Похідні гідантоїну








Формула / Реферат
1. Сполуки, представлені наведеною нижче загальною формулою (1), або їх фармакологічно прийнятні солі:
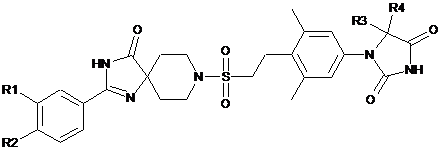 , (1)
, (1)
де,
коли R1 і R2 не є обидва атомами водню, R1 і R2 являють собою незалежно:
1) атом водню;
2) атом галогену;
3) алкільну групу, яка містить один або два атоми вуглецю, яка може бути заміщеною від одного до п'яти атомами фтору; або
4) алкоксигрупу, яка містить один або два атоми вуглецю, яка може бути заміщеною від одного до п'яти атомами фтору; або
R1 і R2 зв'язані один з одним, утворюючи групу, представлену наведеною нижче формулою:
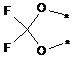
(де кожний символ * означає положення зв'язування з фенільною частиною); і
R3 і R4 являють собою незалежно метильну групу, що може бути заміщеною від одного до трьох атомами фтору; або
R3 і R4 разом із зв'язаним атомом вуглецю утворюють три-шестичленне карбоциклічне кільце (де один з атомів вуглецю, що утворює кільце, може бути замінений атомом кисню, атомом сірки або метилзаміщеним або незаміщеним атомом азоту).
2. Сполука або її фармакологічно прийнятна сіль за п. 1, де R1 і R2 вибрані з наведених нижче комбінацій:
1) R1 являє собою атом водню або атом галогену, і R2 являє собою атом водню, трифторометильну групу або трифторометоксигрупу (за умови, що R1 і R2 не є обидва атомами водню);
2) R1 являє собою трифторометильну групу або трифторометоксигрупу, і R2 являє собою атом водню або атом галогену;
3) R1 i R2 зв'язані один з одним, утворюючи групу, представлену наведеною нижче формулою:
(де кожний символ * означає положення зв'язування з фенільною частиною); і
R3 і R4 являють собою метильні групи; або
R3 і R4 разом із зв'язаним атомом вуглецю утворюють кільце, вибране з наведених нижче:
![]() ,
,
 ,
,
 ,
,
 ,
,
 ,
,
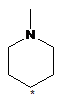
(де * означає положення зв'язування з частиною "імідазолідин-2,4-діон").
3. Сполука або її фармакологічно прийнятна сіль за п. 1, де R1 і R2 вибрані з наведених нижче комбінацій:
1) R1 являє собою трифторометоксигрупу, і R2 являє собою атом фтору;
2) R1 являє собою атом брому, і R2 являє собою атом водню;
3) R1 являє собою трифторометоксигрупу, і R2 являє собою атом фтору;
4) R1 являє собою атом фтору, і R2 являє собою трифторометоксигрупу;
5) R1 являє собою трифторометильну групу, і R2 являє собою атом водню;
6) R1 являє собою атом водню, і R2 являє собою трифторометоксигрупу;
7) R1 і R2 зв'язані один з одним, утворюючи групу, представлену наведеною нижче формулою:
(де кожний символ * означає положення зв'язування з фенільною частиною); і
R3 і R4 являють собою метильні групи; або
R3 і R4 разом із зв'язаним атомом вуглецю утворюють кільце, вибране з наведених нижче:
![]() ,
,
,
,
(де * означає положення зв'язування з частиною "імідазолідин-2,4-діон").
4. Сполука або її фармакологічно прийнятна сіль за п. 1, де R3 і R4 являють собою метильні групи.
5. Сполука або її фармакологічно прийнятна сіль за п. 1, де R3 і R4 разом із зв'язаним атомом вуглецю утворюють кільце, вибране з наведених нижче:
![]() ,
,
,
,
(де * означає положення зв'язування з частиною "імідазолідин-2,4-діон").
6. Сполука або її фармакологічно прийнятна сіль за п. 1, де сполука вибрана з групи, що складається з:
1-(4-(2-((2-(4-фторо-3-(трифторометокси)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону;
1-(4-(2-((2-(3-бромофеніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону;
1-(4-(2-((2-(4-фторо-3-(трифторометил)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону;
1-(4-(2-((2-(3-фторо-4-(трифторометокси)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону;
1-(4-(2-((2-(2,2-дифторобензо[d][1,3]діоксол-5-іл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону;
1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діону;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діону;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діону;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-8-метил-1,3,8-триазаспіро[4.5]декан-2,4-діону;
5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-2-окса-5,7-діазаспіро[3.4]октан-6,8-діону і
4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-4,6-діазаспіро[2.4]гептан-5,7-діону.
7. Сполука або її фармакологічно прийнятна сіль за п. 1, де сполука являє собою 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон.
8. Сполука або її фармакологічно прийнятна сіль за п. 1, де сполука являє собою 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон.
9. Сполука або її фармакологічно прийнятна сіль за п. 1, де сполука являє собою 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діон.
10. Фармацевтична композиція, яка містить сполуку або її фармакологічно прийнятну сіль за будь-яким з пп. 1-9 як активний інгредієнт.
11. Фармацевтична композиція за п. 10, яка є композицією для перорального використання.
12. Фармацевтична композиція для активації внутрішньоклітинної цАМФ-реакції, яка містить сполуку або її фармакологічно прийнятну сіль за будь-яким з пп. 1-9 як активний інгредієнт.
13. Агент активації стовбурових клітин або агент для профілактики або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу, який містить сполуку або її фармакологічно прийнятну сіль за будь-яким з пп. 1-9 як активний інгредієнт.
14. Спосіб профілактики або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу або спосіб активації стовбурових клітин, де спосіб включає введення фармацевтично ефективної кількості композиції, яка включає сполуку або її фармакологічно прийнятну сіль за будь-яким з пп. 1-9, пацієнтові, який потребує профілактики або лікування хвороби або активації стовбурових клітин.
15. Застосування сполуки або її фармакологічно прийнятної солі за будь-яким з пп. 1-9 для виробництва агента активації стовбурових клітин або агента для профілактики або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу.
16. Сполука або її фармакологічно прийнятна сіль за будь-яким з пп. 1-9 для профілактики або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу або для активації стовбурових клітин.
Текст
Реферат: Цим винаходом запропоновані сполуки, представлені наведеною нижче формулою (1), і їх фармакологічно прийнятні солі: R3 O O HN R1 N N R4 N NH S O O O R2 де R1, R2, R3 і R4 є такими, як визначено у формулі винаходу. , (1) UA 115072 C2 (12) UA 115072 C2 UA 115072 C2 5 10 15 20 25 30 35 40 45 50 Галузь техніки Цей винахід стосується фармацевтичних препаратів, які містять похідне гідантоїну як активний інгредієнт, що має високу стійкість до інактивації в процесі метаболізму і виявляє сильний ПТГ-подібний ефект. Попередній рівень техніки Паратиреоїдний гормон (ПТГ) зв'язується з рецептором ПТГ1 (РТН1R), який є рецептором, зв'язаним з G-білком, (GPCR), щоб активізувати G-білок, а потім він викликає активацію принаймні одного сигнального каскаду, такого як каскад циклічного аденозинмонофосфату (цАМФ) / протеїнкінази А. Відомо, що ПТГ є гормоном, який діє на клітини-мішені у нирці та кістках, регулюючи гомеостаз кальцію (Са) та фосфору (Рі) (непатентний документ 1). Рівень концентрації Са у сироватці підтримується завдяки ПТГ здебільшого через пряму та непряму дії на шлунково-кишковий тракт, кістки та нирку. ПТГ сприяє резорбції Са з ниркових канальців та тим самим пригнічує екскрецію Са з організму назовні. Він також підсилює синтез ферменту, який перетворює вітамін D в активний вітамін D у нирці, та тим самим сприяє полегшенню опосередкованій активним вітаміном D абсорбції Са з кишково-шлункового тракту. Крім того, ПТГ підсилює диференціацію остеокластів, опосередковану остеобластами, та сприяє вивільненню Са з кісток. Вважають, що ці дії ПТГ виникають здебільшого внаслідок підвищення рівня циклічного аденозин 3',5'-монофосфату (цАМФ) та/або активації фосфоліпази С (PLC), які відбуваються, коли ПТГ зв'язується з рецептором ПТГ1 (PTH1R). У людей препарати ПТГ [PTH (1-34) та PTH (1-84)] мають сильний остеогенний ефект та викликають суттєве підвищення мінеральної щільності кісток (BMD) та міцності кісток. У наш час більшість ліків для лікування остеопорозу, які є доступними для людей, є інгібіторами резорбції кісток, та єдиним типом остеогенних ліків, які активно підвищують мінеральну щільність кісток, є препарати ПТГ. Вважають, що препарат ПТГ є одним з найефективніших засобів лікування остеопорозу (непатентний документ 2); проте, оскільки він є пептидом, його слід вводити інвазивним способом. Отже, сподіваються на виробництво фармакологічного агента, який має ПТГ-подібні ефекти та який можна вводити неінвазивним способом. Гіпопаратиреоз – це метаболічна хвороба, яка демонструє гіпокальцемію та гіперфосфатемію, спричинені недостатністю ПТГ, який виділяється з паращитовидної залози, та ряд пов'язаних симптомів. Препарати активного вітаміну D та агенти Са застосовуються для лікування гіпопаратиреозу; проте, оскільки ПТГ-опосередкований регуляторний механізм не працює, достатній терапевтичний ефект не досягається. Крім того, оскільки фармацевтичні склади з активним вітаміном D підсилюють виведення кальцію з сечею, довготривале лікування може припускати підвищений ризик нефропатії. Для того, щоб розв'язати ці проблеми, здійснюється дослідження замісної терапії, яка застосовує препарати ПТГ проти цієї хвороби; та було зроблено спробу здійснити декілька інвазивних введень на добу або безперервне введення із застосуванням помпи, щоб досягти достатньої ефективності (непатентний документ 3). Отже, для лікування гіпопаратиреозу бажано отримати покоління фармакологічних агентів, які мають ПТГ-подібні ефекти та які можна також вводити неінвазивним способом. Крім того, фармацевтичний агент, який має ПТГ-подібні ефекти, який також можна вводити неінвазивним способом, є бажаним для лікування хвороб, таких як перелом кісток, адинамічна хвороба кістки, ахондроплазія, гіпохондроплазія, розм'якшення кісток, остеоартрит, артрит, тромбоцитопенія, гіперфосфатемія та пухлинний кальциноз. На тлі цих обставин автори цього винаходу подали патентну заявку заздалегідь на підставі їх відкриття, що сполука, представлена формулою (А): [на патентний документ 1 можна посилатися для W, X, Y, m, n, R 1, R2, R33 та R34 у цій формулі] та її фармакологічно прийнятні солі є корисними як сполуки, що мають ПТГ-подібні ефекти, або більш переважно, як агоністи рецептора ПТГ1, та є корисними для профілактики та/або лікування остеопорозу, перелому кісток, розм'якшення кісток, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу або для активації стовбурових клітин (патентний документ 1). Для того, щоб отримати фармацевтичні агенти, які мають високу клінічну цінність та які можна вводити інвазивно, необхідно враховувати кінетичні показники in vivo, такі як абсорбція, 1 UA 115072 C2 5 10 15 20 25 30 35 40 45 50 розповсюдження, метаболізм та екскреція ліків, окрім їх прямої дії на мішень. Для того, щоб можна було здійснювати пероральне введення, особливо бажано мати фармацевтичний агент, який має ПТГ-подібні ефекти, такі як висока стійкість до інактивації в процесі метаболізму проти печінкових мікросом людини та сильну опосередковану рецептором ПТГ1 людини спроможність виробляти АМФ. Для того, щоб отримати фармацевтичний агент, який можна вводити людям перорально, зазвичай у способі підтвердження ефектів перорального введення за допомогою випробувань in vivo застосовується тваринна модель. Наприклад, щур з видаленою паращитовидною залозою (ТРТХ) відомий як тваринна модель гіпопаратиреозу. Для того, щоб отримати терапевтичний агент, який має сильні ПТГ-подібні ефекти та високу стійкість до інактивації в процесі метаболізму та який діє проти гіпопаратиреозу, коли його вводять перорально, ефективно застосовувати спосіб отримання сполуки, яка діє на рецептор ПТГ1 щурів та яка є стійкою до метаболічних ферментів щура, з наступним дослідженням її дії, коли її вводять перорально щурячій моделі ТРТХ (з видаленою паращитовидною залозою). У сучасній терапії гіпопаратиреозу терапевтичний цільовий діапазон концентрації Са у сироватці відповідає незначно нижчому діапазону, ніж нижча границя нормального діапазону від 7,6 до 8,8 мг/дл (непатентний документ 4). Оскільки нормальний діапазон концентрації Са у сироватці щурів є таким самим рівнем, що і для людини, 10 мг/дл, або йому подібним, то для того, щоб перевірити терапевтичний ефект, важливо досягти концентрації Са у сироватці у щурячій моделі хвороби у межах діапазону від терапевтичного цільового діапазону у людей (7,6 – 8,8 мг/дл) до нижчої границі при гіперкальцемії у людей (приблизно 11,2 мг/дл). Документи попереднього рівня техніки Патентні документи [Патентний документ 1] WO 2010/126030. Непатентні документи [Непатентний документ 1] Kronenberg, H.M., et al., In Handbook of Experimental Pharmacology, Mundy, G.R., and Martin, T.J., (eds), pp.185-201, Springer-Verlag, Heidelberg (1993). [Непатентний документ 2] Tashjian and Gagel, J. Bone Miner. Res. 21:354-365 (2006). [Непатентний документ 3] Rejnmark et al., Osteoporosis Int. Published Online: 27 November 2012. [Непатентний документ 4] Winer KK et al., J. Clin.Endocrinol.Metab. 88(9):4214-4220(2003). Суть винаходу Задачі, які слід розв'язати за допомогою винаходу Метою цього винаходу є відкриття сполук з сильними ПТГ-подібними ефектами та високою стійкістю до інактивації у процесі метаболізму та запропонувати фармацевтичні композиції, які містять такі сполуки, спроможні здійснювати лікування станів, які можуть лікуватися ПТГподібними діями, таких як гіпопаратиреоз. Засоби для розв'язання цих задач За таких обставин автори цього винаходу продовжували здійснювати дослідження та визначили, що нещодавно відкриті похідні гідантоїну цього винаходу демонструють сильну спроможність виробляти цАМФ у клітинах, які експресують рецептор ПТГ1 людини, та мають високу стійкість у мікросомах печінки людини. Автори цього винаходу також визначили, що сполуки цього винаходу демонструють сильну спроможність виробляти цАМФ у клітинах, що експресують рецептор ПТГ1 щура, та мають високу стійкість у гепатоцитах щурів. Крім того, на щурячих моделях ТРТХ, яких піддали пероральному введенню сполук, було нещодавно визначено, що доза у 30 мг/кг відновила концентрацію Са у сироватці до терапевтичного цільового діапазону 7,6 – 8,8 мг/дл. Результати, отримані на цих тваринних моделях, дозволяють припустити, що представлені формулою (1) сполуки, які демонструють сильну дію на рецептор ПТГ1 людини та високу стійкість у мікросомах печінки людини, є корисними як терапевтичні агенти для лікування гіпопаратиреозу. Цей винахід стосується наступного. [1] Сполуки, представлені наведеною нижче загальною формулою (1), або їх фармакологічно прийнятні солі: 2 UA 115072 C2 5 10 15 20 25 30 35 де, коли R1 і R2 не є обидва атомами водню, R1 і R2 являють собою незалежно: 1) атом водню; 2) атом галогену; 3) алкільну групу, яка містить один або два атоми вуглецю, яка може бути заміщеною від одного до п'яти атомами фтору; або 4) алкоксигрупу, яка містить один або два атоми вуглецю, яка може бути заміщеною від одного до п'яти атомами фтору; або R1 і R2 зв'язані один з одним, утворюючи групу, представлену наведеною нижче формулою: (де кожний символ * означає положення зв'язування з фенільною частиною); і R3 і R4 являють собою незалежно метильну групу, що може бути заміщеною від одного до трьох атомами фтору; або R3 і R4 разом із зв'язаним атомом вуглецю утворюють три- – шестичленне карбоциклічне кільце (де один з атомів вуглецю, що утворює кільце, може бути замінений атомом кисню, атомом сірки або метил-заміщеним або незаміщеним атомом азоту). В цьому винаході сполука, в якій комбінація R1 і R2 являє собою трифторометильну групу і атом водню, і де R3 і R4, разом із зв'язаним атомом атом вуглецю, утворюють циклопентильне кільце, піддається виключенню з вищезгаданих сполук, представлених формулою (1). [2] Сполука або її фармакологічно прийнятна сіль за [1], де R1 і R2 вибрані з наведених нижче комбінацій: 1) R1 являє собою атом водню або атом галогену, і R2 являє собою атом водню, трифторометильну групу або трифторометокси-групу (за умови, що R1 і R2 не є обидва атомами водню); 2) R1 являє собою трифторометильну групу або трифторометокси-групу, і R2 являє собою атом водню або атом галогену; 3) R1 і R2 зв'язані один з одним, утворюючи групу, представлену наведеною нижче формулою: (де кожний символ * означає положення зв'язування з фенільною частиною); і R3 і R4 являють собою метильні групи; або R3 і R4 разом із зв'язаним атомом вуглецю утворюють кільце, вибране з наведених нижче: (де * означає положення зв'язування з частиною "імідазолідин-2,4-діон"). [3] Сполука або її фармакологічно прийнятна сіль за [1], де R1 і R2 вибрані з наведених нижче комбінацій: 1) R1 являє собою трифторометокси-групу, і R2 являє собою атом фтору; 3 UA 115072 C2 5 10 15 20 25 30 35 40 2) R1 являє собою атом брому, і R2 являє собою атом водню; 3) R1 являє собою трифторометокси-групу, і R2 являє собою атом фтору; 4) R1 являє собою атом фтору, і R2 являє собою трифторометокси-групу; 5) R1 являє собою трифторометильну групу, і R2 являє собою атом водню; 6) R1 являє собою атом водню, і R2 являє собою трифторометокси-групу; 7) R1 і R2 зв'язані один з одним, утворюючи групу, представлену наведеною нижче формулою: (де кожний символ * означає положення зв'язування з фенільною частиною); і R3 і R4 являють собою метильні групи; або R3 і R4 разом із зв'язаним атомом вуглецю утворюють кільце, вибране з наведених нижче: (де * означає положення зв'язування з частиною "імідазолідин-2,4-діон"). [4] Сполука або її фармакологічно прийнятна сіль за [1], де R3 і R4 являють собою метильні групи. [5] Сполука або її фармакологічно прийнятна сіль за [1], де R3 і R4 разом із зв'язаним атомом вуглецю утворюють кільце, вибране з наведених нижче: (де * означає положення зв'язування з частиною "імідазолідин-2,4-діон"). [6] Сполука або її фармакологічно прийнятна сіль за [1], де сполука вибрана з групи, що складається з: 1-(4-(2-((2-(4-фторо-3-(трифторометокси)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону; 1-(4-(2-((2-(3-бромофеніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)-3,5диметилфеніл)-5,5-диметилімідазолідин-2,4-діону; 1-(4-(2-((2-(4-фторо-3-(трифторометил)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону; 1-(4-(2-((2-(3-фторо-4-(трифторометокси)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону; 1-(4-(2-((2-(2,2-дифторобензо[d][1,3]діоксол-5-іл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону; 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діону; 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діону; 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діону; 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-8-метил-1,3,8-триазаспіро[4.5]декан-2,4-діону; 5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-2-окса-5,7-діазаспіро[3.4]октан-6,8-діону і 4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-4,6-діазаспіро[2.4]гептан-5,7-діону. [7] Сполука або її фармакологічно прийнятна сіль за [1], де сполука являє собою 1-(3,5 4 UA 115072 C2 5 10 15 20 25 30 35 40 45 50 55 60 диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон. [8] Сполука або її фармакологічно прийнятна сіль за [1], де сполука являє собою 1-(3,5диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон. [9] Сполука або її фармакологічно прийнятна сіль за [1], де сполука являє собою 1-(3,5диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діон. [10] Фармацевтична композиція, яка містить сполуку або її фармакологічно прийнятну сіль за будь-яким одним пунктом від [1] до [9] як активний інгредієнт. [11] Фармацевтична композиція за [10], яка є композицією для перорального використання. [12] Фармацевтична композиція для активації внутрішньоклітинної цАМФ-реакції, яка містить сполуку або її фармакологічно прийнятну сіль за будь-яким одним пунктом від [1] до [9] як активний інгредієнт. [13] Агент активації стовбурових клітин або агент для профілактики або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу, який містить сполуку або її фармакологічно прийнятну сіль за будьяким одним пунктом від [1] до [9] як активний інгредієнт. [14] Спосіб профілактики або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу або спосіб активації стовбурових клітин, де спосіб включає введення фармацевтично ефективної кількості композиції, яка містить сполуку або її фармакологічно прийнятну сіль за будь-яким одним пунктом від [1] до [9], пацієнтові, який потребує профілактики або лікування цієї хвороби або активації стовбурових клітин. [15] Застосування сполуки або її фармакологічно прийнятної солі за будь-яким одним пунктом від [1] до [9] для виробництва агента активації стовбурових клітин або агента для профілактики або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу. [16] Сполука або її фармакологічно прийнятна сіль за будь-яким одним пунктом від [1] до [9] для профілактики або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу або для активації стовбурових клітин. Крім того, цим винаходом пропонуються способи лікування патологічних станів, які можна лікувати ПТГ-подібними діями, таких як гіпопаратиреоз, шляхом введення сполуки за формулою (1) або її солі. Ефекти винаходу Цим винаходом пропонуються похідні гідантоїну із сильними ПТГ-подібними ефектами та високою стійкістю до інактивації у процесі метаболізму. Застосування похідних гідантоїну дозволяє лікувати патологічні стани, спричинені ПТГ-подібними діями, такі як гіпопаратиреоз. Стислий опис ілюстративного матеріалу Фігура 1 демонструє графік, на якому зображено середню зміну рівня концентрації Ca в сироватці для кожної сполуки за період аж у 24 години після введення, коли сполуку вводять перорально щурячій моделі з видаленою паращитовидною залозою (ТРТХ) в дозі 30 мг/кг. Варіант здійснення винаходу Цей винахід стосується похідних гідантоїну та їх застосування. Автори цього винаходу вперше синтезували сполуки, представлені вище формулою (1), або їх фармакологічно прийнятні солі та визначили, що сполука або її сіль – це сполука, яка має сильний ефект, подібний до ефекту паратиреоїдного гормону (ПТГ), та високу стійкість до інактивації у процесі метаболізму. Термін "алкіл" у цьому описі винаходу означає одновалентну групу, отриману внаслідок видалення одного атома водню з аліфатичного вуглеводню, та охоплює підгрупу структур гідрокарбілових груп або вуглеводневих груп, які не містять гетероатом або ненасичений вуглець-вуглецевий зв'язок, а містять атоми водню та вуглецю у каркасі. Приклади алкілових груп включають приклади з лінійною та розгалуженою структурами. Алкільна група – це переважно алкільна група, яка включає один або більше атомів вуглецю. Алкільна група – це специфічно, наприклад, метильна група або етильна група, і переважно метильна група. 5 UA 115072 C2 5 10 15 20 25 30 35 40 45 50 55 60 Термін "алкокси", як застосовується у цьому описі винаходу, означає окси-групу, до якої приєднаний визначений вище "алкіл", та переважно означає алкокси-групу, яка включає один або два атоми вуглецю. Специфічні приклади включають метокси-групу та етокси-групу, та переважний приклад – це метокси-група. Вираз "В, необов'язково заміщений А", як застосовується у цьому описі винаходу, вказує на те, що будь-який атом (атоми) водню у В можуть бути заміщеними будь-якою кількістю А. У цьому винаході кількість замісників не обмежується, якщо інше не вказано. Наприклад, кількість замісників може становити від 1 до 5, від 1 до 4, від 1 до 3, від 1 до 2 або 1. "Атом галогену" у цьому описі винаходу означає атом фтору, атом хлору, атом брому або атом йоду. У цьому описі винаходу символ "*" у хімічній формулі позначає положення зв'язку. Сполуки цього винаходу, представлені формулою (1), мають сильні ПТГ-подібні ефекти та високу стійкість до інактивації в процесі метаболізму. Вираз "ПТГ-подібний ефект" у цьому описі винаходу означає активність генерувати внутрішньоклітинний цАМФ (цАМФ - це циклічний аденозинмонофосфат) внаслідок дії на рецептор ПТГ або на шлях сигнальної трансдукції через рецептор ПТГ. У цьому винаході, чи існує "сильний ПТГ-подібний ефект" або чи є "ПТГ-подібний ефект сильним" можна підтвердити шляхом вимірювання цАМФ-сигнальної активності, аналізуючи активність цАМФ-сигнального шляху, наприклад, згідно зі способом, описаним у J. Bone. Miner. Res. 14:11-20, 1999. Специфічно, наприклад, згідно зі способом, описаним у "Тестовому прикладі 1", кількість цАМФ, виробленого у клітинах, примушених експресувати рецептор ПТГ1 людини, визначається із застосуванням комерційно доступного набору cAMP EIA (наприклад, BiotrackcAMP EIA system, GE health care), щоб вимірити концентрацію кожної сполуки при цАМФ-сигнальній активності у 20 % (ЕС20) або їх концентрацію при цАМФ-сигнальній активності у 50 % (ЕС50), при цьому цАМФ-сигнальна активність, отримана після введення 100 нМ PTH (134) людини, визначається як 100 %. У цьому винаході для "сильного ПТГ-подібного ефекту" або для того, щоб "ПТГ-подібний ефект був сильним", наприклад, значення ЕС20 (мкМ), виміряне за вищезгаданим способом, становить переважно 5,0 або менше, більш переважно 3,0 або менше та навіть більш переважно 2,0 або менше. Для ЕС50 значення (мкМ), виміряне за вищезгаданим способом, становить, наприклад, переважно 25,0 або менше, більш переважно 15,0 або менше та навіть більш переважно 10,0 або менше. Чи існує "висока стійкість до інактивації в процесі метаболізму" або чи "стійкість до інактивації в процесі метаболізму є високою" можна підтвердити, застосовуючи загальний спосіб вимірювання. Наприклад, клітини печінки, клітини тонкого кишечника, мікросоми печінки, мікросоми тонкого кишечника, S9 печінки та їм подібні можна застосовувати для такого підтвердження. Специфічно, наприклад, стійкість сполуки у мікросомах печінки можна підтвердити, зробивши вимірювання згідно з описом у роботі T. Kronbach et al. (Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol. Pharmacol, 1989, 36(1), 89-96). Більш специфічно, стійкість можна підтвердити згідно зі способом, описаним у "Тестовому прикладі 3". У цьому винаході "висока стійкість до інактивації в процесі метаболізму" існує або "стійкість до інактивації в процесі метаболізму є високою", коли значення кліренсу (мкл/хвилину/мг) у тесті стійкості до інактивації у процесі метаболізму, при якому застосовуються мікросоми печінки людини, та який описано у вищезгаданому тестовому прикладі, становить переважно 60 або менше, більш переважно 40 або менше та навіть більш переважно 35 або менше. Специфічно, високу стійкість до інактивації в процесі метаболізму можна отримати щодо сполук за вищезгаданою формулою (1), за виключенням, коли комбінація R1 та R2 – це трифторметильна група та атом водню, а R3 та R4 разом зі зв'язаним атомом вуглецю утворюють циклопентильне кільце. Сполуки згідно з цим винаходом, або вільні форми, або фармакологічно прийнятні солі, включено до цього винаходу. Приклади таких "солей" включають солі неорганічних кислот, солі органічних кислот, солі неорганічних лугів, солі органічних основ та солі кислотних або лужних амінокислот. Переважні приклади солей неорганічних кислот включають гідрохлориди, гідроброміди, сульфати, нітрати та фосфати. Переважні приклади солей органічних кислот включають ацетати, сукцинати, фумарати, малеати, тартрати, цитрати, лактати, стеарати, бензоати, метансульфонати, бензолсульфонати та р-толуолсульфонати. Переважні приклади солей неорганічних лугів включають солі лужних металів, такі як солі натрію та солі калію, солі лужноземельних металів, такі як солі кальцію та солі магнію, солі алюмінію та солі амонію. Переважні приклади солей органічних основ включають солі діетиламіну, діетаноламіну, солі меглуміну та солі N, N-дибензилетилендіаміну. 6 UA 115072 C2 5 10 15 20 25 30 35 40 45 50 Переважні приклади солей кислотних амінокислот включають аспартати та глутамати. Переважні приклади солей основних амінокислот включають солі аргініну, солі лізину та солі орнітину. Сполуки цього винаходу можуть абсорбувати вологу, мати абсорбовану воду або утворювати гідрати, коли залишаються на повітрі. Такі гідрати також включено до солей цього винаходу. Крім того, сполуки цього винаходу можуть абсорбувати певні інші розчинники, утворюючи сольвати. Такі солі також охоплюються у цьому винаході як солі сполук за формулою (1). У цьому описі винаходу структурна формула сполуки може представляти певний ізомер заради зручності. Проте, сполуки цього винаходу включають усі ізомери, такі як геометричні ізомери, оптичні ізомери на основі асиметричних вуглеців, стереоізомери та таутомери, а також суміші цих ізомерів, які виникають внаслідок структур сполук, при цьому вони не обмежуються формулами, описаними заради зручності, та можуть бути або одним із ізомерів, або їх сумішшю. Отже, сполуки цього винаходу можуть мати асиметричний атом вуглецю у молекулі та можуть бути присутніми як оптично активні форми та рацемати, проте цей винахід не обмежується будь-яким з них та включає обидва з них. Цей винахід включає усі ізотопи сполук, представлених формулою (1). В ізотопах сполук цього винаходу принаймні один атом замінений атомом, який має такий самий атомний номер (протонне число), але має відмінне масове число (сума кількості протонів та кількості нейтронів). Приклади ізотопів, які містяться у сполуках цього винаходу, включають атом водню, атом вуглецю, атом азоту, атом кисню, атом фосфору, атом сірки, атом фтору та атом хлору, 2 3 13 14 15 17 18 31 32 35 18 36 включаючи H, H, C, C, N, O, O, P, P, S, F та Cl, відповідно. Зокрема, 3 14 радіоізотопи, які розпадаються, випромінюючи радіоактивність, такі як H та C, є корисними у тестах розповсюдження у тканинах організму для фармацевтичних агентів або сполук. Стійкі ізотопи не розпадаються, є майже однаковими у великій кількості та не випромінюють радіоактивність, та, отже, вони є безпечними для застосування. Ізотопи сполук цього винаходу можна перетворити згідно з традиційними способами шляхом заміни реагентом, який містить відповідний ізотоп, реагенту, який застосовується для синтезу. Сполуки згідно з цим винаходом можуть демонструвати кристалічний поліморфізм, але вони особливо не обмежуються будь-якою поліморфною формою, проте вони можуть бути у будьякій одній з цих кристалічних форм або можуть існувати як суміш двох або більше кристалічних форм. Сполуки згідно з цим винаходом включають їх проліки. Проліки – це похідні сполук цього винаходу, які мають хімічно або метаболічно розщеплюванні групи та які перетворюються назад до оригінальних сполук після введення in vivo, демонструючи їхню первинну ефективність, включаючи комплекси, які не утворюються ковалентними зв'язками, та солі. Сполуки, представлені наведеною вище формулою (1), відповідно до цього винаходу, є переважно наступними. У формулі R1 і R2 вибрані з наведених нижче комбінацій: 1) R1 являє собою атом водню або атом галогену, і R2 являє собою атом водню, трифторометильну групу або трифторометокси-групу (за умови, що R1 і R2 не є обидва атомами водню); 2) R1 являє собою трифторометильну групу або трифторометокси-групу, і R2 являє собою атом водню або атом галогену; 3) R1 і R2 зв'язані один з одним, утворюючи групу, представлену наведеною нижче формулою: (де кожний символ * означає положення зв'язування з фенільною частиною); і R3 і R4 являють собою метильні групи; або R3 і R4 разом із зв'язаним атомом вуглецю утворюють кільце, вибране з наведених нижче: 7 UA 115072 C2 5 10 15 20 25 30 35 40 45 50 (де * означає положення зв'язування з частиною "імідазолідин-2,4-діон"). Сполуки, представлені наведеною вище формулою (1), відповідно до цього винаходу, є більш переважно наступними. В формулі R1 і R2 вибрані з наведених нижче комбінацій: 1) R1 являє собою трифторометокси-групу, і R2 являє собою атом фтору; 2) R1 являє собою атом брому, і R2 являє собою атом водню; 3) R1 являє собою трифторометокси-групу, і R2 являє собою атом фтору; 4) R1 являє собою атом фтору, і R2 являє собою трифторометокси-групу; 5) R1 являє собою трифторометильну групу, і R2 являє собою атом водню; 6) R1 являє собою атом водню, і R2 являє собою трифторометокси-групу; 7) R1 і R2 зв'язані один з одним, утворюючи групу, представлену наведеною нижче формулою: (де кожний символ * означає положення зв'язування з фенільною частиною); і R3 і R4 являють собою метильні групи; або R3 і R4 разом із зв'язаним атомом вуглецю утворюють кільце, вибране з наведених нижче: (де * означає положення зв'язування з частиною "імідазолідин-2,4-діон"). Сполуки, представлені вищенаведеною формулою (1), відповідно до цього винаходу, являють собою переважно також сполуку, вибрану з групи, що складається з наступних сполук, або її фармакологічно прийнятні солі. Сполука 1: 1-(4-(2-((2-(4-фторо-3-(трифторометокси)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діон; Сполука 2: 1-(4-(2-((2-(3-бромофеніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)-3,5диметилфеніл)-5,5-диметилімідазолідин-2,4-діон; Сполука 3: 1-(4-(2-((2-(4-фторо-3-(трифторометил)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діон; Сполука 4: 1-(4-(2-((2-(3-фторо-4-(трифторометокси)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діон; Сполука 5: 1-(4-(2-((2-(2,2-дифторобензо[d][1,3]діоксол-5-іл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діон; Сполука 6: 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон; Сполука 7: 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон; Сполука 8: 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діон; Сполука 9: 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-8-метил-1,3,8-триазаспіро[4.5]декан-2,4-діон; Сполука 10: 5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8 8 UA 115072 C2 5 10 15 20 25 30 35 40 45 50 55 60 іл)сульфоніл)етил)феніл)-2-окса-5,7-діазаспіро[3.4]октан-6,8-діон; і Сполука 11: 4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-4,6-діазаспіро[2.4]гептан-5,7-діон. Із вказаних вище сполук від 1 до 11 сполуки 6, 7 і 8 є більш переважними. Такі сполуки, представлені вищезгаданою формулою (1), або їх фармакологічно прийнятні солі згідно з цим винаходом є корисними як сполуки, що мають ПТГ-подібний ефект, переважно як агоністи рецептора ПТГ1, та є корисними для профілактики та/або лікування остеопорозу, перелому кісток, адинамічної хвороби кістки, ахондроплазії, гіпохондроплазії, розм'якшення кісток, остеоартриту, артриту, тромбоцитопенії, гіпопаратиреозу, гіперфосфатемії або пухлинного кальцинозу або їм подібного, або для активації стовбурових клітин. Зі сполуками або їх солями згідно з цим винаходом можна виготовити фармацевтичні склади за традиційними способами у формі таблеток, порошків, дрібних гранул, гранул, таблеток з покриттям, капсул, сиропів, пастилок, інгаляцій, супозиторіїв, ін'єкцій, мазей, офтальмологічних мазей, офтальмологічних препаратів, препаратів для носа, препаратів для вуха, припарок, лосьйонів та їм подібного. Ексципієнти, зв'язувальні агенти, змащувальні агенти, барвники, коригенти та, якщо необхідно, стабілізатори, емульгатори, агенти, що сприяють абсорбції, поверхнево-активні речовини, регулятори рН, консерванти, антиоксиданти та їм подібні, які зазвичай застосовуються, можна застосовувати для виготовлення фармакологічного складу, та вони змішуються з інгредієнтами, які зазвичай застосовуються як сирі матеріали фармацевтичних препаратів та виготовляються за традиційними способами. Наприклад, пероральні препарати виробляються шляхом додавання до сполуки або її фармакологічно прийнятної солі згідно з цим винаходом ексципієнту та, якщо необхідно, зв'язувального агента, розпушувача, змащувального агента, барвника, коригента та їм подібного, з наступним виготовленням їх у вигляді порошку, дрібних гранул, гранул, таблеток, таблеток з покриттям, капсул та їм подібного за допомогою традиційного способу. Приклади цих інгредієнтів включають тваринні масла та рослинні олії, такі як соєва олія, яловиче сало та синтетичний гліцерид; вуглеводні, такі як рідкий парафін, сквалан та твердий парафін; ефірні масла, такі як октилдодецилміристат та ізопропілміристат; вищі спирти, такі як цетостеариловий спирт та бегеніловий спирт; силіконову смолу; силіконове масло; поверхневоактивні речовини, такі як поліоксиетиленовий естер жирної кислоти, естер сорбіту та жирної кислоти, естер гліцерину та жирної кислоти, поліоксиетиленовий естер сорбіту та жирної кислоти, поліоксиетиленоване гідрогенізоване касторове масло та блок-сополімер поліоксиетилену-поліоксипропілену; розчинні у воді полімери, такі як гідроксиетилцелюлоза, поліакрилова кислота, карбоксивініловий полімер, поліетиленгліколь, полівінілпіролідон та метилцелюлоза; нижчі спирти, такі як етанол та ізопропанол; багатоатомні спирти, такі як гліцерин, пропіленгліколь, дипропіленгліколь та сорбіт; цукри, такі як глюкоза та цукроза; неорганічні порошки, такі як кремнієвий ангідрид, алюмосилікат магнію та алюмосилікат; та очищену воду. Приклади ексципієнтів включають лактозу, кукурудзяний крохмаль, білий м'який цукор, глюкозу, маніт, сорбіт, мікрокристалічну целюлозу та діоксид кремнію. Приклади зв'язувальних агентів включають полівініловий спирт, полівініловий етер, метилцелюлозу, етилцелюлозу, аравійську камедь, трагакант, желатин, шелак, гідроксипропілметилцелюлозу, гідроксипропілцелюлозу, полівінілпіролідон, блок-полімер поліпропіленгліколю-поліоксиетилену та меглумін. Приклади розпушувачів включають крохмаль, агар, желатиновий порошок, мікрокристалічну целюлозу, кальцію карбонат, натрію бікарбонат, кальцію цитрат, декстрин, пектин та кальційкарбоксиметилцелюлозу. Приклади змащувальних агентів включають магнію стеарат, тальк, поліетиленгліколь, двоокис кремнію та гідрогенізовану рослинну олію. Барвники, що застосовуються, - це ті барвники, які ухвалено як домішки до фармацевтичних препаратів. Коригенти, що застосовуються, - це порошок какао, ментол, емпазм, м'ятна олія, борнеол, порошок кориці та їм подібне. Очевидно, що ці таблетки та гранули можуть мати покриття з цукру або можуть мати інше покриття, якщо необхідно. Рідкі препарати, такі як сиропи та препарати для ін'єкцій, виробляються шляхом додавання регулятора рН, солюбілізатора, регулютра тонічності та їм подібного, та, якщо необхідно, солюбілізуючого агента, стабілізатора та їм подібного, до сполуки або її фармакологічно прийнятної солі згідно з цим винаходом, та виготовлення з них фармацевтичних складів згідно з традиційним способом. Спосіб виробництва препаратів для зовнішнього застосування не обмежується, та їх можна 9 UA 115072 C2 5 10 15 20 25 30 35 40 45 50 55 60 виробляти за традиційними способами. Специфічно, різні матеріали-сировину, які зазвичай застосовуються для фармацевтичних препаратів, квазі-ліків (препаратів загального впливу), косметики та їм подібного, можна застосовувати як матеріал основи для фармакологічного складу. Специфічні приклади матеріалів основи, що застосовуються, включають матеріалисировину, такі як тваринні масла та рослинні олії, мінеральні оливи, ефірні масла, віск, вищі спирти, жирні кислоти, силіконове масло, поверхнево-активні речовини, фосфоліпіди, спирти, багатоатомні спирти, водорозчинні полімери, глинисті мінерали та очищену воду. Крім цього, якщо необхідно, можна додати регулятори рН, антиоксиданти, хелатори, консерванти та фунгіциди, барвники, ароматизатори та їм подібне. Матеріали основи для препаратів для зовнішнього застосування згідно з цим винаходом не обмежуються цими матеріалами. Якщо необхідно, можна також примішувати інгредієнти, такі як інгредієнти, які мають ефект індукування диференціювання, промотори кровотоку, бактерициди, протизапальні агенти, активатори клітин, вітаміни, амінокислоти, зволожувачі, кератолітичні агенти. Вищезгадані матеріали основи додаються у кількості, що відповідає концентрації, яка зазвичай вибирається для виробництва препаратів для зовнішнього застосування. Спосіб введення сполук або їх солей, або гідратів сполук або солей згідно з цим винаходом особливо не обмежується, та їх можна вводити перорально або парентерально згідно зі способами, що зазвичай застосовуються. Наприклад, їх можна виготовити у вигляді препаратів, таких як таблетки, порошки, гранули, капсули, сиропи, пастилки, препарати для інгаляції, супозиторії, ін'єкції, мазі, офтальмологічні мазі, офтальмологічні препарати, препарати для носа, препарати для вуха, припарки та лосьйони, та вводити. Сполуки цього винаходу є особливо придатними для виготовлення у вигляді пероральних агентів, оскільки вони демонструють відмінну цАМФ-сигнальну активність та мають стійкість до інактивації в процесі метаболізму. Дозу ліків згідно з цим винаходом можна відповідним чином вибрати залежно від суворості симптому, віку, статі, ваги тіла, способу введення, типу солі, специфічного типу хвороби та їм подібного. Незважаючи на те, що доза суттєво варіюється відповідно до типу хвороби та суворості симптому пацієнта, віку пацієнта, різниці у статі та різниці у чутливості до ліків серед пацієнтів та їм подібного, доза зазвичай становить приблизно від 0,03 мг до 1000 мг, переважно від 0,1 мг до 500 мг та більш переважно від 0,1 мг до 100 мг на добу для дорослих, та її вводять один раз на добу або розподіляють на декілька введень на добу. При виробництві сполук цього винаходу, представлених вищезгаданою формулою (1), сполуки матеріалу-сировини та різні реагенти можуть утворювати солі, гідрати та сольвати, при цьому усі варіюються залежно від початкового матеріалу, розчинника, що застосовується, та їм подібного, та вони особливо не обмежуються, доки вони не інгібують реакцію. Розчинник, що застосовується, також варіюється залежно від початкового матеріалу, реагенту та їм подібного, та він особливо не обмежується, доки він не інгібує реакцію та розчинює початковий матеріал до певної міри, вочевидь. Різні ізомери (наприклад, геометричні ізомери, оптичні ізомери на основі асиметричних вуглеців, ротамери, стереоізомери та таутомери) можна очистити та виділити, застосовуючи звичайні способи розділення, наприклад, рекристалізацію, способи діастереомерних солей, способи ферментативного розділення та різні хроматографічні способи (наприклад, тонкошарову хроматографію, хроматографію на колонках, високоефективну рідинну хроматографію та газову хроматографію). Сполуки згідно з цим винаходом, отримані як вільні форми, можна перетворити у солі, які можуть утворюватися цими сполуками, або у гідрати сполук згідно з традиційними способами. Сполуки згідно з цим винаходом, отримані як солі або гідрати сполук, можна також перетворити у вільні форми сполук згідно з традиційними способами. Сполуки згідно з цим винаходом можна виділити та очистити, застосовуючи звичайні хімічні операції, такі як екстракція, концентрація, випаровування, кристалізація, фільтрація, перекристалізація та різні способи хроматографії. Усі процитовані у цьому описі винаходу документи з попереднього рівня техніки включено до цього опису винаходу шляхом посилання. Загальні способи синтезу Сполуки цього винаходу можна синтезувати за різними способами, деякі з яких будуть описані з посиланням на наступні схеми. Схеми мають ілюстративний характер, та цей винахід не обмежується тільки хімічними реакціями та умовами, на які особливо вказано. Незважаючи на те, що деякі замісники виключені у наступних схемах заради ясності, таке виключення не призначено обмежувати опис схем. Характерні сполуки цього винаходу можна синтезувати, 10 UA 115072 C2 5 10 15 20 25 30 застосовуючи відповідні проміжні речовини, відомі сполуки та реагенти. R 1, R2, R3 та R4 у формулах у наступних загальних способах синтезу є такими, як визначено для R 1, R2, R3 та R4 для сполук, представлених вищезгаданою загальною формулою (1) (сполуки представлені формулою 1 у наступних загальних способах синтезу). Сполуки цього винаходу (Формула 1) можна синтезувати за допомогою способів виробництва (Способи А та В), наведених нижче. Схема 1 (Спосіб А) Схема 1 демонструє спосіб отримання похідного гідантоїну (Формула 1) шляхом амідування карбоново-кислотного похідного (1) та аміно-амідного похідного (2) з метою отримання амідамідного похідного (3) з наступним будуванням кільця спіроімідазолону шляхом внутрішньомолекулярної циклізації. Етап 1 – це спосіб амідування карбоново-кислотного похідного (1) та аміно-амідного похідного (2). Приклади зв'язувального реагенту включають N, N'-дициклогексилкарбодіімід (DCC), 1-етил-3-(3-диметиламінопропіл)карбодііміду гідрохлорид (EDC), О-(7-азабензотриазол1-іл)-1,1,3,3-тетраметилуроній гексафторфосфат (HATU) та 4-(4,6-диметокси-1,3,5-триазин-2іл)-4-метилморфоліній хлорид n-гідрат (DMT-MM). Приклади основи включають триетиламін або N, N-діізопропілетиламін. Якщо необхідно, можна використати 4-(диметиламіно)піридин (DMAP) як каталізатор. Приклади відповідного розчинника включають дихлорометан або N, Nдиметилфорамід. Приклади відповідного розчинника реакції, коли застосовується DMT-MM, включають метанол, етанол та ацетонітрил. Температура реакції становить, наприклад, від 0 °C до кімнатної температури, та реакція триває протягом від 0,5 до 24 годин. Отримане амідамідне похідне (3) виділяють за звичайним способом та, якщо необхідно, його можна очистити шляхом перекристалізації або хроматографії. Етап 2 – це спосіб циклізації амід-амідного похідного (3) у присутності придатного лугу, такого як водний розчин гідроксиду натрію або t-бутоксид калію, у придатному розчиннику, такому як етанол, трет-бутанол або диметилсульфоксид. Реакція здійснюється, наприклад, при температурі від кімнатної температури до температури кипіння зі зворотним холодильником протягом від 1 до 24 годин. Отримане похідне гідантоїну (Формула 1) виділяють за звичайними способами, та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Аміно-амідне похідне (2), вказане на Схемі 1, можна синтезувати з похідного піперидину (4). Спосіб синтезу для аміно-амідного похідного (2) зображено у Схемі 2. Схема 2 11 UA 115072 C2 5 10 15 20 25 30 35 40 Етап 3 – це синтез за Стрекером (Strecker): перетворення похідного піперидинону (4) на похідне аміно-нітрилу (5). Специфічно, це спосіб реакції піперидинону (4) з ціанідом натрію або ціанідом калію та хлоридом амонію або ацетатом амонію у відповідному розчиннику, такому як метанол, етанол або тетрагідрофуран, у присутності / відсутності води. Температура реакції становить, наприклад, від кімнатної температури до 80 °C, та тривалість реакції становить від 2 до 72 годин. Отримане похідне аміно-нітрилу (5) виділяють за звичайним способом та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Етап 4 – це спосіб перетворення нітрильної групи на амідогрупу в умовах гідролізу в лужному середовищі у присутності пероксиду водню. Цю реакцію можна здійснювати, наприклад, з посиланням на Chemistry-A European Journal (2002), 8(2), 439-450. Етап 5 – це спосіб гідрогенізації олефінової сполуки (6) в інертному розчиннику, такому як метанол, етанол, трифторетанол, диметилформамід або диметилацетамід, у присутності каталізатора, такого як паладій на вуглецю або гідроксид паладію на вуглецю, відповідно, в атмосфері Н2. Температура реакції становить від кімнатної температури до 80 °C, та реакцію можна здійснювати під тиском. Отримане аміно-амідне похідне (2) виділяють за звичайним способом та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Похідне піперидинону (4), зображене на Схемі 2, можна синтезувати з відомого похідного кетальвінілсульфонілу (7) та похідного гідантоїнарилброміду (8). Спосіб синтезу похідного піперидину (4) зображено на Схемі 3. Схема 3 Етап 6 – це спосіб синтезу похідного кетальарилвінілсульфонілу (9) шляхом сполучання похідного кетальвінілсульфонілу (7) та похідного гідантоїнарилброміду (8) в атмосфері N 2 у присутності паладієвого каталізатора, такого як трис(дибензилідинацетон)-паладій (0) або біс(дибензилідинацетон)паладій, та шляхом додавання фосфінового ліганду, такого як три-третбутилфосфінтетрафторборна кислота, та придатної основи, такої як метилдициклогексиламін, у придатному розчиннику, такому як N-метил-2-піперидон (NMP). Температура реакції становить від 90 °C до температури кипіння зі зворотним холодильником. Отримане похідне кетальарилвінілсульфонілу (9) виділяють за звичайним способом та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Етап 7 – це спосіб перетворення кеталю похідного кетальарилвінілсульфонілу (9) на кетон у придатному розчиннику, такому як водний тетрагідрофуран, у присутності кислоти, такої як хлористоводнева кислота. Температура реакції становить, наприклад, точку кипіння розчинника, а тривалість реакції становить приблизно від 1 до 24 годин. Отримане похідне піперидину (4) виділяють за звичайними способами, та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Похідне гідантоїнарилброміду (8), зображене на Схемі 3, можна синтезувати з 4-бром-3,5диметилаланіну (10) та похідного бромоцтової кислоти (11), або з 2-бром-5-йод-1,3диметилбензолу (13) та похідного амінокислоти (14). Спосіб синтезу похідного гідантоїнарилброміду (8) зображено на Схемі 4. Схема 4 12 UA 115072 C2 5 10 15 20 25 30 35 Етап 8 – це спосіб алкілування 4-бром-3,5-диметилаланіну (10) похідним бромоцтової кислоти (11) у присутності придатної основи, такої як діізопропілетиламін, та у придатному розчиннику, такому як N-метил-2-піперидон (NMP). Температура реакції становить, наприклад, від кімнатної температури до 100 °C, а реакція триває від 1 до 24 годин. Отримане похідне арилбромідамінокислоти (12) виділяють за звичайними способами, та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Етап 9 – це спосіб синтезу похідного арилбромідамінокислоти (12) шляхом сполучання 2бром-5-йод-1,3-диметилбензолу (13) та похідного амінокислоти (14) у присутності металевого каталізатора, такого як йодид міді(І). Реакцію можна здійснювати у присутності придатної основи, такої як діазабіциклоундецен (DBU), та у придатному розчиннику, такому як N, Nдиметилацетамід (DMA), при температурі реакції приблизно від 80 °C до 120 °C. Отримане похідне арилбромідамінокислоти (12) виділяють за звичайними способами, та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Етап 10 – це спосіб синтезу похідного гідантоїнарилброміду (8) шляхом реакції похідного арилбромідамінокислоти з ціанатом натрію у кислотних умовах. Розчинник - це, наприклад, змішаний розчинник, такий як оцтова кислота-дихлорометан; температура реакції становить від кімнатної температури до 60 °C; а тривалість реакції становить від 1 до 24 годин. Отримане похідне гідантоїн-арилброміду (8) можна виділити за звичайними способами, та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Похідне гідантоїнарилброміду (8), зображене на Схемі 3, можна також синтезувати з 4-бром3,5-диметилаланіну (10) та похідного кетону (15). Спосіб синтезу похідного гідантоїнарилброміду (8) зображено на Схемі 5. Схема 5 Етап 11 – це синтез за Стрекером (Strecker), який спрямовує похідне кетону (15) на те, щоб воно стало похідним ариламінонітрилу (16). Більш специфічно, це спосіб, при якому відбувається реакція похідного кетону (15) з 4-бром-3,5-диметилаланіном (10) та триметилсилілціанідом у придатному розчиннику, такому як оцтова кислота. Температура реакції може бути кімнатною температурою, а реакція триває від 1 до 3 годин або біля цього. Отримане похідне ариламінонітрилу (16) виділяють за звичайними способами та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Етап 12 – це спосіб, при якому відбувається реакція похідного ариламінонітрилу (16) з 2,2,2трихлорацетилізоціанатом у придатному розчиннику, такому як дихлорометан, з наступним синтезуванням похідного іміногідантоїну (17) шляхом додавання реагентів, таких як метанол, вода та триетиламін, потім їх залишають реагувати в умовах нагрівання. Отримане похідне іміногідантоїну (17) виділяють за звичайними способами та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. 13 UA 115072 C2 5 10 15 20 25 30 35 Етап 13 – це спосіб перетворення похідного іміногідантоїну (17) у похідне гідантоїнарилброміду (8) у кислотних умовах. Наприклад, синтез можна здійснювати у розчиннику оцтова кислота-вода при нагріванні при приблизно 65 °C протягом від 1 до 6 годин або біля цього. Отримане похідне гідантоїнарилброміду (8) виділяють за звичайними способами та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Схема 6 – це спосіб реакції Хека похідного вінілсульфонаміду (18) та похідного гідантоїнарилброміду (8) у присутності металевого каталізатора з наступною гідрогенізацією олефінової сполуки (19), внаслідок чого отримують похідне гідантоїну (Формула 1). Схема 6 (Спосіб В) Похідне гідатноїну (Формула 1) можна синтезувати шляхом здійснення реакції за Етапом 14 згідно зі способом Етапу 6 та здійснення реакції за Етапом 15 згідно зі способом Етапу 5. Отримане похідне гідантоїну (Формула 1) виділяють за звичайними способами та, якщо необхідно, його можна очистити шляхом кристалізації або хроматографії. Похідне вінілсульфонаміду (18), яке застосовується на Етапі 14, можна синтезувати, звернувшись до Схем 2, 3 та 12 міжнародної патентної публікації WO2010/126030(A1). Усі документи з попереднього рівня техніки, процитовані у цьому описі, включено до цього опису винаходу шляхом посилання. Приклади Зміст цього винаходу буде описано більш докладно за допомогою наступних прикладів та тестового прикладу; проте, цей винахід не обмежується змістом прикладів та тестового прикладу. Усі початкові матеріали та реагенти отримали від комерційних постачальників або 1 синтезували, застосовуючи відомі способи. Спектри H-ЯМР вимірювали, використовуючи Mercury300 (виготовлений компанією Varian), ECP-400 (виготовлений компанією JEOL) або 400MR (виготовлений компанією Varian) з Me4Si як внутрішнім стандартом або без нього (s = синглет, d = дублет, t = триплет, brs = широкий синглет, m = мультиплет). Мас-спектрометричні вимірювання здійснювали, використовуючи мас-спектрометр ZQ2000 (виготовлений компанією Waters), SQD (виготовлений компанією Waters) або 2020 (виготовлений компанією Shimazu). Приклад 1 1-(4-(2-((2-(4-фторо-3-(трифторометокси)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діон (Сполука 1) Реакція 1-1 До розчину 4-бромо-3,5-диметиланіліну (3,47 г, 17,4 ммоль) і діізопропілетиламіну (5,3 мл, 30,4 ммоль) в DMI (13 мл) додавали при кімнатній температурі 2-бромоізомасляну кислоту (3,86 г, 23,1 ммоль). Суміш перемішували при 100 °C протягом однієї години. І потім додавали 2бромоізобутират (496 мг, 2,97 ммоль) і діізопропілетиламін (0,8 мл, 4,59 ммоль) і суміш перемішували при 100 °C протягом однієї години. Додавали до реакційної суміші при кімнатній температурі метанол (52 мл) і 5 N водний 14 UA 115072 C2 5 10 15 20 25 30 розчин гідроксиду натрію (52 мл, 260 ммоль), а потім цю суміш перемішували при 75 °C протягом 1,5 години. Реакційну суміш охолоджували, після чого додавали воду і доводили pH до 5, використовуючи 1 N водний розчин гідросульфату калію, а потім екстрагували, використовуючи етилацетат. Органічний шар промивали водою, потім висушували над безводним сульфатом магнію і концентрували, одержуючи 2-((4-бромо-3,5диметилфеніл)аміно)-2-метилпропіонову кислоту як неочищений продукт (5,79 г). MS(ESI) m/z=286, 288 (M+H)+ Реакція 1-2 До суміші 2-((4-бромо-3,5-диметилфеніл)аміно)-2-метилпропіонової кислоти (5,79 г цієї сполуки одержали з Реакції 1-1) в дихлорометані (62 мл) і оцтової кислоти (62 мл) додавали при кімнатній температурі ціанат натрію (5,03 г, 59,8 ммоль). Суміш перемішували при кімнатній температурі протягом трьох годин. Додавали насичений розчин бікарбонату натрію (400 мл), щоб довести pH до 7-8 використовували 5 N водний гідроксид натрію, і цю суміш екстрагували етилацетатом. Органічний шар висушували над безводним сульфатом магнію і потім концентрували при зниженому тиску. Одержану тверду речовину промивали послідовно етилацетатом-гексаном і потім дихлорометаном-гексаном, одержуючи 1-(4-бромо-3,5диметилфеніл)-5,5-диметилімідазолідин-2,4-діон (3,80 г, 66 %). MS(ESI) m/z=311, 313 (M+H)+ Реакція 1-3 Суміш 8-(вінілсульфоніл)-1,4-діокса-8-азаспіро[4.5]декану (431 мг, 1,85 ммоль), 1-(4-бромо3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону (575 мг, 1,85 ммоль), трис(дибензиліденацетон)паладію(0) (508 мг, 0,55 ммоль), три-трет-бутилфосфінтетрафтороборної кислоти (165 мг, 0,55 ммоль) і метилдициклогексиламіну (2,1 мл, 9,25 ммоль) в Nметил-2-піролідоні (18,5 мл) перемішували в атмосфері азоту при 110 °C протягом двох годин. Реакційну суміш охолодили, загасили водою, а потім екстрагували етилацетатом. Органічний шар промивали водою і сольовим розчином, висушували над безводним сульфатом магнію і потім концентрували при зниженому тиску. Залишок очищали шляхом колоночної хроматографії на аміносилікагелі (дихлорометан - метанол), одержуючи (E)-1-(4-(2-(1,4-діокса-8азаспіро[4.5]декан-8-ілсульфоніл)вініл)-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діон (584 мг, 68 %). MS(ESI) m/z=464 (M+H)+ Реакція 1-4 35 15 UA 115072 C2 5 10 15 20 25 30 35 40 45 До розчину (E)-1-(4-(2-(1,4-діокса-8-азаспіро[4.5]декан-8-ілсульфоніл)вініл)-3,5диметилфеніл)-5,5-диметилімідазолідин-2,4-діону (1,2 г, 2,58 ммоль) в тетрагідрофурані (THF) (26 мл) додавали крапля за краплею 2 N водний розчин хлористоводневої кислоти (26 мл, 52 ммоль) протягом десяти хвилин. Суміш перемішували при 60 °C протягом двох годин. Реакційну суміш охолоджували, після чого доводили її pH до 7, використовуючи 2 N водний розчин гідроксиду натрію, і цю суміш екстрагували етилацетатом. Органічний шар промивали сольовим розчином, висушували над безводним сульфатом магнію і потім концентрували при зниженому тиску. Залишок очищали шляхом колоночної хроматографії на силікагелі (дихлорометан етилацетат), одержуючи (E)-1-(3,5-диметил-4-(2-((4-оксопіперидин-1-іл)сульфоніл)вініл)феніл)5,5-диметилімідазолідин-2,4-діон (998 мг, 92 %). MS(ESI) m/z=420 (M+H)+ Реакція 1-5 До розчину (E)-1-(3,5-диметил-4-(2-((4-оксопіперидин-1-іл)сульфоніл)вініл)феніл)-5,5диметилімідазолідин-2,4-діону (994 мг, 2,37 ммоль) в метанолі (24 мл) додавали при кімнатній температурі ціаністий калій (188 мг, 2,84 ммоль) і ацетат амонію (237 мг, 3,08 ммоль). Суміш перемішували при 60-70 °C протягом трьох годин. Реакційну суміш охолоджували, концентрували при зниженому тиску і потім розбавили етилацетатом. Органічний шар промивали водою і сольовим розчином, висушували над безводним сульфатом магнію і потім концентрували при зниженому тиску. Залишок очищали шляхом колоночної хроматографії на силікагелі (дихлорометан - етилацетат), одержуючи (E)-4-аміно-1-((4-(5,5-диметил-2,4діоксоімідазолідин-1-іл)-2,6-диметилстирил)-сульфоніл)піперидин-4-карбонітрил (681 мг, 68 %). 1 H-ЯМР (300 МГц, DMSO(ДМСО)-d6) δ: 1,3 (6H, s), 1,7 (2H, m), 2,0 (2H, m), 2,3 (6H, s), 2,7 (2H, s), 2,9 (2H, m), 3,4 (2H, m), 6,9 (1H, d, J=15,9 Гц), 7,1 (2H, s), 7,4 (1H, d, J=15,9 Гц), 11,2 (1H, brs) Реакція 1-6 До розчину (E)-4-аміно-1-((4-(5,5-диметил-2,4-діоксоімідазолідин-1-іл)-2,6диметилстирил)сульфоніл)піперидин-4-карбонітрилу (675 мг, 1,50 ммоль) в метанолі (7,5 мл) і диметилсульфоксиді (0,195 мл) при кімнатній температурі додали 2 N водний розчин гідроксиду натрію (1,6 мл, 1,6 ммоль) і потім повільно додавали крапля за краплею 30 % водний розчин пероксиду водню (0,2 мл, 1,95 ммоль). Суміш перемішували при кімнатній температурі протягом однієї години. Додавали до реакційної суміші етилацетат, гексан і насичений водний розчин хлориду амонію. Тверду речовину збирали шляхом фільтрування, промивали і висушували, одержуючи (E)-4-аміно-1-((4-(5,5-диметил-2,4-діоксоімідазолідин-1-іл)-2,6диметилстирил)сульфоніл)піперидин-4-карбоксамід (498 мг, 72 %). MS(ESI) m/z=464 (M+H)+ Реакція 1-7 Суміш (E)-4-аміно-1-((4-(5,5-диметил-2,4-діоксоімідазолідин-1-іл)-2,6диметилстирил)сульфоніл)піперидин-4-карбоксаміду (1,3 г, 2,8 ммоль) і гідроксиду паладію на вуглецю (20 % Pd) (зволоженого приблизно на 50 % водою) (1,3 г) в метанолі (21 мл) і диметилформаміді (DMF) (7 мл) перемішували в атмосфері водню при кімнатній температурі протягом чотирьох годин. Реакційну суміш фільтрували і промивали, а потім фільтрат концентрували при зниженому тиску, одержуючи 4-аміно-1-((4-(5,5-диметил-2,4діоксоімідазолідин-1-іл)-2,6-диметилфенетил)сульфоніл)піперидин-4-карбоксамід (998 мг, 16 UA 115072 C2 77 %). MS(ESI) m/z=466 (M+H)+ Реакція 1-8 5 10 15 20 25 30 35 До розчину 4-аміно-1-((4-(5,5-диметил-2,4-діоксоімідазолідин-1-іл)-2,6диметилфенетил)сульфоніл)піперидин-4-карбоксаміду (120 мг, 0,258 ммоль), 4-фторо-3(трифторометокси)бензойної кислоти (69 мг, 0,309 ммоль) і діізопропілетиламіну (DIEA) (0,09 мл, 0,516 ммоль) в диметилформаміді (2,5 мл) додавали O-(7-азабензотриазол-1-іл)-1,1,3,3тетраметилуроніюгексафторофосфат (HATU) (118 мг, 0,309 ммоль).Суміш перемішували при кімнатній температурі протягом 1,5 години. Реакційну суміш загасили водою і потім екстрагували дихлорометаном. Органічний шар промивали сольовим розчином, промивали безводним сульфатом натрію і потім концентрували при зниженому тиску, одержуючи 1-((4-(5,5диметил-2,4-діоксоімідазолідин-1-іл)-2,6-диметилфенетил)-сульфоніл)-4-(4-фторо-3(трифторометокси)бензамід)піперидин-4-карбоксамід (150 мг, 67 %). MS(ESI) m/z=672 (M+H)+ Реакція 1-9 До перемішаного розчину 1-((4-(5,5-диметил-2,4-діоксоімідазолідин-1-іл)-2,6диметилфенетил)сульфоніл)-4-(4-фторо-3-(трифторометокси)бензамід)піперидин-4карбоксаміду (150 мг, 0,223 ммоль) в трет-бутанолі (2,5 мл) і етанолі (2,5 мл) додавали при 0 °C трет-бутоксид калію (75 мг, 0,670 ммоль). Суміш перемішували в атмосфері азоту при 50 °C протягом 1,5 години. Реакційну суміш охолодили, розбавили водою, загасили насиченим водним розчином хлориду амонію, а потім екстрагували дихлорометаном. Органічний шар промивали водою і сольовим розчином, висушували над безводним сульфатом натрію і потім концентрували при зниженому тиску. Одержаний залишок очищали шляхом колоночної хроматографії на силікагелі (дихлорометан - метанол), одержуючи 1-(4-(2-((2-(4-фторо-3(трифторометокси)феніл)-4-оксо-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)-3,5диметилфеніл)-5,5-диметилімідазолідин-2,4-діон (118 мг, 81 %). 1 MS(ESI) m/z=654 (M+H)+. H-ЯМР (400 МГц, CD3OD) δ: 1,40 (6H, s), 1,71-1,80 (2H, m), 2,002,08 (2H, m), 2,43 (6H, s), 3,22 (4H, s), 3,47-3,57 (2H, m), 3,80-3,88 (2H, m), 7,01 (2H, s), 7,50-7,57 (1H, m), 7,97-8,04 (1H, m), 8,05-8,12 (1H, m) Наступні сполуки Прикладів були синтезовані шляхом операцій, подібних до операцій в Реакціях 1-8 і 1-9 з Прикладу 1, використовуючи відповідні карбонові кислоти як початкові матеріали, реагенти і розчинники. Сполуки 2-5 17 UA 115072 C2 Таблиця 1 18 UA 115072 C2 5 10 15 20 25 30 35 Приклад 2 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон (Сполука 6) Реакція 2-1 Суміш 2-(3-(трифторометил)феніл)-8-(вінілсульфоніл)-1,3,8-триазаспіро[4.5]дека-1-ен-4-ону (150 мг, 0,387 ммоль), синтезованого відповідно до способу, наведеного на Схемах 2, 3 і 12 у WO2010/126030 (A1), 1-(4-бромо-3,5-диметилфеніл)-5,5-диметилімідазолідин-2,4-діону (169 мг, 0,542 ммоль), біс(дибензиліденацетон)паладію (45 мг, 0,077 ммоль), три-третбутилфосфінтетрафтороборної кислоти (22 мг, 0,077 ммоль) і метилдициклогексиламіну (0,123 мл, 0,581 ммоль) в N-метил-2-піролідоні (0,97 мл) перемішували при 100 °C протягом однієї години в атмосфері азоту. Реакційну суміш охолоджували, загасили водою і потім екстрагували етилацетатом. Органічний шар промивали водою і сольовим розчином, висушували над безводним сульфатом натрію і потім концентрували при зниженому тиску. Одержаний залишок очищали шляхом колоночної хроматографії на силікагелі (етилацетат - гексан), одержуючи (E)1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)вініл)феніл)-5,5-диметилімідазолідин-2,4-діон (197 мг, 82 %). MS(ESI) m/z=618 (M+H)+ Реакція 2-2 Суміш (E)-1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)вініл)феніл)-5,5-диметилімідазолідин-2,4-діону (195 мг, 0,316 ммоль) і гідроксиду паладію / вуглецю (20 % Pd) (зволоженого на приблизно 50 % водою) (195 мг, 0,139 ммоль) в 2,2,2-трифтороетанолі (6 мл) перемішували при кімнатній температурі протягом 14 годин в атмосфері водню. Суміш профільтрували і фільтрат концентрували при зниженому тиску. Одержаний залишок очищали шляхом колоночної хроматографії на силікагелі (етилацетат - гексан), одержуючи 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон (121 мг, 62 %). 1 MS(ESI) m/z=620 (M+H)+. H-ЯМР (400 МГц, CD3OD) δ: 1,40 (6H, s), 1,72-1,81 (2H, m), 2,002,10 (2H, m), 2,44 (6H, s), 3,22 (4H, s), 3,50-3,58 (2H, m), 3,80-3,88 (2H, m), 7,01 (2H, s), 7,72-7,79 (1H, m), 7,88-7,94 (1H, m), 8,16-8,23 (1H, m), 8,31 (1H, s) Приклад 3 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон (Сполука 7) Реакція 3 19 UA 115072 C2 5 10 15 20 25 30 Використовуючи відповідні початкові матеріали і розчинники, синтезували 1-(3,5-диметил-4(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-5,5-диметилімідазолідин-2,4-діон (Сполука 7) шляхом операцій, подібних до операцій, описаних в Прикладі 2. 1 MS(ESI) m/z=636 (M+H)+. H-ЯМР (400 МГц, CDCl3) δ: 1,47 (6H, s), 1,70-1,78 (2H, m), 2,102,19 (2H, m), 2,40 (6H, s), 3,00-3,07 (2H, m), 3,19-3,25 (2H, m), 3,45-3,53 (2H, m), 3,81-3,88 (2H, m), 6,94 (2H, s), 7,35 (2H, d, J=8,0 Гц), 7,73 (1H, brs), 7,93 (2H, d, J=8,0 Гц), 9,37 (1H, brs) Приклад 4 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діон (Сполука 8) Реакція 4-1 До суміші циклопентанону (42 мг, 0,500 ммоль) і 4-бромо-3,5-диметиланіліну (100 мг, 0,500 ммоль) в оцтовій кислоті (0,5 мл) додавали при кімнатній температурі триметилсилілціанід (0,063 мл, 0,500 ммоль). Суміш перемішували при кімнатній температурі протягом 1,5 години в атмосфері азоту. Реакційну суміш загасили 28 % водним аміаком (1 мл), розбавили водою і екстрагували дихлорометаном. Органічний шар промивали водою і сольовим розчином, висушували над безводним сульфатом натрію і потім концентрували при зниженому тиску, одержуючи 1-((4-бромо-3,5-диметилфеніл)аміно)циклопентанкарбонітрил як неочищений продукт (152 мг). 1 H-ЯМР (400 МГц, CDCl3) δ: 1,83-1,92 (4H, m), 2,07-2,15 (2H, m), 2,33-2,42 (2H, m), 2,37 (6H, m), 3,71 (1H, brs), 6,56 (2H, s) Реакція 4-2 До розчину 1-((4-бромо-3,5-диметилфеніл)аміно)циклопентанкарбонітрилу (145 мг, 0,495 ммоль) в дихлорометані (5 мл) додавали при кімнатній температурі 2,2,2трихлороактилізоціанат (0,070 мл, 0,593 ммоль). Суміш перемішували при кімнатній температурі протягом однієї години в атмосфері азоту. Додавали триетиламін (0,103 мл, 0,742 ммоль), воду (0,045 мл) і метанол (0,10 мл) і суміш кип'ятили із зворотним холодильником протягом 1,5 години в атмосфері азоту. Реакційну суміш охолоджували, після чого розводили водою і доводили її pH до 5, використовуючи 1 N водний розчин хлористоводневої кислоти, а потім екстрагували дихлорометаном. Органічний шар промивали водою і сольовим розчином, висушували над безводним сульфатом натрію і потім 20 UA 115072 C2 концентрували при зниженому тиску, одержуючи 1-(4-бромо-3,5-диметилфеніл)-4-іміно-1,3діазаспіро[4.4]нонан-2-он як неочищений продукт. MS(ESI) m/z=336, 338 (M+H)+ Реакція 4-3 5 10 15 20 25 Суміш 1-(4-бромо-3,5-диметилфеніл)-4-іміно-1,3-діазаспіро[4.4]нонан-2-ону (неочищений продукт одержаний в попередній реакції) в оцтовій кислоті (1,0 мл) і води (0,25 мл) перемішували протягом 1,5 години при 65 °C в атмосфері азоту. Після наступного додавання оцтової кислоти (1,0 мл) і води (0,25 мл) суміш перемішували протягом 17 годин при 65 °C в атмосфері азоту. Реакційну суміш охолоджували, після чого розводили водою та доводили її pH до 8, використовуючи насичений водний розчин бікарбонату натрію, і екстрагували етилацетатом. Органічний шар промивали водою і сольовим розчином, висушували над безводним сульфатом натрію і потім концентрували при зниженому тиску. Залишок очищали шляхом колоночної хроматографії на силікагелі (етилацетат - гексан), одержуючи 1-(4-бромо3,5-диметилфеніл)-1,3-діазаспіро[4.4]нонан-2,4-діон (121 мг). MS(ESI) m/z=337, 339 (M+H)+ Реакція 4-4 Використовуючи відповідні початкові матеріали і розчинники, одержували 1-(3,5-диметил-4(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діон (Сполука 8) шляхом операцій, подібних до операцій, описаних в Прикладі 2. 1 MS(ESI) m/z=662 (M+H)+. H-ЯМР (400 МГц, ДМСО-d6) δ: 1,36-1,44 (2H, m), 1,60-1,70 (4H. m), 1,82-1,91 (2H, m), 1,91-2,06 (4H, m), 2,38 (6H, s), 3,01-3,09 (2H, m), 3,22-3,30 (2H, m), 3,30-3,42 (2H, m), 3,70-3,77 (2H, m), 7,03 (2H, s), 7,57 (2H, d, J=8,4 Гц), 8,14 (2H, d, J=8,4 Гц) Приклад 5 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-8-метил-1,3,8-триазаспіро[4.5]декан-2,4-діон (Сполука 9) Реакція 5-1 21 UA 115072 C2 5 10 15 Використовуючи трет-бутиловий естер 4-оксопіперидин-1-карбонової кислоти як початковий матеріал і використовуючи відповідний розчинник, одержували трет-бутиловий естер 1-(3,5диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-2,4-діоксо-1,3,8-триазаспіро[4.5]декан-8-карбонової кислоти шляхом операцій, подібних до операцій, описаних в Прикладі 4. MS(ESI) m/z=777 (M+H)+. Реакція 5-2 До перемішаного розчину трет-бутилового естеру 1-(3,5-диметил-4-(2-((4-оксо-2-(4(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-2,4-діоксо1,3,8-триазаспіро[4.5]декан-8-карбонової кислоти (11,7 мг, 0,015 ммоль) в дихлорометані (0,13 мл) додавали при кімнатній температурі трифторооцтову кислоту (0,05 мл, 0,673 ммоль). Суміш поміщали під струм азоту і перемішували при кімнатній температурі протягом однієї години. Реакційну суміш концентрували при зниженому тиску, одержуючи сіль 1-(3,5-диметил-4-(2-((4оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)1,3,8-триазаспіро[4.5]декан-2,4-діону 2 трифторооцтової кислоти (13,6 мг). MS(ESI) m/z=677 (M+H)+. Реакція 5-3 22 UA 115072 C2 5 10 15 20 25 До суміші солі 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-1,3,8-триазаспіро[4.5]декан-2,4-діону 2 трифторооцтової кислоти (21,1 мг, 0,022 ммоль) і мурашиної кислоти (0,033 мл) додавали 37 % водний розчин формальдегіду (0,055 мл). Суміш поміщали під струм азоту і перемішували протягом трьох годин, одночасно нагріваючи при 80 °C. Реакційну суміш концентрували і одержаний осад розбавили етилацетатом. Органічний шар промили розбавленим водним розчином гідроксиду натрію, висушували над безводним сульфатом магнію і потім концентрували при зниженому тиску. Одержаний залишок піддали колоночній хроматографії (дихлорометан - метанол) для очищення, одержуючи 1-(3,5-диметил-4-(2-((4-оксо-2-(4(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл-8-метил1,3,8-триазаспіро[4.5]декан-2,4-діон (4,5 мг, 30 %). 1 MS(ESI) m/z=691 (M+H)+. H-ЯМР (400 МГц, CD3OD) δ: 1,76-1,84 (2H, m), 1,92-2,02 (2H, m), 2,02-2,12 (4H, m), 2,38 (3H, s), 2,46 (6H, s), 2,81-2,88 (2H, m), 2,92-3,02 (2H, m), 3,23 (4H, s), 3,513,60 (2H, m), 3,72-3,80 (2H, m), 7,01 (2H, s), 7,48 (2H, d, J=8,0 Гц), 8,10 (2H, d, J=8,0 Гц) Приклад 6 5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-2-окса-5,7-діазаспіро[3.4]октан-6,8-діон (Сполука 10) Реакція 6 Використовуючи оксетан-3-он як початковий матеріал і використовуючи відповідні розчинники, одержували 5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8триазаспіро[4.5]дека-1-єн-8-іл)сульфоніл)етил)феніл)-2-окса-5,7-діазаспіро[3.4]октан-6,8-діон шляхом операцій, подібних до операцій з Прикладу 4. 1 MS(ESI) m/z=650 (M+H)+. H-ЯМР (400 МГц, CDCl3) δ: 1,69-1,77 (2H, m), 2,12-2,22 (2H, m), 2,45 (6H, s), 3,03-3,11 (2H, m), 3,22-3,29 (2H, m), 3,46-3,53 (2H, m), 3,84-3,91 (2H, m), 4,86 (2H, d, J=7,2 Гц), 5,03 (2H, d, J=7,2 Гц), 7,07 (2H, s), 7,35 (2H, d, J=8,4 Гц), 7,98 (2H, d, J=8,4 Гц), 8,56 (1H, s), 10,34 (1H, s) Приклад 7 23 UA 115072 C2 4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-4,6-діазаспіро[2.4]гептан-5,7-діон (Сполука 11) Реакція 7-1 5 10 15 20 25 30 35 Суміш 2-бромо-5-йодо-1,3-диметилбензолу (300 мг, 0,965 ммоль), 1аміноциклопропанкарбонової кислоти (195 мг, 1,93 ммоль), йодиду міді(I) (37 мг, 0,194 ммоль) і діазабіциклоундецену (0,50 мл, 3,35 ммоль) в диметилацетаміді (2,6 мл) перемішували при 120 °C протягом трьох годин в атмосфері азоту. Реакційну суміш очищали шляхом колоночної хроматографії на силікагелі (Wakosil C18, ацетонітрил - вода (0,1 % мурашиної кислоти)), одержуючи 1-((4-бромо-3,5-диметилфеніл)аміно)циклопропанкарбонову кислоту (219 мг, 80 %). MS(ESI) m/z=284, 286 (M+H)+. Реакція 7-2 До суміші 1-((4-бромо-3,5-диметилфеніл)аміно)циклопропанкарбонової кислоти (198 мг, 0,697 ммоль) в оцтовій кислоті (3 мл) і дихлорометані (1,5 мл) додавали при кімнатній температурі ціанат калію (424 мг, 5,23 ммоль). Суміш перемішували при кімнатній температурі протягом однієї години, а потім перемішували при 60 °C протягом двох годин. Додавали насичений водний розчин бікарбонату натрію, щоб довести pH до 8, і цю суміш екстрагували етилацетатом. Органічний шар промивали водою і сольовим розчином, висушували над безводним сульфатом натрію і потім концентрували при зниженому тиску. Залишок очищали шляхом колоночної хроматографії на силікагелі (етилацетат - гексан), одержуючи 4-(4-бромо3,5-диметилфеніл)-4,6-діазаспіро[2.4]гептан-5,7-діон (49 мг, 23 %). MS(ESI) m/z=309, 311 (M+H)+. Реакція 7-3 Використовуючи відповідні початкові матеріали і розчинники, одержували 4-(3,5-диметил-4(2-((4-оксо-2-(4-(трифторометокси)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-4,6-діазаспіро[2.4]гептан-5,7-діон (Сполука 11) шляхом операцій, подібних до операцій з Прикладу 2. 1 MS(ESI) m/z=634 (M+H)+. H-ЯМР (400 МГц, ДМСО-d6) δ: 0,99-1,03 (2H, m), 1,19-1,27 (4H, m), 1,58-1,64 (2H, m), 1,81-1,90 (2H, m), 2,35 (6H, s), 2,99-3,04 (2H, m), 3,22-3,29 (2H, m), 3,67-3,73 (2H, m), 6,95 (2H, s), 7,56 (2H, d, J=8,4 Гц), 8,12 (2H, d, J=8,4 Гц) Приклад 8 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діон (Сполука 12) Реакція 8 24 UA 115072 C2 5 10 15 Використовуючи відповідні початкові матеріали і розчинники, одержували 1-(3,5-диметил-4(2-((4-оксо-2-(3-(трифторометил)феніл)-1,3,8-триазаспіро[4.5]дека-1-єн-8іл)сульфоніл)етил)феніл)-1,3-діазаспіро[4.4]нонан-2,4-діон шляхом операцій, подібних до операцій з Прикладу 2. 1 MS(ESI) m/z=646 (M+H)+. H-ЯМР (400 МГц, ДМСО-d6) δ: 1,40-1,48 (2H, m), 1,62-1,71 (4H, m), 1,88-1,97 (2H, m), 1,97-2,08 (4H, m), 2,41 (6H, s), 3,03-3,10 (2H, m), 2,29-3,34 (2H, m), 3,38-3,47 (2H, m), 3,72-3,79 (2H, m), 7,06 (2H, s), 7,84 (1H, dd, J=7,6, 7,6 Гц), 8,02 (1H, d, J=7,6 Гц), 8,33 (1H, d, J=7,6 Гц), 8,38 (1H, s). Тестові приклади Для сполук цього винаходу в Тестових прикладах від 1 до 5 у вказаному порядку показані результати випробувань на активність виробництва цАМФ за участі рецептора ПТГ1 (РТН1R) людини, на активність виробництва цАМФ за участі рецептора ПТГ1 (РТН1R) щура, на стійкість до інактивації в процесі метаболізму з використанням мікросом печінки людини, на стійкість до інактивації в процесі метаболізму з використанням гепатоцитів щура і на кальцемічну дію на щурячій моделі ТРТХ. Сполуки, описані в WO2010/126030A1, які показані в Таблиці 2, використовували як порівняльні сполуки. 25 UA 115072 C2 Таблиця 2 5 10 15 Тестовий приклад 1: Вимірювання in vitro цАМФ-сигнальної активності сполук за участі рецептора ПТГ1 (PTH1R). Пептиди PTH (1-34) людини та кальцитонін придбали у Peptide Institute, Inc. (Осака, Японія), розчинили у 10 мМ оцтовій кислоті до 1 мМ та зберігали у морозильнику при -80 °C. Клітинна культура Клітини культивували у модифікованому за способом Дульбеко середовищі Ігла (DMEM), до якого було додано 10 % фетальної бичачої сироватки (Hyclone), 100 одиниць/мл пеніциліну G та 100 мкг/мл стрептоміцину сульфату (Invitrogen Corp), при 37 °C у зволоженій атмосфері, яка містить 5 % CO2. При аналізі цАМФ-сигнальної трансдукції застосовувалися клітини LLC-PK1, які не експресували рецептор ПТГ1, та клітини HKRK-B7, тобто клітини LLC-PK1, які надмірно 5 експресували рецептор ПТГ1 людини, при 9,5 10 рецепторів/клітину (Takasu et al., J. Bone. 26 UA 115072 C2 5 10 15 20 Miner. Res. 14:11-20, 1999). Стимуляція цАМФ 5 Клітини HKRK-B7 або LLC-PK1 посіяли у планшет з 96 комірками по 1 × 10 клітин/комірку та інкубували протягом ночі. Наступного дня додали 50 мкл буферу для аналізу цАМФ (DMEM, 3 мМ ІВМХ, 0,2 мг/мл альбуміну бичачої сироватки, 35 мМ Hepes-NaOH, рН 7,4), який містив PTH (1-34) людини або кожну сполуку, та планшет розташували в інкубаторі при 37 °C. Клітини інкубували протягом 20 хвилин. Після видалення середовища клітини один раз промили 100 мкл буферу для аналізу цАМФ. Планшет розташували на порошку сухого льоду, щоб заморозити клітини, а потім забрали з сухого льоду. Клітини лізували за допомогою 40 мкл 50 мМ HCl та знов заморозили на сухому льоді. Кількість виробленого внутрішньоклітинного цАМФ вимірили, застосовуючи комерційно доступний набір cAMP EIA (Biotrack cAMP EIA system, GE health care). Розрахунок 20 % ефективної концентрації (ЕС20) та 50 % ефективної концентрації (ЕС50) при вимірюванні спроможності індукувати цАМФ in vitro Аналізи здійснювали із застосуванням для змінної градієнтного рівняння S-подібної кривої залежності "доза-ефект". цАМФ-сигнальну активність PTH (1-34) людини при 100 нМ визначили як 100 %-ву, а концентрацію, при якій кожна сполука демонструє 20 % або 50 % цАМФсигнальну активність визначили як ЕС20 або ЕС50. Результати, отримані для клітин HKRK-B7, наведено у Таблиці 3. Ступінь цАМФ-реакції у клітин LLC-PK1 була нижчою, ніж ступінь у клітин HKRK-B7. Таблиця 3 Сполука Сполука 1 Сполука 2 Сполука 3 Сполука 4 Сполука 5 Сполука 6 Сполука 7 Сполука 8 Сполука 9 25 30 35 EC50 (мкМ) 5,8 14 7,2 7,4 8,1 9,0 4,1 3,6 12 Сполука Сполука 10 Сполука 11 Порівняльний приклад 1 Порівняльний приклад 2 Порівняльний приклад 3 Порівняльний приклад 4 Порівняльний приклад 5 Порівняльний приклад 6 EC20 (мкМ) 5,0 1,5 1,5 3,1 2,0 >505 3,1 3,6 EC50 (мкМ) 21 11 4,8 13 9,0 >1000 25 32 Тестовий приклад 2: Вимірювання in vitro цАМФ-сигнальної активності сполук за участі рецептора ПТГ1 щурів. Замість клітин HKRK-B7 використовували клітини LLC-PK46_RATO_PTH1R, які надмірно експресують рецептор ПТГ1 щурів та які запровадили у Chugai Pharmaceutical, для вимірювань за способом, подібним до способу з Тестового прикладу 1. Результати, отримані внаслідок застосування клітин LLC-PK46_RATO_PTH1R, наведено у Таблиці 4. Значення ЕС20 цАМФ-сигнальної активності in vitro рецептора ПТГ1 щурів добре корелювалися зі значеннями для рецептора ПТГ1 людини. Гарну кореляцію між рецепторами щурів та людини також спостерігали для значень ЕС50. Таблиця 4 Сполука Сполука 7 Сполука 8 Сполука 10 40 EC20 (мкМ) 1,3 2,4 1,5 1,6 1,7 2,0 1,1 1,0 2,6 EC20 (мкМ) 0,5 0,4 3,0 EC50 (мкМ) 2,4 1,9 12 Сполука Сполука 11 Порівняльний приклад 1 EC20 (мкМ) 0,8 0,8 EC50 (мкМ) 3,2 2,3 Тестовий приклад 3: Дослідження стійкості до інактивації в процесі метаболізму із застосуванням мікросом печінки людини У 0,1 М фосфатному буфері (рН 7,4) мікросоми печінки людини інкубували зі сполукою або порівняльним зразком разом з NADPH при 37 °C протягом специфічного періоду часу. Концентрацію первинної сполуки у кожній часовій точці реакції вимірювали, застосовуючи РХ/МС/МС, та притаманний кліренс (мкл/хвилину/мг білка) розрахували за похилою прямою відношення часу реакції до кількості залишку. 27 UA 115072 C2 5 Умови аналізу Концентрація сполуки: 1мкМ Мікросома: 0,5 мг/мл NADPH: 1 мМ Час реакції: 0, 5, 15 та 30 хвилин Результати наведено у Таблиці 5. Сполуки 1 – 11 демонстрували високу стійкість до інактивації в процесі метаболізму до мікросом печінки людини порівняно з Порівняльними прикладами 1 – 6. Таблиця 5 10 Сполука Сполука 1 Сполука 2 Сполука 3 Сполука 4 Сполука 5 Сполука 6 Сполука 7 Сполука 8 Сполука 9 15 20 25 Кліренс (мкл/хв./мг) 21 38 29 27 37 29 30 35 28 Сполука Сполука 10 Сполука 11 Сполука 12 Порівняльний приклад 1 Порівняльний приклад 2 Порівняльний приклад 3 Порівняльний приклад 4 Порівняльний приклад 5 Порівняльний приклад 6 Кліренс (мкл/хв./мг) 29 19 63 84 61 74 74 112 154 Тестовий приклад 4: Дослідження стійкості до інактивації в процесі метаболізму із застосуванням гепатоцитів щурів Клітини печінки отримали з печінки щурів (SD, самки) за способом перфузії колагенази. Додали сполуку з Прикладів або Порівняльного прикладу, та цю суміш інкубували при 37 °C протягом специфічного періоду часу, після цього додали розчин, що припиняє реакцію. Концентрацію первинної сполуки у кожній часовій точці реакції вимірювали, застосовуючи 6 РХ/МС/МС, та притаманний кліренс (мкл/10 клітин/хвилину) розрахували за похилою прямою відношення часу реакції до кількості залишку. Умови аналізу 6 Концентрація клітин: 1 × 10 клітин/мл Концентрація сполуки: 1 мкМ Середовище: середовище Е за Вільямсом (Williams) Час реакції: 0, 15, 30, 60, 120 та 240 хвилин Розчин, що припиняє реакцію: ацетонітрил/2-пропанол (4/6, об./об.) Результати наведено у Таблиці 6. Стійкість Сполук 2, 4, 5, 7, 8, 9, 10 та 11 до інактивації в процесі метаболізму у гепатоцитах щурів є підвищеною, порівняно з Порівняльними прикладами 1, 2, 3, 5 та 6. Таблиця 6 30 Сполука Сполука 1 Сполука 2 Сполука 3 Сполука 4 Сполука 5 Сполука 6 Сполука 7 Сполука 8 35 Кліренс 6 (мкл/10 клітин/хв.) Сполука 7,6 3,0 17 2,2 1,0 1,4 0,9 3,0 Сполука 9 Сполука 10 Сполука 11 Порівняльний приклад 1 Порівняльний приклад 2 Порівняльний приклад 3 Порівняльний приклад 5 Порівняльний приклад 6 Кліренс 6 (мкл/10 клітин/хв.) 1,8 0,3 -0,6 5,8 5,9 22 22 22 Тестовий приклад 5: Кальцемічна дія на щурячій моделі ТРТХ Щурів-самок Crl:CD(SD) віком у 4 тижні отримали від Charles River Japan (Atsugi Breeding Center) та протягом одного тижня їх акліматизували до стандартних лабораторних умов: 2026 °C та 35-75 % вологості. Щурів поїли водопровідною водою та необмежено годували стандартним кормом для гризунів (CE-2)(CLEA Japan, Inc.), що містив 1,1 % кальцію, 1,0 % фосфорної кислоти та 250 МО/100 г вітаміну D3. 28
ДивитисяДодаткова інформація
Назва патенту англійськоюHydantoin derivative
Автори англійськоюNishimura, Yoshikazu, Esaki, Toru, Tamura, Tatsuya
Автори російськоюНисимура Йосикадзу, Эсаки Тору, Тамура Тацуя
МПК / Мітки
МПК: C07D 471/10, C07D 519/00, A61K 31/438
Мітки: похідні, гідантоїну
Код посилання
<a href="https://ua.patents.su/34-115072-pokhidni-gidantonu.html" target="_blank" rel="follow" title="База патентів України">Похідні гідантоїну</a>
Похідні гідантоїну для лікування обструктивних хвороб дихальних шляхів
Номер патенту: 86977
Опубліковано: 10.06.2009
Автори: абос Балінт, Стенвалль Крістіна, Ріпа Лена
МПК: A61K 31/506, C07D 401/14, A61P 11/00
Мітки: хвороб, похідні, шляхів, обструктивних, гідантоїну, лікування, дихальних
Формула / Реферат:
1. Сполука формули (І) або її фармацевтично прийнятна сіль , (I)де:R1 представляє С1-2алкіл, циклопропіл, ОСН3, SCH3 або OCF3; вказані алкіл або циклопропіл, як варіант, крім того заміщені одним або більше атомами флуору; аR2 представляє С1-3алкіл.2. Сполука за п. 1, де R1 представляє С1-2алкіл або циклопропіл; вказані алкіл або...
Похідні гідантоїну як інгібітори металопротеїназ

Номер патенту: 89801
Опубліковано: 10.03.2010
Автори: Лундквіст Мікаель, абос Балінт, Шамовскі Іґор, Златоідскі Паволь, Мунк Аф Розеншельд Маґнус
МПК: C07D 401/14, A61P 19/02, A61K 31/506, A61P 9/10, C07D 471/04, C07D 401/12, A61K 31/4375, A61P 11/06, A61P 35/00, A61K 31/4725
Мітки: інгібітори, гідантоїну, похідні, металопротеїназ
Формула / Реферат:
1. Сполука формули (І) або її фармацевтично прийнятна сіль, (I)деR1 представляє Н, галоген, CF3 або CH2CN;R2 представляє С1-С3алкіл; іА, А1 і В, кожен незалежно, представляють СН або N;2. Сполука за пунктом 1, де R1 представляє хлор.3. Сполука за пунктом 1, де R1 представляє CF3.4. Сполука за будь-яким з пунктів 1-3,...
Похідні гідантоїну, корисні як інгібітори металопротеїназ

Номер патенту: 89067
Опубліковано: 25.12.2009
Автори: Перссон Давід Йонас, Вотерсон Дейвід
МПК: A61K 31/4166, C07D 401/12, A61K 31/445, A61P 19/02
Мітки: гідантоїну, металопротеїназ, корисні, інгібітори, похідні
Формула / Реферат:
1. Сполука формули (І), (І)деR1 - (2-4С)алкіл, та є заміщеним двома або більше атомами флуору; аR2 - метил або етил;або її фармацевтично прийнятна сіль.2. Сполука формули (І) за п. 1, де R1 - етил, пропіл або бутил, та є заміщеним двома або більше атомами флуору.3. Сполука формули (І) за п. 1 або за п. 2, де R1 - етил,...
Похідні гідантоїну, корисні як інгібітори металопротеїназ

Номер патенту: 90285
Опубліковано: 26.04.2010
Автори: Златоідскі Паволь, Мунк Аф Розеншельд Маґнус, абос Балінт, Шамовскі Іґор, Лундквіст Мікаель
МПК: A61P 9/10, A61K 31/444, C07D 471/04, A61P 19/02, C07D 401/14, A61K 31/506, A61P 35/00, A61P 11/06
Мітки: гідантоїну, корисні, інгібітори, металопротеїназ, похідні
Формула / Реферат:
1. Сполука формули (І) або її фармацевтично прийнятна сіль, (I)деR1 - циклобутил або циклопропіл; вказаний циклопропіл, як варіант, крім того заміщено СН3, CN або одним або двома атомами флуору;R2 - (С1-3)-алкіл або циклопропіл; а А, А1 та В, незалежно, - СН або N;2. Сполука за п. 1, де R1 - циклопропіл.3. Сполука за п. 1...
Похідні гідантоїну, що застосовуються як mmp інгібітори
Номер патенту: 93425
Опубліковано: 10.02.2011
Автори: Мунк Аф Розеншольд Маґнус, Чапмен Девід, абос Балі
МПК: A61K 31/506, A61P 19/00, A61P 11/06, A61P 19/02, C07D 403/12, A61P 11/00, C07D 239/34
Мітки: застосовуються, похідні, гідантоїну, інгібітори
Формула / Реферат:
1. Сполука формули (І) або її фармацевтично прийнятна сіль, (I) де R1 - Н, СН3, СН3СН2, CF3 або циклопропіл; а R2 - Н або СН3. 2. Сполука за п. 1, яка має (4S)-стереохімію, яку показано нижче:
Попередній патент: Пристрій для безперервного діагностування технічного стану нерознімних і рознімних з’єднань елементів тіл обертання
Наступний патент: Стероїдна композиція для місцевого використання, спосіб її виготовлення й спосіб лікування
Випадковий патент: Програмно-технічний тренажерно-імітаційний комплекс підготовки радіотелеграфіста "радист-к"