Сполуки оксазол[5,4-b]піридин-5-ілу та їх застосування у лікуванні раку
Номер патенту: 109677
Опубліковано: 25.09.2015
Автори: Коутс Дейвід Ендрю, Мартін Хосе Альфредо, Мартін де ла Нава Ева Марія, Гілмор Раймонд








Формула / Реферат
1. Сполука формули:
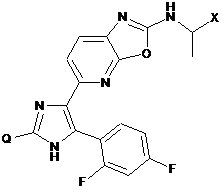 ,
,
де:
X - метоксіетил або етоксиметил;
Q - циклопропіл, 2-метилпропанол-2-іл, 3-метилоксетан-3-іл або 1-гідроксиметил-1-циклопропіл;
або її фармацевтично прийнятна сіль.
2. Сполука за п. 1, яка являє собою 2-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-3-метокси-1-метилпропіл]аміно]оксазол[5,4-b]піридин-5-іл]-1H-імідазол-2-іл]-2-метилпропан-1-ол, або її фармацевтично прийнятна сіль.
3. Сполука за п. 1 або п. 2, яка являє собою кристалічний 2-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-3-метокси-1-метилпропіл]аміно]оксазол[5,4-b]піридин-5-іл]-1Н-імідазол-2-іл]-2-метилпропан-1-ол, який характеризується порошковою рентгенограмою (випромінювання Cu-анода, l=1,54060 Ǻ), що містить пік при 15,06 і один або декілька піків при 19,94, 10,31 і 20,78 (2θ±0,2°).
4. Сполука за п. 1 або п. 2, яка являє собою кристалічний 2-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-3-метокси-1-метилпропіл]аміно]оксазол[5,4-b]піридин-5-іл]-1Н-імідазол-2-іл]-2-метилпропан-1-ол, який характеризується порошковою рентгенограмою (випромінювання Си-анода, l=1,54060 Ǻ), що містить пік при 13,73 і один або декілька піків при 16,54, 22,87 і 18,57(2q±0,2°).
5. Сполука за п. 1, яка являє собою 5-[2-циклопропіл-5-(2,4-дифторфеніл)-1Н-імідазол-4-іл]-N-[(1S)-3-метокси-1-метилпропіл]оксазол[5,4-b]піридин-2-амін, або її фармацевтично прийнятна сіль.
6. Сполука за п. 1, яка являє собою 5-[5-(2,4-дифторфеніл)-2-(3-метилоксетан-3-іл)-1Н-імідазол-4-іл]-N-[(1S)-3-метокси-1-метилпропіл]оксазол[5,4-b]піридин-2-амін, або її фармацевтично прийнятна сіль.
7. Сполука за п. 1, яка являє собою [1-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-2-етокси-1-метилетил]аміно]оксазол[5,4-b]піридин-5-іл]-1Н-імідазол-2-іл]циклопропіл]метанол, або
її фармацевтично прийнятна сіль.
8. Фармацевтична композиція, яка містить сполуку або сіль за будь-яким із пп. 1-7 в комбінації з одним(ією) або декількома фармацевтично прийнятними носіями, розріджувачами або допоміжними речовинами.
9. Фармацевтична композиція за п. 8, яка додатково містить один або декілька терапевтичних агентів.
10. Сполука або сіль за будь-яким із пп. 1-7 для застосування у терапії.
11. Сполука або сіль за будь-яким із пп. 1-7 для застосування у лікуванні раку.
12. Сполука або сіль для застосування за п. 11, де раком є рак яєчників.
13. Сполука або сіль для застосування за п. 11, де раком є множинна мієлома.
14. Сполука або сіль за будь-яким із пп. 1-7 для застосування в одночасній, роздільній або послідовній комбінації з сунітинібом у лікуванні раку.
15. Сполука або сіль для застосування за п. 14, де раком є рак нирок.
16. Застосування сполуки або солі за будь-яким із пп. 1-7 для виготовлення лікарського засобу для лікування раку яєчників.
17. Застосування сполуки або солі за будь-яким із пп. 1-7 для виготовлення лікарського засобу для лікування множинної мієломи.
Текст
Реферат: Цей винахід пропонує сполуки оксазол[5,4-b]піридин-5-ілу, придатні для лікування раку. UA 109677 C2 (12) UA 109677 C2 UA 109677 C2 5 10 15 20 25 30 35 40 45 р38 MAP-кіназа являє собою мітоген-активовану протеїнкіназу (MAP), яка належить до суперродини серин/треонінкіназ. Ця кіназа активується позаклітинними стресами, такими як тепло, ультрафіолетове випромінювання і осмотичний стрес, а також запальними стимулами, такими як ліпополісахариди. Після активації р38 МАР-кіназа фосфорилує внутрішньоклітинні білкові субстрати, які регулюють біосинтез прозапальних цитокінів фактор некрозу пухлин α (TNFα), інтерлейкін-1β (IL-1β), інтерлейкін-6 (IL-6) та інтерлейкін-8 (IL-8). Припускають причетність цих цитокінів до патології ряду хронічних запальних захворювань. Хронічне запалення є ключовим фактором ризику для розвитку раку. Наприклад, каскад реакцій р38 МАР-кінази є мішенню асоційованого із саркомою Капоші (KSHV) вірусу герпесу, що призводить до хронічного запалення і розвитку саркоми. Крім того, припускають причетність цитокінів, регульованих р38 МАР-кіназою, наприклад, IL-8, до стимулювання ангіогенезу, пов'язаного з ростом пухлин. Фосфорилована форма мітоген-активованої протеїнкінази протеїнкіназа 2 (або pMAPKAPK2) також є кіназою у каскаді реакцій р38 MAP-кінази і може безпосередньо активуватись р38 МАР-кіназою. Дослідження на мишах з нокаутованою MAPKAPK2 демонструють зниження продукування цитокінів, що дозволяє висунути припущення про те, що MAPKAPK2 може бути ключовим регулятором запальної реакції, а також може бути потенційною мішенню для протизапальної і/або ракової терапії (WO2005120509). Як відомо, для застосування у лікуванні запальних захворювань сполуки азабензотіазолілу були розкриті як інгібітори р38 MAP-кінази (наприклад, WO2007016392). Крім того, як відомо для застосування у лікуванні раку сполуки азабензімідазолілу були розкриті як інгібітори р38 MAPкінази (наприклад, WO2005075478). У WO200917822 розкриті сполуки імідазолілоксазолу і оксазол[4,5-b]піридин-6-ілу, що є придатними як інгібітори кінази PI3. Однак деякі інгібітори р38 МАР-кінази або інгібітори цитокінів можуть мати проблеми з біологічною доступністю і абсорбуванням, які обмежують їх in vivo вплив і терапевтичне застосування. Крім того, деякі інгібітори р38 МАР-кінази можуть вчиняти негативні токсичні впливи на пацієнта (особливо шлунково-кишкову токсичність) і спричинюють ризик негативної взаємодії лікарських речовин. Таким чином, існує потреба в альтернативних лікарських засобах для пригнічення цитокінів. За варіантом, якому віддають перевагу, такі сполуки здатні з підвищеною ефективністю пригнічувати р38 МАР-кіназу і мають більшу біологічну доступність. За варіантом, якому віддають перевагу, такі сполуки також мають поліпшений токсикологічний профіль (особливо шлунково-кишкової токсичності) і знижують ризик негативної взаємодії лікарських речовин. Даний винахід пропонує нові сполуки оксазол[5,4-b]піридин-5-ілу, які можуть мати клінічне застосування як окремий агент для лікування раку і, зокрема, раку яєчників і/або множинної мієломи. Крім того, цей винахід пропонує нові сполуки оксазол[5,4-b]піридин-5-ілу, які можуть мати клінічне застосування в комбінації з іншим терапевтичним агентом, таким як сунітиніб (sunitinib), для лікування раку і, зокрема, раку нирок. Крім того, сполуки за цим винаходом є ефективними інгібіторами р38 МАР-кінази (передачі сигналів MAP-кіназ р38α, р38β і р38 в ракових клітинах), і можуть мати поліпшений токсикологічний профіль (особливо шлунковокишкової токсичності) і знижений рівень ризику негативної взаємодії лікарських речовин у порівнянні з певними раніше відомими інгібіторами р38 МАР-кінази. Даний винахід пропонує сполуки Формули I: де: X – метоксиетил або етоксиметил; Q – циклопропіл, 2-метилпропанол-2-іл, циклопропіл; 1 3-метилоксетан-3-іл, 1-гідроксиметил-1 UA 109677 C2 5 10 15 20 25 30 35 40 45 50 55 60 або їх фармацевтично прийнятну сіль. Цей винахід також пропонує кристалічний 2-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-3-метокси-1метилпропіл]аміно]оксазол[5,4-b]піридин-5-іл]-1H-імідазол-2-іл]-2-метилпропан-1-ол, який характеризується порошковою рентгенограмою (випромінювання Cu-анода, λ=1,54060 Е), що містить пік при 15,06 і один або декілька піків при 19,94, 10,31 і 20,78 (2θ0,2). Цей винахід також пропонує кристалічний 2-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-3-метокси-1метилпропіл]аміно]оксазол[5,4-b]піридин-5-іл]-1H-імідазол-2-іл]-2-метилпропан-1-ол, який характеризується порошковою рентгенограмою (випромінювання Cu-анода, λ=1,54060 Е), що містить пік при 13,73 і один або декілька піків при 16,54, 22,87 і 18,57 (2θ0,2). Цей винахід пропонує сполуку, яка являєсобою 2-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-3метокси-1-метилпропіл]аміно]оксазол[5,4-b]піридин-5-іл]-1H-імідазол-2-іл]-2-метилпропан-1-ол, або її фармацевтично прийнятну сіль. Цей винахід пропонує сполуку, яка являє собою 5-[2-циклопропіл-5-(2,4-дифторфеніл)-1Hімідазол-4-іл]-N-[(1S)-3-метокси-1-метилпропіл]оксазол[5,4-b]піридин-2-амін, або її фармацевтично прийнятну сіль. Цей винахід пропонує сполуку, яка являє собою 5-[5-(2,4-дифторфеніл)-2-(3-метилоксетан3-іл)-1H-імідазол-4-іл]-N-[(1S)-3-метокси-1-метилпропіл]оксазол[5,4-b]піридин-2-амін, aбо її фармацевтично прийнятну сіль. Цей винахід пропонує сполуку, яка являє собою [1-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-2-етокси1-метилетил]аміно]оксазол[5,4-b]піридин-5-іл]-1H-імідазол-2-іл]циклопропіл]метанол, aбо її фармацевтично прийнятну сіль. Цей винахід пропонує спосіб лікування раку яєчників у ссавця, що включає введення ссавцю, який потребує такого лікування, ефективної кількості сполуки або солі за цим винаходом. Цей винахід пропонує спосіб лікування множинної мієломи у ссавця, що включає введення ссавцю, який потребує такого лікування, ефективної кількості сполуки або солі за цим винаходом. Цей винахід пропонує спосіб лікування раку, зокрема раку нирок, у ссавця, що включає введення ссавцю, який потребує такого лікування, ефективної кількості сполуки або солі за цим винаходом в одночасній, роздільній або послідовній комбінації з сунітинібом. Цей винахід також пропонує фармацевтичні композиції, що містять сполуку або сіль за цим винаходом в комбінації з одним(-ією) або декількома фармацевтично прийнятними носіями, розріджувачами або допоміжними речовинами. За конкретним варіантом здійснення винаходу згадана композиція також містить один або декілька інших терапевтичних агентів. Більш конкретно, згаданим іншим терапевтичним агентом є сунітиніб. Цей винахід також пропонує сполуку або сіль за цим винаходом для застосування в терапії. Винахід також пропонує сполуку або сіль за цим винаходом для застосування у лікуванні раку. Крім того, цей винахід пропонує застосування сполуки або солі за цим винаходом для виробництва лікарського засобу для лікування раку. Крім того, цей винахід пропонує застосування сполуки або солі за цим винаходом для застосування у лікуванні раку. Зокрема, цим раком є рак яєчників. Крім того, цим раком є множинна мієлома. Цей винахід також пропонує сполуку за цим винаходом або її фармацевтично прийнятну сіль і сунітиніб у вигляді комбінованої фармацевтичної композиції для одночасного, роздільного або послідовного застосування в терапії. Цей винахід також пропонує сунітиніб для застосування в одночасній, роздільній або послідовній комбінації зі сполукою за цим винаходом або її фармацевтично прийнятною сіллю у лікуванні раку. Альтернативно, цей винахід пропонує сполуку за цим винаходом або її фармацевтично прийнятну сіль для застосування в одночасній, роздільній або послідовній комбінації з сунітинібом у лікуванні раку. Більш конкретно, вказаним раком є рак нирок. Фахівцям в цій галузі буде зрозуміло, що сполуки Формули I можуть утворювати солі. Сполуки за цим винаходом містять основні гетероцикли і, відповідно, реагують з будь-якою з цілого ряду неорганічних і органічних кислот з утворенням фармацевтично прийнятних солей, які одержують шляхом додання кислоти. Такі фармацевтично прийнятні солі, які одержують шляхом додання кислоти, і загальна методика їх одержання є добре відомими у цій галузі. Дивись, наприклад, P. Stahl, et al., HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE, (VCHA/Wiley-VCH, 2008); S.M. Berge, et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, Vol 66, No. 1, January 1977. Фахівцю в цій галузі буде зрозуміло, що сполуки за цим винаходом містять щонайменше один хіральний центр. У цьому винаході розглядаються всі індивідуальні енантіомери або діастереомери, а також суміші енантіомерів і діастереомерів вказаних сполук, в тому числі 2 UA 109677 C2 5 10 15 20 25 30 35 рацемати. За варіантом, якому віддають перевагу, сполуки за цим винаходом, які містять щонайменше один хіральний центр, існують у вигляді окремих енантіомерів або діастереомерів. Окремі енантіомери або діастереомери можуть бути одержані, починаючи з хіральних реагентів або за допомогою методів стереоселективного або стереоспецифічного синтезу. Альтернативно, окремі енантіомери або діастереомери можуть бути виділені із сумішей стандартними хроматографічними методами із застосуванням "хіральних" колонок або кристалізаційними методами. Сунітиніб, наявний у продажу під торгівельною маркою SUTENT, являє собою дрібномолекулярний, багатоцільовий інгібітор тирозинкіназних рецепторів для перорального введення, який був схвалений FDA (Управління з санітарного нагляду за харчовими продуктами і медикаментами (США)) для лікування гіпернефроїдного раку та стромальних пухлин шлунково-кишкового тракту, стійких до іматинібу (imatinib). Сунітиніб розкритий у WO200160814. Схема I Одержання сполук Формули I, де R – циклопропіл або 3-метилоксетан-3-іл; X відповідає визначенню, наведеному вище. Орто-гідроксипіридил-3-аміни (А) обробляють ізотіоціанатами (B), які можуть являти собою рацемічну суміш або окремий енантіомер, в етанолі при нагріванні. Для видалення сірководню під час нагрівання періодично додають надлишок N, N'-дизаміщеного-карбодиіміду. Наприклад, можуть бути застосовані N, N'-дициклогексилкарбодиімід, N, N'-диізопропілкарбодиімід або 1етил-3-(3-диметиламінопропіл)карбодиіміду гидрохлорид. Синтез ізотіоціанатів (B), які можуть являти собою рацемічну суміш або окремий енантіомер, описаний у наведених нижче препаративних методиках. Схема II Одержання сполук Формули I, де R – метил-2-метилпропанкарбоксилат-2-іл або метилциклопропанкарбоксилат-1-іл; X відповідає визначенню, наведеному вище. 5-(1H-імідазол-4-іл)оксазол[5,4-b]піридини (С) відновлюють борогідридом літію у простому ефірі з одержанням сполук формули I. Проміжні сполуки (С) аналогічним чином одержують з відповідних гідроксипіридил-3-амінів (A) (Схема I), де R являє собою метил-2метилпропанкарбоксилат-2-іл або метилциклопропанкарбоксилат-1-іл з ізотіоціанатами (B), як показано на схемі I. Схема III Синтез проміжних хімічних сполук (А), де R – метил-2-метилпропанкарбоксилат-2-іл, метилциклопропанкарбоксилат-1-іл, циклопропіл або 3-метилоксетан-3-іл 3 UA 109677 C2 NO2 NH2 N (A) N R N H F F 5 10 15 20 25 (D) На піридиновому кільці 6-(1H-імідазол-4-іл)-3-нітропіридин-2-аміну (D) здійснюють перетворення функціональних груп, що залучають діазотування аміногрупи 2-піридила з швидким припиненням реакції холодною водою, з подальшою гідрогенізацією нітрогрупи 3піридила з одержанням проміжних хімічних сполук (А). Схема IV Синтез проміжних хімічних сполук (D), де R відповідає визначенню, наведеному в описі Схеми III. Проміжні сполуки (D) одержують з 1-(6-аміно-5-нітро-2-піридил)-2-(2,4-дифторфеніл)етан1,2-діону (F) і відомих альдегідів (Е) шляхом нагрівання у діоксані з ацетатом амонію. Синтез проміжної сполуки (F) описаний в наведених нижче препаративних методиках. Сполуки за цим винаходом одержують по суті як показано на Схемах і у наведених нижче Препаративних методиках і Прикладах. Реагенти і вихідні матеріали є легко доступними для фахівця у цій галузі або можуть бути одержані за допомогою способів, які вибирають з-посеред стандартних методів органічної та гетероциклічної хімії, методик, що є аналогічними синтезу відомих структурно подібних сполук, а також методик, описаних у наведених нижче Прикладах, в тому числі будь-яких нових методик. Слід розуміти, що вказані Препаративні методики та Приклади наведені з ілюстративною, а не обмежувальною метою, і що різні модифікації можуть бути здійснені будь-яким фахівцем у цій галузі. Найменування, що застосовуються у наведених нижче Препаративних методиках та Прикладах, як правило, наводяться за номенклатурою ІЮПАК, представленою у програмі Symyx Draw, версія 3.2.NET. Препаративна методика 1 Трет-бутил(3S)-3-[бензил-[(1S)-1-фенілетил]аміно]бутаноат N CO2t-Bu 30 35 Препаративні методики 1 і 2 були описані в WO 2006/076595 для R, R енантіомеру. Дивись також у Davies, S.G. and Ichihara, O. Tetrahedron: Asymmetry 1991, 2, 183-186 методику для асиметричного синтезу 3-амінобутаноатів з (Е)-бут-2-еноатів (кротонатів). (1S)-N-бензил-1-фенілетанамін (28,53 г, 135 ммоль) розчиняють в безводному тетрагідрофурані (THF), і одержаний розчин охолоджують до температури 0С в атмосфері аргону. Розчин N-бутиллітію (2,5 М в гексанах, 54 мл, 135 ммоль) додають краплями протягом 30 хв. Реакційну суміш перемішують протягом 20 хв при температурі 0С, після чого охолоджують до температури -78С. Розчин трет-бутил-(E)-бут-2-еноату (10 г, 70,32 ммоль) у безводному THF (75 мл) додають до реакційної суміші протягом 20 хв. Через 75 хв реакційну суміш гасять доданням насиченого розчину NH4Cl (175 мл) і насиченого водного розчину NaCl (розсолу, 100 мл). Шари відокремлюють, і водний шар екстрагують діетиловим ефіром (2125 мл). Об'єднані органічні шари сушать безводним MgSO 4, фільтрують, і концентрують до одержання масла жовтого кольору. Неочищений продукт розчиняють у гексанах (250 мл), і 4 UA 109677 C2 5 промивають 10 % водним розчином лимонної кислоти (375 мл). Органічний шар сушать MgSO4, фільтрують, і концентрують до одержання вказаної в заголовку сполуки у вигляді масла жовтого кольору (24,12 г, 97 %). LC-ES/MS m/z 354 (М+1). Препаративна методика 2 (3S)-3-[бензил-[(1S)-1-фенілетил]аміно]бутан-1-ол N OH 10 15 трет-бутил(3S)-3-[бензил-[(1S)-1-фенілетил]аміно]бутаноат (24 г, 67,9 ммоль) розчиняють в безводному THF (237 мл), і охолоджують до температури 0С в атмосфері аргону. 1 М розчин літійалюмінійгідріду в тетрагідрофурані (237 мл, 237 ммоль) додають краплями протягом 10 хв. Реакційну суміш перемішують при температурі 0С протягом 1 год., і потім при температурі 60С протягом 1 год. Суміш охолоджують до кімнатної температури (RT), і розбавляють діетиловим ефіром (500 мл). Реакцію гасять сумішшю CELITE і Na2SO410 H2O (1:1), яку додають порціями протягом 15 хв. Суміш фільтрують, і концентрують у вакуумі до одержання вказаної в заголовку сполуки у вигляді безбарвного масла (17,54 г, 90 %). LC-ES/MS m/z 284 (М+1). Препаративна методика 3 (2S)-N-бензил-4-метокси-N-[(1S)-1-фенілетил]бутан-2-амін N O 20 25 30 (3S)-3-[бензил-[(1S)-1-фенілетил]аміно]бутан-1-ол (17,54 г, 61,9 ммоль) розчиняють в безводному THF (186 мл), і охолоджують до температури 0С в атмосфері аргону. Гідрид натрію (4,95 г, 60 % суспензія у мінеральному маслі, 123,8 ммоль) додають порціями протягом 10 хв. Одержану суміш перемішують при температурі 0С протягом 15 хв, після чого витримують для нагрівання до кімнатної температури. Метилйодид (10,54 г, 74,28 ммоль) додають краплями протягом 30 хв. Після перемішування протягом додаткових 30 хв реакційну суміш гасять доданням насиченого розчину NH4Cl у воді. Шари відокремлюють, і водний шар екстрагують діетиловим ефіром (2100 мл). Об'єднані органічні шари сушать безводним MgSO 4, концентрують, і неочищений продукт очищають шляхом хроматографування з нормальною фазою (два 120 г силікагелеві картриджі, 10 % розчин метил-трет-бутилового простого ефіру у гексанах) з одержанням вказаної в заголовку сполуки у вигляді безбарвного масла (14,96 г, 81 %). LC-ES/MS m/z 298 (М+1). Препаративна методика 4 (S)-4-метоксибутан-2-аміну гідрохлорид NH3Cl O 35 40 45 (2S)-N-бензил-4-метокси-N-[(1S)-1-фенілетил]бутан-2-амін (14,96 г, 50,29 ммоль) розчиняють в метанолі (400 мл). Розчин дезоксигенують шляхом барботування азоту. До вказаного розчину додають 20 % гідроксид паладію на вуглеці (1,50 г), одержану суспензію насичують воднем, і перемішують в атмосфері водню протягом 16 год. Основним продуктом, присутнім на цей момент, є монодебензилований продукт. Суспензію фільтрують через шар CELITE, і до одержаного розчину додають 1,1 г 20 % гідроксиду паладію на вуглеці. Суспензію перемішують протягом 24 год. в атмосфері водню. Суспензію фільтрують через шар CELITE, до суміші додають 2н розчин HCl в діетиловому ефірі (60 мл), і перемішують протягом 30 хв. Розчин концентрують при зниженому тиску до одержання вказаної в заголовку сполуки у вигляді 1 твердої речовини білого кольору (7,01 г, 99 %). H-ЯМР (400 МГц, CDCl3); δ 1,48 (3H, d, J=6,8 Гц), 1,8-1,9 (1Н, m), 2,0-2,1 (1Н, m), 3,37 (3Н, s), 3,5-3,7 (3Н, m), 8,3 (3Н, br). Препаративна методика 5 (R)-N-[(1S)-3-метокси-1-метилпропіл]-2-метилпропан-2-сульфінамід 5 UA 109677 C2 HN S O O 5 10 15 20 25 30 35 Наведена нижче методика адаптована з Ellman, J. A. et al J. Org. Chem. 2007, 72, 626-629. До 1н розчину HCl (7,0 мл, 7,00 ммоль) краплями додають 1,3,3-триметоксибутан (53,19 мл, 337,38 ммоль), одержаний розчин нагрівають до температури 50С, і перемішують протягом 30 хв. До заздалегідь охолодженої до кімнатної температури вказаної суміші додають спочатку бікарбонат натрію (16,50 г, 196,41 ммоль), потім діетиловий ефір і MgSO4. Після фільтрування з подальшим випарюванням розчинника одержують кетоефірну проміжну сполуку, 4метоксибутан-2-он, у вигляді масла жовтого кольору. Вказане масло додають до розчину (R)(+)-2-метил-2-пропансульфінаміду (36,80 г, 303,64 ммоль) і етоксиду титану (IV) (123,14 г, 539,80 ммоль) у THF (482 мл) при температурі 25С в атмосфері азоту. Одержану суспензію жовтого кольору нагрівають до температури 60С, і перемішують при цій температурі протягом 16 год. Реакційну суміш охолоджують до кімнатної температури, і потім до температури -48С. Краплями додають 1,0 М розчин три(втор-бутил)борогідриду літію в THF (539,80 мл, 539,80 ммоль). Реакційну суміш витримують для нагрівання до кімнатної температури. Через 1 год. реакційну суміш охолоджують до температури 0С, і додають метанол (1100 мл) з одночасним швидким перемішуванням до припинення виділення газу. Одержану суспензію фільтрують через шар CELITE, і відфільтрований осад промивають етилацетатом. Фільтрат промивають розсолом, і шар розсолу двічі екстрагують етилацетатом. Об'єднані органічні шари сушать Na2SO4, фільтрують, і упарюють до одержання масла жовтого кольору. Неочищений продукт адсорбують на силікагелі, і очищають на колонці з силікагелем із застосуванням градієнту гексан/етилацетат (від 7:1 до 100 % етилацетату) з одержанням цільового продукту. Інші фракції, які містять неполярні домішки і цільовий продукт, збирають, і знову очищають хроматографією на силікагелі. Неполярні домішки видаляються сумішшю гексан/етилацетат, 4:1. Цільовий продукт елююють сумішшю дихлорметан/метанол, 95:5, з одержанням додаткового матеріалу. Дві порції матеріалу об'єднують з одержанням 38 г (54 %), які, за даними LCMS (рідинна хроматографія/мас-спектрометрія), являють собою суміш цільового/нецільового діастереомерів у співвідношенні приблизно 3:1. Вказаний матеріал (38 г) змішують з іншою партією матеріалу (23 г), яку одержують за тією ж самою загальною методикою, і діастереомери (3:1, 61 г) розділяють засобами хіральної високоефективної рідинної хроматографії (Нерухома фаза: OD-H; Розмір колонки: (20 мкм, 80250 мм); Режим елюювання: ізократичний; Рухома фаза: гексан/ізопропанол; Швидкість потоку: 300 мл/хв; УФ-детекція: 215,16 нм; Навантаження: 4 г/6 хв. Перший елюційний пік являє собою мінорний діастереомер, T R=4,75 хв. Другий елюційний пік являє собою основний діастереомер (вказану в заголовку сполуку), T R=6,61 хв. Вказану в заголовку сполуку одержують у вигляді масла світло-жовтого кольору (43,5 г) після хіральної хроматографії. ES/MS m/z 208 (М+1); енантіомерний надлишок >98 % ee. Препаративна методика 6 (S)-4-метоксибутан-2-аміна гідрохлорид NH3Cl O 40 45 50 Розчин хлористого водню (4,0 М) в діоксані (110,15 г, 419,61 ммоль) додають до розчину (R)N-[(1S)-3-метокси-1-метилпропіл]-2-метилпропан-2-сульфінаміду (43,5 г, 209,80 ммоль) в 1,4діоксані (109 мл) при температурі 0C, і перемішують реакційну суміш протягом 1 год. при кімнатній температурі. Розчинник концентрують при зниженому тиску, залишок повторно суспендують в толуолі, і розчинник випарюють під зниженим тиском. Залишок сушать у вакуумі протягом 15 хв. Додають THF, і в осад випадає тверда речовина білого кольору. Вказану тверду речовину відфільтровують, промивають THF, дають висохнути, і збирають з одержанням вказаної в заголовку сполуки (25,4 г, 87 %). Абсолютна конфігурація аміну може бути підтверджена шляхом дериватизації (S)-(-)-αметокси-α-трифторметилфенілоцтовою кислотою і порівняння ЯМР з такою ж самою похідною (S)-4-метоксибутан-2-аміну Препаративної методики 4, одержаною з хірального шляху. Препаративна методика 7 (3S)-3-ізотіоціанат-1-метоксибутан 6 UA 109677 C2 S N 5 10 O (S)-4-метоксибутан-2-аміна гідрохлорид (25,4 г, 181,92 ммоль) суспендують у THF (609 мл), і додають триетиламін (TEA, 32,17 мл, 230,78 ммоль). До суспензії білого кольору додають 1,1'тіокарбонілдиімідазол (46,74 г, 251,76 ммоль) (злегка екзотермічна реакція), і одержану суспензію жовтого кольору перемішують в атмосфері азоту протягом ночі. До вказаної суспензії жовтого кольору додають спочатку етилацетат (500 мл), потім 1н розчин HCl (500 мл). Органічну фазу відокремлюють, і промивають 1н розчином HCl (3200 мл), водою (200 мл) і розсолом (200 мл), сушать Na2SO4, фільтрують, і концентрують при зниженому тиску до 1 одержання вказаної в заголовку сполуки (25,3 г; 83 %) у вигляді масла жовтого кольору. H ЯМР (400 МГц, CDCl3); δ 1,37 (3H, d, J=6,6 Гц), 1,83 (2Н, q, J=6,6 Гц), 3,35 (3Н, s), 3,6-3,4 (2Н, m); 3,99 (1H, six, J=6,6 Гц). Препаративна методика 8 Метил-2,2-диметил-3-оксопропаноат O O O 15 20 25 H Метил-3-гідрокси-2,2-диметилпропаноат (52,4 г, 396,49 ммоль) розчиняють у дихлорметані (495 мл), і охолоджують суміш на бані (суміш води з льодом). Порціями додають спочатку трихлорізоціанурову кислоту (101,36 г, 436,14 ммоль), потім 2,2,6,6-тетраметилпіперидин-Nоксид (6,20 г, 39,65 ммоль). Одержану суміш перемішують при температурі 0С протягом 15 хв, після чого витримують для нагрівання до кімнатної температури, і додатково перемішують протягом 60 хв. Після цього тверду речовину відфільтровують через CELITE, і промивають дихлорметаном (300 мл). Фільтрат промивають насиченим розчином Na2CO3 у воді. Органічну фазу сушать Na2SO4, фільтрують, і концентрують при зниженому тиску до одержання вказаної в заголовку сполуки (41,24 г, 80 %) у вигляді масла зеленуватого кольору. Вказаний продукт 1 використовують без подальшого очищення на наступній стадії реакції. H ЯМР (400 МГц, CDCl3); δ 1,34 (6Н, s), 1,34 (6Н, s), 3,74 (3Н, s), 9,64 (1Н, s). Препаративна методика 9 6-[2-(2,4-дифторфеніл)етиніл]-3-нітропіридин-2-амін NO2 F N NH2 F 30 35 40 6-хлор-3-нітропіридин-2-іламін (1254 г, 7,23 моль), триетиламін (1510 мл, 10,84 ммоль) і ацетонітрил (10 л) завантажують в атмосфері азоту у 20 л 4-горлу круглодонную колбу, споряджену механічною мішалкою. До одержаної суспензії жовтого кольору додають йодид міді (I) (13,9 г, 72,3 ммоль) і хлорид біс(трифенілфосфін)паладію (II) (50,72 г, 72,3 ммоль). Одержану суспензію блідо-оранжевого кольору охолоджують до температури 0-5С, після чого знегажують азотом протягом 10 хв. Краплями впродовж 60 хв додають розчин 1-етиніл-2,4-дифторбензолу (1100 г, 7,95 моль) в ацетонітрилі (2,5 л). Одержану суміш перемішують при кімнатній температурі (30C) протягом ночі. Суміш охолоджують до температури 0-5С. До суспензії додають толуол (6 л), суміш перемішують протягом 45 хв, і фільтрують через пористий скляний фільтр. Тверду речовину промивають толуолом (33 л), водою (23 л), і сушать протягом ночі у вакуумній печі з одержанням вказаної в заголовку сполуки у вигляді твердої речовини жовтого кольору (1750 г, 92 %). LC-ES/MS m/z 276 (М+1). Препаративна методика 10 1-(6-аміно-5-нітро-2-піридил)-2-(2,4-дифторфеніл)етан-1,2-діон 7 UA 109677 C2 F NO2 O N F 5 10 15 NH2 O До холодної суспензії (0-10С) 6-[2-(2,4-дифторфеніл)етиніл]-3-нітропіридин-2-аміну (500 г, 1,82 моль) в ацетоні (10 л) додають холодний буферний розчин [NaH 2PO4 (0,8 M)/Na2HPO4 (0,8 М)=85/15 (в об'ємному відношенні)] (рН=6,0; 0-10С, 10 л). Підтримують температуру біля 15С. Порціями (3 порції) додають перманганат калію (1035 г, 6,55 моль). Суміш перемішують протягом 4 год. при температурі 15С. рН доводять до рН 5,0, і підтримують температуру нижчою ніж 15С. Повільно додають 28 % розчин тіосульфату натрію (2054 мл, 3,64 моль), та підтримують температуру нижчою ніж 15С і рН нижчим 7,5. До суспензії додають розсіл (7,5 л) і суміш метил-трет-бутилового простого ефіру (3,75 л) і етилацетату (3,75 л). Суміш перемішують протягом 15 хв при температурі 13С. Дві фази розділяють, і водну суспензію брунатного кольору двічі екстрагують метил-трет-бутиловим простим ефіром (3,5 л). Об'єднані органічні шари збирають, і промивають розсолом (23 л), сушать Na2SO4, фільтрують, і випарюють розчинник при зниженому тиску до одержання вказаної в заголовку сполуки у вигляді твердої речовини (375 г) жовтого кольору. Експеримент повторюють за тих же умов ще два рази. Одержані партії об'єднують з одержанням 1080 г. LC-ES/MS m/z 308 (М+1). Препаративна методика 11 Метил-2-[4-(6-аміно-5-нітро-2-піридил)-5-(2,4-дифторфеніл)-1H-імідазол-2-іл]-2метилпропаноат NO2 N O O N N H F 20 25 30 NH2 F У круглодонну колбу з парціальним конденсатором гарячого зрошення завантажують 1-(6аміно-5-нітро-2-піридил)-2-(2,4-дифторфеніл)етан-1,2-діон (50 г, 162,75 ммоль), ацетат амонію (126,72 г, 1,63 моль) метил-2,2-диметил-3-оксопропаноат (42,36 г, 325,51 ммоль) і 1,4-діоксан (163 мл). Реакційну суміш нагрівають до температури 80С протягом 1,5 год. Розчин початкового помаранчевого кольору тьмянішає з підігрівом. Реакційну суміш концентрують при зниженому тиску для видалення діоксану, і залишок сушать під високим вакуумом протягом ночі. Залишок повторно розчиняють в етилацетаті (800 мл), і екстрагують 2 М розчином Na 2CO3 у воді. Органічну фазу сушать MgSO4, концентрують, і сушать під високим вакуумом протягом ночі з одержанням вказаної в заголовку сполуку у вигляді неочищеної твердої речовини помаранчевого кольору (80 г), яку використовують на наступній стадії без додаткового очищення. LC-ES/MS m/z 418 (М+1). Препаративна методика 12 Метил-2-[5-(2,4-дифторфеніл)-4-(6-гідрокси-5-нітро-2-піридил)-1H-імідазол-2-іл]-2метилпропаноат NO2 N O O N N H F 35 40 OH F До розчину метил-2-[4-(6-аміно-5-нітро-2-піридил)-5-(2,4-дифторфеніл)-1H-імідазол-2-іл]-2метилпропаноату (56 г, 134,17 ммоль) у диметилсульфоксиді (DMSO, 400 мл) і воді (320 мл) краплями додають сірчану кислоту (95-97 %, 80 мл). Потім вказану суміш охолоджують до температури 0С. До вказаної вище суміші краплями протягом 15 хв при температурі 0С додають розчин нітриту натрію (18,70 г, 268,35 ммоль) у воді (80 мл). Реакційну суміш перемішують протягом 20 хв при вказаній температурі, після чого охолоджувальну баню видаляють з підвищенням температури вказаної суміші до кімнатної. До реакційної суміші додають 0,8 М забуференого водного розчину дигідрофосфату натрію (1200 мл, рН=6). 8 UA 109677 C2 5 Одержують суспензію жовтого кольору. Цю суспензію перемішують при кімнатній температурі протягом 1 год. Тверду речовину відфільтровують, промивають водою, і сушать у печі до одержання вказаної в заголовку сполуки у вигляді твердої речовини помаранчевого кольору (49,5 г, 88 %). LC-ES/MS m/z 419 (М+1). Препаративна методика 13 Метил-2-[4-(5-аміно-6-гідрокси-2-піридил)-5-(2,4-дифторфеніл)-1H-імідазол-2-іл]-2метилпропаноат NH2 N O O N OH N H F 10 15 20 25 F Суміш метил-2-[5-(2,4-дифторфеніл)-4-(6-гідрокси-5-нітро-2-піридил)-1H-імідазол-2-іл]-2метилпропаноату (49,5 г, 118,32 ммоль) і 5 % (мас.) (по сухій речовині) паладію на активованому вугіллі (4,95 г, 2,33 ммоль) у метанолі (1,18 л) перемішують в атмосфері водню (еластична камера з газом під тиском) при кімнатній температурі протягом ночі. Суспензію фільтрують через CELITE, промивають метанолом, і концентрують фільтрат при зниженому тиску до одержання неочищеної вказаної в заголовку сполуки (39 г) у вигляді твердої речовини брунатного кольору. Неочищений продукт (90 г, 231,74 ммоль), одержаний здійсненням множини таких самих синтезів, очищають таким чином. Вказаний матеріал суспендують в суміші (1:1) дихлорметану (450 мл) і етилацетату (450 мл). Суспензію перемішують при кімнатній температурі протягом ночі. Відфільтровують суспензію, і тверду речовину промивають сумішшю (1:1) дихлорметан/етилацетат. Твердій речовині брунатного кольору дають висохнути, і збирають з одержанням 67 г вказаної в заголовку сполуки з чистотою>98 %, яку визначали засобами рідинної хроматографії/мас-спектрометрії. LC-ES/MS m/z 389 (М+1). Препаративна методика 14 Метил-2-[5-(2,4-дифторфеніл)-4-[2-[[(1S)-3-метокси-1-метилпропіл]аміно]оксазол[5,4b]піридин-5-іл]-1H-імідазол-2-іл]-2-метилпропаноат N N O O 35 40 O O N N H F 30 H N F (3S)-3-ізотіоціанат-1-метокси-бутан (24,68 г, 169,94 ммоль) додають до суспензії метил-2-[4(5-аміно-6-гідрокси-2-піридил)-5-(2,4-дифторфеніл)-1H-імідазол-2-іл]-2-метилпропаноату (55 г, 141,62 ммоль) в етанолі (550 мл) при кімнатній температурі. Реакційну суміш перемішують із кип'ятінням зі зворотним холодильником протягом ночі і потім охолоджують до температури 50С. До суміші додають дициклогексилкарбодиімід (37,99 г, 184,10 ммоль), і перемішують одержану суспензію із кип'ятінням зі зворотним холодильником протягом 20 год. Вказану реакційну суміш витримують до досягнення кімнатної температури, і випарюють розчинник при зниженому тиску. Залишок абсорбують на силікагелі, і очищають на колонці з силікагелем (спочатку застосовуючи дихлорметан як елюент для видалення більшості неполярних домішків, потім суміш дихлорметан/метанол (95:5) для елюювання цільового продукту) з одержанням вказаної в заголовку сполуки (52 г, 74 %) у вигляді піни темно-брунатного кольору. LC-ES/MS m/z 500 (М+1). Препаративна методика 15 2-[(1S)-2-гідрокси-1-метилетил]ізоіндолін-1,3-діон O N OH O Суміш (2S)-2-амінопропан-1-олу (26 мл, 333 ммоль) і фталевого ангідриду (51,7 г, 349,4 ммоль) нагрівають при температурі 140С протягом ночі. Протягом цього часу тверда речовина 9 UA 109677 C2 5 перетворюється на рідину помаранчевого кольору. Реакційну суміш охолоджують до кімнатної температури і розбавляють етилацетатом (10 мл/г). Органічну фазу промивають насиченим розчином NaHCO3 і 10 % лимонною кислотою, сушать над MgSO4, фільтрують, і концентрують до одержання вказаної в заголовку сполуки (68,3 г, 98 %) у вигляді твердої речовини білого кольору, яку використовують без подальшого очищення. LC-ES/MS m/z 206 (М+1). Препаративна методика 16 2-[(1S)-2-етокси-1-метилетил]ізоіндолін-1,3-діон O N O O 10 15 До розчину 2-[(1S)-2-гідрокси-1-метилетил]ізоіндолін-1,3-діону (47 г, 229 ммоль) і йодетану (89,3 г, 572,5 ммоль) у THF (376 мл) однією порцією додають трет-бутоксид калію (64,25 г, 572,5 ммоль). Вказану суміш перемішують в атмосфері азоту протягом 15 год. Суміш розбавляють етилацетатом (200 мл), і промивають розсолом (200 мл). Водну фазу екстрагують етилацетатом (2100 мл). Об'єднані органічні екстракти сушать MgSO 4, фільтрують, концентрують при зниженому тиску, і потім сушать під високим вакуумом до одержання вказаної в заголовку сполуки (39,4 г, 74 %) у вигляді твердої речовини помаранчевого кольору, яку використовують без подальшого очищення. LC-ES/MS m/z 234 (М+1). Препаративна методика 17 (2S)-1-етоксипропан-2-аміну гідрохлорид HCl-H2N O 20 25 30 2-[(1S)-2-етокси-1-метилетил]ізоіндолін-1,3-діон (12,84 г, 55 ммоль) розчиняють у метанолі (120 мл). Повільно додають моногідрат гідразину (6,9 мл, 138 ммоль), і перемішують суміш при температурі 40C протягом 4 год. (утворюється тверда речовина білого кольору). Додають розчин NaOH (1 мл), і рН зростає до 13-14. Тверду речовину відфільтровують, і промивають дихлорметаном. Шари фільтрату розділяють, і водний шар додатково екстрагують дихлорметаном. Об'єднані органічні шари сушать MgSO 4, і фільтрують. До вказаного розчину додають 2н розчин HCl в ефірі (70 мл, 140 ммоль). Суміш перемішують протягом 15 хв, і розчинник випарюють при зниженому тиску з одержанням вказаної в заголовку сполуки у 1 вигляді твердої речовини білого кольору (6,61 г, 86 %). H ЯМР (400 МГц, CD3OD); δ 1,22 (t, 3Н, J=7,02 Гц), 1,28 (d, 3Н, J=6,52 Гц), 3,42 (m, 2Н), 3,58 (m, 3H). Препаративна методика 18 (2S)-1-етокси-2-ізотіоціанатпропан S O N 35 40 До розчину (2S)-1-етоксипропан-2-аміна гідрохлориду (2 г, 12,03 ммоль) у диметилформаміді (DMF, 20 мл) і ТЕА (1,85 мл, 13,24 ммоль) додають 1,1'тіокарбонілдиімідазол (2,36 г, 13,24 ммоль). Вказану суміш перемішують в атмосфері азоту протягом 16 год. Суміш розбавляють етилацетатом, і ретельно промивають 1н розчином HCl, водою і розсолом, сушать MgSO4, і концентрують при зниженому тиску (температура бані не повинна перевищувати 20C, щоб уникнути випаровування продукту) до одержання неочищеного продукту (1,84 г), що містить вказану в заголовку сполуку, яку використовують без 1 подальшого очищення. H ЯМР (400 МГц, CDCl3) δ 1,26 (t, 3Н, J=7,13 Гц), 1,33 (d, 2H, J=6,63 Гц), 3,46 (dd, J=5,86 Гц, 1,62 Гц, 2Н), 3,55 (dd, J=13,98 Гц, 6,97 Гц, 2Н), 3,93 (m, 1H). Препаративна методика 19 3-метилоксетан-3-карбальдегід O O H 10 UA 109677 C2 5 10 15 20 (3-метилоксетан-3-іл)метанол (6,0 г, 58,75 ммоль) розчиняють в дихлорметані (117 мл). Порціями при температурі -5C додають спочатку трихлорізоціанурову кислоту (13,93 г, 59,92 ммоль), потім 2,2,6,6-тетраметилпіперидин-1-оксил (TEMPO) (0,92 г, 5,87 ммоль). Реакційну суміш перемішують при температурі -5С протягом 20 хв, витримують для нагрівання до кімнатної температури, і перемішують протягом додаткових 20 хв. Суміш фільтрують через шар СELITE, розбавляють дихлорметаном (200 мл), і промивають насиченим водним розчином Na2CO3 (100 мл), 1н розчином HCl (100 мл) і розсолом (50 мл). Органічну частину концентрують до одержання вказаної в заголовку сполуки у вигляді масла помаранчевого кольору (4,17 г, 1 71 %), яке використовують без подальшого очищення. H ЯМР (400 МГц, CDCl3); δ 1,48 (s, 3H), 4,50 (d, 2H, J=6,34 Гц), 4,88 (d, 2H, J=6,34 Гц), 9,95 (s, 1H). Альтернативна препаративна методика: Бромід калію (11,65 г, 0,098 моль) додають до суміші (3-метилоксетан-3-іл)метанолу (200 г, 1,96 моль) і TEMPO (3,06 г, 0,019 моль) у дихлорметані (2 л) при температурі 0C. Потім до вищевказаного розчину краплями додають водний 10 % розчин гіпохлориту натрію (1,6 л, 2,35 моль), доведеного до рН 9 за допомогою твердого NaHCO 3 (додавання впродовж 3 год. з підтримуванням внутрішньої температури
ДивитисяДодаткова інформація
Назва патенту англійськоюOxazolo[5,4-b]pyridin-5-yl compounds and their use for the treatment of cancer
Автори англійськоюCoates, David, Andrew, Gilmour, Raymond, Martin, Jose, Alfredo, Martin de la Nava, Eva, Maria
Автори російськоюКоутс Дэйвид Эндрю, Гилмор Раймонд, Мартин Хосе Альфредо, Мартин дэ ла Нава Ева Мария
МПК / Мітки
МПК: C07D 498/04, A61K 31/424, A61P 35/00
Мітки: лікуванні, раку, оксазол[5,4-b]піридин-5-ілу, сполуки, застосування
Код посилання
<a href="https://ua.patents.su/33-109677-spoluki-oksazol54-bpiridin-5-ilu-ta-kh-zastosuvannya-u-likuvanni-raku.html" target="_blank" rel="follow" title="База патентів України">Сполуки оксазол[5,4-b]піридин-5-ілу та їх застосування у лікуванні раку</a>
Ізоіндолінові сполуки для застосування при лікуванні раку

Номер патенту: 107783
Опубліковано: 25.02.2015
Автори: Мюллер Джордж В., Рачелмен Алєксандер Л.
МПК: C07D 413/14, C07D 401/04, C07D 417/14, C07D 401/14, C07D 405/14, A61K 31/45, A61P 35/00
Мітки: ізоіндолінові, сполуки, раку, застосування, лікуванні
Формула / Реферат:
1. Сполука Формули І (І)або її фармацевтично прийнятна сіль, сольват або стереоізомер, де X являє собою СН2; Y являє собою О; m являє собою ціле число 0, 1, 2 або 3; R1 являє собою водень або C1-6алкіл; R2 являє собою водень, -NO2, C1-10алкіл,...
Лікарські сполуки, що модулюють активність піруваткінази-м2, композиції на їх основі та застосування при лікуванні раку

Номер патенту: 107667
Опубліковано: 10.02.2015
Автори: Салітуро Франческо Дж., Сондерз Джеффрі О., Янь Шуньци
МПК: A61P 35/00, A61K 31/497
Мітки: композиції, модулюють, застосування, піруваткінази-м2, активність, сполуки, основі, лікарські, раку, лікуванні
Формула / Реферат:
1. Сполука формули (I) (I) або її фармацевтично прийнятна сіль, де W, X, Y і Z незалежно вибрані з CH або N;D і D1 незалежно вибрані зі зв'язку або NRb;A представляє необов'язково заміщений...
2-(піридин-2-іл)піримідинові сполуки та їх застосування для боротьби зі шкідливими грибами

Номер патенту: 83314
Опубліковано: 25.06.2008
Автори: Штратманн Зігфрид, Хюнгер Удо, Шівек Франк, Грамменос Вассіліос, Кьоле Харальд, Мюллер Бернд, Шерер Маріа, Шьофль Ульріх, Гроте Томас, Райнхаймер Йоахим, Блеттнер Карстен, Швьоглер Аня, Шефер Петер, Ретер Ян, Гевер Маркус, Штірль Райнхард
МПК: C07D 401/04, A01N 43/54, A01P 3/00
Мітки: застосування, 2-(піридин-2-іл)піримідинові, шкідливими, боротьби, сполуки, грибами
Формула / Реферат:
1. 2-(піридин-2-іл)піримідинові сполуки загальної формули І , (І)у якійk являє собою 0, 1, 2 або 3;m являє собою 0, 1, 2, 3, 4 або 5;n являє собою 1, 2, 3, 4 або 5;R1 незалежно один від іншого являють собою галоген, ОН, CN, NO2, С1-С4-алкіл, С1-С4-галоалкіл, С1-С4-алкоксигрупу, С1-С4-галоалкоксигрупу, С2-С4-алкеніл, С2-С4-алкініл,...
Сульфонілетилфосфородіамідати, призначені для застосування при лікуванні раку

Номер патенту: 88468
Опубліковано: 26.10.2009
Автори: Вік Майкл М., Шоу Стівен Р., Ші Сонюань, Аллен Девід Р., Вебер Кевін Т., Саймон Рейна Дж., Робінсон Луіз, Пітерсон Брайан Т., Ма Веньлі
МПК: A61P 35/04, A61K 31/664, C07F 9/24
Мітки: раку, призначені, сульфонілетилфосфородіамідати, лікуванні, застосування
Формула / Реферат:
1. Сполука сульфонілетилфосфородіамідату формули А, формули В або формули С:,у якій:кожний R незалежно означає водень, С1-С6алкіл або -СН2СН2Х, де кожний X незалежно означає Сl, Вr, С1-С6алкансульфонілоксигрупу, галоген-С1-С6алкансульфонілоксигрупу або бензолсульфонілоксигрупу, яка необов'язково містить до 3 замісників, вибраних з групи, яка включає...
Сполуки 2-метил-1-феніл-2-(піридин-2-іл)пропан-1-аміну та їх застосування для лікування депресії

Номер патенту: 106056
Опубліковано: 25.07.2014
Автори: Нуґіель Давід, Балестра Майкл, Шен Ліон, Фрітц Вілліам, Ернст Глен Е, Бернстайн Пітер, Маккаулей Джон П.
МПК: A61P 25/24, A61K 31/4402, C07D 213/38
Мітки: депресії, лікування, сполуки, 2-метил-1-феніл-2-(піридин-2-іл)пропан-1-аміну, застосування
Формула / Реферат:
1. 2-Метил-1-феніл-2-(піридин-2-іл)пропан-1-амін або його фармацевтично прийнятна сіль.2. Сполука за п. 1, якою є (S)-2-метил-1-феніл-2-(піридин-2-іл)пропан-1-амін або його фармацевтично прийнятна сіль.3. Сполука за п. 2, де фармацевтично прийнятною сіллю є сіль, утворена з хлоридної, бромідної, сульфітної, фосфатної, лимонної, виннокам’яної, молочної, піровиноградної, оцтової, бурштинової, фумарової, малеїнової,...
Попередній патент: Протигрибкові 5,6-дигідро-4н-піроло[1,2-а][1,4]бензодіазепіни і 6н-піроло[1,2-а][1,4]бензодіазепіни, заміщені фенільними похідними
Наступний патент: Одношнековий екструдер
Випадковий патент: Синергічна інсектицидна композиція