Похідні гідантоїну, корисні як інгібітори металопротеїназ
Номер патенту: 90285
Опубліковано: 26.04.2010
Автори: абос Балінт, Шамовскі Іґор, Лундквіст Мікаель, Мунк Аф Розеншельд Маґнус, Златоідскі Паволь






Формула / Реферат
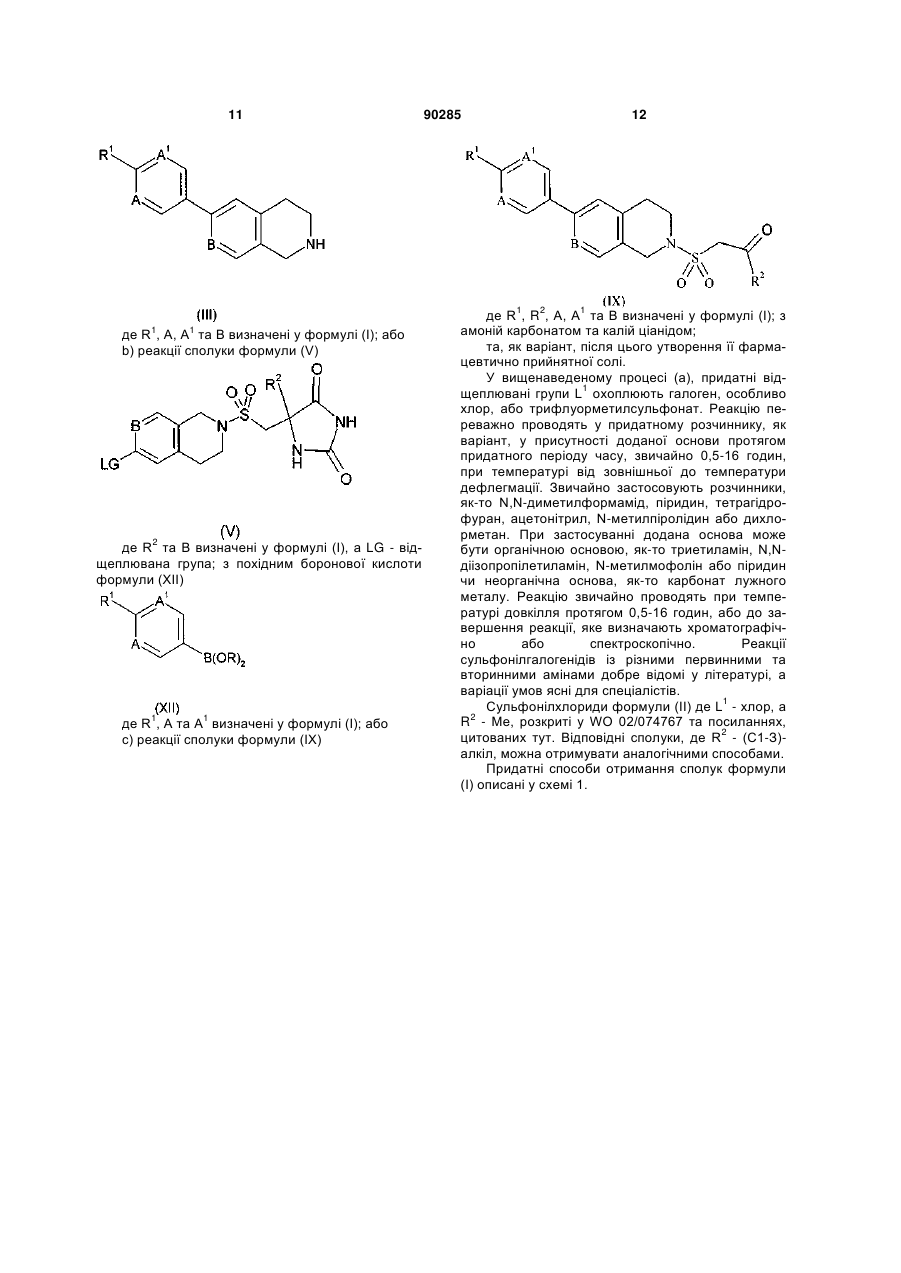
1. Сполука формули (І) або її фармацевтично прийнятна сіль
 , (I)
, (I)
де
R1 - циклобутил або циклопропіл; вказаний циклопропіл, як варіант, крім того заміщено СН3, CN або одним або двома атомами флуору;
R2 - (С1-3)-алкіл або циклопропіл; а
А, А1 та В, незалежно, - СН або N;
2. Сполука за п. 1, де R1 - циклопропіл.
3. Сполука за п. 1 або п. 2, де R2 - метил або етил.
4. Сполука за будь-яким з пп. 1-3, де В - СН.
5. Сполука за будь-яким з пп. 1-4, де А та А1, кожний, - N.
6. Сполука за п. 1, котру вибрано з групи:
(5S)-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4-дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон;
(5S)-5-({[6-(6-циклопропілпіридин-3-іл)-3,4-дигідро-2,7-нафтиридин-2(1H)-іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон;
(5S)-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4-дигідро-2,7-нафтиридин-2(1H)-іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон;
(5S)-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4-дигідро-2,7-нафтиридин-2(1H)-іл]сульфоніл}метил)-5-етилімідазолідин-2,4-діон;
(5S)-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4-дигідроізохінолін-2(1H)-іл]сульфоніл}метил)-5-етилімідазолідин-2,4-діон;
(5S)-5-({[6-(2-циклобутилпіримідин-5-іл)-3,4-дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон;
(5S)-5-метил-5-({[6-[2-(1-метилциклопропіл)піримідин-5-іл]-3,4-дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)імідазолідин-2,4-діон;
(5S)-5-циклопропіл-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4-дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)імідазолідин-2,4-діон;
та її фармацевтично прийнятні солі.
7. Фармацевтична композиція, що містить сполуку формули (І) або її фармацевтично прийнятну сіль за будь-яким з пп. 1-6 у поєднанні з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм.
8. Застосування сполуки формули (І) або її фармацевтично прийнятної солі за будь-яким з пп. 1-6 у виробництві медикаменту для застосування у лікуванні обструктивної хвороби дихальних шляхів.
9. Застосування за п. 8, де обструктивною хворобою дихальних шляхів є астма або хронічна обструктивна хвороба легень.
10. Спосіб лікування обструктивної хвороби дихальних шляхів, в якому пацієнту вводять терапевтично ефективну кількість сполуки формули (І) або її фармацевтично прийнятної солі за будь-яким з пп. 1-6.
Текст
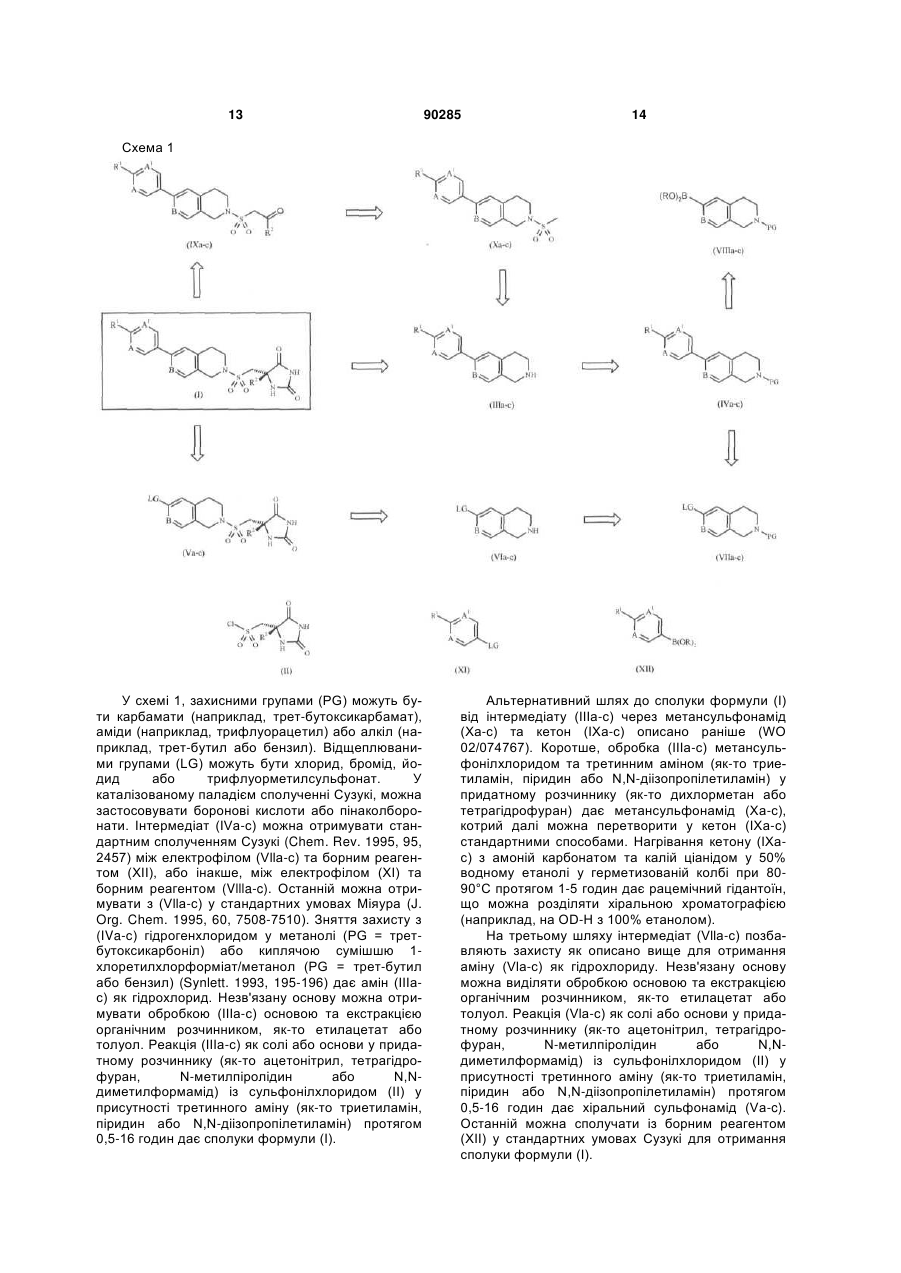
1. Сполука формули (І) або її фармацевтично прийнятна сіль C2 2 (19) 1 3 90285 4 9. Застосування за п. 8, де обструктивною хворобою дихальних шляхів є астма або хронічна обструктивна хвороба легень. 10. Спосіб лікування обструктивної хвороби дихальних шляхів, в якому пацієнту вводять терапев тично ефективну кількість сполуки формули (І) або її фармацевтично прийнятної солі за будь-яким з пп. 1-6. Заявлений винахід стосується деяких похідних гідантоїну, корисних в інгібуванні металопротеїназ, способів їх отримання, фармацевтичних композицій, що їх містять, та їх застосування у терапії. Металопротеїнази є надродиною протеїназ (ферментів), кількість відомостей про які разюче збільшилась в останні роки. Спираючись на структурні та функціональні міркування, ці ферменти класифікували на родини та надродини, як описано Ν.Μ Hooper (1994) FEBS Letters 354:1-6. Приклади металопротеїназ залучають родину матриксних металопротеїназ (ММР), як-то колагенази (ММР1, ММР8, ММР13, ММР18), желатинази (ММР2, ММР9), стромелізини (ММР3, ММР10, ММР11), матрилізини (ММР7, ММР26), металоеластаза (ММР12), енамелізин (ММР19), типів мембран МТ-ММР (ММР14, ММР15, ММР16, ММР17, ММР24, ММР25) та інші (ММР20, ММР21, ММР22, MMP23a/b, MMP28); родину ADAM (дезінтегрин, металопротеїназа, також відомі як репролізин, адамалізин або MDC), яке тепер охоплює 32 відомі ADAM з активністю секретази та шедази, якто TNF-перетворювальний фермент (ADAM17), та 18 відомих ADAMTS (дезінтегрин тромбоспондин металопротеїнази), що охоплюють агреканази (ADAMTS4, ADAMTS5); родину астацину, яке залучає ферменти, як-то проколаген-оброблювальна протеїназа (РСР); та інші металопротеїнази, як-то родину ендотелін-перетворювальних ферментів та родину ангіотензин-перетворювальних ферментів. Металопротеїнази вважають важливими у великому надлишку фізіологічних процесів хвороби, що залучає перетворення тканин, як-то ембріональний розвиток, остеогенез та маточне перетворення при менструації. Це базовано на здатності металопротеїназ розщеплювати широкий діапазон матриксних субстратів, як-то колаген, протеоглікан та фібронектин. Металопротеїнази також вважають важливими в процесінгу або секреції біологічно важливих медіаторів клітин, як-то фактор некрозу пухлин (TNF); та процесингу посттрансліційного протеолізу або втраті біологічно важливих білків мембран, як-то lgE-рецептор низької спорідненості CD23 (для більш повного переліку дивись N. М. Hooper etal., (1997) Biochem J. 321:265-279). Металопротеїнази асоційовано з багатьма станами хвороб. Інгібування активності одної або більше металопротеїназ може бути корисним у станах хвороб, наприклад з групи: різні запальні та алергічні хвороби, як-то запалення суглобів (головним чином ревматоїдний артрит, остеоартрит та подагра), запалення шлунково-кишкового тракту (головним чином, запальна хвороба кишечнику, виразковий коліт та гастрит), запалення шкіри (го ловним чином, псоріаз, екзема, дерматит); метастаз або інвазія пухлини; в асоційованих хворобах, де деградація випереджає синтез позаклітинного матриксу, як-то остеоартрит; хвороба резорбції кісток (як-то остеопороз та хвороба Педжета); хвороби, асоційовані з аберантним ангіогенезом; посилене перетворення колагену, асоційоване з діабетом, періодонтальна хвороба (як-то періодонтит), укривання виразками рогівки, укривання виразками шкіри, післяопераційні стани (якто анастомоз товстої кишки) та загоювання ран на шкірі; демієлінізуюча хвороби центральної та периферійної нервових систем (як-то розсіяний склероз); хвороба Альцгеймера; перетворення позаклітинного матриксу, спостережене в серцевосудинних хворобах, як-то рестеноз та атеросклероз; та хронічні обструктивні хвороби легенів, COPD. ММР12, також відома як еластаза макрофагів або металоеластаза, було спочатку клоновано у миші Shapiro et al [1992, Journal of Biological Chemistry 267: 4664] та у людині тою ж групою у 1995. ММР12 переважно експресується в активованих макрофагах, та показана як секретована альвеолярними макрофагами курців [Shapiro et al, 1993, Journal of Biological Chemistry, 268: 23824], а також у пінистих клітинах у атеросклеротичних ураженнях [Matsumoto et al, 1998, Am. J. Pathol. 153: 109]. Мишача модель COPD має основою провокаційну пробу миші димом сигарет протягом 6 місяців, дві сигарети на добу 6 діб на тиждень. У мишей дикого типу розвивалася легенева емфізема після цього поводження. Коли ММР12нокаутних мишей тестували у цій моделі, у них не розвивалася значна емфізема, показуючи, що ММР12 є ключовим ферментом у патогенезі COPD. Роль ММР, як-то ММР12. у COPD (емфізема та бронхіт) обговорено Anderson та Shinagawa, 1999, Current Opinion у Anti-inflammatory та Immunomodulatory Investigational Drugs 1(1): 29-38. Зараз виявлено, що паління посилює інфільтрацію макрофагів та експресію макрофагами ММР-12 у бляшках сонної артерій людини Kangavari [Matetzky S, Fishbein MC etal., Circulation 102:(18), 36-39 Suppl. S, Oct31, 2000]. MMP9 (Желатиназа В; Колагеназа 92кДа тип IV; Желатиназа 92кДа) є секретованим білком, котрий спершу очищали, тоді клонували та секвенсували, у 1989 [S.M. Wilhelm et al (1989) J. Biol. Chem. 264 (29): 17213-17221; опубліковано у J. Biol. Chem. (1990) 265 (36): 22570]. Нещодавний огляд ММР9 дає чудову інформацію та посилання стосовно цієї протеази: Т.Н. Vu & Ζ. Werb (1998) (Υ: Matrix Metalloproteinases, 1998, edited by W.C. Parks & R.P. Mecham, pp. 115-148, Academic Press. 5 ISBN 0-12-545090-7). Наступні посилання зроблено з цього огляду Т.Н. Vu & Ζ. Werb (1998). Експресія ММР9 є обмеженою звичайно кількома типами клітин, як-то трофобласти, остеокласти, нейтрофіли та макрофаги. Однак, експресію можна індукувати у цих же клітинах та в інших типах клітин кількома медіаторами, як-то піддаванням клітин дії факторів росту або цитокінів. Це є тими ж медіаторами, що часто залучені в ініціацію запальної реакції. Як і інші секретовані ММР, ММР9 виділяється як неактивний профермент, котрий далі розщеплюється з утворенням ферментативно активного ферменту. Протеази, потрібні для цієї активації in vivo, невідомі. Співвідношення активної ММР9 з неактивним ферментом є далі регульованим in vivo взаємодією із ТІМР-1 (тканинний нгібітор металопротеїнази -1), природно існуючим білком. ТІМР-1 приєднується до Стермінального регіону ММР9, призводячи до інгібування каталітичного домену ММР9. Співвідношення індукованої експресії проММР9, розщеплення проММР9 до активної ММР9 та присутність ТІМР-1 комбінуються для визначення кількості каталітично активної ММР9, котра є присутньою на локальній ділянці. Протеолітично активна ММР9 атакує субстрати, а саме желатин, еластин, та нативні колагени типу IV та типу V; вона не має активність, проти нативного колагену типу І, протеогліканів або ламінінів. Є зростаюча кількість даних стосовно ролі ММР9 у різних фізіологічних та патологічних процесах. Фізіологічні ролі охоплюють інвазію трофобластів ембріону через епітелій матки на ранніх стадіях імплантації ембріону; деяку роль у рості та розвитку кісток; та міграцію запальних клітин з судинної мережі у тканини. Виділення ММР9, виміряне ферментним імуноаналізом, було дуже посиленим у рідинах та у АМ-надосадкових рідинах від нелікованих астматиків порівняно з іншими популяціями [Am. J. Resp. Cell & Моl. Biol., Nov 1997, 17 (5): 583-591]. Також, посилену експресію ММР9 спостережено у деяких інших патологічних станах, тим залучаючи ММР9 у процеси хвороб, як-то COPD, артрит, метастази пухлин, хвороба Альцгеймера, розсіяний склероз, та руйнування бляшок в атеросклерозі, призводячи до гострих коронарних станів, як-то інфаркт міокарда. Ряд інгібіторів металопротеїназ відомі (дивись, наприклад, огляди інгібіторів ММР Beckett R.P. та Whittaker Μ., 1998, Exp. Opin. Ther. Patents, 8(3): 259-282; та by Whittaker M. etal, 1999, Chemical Reviews 99(9): 2735-2776). WO 02/074767 розкриває похідні гідантоїну формули 90285 6 що є корисними як інгібітори ММР, особливо як потужні інгібітори ММР12. Нами розкрито наступну групу похідних гідантоїну, що є інгібіторами металопротеїназ та представляють особливий інтерес в інгібуванні ММР, як-то ММР12 та ММР9. Сполуки заявленого винаходу мають корисну ефективність, селективність та/або фармакокінетичні властивості. Згідно із заявленим винаходом запропоновані сполуки формули (І) де R1 - циклобутил або циклопропіл; вказаний циклопропіл, як варіант, крім того заміщено СН3, CN або одним або двома атомами флуору; R2 - (С1-3)-алкіл або циклопропіл; а А, А1 та В, незалежно, - СН або N; та їх фармацевтично прийнятні солі. Сполуки формули (І) можуть бути у формі енантіомерів. Треба розуміти, що усі енантіомери, діастереомери, рацемати та їх суміші охоплені рамками винаходу. Сполуки формули (І) можуть також бути у формі різних таутомерів. Усі можливі таутомерні форми та їх суміші охоплені рамками винаходу. В одному втіленні R1 - циклопропіл; вказаний циклопропіл, як варіант, крім того заміщено одним або двома атомами флуору. В одному втіленні R1 - циклопропіл. В одному втіленні R2 - метил або етил. В одному втіленні R2 - метил. В одному втіленні А та А1, кожний, - N. У ще одному втіленні А - N, а А1 - СН. В одному втіленні В - N. У ще одному втіленні В - СН. В одному втіленні R1 - циклопропіл; R2 - метил або етил; А та А1, кожний, - N; а В -СН. В одному втіленні R1 - циклопропіл; R2 - метил або етил; А та А1, кожний, - N; а В - N. В одному втіленні R1 - циклопропіл; R2 - метил або етил; А - N, А1 - СН; а В - N. В одному втіленні R1 - циклопропіл; R2 - метил або етил; А - N, а А1 - СН; а В - СН. В одному втіленні R1 - циклопропіл; вказаний циклопропіл, як варіант, крім того заміщено СН3, CN або одним або двома атомами флуору; R2 (С1-3)-алкіл; а А, А1 та В, незалежно, - СН або N. Якщо не вказане інше, термін "(С1-3)-алкіл" стосується алкілу з лінійним або розгалуженим ланцюгом, що має від 1 до 3 атомів карбону, приклади таких груп охоплюють метил, етил, н-пропіл та і-пропіл. Приклади циклопропільного кільця, як варіант, крім того заміщеного одним або двома атомами флуору, охоплюють 1-флуор-1-циклопропіл, 2,2 7 дифлуор-1-циклопропіл циклопропіл: 90285 та 2,3-дифлуор-1 Приклади сполук винаходу охоплюють: (5S)-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)-5метилімідазолідин-2,4-діон; (5S)-5-({[6-(6-циклопропілпіридин-3-іл)-3,4дигідро-2,7-нафтиридин-2(1H)іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон; (5S)-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4дигідро-2,7-нафтиридин-2(1Н)іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон; (5S)-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4дигідро-2,7-нафтиридин-2(1H)іл]сульфоніл}метил)-5-етилімідазолідин-2,4-діон; (5S)-5-({[6-(2-циклопропілпіримідин-5-іл)-3,4дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)-5етилімідазолідин-2,4-діон; (5S)-5-({[6-(2-циклобутилпіримідин-5-іл)-3,4дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)-5метилімідазолідин-2,4-діон; (5S)-5-метил-5-({[6-[2-(1метилциклопропіл)піримідин-5-іл]-3,4дигідроізохінолін-2(1Н)іл]сульфоніл}метил)імідазолідин-2,4-діон; (5S)-5-Циклопропіл-5-({[6-(2циклопропілпіримідин-5-іл)-3,4-дигідроізохінолін2(1Н)-іл]сульфоніл}метил)імідазолідин-2,4-діон; та їх фармацевтично прийнятні солі. Кожна ілюстрована сполука - особливий та незалежний аспект винаходу. Сполуки формули (І) можуть бути у формі енантіомерів. Тому усі енантіомери, діастереомери, рацемати та їх суміші охоплені рамками винаходу. Різні оптичні ізомери можна виділяти розділенням рацемічної суміші сполук звичайними способами, наприклад, фракційною кристалізацією або РХВТ. Альтернативно оптичні ізомери можна отримувати асиметричним синтезом, або синтезом з оптично активних вихідних матеріалів. Коли є оптичні ізомери сполуки винаходу, нами розкрито усі індивідуальні оптично активні форми та їх комбінації як індивідуальні конкретні втілення винаходу, а також їх відповідні рацемати. Переважно сполуки формули (І) мають (5S)стереобудову, яку показано нижче: Коли є таутомери сполуки винаходу, нами розкрито усі індивідуальні таутомерні форми та їх комбінації як індивідуальні конкретні втілення винаходу. 8 Заявлений винахід охоплює сполуки формули (І) у формі солей. Придатні солі охоплюють солі, утворені з органічних або неорганічних кислот або органічних або неорганічних основ. Такі солі звичайно є фармацевтично прийнятними солями, хоча фармацевтично неприйнятні солі можуть бути корисними при отриманні та очистці конкретних сполук. Так солі охоплюють кислотно-адитивні солі, як-то гідрохлорид, гідробромід, цитрат, тозилат та малеат та солі утворені фосфатною кислот або сульфатною кислотами. Згідно з ще одним аспектом придатні солі є солями основ, як-то сіль лужного металу, наприклад, натрію або калію, сіль лужно-земельного металу, наприклад, кальцію або магнію, або сіль органічного аміну, наприклад, триетиламіну. Солі сполук формули (І) можуть бути утворені реакцією незв'язаної основи або іншої її солі з одним або більше еквівалентами прийнятної кислоти або основи. Сполуки формули (І) корисні, тому що вони виявляють фармакологічну активність у тваринах та є таким чином можливо корисними як фармацевтичні. Зокрема, сполуки винаходу є інгібіторами металопротеїназ та їх можна таким чином застосовувати у лікуванні хвороб або станів, опосередкованих ММР12 та/або ММР9, як-то астма, риніт, хронічні обструктивні хвороби легень (COPD), артрит (як-то ревматоїдний артрит та остеоартрит), атеросклероз та рестеноз, рак, інвазія та метастаз, хвороби, що стосуються руйнування тканин, ослаблення заміщень тазостегнового суглоба, періодонтальна хвороба, фіброзна хвороб, інфаркт та серцева хвороба, фіброз нирок та печінки, ендометріоз, хвороби, пов'язані з ослабленням зовнішньоклітинного матриксу, серцева недостатність, аневризми аорти, хвороби ЦНС, як-то хвороба Альцгеймера та розсіяний склероз (MS) та гематологічні розлади. Загалом, сполуки заявленого винаходу є потужними інгібіторами ММР9 та ММР12. Сполуки заявленого винаходу також виявляють гарну селективність стосовно відносної втрати інгібування різних інших ММР, як-то ММР8, ММР14 та ММР19. Відповідно, заявлений винахід стосується сполуки формули (І), або її фармацевтично прийнятної солі, що визначено вище, для застосування у терапії. Згідно з ще одним аспектом, винахід стосується застосування сполуки формули (І), або її фармацевтично прийнятної солі, що визначено вище, у виробництві медикаменту для застосування у терапії. Згідно з ще одним аспектом, винахід стосується застосування сполуки формули (І), або її фармацевтично прийнятної солі, що визначено вище, у виробництві медикаменту для застосування у лікуванні хвороб або станів, у котрих інгібування ММР12 та/або ММР9 є корисним. Згідно з ще одним аспектом, винахід стосується застосування сполуки формули (І), або її фармацевтично прийнятної солі, що визначено вище, у виробництві медикаменту для застосування у лікуванні запальної хвороби. 9 Згідно з ще одним аспектом, винахід стосується застосування сполуки формули (І), або її фармацевтично прийнятної солі, що визначено вище, у виробництві медикаменту для застосування у лікуванні обструктивної хвороби дихальних шляхів, як-то астма або COPD. Згідно з ще одним аспектом, винахід стосується застосування сполуки формули (І), або її фармацевтично прийнятної солі, що визначено вище, у виробництві медикаменту для застосування у лікуванні ревматоїдного артриту, остеоартриту, атеросклерозу, раку або розсіяного склерозу. У контексті опису термін "терапія" також охоплює "профілактику" якщо не вказане інше. Терміни "терапевтичний" та "терапевтично" слід вживати відповідно. Профілактика, як очікують, буде особливо важливою для лікування осіб, які потерпали від попереднього приступу, або вважаються такими, що мають посилений ризик хвороби або стану, що розглядають. Особи при ризику розвинення конкретної хвороби або стани загалом охоплюють тих, хто має родинну історію хвороби або стану, або тих, які ідентифіковані генетичним тестуванням або скринінгом як особливо сприйнятливих для розвинення хвороби або стану. Винахід крім того стосується способу лікування хвороби або стану, у котрих інгібування ММР12 та/або ММР9 є корисним, котрий полягає у призначенні пацієнту терапевтично ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі, що визначено вище. Винахід також стосується способу лікування обструктивної хвороби дихальних шляхів, наприклад, астми або COPD, котрий полягає у призначенні пацієнту терапевтично ефективної кількості сполуки формули (І) або її фармацевтично прийнятної солі, що визначено вище. Для вищезгаданого терапевтичного застосування дозування, безумовно, залежатиме від застосовуваної сполук, режиму застосування, потрібного лікування та розладу, який лікують. Добове дозування сполуки формули (І)/сіль (активний інгредієнт) може бути у межах від 0,001мг/кг до 75мг/кг, зокрема від 0,5 мг/кг до 30мг/кг. Ця добова доза може бути наданою окремими дозами, коли необхідно. Звичайно окремі дози містять приблизно 1мг - 500мг сполуки цього винаходу. Сполуки формули (І) та їх фармацевтично прийнятні солі можна застосовувати як такі але загалом можна застосовувати у формі фармацевтичної композиції, у котрій сполуку/сіль формули (І) (активний інгредієнт) поєднано з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм. Залежно від режиму застосування, фармацевтична композиція переважно міститиме від 0,05 до 99 мас.%, більш переважно від 0,10 до 70мас.% активного інгредієнту, та від 1 до 99,95мас.%, більш переважно від 30 до 99,90мас.% фармацевтично прийнятного ад'юванту, розріджувачу або 90285 10 носія, усі мас-проценти надані на основі загальної маси композиції. Звичайні способи селекції та отримання придатних фармацевтичних композицій описані, наприклад, у "Pharmaceuticals - The Science of Dosage Form Designs", Μ. Ε. Aulton, Churchill Livingstone, 1988. Таким чином, заявлений винахід також стосується фармацевтичної композиції, що містить сполуку формули (І) або її фармацевтично прийнятну сіль, що визначено вище, у поєднанні з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм. Винахід крім того стосується способу отримання фармацевтичної композиції винаходу, котрий полягає у змішуванні сполуки формули (І) або її фармацевтично прийнятної солі, що визначено вище, із фармацевтично прийнятним ад'ювантом, розріджувачем або носієм. Фармацевтичні композиції цього винаходу можна застосовувати стандартним чином для хвороби або стану, що потрібно лікувати, наприклад пероральним, місцевим, парентеральним, букальним, назальним, вагінальним або ректальним застосуванням або інгаляцією. Для цього сполуки цього винаходу можна формувати засобами, відомими у рівні техніки у форму, наприклад, таблеток, капсул, водних або олійних розчинів, суспензій, емульсій, кремів, мазей, гелів, назальних спреїв, супозиторіїв, мілко подрібнених порошків чи аерозолів для інгаляції, та для парентерального застосування (у т. ч. внутрішньовенно, внутрішньом'язово або вливанням) стерильних водних або олійних розчинів або суспензій або стерильних емульсій. На додаток до сполуки заявленого винаходу фармацевтична композиція цього винаходу може також містити, або бути співзастосованою (одночасно або послідовно) з одним або більше фармакологічних засобів у лікуванні одної або більше хвороб або станів, розглянутих вище, як-то "Symbicort" (торговий знак). Заявлений винахід крім того стосується способу отримання сполуки формули (І) або її фармацевтично прийнятної солі, що визначено вище, котрий полягає у: a) реакції сполуки формули (II) де R2 визначено у формулі (І), a L1 - відщеплювана група, із сполукою формули (III) (або її сіллю) 11 де R1, А, А1 та В визначені у формулі (І); або b) реакції сполуки формули (V) де R2 та В визначені у формулі (І), a LG - відщеплювана група; з похідним боронової кислоти формули (XII) де R1, А та А1 визначені у формулі (І); або с) реакції сполуки формули (IX) 90285 12 де R1, R2, А, А1 та В визначені у формулі (І); з амоній карбонатом та калій ціанідом; та, як варіант, після цього утворення її фармацевтично прийнятної солі. У вищенаведеному процесі (а), придатні відщеплювані групи L1 охоплюють галоген, особливо хлор, або трифлуорметилсульфонат. Реакцію переважно проводять у придатному розчиннику, як варіант, у присутності доданої основи протягом придатного періоду часу, звичайно 0,5-16 годин, при температурі від зовнішньої до температури дефлегмації. Звичайно застосовують розчинники, як-то Ν,Ν-диметилформамід, піридин, тетрагідрофуран, ацетонітрил, N-метилпіролідин або дихлорметан. При застосуванні додана основа може бути органічною основою, як-то триетиламін, Ν,Νдіізопропілетиламін, N-метилмофолін або піридин чи неорганічна основа, як-то карбонат лужного металу. Реакцію звичайно проводять при температурі довкілля протягом 0,5-16 годин, або до завершення реакції, яке визначають хроматографічно або спектроскопічно. Реакції сульфонілгалогенідів із різними первинними та вторинними амінами добре відомі у літературі, а варіації умов ясні для спеціалістів. Сульфонілхлориди формули (II) де L1 - хлор, a 2 R - Me, розкриті у WO 02/074767 та посиланнях, цитованих тут. Відповідні сполуки, де R2 - (С1-З)алкіл, можна отримувати аналогічними способами. Придатні способи отримання сполук формули (І) описані у схемі 1. 13 90285 14 Схема 1 У схемі 1, захисними групами (PG) можуть бути карбамати (наприклад, трет-бутоксикарбамат), аміди (наприклад, трифлуорацетил) або алкіл (наприклад, трет-бутил або бензил). Відщеплюваними групами (LG) можуть бути хлорид, бромід, йодид або трифлуорметилсульфонат. У каталізованому паладієм сполученні Сузукі, можна застосовувати боронові кислоти або пінаколборонати. Інтермедіат (IVa-c) можна отримувати стандартним сполученням Сузукі (Chem. Rev. 1995, 95, 2457) між електрофілом (Vlla-c) та борним реагентом (XII), або інакше, між електрофілом (XI) та борним реагентом (Vllla-c). Останній можна отримувати з (Vlla-c) у стандартних умовах Міяура (J. Org. Chem. 1995, 60, 7508-7510). Зняття захисту з (IVa-c) гідрогенхлоридом у метанолі (PG = третбутоксикарбоніл) або киплячою сумішшю 1хлоретилхлорформіат/метанол (PG = трет-бутил або бензил) (Synlett. 1993, 195-196) дає амін (ІІІас) як гідрохлорид. Незв'язану основу можна отримувати обробкою (ІІІа-с) основою та екстракцією органічним розчинником, як-то етилацетат або толуол. Реакція (ІІІа-с) як солі або основи у придатному розчиннику (як-то ацетонітрил, тетрагідрофуран, N-метилпіролідин або N,Nдиметилформамід) із сульфонілхлоридом (II) у присутності третинного аміну (як-то триетиламін, піридин або N,N-діізопропілетиламін) протягом 0,5-16 годин дає сполуки формули (І). Альтернативний шлях до сполуки формули (І) від інтермедіату (ІІІа-с) через метансульфонамід (Ха-с) та кетон (ІХа-с) описано раніше (WO 02/074767). Коротше, обробка (ІІІа-с) метансульфонілхлоридом та третинним аміном (як-то триетиламін, піридин або N,N-діізопропілетиламін) у придатному розчиннику (як-то дихлорметан або тетрагідрофуран) дає метансульфонамід (Ха-с), котрий далі можна перетворити у кетон (ІХа-с) стандартними способами. Нагрівання кетону (ІХас) з амоній карбонатом та калій ціанідом у 50% водному етанолі у герметизованій колбі при 8090°С протягом 1-5 годин дає рацемічний гідантоїн, що можна розділяти хіральною хроматографією (наприклад, на OD-H з 100% етанолом). На третьому шляху інтермедіат (Vlla-c) позбавляють захисту як описано вище для отримання аміну (Vla-c) як гідрохлориду. Незв'язану основу можна виділяти обробкою основою та екстракцією органічним розчинником, як-то етилацетат або толуол. Реакція (Vla-c) як солі або основи у придатному розчиннику (як-то ацетонітрил, тетрагідрофуран, N-метилпіролідин або N,Nдиметилформамід) із сульфонілхлоридом (II) у присутності третинного аміну (як-то триетиламін, піридин або N,N-діізопропілетиламін) протягом 0,5-16 годин дає хіральний сульфонамід (Va-c). Останній можна сполучати із борним реагентом (XII) у стандартних умовах Сузукі для отримання сполуки формули (І). 15 Інтермедіати (Vlla-b) зручно отримувати наступними способами. 1,2,3,4-тетрагідроізохінолін-інтермедіат (Vila) Способи синтезу 1,2,3,4-тетрагідроізохінолінів добре відомі у літературі. Класичним шляхом є реакція Померанца-Фритца бензальдегідів із захищеним діацеталем аміноацетальдегідом (Org. React. 1951, 6, 191), що дає ізохінолінове ядро, котре при каталітичному відновленні дає 1,2,3,4тетрагідро-ізохіноліни. Ще одним шляхом є реакція Бішлера-Напіранскі (Org. React. 1951, 6, 74) карбамат 2-фенілетанамінів із фосфорилхлоридом киплячому толуолі або ксилолах. Відновлення утвореного циклічного бензаміду літій алюмогід 90285 16 ридом у тетрагідрофурані (J. Med. Chem. 1987, 30(12), 2208-2216) або дибораном у тетрагідрофурані (J. Med. Chem. 1980, 23(5), 506-511) дає 1,2,3,4-тетрагідроізохінолін. Варіацією реакції Бішлера-Напіранскі є синтез Піктета-Спенглера (Org. React. 1951, 6, 151). У цій реакції аміди, карбамати або сульфонаміди 2-фенілетанамінів гріють із параформальдегідом та сильними протонними кислотами (як-то трифлуороцтова кислота, сульфатна кислота) або кислотами Льюіса у розчиннику (як-то дихлорметан, толуол, мурашина кислота) для отримання 1,2,3,4-тетрагідроізохіноліну одною стадією (Tetrahedron 2002, 58(8), 1471-1478). Схема 2 Переважно 1,2,3,4-тетрагідроізохіноліновий інтермедіат (Vlla) синтезують шляхом А, показаним у схемі 2. Це шлях є реакцією типу ФриделяКрафта N-[2-(3-бромфеніл)етил]-2,2,2трифлуорацетаміду із формальдегідом та сульфатною кислотою в оцтовій кислоті (Tetrahedron Lett. 1996, 37(31), 5453-5456), що дає суміш 6-бром- та 8-бромізомерів у співвідношенні 3:1. Заміщення трифлуорацетамідної групи ВОС-групою дає (Vlla). Регіоізомери розділити на цій стадії нелегко. 1,2,3,4-тетрагідро-2,7-нафтиридиновий інтермедіат (VIІb) На відміну від 1,2,3,4-тетрагідроізохінолінів, є кілька прикладів способів синтезу протягом 1,2,3,4-тетрагідро-2,7-нафтиридин у літературі. Одним важливим способом отримання 1,2,3,4тетрагідро-2,7-нафтиридин є регіоселективне каталітичне відновлення 2,7-нафтиридину (Eur. J. Схема 3 Med. Chem. Ther. 1996, 31(11), 875-888). Синтез 2,7-нафтиридину та деяких його похідних описано у літературі. Один класичний шлях має кілька етапів та починається з каталізованої кислотою конденсації малононітрилу із діетил 1,3ацетондикарбоксилатом (J. Chem. Soc. 1960, 35133515; дивись також J. Heterocycl. Chem. 1970, 7, 419-421). Трохи відмінний шлях до 2,7нафтиридину стосується окиснення 4-форміл-2,7нафтиридин для отримання 2,7-нафтиридин-4карбонової кислоти, а потім декарбоксилування (Synthesis 1973, 46-47). Повністю відмінний спосіб має основою внутрішню реакцію Дильса-Адлера N-(етоксикарбоніл)-N-(бут-3-иніл)амінометилпіразин та дає суміш 1,2,3,4-тетрагідро-2,7нафтиридину та 5,6,7,8-тетрагідро-1,7нафтиридину після гідролізу карбаматної групи (WO 02/064574). 17 Переважно 1,2,3,4-тетрагідро-2,7нафтиридиновий інтермедіат (Vllb) можна синтезувати як показано у схемах 3 та 4. У шляху В комерційно доступний 6-метоксинікотинальдегід обробляють сіллю літію Ν,N,N триметилетилендіаміну, тоді н-BuLi у гексанах, а потім йодом для отримання 4-йодо-6метоксинікотинальдегіду (Tetrahedron Lett. 1993, 34(39), 6173-6176). Йодосполуку сполучають із триметилсилілацетиленом у звичайних умовах Сонагашира-Хагіхара (Synthesis 1980, 627-630) та утворений 6-метокси-4 90285 18 [(триметилсиліл)етиніл]нікотинальдегід конденсують із амоній гідроксидом в етанолі для отримання 3-метокси-2,7-нафтиридину (Synthesis 1999, 2, 306-311). Регіоселективне каталітичне відновлення (Eur. J. Med. Chem. Ther. 1996, 31(11), 875-888) дає 6-метокси-1,2,3,4-тетрагідро-2,7-нафтиридин. Деметилування та N-захист ВОС-ангідридом та під кінець обробка утвореного треn-бутил 6-гідрокси3,4-дигідро-2,7-нафтиридин-2(1Н)-карбоксилату трифлатним ангідридом у двофазній системі дає (Vllb). Схема 4 У шляху С, комерційно доступний 5-бром-2метокси-4-метилпіридин у безводному тетрагідрофурані металізують = н-BuLi та тоді обробляють N,N-диметилформамідом для отримання 6метокси-4-метилнікотинальдегіду. Це перетворювали у трет-бутилімін із трет-бутиламін у дихлорметан. Металування літій 2,2,6,6тетраметилріреridide (Li-TMP) (J. Org. Chem. 1993, 58, 2463-2467) та додавання N,Nдиметилформамід дає іміноацетальдегід, котрий відновлюють натрій ціаноборогідрид у метанолі для отримання 2-трет-бутил-6-метокси-1,2,3,4тетрагідро-2,7-нафтиридин. Відщеплення метилу киплячою 48% гідробромідною кислотою та обробка трифлатним ангідридом у присутності основи дає (Vllb), захищений як трет-бутиламін. Спеціалістам буде ясно, що у процесах заявленого винаходу деякі можливо реакційні функціональні групи, як-то гідрокси- або аміногрупи у вихідних реагентах або інтермедіатах можуть потребувати захисту придатними захисними групами. Таким чином, отримання сполук винаходу може полягати на різних стадіях у додаванні та видаленні одної або більше захисних груп. Придатні захисні групи та способи їх додавання та видалення описані у 'Protective Groups in Organic Chemistry', edited by J.W.F. McOmie, Plenum Press (1973) та 'Protective Groups in Organic Synthesis', 3rd edition, T.W. Greene and P.G.M. Wuts, Wiley-lnterscience (1999). Сполуки винаходу та їх інтермедіати можна виділяти з їх реакційних сумішей та, якщо необхідно, очищати стандартними способами. Заявлений винахід далі пояснено наступними ілюстративними прикладами. Загальні способи 1 Н ЯМР та 13С ЯМР-спектри реєстрували на приладах Varian Inova 400МГц або Varian MercuryVX 300МГц. Центральні піки хлороформу-d ( Η 7,27чнм-1), диметилсульфоксиду-d6 ( Η 2,50чнм-1), ацетонітрилу-d4 ( Η 1,95чнм-1) або метанолу-d4 ( Η 3,31чнм-1) застосовували як внутрішній стандарт. Хроматографію на колонці проводили із силікагелем (0,040-0,063мм, Merck) із слабким надлишковим тиском (0,2-0,4бар). Колонку Kromasil KR-1005-C18(250 20мм, Akzo Nobel) та суміш ацетонітрил/вода із 0,1% ТФК при швидкості потоку 10мл/хвил застосовували для препаративної РХВТ. Якщо не вказане інше, вихідні матеріали були комерційно доступними. Усі розчинники та комерційні реагенти були лабораторного ґатунку та їх застосовували як отримані. Органічні фази від екстракції сушили безводним магній сульфатом якщо не встановлено інше. Органічні фази або розчини концентрували роторним випарюванням. Виходи не оптимізовано. Наступний спосіб застосовували для РХ-МС аналізу: Прилад Agilent 1100; Колонка Waters Symmetry 2,1 30мм; Mass APCI; швидкість потоку 0,7мл/хвил; Довжина хвилі 254 або 220нм; Роз 19 чинник А: вода + 0,1% ТФК; Розчинник В: ацетонітрил + 0,1% ТФК ; градієнт 15-95% /В 2,7 хвилин, 95% В 0,3 хвилин. Наступний спосіб застосовували для ГХ-МСаналізу: Прилад Hewlett Packard 5890 Series //; Колонка Agilent НР-5 (30м 0,32мм ID); Mac-селективний детектор Hewlett Packard 5971 Series ; Тиск 55кПа Не; Програма шафи 100°С (3 хвилини) до 300°С, 25°С/хвилин. Скорочення: ВОС-ангідрид ді-трет-бутил дикарбонат n-BuLi н-бутил літій ДХМ дихлорметан DIPEA N,N-діізопропілетиламін ДМФ N,N-диметилформамід ДМСО диметилсульфоксид EtOAc етилацетат ЕtOН етанол ГХ-МС газова хроматографія-мас спектрометрія LDA літій діізопропіламід МеОН метанол РХ-МС рідинна хроматографі-масспектроскопія PdCI2 х dppf 1,1 -біс(дифенілфосфіно)фероцен паладій(ІІ)дихлорид TEA триетиламін ТГФ тетрагідрофуран ТВМЕ трет-бутил-метил-етер ТФК трифлуороцтова кислота Трифлатний ангідрид - трифлуорметансульфоновий ангідрид (Tf2O) Приклад 1 (5S)-5-({[6-(2-Циклопропілпіримідин5-іл)-3,4-дигідроізохінолін-2(1Н)іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон [(4S)-4-Метил-2,5-діоксоімідазолідин-4іл]метансульфонілхлорид (0,020г, 0,087ммоль) у безводному тетрагідрофурані (0,40мл) додавали краплями при перемішуванні до розчину 6-[2(циклопропіл)піримідин-5-іл]-1,2,3,4тетрагідроізохіноліну (0,023г, 0,091ммоль), DIPEA (0,022мл, 0,13ммоль) та сухого ТГФ (0,50мл) при кімнатній температурі. Після завершення додавання розчин перемішували при кімнатній температурі протягом 2 годин та тоді переносили у воду з розсолом та екстрагували двічі EtOAc. Комбіновані органічні фази промивали розсолом, сушили, фільтрували та концентрували для отримання сирого продукту. Очистка препаративною РХВТ дала 0,021г (50%) заголовної сполуки як білий твердий продукт. PX-MC m/z 442 (M+1); 1 Н ЯМР (CD3CN) 8,97 (s, 2Н), 8,62 (br s, 1H), 7,52 (s, 1H), 7,51 (dd, 1H), 7,30 (d, 1H), 6,40 (br s, 1H), 4,48 (s, 2H), 3,54 (t, 2H), 3,51 (d, 1H), 3,42 (d, 90285 20 1H), 3,01 (t, 2H), 2,38 (m, 1H), 1,48 (s, 3H) та 1,23 (m, 4H) чнм-1. Вихідні матеріали отримували таким чином: 6-[2-(Циклопропіл)піримідин-5-іл]-1,2,3,4тетрагідроізохінолін трет-Бутил 6-[2-(циклопропіл)піримідин-5-іл]3,4-дигідроізохінолін-2(1Н)-карбоксилат (0,034г, 0,13ммоль) перемішували у ТФК (1,0мл) та ДХМ (1,0мл) при кімнатній температурі протягом ночі, тоді концентрували двічі, другий раз з доданим толуолом (5мл), для отримання трифлуорацетату заголовного продукту. 1 Н ЯМР (CD3OD) 8,87 (s, 2H), 7,60 (d, 1H), 7,59 (s, 1H), 7,37 (d, 1H), 4,43 (s, 2H), 3,55 (t, 2Н), 3,21 (t, 2Н), 2,27 (m, 1Н), та 1,14 (m, 4H) чим-1. Сирий продукт переносили у 1Μ розчин натрій карбонату (10мл) та екстрагували двічі ЕtOАс. Комбіновані органічні фази промивали розсолом, сушили, фільтрували та концентрували для отримання 0,023г (94%) заголовного білого твердого продукту. PX-MC m/z 252(M+1). 5-Бром-2-циклопропілпіримідин Заголовну сполуку отримували згідно з Hickey et al. (WO 00/066566). PX-MC m/z 199/201 (М+1); 1 Н ЯМР (CDCI3) 8,61 (s, 2Н), 2,30-2,18 (m, 1Н) та 1,15-1,10 (m, 4H) чим-1. трет-Бутил 6-[2-(циклопропіл)піримідин-5-іл]3,4-дигідроізохінолін-2(1Н)-карбоксилат 4:1 суміш (0,097г, 0,27ммоль) трет-бутил 6(4,4,5,5-тетраметил-1,3,2-діоксаборолан-2-іл)-3,4дигідроізохінолін-2(1H)-карбоксилату та трет-бутил 8-(4,4,5,5-тетраметил-1,3,2-діоксаборолан-2-іл)3,4-дигідроізохінолін-2(1Н)-карбоксилату, 5-бром2-циклопропілпіримідин (0,054г, 0,27ммоль), PdCI2 x dppf (0,0045г), 2М натрій карбонату (1,0мл), толуолу (4,0мл) та ЕtOН (1,0мл) продували сухим аргоном протягом 10 хвилин, тоді гріли у герметизованій колбі при 81°С протягом 6 годин. Чорний розчин фільтрували через скляне волокно, переносили у воду з розсолом та промивали двічі ЕtOАс. Комбіновані органічні фази сушили, фільтрували та концентрували із силіцій оксидом (5г). Хроматографія на колонці з ЕtOАс-гептанами (1:5 через 1:2) дала 0,034г (36%) заголовного білого твердого продукту. PX-MC m/z 352 (M+1); 1 Н ЯМР (CDCI3) 8,74 (s, 2H), 7,35 (dd, 1H), 7,29 (s, 1H), 7,22 (d, 1H), 4,62 (s, 2H), 3,68 (t, 2Н), 2,90 (t, 2Н), 2,30 (m, 1Н), 1,50 (s,9H), 1,18 (m, 2H) та 1,11 (m, 2Н)чнм-1. трет-Бутил 6-(4,4,5,5-тетраметил-1,3,2діоксаборолан-2-іл)-3,4-дигідроізохінолін-2(1Н)карбоксилат 3:1 суміш (0,49г, 1,6ммоль) трет-бутил 6-бром3,4-дигідроізохінолін-2(1Н)-карбоксилату та третбутил 8-бром-3,4-дигідроізохінолін-2(1Н)карбоксилату, біс(пінаколато)диборану (0,45г, 1,8ммоль), PdCI2 x dppf (0,039г, 0,048ммоль), калій ацетату (0,48г, 4,8ммоль) та ДМФ (8,0мл) гріли при 81°С протягом ночі. Розчинник випарювали, залишок переносили у воду з розсолом та промивали двічі ЕtOАс. Органічну фазу сушили, фільтрували та концентрували. Хроматографія на колонці з 21 90285 22 ЕtOАс-гептанами (1:10 через 1:4) дала 0,24г 4:1 суміш заголовного продукту та трет-бутил 8(4,4,5,5-тетраметил-1,3,2-діоксаборолан-2-іл)-3,4дигідроізохінолін-2(1Н)-карбоксилату. 1 Н ЯМР (CDCI3) 7,62 (d, 1H), 7,60 (s, 1H), 7,13 (d, 1H), 4,59 (s, 2H), 3,64 (t, 2H), 2,85 (t, 2Н), 1,50 (s, 9Н) та 1,35 (s, 12Н) чнм-1 (6-ізомер). 1 Н ЯМР (CDCI3) 7,69 (d, 1H), 7,24-7,14 (m's, 2H), 4,88 (s, 2H), 3,64 (t, 2H), 2,85 (t, 2Н), 1,50 (s, 9Н) та 1,35 (s, 12Н) чнм-1 (8-ізомер). трет-Бутил 6-бром-3,4-дигідроізохінолін-2(1Н)карбоксилат 6-Бром-2-(трифлуорацетил)-1,2,3,4тетрагідроізохінолін отримували двома етапами з [2-(3-бромфеніл)етил]аміну (4,0г, 20ммоль) наступним способом Stokker (Tetrahedron Lett. 1996, 37(31), 5453-5456). Хроматографія на колонці з ЕtOАс-гептанами (1:10 - 1:6) дала 2,3г (7,5ммоль) 3:1 суміші 6-бром-2-(трифлуорацетил)-1,2,3,4тетрагідроізохіноліну та 8-бром-2(трифлуорацетил)-1,2,3,4-тетрагідроізохіноліну. 1 Н ЯМР (CDCI3) 7,62 (d, 1H), 7,60 (s, 1H), 7,13 (d, 1H), 4,59 (s, 2H), 3,64 (t, 2H), 2,85 (t, 2H) та 1,50 (s, 9Н) та 1,35 (s, 12Н) чнм-1 (6-ізомер). 1 Н ЯМР (CDCI3) 7,69 (d, 1H), 7,24-7,14 (m, 2H), 4,88 (s, 2H), 3,64 (t, 2H), 2,85 (t, 2H) та 1,50 (s, 9Н) та 1,35 (s, 12Н) чнм-1 (8-ізомер). Цю суміш перемішували з абсолютним ЕtOН (100мл) та 25% амоній гідроксидом (10мл) при 60°С протягом 4 годин. Ще додавали 25% амоній гідроксид (15мл) та перемішування продовжували при кімнатній температурі протягом ночі. Летючі домішки випарювали до сирого аміну як білого твердого продукту. РХ-МС m/z 212/214 (М+1). Сухий ТГФ (50мл) та DIPEA (1,3мл, 7,5ммоль) додавали, а потім ВОС-ангідрид (1,8г, 8,2ммоль). Суміш перемішували протягом ночі при кімнатній температурі. Летючі домішки випарювали та за лишок переносили у воду. рН доводили до 2 1 Μ фосфатною кислотою та продукт екстрагували двічі ЕtOАс. Комбіновані органічні фази промивали розсолом, трохи підлужували насиченим натрій гідрокарбонатом, сушили, фільтрували та концентрували. Сирий продукт очищали хроматографією на колонці з ЕtOАс-гептанами (1:50 через 1:20) для отримання 2,24г (96%) 3:1 суміші заголовного продукту та трет-бутил 8-бром-3,4дигідроізохінолін-2(1Н)-карбоксилату. РХ-МС m/z 256/258 (М-56); 1 Н ЯМР (CDCI3) 7,31 (dd, 1Н), 7,30 (br s, 1H), 6,98 (d, 1H), 4,52 (s, 2H), 3,63 (t, 2H), 2,81 (t, 2H) та 1,50 (s, 9H) чнм-1 (6-ізомер). 1 Н ЯМР (CDCI3) 7,42 (dd, 1H), 7,12-7,01 (m's, 2H), 4,55 (s, 2H), 3,64 (t, 2H), 2,84 (t, 2Н) та 1,51 (s, 9H) чнм-1 (8-ізомер). Альтернативно, 6-(2-циклопропіл-піримідин-5іл)-1,2,3,4-тетрагідро-ізохінолін можна отримувати таким чином: a) 1,2,3,4-Тетрагідро-ізохінолін-6-ол гідробромід 6-Метокси-1,2,3,4-тетрагідро-ізохінолін гідрохлорид, отриманий як у WO 2004/26305, (18,9г, 94ммоль) у 48% водній гідробромідній кислоті гріли при 100°С протягом 12 годин та тоді охолоджували до 0°С. Твердий продукт відфільтровували, промивали трет-бутил-метил-етером та сушили. Вихід = 17,1г (79%) APCI-MC m/z: 150 [М+Н+]; 1 Н ЯМР (400МГц, ДМСО-d6) 2,91 (t, 2Н), 3,273,35 (m, 2Н), 4,13 (t, 2Н), 4,52 (s, 1Н), 6,59 (d, 1Н), 6,66 (dd, 1Н), 7,00 (d, 1Н), 9,07 (s, 2Н) чнм-1. b) 6-Трифлуорметансульфонілокси-3,4дигідро-1Н-ізохінолін-2-карбонової кислоти третбутил-естер Вищенаведені два етапи проводили як описано у Synthetic Communications, 25(20), 3255-3261, (1995). c) 6-(2-Циклопропіл-піримідин-5-іл)-3,4дигідро-1Н-ізохінолін-2-карбонової кислоти третбутил-естер 6-Трифлуорметансульфонілокси-3,4-дигідро1Н-ізохінолін-2-карбонової кислоти трет-бутилестер (11,51г, 30ммоль) розчиняли у ДМФ (250мл) та жовтий розчин продували аргоном (г). Калій ацетат (8,83г, 90ммоль), біс(пінаколато)дибор (8,38г, 33ммоль), PdCI2 dppf (1,22г, 1,5ммоль) та dppf (0,83г, 1,5ммоль) додавали та суміш знов продувалиаргоном. Суміш тоді гріли до 90°С протягом 2 годин. Трикалій фосфат моногідрат (18г, 78ммоль) додавали, а потім 2-циклопропіл-5-бромпіримідин (7,76г, 39ммоль) та перемішування продовжували протягом 5 годин при 90°С. Реакційну суміш виливали у насичений розчин натрій гідрокарбонату та екстрагували кілька разів етилацета том. Етилацетатний розчин сушили магній сульфатом, сушильний засіб відфільтровували та фільтрат випарювали. Залишок очищали флешхроматографією, елюючи сумішшю етилацетат: гептан (1:3) для отримання 8,1г (76%) заголовної сполуки як безбарвного твердого продукту. АРСІ-МС m/z: 352 [М+Н+]; 1 Н-ЯМР(СОСІ3): 8,77 (2Н, s), 7,36 (1Н, d), 7,31 (1Н, brs), 7,24 (1Н, d), 4,63 (2Н, s), 3,70 (2Н, brt), 2,92 (2Н, brt), 2,35 (1Н, m), 1,51 (9Н, s), 1,241,10 (4Н, m) чнм-1. d) 6-(2-Циклопропіл-піримідин-5-іл)-1,2,3,4тетрагідро-ізохінолін 6-(2-Циклопропіл-піримідин-5-іл)-3,4-дигідро1Н-ізохінолін-2-карбонової кислоти трет-бутилестер (9,49г, 27ммоль) розчиняли в етилацетаті (100мл) при 50°С, та додавали 1,5М гідрогенхлорид в етилацетаті (200мл). Через 1 годину суміш охолоджували до кімнатної температури та твердий продукт відфільтровували та сушили. 23 АРСІ-МС m/z: 252 [М+Н+]; 1 H-ЯMP(CD3OD): 9,35 (2Н, s), 7,76-7,70 (2Н, brs+brdd), 7,46 (1Н, d), 4,47 (2Н, s), 3,57 (2Н, t), 3,25 (2Н, t), 2,46 (1Н, m), 1,51-1,45 (4Н, m) чнм-1. 13 С-ЯМР(СО3ОD): 168,39, 155,82, 134,34, 132,65, 132,52, 131,32, 129,22, 128,74, 126,69, 45,56, 42,69, 26,17, 16,51, 14,11чнм-1. Дигідрохлорид (8,82г, 27ммоль) суспендували у воді (100мл) та додавали 2М NaOH (300мл). Суміш тоді екстрагували сумішшю 4:1 етилацетат / діетил-етер (4 300мл). Комбіновані органічні фази сушили безводним калій карбонатом, фільтрували та випарювали для отримання заголовної сполуки як незв'язаної основи (6,65г). АРСІ-МС m/z: 252 [М+Н+]; 1 H^MP(CD3OD): 8,81 (2Н, s), 7,43-7,38 (2Н, d+s), 7,18 (1Н, d), 3,99 (2Н, s), 3,10 (2Н, t), 2,90 (2Н, t), 2,25 (1Н, m), 1,18-1,06 (4Н, m) чнм-1. 13 С-ЯМР(СО3СD): 171,53, 155,83, 137,05, 137,01, 133,50, 132,32, 128,51, 128,36, 125,20, 48,35, 44,28, 29,49, 18,38, 11,16 чнм-1. Приклад 2 (5S)-5-({[6-(6-Циклопропілпіридин-3іл)-3,4-дигідро-2,7-нафтиридин-2(1Н)іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон Заголовну сполуку отримували з 6-(6циклопропілпіридин-3-іл)-1,2,3,4-тетрагідро-2,7нафтиридин гідрохлориду (0,63ммоль) та [(4S)-4метил-2,5-діоксоімідазолідин-4іл]метансульфонілхлориду (0,70ммоль) загальним способом прикладу 1. Хроматографія на колонці ЕtOАс та ЕtOАс-МеОН (9:1) як елюентами дала 0,060г майже чистого продукту. Перекристалізація з 99% ЕtOН дала 0,019г (7,0%) заголовної сполуки як білий твердий продукт. РХ-МС m/z 442 (М+1); 1 Н ЯМР (ДМСО-d6) 10,8 (s, 1H), 9,06 (d, 1H), 8,49 (s, 1H), 8,26 (dd, 1H), 8,06 (s, 1H), 7,83 (s, 1Н), 7,39 (d, 1Н), 4,45 (s, 2Н), 3,61 (d, 1Н), 3,48 (d, 1Н), 3,46 (m, 2Н), 2,97 (m, 2Н), 2,16 (m, 1Н), 1,34 (s, ЗН) та 1,02-0,94 (m, 4Н) чнм-1. Вихідні матеріали отримували таким чином: 6-(6-Циклопропілпіридин-3-іл)-1,2,3,4тетрагідро-2,7-нафтиридин гідрохлорид трет-Бутил 6{[(трифлуорметил)сульфоніл]окси}-3,4-дигідро-2,7нафтиридин-2(1Н)-карбоксилат (0,34г, 0,90ммоль), 2-циклопропіл-5-(4,4,5,5-тетраметил-1,3,2діоксаборолан-2-іл)піридин (0,20г, 0,82ммоль), PdCI2 x dppf (0,050г), насичений натрій карбонат (2мл), ЕtOН (4мл) та толуол (4мл) перемішували при 80°С протягом 2 годин. Розчин охолоджували до кімнатної температури, переносили у воду (15мл) та екстрагували три рази EtOAc-Et2O. Комбіновані органічні фази сушили, фільтрували та концентрували. Очистка хроматографією на колонці з ЕtOАс-гептанами (1:1 через 3:1) та ЕtOАс 90285 24 МеОН (9:1) як елюенти дала 0,22г (70%) третбутил 6-(6-циклопропілпіридин-3-іл)-3,4-дигідро2,7-нафтиридин-2(1Н)-карбоксилату як білий твердий продукт. PX-MC m/z 352 (M+1). Цей матеріал розчиняли у ЕtOАс (5мл) та перемішували із 1,5М гідрогенхлоридом у ЕtOАс (5мл) при 50°С протягом 4 годин. Розчинник випарювали до сирої заголовної сполуки (0,63ммоль) з кількісним виходом. 2-Циклопропіл-5-(4,4,5,5-тетраметил-1,3,2діоксаборолан-2-іл)піридин 0,5М Цинк хлорид у ТГФ (5,5мл, 2,8ммоль) додавали до розчину 0,5М циклопропілмагній броміду у ТГФ (5,5мл, 2,8ммоль) під аргоном. Розчин перемішували при кімнатній температурі протягом 2 годин з утворенням кашки. До неї додавали одною порцією 2,5-дибромпіридин (0,65г, 2,8ммоль) та PdCI2 x dppf (0,041г, 0,050ммоль). Через кілька хвилин спостерігали виділення теплоти та кашка гусла, виділення теплоти знижувалося та кашку перемішували при кімнатній температурі протягом ночі. Реакційну суміш виливали у насичений розчин натрій гідрокарбонату та екстрагували етером. Етерну фазу сушили, фільтрували та концентрували, тоді знов розчиняли у ДХМ та переносили на тонкий шар силікагелю, який промивали ДХМ та промивки концентрували. Залишок переносили в етер та промивали 1,0М гідрохлоридною кислотою. Кислотну водну фазу підлужували 2,0М натрій гідроксидом та продукт знов екстрагували етером. Комбіновані етерні фази промивали розсолом, сушили, фільтрували та концентрували для отримання 0,28г (50%) 5-бром-2циклопропілпіридину як жовтого масла. РХ-МС m/z 197,9/199,9 (М+1); 1 Н-ЯМР(СОСІ3) 8,48 (d, 1H), 7,63 (dd, 1H), 7,04 (d, 1H), 1,99 (m, 1H), 1,03-0,98 (m, 4Н) чнм-1. 5-Бром-2-циклопропілпіридин (0,21г, 1,1ммоль), біс(пінаколато)дибор (0,31г, 1,2ммоль) та калій ацетат (0,32г, 3,2ммоль) суспендували у діоксані (10мл). Кашку дегазували аргоном протягом 10 хвилин та тоді додавали PdCI2 x dppf (0,026г). Реакційну суміш гріли до 80°С протягом 15 годин та тоді, після охолодження до кімнатної температури, фільтрували через шар броунмілериту. Фільтрат концентрували для отримання чорного масла, що розчиняли в етері та екстрагували чотири рази 1,0М натрій гідроксидом. Комбіновані жовті водні фази охолоджували до 10°С, підкислювали 2,5М гідрохлоридною кислотою до рН 6,5 та тоді екстрагували етером. Комбіновані органічні фази сушили безводним магній сульфатом магній, фільтрували та концентрували для отримання 0,27г (103%) заголовного продукту як жовтого масла, що повільно тверднуло. 1Н-ЯМР показав чистоту потрібного продукту приблизно 60-65%, з пінаколбораном як головним забрудненням. Сирий матеріал застосовували без наступної очистки. ГХ-МС m/z 245,2 (М+), 244,2 (М-1); 1 Н-ЯМР(СОСІ3) 8,78 (brs, 1H), 7,93 (dd, 1H), 7,10 (d, 1H), 2,10 (m, 1H), 1,34 (s, 12H), 1,10-1,00 (т, 4Н)чнм-1. 25 трет-Бутил 6{[(трифлуорметил)сульфоніл]окси}-3,4-дигідро-2,7нафтиридин-2(1Н)-карбоксилат Сирий 3-метокси-2,7-нафтиридин (отримано з 4,4ммоль 6-метокси-4[(триметилсиліл)етилніл]нікотинальдегіду) гідрогенували (тиск 30фунт/кв.дюйм) при кімнатній температурі з РtO2 (приблизно 0,1г) у НОАс (25мл) протягом 2,5 годин. Розчин фільтрували через шар броунмілериту та прозорий фільтрат концентрували сублімаційною сушкою для отримання сирого 6-метокси-1,2,3,4-тетрагідро-2,7нафтиридин ацетату. РХ-МС m/z 165 (М+1). Цей матеріал гріли з дефлегмацією у 48% гідробромідній кислоті протягом 10 годин. Летючі домішки випарювали та залишок сушили під вакуумом при 45°С для отримання сирого 5,6,7,8тетрагідро-2,7-нафтиридин-3-ол гідроброміду (приблизно 0,70г). РХ-МС m/z 151 (M+1). Цей матеріал (приблизно 4,8ммоль) розчиняли у воді (13мл) та обробляли із ТГФ (33мл), Et3N (0,85мл, 6,0ммоль) та ВОС-ангідрид (1,6г, 7,3ммоль) при кімнатній температурі. Після перемішування при цій температурі протягом 6 годин розчин концентрували до одної треті його вихідного об'єму та залишок переносили у воду та екстрагували три рази ЕtOАс. Комбіновані органічні фази сушили, фільтрували та концентрували для отримання 0,80г (67% сир вихід) трет-бутил 6-гідрокси3,4-дигідро-2,7-нафтиридин-2(1Н)-карбоксилату як білого твердого продукту. РХ-МС m/z 251 (М+1), 195 (М-55). Цей матеріал (приблизно 5,4ммоль) розчиняли у двофазній системі толуолу (20мл) та 30% водного трикалій ортофосфату (20мл), та обробляли трифлатним ангідридом (1,6мл, 6,8ммоль) при 4°С [Org. Lett. 2002, 4(26), 4717-4718]. Льодяну баню видаляли, перемішування продовжували протягом 2 годин при кімнатній температурі, після чого дві фази були розділені. Водну фазу промивали толуолом. Комбіновані органічні фази промивали розсолом, сушили та концентрували. Очистка хроматографією на колонці з ЕtOАс-гептанами (2:1) як елюентом дала 0,45г (17% виходу) заголовного продукту. РХ-МС m/z 383 (М+1), 283 (М-99). 3-Метокси-2,7-нафтиридин При перемішуванні до розчину Ν,N,N'триметилетилендіаміну (1,9мл, 15ммоль) у безводному тетрагідрофурані (65мл) під аргоном при 70°С повільно додавали 1,6М н-BuLi у гексанах (9,0мл, 14ммоль). Після перемішування при -70°С протягом 15 хвилин додавали краплями 6метокси-нікотинальдегід (1,3г, 9,8ммоль). Після завершення додавання перемішування продовжували при -70°С протягом ще 15 хвилин. Тоді 1,6М н-BuLi у гексанах (10мл, 16ммоль) додавали краплями та перемішування продовжували при -45°С протягом 4 годин. Розчин охолоджували до -70°С та тоді розчин йоду (3,0г, 12ммоль) у безводному тетрагідрофурані (25мл) додавали краплями. Після завершення додавання перемішування продовжували при -70°С протягом 30 хвилин та тоді при 90285 26 кімнатній температурі протягом 3 годин. Сирий продукт переносили в етер (40мл) та промивали насиченим амоній хлоридом (2 40мл) та 5% натрій тіосульфатом (2 20мл). Органічну фазу сушили, фільтрували та концентрували. Очистка хроматографією на колонці з ЕtOАс-гептанами (1:1) як елюентом дала 0,41г (15% виходу) 4-йодо-6метоксинікотинальдегіду. PX-MC m/z 264 (M+1); 1 Н ЯМР (CDCI3) 9,95 (s, 1Н), 8,53 (s, 1Н), 7,32 (s, 1Н) та 3,98 (s, 3Н) чнм-1. 4-Йодо-6-метоксинікотинальдегід (0,41г, 1,6ммоль), триметилсилілацетилен (0,35мл, 2,8ммоль), PdCI2(PPh3)2 (каталітична кількість), Cul (каталітична кількість), триетиламін (2мл) та ТГФ (10мл) перемішували при 60°С протягом 2 годин. Летючі домішки випарювали та залишок переносили у воду та екстрагували етером. Органічну фазу сушили, фільтрували та концентрували. Очистка хроматографією на колонці з ЕtOАсгептанами (1:3) як елюентом дала 0,25г (68% виходу) 6-метокси-4-[(триметилсиліл)етилніл]нікотинальдегіду. PX-MC m/z 234 (M+1); 1 Н ЯМР (CDCI3) 10,4 (s, 1Н), 8,73 (s, 1Н), 6,84 (s, 1Н), 4,03 (s, 3Н) та 0,30 (s, 9H) чнм-1. 6-Метокси-4-[(триметилсиліл)етилніл]нікотинальдегід (0,25г, 1,1ммоль) та 7М аміак у МеОН (5мл) перемішували у герметизованій колбі при 80°С протягом ночі. Розчин концентрували, переносили у насичений натрій карбонат та екстрагували етером. Органічну фазу сушили, фільтрували та концентрували для отримання 0,20г заголовного продукту. ГX-MC m/z 160 (M+); 1 Н ЯМР (CDCI3) 9,41 (s, 1Н), 9,27 (s, 1Н), 8,47 (d, 1Н), 7,64 (d, 1Н), 7,03 (s, 1Н) та 4,12 (s, 3Н)чнм-1. Приклад 3 (5S)-5-({[6-(2-Циклопропілпіримідин5-іл)-3,4-дигідро-2,7-нафтиридин-2(1Н)іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон При перемішуванні до розчину 6-(2циклопропілпіримідин-5-іл)-1,2,3,4-тетрагідро-2,7нафтиридин гідрохлориду (0,12г, 0,42ммоль) у ДХМ (10мл) додавали TEA (0,12мл, 0,84ммоль), а потім краплями [(4S)-4-метил-2,5діоксоімідазолідин-4-іл]метансульфонілхлорид (0,090г, 0,40ммоль) у ТГФ (10мл) при -10°С. Суміш перемішували при кімнатній температурі протягом ночі, концентрували, переносили у воду (10мл) та екстрагували чотири рази ЕtOАс. Комбіновані органічні фази сушили, фільтрували та концентрували. Очистка препаративною РХВТ дала 0,12г (64%) заголовної сполуки як білий твердий продукт. PX-MC m/z 442,9 (М+1); 27 1 Н ЯМР (CD3OD) 9,05 (s, 2H), 8,43 (s, 1H), 7,81 (s, 1H), 4,63 (s, 2H), 3,40 (t, 2H), 3,38(q, 2Н), 3,00 (t, 2Н), 2,20 (m, 1 Η), 1,40 (s, 3Н) та 1,05 (m, 4H) чнм-1. Приклад 4 (5S)-5-({[6-(2-Циклопропілпіримідин5-іл)-3,4-дигідро-2,7-нафтиридин-2(1Н)іл]сульфоніл}метил)-5-етилімідазолідин-2,4-діон Заголовну сполуку отримували загальним способом прикладу 3, але застосовуючи [(4S)-4-етил2,5-діоксоімідазолідин-4-іл]метансульфонілхлорид. PX-MC m/z 457 (M+1). Вихідні матеріали отримували таким чином: 6-(2-Циклопропілпіримідин-5-іл)-1,2,3,4тетрагідро-2,7-нафтиридин гідрохлорид Суміш 2-трет-бутил-6-(2циклопропілпіримідин-5-іл)-1,2,3,4-тетрагідро-2,7нафтиридину (0,12г, 0,39ммоль), 1-хлоретил хлорформіату (1,0мл, 5,8ммоль) та толуолу (10мл) гріли з дефлегмацією протягом 4 годин із захистом від вологи (туба з кальцій хлоридом). Після концентрації до сухого стану темний залишок переносили у МеОН (10мл) та гріли з дефлегмацією протягом більше 3 годин. Активоване вугілля (1г) додавали та дефлегмацію продовжували протягом 20 хвилин. Тоді суміш фільтрували через броунмілерит та прозорий фільтрат концентрували для отримання заголовної сполуки (0,12г) як твердого продукту. PX-MC m/z 253 (M+1); 1 H ЯМР (СОСІ3) 9,22 (s, 2H), 8,57(s, 1H), 7,98 (s, 1H), 4,41 (s, 2H), 3,45 (t, 2H), 2,44 (m, 2H), 2,32 (m, 1H) та 1,21 (m, 4Н)чнм-1. 2-трет-Бутил-6-(2-циклопропілпіримідин-5-іл)1,2,3,4-тетрагідро-2,7-нафтиридин При перемішуванні до холодного (4°С) розчину 2-трет-бутил-6-гідрокси-1,2,3,4-тетрагідро-2,7нафтиридину (0,15г, 0,73ммоль) у піридині (5,0мл) повільно додавали трифлатний ангідрид (0,14мл, 0,80ммоль). Після завершення додавання суміш перемішували при 4°С протягом 30хвилин, гасили 5% розчином калій карбонату (10мл) та екстрагували чотири рази ДХМ. Комбіновані органічні фази сушили, фільтрували та концентрували для отримання сирого продукту. Хроматографія на колонці із ЕtOН-ТВМЕ (1:9) як елюентом дала 0,30г сирого трифлату як масла. РХ-МС m/z 339,2 (М+1). Трифлат розчиняли у діоксані (10мл) та додавали безводний калій ацетат (0,43г, 4,5ммоль), 2циклопропілпіримідин-4-боронову кислоту (0,14г, 0,89ммоль) та PdCI2 x dppf (0,0050г). Суміш дегазували аргоном, герметизували та перемішували при 90°С протягом ночі. Після охолодження розчин переносили у воду (20мл) та екстрагували три рази ЕtOАс. Комбіновані органічні фази промивали 90285 28 розсолом, сушили, фільтрували та концентрували. Хроматографія на колонці ЕtOН-ТВМЕ (1:9) та ТВМЕ-ЕtOН-ТЕА (20:2:1) дала 0,12г (53% за два етапи) заголовної сполуки як світло-коричневого твердого продукту. РХ-МС m/z 309 (М+1); 1 Н ЯМР (CDCI3) 9,05 (s, 2H), 8,45 (s, 1H), 7,38 (s, 1H), 3,97 (m, 2H), 2,95 (m, 4H), 2,00 (m, 1H), 1,21 (s, 9H), 1,11 (dt, 2H) та 1,09 (dt, 2H) чнм-1. 2-трет-Бутил-6-гідрокси-1,2,3,4-тетрагідро-2,7нафтиридин Розчин 2-трет-бутил-6-метокси-1,2,3,4тетрагідро-2,7-нафтиридину (5,1г, 23ммоль) та 45% гідробромідної кислоти в оцтовій кислоті (70мл) гріли герметизованій тубі при 100°С протягом 1 годин, охолоджували до кімнатної температури та концентрували. Залишок обережно розчиняли у 20% розчин калій карбонату (100мл) та екстрагували чотири рази ЕtOАс. Комбіновані органічні фази сушили, фільтрували та концентрували. Перекристалізація з ТВМЕ-гексанів дала 3,7г (77%) заголовної сполуки як білого твердого продукту. РХ-МС m/z 207 (М+1); 1 Н ЯМР (CDCI3) 7,21 (s, 1Н), 6,35 (s, 1Н), 4,77 (m, 2Н), 4,11 (m's, 4Н) та 1,31 (s, 9H) чнм-1. 2-Циклопропілпіримідин-4-боронова кислота Заголовну сполуку отримували з 4-бром-2циклопропілпіримідину (WO 00/066566) у 90% виходу (25ммоль) способом Li et al. (J. Org. Chem. 2002, 67, 5394-5397). РХ-МС засвідчила, що продукт складався з боронової кислоти та тримерного ангідриду (сим-бороксин). РХ-МС m/z 165 (М+1) та 439 (М+1). 2-трет-Бутил-6-метокси-1,2,3,4-тетрагідро-2,7нафтиридин При перемішуванні до розчину 2,2,6,6тетраметилпіперидину (9,0мл, 60ммоль) у сухому ТГФ (300мл) під аргоном при -20°С повільно додавали 1,6М н-BuLi у гексанах (40мл, 60ммоль) при 20°С. Після завершення додавання перемішування продовжували при -20°С протягом 40 хвилин. Тоді додавали краплями при -20°С розчин третбутил-[(6-метокси-4-метилпіридин-3іл)метилен]аміну (6,3г, 30ммоль) у сухому ТГФ (100мл). Суміш перемішували при -15 - -10°С протягом 1,5 годин, а тоді охолоджували до -20°С. Безводний ДМФ (6,5мл, 70ммоль) додавали краплями протягом 5 хвилин та перемішування продовжували при -10°С протягом 1,5 годин. Тоді додавали льодяну оцтову кислоту (60мл) у МеОН (250мл), а потім порціями натрій ціаноборогідрид (2,3г, 40ммоль) протягом 5 хвилин. Після перемішування протягом ночі розчинник випарювали та додавали повільно 20% розчин калій карбонату до рН 9. Суміш екстрагували чотири рази ТВМЕ. Комбіновані органічні фази промивали розсолом, сушили та концентрували для отримання сирого масла. Перегонка під вакуумом дала 5,2г (77%) заголовної сполуки як безбарвне масло, тчк.кип. 105-106°С/0,5мм Hg. ГХ-МС m/z 220,1 (М+); 1 Н ЯМР (CDCI3) 7,90 (s, 1Н), 6,55 (s, 1Н), 3,95 (s, 3Н), 3,81 (m, 3Н), 2,95-2,90 (m, 3Н) та 1,11 (s, 9Н)чнм-1. 29 трет-Бутил-[(6-метокси-4-метилпіридин-3іл)метилен]амін 2-Метокси-4-метилнікотинальдегід (1,8г, 12ммоль), трет-бутиламін (15мл), 3Å молекулярні сита (8г) та сухий ДХМ (10мл) змішували та тримали при кімнатній температурі із захистом від вологи (туба з кальцій хлоридом). Після двох діб суміш фільтрували та молекулярні сита промивали кілька разів сухим ДХМ. Комбіновані промивки концентрували для отримання 2,2г (89%) заголовної сполуки як сирого масла, що застосовували у наступному етапі. 1 Н ЯМР (CDCI3) 8,55 (br s, 1 Η), 8,50 (s, 1 Η), 6,50 (s, 1 Η), 3,96 (s, 3Н), 2,50 (s, 3Н) та 1,30 (s, 9Н)чнм-1. 2-Метокси-4-метилнікотинальдегід При перемішуванні до розчину 5-бром-2метокси-4-метилпіридину (2,6г, 13ммоль) у сухому ТГФ (40мл) під аргоном при -70°С додавали 1,6М н-BuLi у гексанах (8,1мл, 14ммоль) протягом 10 хвилин. Суміш перемішували при -70°С протягом 30 хвилин та тоді додавали порціями безводний ДМФ (1,2мл, 15ммоль) так, щоб тримати температуру -70°С. Після завершення додавання суміш перемішували при -70°С протягом 30 хвилин та тоді при кімнатній температурі протягом ночі. Реакцію гасили 1М гідрохлоридною кислотою (40мл) та тоді екстрагували три рази ТВМЕ. Комбіновані органічні фази промивали розсолом, сушили, фільтрували та концентрували. Хроматографія на колонці із ТВМЕ-світлим петролейним етером (1:1) як елюентом дала 1,8г (91%) заголовної сполуки як блідо-жовтого твердого продукту. PX-MC m/z 152 (M+1); 1 Н ЯМР (CDCI3) 10,1 (s, 1Н), 8,55 (s, 1Н), 6,61 (s, 1 Η), 4,05 (s, 3Н), 2,60 (s, ЗН) чнм-1. [(4S)-4-Етил-2,5-діоксоімідазолідин-4іл]метансульфонілхлорид Отримано, як описано у WO 02/074767 для [(4S)-4-метил-2,5-діоксоімідазолідин-4іл]метансульфонілхлориду. Приклад 5 (5S)-5-({[6-(2-Циклопропілпіримідин5-іл)-3,4-дигідроізохінолін-2(1H)іл]сульфоніл}метил)-5-етилімідазолідин-2,4-діон Заголовну сполуку отримували розділенням хіральною хроматографією (±)-5-({[6-(2циклопропілпіримідин-5-іл)-3,4-дигідроізохінолін2(1H)-іл]сульфоніл}метил)-5-етилімідазолідин-2,4діону способом, описаним у WO 02/074767. Препаративно-хроматографічні дані: Колонка Chiracel OD-H (L 25см, 2см) Daicel Chemical Industries Ltd. Елюент: 100% EtOH потік: 15мл/ хвилин Визначення УФ 254нм. Аналітичні хроматографічні дані 90285 30 Колонка Chiralcel OD-H (L 15см, 0,46см) Daicel Chemical Industries Ltd. Елюент: 100% EtOH потік: 0,30мл/ хвилин Визначення УФ 254/220нм. Час утримування (tR) - дивись нижче (S)-енантіомер (tR 13,0 хвилин) 1 Н ЯМР (ДМСО-d6) 10,79 (br s, 1Η), 8,94 (s, 2H), 7,97 (br s, 1H), 7,60 - 7,56 (m, 2H), 7,33 - 7,28 (m, 1H), 4,41 (s, 2H), 3,59 - 3,40 (m, 4H), 2,96 (t, J = 6,2 Гц, 2Н), 2,28 - 2,21 (m, 1H), 1,65 (q, J= 7,6 Гц, 2H), 1,11-1,00 (m, 4H) та 0,78 (t, J= 7,5 Гц, 3H) чнм1 . (R)-енантіомер (tR 18,3 хвилин) 1 H ЯМР (ДМСО-d6) 10,79 (br s, 1H), 8,94 (s, 2H), 7,97 (br s, 1H), 7,60-7,56 (m, 2H), 7,32-7,28 (m, 1H), 4,41 (s, 2H), 3,58-3,40 (m, 4H), 2,96 (t, J = 6,2 Гц, 2Н), 2,28-2,21 (m, 1H), 1,65 (q, J = 7,6Гц, 2Н), 1,11-1,00 (m, 4H) та 0,78 (t, J = 7,5 Гц, 3Н) чнм-1. (±)-5-({[6-(2-Циклопропілпіримідин-5-іл)-3,4дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)-5етилімідазолідин-2,4-діон) Заголовну сполуку отримували з 6-(2циклопропілпіримідин-5-іл)-1,2,3,4-тетрагідроізохінолін способом, описаним у WO 02/074767. PX-MC m/z 456 (M+1); 1 Н ЯМР (ДМСО-d6) 10,78 (br s, 1Н), 8,94 (s, 2Н), 8,03 (br s, 1H), 7,61-7,56 (m, 2H), 7,30 (d, J = 8,5 Гц, 1H), 4,42 (s, 2H), 3,60-3,40 (m, 4H), 2,96 (t, J = 6,2 Гц, 2Н), 2,26-2,20 (m, 1H), 1,65 (q, J = 7,2Гц, 2Н), 1,10-1,01 (m, 4H) та 0,78 (t, J= 7,5 Гц, 3Н)чнм-1. Приклад 6 (5S)-5-({[6-(2-Циклобутилпіримідин5-іл)-3,4-дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)-5-метилімідазолідин-2,4-діон Заголовну сполуку отримували способом, описаним у прикладі 1. PX-MC m/z 456 (M+1); 1 Н ЯМР (300МГц, ДМСО-d6) 1,31 (s, 3Н), 1,83-2,12 (m, 2H), 2,26-2,45 (m, 4H), 2,96 (s, 2Н), 3,19-3,55 (m, 4Н), 3,78 (q, 1Н), 4,43 (s, 2Н), 6,92 (s, 1Н), 7,32 (d, 1Н), 7,60 (s, 2Н), 9,04 (s, 2Н), 10,81 (s, 1Н) чнм-1. Потрібні вихідні матеріали були також отримані загальними способами, описаними у прикладі 1: 1,1-Диметилетил 6-(2-циклобутилпіримідин-5іл)-3,4-дигідроізохінолін-2(1Н)-карбоксилат PX-MC m/z 366 (M+1); 1 Н ЯМР (300 МГц, CDCI3) 8,81 (s, 2H), 7,137,37 (m, 3Н), 4,55 (d, 2H), 3,64 (t, 2H), 2,82-2,90 (m, 2H), 2,30-2,50 (m, 6H), 1,84-2,14 (m, 1H), 1,45 (s, 9H) чнм-1. 6-(2-Циклобутилпіримідин-5-іл)-1,2,3,4тетрагідроізохіноліній хлорид PX-MC m/z 266 (M+1); Циклобутил(іміно)метанаміній хлорид 31 1 Н ЯМР (300 МГц, ДМСО-d6) 1,69-1,84 (m, 1H), 1,86-2,05 (m, 2H), 2,08-2,32 (m, 3Н), 3,29-3,42 (m, 1H), 8,85 (s, 4H) чнм-1. 5-Бром-2-циклобутилпіримідин ГX-MC m/z 211/213 (M); 1 Н ЯМР (300МГц, CDCI3) 1,88-2,17 (m, 2H), 2,36-2,46 (m, 4H), 3,78 (td, 1H), 8,72 (s, 2Н)чнм-1. Приклад 7 (5S)-5-Метил-5-({[6-[2-(1метилциклопропіл)піримідин-5-іл]-3,4-дигідроізохінолін-2(1Н)-іл]сульфоніл}метил)імідазолідин-2,4діон Заголовну сполуку отримували способом, описаним у прикладі 1. PX-MC m/z 456 (M+1); 1 Н ЯМР (300МГц, ДMCO-d6) 0,94 (q, 2H), 1,30 (d, 2H), 1,34 (s, 3Н), 1,54 (s, 3Н), 2,96 (t, 2H), 3,403,62 (m, 4H), 4,42 (s, 2H), 7,31 (d, 1H), 7,58 (d, 2H), 8,06 (s, 1H), 8,97 (s, 2H), 10,77 (s, 1Н)чнм-1. Потрібні вихідні матеріали були також отримані загальними способами, описаними у прикладі 1: 6-[2-(1-Метилциклопропіл)піримідин-5-іл]1,2,3,4-тетрагідроізохіноліній хлорид PX-MC m/z 266 (M+1); 1 Н ЯМР (300МГц, ДMCO-d6) 0,94 (q, 2Н), 1,30 (q, 2Н), 1,53 (s, 3Н), 2,96-3,14 (m, 2Н), 3,29-3,42 (m, 2H), 4,18-4,32 (m, 2H), 5,81 (s, 1H), 7,15-7,27 (m, 1H), 7,36 (t, 1H), 7,63 (d, 1Н), 8,98 (s, 2Н), 9,85 (s, 1Н) чнм-1. Іміно(1-метилциклопропіл)метанаміній хлорид 1 Н ЯМР (300МГц, ДМСО-d6) 0,46 (dd, 2Н), 0,91 (q, 2Н), 1,21 (s, 3Н), 7,35 (s, 4H) чнм-1. 5-Бром-2-(1-метил циклопропіл)піримідин PX-MC m/z 213/215 (М+1); 1 Н ЯМР (399,988 МГц, CDCI3) 0,93 (dd, 2Н), 1,35 (dd, 2Н), 1,54 (s, 3Н), 8,59 (s, 2H) чнм-1. Приклад 8 (5S)-5-Циклопропіл-5-({[6-(2циклопропілпіримідин-5-іл)-3,4-дигідроізохінолін2(1Н)-іл]сульфоніл}метил)імідазолідин-2,4-діон Заголовну сполуку отримували з 6-[2(циклопропіл)піримідин-5-іл]-1,2,3,4тетрагідроізохіноліну та (4S)-(4-циклопропіл-2,5 90285 32 діоксоімідазолідин-4-іл)метансульфонілхлориду загальним способом, описаним у прикладі 1. PX-MC m/z 468 (M+1); 1 Н ЯМР (400МГц, ДМСО-d6) 0,18 (q, 1Н), 0,33-0,56 (m, 3Н), 1,02-1,17 (m, 5H), 2,24 (dd, 1Н), 2,96 (t, 2Н), 3,40-3,82 (m, 4Н), 4,43 (s, 2Н), 7,31 (t, 1Н), 7,58 (d, 2Н), 7,95 (s, 1Н), 8,94 (s, 2Н), 10,74 (s, 1Н) чнм-1. Потрібні вихідні матеріали отримували таким чином: 2-Бензилсульфаніл-1-циклопропіл-етанон Бензилмеркаптан (15,6мл, 0,133моль) перемішували у ДХМ (100мл), додавали триетиламін (20,5мл, 0,146моль), суміш охолоджували у бані лід/ацетон та 2-бром-1-циклопропіл-етанон, отриманий як у WO 03/074495, (21,77г, 0,133моль) у ДХМ (100мл) додавали краплями. Суміш перемішували протягом 48 годин, промивали водою, тоді розсолом, сушили натрій сульфатом та випарювали. ГХ-МС m/z 206 (М); 1 Н ЯМР (400МГц, CDCI3) 0,86-0,91 (m, 2H), 0,99-1,03 (m, 2H), 2,05-2,16 (m, 1Н), 3,22 (s, 2H), 3,64 (s, 2H), 7,18-7,31 (m, 5H) чнм-1. Цей матеріал застосовували без наступної очистки. Бензилсульфанілметил-5-циклопропілімідазолідин-2,4-діон 2-Бензилсульфаніл-1-циклопропіл-етанон (27,55г, 0,133моль) розчиняли в етанолі (250мл) та розподіляли у колби 20 40мл. Натрій ціанід (6,52г, 0,133моль) та амоній карбонат (64г, 0,667моль) розчиняли у воді (250мл) та розподіляли у колби, котрі далі герметизували та гріли при 90°С протягом 5 годин. Після охолодження до кімнатної температури вміст колб поєднували, додавали ТВМЕ та суміш промивали водою ( 2), розсолом ( 1), а тоді сушили натрій сульфатом. Випарювання тоді дало сирий продукт (16,5г, 45%). Цей матеріал абсорбували на силіцій діоксид та хроматографували (5 9,5см колонка силіцій діоксиду), елюючи ізо-гексаном - суміш 50% етилацетат : ізо-гексан для отримання заголовної сполуки (11,81г, 32,1%). PX-MC m/z 277 (M+1); 1 НЯМР (400МГц, CDCI3) 0,23-0,59 (m, 4Н), 1,12-1,19 (m, 1Н), 2,87 (dd, 2H), 3,67-3,74 (m, 2Н), 6,06 (s, 1Н), 7,15-7,33 (m, 5Н), 8,66 (s, 1Н)чнм-1. Ізомери розділяли на напів-препаративній колонці Chiralpak AD. Елюент: 65% Етанол / 35% ізо-гексан Концентрація: 50мг/мл Об'єм уведення: 2мл Час виконання: 21хвил. Хіральний аналіз на колонці Chiralpak AD 25 0,46см, 0,7мл / хвилину дала часи утримування 8,9 та 11,5 хвилин. Пізніший ізомер застосовували для наступної реакції. 1 Н ЯМР (400МГц, CDCI3) 0,19-0,58 (m, 4H), 1,10-1,24 (m, 1H), 2,86 (dd, 2H), 3,62-3,78 (m, 2H), 5,87 (s, 1H), 7,16-7,34 (m, 5H), 8,51 (s, 1H) чнм-1. (4S)-(4-Циклопропіл-2,5-діоксоімідазолідин-4іл)метансульфонілхлорид 33 (5S)-Бензилсульфанілметил-5-циклопропілімідазолідин-2,4-діон (770мг, 2,78ммоль) розчиняли у 90% оцтовій кислоті (100мл) та охолоджували у бані лід/вода і продували газуватий хлор протягом 10 хвилин. Реакційну суміш сушили сублімацією для отримання заголовної сполуки як білого твердого продукту (640мг, 91%). 1 Н ЯМР (400МГц, ТГФ) 0,37-0,65 (m, 4Н), 1,25-1,33 (m, 1 Η), 4,62 (dd, 2H), 7,39 (s, 1H), 9,86 (s, 1Н)чнм-1. Аналізи виділених ферментів ММР12 Рекомбінантний каталітичний домен ММР12 людини можна експресувати та очищати як описано Parkar A.A. et al, (2000), Protein Expression and Purification, 20, 152. Очищений фермент можна застосовувати для дослідження інгібіторної активності таким чином: ММР12 (50нг/мл кінцева концентрація) інкубують протягом 60 хвилин при кімнатній температурі з синтетичним субстратом McaPro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (10мкМ) у буфері для аналізу (0,1Μ "Tris-HCI" (торговий знак), рН 7,3, що містить 0,1Μ NaCI, 20мМ СаСІ2, 0,020мМ ZnCI та 0,05% (маса/об'єм) детергенту "Brij 35" (торговий знак)) у присутності (10 концентрацій) або відсутності інгібіторів. Активність визначають виміром флуоресценції при випр 320нм та збудж 405нм. Процент інгібування розраховують таким чином: % Інгібування = [Флуоресценція+інгібітор - Флуоресценціяфону] / [Флуоресценціябез інгібітору - Флуоресценціяфону]. ММР8 Очищений про-ММР8 отримували від Calbiochem. Фермент (при 10мкг/мл) активують паміно-феніл-меркурій ацетатом (АРМА) при 1мМ протягом 2,5 годин, 35°С. Активований фермент можна застосовувати для дослідження інгібіторної активності таким чином: ММР8 (200нг/мл кінцева концентрація) інкубують протягом 90 хвилин при 35°С (80% Н2О) з синтетичним субстратом McaPro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (12,5мкМ) у буфері для аналізу (0,1Μ "Tris-HCI" (торговий знак), рН 7,5, що містить 0,1Μ NaCI, 30мМ СаСІ2, 0,040мМ ZnCI та 0,05% (маса/об'єм) детергенту "Brij 35" (торговий знак)) у присутності (10 концентрацій) або відсутності інгібіторів. Активність визначають виміром флуоресценції при випр 320нм та збудж 405нм. Процент інгібування розраховують таким чином: % Інгібування = [Флуоресценція+інгібітор - Флуоресценціяфону] / [Флуоресценціябез інгібітору - Флуоресценціяфону]. ММР9 Рекомбінантний каталітичний домен ММР9 людини експресували та очищали хроматографією 90285 34 на колонці з хелатом Zn, а потім хроматографією на колонці з афінітетом до гідроксамату. Фермент можна застосовувати для дослідження інгібіторної активності таким чином: ММР9 (5нг/мл кінцева концентрація ) інкубують протягом 30 хвилин при кімнатній температурі з синтетичним субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (5мкМ) у буфері для аналізу (0,1Μ "Tris-HCI" (торговий знак), рН 7,3, що містить 0,1Μ NaCI, 20мМ СаСІ2, 0,020мМ ZnCI та 0,05% (маса/об'єм) детергенту "Brij 35" (торговий знак)) у присутності (10 концентрацій) або відсутності інгібіторів. Активність визначають виміром флуоресценції при випр 320нм та збудж 405нм. Процент інгібування розраховують таким чином: % Інгібування = [Флуоресценція+інгібітор - Флуоресценціяфону] / [Флуоресценціябез інгібітору - Флуоресценціяфону]. ММР14 Рекомбінантний каталітичний домен ММР14 людини можна експресувати та очищати як описано Parkar A.A. et al, (2000), Protein Expression and Purification, 20, 152. Очищений фермент можна застосовувати для дослідження інгібіторної активності таким чином: ММР14 (10нг/мл кінцева концентрація ) інкубують протягом 60 хвилин при кімнатній температурі з синтетичним субстратом McaPro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (10мкМ) у буфері для аналізу (0,1Μ "Tris-HCI" (торговий знак), рН 7,5, що містить 0,1Μ NaCI, 20мМ СаСІ2, 0,020мМ ZnCI та 0,05% (маса/об'єм) детергенту "Brij 35" (торговий знак)) у присутності (5 концентрацій) або відсутності інгібіторів. Активність визначають виміром флуоресценції при випр 320нм та збудж 405нм. Процент інгібування розраховують таким чином: % Інгібування = [Флуоресценція+інгібітор - Флуоресценціяфону] / [Флуоресценціябез інгібітору - Флуоресценціяфону]. Протокол тестування проти інших матриксних металопротеїназ, у т.ч. ММР9, застосовуючи експресовану та очищену проММР, описано, наприклад, С. Graham Knight et аl., (1992) FEBS Lett., 296(3), 263-266. MMP19 Рекомбінантний каталітичний домен ММР19 людини можна експресувати та очищати як описано Parkar A.A. et аl, (2000), Protein Expression and Purification, 20:152. Очищали фермент можна застосовувати для дослідження інгібіторної активності таким чином: ММР19 (40нг/мл кінцева концентрація ) інкубують протягом 120 хвилин при 35°С з синтетичним субстратом Mca-Pro-Leu-Ala-NvaDpa-Ala-Arg-NH2 (5мкМ) у буфері для аналізу (0,1Μ "Tris-HCI" (торговий знак), рН 7,3, що містить 0,1Μ NaCI, 20мМ СаСІ2, 0,020мМ ZnCI та 0,05% (маса/об'єм) детергенту "Brij 35" (торговий знак)) у 35 90285 присутності (5 концентрації) або відсутності інгібіторів. Активність визначають виміром флуоресценції при випр 320нм та збудж 405нм. Процент інгібування розраховують таким чином: 36 % Інгібування = [Флуоресценція+інгібітор - Флуоресценціяфону] / [Флуоресценціябез інгібітору - Флуоресценціяфону]. Наступна таблиця показує дані стосовно селекції сполук заявленого винаходу. Таблиця Сполука приклад 1 приклад 2 приклад 5 hММР12 ІК50 (нМ) 8,78 26,6 6,9 Комп’ютерна верстка М. Ломалова hММР9 ІК50 (нМ) 10,1 21,5 3,8 hММР8 ІК50 (нМ) 3050 2470 1310 Підписне hММР14 ІК50 (нМ) >10000 >10000 8810 hММР19 IK50 (hM) >10000 >10000 3760 Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюHydantoin derivatives useful as mmp inhibitors
Автори англійськоюGabos Balint, Lundkvist Michael, Munck Af Rosenschoeld Magnus, Shamovsky Igor, Zlatoidsky Pavol
Назва патенту російськоюПроизводные гидантоина, полезные как ингибиторы металлопротеиназ
Автори російськоюГабос Балинт, Лундквист Микаэль, Мунк Аф Розеншэльд Магнус, Шамовски Игор, Златоидски Павол
МПК / Мітки
МПК: A61P 9/10, C07D 471/04, A61P 35/00, A61K 31/506, C07D 401/14, A61P 11/06, A61K 31/444, A61P 19/02
Мітки: корисні, гідантоїну, похідні, інгібітори, металопротеїназ
Код посилання
<a href="https://ua.patents.su/18-90285-pokhidni-gidantonu-korisni-yak-ingibitori-metaloprotenaz.html" target="_blank" rel="follow" title="База патентів України">Похідні гідантоїну, корисні як інгібітори металопротеїназ</a>
Похідні гідантоїну, корисні як інгібітори металопротеїназ

Номер патенту: 89067
Опубліковано: 25.12.2009
Автори: Вотерсон Дейвід, Перссон Давід Йонас
МПК: A61P 19/02, A61K 31/4166, C07D 401/12, A61K 31/445
Мітки: корисні, гідантоїну, інгібітори, металопротеїназ, похідні
Формула / Реферат:
1. Сполука формули (І), (І)деR1 - (2-4С)алкіл, та є заміщеним двома або більше атомами флуору; аR2 - метил або етил;або її фармацевтично прийнятна сіль.2. Сполука формули (І) за п. 1, де R1 - етил, пропіл або бутил, та є заміщеним двома або більше атомами флуору.3. Сполука формули (І) за п. 1 або за п. 2, де R1 - етил,...
Похідні гідантоїну як інгібітори металопротеїназ

Номер патенту: 89801
Опубліковано: 10.03.2010
Автори: Лундквіст Мікаель, Златоідскі Паволь, Шамовскі Іґор, абос Балінт, Мунк Аф Розеншельд Маґнус
МПК: A61P 19/02, A61K 31/506, C07D 401/12, C07D 471/04, A61P 11/06, A61P 9/10, A61K 31/4725, A61K 31/4375, C07D 401/14, A61P 35/00
Мітки: похідні, гідантоїну, інгібітори, металопротеїназ
Формула / Реферат:
1. Сполука формули (І) або її фармацевтично прийнятна сіль, (I)деR1 представляє Н, галоген, CF3 або CH2CN;R2 представляє С1-С3алкіл; іА, А1 і В, кожен незалежно, представляють СН або N;2. Сполука за пунктом 1, де R1 представляє хлор.3. Сполука за пунктом 1, де R1 представляє CF3.4. Сполука за будь-яким з пунктів 1-3,...
Похідні гідроксипіпеколат гідроксамової кислоти як інгібітори матричних металопротеїназ

Номер патенту: 59453
Опубліковано: 15.09.2003
Автори: Летавік Майкл Ентоні, Чупак Луїз Стенлі, Макклур Кім Френсіс, Ноу Марк Карл
МПК: C07D 409/12, A61P 43/00, C07D 401/12, C07D 211/96, A61P 9/08, A61P 31/18, C07D 211/66, C07D 405/12, C07D 211/60, A61P 11/06, C07D 413/12, A61P 25/28, A61P 17/00, A61K 31/445, A61P 37/08, A61P 19/02, A61P 11/00, A61P 3/10, A61P 35/00, A61K 31/454, A61K 31/4525, A61P 31/04, A61P 25/00, A61P 27/02, A61P 19/10, A61P 1/04, A61P 37/06, A61K 31/443, A61P 1/02
Мітки: кислоти, інгібітори, гідроксамової, матричних, похідні, гідроксипіпеколат, металопротеїназ
Формула / Реферат:
1. Похідні гідроксипіпеколат гідроксамової кислоти формули (I), (I)де R1-R8 вибирають з групи, що складається з гідроксилу, гідрогену, галогену, -CN, (С1-С6)алкілу, (С2-С6)алкенілу, (С6-С10)арил(С2-С6)алкенілу, (С2-С9)гетероарил(С2-С6)алкенілу, (С2-С6)алкінілу, (С6-С10)арил(С2-С6)алкінілу, (С2-С9)гетероарил(С2-С6)алкінілу, (С1-С6)алкіламіногрупи,...
Похідні b-карболіну, корисні як інгібітори фосфодіестерази

Номер патенту: 74826
Опубліковано: 15.02.2006
Автори: Мейсілаг Марк Дж., Суі Жіхуа
МПК: C07D 491/048, A61P 5/24, A61P 9/00, A61P 9/10, C07D 471/04, A61P 15/00, A61P 9/12, A61K 31/4365, A61P 7/02, A61K 31/437, A61P 9/04, A61K 31/444, A61K 31/5377, C07D 495/04, A61K 31/506, A61P 15/10, A61P 11/06, A61P 43/00
Мітки: фосфодіестерази, b-карболіну, похідні, корисні, інгібітори
Формула / Реферат:
1. Сполука формули (І):,(I),деR1 є незалежно вибраним з групи, що складається з галогену, нітрогрупи, гідроксигрупи, С1-С8алкілу, С1-С8алкоксигрупи, -NH2, -NHRА-, -N(RА)2, -O-RА, -С(О)NH2, -C(О)NHRA, -C(О)N(RA)2, -NС(O)-RA, -SO2NHRA, -SO2N(RA)2, фенілу (заміщеного, при потребі, RB у кількості від одного до трьох) та гетероарилу (заміщеного, при...
Похідні заміщеного піролопіридинону, корисні як інгібітори фосфодіестерази

Номер патенту: 72611
Опубліковано: 15.03.2005
Автори: Суі Жіхуа, Мейсілаг Марк Дж., Джіанг Вейквін, Гуан Джіхуа, Лантер Джеймс
МПК: A61P 15/00, A61P 9/12, A61P 9/00, A61P 15/10, A61P 15/02, A61P 9/04, A61K 31/506, A61P 15/08, A61K 31/4709, A61P 43/00, A61P 9/10, A61K 31/5377, A61P 3/10, A61K 31/496, A61K 31/437, C07D 471/04, A61P 7/02, A61P 11/06, A61P 15/06
Мітки: заміщеного, фосфодіестерази, інгібітори, похідні, піролопіридинону, корисні
Формула / Реферат:
1. Сполука формули (І) або (II):(I) або(II),деR1 є вибраним з групи, що складається з гідрогену, карбоксигрупи, -С(О)-С1-С6-алкілу, -С(О)-С1-С6-алкоксигрупи, -С(О)-NН-С1-С6-алкіл-NН2, -С(О)-NH-С1-С6-алкіл-NHRA, -С(О)-NН-С1-С6-алкіл-N(RA)2, -С(О)-NH2, -С(О)-NHRA,...
Попередній патент: Пресований структурний елемент (варіанти) і спосіб його виготовлення, зварний структурний елемент і спосіб його виготовлення
Наступний патент: Алюмотермічний спосіб одержання кремнію високого ступеня чистоти
Випадковий патент: Сушильна установка