Похідні оксадіазолу, активні до сфінгозин-1-фосфату (s1p)







Формула / Реферат
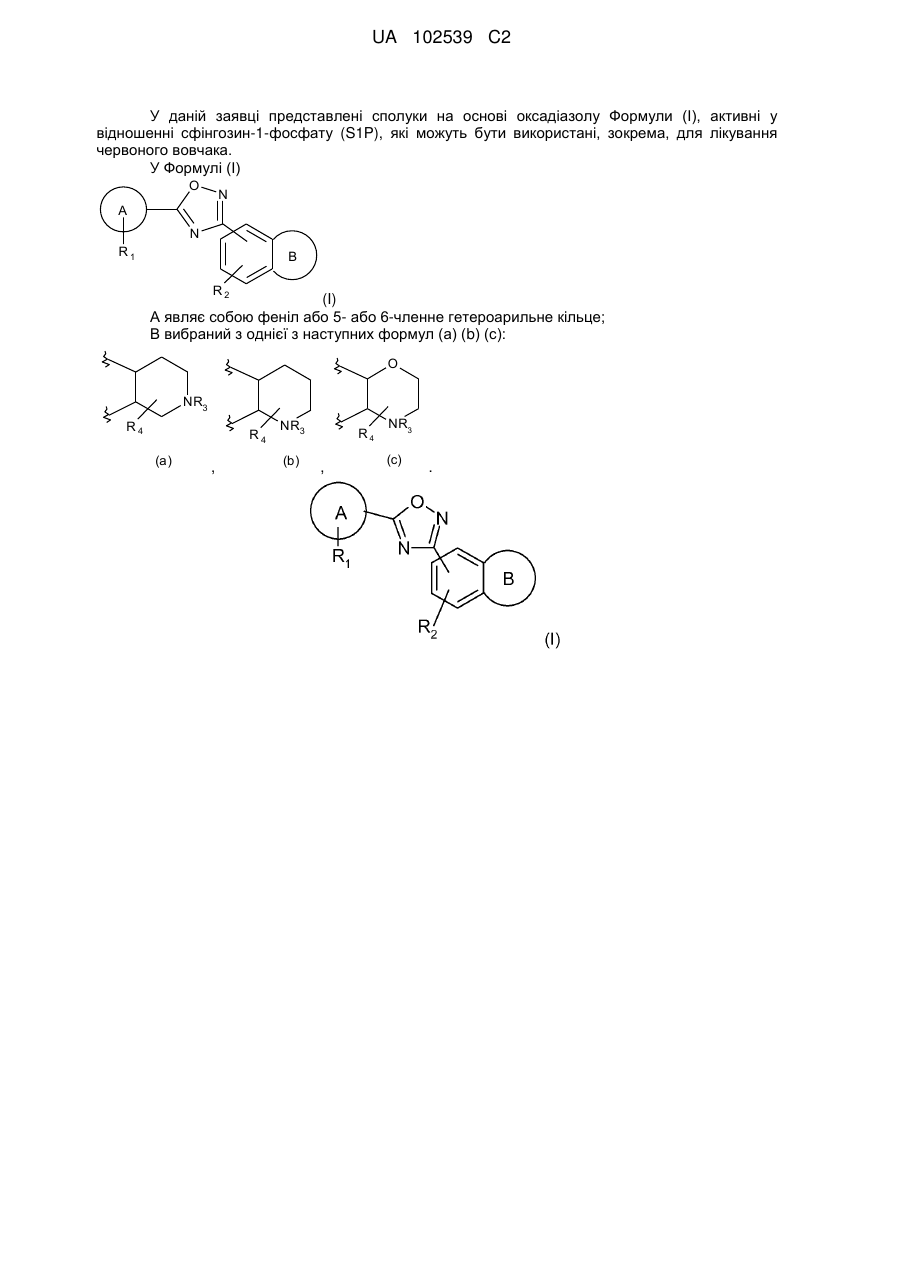
1. Сполука формули (І) або її фармацевтично прийнятна сіль:
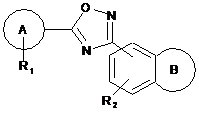 , (I)
, (I)
у якій:
А являє собою феніл;
R1 являє собою до двох замісників, незалежно вибраних з хлору, ізопропокси й ціано;
R2 являє собою водень або метил;
В являє собою:
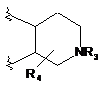 , (а)
, (а)
R3 являє собою водень, метил або (СН2)1-3СО2Н;
R4 являє собою водень.
2. Сполука, вибрана з групи наступних сполук:
5-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-1,2,3,4-тетрагідроізохінолін;
2-[(1-метилетил)окси]-5-[3-(1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5-іл]бензонітрил;
[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1Н)-ізохінолініл]оцтова кислота;
3-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1Н)-ізохінолініл]пропанова кислота;
4-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1Н)-ізохінолініл]бутанова кислота;
2-[(1-метилетил)окси]-5-[3-(2-метил-1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5-іл]бензонітрил;
[8-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2Н-хромен-4-іл]оцтова кислота;
3-[6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)-ізохінолініл]пропанова кислота;
[6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)-ізохінолініл]оцтова кислота;
6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-1,2,3,4-тетрагідроізохінолін;
2-[(1-метилетил)окси]-5-[3-(5-метил-1,2,3,4-тетрагідро-6-ізохінолініл)-1,2,4-оксадіазол-5-іл]бензонітрил;
[6-(5-{3-ціано4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)-ізохінолініл]оцтова кислота;
3-[6-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)-ізохінолініл]пропанова кислота
та їх фармацевтично прийнятні солі.
3. Застосування сполуки відповідно до будь-якого з пп. 1, 2 для лікування станів або порушень, опосередкованих рецептором S1P1.
4. Застосування за п. 3, в якому стан або порушення являє собою розсіяний склероз, аутоімунні захворювання, хронічні запальні порушення, астму, запальні невропатії, артрит, трансплантацію, хворобу Крона, неспецифічний виразковий коліт, червоний вовчак, псоріаз, пошкодження внаслідок ішемії-реперфузїї, солідні пухлини і метастази пухлини, захворювання, пов'язані з ангіогенезом, судинні захворювання, больові стани, гострі вірусні захворювання, запальні стани кишечнику, інсулінозалежний та інсулінонезалежний діабет.
5. Застосування за п. 3, в якому стан являє собою червоний вовчак.
6. Застосування сполуки відповідно до будь-якого з пп. 1, 2 для одержання лікарського засобу для використання в лікуванні станів або порушень, опосередкованих рецептором S1P1.
7. Застосування за п. 6, в якому стан або порушення являють собою розсіяний склероз, аутоімунні захворювання, хронічні запальні порушення, астму, запальні невропатії, артрит, трансплантацію, хворобу Крона, неспецифічний виразковий коліт, червоний вовчак, псоріаз, пошкодження внаслідок ішемії-реперфузії, солідні пухлини і метастази пухлини, захворювання, пов'язані з ангіогенезом, судинні захворювання, больові стани, гострі вірусні захворювання, запальні стани кишечнику, інсулінозалежний та інсулінонезалежний діабет.
8. Застосування за п. 6, в якому стан являє собою червоний вовчак.
9. Фармацевтична композиція, що містить сполуку відповідно до будь-якого з пп. 1, 2.
10. Спосіб лікування станів або порушень у ссавців, включаючи людину, які можуть бути опосередковані рецептором S1P1, який включає введення пацієнтові терапевтично безпечної й ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі.
11. Спосіб лікування за п. 10, в якому стан являє собою червоний вовчак.
Текст
Реферат: UA 102539 C2 (12) UA 102539 C2 У даній заявці представлені сполуки на основі оксадіазолу Формули (І), активні у відношенні сфінгозин-1-фосфату (S1P), які можуть бути використані, зокрема, для лікування червоного вовчака. У Формулі (І) O N A N R1 B R2 (I) А являє собою феніл або 5- або 6-членне гетероарильне кільце; В вибраний з однієї з наступних формул (а) (b) (с): O NR3 R4 R4 (a) , NR3 (b) R4 , NR3 (c) . UA 102539 C2 5 10 15 20 25 30 35 40 45 50 55 60 Даний винахід стосується нових похідних оксадіазолу, що мають фармакологічну активність, способів їх одержання, фармацевтичних композицій, що їх містять та їх застосування в лікуванні різних порушень. Сфінгозин-1-фосфат (S1P) являє собою біоактивний ліпідний медіатор, утворений у результаті фосфорилування сфінгозину сфінгозинкиназами, і виявляється у високих рівнях в крові. Він продукується й секретується безліччю типів клітин, включаючи клітини гематопоетичного походження, як-от тромбоцити і лаброцити (Okamoto et al 1998 J Biol Chem 273(42):27104; Sanchez and HIa 2004, J Cell Biochem 92:913). Він здійснює різноманітні біологічні дії, включаючи регуляцію проліферації, диференціювання, рухливості клітин, васкуляризації й активації запальних клітин і тромбоцитів (Pyne and Pyne 2000, Biochem J. 349: 385). Було описано п'ять підтипів S1P-чутливих рецепторів: S1P1 (Edg-1), S1P2 (Edg-5), S1P3 (Edg-3), S1P4 (Edg-6) і S1P5 (Edg-8), утворюючих частину сімейства рецепторів кон'югованого з Gбілком гена диференціювання ендотелію (Chun et al 2002 Pharmacological Reviews 54:265, Sanchez and HIa 2004 J Cellular Biochemistry, 92:913). Ці 5 рецепторів демонструють диференціальну експресію мРНК, причому S1P1-3 широко експресується, S1P4 експресується на лімфоїдних і гематопоетичних тканинах і S1P5 – перш за все в мозку й у меншій мірі в селезінці. Вони сигналізують через різні субпопуляції G-білків, промотуючи різні біологічні реакції (Kluk and HIa 2002 Biochem et Biophysica Acta 1582:72, Sanchez and HIa 2004, J Cellular Biochem 92:913). Запропоновані ролі для рецептора S1P1 включають трафік лімфоцитів, індукцію/супресію цитокінів і ефекти відносно ендотеліальних клітин (Rosen and Goetzl 2005 Nat Rev Immunol. 5:560). Агоністи рецептора S1P1 використовувалися в безлічі автоімунних і трансплантаційних моделей тварин, включаючи моделі MS Експериментального Автоімунного Енцефаломеліту (EAE), для зменшення серйозності викликаного захворювання (Brinkman et al 2003 JBC 277:21453; Fujino et al 2003 J Pharmacol Exp Ther 305:70; Webb et al 2004 J Neuroimmunol 153:108; Rausch et al 2004 J Magn Reson Imaging 20:16). Ця активність, як повідомлялося, опосередкує ефектом агоністів S1P1 на циркуляцію лімфоцитів через лімфатичну систему. Лікування агоністами S1P1 приводить до секвестрації лімфоцитів у межах вторинних лімфоїдних органів, таких як лімфатичні вузли, викликаючи оборотну периферичну лімфопенію на моделях тварин (Chiba et al 1998, J Immunology 160:5037, Forrest et al 2004 J Pharmacol Exp Ther 309:758; Sanna et al 2004 JBC 279:13839). Опубліковані дані відносно агоністів дозволяють передбачити, що лікування сполукою викликає зникнення рецепторів S1P1 з поверхні клітини в результаті інтерналізації (Graler and Goetzl 2004 FASEB J 18:551; Matloubian et al 2004 Nature 427:355; Jo et al 2005 Chem Biol 12:703), і саме це скорочення рецепторів S1P1 на імунних клітинах робить свій внесок до скорочення руху Т-лімфоцитів з лімфатичних вузлів назад у кровотік. Делеція гена S1P1 викликає ембріональну летальність. Експерименти з дослідження ролі рецептора S1P1 в міграції й трафіку лімфоцитів включали адоптивне перенесення мічених S1P1-дефіцитних Т-лімфоцитів у організм опромінених мишей дикого типу. Ці клітини показали понижений вихід з вторинних лімфоїдних органів (Matloubian et al 2004 Nature 427:355). S1P1 також відводили роль у модуляції злиття ендотеліальних клітин (Allende et al 2003 102:3665, Blood Singelton et al 2005 FASEB J 19:1646). Відносно цієї ендотеліальної дії повідомлялося, що агоністи S1P1 мали ефект на ізольовані лімфатичні вузли, що може робити внесок до ролі в корекції імунних порушень. Агоністи S1P1 викликають закриття ендотеліальних стромальних "воріт" лімфатичних пазух, які дренують лімфатичні вузли й запобігають виходу лімфоцитів (Wei еt al 2005, Nat. Immunology 6:1228). Було показано, що імуносупресивна сполука FTY720 (JP11080026-A) знижує кількість циркулюючих лімфоцитів у тварин і людини, має модулюючу захворювання активність при імунних порушеннях на моделях тварин і зменшує швидкість ремісії в рецидивно-ремітивному розсіяному склерозі (Brinkman et al 2002 JBC 277:21453, Mandala et al 2002 Science 296:346, Fujino et al 2003 J Pharmacology and Experimental Therapeutics 305:45658, Brinkman et al 2004 American J Transplantation 4:1019, Webb et al 2004 J Neuroimmunology 153:108, Morris et al 2005 EurJ Immunol 35:3570, Chiba 2005 Pharmacology and Therapeutics 108:308, Kahan et al 2003, Transplantation 76:1079, Kappos et al 2006 New Eng J Medicine 335:1124). Ця сполука є проліками, які фосфорилують in vivo сфінгозинкиназами з утворенням молекули, яка має агоністичну активність відносно рецепторів S1P1, S1P3, S1P4 і S1P5. Клінічні дослідження показали, що лікування з використанням FTY720 приводить до брадикардії в перші 24 години лікування (Kappos et al 2006 New Eng J Medicine 335:1124). На основі ряду експериментів на основі клітин і на тваринах вважається, що брадикардія є наслідком агонізму до рецептора S1P3. Ці експерименти включають використання S1P3 нокаут-тварин, які, на відміну від мишей 1 UA 102539 C2 5 10 15 20 25 30 35 40 45 дикого типу, не демонструють брадикардію після введення FTY720 і використання S1P1селективних сполук (Hale et al 2004 Bioorganic & Medicinal Chemistry Letters 14:3501, Sanna et al 2004 JBC 279:13839, Koyrakh et al 2005 American J Transplantation 5:529). Отже, існує потреба в сполуках-агоністах рецептора S1P1 з селективністю по S1P3, відносно яких можна було б чекати понижену тенденцію до індукції брадикардії. У наступних заявках на патент описані похідні оксадіазолу як агоністи S1P1: WO03/105771, WO05/058848, WO06/047195, WO06/100633, WO06/115188, WO06/131336, WO07/024922 і WO07/116866. У наступних заявках на патент описані похідні тетрагідроізохінолініл-оксадіазолу як агоністи рецептора S1P: WO06/064757, WO06/001463, WO04/113330. В даний час був виявлений структурно новий клас сполук, які являють собою агоністи рецептора S1P1. Даний винахід тому відноситься до сполук формули (I) або до їх фармацевтично прийнятних солей: A являє собою феніл або 5- або 6-членне гетероарильне кільце; R1 являє собою до двох замісників, незалежно вибраних з галогену, C (1-3) алкокси, C(1-3) фторалкілу, ціано, C(1-3) фторалкокси, C(1-6) алкілу і С(3-6) циклоалкілу; R2 являє собою водень, галоген або C(1-4) алкіл; B вибраний з однієї з наступних груп: R3 являє собою водень, C(1-3) алкіл або (CH2)1-3CO2H; R4 являє собою водень або C(1-3) алкіл; коли R2 або R4 позначає алкіл, він може бути перерваний киснем. У одному варіанті здійснення винаходу, A являє собою феніл; та/або R1 являє собою до двох замісників, незалежно вибраних з хлору, ізопропокси й ціано; та/або R2 являє собою водень; та/або B являє собою (a); та/або R3 являє собою водень, метил або (CH2)1-3CO2H; та/або R4 являє собою водень. A являє собою феніл; R1 являє собою до двох замісників, незалежно вибраних з хлору, ізопропокси й ціано; R2 являє собою водень або метил; B являє собою (a); R3 являє собою водень, метил або (CH2)1-3CO2H; R4 являє собою водень. У одному варіанті здійснення A являє собою феніл. У іншому варіанті здійснення A являє собою 3,4-двозаміщений феніл. У одному варіанті здійснення R1 являє собою двох замісників, один з яких являє собою C (1-3) алкокси, інший вибраний з галогену або ціано. В іншому варіанті здійснення R 1 позначає два замісники, один з яких являє собою ізопропокси, а інший вибраний з хлору або ціано. В іншому варіанті здійснення R1 являє собою два замісники, вибрані з хлору, ізопропокси й ціано. В іншому варіанті здійснення R1 являє собою хлор і ізопропокси. В іншому варіанті здійснення R 1 являє собою хлор в положенні 3 і ізопропокси в положенні 4, коли A являє собою феніл. У 2 UA 102539 C2 5 10 15 20 25 30 35 40 45 50 55 60 іншому варіанті здійснення R1 являє собою ізопропокси й ціано. В іншому варіанті здійснення R1 являє собою ціано в положенні 3 і ізопропокси в положенні 4, коли A являє собою феніл. У одному варіанті здійснення R2 являє собою водень або метил. У одному варіанті здійснення B являє собою (a). У одному варіанті здійснення R3 являє собою водень, метил або (CH2)1-3CO2H. У одному варіанті здійснення R4 являє собою водень. Термін "алкіл" як група або частина групи, наприклад, алкокси або гідроксіалкілу відноситься до прямої або розгалуженої алкільної групи в усіх ізомерних формах. Термін "C(1-6) алкіл" відноситься до алкільної групи, як визначено вище, що містить щонайменше 1 і щонайбільше 6 атомів вуглецю. Приклади таких алкільних груп включають метил, етил, пропіл, ізопропіл, нбутил, ізобутил, втор-бутил або трет-бутил. Приклади таких алкокси-груп включають метокси, етокси, пропокси, ізопропокси, бутокси, ізобутокси, втор-бутокси і трет-бутокси. Відповідні C(3-6) циклоалкільні групи включають циклопропіл, циклобутил, циклопентил і циклогексил. В рамках винаходу термін "галоген" відноситься до фтору (F), хлору (Cl), брому (Br) або йоду (I). Термін "гетероарил" позначає ненасичене кільце, яке включає один або більш гетероатомів, вибраних з O, N або S. Приклади 5- або 6-членних гетероарильних кілець включають піроліл, триазоліл, тіадіазоліл, тетразоліл, імідазоліл, піразоліл, ізотіазоліл, тіазоліл, ізоксазоліл, оксазоліл, оксадіазоліл, фуразаніл, фураніл, тієніл, піридил, піримідиніл, піразиніл, піридазиніл і триазиніл. У деяких із сполук формули (I), залежно від природи замісника, є хіральні атоми вуглецю, і тому сполуки формули (I) можуть існувати як стереоізомери. Винахід охоплює всі оптичні ізомери, такі як стереоізомерні форми сполук формули (I), включаючи енантіомери, діастереоізомери та їх суміші, такі як рацемати. Різні стереоізомерні форми можуть бути розділені або відокремлені одна від одної звичайними способами, або будь-який даний ізомер може бути одержаний звичайним стереоселективним або асиметричним синтезом. Деякі зі сполук, описаних тут, можуть існувати в різних таутомерних формах, і слід розуміти, що винахід охоплює всі такі таутомерні форми. Слід розуміти, що деякі сполуки за винаходом містять як кислотні, так і основні групи і можуть тому існувати як цвітеріони при деяких значеннях рН. Відповідними сполуками за винаходом є: 5-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-1,2,3,4-тетрагідроізохінолін 2-[(1-метилетил)окси]-5-[3-(1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрил [5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1Н)ізохінолініл]оцтова кислота 3-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1Н)ізохінолініл]пропанова кислота 4-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1Н)ізохінолініл]бутанова кислота 2-[(1-метилетил)окси]-5-[3-(2-метил-1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрил [8-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2H-хромен-4іл]оцтова кислота 3-[6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолініл]пропанова кислота [6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолініл]оцтова кислота 6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-1,2,3,4тетрагідроізохінолін 2-[(1-метилетил)окси]-5-[3-(5-метил-1,2,3,4-тетрагідро-6-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрил [6-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолініл]оцтова кислота 3-[6-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолініл]пропанова кислота або їх фармацевтично прийнятні солі. Фармацевтично прийнятні похідні сполук формули (I) включають будь-яку фармацевтично прийнятну сіль, складний ефір або сіль такого складного ефіру сполуки формули (I), яка, при 3 UA 102539 C2 5 10 15 20 25 30 35 40 45 50 55 введенні реципієнтові, здатна приводити (прямо або побічно) до сполуки формули (I) або її активному метаболіту або залишку. Сполуки формули (I) можуть утворювати солі. Слід розуміти, що для використання в медицині солі сполуки формули (I) мають бути фармацевтично прийнятними. Відповідні фармацевтично прийнятні солі є очевидними для фахівця й включають описані в J. Pharm. SCL, 1977, 66, 1-19, як-от солі додання з кислотою, утворені з неорганічними кислотами, наприклад, хлористоводневу, бромистоводневу, сірчану, азотну або фосфорну кислоту; й органічні кислоти, наприклад, бурштинову, малеїнову, оцтову, фумарову, лимонну, винну, бензойну, птолуолсульфонову, метансульфонову або нафталісульфонову кислоту. Деякі зі сполук формули (I) можуть утворювати солі додання з кислотою з одним або більш еквівалентами кислоти. Даний винахід включає всі можливі стехіометричні і нестехіометричні форми. Солі можуть також бути одержані з фармацевтично прийнятних основ, включаючи неорганічні основи й органічні основи. Солі, одержані з неорганічних основ, включають солі алюмінію, амонію, кальцію, міді, тривалентного заліза, двовалентного заліза, літію, магнію, тривалентного марганцю, двовалентного марганцю, калію, натрію, цинку й т.п. Солі, одержані з фармацевтично прийнятних органічних основ, включають солі первинних, вторинних і третинних амінів; заміщені аміни, включаючи природні заміщені аміни; й циклічні аміни. Окремі випадки фармацевтично прийнятних органічних основ включають аргінін, бетаїн, кофеїн, холін, N, N'-дибензилетилендіамін, діетиламін, 2-діетиламіноетанол, 2-диметиламіноетанол, етаноламін, етилендіамін, N-етил-морфолін, N-етилпіперидин, глюкамін, глюкозамін, гістидин, гідрабамін, ізопропіламін, лізин, метилглюкамін, морфолін, піперазин, піперидин, прокаїн, пурини, теобромін, триетиламін, триметиламін, трипропіламін, трис(гідроксиметил) амінометан (TRIS, трометамол) і т.п. Солі можна також утворити з катіонообмінних смол, наприклад поліамінних смол. Коли сполука згідно з даним винаходом є основною, солі можуть бути одержані з фармацевтично прийнятних кислот, включаючи неорганічні й органічні кислоти. Такі кислоти включають оцтову, бензолсульфонову, бензойну, камфорсульфонову, лимонну, етансульфонову, етандисульфонову, фумарову, глюконову, глутамінову, бромистоводневу, хлористоводневу, ізетинову, молочну, малеїнову, яблучну, мигдальну, метансульфонову, муцинову, памову, пантотенову, фосфорну, пропіонову, бурштинову, сірчану, винну, птолуолсульфонову кислоту і т.п. Фармацевтично прийнятні солі додавання можуть бути одержані звичайним способом реакцією з відповідною кислотою або похідною кислоти. Фармацевтично прийнятні солі з основами можуть бути одержані звичайним способом реакцією з відповідною неорганічною або органічною основою. Сполуки формули (I) можуть бути одержані в кристалічній або некристалічній формі, й, у разі кристалічної форми, можуть бути гідратовані або сольватовані. Цей винахід включає стехіометричні гідрати або сольвати, а також сполуки, що містять змінні кількості води та/або розчинника. У рамки винаходу входять усі солі, сольвати, гідрати, комплекси, поліморфи, проліки, мічені радіоактивним ізотопом похідні, стереоізомери й оптичні ізомери сполук формули (I). Потенціал і ефективність сполук за винаходом відносно рецептора S1P1 можуть бути визначені тестом ГТФS, що проводиться на клонованому рецепторі людини, як описано тут. Сполуки формули (I) продемонстрували агоністичну активність відносно рецептора S1P1 при використанні функціональних тестів, описаних тут. Сполуки формули (I) і їх фармацевтично прийнятні солі можуть тому бути використані в лікуванні станів або порушень, опосередкованих рецептором S1P1. Зокрема, сполуки формули (I) і їх фармацевтично прийнятні солі можуть бути використані в лікуванні розсіяного склерозу, автоімунних захворювань, хронічних запальних порушень, астми, запальних невропатій, артриту, трансплантації, хвороби Крону, неспецифічного виразкового коліту, червоного вовчаку, псоріазу, пошкодження унаслідок ішемії-реперфузії, солідних пухлин і метастазів пухлини, захворювань, пов'язаних з ангіогенезом, судинних захворювань, больових станів, гострих вірусних захворювань, запальних станів кишечнику, інсулінозалежного й інсулінонезалежного діабету (далі званих "Порушеннями за винаходом"). Сполуки формули (I) і їх фармацевтично прийнятні солі можуть тому бути використані в лікуванні червоного вовчаку. Сполуки формули (I) і їх фармацевтично прийнятні солі можуть тому бути використані в лікуванні псоріазу. Сполуки формули (I) і їх фармацевтично прийнятні солі можуть тому бути використані в лікуванні розсіяного склерозу. 4 UA 102539 C2 5 10 15 20 25 30 35 40 45 50 55 60 Слід розуміти, що "лікування" в рамках винаходу включає профілактику, а також полегшення встановлених симптомів. Таким чином, винахід також відноситься до сполук формули (I) або до їх фармацевтично прийнятних солей для вживання в якості терапевтичних речовин, зокрема, в лікуванні станів або порушень, опосередкованих рецептором S1P1. Зокрема, винахід відноситься до сполуки формули (I) або до її фармацевтично прийнятної солі для застосування як терапевтичної речовини в лікуванні розсіяного склерозу, автоімунних захворювань, хронічних запальних порушень, астми, запальних невропатій, артриту, трансплантації, хвороби Крону, неспецифічного виразкового коліту, червоного вовчаку, псоріазу, пошкодження унаслідок ішеміїреперфузії, солідних пухлин і метастазів пухлини, захворювань, пов'язаних з ангіогенезом, судинних захворювань, больових станів, гострих вірусних захворювань, запальних станів кишечнику, інсулінозалежного й інсулінонезалежного діабету. Сполуки формули (I) і їх фармацевтично прийнятні солі можуть тому бути використані як терапевтичні речовини в лікуванні червоного вовчаку. Сполуки формули (I) і їх фармацевтично прийнятні солі можуть тому бути використані як терапевтичні речовини в лікуванні псоріазу. Сполуки формули (I) і їх фармацевтично прийнятні солі можуть тому бути використані як терапевтичні речовини в лікуванні розсіяного склерозу. Винахід також відноситься до способу лікування станів або порушень у ссавців, включаючи людину, які можуть бути опосередковані рецептором S1P1, який включає введення пацієнтові терапевтично безпечної й ефективної кількості сполуки формули (I) або її фармацевтично прийнятної солі. Зокрема, винахід відноситься до способу лікування розсіяного склерозу, автоімунних захворювань, хронічних запальних порушень, астми, запальних невропатій, артриту, трансплантації, хвороби Крону, неспецифічного виразкового коліту, червоного вовчаку, псоріазу, пошкодження внаслідок ішемії-реперфузії, солідних пухлин і метастазів пухлини, захворювань, пов'язаних з ангіогенезом, судинних захворювань, больових станів, гострих вірусних захворювань, запальних станів кишечнику, інсулінозалежного й інсулінонезалежного діабету, який включає введення пацієнтові терапевтично безпечної й ефективної кількості сполуки формули (I) або її фармацевтично прийнятної солі. Винахід відноситься до способу лікування червоного вовчаку, який включає введення пацієнтові терапевтично безпечної й ефективної кількості сполуки формули (I) або її фармацевтично прийнятної солі. Винахід відноситься до способу лікування псоріазу, який включає введення пацієнтові терапевтично безпечної й ефективної кількості сполуки формули (I) або її фармацевтично прийнятної солі. Винахід відноситься до способу лікування розсіяного склерозу, який включає введення пацієнтові терапевтично безпечної і ефективної кількості сполуки формули (I) або її фармацевтично прийнятної солі. У іншому аспекті винахід відноситься до застосування сполуки формули (I) або її фармацевтично прийнятної солі в одержанні лікарського засобу для використання в лікуванні станів або порушень, опосередкованих рецептором S1P1. Зокрема, винахід відноситься до сполуки формули (I) або до її фармацевтично прийнятної солі для застосування при одержанні лікарського засобу для використання в лікуванні розсіяного склерозу, автоімунних захворювань, хронічних запальних порушень, астми, запальних невропатій, артриту, трансплантації, хвороби Крону, неспецифічного виразкового коліту, червоного вовчаку, псоріазу, пошкодження внаслідок ішемії-реперфузії, солідних пухлин і метастазів пухлини, захворювань, пов'язаних з ангіогенезом, судинних захворювань, больових станів, гострих вірусних захворювань, запальних станів кишечнику, інсулінозалежного й інсулінонезалежного діабету. Сполуки формули (I) та їх фармацевтично прийнятні солі можуть бути використані в одержанні лікарського засобу для використання в лікуванні червоного вовчаку. Сполуки формули (I) і їх фармацевтично прийнятні солі можуть бути використані в одержанні лікарського засобу для використання в лікуванні псоріазу. Сполуки формули (I) і їх фармацевтично прийнятні солі можуть бути використані в одержанні лікарського засобу для використання в лікуванні розсіяного склерозу. Для застосування сполук формули (I) і їх фармацевтично прийнятних солей у терапії, їх зазвичай складають у фармацевтичну композицію відповідно до звичайної фармацевтичної практики. Даний винахід також відноситься до фармацевтичної композиції, яка включає сполуку формули (I) або її фармацевтично прийнятну сіль і фармацевтично прийнятний носій або ексципієнт. 5 UA 102539 C2 5 10 15 20 25 30 35 40 45 50 55 У іншому аспекті даний винахід відноситься до способу одержання фармацевтичної композиції, що включає змішування сполуки формули (I) або її фармацевтично прийнятної солі й фармацевтично прийнятного носія або ексципієнта. Фармацевтичну композицію за винаходом, яка може бути одержана шляхом змішування, переважно при температурі довкілля й атмосферному тиску, зазвичай адаптують для перорального, парентерального або ректального введення, і вона також може бути у формі пігулок, капсул, пероральних рідких препаратів, порошків, гранул, пігулок для розсмоктування, відновлюваних порошків, розчинів або суспензій для ін'єкції або інфузії або супозиторіїв. У цілому переважними є перорально введені композиції. Пігулки й капсули для перорального введення можуть бути в стандартній лікарській формі й можуть містити звичайні ексципієнти, такі як сполучні речовини (наприклад, заздалегідь желатований кукурудзяний крохмаль, полівінілпіролідон або гідроксипропілметилцелюлозу); наповнювачі (наприклад, лактозу, мікрокристалічну целюлозу або гідрофосфат кальцію); лубриканти для таблетування (наприклад, стеарат магнію, тальк або діоксид кремнію); дезінтегратори (наприклад, картопляний крохмаль або гліколят крохмалю натрію); і прийнятні змочувальні речовини (наприклад, лаурилсульфат натрію). Пігулки можуть бути забезпечені покриттям згідно зі способами, відомими в звичайній фармацевтичній практиці. Пероральні рідкі препарати можуть бути у формі, наприклад, водної або масляної суспензії, розчинів, емульсій, сиропів або еліксирів, або можуть бути у формі сухого продукту для відновлення водою або іншим відповідним носієм перед використанням. Такі рідкі препарати можуть містити звичайні добавки, такі як суспендуючі агенти (наприклад, сироп сорбіту, похідні целюлози або гідровані харчові жири), емульгатори (наприклад, лецитин або гуміарабік), неводні носії (які можуть включати харчові масла, наприклад, мигдалеве масло, складні ефіри масел, етиловий спирт або фракціоновані рослинні олії), консерванти (наприклад метил або пропіл-п-гідроксибензоати або сорбінову кислоту) і, якщо бажано, звичайні ароматизатори або фарбники, буферні солі й підсолоджувачі. Препарати для перорального введення можуть бути переважно складені для забезпечення контрольованого вивільнення активної сполуки. Для парентерального введення рідкі стандартні лікарські форми одержують, використовуючи сполуку за винаходом або її фармацевтично прийнятні солі й стерильний носій. Склади для ін'єкції можуть бути представлені в стандартній лікарській формі, наприклад, в ампулах або в мультидозі, з використанням сполуки за винаходом або її фармацевтично прийнятних похідних і стерильного носія, в разі потреби з додаванням консерванту. Композиції можуть приймати такі форми, як суспензії, розчини або емульсії в масляних або водних носіях і можуть містити засоби для складання, як-то суспендуючі, стабілізуючі та/або диспергуючі агенти. Альтернативно, активний інгредієнт може бути в порошковій формі для відновлення з використанням відповідного носія, наприклад, стерильної апірогенної води, перед використанням. Сполука, залежно від носія й використовуваної концентрації, може бути або суспендована, або розчинена в носієві. При одержанні розчинів сполука може бути розчинена для ін'єкції й стерилізована фільтрацією перед заповненням у відповідну пляшечку або ампулу і закупорюванням. Переважно, ад'юванти, такі як місцевий анестетик, консерванти й буферні агенти, розчиняють у носієві. Для збільшення стабільності композиція може бути заморожена після заповнення в пляшечку, і вода може бути видалена під вакуумом. Парентеральні суспензії одержують в основному тим самим чином, за винятком того, що сполука суспендує в носієві замість розчинення, й стерилізація не може бути здійснена фільтрацією. Сполука може стерилізуватися дією етиленоксиду перед суспендуванням у стерильному носієві. Переважно, поверхнево-активну речовину або змочувальну речовину включають у композицію, щоб полегшити однорідний розподіл сполуки. Лосьйони можуть бути складені з водною або масляною основою й зазвичай також містять один або більше емульгаторів, стабілізуючих агентів, диспергуючих агентів, суспендуючих агентів, загусників або фарбників. Краплі можуть бути складені з водною або неводною основою, що також включає один або більш диспергуючих агентів, стабілізуючих агентів, солюбілізуючих агентів або суспендуючих агентів. Вони можуть також містити консервант. Сполуки формули (I) або їх фармацевтично прийнятні солі можуть також бути складені в ректальних композиціях, таких як супозиторії або клізми, що наприклад містять звичайну основу для супозиторіїв, таку як масло какао або інші гліцериди. Сполуки формули (I) або їх фармацевтично прийнятні солі можуть також бути складені як препарати депо. Такі суміші тривалої дії можуть вводитися імплантацією (наприклад, підшкірно або внутрішньом'язово) або внутрішньом'язовою ін'єкцією. Таким чином, наприклад, сполуки за винаходом можуть бути складені з відповідними полімерними або гідрофобними матеріалами 6 UA 102539 C2 5 10 15 20 25 30 35 40 45 50 (наприклад, у формі емульсії в прийнятному маслі) або іонообмінними смолами, або у формі помірно розчинних похідних, наприклад, як помірно розчинна сіль. Для внутрішньоносового введення сполуки формули (I) або їх фармацевтично прийнятні солі можуть бути складені у формі розчинів для введення через відповідний пристрій з відміряною дозою або пристрій для введення однієї дози, або, альтернативно, як порошкову сполуку з відповідним носієм для введення з використанням відповідного пристрою для доставки. Таким чином, сполуки формули (I) або їх фармацевтично прийнятні солі можуть бути складені для перорального, щічного, парентерального, топічного (включаючи очне і носове) введення, введення у формі депо або ректального введення, або у формі, придатній для введення інгаляцією або вдуванням (через рот або ніс). Сполуки формули (I) або їх фармацевтично прийнятні солі можуть бути складені для топічного введення у формі мазей, кремів, гелів, лосьйонів, песаріїв, аерозолів або крапель (наприклад, очних, вушних або носових крапель). Мазі й креми можуть, наприклад, бути складені з водною або масляною основою з додаванням відповідних загусників та/або желеутвірних засобів. Мазі для введення в око можуть бути одержані стерильним чином з використанням стерилізованих компонентів. Композиція може містити від 0,1 % до 99 мас. %, переважно від 10 до 60 мас. %, активної речовини, залежно від способу введення. Доза сполуки, використовуваної в лікуванні вказаних порушень, варіює звичайним чином залежно від серйозності порушень, маси тіла пацієнта та інших подібних чинників. Проте, як приклад, відповідні разові дози можуть складати від 0,05 до 1000 мг, від 1,0 до 500 мг або від 1,0 до 200 мг, і такі разові дози можна вводити частіш, ніж один раз на добу, наприклад, два або три рази на добу. Сполуки формули (I) або їх фармацевтично прийнятні солі можуть використовуватися в комбінованих препаратах. Наприклад, сполуки за винаходом можуть використовуватися в комбінації з циклоспорином A, метотрексатом, стероїдами, рапаміцином, інгібіторами прозапальних цитокінів, імуномодуляторами, включаючи біологічні або інші терапевтично активні сполуки. Заявлений винахід також включає ізотопно-мічені сполуки, які є ідентичними вказаним у формулі I і подальших, але в яких один або більше атомів замінені атомом, що має атомну масу або масове число, відмінне від атомної маси або масового числа, що зазвичай зустрічаються в природі. Приклади ізотопів, які можуть бути включені в сполуки за винаходом, включають 3 11 14 18 123 ізотопи водню, вуглецю, азоту, кисню, фосфору, фтору, йоду і хлору, такі як H, C, C, F, I 125 та I. Сполуки згідно з даним винаходом і фармацевтично прийнятні солі вказаних сполук, які містять вказані ізотопи та/або інші ізотопи інших атомів, знаходяться в рамках даного винаходу. Ізотопно-мічені сполуки згідно з даним винаходом, наприклад, ті, в які включені радіоактивні 3 14 ізотопи, такі як H, C, можуть бути використані в тестах на розподіл лікарського засобу та/або 3 14 субстрату в тканині. Ізотопи тритій, тобто, H, і вуглець-14, тобто, C, є особливо переважними 11 8 через простоту їх одержання й детекції. Ізотопи C і F особливо корисні в РЕТ (позитронна 125 емісійна томографія), і ізотопи I особливо корисні в SPECT (однофотонна емісійна комп'ютеризована томографія), які використовуються у візуалізації мозку. Далі, заміщення 2 важчими ізотопами, такими як дейтерій, тобто, H, може забезпечити деякі терапевтичні переваги, що є наслідком більшої метаболічної стабільності, наприклад, збільшеного періоду напівжиття in vivo, або знижених вимог відносно дозування й, отже, може бути переважним при деяких обставинах. Ізотопно-мічені сполуки формули (I) за винаходом можуть у цілому бути одержані шляхом здійснення процедур, розкритих у Схемах та/або в Прикладах, представлених нижче, із заміною не міченого ізотопом реактиву легкодосяжним ізотопно-міченим реактивом. У іншому аспекті цей винахід відноситься до способів одержання сполуки формули (I). Сполуки формули (I), в якій B являє собою і R3 являє собою (CH2)1-3CO2H, може бути одержані згідно зі Схемою 1, на якій R1, R2, R4 і A мають значення, визначені для формули (I), n=3, hal являє собою хлор, бром або йод, P являє собою захисну групу, і R являє собою алкільну групу (наприклад, етил або трет-бутил). 7 UA 102539 C2 Схема 1 5 10 15 20 25 Сполуки формули (ii)(наприклад, одержані як описано в Tetrahedron (2006), 62(29), 68696875) можуть бути перетворені на відповідну захищену похідну (iii), де P являє собою, наприклад, трет-бутоксикарбоніл, з використанням відповідного захисного реагенту, такого як біс(1,1-диметилетил) бікарбонат. Сполуки формули (iii) можуть бути перетворені в сполуки формули (iv) реакцією з гідроксиламіном у присутності відповідної основи, такої як бікарбонат натрію. Сполуки формули (iv) можуть бути перетворені на сполуки формули (vi) реакцією з хлорангідридом кислоти формули (v) у присутності відповідної основи, такої як N, Nдіізопропілетиламін, у разі потреби у присутності каталізатора, такого як 4-діметиламінопіридин. Реакція може бути здійснена спочатку при температурі довкілля з подальшим нагріванням для здійснення циклізації в оксадіазол. Сплолуки формули (vi) можуть бути піддані реакції видалення захисної групи з утворенням сполук формули (vii), наприклад, з відповідною кислотою, такій як соляна кислота, де P являє собою трет-бутоксикарбоніл. Сполуки формули (vii) можуть бути перетворені в сполуки формули (ix) реакцією з алкілувальним агентом формули (viii) у присутності відповідної основи, такої як карбонат цезію. Сполуки формули (ix) можуть бути перетворені на деякі сполуки формули (I) гідролізом з відповідною основою, такою як гідроксид літію, або (коли R=трет-бутил) відповідною кислотою, такою як соляна кислота або трифтороцтова кислота. Сполуки формули (v) є відомими сполуками або можуть бути одержані звичайними засобами з відповідних кислот, які самі є відомими сполуками або можуть бути одержані звичайними засобами. Сполуки формули (iii), в якій R2 знаходиться в положенні 5, і ціаногрупа знаходиться в положенні 6 тетрагідроізохінолінової кільцевої системи, можуть бути одержані, як показано на Схемі 2, на якій R1 має значення, визначені для формули (I), R4 являє собою водень, і P являє собою захисну групу. Схема 2 8 UA 102539 C2 5 10 15 20 25 30 Сполуки формули (x) можуть бути перетворені на сполуки формули (xii) реакцією з нітрометаном у присутності відповідної основи, такої як ацетат амонію. Сполуки формули (xi) можуть бути перетворені на сполуки формули (xii) відновленням з використанням відповідного відновника, такого як борогідрид літію у присутності хлортриметилсілану. Сполуки формули (xii) можуть бути перетворені на сполуки формули (xiii) реакцією з відповідним формілуучим реагентом, як-от етилформіат. Сполуки формули (xiii) можуть бути перетворені на сполуки формули (xiv) реакцією з відповідним джерелом формальдегіду, таким як параформальдегід, у присутності відповідного кислотного каталізатора, такого як мурашина кислота. Сполуки формули (xiv) можуть бути перетворені на сполуки формули (xv) реакцією з відповідним деметилуучим агентом, таким як трибромід бору. Сполуки формули (xv) можуть бути перетворені на відповідні захищені похідні формули (xvi), де P являє собою, наприклад, третбутоксикарбоніл, з використанням відповідного захисного реагенту, такого як біс(1,1диметилетил) бікарбонат. Сполуки формули (xvi) можуть бути перетворені на сполуки формули (xvii) реакцією з відповідним трифторметансульфонілуучим агентом, таким як трифторметансульфоновий ангідрид. Сполуки формули (xvii) можуть бути перетворені на сполуки формули (iii) (у якій R2 знаходиться в положенні 5, R4 позначає водень і ціаногрупа знаходиться в положенні 6) реакцією з відповідним джерелом ціаніду, таким як ціанід цинку, у присутності відповідного каталізатора, такого як тетракис(трифенілфосфін) паладій (0). Всі публікації, включаючи, але не обмежуючись ними, патенти і заявки на патент, процитовані в цьому описі, включені шляхом посилання, неначебто кожна індивідуальна публікація була спеціально й індивідуально позначена як включена у даний опис шляхом посилання. Наступні Описи й Приклади ілюструють одержання сполук за винаходом. Абревіатури: г – грами мг – міліграми мл – мілілітри мкл – мікролітри MеCN – ацетонітріл MеOH – метанол EtOH – етанол Et2O – простий діетиловий ефір EtOAc – етилацетат 9 UA 102539 C2 5 10 15 20 25 30 35 40 45 50 DCM – дихлорметан DIAD – діізопропіл азодикарбоксилат DIPEA – N, N-діізопропілетиламін DMAP – N, N-диметил-4-піридинамін DME – 1,2-біс(метилоксі)етан DMF – N, N-диметилформамід DMSO – диметилсульфоксид EDAC – N-(3-диметиламінопропіл)-N'-етилкарбодиімід гідрохлорид EDC – N-(3-диметиламінопропіл)-N'-етилкарбодиімід гідрохлорид EDCI-N-(3-диметиламінопропіл)-N'-етилкарбодиімід гідрохлорид HOBT/HOBt – гідроксибензотриазол IPA – ізопропіловий спирт NCS – N-хлорсукцинімід PYBOP – бензотриазол-1-іл-окситрипірролідинофосфоний гексафторфосфат THF – тетрагідрофуран Dba – дибензиліден ацетон RT – температура довкілля °C – градуси Цельсія M – моль H – протон s – синглет d – дублет t – триплет q – квартет MГц – мегагерц MEOD – мічений дейтерієм метанол LCMS – Рідинна хроматографія з мас-спектрометрією LC/MS – Рідинна хроматографія з мас-спектрометрією MS – мас-спектрометрія ES – Електророзпилення + MH+ – маса іон+H MDAP – мас-направлена автоматизована препаративна рідинна хроматографія насич. – насичений Boc – трет-бутилоксикарбоніл SCX або SCX-3 – екстракційна твердофазна (SPE) колонка із залишками бензолсульфонової кислоти, іммобілізованими на твердій фазі (наприклад, колонки IST Isolute™). Розділ загальної хімії Способи, описані нижче, наведені в ілюстративних цілях, проміжні сполуки в описаному в прикладах одержанні, не обов'язково були одержані від конкретних описаних завантажень. Приклад одержання 1 1,1-диметилетил-5-[(гідроксіаміно)(іміно)метил]-3,4-дигідро-2(1H)-ізохінолінкарбоксилату 1,2,3,4-тетрагідро-5-ізохінолінкарбонітрил гідрохлорид (5,0 г/25,7 ммоль, від Fluorchem) розділяли між DCM (100 мл) і 2н NаOH. Шар DCM збирали, висушували (гідрофобна фрита) й упарювали. Вільна основа, 1,2,3,4-тетрагідро-5-ізохінолінкарбонітрил, розчиняли в сухому DCM (100 мл) і обробляли біс(1,1-диметилетил)бікарбонатом (1,1 екв., 6,17 г). Реакційну суміш перемішували при температурі довкілля під аргоном протягом 18 годин, промивали 2н NаOH (100 мл), 2н HCl (100 мл), висушували (гідрофобна фрита) й упарювали, одержуючи сирий 1,1диметилетил-5-ціано-3,4-дигідро-2(1Н)-ізохінолінкарбоксилат, який використовували без 1 подальшого очищення (LCMS 100 %, ЯМР суміш 2:1 з tBuOH). Ізольований вихід 7,58 г. H ЯМР (400 Мгц, CDCl3) δ (серед іншого) 7,52 (1Н, д), 7,35-7,16 (2H, м), 4,60 (2H, ушир.с), 3,71 (2H, т), + 3,04 (2H, т), 1,5 (9H, с); m/z (API-ES) 203 [M+H-56] . 10 UA 102539 C2 5 10 15 20 25 30 35 40 Сирий матеріал, одержаний вище, гідроксиламін.HCl (14,31 г, 206 ммоль) і бікарбонат натрію (21,63 г, 257 ммоль) додавали в круглодонну колбу на 500 мл, що містить етанол (200 мл). Реакційну суміш нагрівали при 65 °C протягом 24 годин. Охолоджену реакційну суміш упарювали і розділяли між DCM (2100 мл) і водою (100 мл). Об'єднані шари DCM збирали і висушували. Аналіз LC/MS показав, що матеріал має чистоту 80 %. Цей матеріал розчиняли в етанолі (100 мл) і фільтрували, щоб видалити нерозчинені домішки. Аналіз LC/MS показав, що матеріал має чистоту 92 %. 1,1-диметилетил-5-[(гідроксіаміно)(іміно)метил]-3,4-дигідро-2(1Н)1 ізохінолінкарбоксилат використовували без подальшого очищення. H ЯМР (400 МГц, CDCl3) δ 7,82 (1Н, ушир.с), 7,36-7,16 (3H, м), 4,80 (2H, ушир.с), 4,58 (2H, ушир.с), 3,59 (2H, ушир.т), 2,98 + (2H, т), 1,49 (9H, с); m/z (API-ES) 292 [M+H] . Приклад одержання 2 3-хлор-4-[(1-метилетил)окси]метилбензоату 3-хлор-4-гідроксиметилбензоат (50 г, 0,27 моль), K2CO3 (74 г, 0,54 моль) і йодопропан (29,5 мл, 0,23 моль) перемішували при температурі довкілля в DMF (100 мл). Через 18 годин розчинник видаляли випаровуванням під вакуумом, і залишок піддавали хроматографії на колонці з силікагелем 60 в суміші EtOAc/гексан (1:1), одержуючи цільову сполуку у формі масла 1 (55 г, 90 %). H ЯМР (400 МГц, CDCl3) δ 8,05 (1Н, д), 7,89 (1Н, дд), 6,94 (1Н, д), 4,60-4,72 (1Н, м), + 3,89 (3H, с), 1,41 (6H, д); m/z (API-ES) 229 [M+H] . Приклад одержання 3 1,1-диметилетил-5-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро2(1Н)-ізохінолінкарбоксилату 1,1-диметилетил-5-[(гідроксіаміно)(іміно)метил]-3,4-дигідро-2(1Н)-ізохінолінкарбоксилат (Приклад одержання 1, 2 г, 6,86 ммоль) розчиняли в тетрагідрофурані (THF)(100 мл) і перемішували з гідридом натрію (60 %-а дисперсія, 0,302 г, 7,55 ммоль) під аргоном при температурі довкілля протягом 30 хвилин. Потім додавали 3-хлор-4-[(1метилетил)окси]метилбензоат (Приклад одержання 2) (2,355 г, 10,30 ммоль), і реакційну суміш нагрівали при температурі кипіння розчинника протягом 1,5 годин. Охолоджену реакційну суміш упарювали й розділяли між DCM (100 мл) і водою (100 мл). Водний шар промивали DCM (50 мл) і об'єднані шари DCM висушували (гідрофобна фрита) й упарювали. Сирий продукт очищали хроматографією через тонкий шар силікагелю, елюючи DCM, одержуючи 1,1-диметилетил-5-(5{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1Н)1 ізохінолінкарбоксилат (2,57 г, 5,47 ммоль, 80 %-ий вихід). H ЯМР (400 Мгц, CDCl3) δ 8,23 (1Н, с), 8,05 (1Н, д), 7,95 (1Н, д), 7,34 (1Н, т), 7,27 (1Н, д), 7,06 (1Н, д), 4,76-4,63 (1Н, м), 4,66 (2H, ушир.с), 3,67 (2H, ушир.т), 3,25 (2H, ушир.т), 1,51 (9H, с), 1,45 (6H, д); m/z (API-ES) 414, 416 + [M+H-56] . Приклад одержання 4 1,1-диметилетил-5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4дигідро-2(1Н)-ізохінолінкарбоксилату До розчину 3-ціано-4-[(1-метилетил)окси]бензойної кислоти (може бути одержана як описано в WO 2005/58848, 737 мг, 3,59 ммоль) і оксалілхлориду (360 мкл) в DCM (20 мл) додавали DMF 11 UA 102539 C2 5 10 15 20 25 30 35 40 45 (20 мкл). Розчин перемішували при температурі довкілля протягом 90 хвилин, потім концентрували у вакуумі й повторно розчиняли в сухому DMF (6 мл). Окремо 1,1-диметилетил5-[(гідроксіаміно)(іміно)метил]-3,4-дигідро-2(1Н)-ізохінолінкарбоксилат (Приклад одержання 1, 1,05 г, 3,60 ммоль), N, N-диметил-4-піридинамін (DMAP) (20 мг) і N, N-діізопропілетиламін (DIPEA) (1,31 мл, 7,50 ммоль) розчиняли в DMF (10 мл). 5 мл першого розчину DMF додавали до другого розчину, і жовтий розчин перемішували при температурі довкілля протягом 1 години й потім при 95 °C протягом 18 годин. Реакційну суміш концентрували у вакуумі, потім розділяли між DCM (50 мл) і насиченим водним розчином NaHCO3 (50 мл). Органічні фракції промивали насиченим водним розчином NaHCO3 (50 мл), потім об'єднані водні екстрагували DCM (20 мл). Об'єднані органічні фракції концентрували у вакуумі, одержуючи сире коричневе масло. Флешхроматографія (градієнт MеOH [0,5-4 %] в DCM) і концентрація у вакуумі дала цільову сполуку у 1 формі твердої речовини світло-жовтого кольору (512 мг, 37 %). H ЯМР (400 МГц, CDCl3) δ 8,42 (1Н, д), 8,34 (1Н, дд), 7,96 (1Н, д), 7,34 (1Н, т), 7,29 (1Н, д), 7,13 (1Н, д), 4,80 (1Н, септет), 4,67 + (2H, с), 3,68 (2H, т), 3,24 (2H, т), 1,51 (9H, с), 1,48 (6H, д); m/z (ES) 361 [M+H-100] . Приклад одержання 5 [5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1H)ізохінолініл]етилацетат сіль мурашиної кислоти Суспензію 2-[(1-метилетил)окси]-5-[3-(1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрил (Приклад 3, 28,8 мг, 0,080 ммоль), етил бромацетату (35,5 мкл, 0,32 ммоль) і Cs2CO3 (130 мг, 0,40 ммоль) у DMF (2 мл) перемішували при температурі довкілля протягом 1 години і потім нагрівали при 60 °C протягом 5 хвилин в мікрохвильовому реакторі. Суспензію додавали в картридж SCX, і продукт елюювали метанольним розчином аміаку. Концентрація у вакуумі дала сирий продукт (43 мг), який був очищений MDAP, одержуючи цільову сполуку як 1 непігментовану плівку (11,6 мг, 32 %). H ЯМР (400 МГц, CDCl3) δ 8,41 (1Н, д), 8,33 (1Н, дд), 8,09 (1Н, ушир.с), 8,01 (1Н, д), 7,34 (1Н, здається т), 7,21 (1Н, д), 7,12 (1Н, д), 4,80 (1Н, септет), 4,24 (2H, кв), 4,00 (2H, с), 3,52 (2H, с), 3,35 (2H, т), 3,07 (2H, т), 1,48 (6H, д), 1,31 (3H, т); m/z (ES) 447 + [M+H] . Приклад одержання 6 3-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1H)ізохінолініл]метилпропаноат сіль мурашиної кислоти Суспензію 2-[(1-метилетил)окси]-5-[3-(1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрилу (Приклад 3, 28,8 мг, 0,080 ммоль), метил-3-бромпропіонату (35 мкл, 0,32 ммоль) і Cs2CO3 (130 мг, 0,40 ммоль) в DMF (2 мл) перемішували при температурі довкілля протягом 1 години і потім нагрівали при 60 °C протягом 5 хвилин у мікрохвильовому реакторі. Додавали додаткову кількість метил-3-бромпропіонату (35 мкл, 0,32 ммоль), і суспензію перемішували при температурі довкілля протягом 3,5 годин і потім нагрівали при збільшуваних температурах у мікрохвильовому реакторі, поки температура не досягла 150 °C за 30 хвилин. Суспензію фільтрували й концентрували у вакуумі, потім очищали MDAP, одержуючи цільову сполуку у 1 формі жовтої плівки (5,9 мг, 17 %). H ЯМР (400 МГц, CDCl3) δ 8,42(1Н, д), 8,33 (1Н, дд), 8,21 (1Н, ушир.с), 8,04 (1Н, д), 7,35 (1Н, т), 7,24 (1Н, д), 7,13 (1Н, д), 4,80 (1Н, септет), 3,99 (2H, с), + 3,72 (3H, с), 3,38 (2H, т), 3,12-3,06 (4H, м), 2,78 (2H, т), 1,48 (6H, д); m/z (ES) 447 [M+H] . Приклад одержання 7 4-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1H)ізохінолініл]етилбутаноат сіль мурашиної кислоти 12 UA 102539 C2 5 10 15 20 25 30 35 40 Суспензію 2-[(1-метилетил)окси]-5-[3-(1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрилу (Приклад 3, 28,8 мг, 0,080 ммоль), етил-4-бромбутирату (45,8 мкл, 0,32 ммоль) і Cs2CO3 (130 мг, 0,40 ммоль) у DMF (2 мл) перемішували при температурі довкілля протягом 1 години й потім нагрівали при 60 °C протягом 5 хвилин у мікрохвильовому реакторі. Додавали додаткову кількість етил-4-бромбутирату (45,8 мкл, 0,32 ммоль), і суспензію перемішували при температурі довкілля протягом 3,5 годин. Потім її фільтрували, концентрували у вакуумі й 1 очищали MDAP, одержуючи цільову сполуку у формі жовтої плівки (8,0 мг, 21 %). H ЯМР (400 МГц, CDCl3) δ 8,42 (1Н, д), 8,34 (1Н, дд), 8,27 (ушир.с), 8,09 (1Н, д), 7,39 (1Н, т), 7,27 (1Н, д), 7,13 (1Н, д), 4,80 (1Н, септет), 4,19 (2H, с), 4,13 (2H, кв), 3,47 (2H, т), 3,28 (2H, т), 2,99 (2H, т), 2,44 + (2H, т), 2,10 (2H, квінтет), 1,48 (6H, д), 1,25 (3H, т); m/z (ES) 475 [M+H] . Приклад одержання 8 3-хлор-4-[(1-метилетил)окси]бензоїлхлориду У круглодонну колбу завантажували 3-хлор-4-[(1-метилетил)окси]бензойну кислоту (від Paragos Product List; 10,2 г, 47,5 ммоль), дихлорметан (158 мл) і оксалілхлорид (8,29 мл, 95 ммоль). Реакційну суміш охолоджували до 0 °C в бані лід/вода, після чого додавали N, Nдиметилформамід (0,158 мл). Розчину давали нагрітися до температури довкілля протягом ночі. Розчинник упарювали, одержуючи цільову сполуку у формі твердої речовини кремового кольору (11,4 г). 1 H ЯМР (ХЛОРОФОРМ-d) δ: 8,14 (д, 1H), 8,00 (дд, 2,5 Гц, 1H), 6,98 (д, 1Н), 4,73 (септет, 1H), 1,44 (д, 6H). Приклад одержання 9 2-метил-1-(метилокси)-3-[(E)-2-нітроетеніл]бензолу До розчину 2-метил-3-(метилокси)бензальдегіду (від Allichem Product List; 20 г; 133 ммоль) у нітрометані (400 мл) додавали ацетат амонію (6,16 г, 80 ммоль), і одержану помаранчеву суміш перемішували протягом 1 години при 100 °C, потім охолоджували до температури довкілля й концентрували у вакуумі. Залишок розділяли між етилацетатом (x2) і сольовим розчином, і органічні шари промивали сольовим розчином, висушували (MgSО4) і концентрували у вакуумі. Розтирання з сумішшю дихлорметан/простий ефір дало цільову сполуку у формі твердої речовини жовтого кольору (6,6 г). 1 H ЯМР (CDCl3) δ: 8,35 (д, 1Н), 7,48 (д, 1Н), 7,22 (т, 1Н), 7,11 (д, 1Н), 6,96 (д, 1Н), 3,86 (с, 3H), 2,33 (с, 3H). Після упарювання маткових розчинів додаткову кількість продукту (7,22 г) одержували розтиранням з простим діетиловим ефіром. Маткові розчини знову упарювали, й залишок очищали хроматографією, елюючи етилацетатом (5-10 %) в циклогексані, одержуючи додаткову кількість продукту (3,45 г) після розтирання з простим ефіром. Приклад одержання 10 {2-[2-метил-3-(метилокси)феніл]етил}аміну 13 UA 102539 C2 5 10 15 20 25 30 35 До борогідриду літію (2M в THF; 2,0 мл, 4,0 ммоль) (каламутний розчин) при температурі довкілля додавали по краплях за 0,5 хв. хлортриметилсилан (1,02 мл, 8,00 ммоль). Утворювався осад, і через приблизно 3 хвилини 2-метил-1-(метилокси)-3-[(E)-2нітроетеніл]бензол (Приклад одержання 9, 193 мг, 1,0 ммоль) у THF (4 мл) додавали по краплях через шприц за 5 хвилин, упевнявшись, що температура залишалася приблизно 25 °C (використання ванни з холодною водою). Розчин перемішували при температурі довкілля протягом ночі. Суміш охолоджували за допомогою ванни з льодом, повільно додавали метанол, і розчинник видаляли у вакуумі. Залишок розділяли між 25 % водним розчином гідроксиду натрію і дихлорметаном. Водну фазу відокремлювали й екстрагували три рази дихлорметаном, і об'єднані органічні шари упарювали. Очищення залишку твердофазною екстракцією (колонка SCX) з елююванням метанолом, потім 2н аміаком в метанолі, з подальшим упарюванням 1 фракції, що містить аміак, дала цільову сполуку (80 мг). H ЯМР (CDCl3) δ: 7,10 (т, 1Н), 6,78 (д, 1Н), 6,73 (д, 1Н), 3,81 (с, 3H), 2,91 (т, 2H), 2,77 (т, 2H), 2,18 (с, 3H), 1,76 (ушир.с, 2H). Приклад одержання 11 {2-[2-метил-3-(метилокси) феніл]етил}формаміду {2-[2-метил-3-(метилокси)феніл]етил}амін (Приклад одержання 10, 1,75 г, 10,6 ммоль) нагрівали із зворотним холодильником з етилформіатом (97 %, 15 мл) протягом 15 г. Видалення розчинника дало сирий продукт (приблизно 2 г), який очищали хроматографією на силікагелі, елюючи градієнтом 13-63 % етилацетату в циклогексані, одержуючи цільову сполуку (1,57 г). MS + m/z 194 [Мн ]. Приклад одержання 12 5-метил-6-(метилокси)-3,4-дигідро-2(1Н)-ізохінолінкарбальдегіду До розчину {2-[2-метил-3-(метилокси)феніл]етил}формаміду (Приклад одержання 11, 1,93 г, 10,0 ммоль) в мурашиній кислоті (20 мл) додавали параформальдегід (0,315 г, 10,5 ммоль), і одержану суміш нагрівали із зворотним холодильником (100 °C) протягом 20 хв. Суміш швидко охолоджували крижаною холодною водою до температури довкілля і розчинник видаляли. Очищення хроматографією на силікагелі з елююванням градієнтом метанолу в дихлорметані дала цільову сполуку у формі твердої речовини білого кольору (1,64 г), який використовували + без подальшого очищення. MS m/z 206 [Мн ]. Приклад одержання 13 5-метил-1,2,3,4-тетрагідро-6-ізохінолінолу До розчину 5-метил-6-(метилокси)-3,4-дигідро-2(1Н)-ізохінолінкарбальдегіду (Приклад одержання 12, 1,49 г, 7,26 ммоль) у дихлорметані (25 мл) при 0 °C під азотом повільно додавали трибромід бору (9,09 г, 36,3 ммоль). Після 85 хв. при 0 °C повільно додавали метанол 14 UA 102539 C2 5 10 15 20 25 30 (25 мл), і одержану суміш залишали при температурі довкілля протягом трьох днів. Видалення розчинника й розтирання з сумішшю метанол/дихлорметан дало білий твердий залишок; маткові розчини концентрували, й залишок завантажували на екстракційний твердофазний картридж SCX (20 г) і елюювали метанолом, потім 2н аміаком у метанолі, елюючи продукт. Після випаровування розчинника цільову сполуку (780 мг) одержували у формі твердої 1 речовини світло-жовтого кольору. H ЯМР (ДМСО-d6) δ: 8,91 (ушир.с, 1Н), 6,62 (д, 1Н), 6,56 (д, 1Н), 3,70 (с, 2H), 2,92 (т, 2H), 2,48 (т, 2H), 1,96 (с, 3H). Приклад одержання 14 1,1-диметилетил-6-гідрокси-5-метил-3,4-дигідро-2(1H)-ізохінолінкарбоксилату До суспензії 5-метил-1,2,3,4-тетрагідро-6-ізохінолінолу (Приклад одержання 13; 163 мг; 1,0 ммоль) у дихлорметані (5 мл) при 0 °C додавали триетиламін (0,21 мл; 1,50 ммоль), потім біс(1,1-диметилетил)бікарбонат (240 мг; 1,10 ммоль). Розчинник упарювали, й залишок розділяли між етилацетатом і сольовим розчином. Водний шар потім екстрагували етилацетатом, і об'єднані органічні шари висушували (MgSO4) і упарювали, одержуючи жовте масло. Очищення флеш-хроматографією з елююванням з градієнтом 5-25 % етилацетату в циклогексані дало цільову сполуку у формі білої піни (232 мг). MS m/z 262 [М-Н]. Приклад одержання 15 1,1-диметилетил-5-метил-6-{[(трифторметил)сульфоніл]окси}-3,4-дигідро-2(1Н)ізохінолінкарбоксилату До розчину 1,1-диметилетил-6-гідрокси-5-метил-3,4-дигідро-2(1Н)-ізохінолінкарбоксилату (Приклад одержання 14; 263 мг; 1,00 ммоль) у дихлорметані (5 мл) при -30 °C під азотом додавали піридин (0,162 мл; 2,00 ммоль) з подальшим повільним додаванням трифторметансульфонового ангідриду (0,186 мл; 1,10 ммоль). Після 0,5 г при температурі від 30 °C до -20 °C розчинник видаляли, і залишок збирали етилацетатом. Розчин промивали 1н соляною кислотою, потім насиченим розчином бікарбонату натрію, висушували (MgSO4) і концентрували у вакуумі, одержуючи цільову сполуку формі світло-жовтого масла (368 мг). MS m/z 394 [М-Н]. Приклад одержання 16 1,1-диметилетил-6-ціано-5-метил-3,4-дигідро-2(1H)-ізохінолінкарбоксилату 15 UA 102539 C2 5 10 15 20 25 Розчин 1,1-диметилетил-5-метил-6-{[(трифторметил)сульфоніл]окси}-3,4-дигідро-2(1Н)ізохінолінкарбоксилату (Приклад одержання 15; 395 мг; 1,00 ммоль) у DMF (4 мл) дегазували під високим вакуумом при перемішуванні протягом 15 хвилин, після чого додавали ціанід цинку (153 мг; 1,30 ммоль) і тетракис(трифенілфосфін)паладій (0) (116 мг; 0,10 ммоль) під азотом. Одержану темно-жовту суміш перемішували при 100 °C протягом 6 годин. Розчинник видаляли, залишок розчиняли в етилацетаті, й розчин промивали насиченим бікарбонатом натрію. Водний шар потім екстрагували етилацетатом, і об'єднані органічні шари промивали сольовим розчином, висушували (MgSO4) і упарювали, одержуючи сирий продукт. Очищення флешхроматографією на силікагелі з елююванням з градієнтом 5-25 % етилацетату в циклогексані 1 дало цільову сполуку (214 мг) у формі твердої речовини білого кольору. H ЯМР (CDCl3) δ: 7,44 (д, 1Н), 7,04 (д, 1Н), 4,60 (с, 2H), 3,69 (т, 2H), 2,75 (т, 2H), 2,46 (с, 3H), 1,49 (с, 9H). Приклад одержання 17 1,1-диметилетил-6-[(гідроксіаміно)(іміно)метил]-5-метил-3,4-дигідро-2(1H)ізохінолінкарбоксилату 1,1-диметилетил-6-ціано-5-метил-3,4-дигідро-2(1Н)-ізохінолінкарбоксилат (Приклад одержання 16; 404 мг; 1,48 ммоль) перемішували протягом ночі при 80 °C з гідрохлоридом гідроксиламіну (619 мг; 8,90 ммоль) і бікарбонатом натрію (748 мг; 8,9 ммоль) в етанолі (10 мл). Додавали додаткову кількість бікарбонату натрію (400 мг) і гідрохлориду гідроксиламіну (300 мг), і нагрівання продовжували протягом приблизно 8 г. Суміш охолоджували до кімнатної температури, й одержану тверду речовину видаляли, промивали етанолом і висушували під високим вакуумом, одержуючи цільову сполуку у формі твердої речовини білого кольору (431 мг). 1 H ЯМР (ДМСО-d6) δ: 7,06 (д, 1Н), 6,99 (д, 1Н), 4,48 (ушир.с, 2H), 3,58 (т, 2H), 2,67 (т, 2H), 2,19 (с, 3H), 1,42 (с, 9H) (обмінювані не вказані; не видні чітко). Приклад одержання 18 1,1-диметилетил-6-[{[({3-хлор-4-[(1-метилетил)окси]феніл}карбоніл)окси]аміно}(іміно)метил]5-метил-3,4-дигідро-2(1H)-ізохінолінкарбоксилату 16 UA 102539 C2 5 10 15 До розчину 1,1-диметилетил-6-[(гідроксіаміно)(іміно)метил]-5-метил-3,4-дигідро-2(1Н)ізохінолінкарбоксилату (Приклад одержання 17; 435 мг; 1,42 ммоль) в дихлорметані (10 мл) додавали триетиламін (0,30 мл; 2,14 ммоль), потім 3-хлор-4-[(1-метилетил)окси]бензоїлхлорид (Приклад одержання 8; 365 мг; 1,57 ммоль) у дихлорметані (5 мл). Через 30 хвилин реакційну суміш концентрували, й залишок розділяли між етилацетатом і насиченим бікарбонатом натрію. Водний шар потім екстрагували етилацетатом, і об'єднані органічні шари промивали сольовим розчином, висушували (MgSO4) і концентрували у вакуумі. Очищення флеш-хроматографією на силікагелі з елююванням з градієнтом 10-50 % етилацетату в циклогексані дало цільову сполуку у формі білої піни (413 мг). MS m/z 500 [М-Н]. Приклад одержання 19 1,1-диметилетил-6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил3,4-дигідро-2(1H)-ізохінолінкарбоксилату Розчин 1,1-диметилетил-6-[{[({3-хлор-4-[(1метилетил)окси]феніл}карбоніл)окси]аміно}(іміно)метил]-5-метил-3,4-дигідро-2(1Н)ізохінолінкарбоксилату (Приклад одержання 18; 413 мг; 0,823 ммоль) в 1,4-діоксані (15 мл) перемішували при 110 °C протягом приблизно 18 г. Суміш охолоджували до температури довкілля й розчинник видаляли. Очищення флеш-хроматографією на силікагелі з елююванням 17 UA 102539 C2 5 10 15 5-25 % етилацетату в циклогексані, потім 13-63 % етилацетатом в циклогексані, дало цільову 1 сполуку у формі білої піни (220 мг). H ЯМР (ХЛОРОФОРМ-d) δ: 8,24 (д, 1Н), 8,05 (дд, 1Н), 7,76 (д, 1Н), 7,10 (д, 1Н), 7,06 (д, 1Н), 4,72 (септет, 1Н), 4,63 (с, 2H), 3,71 (т, 2H), 2,84 (т, 2H), 2,52 (с, 3H), 1,51 (с, 9H), 1,45 (д, 6H). Приклад одержання 20 3-[6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1H)ізохінолініл]етилпропаноату До суспензії 6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-1,2,3,4тетрагідроізохіноліну трифторацетату (Приклад 10; 90 мг; 0,18 ммоль) в ацетонітрилі (3 мл) додавали 1,8-діазабіцикло[5.4.0]ундец-7-ен (DBU; 0,16 мл; 1,09 ммоль). До розчину додавали етилакрилат (0,03 мл; 0,27 ммоль), і одержану суміш перемішували при 70 °C протягом 0,5 г. Суміш охолоджували до температури довкілля і розчинник видаляли. Залишок розділяли між етилацетатом і сольовим розчином, і водний шар потім екстрагували етилацетатом. Об'єднані органічні шари промивали сольовим розчином, висушували (MgSO4) і концентрували у вакуумі, + одержуючи цільову сполуку (90 мг). MS m/z 484 [МН ]. Приклад одержання 21 [6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1H)ізохінолініл]етилацетату 18 UA 102539 C2 5 10 15 20 25 До суспензії 6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-1,2,3,4тетрагідроізохінолін трифторацетату (Приклад 10; 90 мг; 0,18 ммоль) у DMF (3 мл) під азотом при температурі довкілля додавали карбонат цезію (177 мг; 0,54 ммоль), потім етилбромацетат (0,026 мл; 0,23 ммоль). Через 35 хв. суміш розбавляли етилацетатом, і осад видаляли. Розчин промивали водою, й водний шар потім екстрагували етилацетатом. Органічний шар промивали сольовим розчином, висушували над MgSO4 і концентрували у вакуумі, одержуючи цільову + сполуку (75 мг). MS m/z 470 [МН ]. Приклад одержання 22 1,1-диметилетил-6-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил3,4-дигідро-2(1H)-ізохінолінкарбоксилату Оксалілхлорид (571 мг, 4,5 ммоль) додавали до розчину 3-ціано-4-[(1метилетил)окси]бензойної кислоти (від AK Scientific Product Catalog; 800 мг, 3,90 ммоль) у дихлорметані (15 мл) з подальшим додаванням DMF (каталітична кількість). Реакційну суміш перемішували протягом 45 хвилин, додавали додаткову кількість оксалілхлориду (0,1 мл) і DMF (1 крапля), і перемішування продовжували протягом 20 хвилин. Розчинник упарювали, залишок упарювали спільно з толуолом, потім висушували під високим вакуумом протягом 30 хвилин. Сирий хлорангідрид кислоти розчиняли в ацетонітрилі (10 мл) і додавали до суспензії 1,1диметилетил 6-[(гідроксіаміно)(іміно)метил]-5-метил-3,4-дигідро-2(1Н)-ізохінолінкарбоксилату (Приклад одержання 17; 916 мг, 3,00 ммоль) і триетиламіну (607 мг, 6,00 ммоль) в ацетонітрилі (15 мл). Реакційну суміш перемішували при температурі довкілля протягом 40 хвилин, потім нагрівали із зворотним холодильником протягом 24 годин. Реакційну суміш охолоджували, й розчинник упарювали. Залишок розділяли між етилацетатом і насиченим розчином бікарбонату натрію. Органічну фазу відокремлювали, промивали сольовим розчином, висушували (MgSO4) і упарювали, одержуючи сирий продукт (1,3 г). Залишок очищали хроматографією, елюючи з 81 38 % етилацетату в циклогексані, одержуючи цільову сполуку (486 мг). H ЯМР (ХЛОРОФОРМd) δ: 8,42 (д, 1Н), 8,33 (дд, 1Н), 7,76 (д, 1Н), 7,12 (д, 1Н), 7,1 1 (д, 1Н), 4,80 (септет, 1Н), 4,64 (с, 2H), 3,72 (т, 2H), 2,84 (т, 2H), 2,52 (с, 3H), 1,51 (с, 9H), 1,48 (д, 6H). 19 UA 102539 C2 Приклад одержання 23 [6-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1H)ізохінолініл]етилацетату 5 10 15 20 25 30 35 Суміш солі трифтороцтової кислоти з 2-[(1-метилетил)окси]-5-[3-(5-метил-1,2,3,4-тетрагідро6-ізохінолініл)-1,2,4-оксадіазол-5-іл]бензонітрилом (Приклад 11; 100 мг, 0,20 ммоль), етилбромацетату (44 мг, 30 мкл, 0,26 ммоль) і карбонату цезію (200 мг, 0,61 ммоль) в сухому DMF (3 мл) перемішували при температурі довкілля протягом 1 години. Реакційну суміш розбавляли водою (10 мл) і екстрагували етилацетатом (3 × 5 мл). Об'єднані екстракти висушували й упарювали. Очищення хроматографією на силікагелі з елююванням з 10 %-им етилацетатом у циклогексані дало цільову сполуку у формі безбарвного масла (85 мг). MS m/z + 461 [МН] . Приклад одержання 24 3-[6-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1H)ізохінолініл]етилпропаноату Суміш солі трифтороцтової кислоти з 2-[(1-метилетил)окси]-5-[3-(5-метил-1,2,3,4-тетрагідро6-ізохінолініл)-1,2,4-оксадіазол-5-іл]бензонітрилом (Приклад 11; 100 мг, 0,20 ммоль), етилакрилату (31 мг, 33 мкл, 0,31 ммоль) і 1,8-діазабіцикло[5.4.0]ундец-7-ена (DBU; 187 мг, 185 мкл, 1,23 ммоль) в ацетонітрилі (3 мл) перемішували при температурі довкілля протягом 3 годин. Розчинник упарювали, й залишок розчиняли в етилацетаті (10 мл); розчин промивали водою (5 мл), висушували й упарювали. Очищення залишку хроматографією на силікагелі з елююванням з 10 %-им етилацетатом у циклогексані дало цільову сполуку у формі безбарвного + масла (86 мг). MS m/z 475 [МН] . Приклад 1 5-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-1,2,3,4-тетрагідроізохінолін гідрохлориду 1,1-диметилетил-5-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро2(1Н)-ізохінолінкарбоксилату (Приклад одержання 3, 2,57 г, 5,47 ммоль) перемішували в 4M HCl в 1,4-діоксані (100 мл). Через 1 годину реакційна суміш стала каламутною й перемішування продовжували протягом ще 16 годин. Упарювання дало 5-(5-{3-хлор-4-[(1метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-1,2,3,4-тетрагідроізохінолін гідрохлориду (2,23 г, 1 5,49 ммоль, 100 %-ий вихід). H ЯМР (400 МГц, d6-ДМСО) δ 9,43 (2H, с), 8,19 (1Н, с), 8,11 (1Н, дд), 8,03-8,00 (1Н, м), 7,51-7,45 (3H, м), 4,89 (1Н, септет), 4,38 (2H, с), 3,43 (2H, т), 3,35-3,32 (2H, + м), 1,37 (6H, д); m/z (API-ES) 370, 372 [M+H] . 20 UA 102539 C2 Приклад 2 2-[(1-метилетил)окси]-5-[3-(1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрил гідрохлориду 5 10 15 20 25 30 35 40 Суміш 1,1-диметилетил-5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4дигідро-2(1Н)-ізохінолінкарбоксилату (Приклад одержання 4; 53,7 мг, 0,117 ммоль) і 4M HCl в діоксані (2 мл) перемішували при температурі довкілля протягом 64 годин. Суміш концентрували у вакуумі, залишок розтирали з Et2O і висушували у вакуумі, одержуючи тверду 1 речовину світло-коричневого кольору (35,1 мг, 76 %). H ЯМР (400 МГц, d6-ДМСО) δ 9,52 (2H, ушир.с), 8,52 (1Н, д), 8,40 (1Н, дд), 8,02 (1Н, дд), 7,57, (1Н, д), 7,51-7,48 (2H, м), 4,99 (1Н, + септет), 4,38 (2H, с), 3,39-3,34 (4H, м), 1,39 (6H, д); m/z (ES) 361 [M+H] . Приклад 3 2-[(1-метилетил)окси]-5-[3-(1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрилу Суміш 1,1-диметилетил-5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4дигідро-2(1Н)-ізохінолінкарбоксилату (Приклад одержання 4, 512 мг, 1,11 ммоль) і 4M HCl в діоксані (20 мл) перемішували при температурі довкілля протягом 18 годин. Суміш концентрували у вакуумі, одержуючи тверду речовину світло-жовтого кольору, яку повторно розчиняли в MeOH. Цей розчин завантажували в картридж SCX-3 (10 г), і продукт елюювали 1 %-им NH3 в MeOH. Концентрація дала жовте масло, яке тверднуло при відстоюванні протягом 1 ночі (366 мг, 91 %). H ЯМР (400 МГц, d6-ДМСО) δ 8,38 (1Н, д), 8,32 (1Н, дд), 7,94 (1Н, д), 7,327,17 (2H, м), 7,13 (1Н, м), 4,80 (1Н, септет), 4,10 (2H, с), 3,20-3,14 (4H, м), 1,48 (6H, д); m/z (ES) + 361 [M+H] . Приклад 4 Гідрохлорид [5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро2(1H)-ізохінолініл]оцтової кислоти Розчин солі мурашиної кислоти з [5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол3-іл)-3,4-дигідро-2(1H)-ізохінолініл]етилацетатом (Приклад одержання 5, 11,6 мг, 0,026 ммоль) і LіOH (0,3 мг) в суміші THF-MeOH-вода (300 мкл, 300 мкл, 200 мкл) нагрівали при 100 °C протягом 3 хвилин у мікрохвильовому реакторі. Додавали 2M водний розчин HCl (1 мл), потім розчин екстрагували DCM (22 мл). Об'єднані органічні фази концентрували у вакуумі, 1 одержуючи цільову сполуку у формі твердої речовини білого кольору (10,1 мгм, 85 %). H ЯМР (400 МГц, d4-MeOH) δ 8,48 (1Н, д), 8,44 (1Н, дд), 8,21 (1Н, д), 7,44 (1Н, т), 7,47 (2H, д, що здається), 4,97 (1Н, септет), 4,70 (2H, с), 4,32 (2H, с), 3,67 (2H, т), 3,57 (2H, т), 1,47 (6H, д); m/z + (ES) 419 [M+H] . Приклад 5 Гідрохлорид 3-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро2(1H)-ізохінолініл]пропіонової кислоти 21 UA 102539 C2 5 10 15 20 25 30 35 Розчин солі мурашиної кислоти з 3-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4оксадіазол-3-іл)-3,4-дигідро-2(1Н)-ізохінолініл]метилпропаноатом (Приклад одержання 6, 5,9 мг, 0,0132 ммоль) і LіOH (0,15 мг) в суміші THF-MeOH-вода (300 мкл, 300 мкл, 100 мкл) нагрівали при 100 °C протягом 3 хвилин у мікрохвильовому реакторі. Додавали послідовно 2M водний розчин HCl (1 мл) і DCM (2 мл), внаслідок чого утворювався осад. Фільтрація дала цільову 1 сполуку у формі твердої речовини білого кольору (6,5 мг, кільк.). H ЯМР (400 МГц, d6-ДМСО) δ 12,71 (1Н, ушир.с), 11,41 (1Н, ушир.с), 8,53 (1Н, д), 8,41 (1Н, дд), 8,05 (1Н, д), 7,57 (1Н, д), 7,53 (1Н, т), 7,46 (1Н, д), 4,99 (1Н, септет), 4,53 (2H, ушир.с), 3,46-3,43 (4H, м), 3,65 (2H, ушир.с), 2,98 + (2H, т), 1,39 (6H, d); m/z (ES) 433 [M+H] . Приклад 6 Гідрохлорид 4-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро2(1H)-ізохінолініл]бутанової кислоти Цільову сполуку одержували подібно до Прикладу 4, замінюючи [5-(5-{3-ціано-4-[(1метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4-дигідро-2(1Н)-ізохінолініл]етилацетат сіллю мурашиної кислоти з 4-[5-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-3,4дигідро-2(1Н)-ізохінолініл]етилбутаноатом (Приклад одержання 7, 8,0 мг). Цільову сполуку 1 одержували у формі твердої речовини білого кольору (10,1 мг). H ЯМР (400 МГц, d4-MeOH) δ 8,36 (1Н, д), 8,33 (1Н, дд), 8,23 (1Н, д), 7,43 (1Н, т), 7,37 (1Н, д), 7,35 (1Н, д), 4,85 (1Н, септет), 4,52 (2H, ушир.с), 3,63-3,54 (4H, ушир.м), 3,30 (2H, дд, що здається), 2,43 (2H, т), 2,06 (2H, + квінтет), 1,36 (6H, д); m/z (ES) 447 [M+H] . Приклад 7 2-[(1-метилетил)окси]-5-[3-(2-метил-1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрил До розчину 2-[(1-метилетил)окси]-5-[3-(1,2,3,4-тетрагідро-5-ізохінолініл)-1,2,4-оксадіазол-5іл]бензонітрилу (Приклад 3, 25,0 мг, 0,069 ммоль) і формальдегіду (37 % вага./об. у H2O, 8,0 мкл, 0,10 ммоль) у DCM (5 мл) додавали NaBH(OAc)3 (29 мг, 0,14 ммоль), і одержаний розчин перемішували при температурі довкілля протягом 20 хв. Додавали сольовий розчин (5 мл), і водний шар екстрагували DCM (5 мл). Об'єднані органічні фази концентрували у вакуумі, одержуючи тверду речовину білого кольору. Цю тверду речовину повторно розчиняли в суміші ДМСО-MeOH, додавали в картридж SCX і промивали MеOH. Продукт елюювали NH3 в MеOH; 1 концентрація у вакуумі дала тверду речовину білого кольору (20,1 мг, 78 %). H ЯМР (400 МГц, CDCl3) δ 8,41 (1Н, д), 8,32 (1Н, дд), 7,98 (1Н, д), 7,29 (1Н, т), 7,20 (1Н, д), 7,12 (1Н, д), 4,79 (1Н, септет), 3,67 (2H, с), 3,30 (2H, т), 2,75 (2H, т), 2,49 (3H, с), 1,47 (6H, д). Приклад 8 3-[6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолініл]пропанова кислота 22 UA 102539 C2 5 10 15 20 25 30 До розчину 3-[6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4дигідро-2(1Н)-ізохінолініл]етилпропаноату (Приклад одержання 20; 90 мг; 0,186 ммоль) в етанолі (2 мл) і THF (2 мл) при температурі довкілля додавали 2н гідроксид натрію (0,186 мл; 0,372 ммоль), і одержану суміш перемішували при температурі довкілля приблизно протягом 2,5 годин. Видалення більшої частини розчинників, розбавлення приблизно 1 мл води з подальшим додаванням оцтової кислоти (приблизно 300 мкл) дало осад. Суміш екстрагували дихлорметаном, одержуючи, після видалення розчинника й розтирання з простим діетиловим 1 ефіром, цільову сполуку у формі твердої речовини білого кольору (40 мг). H ЯМР (ДМСО-d6) δ: 8,15 (д, 1Н), 8,09 (дд, 1Н), 7,64 (д, 1Н), 7,43 (д, 1Н), 7,09 (д, 1Н), 4,88 (септет, 1Н), 3,64 (с, 2H), + 2,76 (ушир.с, 4H), 2,72 (т, 2H), 2,44 (т, 2H), 2,41 (с, 3H), 1,36 (д, 6H). MS m/z 456 [МH ]. Приклад 9 [6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолініл]оцтова кислота До розчину [6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4дигідро-2(1Н)-ізохінолініл]етилацетату (Приклад одержання 21; 75 мг; 0,16 ммоль) в етанолі (2 мл) і THF (2 мл) при температурі довкілля додавали 2н гідроксид натрію, й одержану суміш перемішували при температурі довкілля протягом 40 хв. Розчинники видаляли, й залишок розчиняли у воді (приблизно 1 мл). Додавали оцтову кислоту (1 мл), всі розчинники видаляли, й залишок упарювали спільно з толуолом, одержуючи тверду речовину світло-жовтого кольору. Цю тверду речовину розтирали з простим діетиловим ефіром, одержуючи цільову сполуку, яке + 1 відфільтрували у формі твердої речовини світло-жовтого кольору (50 мг). MS m/z 442 [МН ]. H ЯМР (ДМСО-d6) δ: 8,16 (ушир.с, 1Н), 8,10 (д, 1Н), 7,62 (д, 1Н), 7,44 (д, 1Н), 7,05 (д, 1Н), 4,88 (септет, 1Н), 3,70 (ушир.с, 2H), 2,91 (ушир.с, 2H), 2,77 (ушир.с, 4H), 2,42 (ушир.с, 3H), 1,36 (д, 6H). Приклад 10 6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-1,2,3,4тетрагідроізохінолін трифторацетат До розчину 1,1-диметилетил-6-(5-{3-хлор-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)5-метил-3,4-дигідро-2(1Н)-ізохінолінкарбоксилату (Приклад одержання 19; 210 мг; 0,434 ммоль) 23 UA 102539 C2 5 10 15 20 25 30 35 у дихлорметані (3 мл) при 0 °C додавали по краплях трифтороцтову кислоту (3 мл). Одержану суміш перемішували при 0 °C протягом 1 години, одержуючи світло-жовтий розчин. Видалення розчинника й випаровування спільне з толуолом дало тверду речовину білого кольору, яку розтирали з простим діетиловим ефіром, одержуючи цільову сполуку у формі твердої речовини 1 білого кольору (206 мг). H ЯМР (ДМСО-d6) δ: 9,08 (ушир.с, 2H), 8,17 (д, 1Н), 8,10 (дд, 1Н), 7,78 (д, 1Н), 7,45 (д, 1Н), 7,28 (д, 1Н), 4,84-4,94 (м, 1Н), 4,38 (ушир.с, 2H), 3,45-3,52 (м, 2H), 2,98 (т, + 2H), 2,46 (с, 3H), 1,36 (д, 6H). MS m/z 384 [МН ]. Приклад 11 Сіль трифтороцтової кислоти і 2-[(1-метилетил)окси]-5-[3-(5-метил-1,2,3,4-тетрагідро-6ізохінолініл)-1,2,4-оксадіазол-5-іл]бензонітрилу Трифтороцтову кислоту (3 мл) додавали до охолодженого льодом розчину 1,1-диметилетил6-(5-{3-ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолінкарбоксилату (Приклад одержання 22; 486 мг, 1,02 ммоль) у дихлорметані (3 мл). Реакційну суміш перемішували при 0 °C протягом 30 хвилин. Розчинник упарювали, й залишок спільно упарювали з толуолу (x2). Розтирання залишку з простим діетиловим ефіром дало цільову сполуку у формі безбарвної твердої речовини, яку фільтрували й висушували (485 мг). 1 H ЯМР (400 Мгц, CDCl3) δ: 1,48 (6H, д), 2,54 (3H, с), 3,09 (2H, м), 3,5 (2H, затінений залишковим розчинником), 4,36 (2H, с), 4,80 (1Н, м), 7,08-7,15 (2H, м), 7,85 (1Н, д), 8,33 (1Н, д), 8,42 (1Н, с), + 10,20 (2H, ушир.с). MS m/z 375 [МН] . Приклад 12 [6-(5-{3-Ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолініл]оцтова кислота 2M гідроксиду натрію (2 мл) додавали до суспензії [6-(5-{3-ціано-4-[(1метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)-ізохінолініл]етилацетату (Приклад одержання 23; 80 мг, 0,17 ммоль) в етанолі (2 мл). Реакційну суміш перемішували при температурі довкілля протягом 2 годин. Розчинник упарювали, й залишок розбавляли водою (5 мл). Розчин підкисляли оцтовою кислотою й екстрагували етилацетатом (3 × 5 мл). Водну фазу упарювали, й залишок очищали MDAP, одержуючи цільову сполуку у формі безбарвної твердої 1 речовини (7 мг). H ЯМР (400 МГц, d4MeOH) δ: 1,46 (6H, д), 2,55 (3H, с), 3,19 (2H, т), 3,35 (2H, с), 3,67 (2H, т), 3,78 (2H, с), 4,53 (2H, с), 4,95 (1Н, м) (частково затінений водним піком), 7,20 (1Н, д), + 7,54 (1Н, д), 7,85 (1Н, d), 8,50-8,60 (2H, м). MS m/z 433 [МН] . Приклад 13 3-[6-(5-{3-Ціано-4-[(1-метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро2(1H)-ізохінолініл]пропанової кислоти натрієва сіль 24 UA 102539 C2 5 10 15 20 25 30 35 40 45 50 2M гідроксиду натрію (2 мл) додавали до розчину 3-[6-(5-{3-ціано-4-[(1метилетил)окси]феніл}-1,2,4-оксадіазол-3-іл)-5-метил-3,4-дигідро-2(1Н)ізохінолініл]етилпропаноату (Приклад одержання 24; 80 мг, 0,17 ммоль) в етанолі (2 мл) при 60 °C. Реакційну суміш перемішували при 60 °C протягом 2 годин, охолоджували до температури довкілля й розбавляли водою (2 мл). Тверду речовину фільтрували, промивали невеликою кількістю води й висушували, одержуючи цільову сполуку у формі безбарвної 1 твердої речовини (55 мг). H ЯМР (400 МГц, d6ДМСО) δ: 1,39 (6H, д), 2,08 (2H, т), 2,44 (3H, с), 2,59-2,78 (6H, м), 3,56 (2H, с), 4,98 (1Н, м), 7,09 (1Н, д), 7,55 (1Н, д), 7,65 (1Н, д), 8,40 (1Н, дд), + 8,50 (1Н, с). MS m/z 447 [МН] . Мембранний препарат для тесту S1P1 ГТФS Для мембранних препаратів всі стадії здійснювали при 4 °C. Клітини гепатоми щура, стабільно експресуючі рецептор S1P1 людини, або клітини базофільного лейкозу щура (RBL), стабільно експресуючі рецептор S1P3 людини, вирощували до 80 % злиття, після чого збирали в 10 мл фосфатного буферного сольового розчину (PBS) і центрифугували при 1200 об/хв. протягом 5 хвилин. Після видалення супернатанту осад повторно суспендували, й клітини гомогенізували в скляній мішалці Waring 2 циклами по 15 секунд в 200 мл буфера (50 мM HEPES, 1 мM лейпептину, 25 мкг/мл бацитрацину, 1 мM EDTA, 1 мM PMSF, 2 мкM пепстатину A). Мішалку занурювали в лід на 5 хвилин після першого циклу й на 10-40 хвилин після завершального циклу, щоб дати піні розсіятися. Матеріал потім центрифугували при 500 g протягом 20 хвилин, і супернатант центрифугували протягом 36 хвилин при 48000 g. Осад повторно суспендували в тому самому буфері, як описано вище, але без PMSF і пепстатину A. Матеріал потім пропускали через голку діаметром 0,6 мм, збирали до необхідного об'єму, (зазвичай x4 об'єм від первинного осаду клітин), розділяли на аліквоти й зберігали замороженим при -80 °C. Альтернативний мембранний препарат для тесту S1P1 ГТФS Всі стадії здійснювали при 4 °C. Клітини гомогенізували у скляній мішалці Waring в ході 2 циклів по 15 секунд у 200 мл буферу (50 мM HEPES, 1 мM лейпептину, 25 мкг/мл бацитрацину, 1 мM EDTA, 1мM PMSF, 2 мкM пепстатину A). Мішалку занурювали в лід на 5 хвилин після першого циклу і на 10-40 хвилин після завершального циклу, щоб дати піні розсіятися. Матеріал потім центрифугували при 500 g протягом 20 хвилин, і супернатант центрифугували протягом 36 хвилин при 48000 g. Осад повторно суспендували в тому самому буфері, як описано вище, але без PMSF і пепстатину A. Матеріал потім пропускали через голку діаметром 0,6 мм, збирали до необхідного об'єму, (зазвичай x4 об'єм від первинного осаду клітин), розділяли на аліквоти й зберігали замороженим при -80 °C. Тест S1P1 ГТФS Мембрани гепатоми щура з S1P1 людини (1,5 мкг/лунка) наліплювали на покриті аглютиніном паростки пшениці (WGA) гранули для тесту сцинтиляційної близькості (SPA) (0,125 мг/лунка) в тестовому буфері (HEPES 20 мM, MgCl2 10 мM, NaCl 100 мM і pH, доведений до 7,4 з використанням KOH 5M, також додавали ГДФ 10 мкM FAC (кінцева тестова концентрація) і сапонін 90 мкг/мл FAC). Після 30 хвилин попередньої сполуки на льоду гранули й мембранну суспензію розподіляли в білі поліпропіленові 384-лункові планшети Greiner LV (5 мкл/лунка), що містять 0,1 мкл 35 сполуки. Потім в планшети з агоністом додавали 5 мкл/лунка [ S]-ГТФS (0,5 нM кінцева концентрація міченого ліганду), складеного в тестовому буфері. Кінцеву тестову суміш (10,1 мкл) потім центрифугували при 1000 об./хв. протягом 5 хвилин, потім негайно зчитували на приладі Viewlux. Всі тестовані сполуки розчиняли в ДМСО в концентрації 10 мM і готували в 100 %-ому ДМСО, використовуючи стадію розведення 1 в 4, щоб одержати 11-точкові криві доза-відповідь. Розведення переносили на тестові планшети, забезпечуючи постійну концентрацію ДМСО в планшетах для всіх тестів. 25 UA 102539 C2 5 10 15 20 25 30 35 40 45 Всі дані піддавали нормалізації до середніх значень з 16 верхніх і 16 нижніх контрольних лунок на кожному планшеті. Потім використовували побудову кривої по 4 параметрах. Альтернативний спосіб для тесту S1P1 ГТФS Мембрани RH7777, експресуючі S1P1 (1,5 мкг/лунка), гомогенізували, пропускаючи через голку калібру 23G. Їх наліплювали на покриті гранули WGA для SPA (0,125 мг/лунка) в тестовому буфері (HEPES 20 мM, MgCl2 10 мM, NaCl 100 мM і pH, доведений до 7,4 з використанням KOH 5M). Також додавали ГДФ 10 мкM FAC і сапонін 90 мкг/мл FAC. Після 30 хвилин попередньої сполуки на льоду гранули і мембранну суспензію розподіляли в білі поліпропіленові 384-лункові планшети Greiner LV (5 мкл/лунка), що містять 0,1 мкл 35 сполуки. Потім в планшети додавали 5 мкл/лунка [ S]-ГТФS (0,5 нM для S1P1 або 0,3 нM для S1P3, кінцева концентрація міченого ліганду), складеного в тестовому буфері. Кінцеву тестову суміш (10,1 мкл) потім герметизували, центрифугували, потім негайно зчитували на приладі Viewlux. Проілюстровані в прикладах сполуки за винаходом мали pEC50>5. Приклади 1-7, 8, 9, 11-13 мали pEC50>7. Приклади 2, 4, 6 і 8 мали pEC50>8. S1P3 Мембрани клітин базофільного лейкозу щура з S1P3 (RBL-2H3) (1,5 мкг/лунка) наліплювали на покриті гранули WGA для SPA (0,125 мг/лунка) в тестовому буфері (HEPES 20 мM, MgCl2 3 мM, NaCl 100 мM і pH, доведений до 7,4 з використанням KOH 5M), також додавали ГДФ 10 мкM FAC (кінцева тестова концентрація) і сапонін 90 мкг/мл FAC). Після 30 хвилин попередньої сполуки на льоду гранули й мембранну суспензію розподіляли в білі поліпропіленові 384-лункові планшети Greiner LV (5 мкл/лунка), що містять 0,1 мкл сполуки. Потім у планшети з агоністом додавали 5 мкл/лунка [35S]-ГТФ(S (0,5 нM кінцева концентрація міченого ліганду), складеного в тестовому буфері. Кінцеву тестову суміш (10.1 мкл) потім центрифугували при 1000 об./хв. протягом 5 хвилин, потім негайно зчитували на приладі Viewlux. Всі тестовані сполуки розчиняли в ДМСО в концентрації 10 мM і готували в 100 %-ому ДМСО, використовуючи стадію розведення 1 в 4, щоб одержати 11-точкові криві доза-відповідь. Розведення переносили на тестові планшети, забезпечуючи постійну концентрацію ДМСО в планшетах для всіх тестів. Всі дані піддавали нормалізації до середніх значень з 16 верхніх і 16 нижніх контрольних лунок на кожному планшеті. Потім використовували побудову кривої по 4 параметрам. Альтернативний спосіб для тесту S1P3 ГТФS Мембрани RBL, експресуючі S1P3 (1,5 мкг/лунка) гомогенізували, пропускаючи через голку калібру 23G. Їх наліплювали на покриті гранули WGA для SPA (0,125 мг/лунка) в тестовому буфері (HEPES 20 мM, MgCl2 10 мM, NaCl 100 мM і pH, доведений до 7,4 з використанням KOH 5M). Також додавали ГДФ 10 мкM FAC і сапонін 90 мкг/мл FAC. Після 30 хвилин попереднього поєднання на льоду гранули й мембранну суспензію розподіляли в білі поліпропіленові 384-лункові планшети Greiner LV (5 мкл/лунка), що містять 35 0,1 мкл сполуки. Потім у планшети додавали 5 мкл/лунка [ S]-ГТФS (0,5 нM для S1P1 або 0,3 нM для S1P3, кінцева концентрація міченого ліганду), складеного в тестовому буфері. Кінцеву тестову суміш (10,1 мкл) потім герметизували, центрифугували, потім негайно зчитували на приладі Viewlux. Проілюстровані в прикладах сполуки, перевірені в цьому тесті, мали pEC50
ДивитисяДодаткова інформація
Назва патенту англійськоюOxadiazole derivatives active on sphingosine-1-phosphate (s1p)
Автори англійськоюHeer, Jag, Paul, Heightman, Thomas, Daniel
Назва патенту російськоюПроизводные оксадиазола, активные в отношении сфингозин-1-фосфата (s1p)
Автори російськоюХир Джаг Пол, Хейгтмен Томас Дэниел
МПК / Мітки
МПК: A61P 37/00, A61P 17/00, C07D 217/16, A61K 31/4725, C07D 217/06, C07D 413/14, C07D 413/04, C07D 217/22
Мітки: активні, s1p, похідні, оксадіазолу, сфінгозин-1-фосфату
Код посилання
<a href="https://ua.patents.su/30-102539-pokhidni-oksadiazolu-aktivni-do-sfingozin-1-fosfatu-s1p.html" target="_blank" rel="follow" title="База патентів України">Похідні оксадіазолу, активні до сфінгозин-1-фосфату (s1p)</a>
Похідні оксадіазолу, активні у відношенні сфінгозин-1-фосфату (s1p)

Номер патенту: 101349
Опубліковано: 25.03.2013
Автори: Хір Джаг Пол, Уолл Йан Девід, Хейгтмен Томас Деніел, Джонсон Крістофер Норберт, Скідмор Джон, Херст Девід Найджел, Демон Емманюель Юбер, Уітерінгтон Джейсон
МПК: C07D 413/04, C07D 271/06, C07D 223/00, A61P 17/00, A61P 37/00, C07D 267/00, C07D 413/14, A61K 31/55
Мітки: s1p, відношенні, активні, сфінгозин-1-фосфату, похідні, оксадіазолу
Формула / Реферат:
1. Сполука формули (І) або її фармацевтично прийнятна сіль:, (I) А являє собою феніл або 5- або 6-членне кільце гетероарилу; R1 являє собою до двох замісників, незалежно вибраних з галогену, С(1-3)алкокси, С(1-3)фторалкілу, ціано, необов'язково заміщеного фенілу, С(1-3)фторалкокси, С(1-6)алкілу і...
Похідні тіооксиндолу, фармацевтична композиція, що містить вказані похідні та активні інгредієнти ліків для стимулювання контрацепції і лікування дисфункціональної кровотечі, карцином та аденокарцином

Номер патенту: 73499
Опубліковано: 15.08.2005
Автори: Теглі Кристофер М., Вробель Джей І., Коко Марчі С., Фенсам Ендрю, Едвардс Джеймс П., Ці Лінь, Меленскі Едвард Дж., Джоунз Тодд К., Цанг Пувен
МПК: A61P 5/36, C07D 209/08, C07D 209/30, C07D 209/40
Мітки: стимулювання, містить, тіооксиндолу, контрацепції, композиція, дисфункціональної, ліків, інгредієнти, фармацевтична, карцином, похідні, активні, кровотечі, аденокарцином, лікування, вказані
Формула / Реферат:
1. Похідне тіооксиндолу формули:,де:R1 та R2 незалежним чином вибирають з групи, що включає Н, С1-С6 алкіл, заміщений С1-С6 алкіл, ОН, О(С1-С6 алкіл), О(заміщений С1-С6 алкіл), Оас, арил, заміщений арил, гетероарил, заміщений гетероарил, (С1-С6 алкіл)арил, (С1-С6 алкіл)гетероарил, 1-пропініл та 3-пропініл;або R1 та R2 сполучені з утворенням кільця, яке містить -CH2(CH2)nCH2-; -CH2CH2C(CH3)2CH2CH2-; -O(CH2)mCH2-;...
Похідні оксадіазолу як інгібітори dgat

Номер патенту: 91033
Опубліковано: 25.06.2010
Автори: Берч Алан Мартін, Плоурайт Еллін, Батлін Роджер Джон, Новак Торстен, Маккулл Вільям, Боукер С'юзанн Саксон, Доналд Крейґ Сем'юел
МПК: C07D 413/12, A61K 31/4245, A61P 3/10, C07D 271/10, A61P 3/04
Мітки: інгібітори, похідні, оксадіазолу
Формула / Реферат:
1. Сполука формули (І) (І)або її фармацевтично прийнятна сіль, де:R1 є фенілом, як варіант, заміщеним 1, 2 або 3 замісниками, незалежно вибраними з галогену, (1-4С)алкілу, етинілу, (1-4С)алкокси, гідрокси, (1-4С)алкоксі(1-4С)алкокси, метоксиметилу, ціанометилу, гідроксі(1-4С)алкілу, трифлуорметилу, дифлуорметокси, трифлуорметокси, трифлуорметилтіо,...
Похідні оксадіазолу та їх застосування як потенціювальних засобів рецепторів глутамату

Номер патенту: 99129
Опубліковано: 25.07.2012
Автори: Клейтон Джошуа, Емпфілд Джеймс, Ма Фупенг, Слассі Адбельмалік, Ісаак Месвін, Фолмер Джеймс, Егле Ян
МПК: A61K 31/496, C07D 413/14, A61P 25/00
Мітки: рецепторів, глутамату, потенціювальних, застосування, похідні, засобів, оксадіазолу
Формула / Реферат:
1. Сполука формули І:,Формула І де R1 - галоген або С1-3галогеналкоксил; Q - або , та R2 - гідроген або С1-3алкіл, або її фармацевтично...
Похідні оксадіазолу та їх застосування як позитивних алостеричних модуляторів метаботропних глутаматних рецепторів

Номер патенту: 92496
Опубліковано: 10.11.2010
Автори: Гагліарді Стефанія, Паломбі Джованні, Бугада П'єргіуліано, Льо Пул Емманюел, Мютєл Венсан, Роше Жан-Філіп
МПК: A61P 25/00, C07D 413/14, C07D 413/04, A61K 31/4245, C07D 417/14
Мітки: глутаматних, алостеричних, оксадіазолу, рецепторів, застосування, позитивних, модуляторів, метаботропних, похідні
Формула / Реферат:
1. Сполука загальної формули І:, де W являє собою (С5-С7)циклоалкільне, (С3-С7)гетероциклоалкільне, (С3-С7)гетероциклоалкіл-(С1-С3)алкільне або (С3-С7)гетероциклоалкенільне кільце; R1 й R2 незалежно один від одного являють собою водень, -(С1-С6)алкіл, -(С2-С6)алкеніл, -(С2-С6)алкініл,...
Попередній патент: Напівнавісний фасад
Наступний патент: П’ятичленні гетероцикли, їх конденсовані похідні та їх застосування в лікуванні діабету
Випадковий патент: Способи і пристрої для кодування і декодування сигналу