідь на комбіновану терапію (Групи 5-8) 3 У Групі 5 Сполука A у подвійній комбінації із Сполукою B привела до одержання T 366 мм , що відповідає 39% Т/С, та недостовірного середнього інгібування росту пухлини. Комбінація привела до незначно кращого результату у порівнянні з монотерапією Сполукою A у Групі 2 та монотерапією Сполукою В у Групі 3. У Групі 6 Сполука A у подвійній комбінації із цетуксимабом привела до одержання T 114 3 мм , що відповідає 12% Т/С, та достовірного інгібування росту пухлини (P 0,01). Комбінація привела до достовірно кращого результату у порівнянні з монотерапією Сполукою A у Групі 2 та монотерапією цетуксимабом у Групі 4 (P 0,01). У Групі 7 Сполука В у подвійній комбінації із цетуксимабом привела до одержання T 459 3 мм , що відповідає 49% Т/С, та недостовірного інгібування. Комбінація привела до недостовірно кращого результату у порівнянні з монотерапією Сполукою В у Групі 3 та монотерапією цетуксимабом у Групі 4. У Групі 8 потрійна комбінація Сполуки A, Сполуки B та цетуксимабу привела до одержання 3 T -3 мм , що відповідає -2% Т/ ТО, та достовірної активності (P 0,001). Комбінація привела до достовірно кращого результату у порівнянні з монотерапією Сполукою A у Групі 2 (P 0,001), монотерапією Сполукою В у Групі 3 (P 0,01) та монотерапією цетуксимабом у Групі 4 (P 0,001). Крім того, вона привела до достовірно кращого результату у порівнянні з подвійними 19 UA 115786 C2 комбінаціями Сполука A/Сполука В у Групі 5 та Сполука В/цетуксимаб у Групі 7 (P 0,01) та до недостовірно кращого результату у порівнянні з подвійною комбінацією Сполука A/цетуксимабом у Групі 6. ФОРМУЛА ВИНАХОДУ 5 1. Фармацевтична комбінація, що містить: (а) інгібітор B-Raf формули O O NH HN N Cl N N 10 15 20 N NH F O S O або його фармацевтично прийнятну сіль, (b) інгібітор EGFR, де інгібітор EGFR являє собою ерлотиніб або цетуксимаб та, необов'язково, (c) інгібітор РІ3K-, де інгібітор РІ3К- являє собою Сполуку В , (В) для одночасного, окремого або послідовного введення. 2. Комбінація за п. 1, у якій інгібітор EGFR являє собою ерлотиніб. 3. Комбінація за п. 1, у якій інгібітор EGFR являє собою цетуксимаб. 4. Застосування комбінації за п. 1 для виробництва лікарського препарату для лікування колоректального раку. 5. Спосіб лікування колоректального раку у пацієнтів-людей, де колоректальний рак характеризується мутацією B-Raf, що включає одночасне, окреме або послідовне введення терапевтично ефективної кількості: (а) інгібітору B-Raf формули 20 UA 115786 C2 O O NH HN N Cl N N N NH F O S O або його фармацевтично прийнятної солі, (b) інгібітору EGFR, де інгібітор EGFR являє собою ерлотиніб або цетуксимаб та, необов'язково, (c) інгібітору РІ3K-, де інгібітор РІ3К- являє собою Сполуку В 5 . (В) 6. Спосіб за п. 5, у якому мутація B-Raf є V600 мутацією. Комп’ютерна верстка А. Крулевський Міністерство економічного розвитку і торгівлі України, вул. М. Грушевського, 12/2, м. Київ, 01008, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 21
ДивитисяДодаткова інформація
Назва патенту англійськоюPharmaceutical combinations comprising a b-raf inhibitor, an egfr inhibitor and optionally a pi3k-alpha inhibitor
Автори англійськоюCaponigro, Giordano, Stuart, Darrin, Moutouh-De Parseval, Laure
Автори російськоюКапонигро Джордано, Стюарт Даррин, Муту-Дэ Парсеваль Лора
МПК / Мітки
МПК: A61K 31/4439, A61K 31/506, A61K 39/395, A61K 31/517
Мітки: містить, фармацевтична, рі3k-a, комбінація, b-raf, необов'язково, та, інгібітор
Код посилання
<a href="https://ua.patents.su/23-115786-farmacevtichna-kombinaciya-shho-mistit-ingibitor-b-raf-ingibitor-egfr-ta-neobovyazkovo-ingibitor-ri3k-a.html" target="_blank" rel="follow" title="База патентів України">Фармацевтична комбінація, що містить інгібітор b-raf, інгібітор egfr та, необов’язково, інгібітор рі3k-a</a>
Фармацевтична комбінація, що містить інгібітор b-raf та інгібітор деацетилази гістонів, та її застосування при лікуванні проліферативних захворювань

Номер патенту: 115151
Опубліковано: 25.09.2017
Автори: Галлахер Стюарт Джон, Херсі Пітер
МПК: A61K 31/4045, A61K 31/506
Мітки: інгібітор, b-raf, застосування, містить, захворювань, проліферативних, комбінація, фармацевтична, гістонів, деацетилази, лікуванні
Формула / Реферат:
1. Фармацевтична комбінація, що містить:(а) інгібітор B-Raf формули (I) (I)або його фармацевтично прийнятну сіль; та(b) інгібітор деацетилази гістонів, який являє собою панобіностат, або його фармацевтично прийнятну сіль,для одночасного, окремого або послідовного введення.2. Фармацевтична комбінація за п. 1 для застосування при...
Фармацевтична комбінація, яка містить тестостерон та інгібітор фде5, для лікування жіночої сексуальної дисфункції

Номер патенту: 112406
Опубліковано: 12.09.2016
Автори: Блумерс Йоханнес Мартінус Марія, Тейтен Ян Йохан Адріаан
МПК: A61P 15/08, A61K 31/519, A61K 31/4985, A61K 31/568
Мітки: тестостерон, лікування, фармацевтична, фде5, містить, сексуальної, дисфункції, жіночої, комбінація, яка, інгібітор
Формула / Реферат:
1. Застосування комбінації тестостерону та інгібітору ФДЕ5, який вибирають з групи, що складається з силденафілу, варденафілу, Е-4021, Е-8010, Е-4010, AWD-12-217 (запринасту), AWD 12-210, UK-346,664, UK-369003, UK-357903, BMS-341400, BMS-223131, FR226807, FR-229934, EMR-6203, Sch-51866, ІС485, ТА-1790, DA-8159, NCX-911 або KS-505a, в одержанні медикаменту для лікування жіночої сексуальної дисфункції, де зазначений тестостерон забезпечується...
Фармацевтична композиція, що містить інгібітор альдегідредуктази та інгібітор ангіотензинперетворюючого ферменту

Номер патенту: 64761
Опубліковано: 15.03.2004
Автори: Кері Френк, Кемерон Норман Юджін, Коттер Мері Енн, Таффін Девід Патрік
МПК: A61K 45/06, A61K 38/55, A61K 31/165
Мітки: композиція, ферменту, альдегідредуктази, фармацевтична, ангіотензинперетворюючого, інгібітор, містить
Формула / Реферат:
1. Фармацевтична композиція, яка відрізняється тим, що включає інгібітор альдегідредуктази та інгібітор ферменту перетворення ангіотензину (АСЕ) разом з фармацевтично прийнятним носієм і/або розріджувачем.2. Фармацевтична композиція за п. 1, яка відрізняється тим, що інгібітор альдегідредуктази вибирають з епалрестату, толрестату, понолрестату, зополрестату, AD-5467, SNK-860, ADN-138, AS-3201, зенарестату, сорбінілу, метосорбінілу,...
Комбінація, що містить інгібітор мек та інгібітор в-raf

Номер патенту: 105064
Опубліковано: 10.04.2014
Автори: Лебовіц Пітер, Дамбл Мелісса, Кумар Ракеш, Лакерр Сільві
МПК: A01N 43/90, A61P 35/00, A61K 31/519
Мітки: мек, містить, інгібітор, комбінація, в-raf
Формула / Реферат:
1. Комбінація, що містить: (і) сполуку за формулою (І)(I)або її фармацевтично прийнятну сіль, або сольват; та (іі) сполуку за формулою (II) (II)або її фармацевтично прийнятну сіль.2. Комбінація за п. 1, де сполука (і) є у вигляді сольвату...
Комбінація, що містить метотрексат та інгібітор дгодг

Номер патенту: 104449
Опубліковано: 10.02.2014
Автори: Годессарт Маріна Нурія, Піскуета Лаланса Марія Пілар
МПК: A61K 45/06, A61K 31/44, A61K 31/519, A61P 29/00
Мітки: комбінація, містить, метотрексат, інгібітор, дгодг
Формула / Реферат:
1. Комбінація, що містить (а) метотрексат і (b) негепатотоксичний інгібітор ДГОДГ формули (І):, у якій: R1 вибраний з групи, що включає атоми водню, атоми галогенів, С1-С4-алкіл, С3-С4-циклоалкіл, -CF3 і -OCF3, R2 вибраний з групи, що включає атоми водню, атоми галогенів і С1-С4-алкільні...
Попередній патент: Пристрій в бігувальній машині і вироби, одержувані при його використанні
Наступний патент: Спосіб одержання антигенів haemophilus influenzae типу b
Випадковий патент: Сполуки імідазо[1,2-а]піридину як інгібітори тирозинкінази рецепторів