Інгібітори бета-секретази
Номер патенту: 113641
Опубліковано: 27.02.2017
Автори: Чжао Йі, Фукс Клаус, Какатіан Сальвасіон, Раст Георг, Йуан Цзин, Сінгх Суреш Б., Чжен Яюн, Венкатраман Шанкар, Рівз Джонатан, Діллард Лоуренс Уейн, Сю Ченронг, Бухтіяров Юрій, Лала Діпак С., Джіа Ланкі, Моралес-Рамос Енджел, Дорнер-Чосек Корнелія





Формула / Реферат
1. Сполука, представлена структурною формулою, вибраною з:
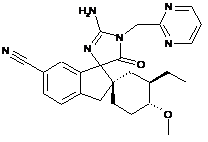
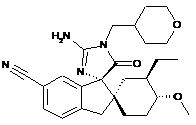
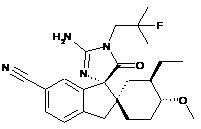
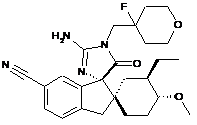
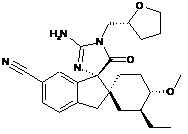
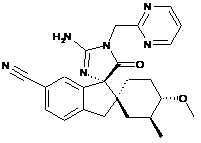
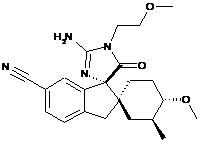
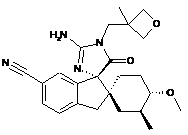
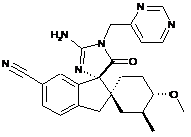

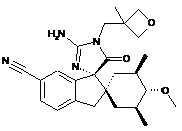
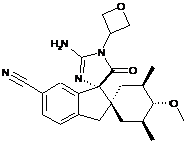

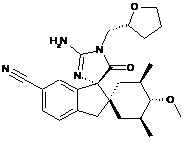
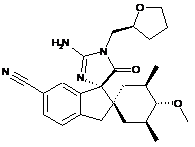
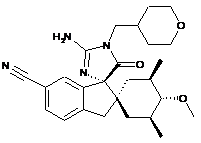
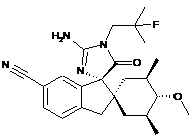



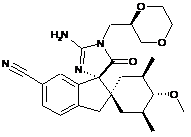
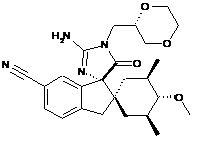
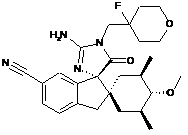
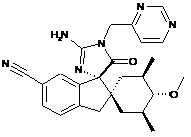

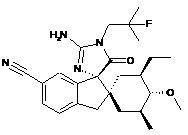
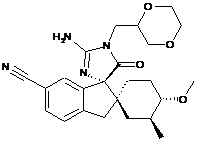
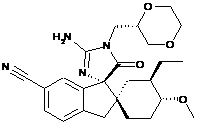
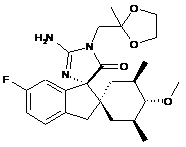
або її фармацевтично прийнятна сіль.
2. Сполука за п. 1 або її фармацевтично прийнятна сіль для застосування як лікарського засобу.
3. Фармацевтична композиція, яка містить щонайменше одну сполуку за п. 1 або її фармацевтично прийнятну сіль у суміші з фармацевтично прийнятним ад'ювантом, розріджувачем і/або носієм.
4. Сполука за п. 1 або її фармацевтично прийнятна сіль для застосування при лікуванні опосередкованого ВАСЕ1 порушення або захворювання.
5. Сполука або її фармацевтично прийнятна сіль для застосування за п. 4, де опосередковане ВАСЕ1 порушення або захворювання вибирають з групи, що складається з нейродегенеративного порушення, зниження когнітивних здатностей, когнітивного порушення, деменції та захворювання, що характеризується формуванням відкладень β-амілоїду або нейрофібрилярних клубків.
6. Сполука або її фармацевтично прийнятна сіль для застосування за п. 5, де порушення або захворювання вибирають з групи, що складається з хвороби Альцгеймера, трисомії за 21 парою хромосом (синдром Дауна), спадкової церебральної геморагії з амілоїдозом голландського типу (НCHWA-D), сенільної деменції, церебральної амілоїдної ангіопатїї, дегенеративної деменції, деменцій змішаного судинного та дегенеративного походження, деменції, асоційованої з хворобою Паркінсона, деменції, асоційованої з прогресуючим супрануклеарним паралічем, деменції, асоційованої з корковою базальною дегенерацією, хвороби Альцгеймера з дифузійними тільцями Леві, сухої вікової макулярної дегенерації (AMD) і глаукоми.
7. Сполука або її фармацевтично прийнятна сіль для застосування за п. 6, де порушення або захворювання являє собою хворобу Альцгеймера.
8. Сполука або її фармацевтично прийнятна сіль для застосування за п. 6, де порушення або захворювання являє собою глаукому.
9. Застосування сполуки за п. 1 або її фармацевтично прийнятної солі для виробництва лікарського засобу для лікування у суб'єкта опосередкованого ВАСЕ1 порушення.
10. Застосування сполуки за п. 9 або її фармацевтично прийнятної солі, де опосередковане ВАСЕ1 захворювання або порушення вибирають з групи, що складається з нейродегенеративного порушення, зниження когнітивних здатностей, когнітивного порушення, деменції та захворювання, що характеризується формуванням відкладень b-амілоїду або нейрофібрилярних клубків.
11. Застосування сполуки за п. 10 або її фармацевтично прийнятної солі, де порушення або захворювання вибирають з групи, що складається з хвороби Альцгеймера, трисомії за 21 парою хромосом (синдром Дауна), спадкової церебральної геморагії з амілоїдозом голландського типу (HCHWA-D), сенільної деменції, церебральної амілоїдної ангіопатії, дегенеративної деменції, деменцій змішаного судинного та дегенеративного походження, деменції, асоційованої з хворобою Паркінсона, деменції, асоційованої з прогресуючим супрануклеарним паралічем, деменції, асоційованої з корковою базальною дегенерацією, хвороби Альцгеймера з дифузійними тільцями Леві, сухої вікової макулярної дегенерації (AMD) і глаукоми.
12. Застосування сполуки за п. 11 або її фармацевтично прийнятної солі, де захворювання або порушення являє собою хворобу Альцгеймера.
13. Застосування сполуки за п. 11 або її фармацевтично прийнятної солі, де захворювання або порушення являє собою глаукому.
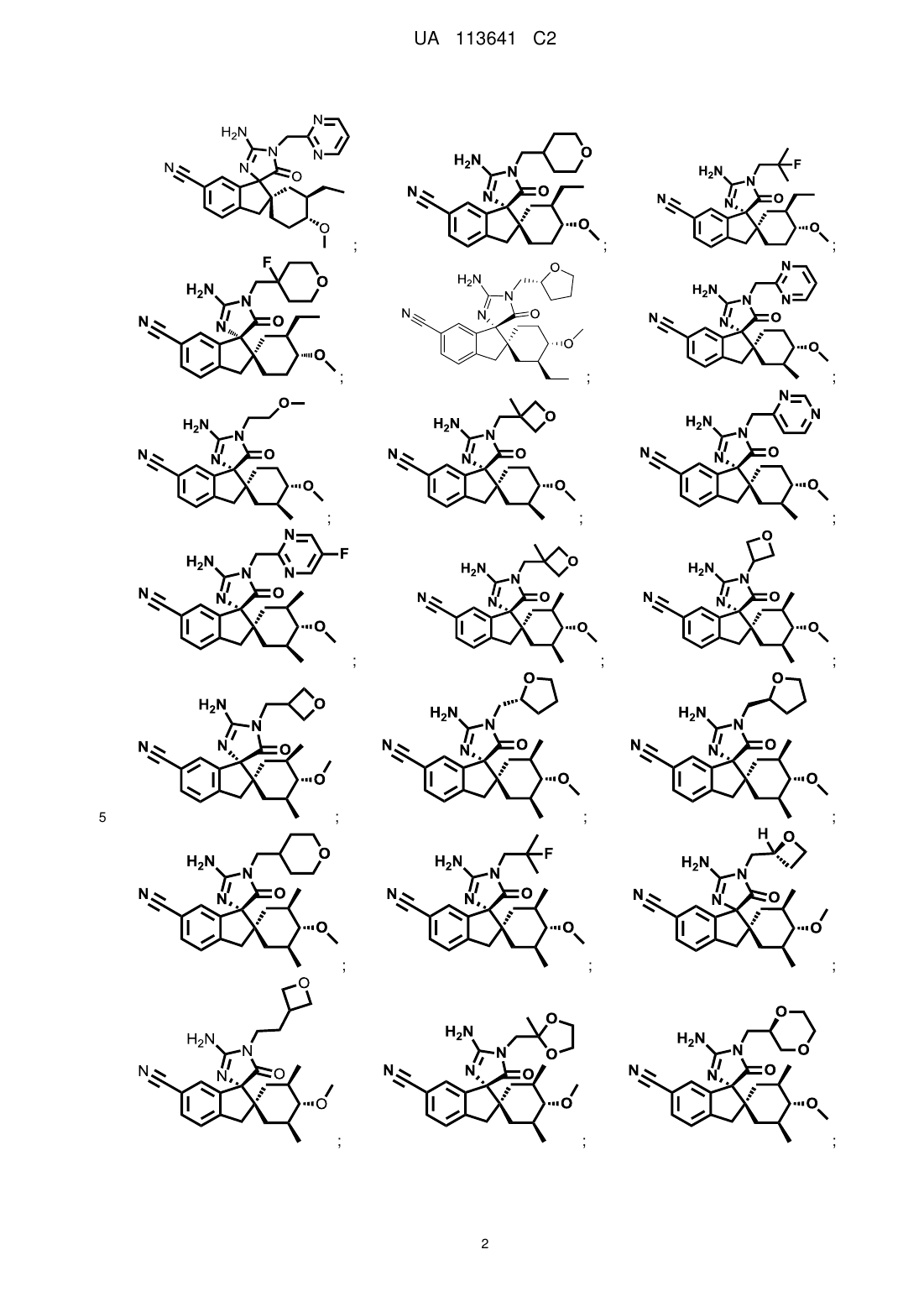
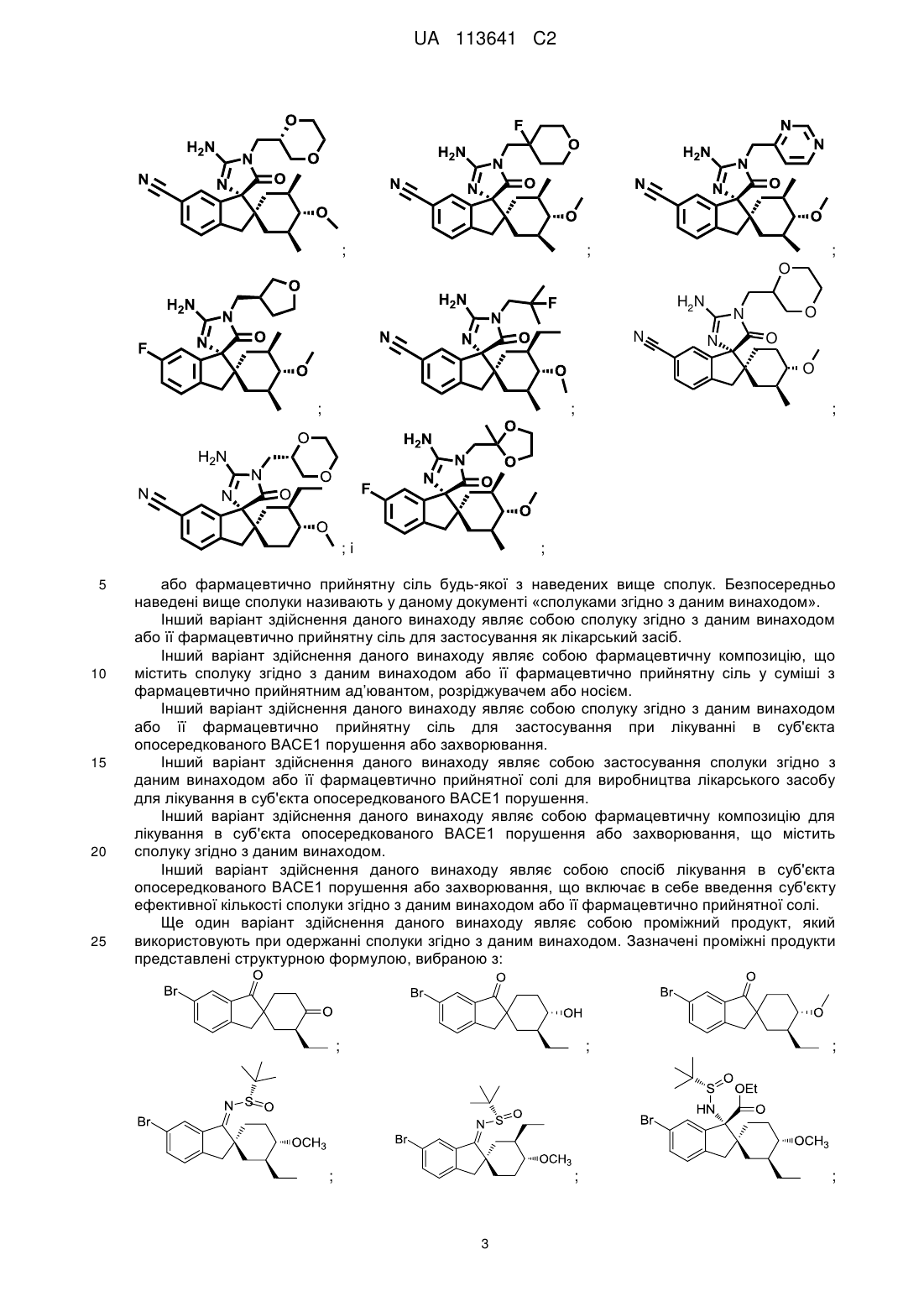
14. Сполука, вибрана з групи, що складається з:
 ;
;
 ;
;
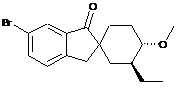 ;
;
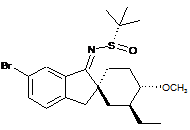 ;
;
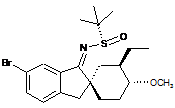 ;
;
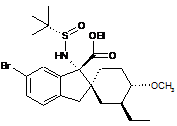 ;
;
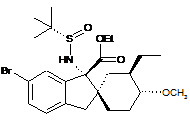 ;
;
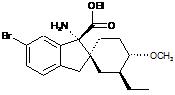 ;
;
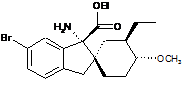 ;
;
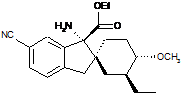 ;
;
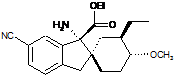 ;
;
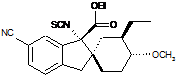 ;
;
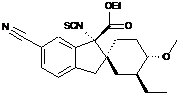 ;
;
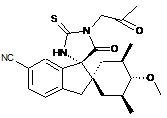 ;
;
 ;
;
 ;
;
 ;
;
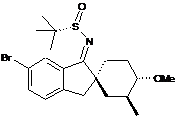 ;
;
 ;
;
 ;
;
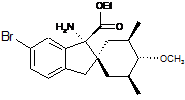 ;
;
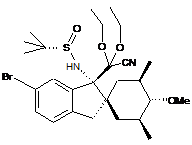 ;
;
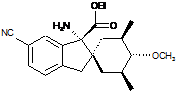 і
і
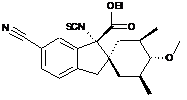 ;
;
або її сіль.
15. Сполука, що має структуру
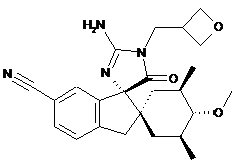 , або її сіль.
, або її сіль.
16. Сполука, що має структуру
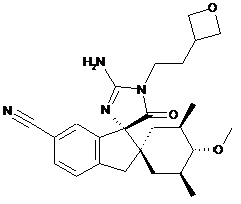 , або її сіль.
, або її сіль.
17. Сполука, що має структуру
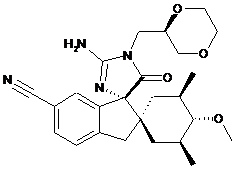 , або її сіль.
, або її сіль.
18. Сполука, що має структуру
 , або її сіль.
, або її сіль.
Текст
Реферат: Даний винахід стосується спіроциклічних ацилгуанідинів і їхнього застосування як інгібіторів активності ферменту -секретази (ВАСЕ1), фармацевтичних композицій, що містять зазначені сполуки, і способів застосування зазначених сполук як терапевтичних засобів для лікування нейродегенеративних порушень, порушень, що характеризуються зниженням когнітивних UA 113641 C2 (12) UA 113641 C2 здатностей, когнітивних порушень, деменції та захворювань, формуванням відкладень -амілоїду або нейрофібрилярних клубків. що характеризуються UA 113641 C2 5 10 15 20 25 30 35 40 45 50 55 I. Родинні заявки Дана заявка претендує на пріоритет відповідно до попередньої заявки на видачу патенту США № 61/606786, поданої 5 березня 2012 року, повний зміст якої включено в даний документ за допомогою посилання. II. Область техніки Даний винахід відноситься до спіроциклічних ацилгуанідинів і їхнього застосування як інгібітори активності ферменту -секретази (BACE1), до фармацевтичних композицій, що містять зазначені сполуки, і до способів застосування зазначених сполук як терапевтичні засоби для лікування нейродегенеративних порушень, порушень, що характеризуються зниженням когнітивних здатностей, когнітивних порушень, деменції та захворювань, що характеризуються формуванням відкладень -амілоїду або нейрофібрилярних клубків. III. Рівень техніки Відкладення -амілоїду (також називаного в даному документі «Абета» або «A») і нейрофібрилярні клубки являють собою дві основні патологічні характеристики, асоційовані з хворобою Альцгеймера (AD), включаючи генетично зв'язані сімейні форми з раннім початком, обумовлені мутаціями в білку-попереднику амілоїду (APP) пресенеліне 1 і 2, а також спорадичну AD із пізнім початком. Клінічно AD характеризується втратою пам'яті, когнітивної здатності, здатності до мислення, судження й орієнтації. У міру прогресування захворювання також страждають рухова, сенсорна та лінгвістична здатності доти, поки не відбувається глобальне порушення складних когнітивних функцій. Такі когнітивні втрати відбуваються поступово, але, як правило, ведуть до важкого порушення та, зрештою, до смерті в межах 4-12 років. Відкладення -амілоїду переважно являють собою агрегат пептиду Абета, що, у свою чергу, являє собою продукт протеолізу APP. Більш конкретно, пептид A виникає внаслідок розщеплення APP на С-кінцях за допомогою однієї або декількох -секретаз, і на N-кінцях за допомогою ферменту -секретази (BACE1), також відомої як аспартилпротеаза та мемапсин 2, як частини -амілоїдогенного шляху. Активність BACE прямо корелює з генерацією пептиду A із APP, і все більше досліджень вказують на те, що інгібування BACE інгібує продукцію пептиду A. Амілоїдогенні бляшки та судинна амілоїдна ангіопатія також характерна для головного мозку пацієнтів із трисомією за 21 парою хромосом (синдром Дауна), спадкоємної церебральної геморрагією з амілоїдозом голандського типу (HCHWA-D), і іншими нейродегенеративними порушеннями. Нейрофібрилярні клубки також виникають при інших нейродегенеративних порушеннях, включаючи порушення, які індукують деменцію. Недавно повідомлялося, що Абета залучений в розвиток апоптозу гангліозних клітин сітківки (RGC) при глаукомі, з наявністю у пацієнтів із глаукомою опосередкованого каспазою-3 аномального процесингу білка-попередника амілоїду, збільшеної експресії Абета в RGC при експериментальній глаукомі та знижених рівнів A у склоподібному тілі (що вказують на відкладення A у сітківці). Відкладення амілоїду також були пов'язані з макулярною дегенерацією у пацієнтів, що страждають сухою віковою макулярною дегенерацією (AMD), і в моделях AMD на тваринах. У WO 2010/021680, WO 2011/106414 і WO 2010/105179 розкриті спіроциклічні ацилгуанідини зі спіроциклічною структурою як інгібітори бета-секретази. IV. Сутність винаходу Даний винахід відноситься до сполук, які є інгібіторами BACE1 і застосовні як терапевтичні засоби для лікування захворювання або порушення, що характеризується підвищеним відкладенням -амілоїду або рівнями -амілоїду в пацієнта. Розкриті інгібітори BACE1 є не тільки дуже потужними інгібіторами ферменту BACE1 (метод аналізу 1), але також демонструють: (1) дуже потужну інгібуючу активність у методі аналізу Abeta на клітинах (метод аналізу 2), (2) селективність відносно каналу hERG серця в методі аналізу на клітинах (метод аналізу 3), а також (3) низьку схильність викликати фосфоліпідоз у методі аналізу фосфоліпідозу на клітинах (метод аналізу 4). Таким чином, даний винахід відноситься до сполук, які демонструють сполучення високої активності як інгібітори BACE1, високої селективності відносно каналу hERG серця та низької фосфоліпідозної активності. Один варіант здійснення даного винаходу являє собою сполуку, представлену структурною формулою вибраною з: 1 UA 113641 C2 ; ; ; ; ; ; ; ; ; 5 ; ; ; ; ; 2 ; ; ; ; ; ; ; UA 113641 C2 ; ; ; O H2 N N O N N O O ; ; ;і 5 10 15 20 25 ; ; або фармацевтично прийнятну сіль будь-якої з наведених вище сполук. Безпосередньо наведені вище сполуки називають у даному документі «сполуками згідно з даним винаходом». Інший варіант здійснення даного винаходу являє собою сполуку згідно з даним винаходом або її фармацевтично прийнятну сіль для застосування як лікарський засіб. Інший варіант здійснення даного винаходу являє собою фармацевтичну композицію, що містить сполуку згідно з даним винаходом або її фармацевтично прийнятну сіль у суміші з фармацевтично прийнятним ад’ювантом, розріджувачем або носієм. Інший варіант здійснення даного винаходу являє собою сполуку згідно з даним винаходом або її фармацевтично прийнятну сіль для застосування при лікуванні в суб'єкта опосередкованого BACE1 порушення або захворювання. Інший варіант здійснення даного винаходу являє собою застосування сполуки згідно з даним винаходом або її фармацевтично прийнятної солі для виробництва лікарського засобу для лікування в суб'єкта опосередкованого BACE1 порушення. Інший варіант здійснення даного винаходу являє собою фармацевтичну композицію для лікування в суб'єкта опосередкованого BACE1 порушення або захворювання, що містить сполуку згідно з даним винаходом. Інший варіант здійснення даного винаходу являє собою спосіб лікування в суб'єкта опосередкованого BACE1 порушення або захворювання, що включає в себе введення суб'єкту ефективної кількості сполуки згідно з даним винаходом або її фармацевтично прийнятної солі. Ще один варіант здійснення даного винаходу являє собою проміжний продукт, який використовують при одержанні сполуки згідно з даним винаходом. Зазначені проміжні продукти представлені структурною формулою, вибраною з: ; ; ; ; 3 ; ; UA 113641 C2 ; ; ; ; ; ; ; ; 5 15 ; ; ; ; ; ; ; ; 10 ; ; і ; або солі будь-якої з наведених вище сполук. V. Докладний опис винаходу Сполуки згідно з даним винаходом мають потужну інгібуючу активність відносно ферменту BACE1 і продукції Абета поряд із селективністю відносно каналу hERG і низькою схильністю викликати фосфоліпідоз. Наприклад, сполуки згідно з даним винаходом демонструють інгібування BACE1 з IC50 < 15 нМ, інгібування продукції Абета в клітинах із IC50 < 2 нМ, інгібування hERG 150 мкМ. Зазначені сполучені властивості роблять сполуки згідно з даним винаходом застосовними для лікування в людей патологічних станів, зокрема, для лікування хвороби Альцгеймера, а також інших порушень та захворювань, опосередкованих BACE1. Як було встановлено Sanguinetti et al. (1995, Cell, Apr. 21, 81(2):299-307) і більшим обсягом 4 UA 113641 C2 5 10 15 20 25 30 35 40 45 50 55 60 наступних даних, інгібування каналу hERG (ген специфічних калієвих каналів людини) ксенобіотиками та наступна вповільнена реполяризація серця асоційовані з підвищеним ризиком специфічної поліморфної шлуночкової тахіаритмії, двонаправленної шлуночкової тахікардії. Щоб уникнути такого ризику на ранніх стадіях, звичайною практикою є скринінг взаємодії з hERG у системі in vitro з використанням гетерологічної експресії каналу hERG, і такий тип аналізу також є важливою частиною подальшого доклінічного профілювання кандидата, рекомендованого в керівництві ICH S7B (International Conference on Harmonization (2005): ICH Topic S 7 B The nonclinical Evaluation of the Potential for delayed Ventricular Repolarization; (QT Interval Prolongation) by Human Pharmaceuticals (www.ich.org/products/guidelines/safety/article/safety-guidelines.html). У цьому зв'язку, слабке інгібування каналу hERG, таке як продемонстроване сполуками згідно з даним винаходом, украй бажане для терапевтичних засобів. Фосфоліпідоз являє собою порушення накопичення ліпідів, при якому надлишок фосфоліпідів накопичують всередині клітин. Індукований ліками фосфоліпідоз являє собою небажану реакцію на ліки. Отже, щоб уникнути шкідливих побічних ефектів, сполуки з низькою фосфоліпідозною активністю переважні для застосування в терапевтичній практиці на людях. Дані, представлені нижче в таблиці 1, демонструють, що сполуки згідно з даним винаходом мають таке сполучення потужної активності у відношенні BACE1 у клітинах, селективності у відношенні hERG серця та низької схильності викликати фосфоліпідоз. Уважається, що сполуки, наведені як приклад у WO 2010/021680 і WO 2010/105179, не мають таке сполучення бажаних властивостей. Крім того, у таблиці 2 представлені дані, які демонструють, що певні сполуки згідно з даним винаходом характеризуються істотно більш низькими інгібуючими значеннями IC50 у методі аналізу ферменту BACE1, а також у методі аналізу Абета на клітинах, у порівнянні з певними сполуками порівняння, описаними у WO 2010/105179. Термінам, конкретно не визначеним у даному документі, варто привласнити значення, які були б привласнені ним фахівцем у даній області техніки у світлі даного розкриття та контексту. Однак якщо не зазначене інше, то використовувані в даному описі наступні терміни характеризуються зазначеним значенням, і дотримуються наступні умови. Якщо сполука згідно з даним винаходом описується за допомогою назви або структури без вказівки всіх таутомерних форм, то варто розуміти, що така сполука та її фармацевтично прийнятні солі охоплюють всі таутомери. Якщо сполука згідно з даним винаходом описується за допомогою назви або структури без вказівки стереохімії, то варто розуміти, що така сполука та її фармацевтично прийнятні солі охоплюють всі стереоізомери, оптичні та геометричні ізомери (наприклад, енантіомери, діастереомери, E/Z ізомери, і т.п.) та їхні рацемати, а також суміші з різними пропорціями окремих енантіомерів, суміші діастереоізомерів або суміші будь-яких із вищевикладених форм. Якщо стереоізомер, оптичний або геометричний ізомер описаний за допомогою назви або структури, то варто розуміти, що стереоізомерна, оптична або геометрична ізомерна чистота пойменованого або зазначеного стереоізомера, оптичного або геометричного ізомеру становить, щонайменше, 60%, 70%, 80%, 90%, 99% або 99,9% за масою чистої речовини. Стереоізомерна, оптична або геометрична ізомерна чистота визначається шляхом розподілу маси пойменованого або зазначеного стереоізомера, оптичного або геометричного ізомеру в суміші на сумарну масу всіх стереоізомерів, оптичних або геометричних ізомерів у суміші. Якщо сполука згідно з даним винаходом або її фармацевтично прийнятна сіль пойменовані або зазначені за допомогою структури, то варто розуміти, що сольвати, гідрати та безводні форми такої сполуки і сольвати, гідрати та безводні форми її фармацевтично прийнятної солі включені в даний винахід. Термін «сольвати» відноситься до кристалічних форм, у яких молекули розчинника включені в кристалічні ґрати у процесі кристалізації. Сольват може містити в собі воду або неводний розчинник, такий як етанол, ізопропанол, DMSO, оцтову кислоту, етаноламін й EtOAc. Сольвати, в яких вода являє собою молекулу розчинника, включену в кристалічні ґрати, зазвичай називають «гідратами». Гідрати містять у собі стехіометричні гідрати, а також композиції, що містять різні кількості води. Термін «безводні форми» відноситься до сполук, що не містять якого-небудь розчинника або води, або по суті не містять розчинника або води, включених у кристалічну структуру (наприклад, молярне співвідношення розчинника або води до сполуки менш ніж 1/10, 1/20; 1/100; або 1/200). Солі Використовуване в даному документі вираження «фармацевтично прийнятний» відноситься до таких сполук, речовин, композицій та/або лікарських форм, які, у рамках здорового медичного судження, є підходящими для застосування в контакті в тканинами людей та тварин без прояву надмірної токсичності, подразнення, алергійної реакції або іншої проблеми або 5 UA 113641 C2 5 10 15 20 25 30 35 40 45 50 55 60 ускладнення, і розмірні з прийнятним співвідношенням користь/ризик. Використовуваний у даному документі термін «фармацевтично прийнятні солі» відноситься до похідних розкритих сполук, у яких вихідна сполука модифікована за допомогою утворення її кислих або лужних солей. Приклади фармацевтично прийнятних солей містять у собі без обмеження солі неорганічних або органічних кислот із залишками основ, таких як аміни; лужні або органічні солі залишків кислот, таких як карбонові кислоти; і т.п. Наприклад, такі солі містять у собі солі амонію, L-аргініну, бетаїну, бенетаміну, бензатину, гідроксиду кальцію, холіну, деанолу, діетаноламіну (2,2'-імінобіс(етанолу)), діетиламіну, 2-(діетиламіно)етанолу, 2-аміноетанолу, етилендіаміну, N-етилглюкаміну, гідрабаміну, 1H-імідазолу, лізину, гідроксиду магнію, 4-(2гідроксіетил)морфоліну, піперазину, гідроксиду калію, 1-(2-гідроксіетил)піролідину, гідроксиду натрію, триетаноламіну (2,2',2“-нітрилотрис(етанолу)), трометаміну, гідроксиду цинку, оцтової кислоти, 2,2-дихлороцтової кислоти, адипінової кислоти, альгінової кислоти, аскорбінової кислоти, L-аспарагінової кислоти, бензолсульфонової кислоти, бензойної кислоти, 2,5дигідроксибензойної кислоти, 4-ацетамідобензойної кислоти, (+)-камфорної кислоти, (+)камфор-10-сульфонової кислоти, карбонової кислоти, коричної кислоти, лимонної кислоти, цикламової кислоти, капринової кислоти, додецилсірчаної кислоти, етан-1,2-дисульфонової кислоти, етансульфонової кислоти, 2-гідроксіетансульфонової кислоти, етилендіамінтетраоцтової кислоти, мурашиної кислоти, фумарової кислоти, галактарової кислоти, гентизинової кислоти, D-глюкогептонової кислоти, D-глюконової кислоти, Dглюкуронової кислоти, глутамінової кислоти, глутарової кислоти, 2-оксоглутарової кислоти, гліцерофосфорної кислоти, гліцину, гліколевої кислоти, капронової кислоти, гіпурової кислоти, бромистоводневої кислоти, соляної кислоти, ізомасляної кислоти, DL-молочної кислоти, лактобіонової кислоти, лауринової кислоти, лізину, малеїнової кислоти, (-)-L-яблучної кислоти, малонової кислоти, DL-мигдальної кислоти, метансульфонової кислоти, галактарової кислоти, нафталін-1,5-дисульфонової кислоти, нафталін-2-сульфонової кислоти, 1-гідрокси-2-нафтойної кислоти, нікотинової кислоти, азотної кислоти, октанової кислоти, олеїнової кислоти, оротової кислоти, щавлевої кислоти, пальмітинової кислоти, памоєвої кислоти (ембонової кислоти), фосфорної кислоти, пропіонової кислоти, (-)-L-піроглутамінової кислоти, саліцилової кислоти, 4аміносаліцилової кислоти, себацинової кислоти, стеаринової кислоти, бурштинової кислоти, сірчаної кислоти, таннину, (+)-L-винної кислоти, тіоціанової кислоти, пара-толуолсульфонової кислоти й ундециленової кислоти. Інші фармацевтично прийнятні солі можуть бути утворені з катіонами металів, таких як алюміній, кальцій, літій, магній, калій, натрій, цинк і т.п. (див. також Pharmaceutical salts, Berge, S.M. et al., J. Pharm. Sci., (1977), 66, 1-19). Фармацевтично прийнятні солі згідно з даним винаходом можуть бути синтезовані з вихідної сполуки, що містить фрагмент основи або кислоти, за допомогою загальноприйнятих хімічних способів. Як правило, такі солі можуть бути отримані шляхом здійснення взаємодії форм вільної кислоти або основи таких сполук із достатньою кількістю відповідної основи або кислоти у воді або в органічному розріджувачі, такому як простий ефір, етилацетат, етанол, ізопропанол або ацетонітрил, або їхня суміш. Солі відмінних від згаданих вище кислот, які застосовні, наприклад, для очищення або виділення сполук згідно з даним винаходом (наприклад, трифторацетати), також становлять частину даного винаходу. Біологічні дані Метод аналізу BACE1 (метод аналізу 1) Інгібуючу активність сполук оцінювали за допомогою аналізу активності BACE1 методом гасіння флуоресценції з використанням комерційно доступного субстрату HiLyte Fluor™488-GluVal-Asn-Leu-Asp-Ala-Glu-Phe-Lys-(QXL™ 520)-OH (SEQ ID NO:1) AnaSpec, San Jose, CA) і процесованої бета-секретази людини BACE1 (амінокислоти 1-454), злитої з myc-Hys-міткою та секретованої HEK293/BACEect. клітинами в OptiMEM™ (Invitrogen). Субстрат розчиняли в DMSO до концентрації 1 мг/мл. Аналіз проводили в 384-ямковому планшеті у присутності OptiMEM™ (супернатант, зібраний через 24 години й очищений від клітинного дебриса шляхом центрифугування), що містить ектодомен BACE1, 25 мкл води з дворазовою щодо бажаної концентрацією тестованої сполуки та 2% DMSO, 1 мкМ пептиду субстрату, 20 мМ NaOAc, pН 4,4 і 0,04% Тритон-X100 у сумарному аналізованому обсязі 50 мкл. Загалом, у планшет додавали 25 мкл розведення сполуки з наступним додаванням 10 мкл BACE1 з OptiMEM™ у розведенні 1:10 у воді з 0,2% Тритона Х-100. Реакцію ініціювали додаванням 15 мкл субстрату в NaOAc буфері. Реакційну ® суміш інкубували при к. т. (у темряві) у рідері з множинним зчитуванням міток Envision (Perkin Elmer) і реєстрували розщеплення субстрату у вигляді кінетики протягом 60 хвилин при довжині хвилі порушення 485 нм і довжині хвилі випущення 538 нм. У кожному планшеті втримувалися 6 UA 113641 C2 5 10 15 20 25 30 35 40 45 50 55 60 контрольні лунки, що не містять ферменту. З метою одержання значень швидкості реакції у всіх 384 лунках визначали збиток інтенсивності флуоресценції в часі. Значення швидкості використовували для обчислення регуляції (в %), приймаючи контроль з 1% DMSO та відсутністю інгібування за 100% та контроль без ферменту за 0%. Значення IC50 обчислювали за допомогою апроксимації регуляції (в %) щодо концентрації тестованої сполуки з використанням Assay Explorer. Метод аналізу на клітинах H4-APPwt (метод аналізу 2) Ефективність сполук згідно з даним винаходом на клітинах оцінювали в методі аналізу з моніторингом продукції Абета1-x пептидів у лінії клітин нейрогліоми Н4 (ATCC, кат.№ HTB-148), стабільно експресуючої APP людини, з використанням імуноаналізу, такого як AlphaLISA (PerkinElmer, кат.№ AL288). Тестовані сполуки розчиняли в DMSO і попередньо розбавляли культуральним середовищем (DMEM із 10% FBS і 1% пеніциліну/стрептоміцину) з одержанням подвійної щодо кінцевої концентрації сполук для аналізу. В 96-ямковий культуральний планшет додавали рівні обсяги дворазових розчинів тестованих сполук і суспензії клітин, так щоб у кожній лунці втримувалося ~10000 клітин у кінцевому обсязі в 200 мкл. Кінцева концентрація DMSO при аналізі становила 0,2%. Планшети інкубували протягом 5 годин при температурі 37 C, 5% CO2, щоб дозволити клітинам прилипнути до дна лунок у присутності тестованих сполук. Потім середовище видаляли та заміняли свіжим середовищем, що містить тестовані сполуки в тій самій кінцевій концентрації. Планшети інкубували протягом 18 годин при температурі 37 C, 5% CO2. Концентрації Абета1-x визначали з використанням імуноаналізу AlphaLISA (PerkinElmer, кат.№ AL288), дотримуючись протоколу виробника. Для обчислення значень інгібування (в %) у кожній лунці з тестованими сполуками як контроль без інгібування та фонового контролю, відповідно, використовували концентрації Абета1-x у лунках, що містять або DMSO, або 10 мкМ інгібітора бета-секретази (BACE інгібітор IV, EMD Bioscience, кат.№ 565788). Значення інгібування (в %) визначали щодо концентрацій сполуки з використанням апроксимації кривої за чотирма параметрами, і обчислювали значення IC 50 (концентрація сполуки, при якій спостерігається ефект інгібування на 50%) як концентрацію сполуки, що відповідає точці перегину кривої. Метод аналізу каналів hERG (метод аналізу 3) Клітини: Клітини HEK 293 (ембріональні клітини нирок людини) були стабільно трансфіковані кДНК hERG. Піпетки та розчини: Клітини переохолоджували омиваючим розчином, що містить (у мМ): NaCl (137), KCl (4,0), MgCl2 (1,0), CaCl2 (1,8), глюкозу (10), HEPES (10), pН 7,4 з NaOH. Петч-піпетки виготовляли з трубок із боросилікатного скла з використанням пристосування для горизонтального витягування та заповнювали піпетованим розчином, що містить (у мМ): K-аспартат (130), MgCl2 (5,0), EGTA (5,0), K2ATP (4,0), HEPES (10,0), pН 7,2 з KOH. Опір мікроелектродів перебував у межах від 2 до 5 МОм. Стимулювання та реєстрація: Мембранні струми реєстрували з використанням петч-кламп ампліфікатора EPC-10 і програмного забезпечення PatchMaster. Опосередковані hERG мембранні струми реєстрували при 35°C з використанням методу петч-кламп у конфігурації «ціла клітина». Вихідний потенціал трансфікованих клітин HEK293 фіксували (-60 мВ) і виявляли опосередковані hERG інактивуючі слідові струми з використанням послідовності імпульсів із фіксованими амплітудами (активація/інактивація: 40 мВ протягом 2000 мс; відновлення: -120 мВ протягом 2 мс; лінійна зміна до 40 мВ протягом 2 мс; інактивуючий слідовий струм: 40 мВ протягом 50 мс) з повторюваними інтервалами в 15 с. Для процедури вирахування P/n витоку протягом кожного інтервалу між імпульсами реєстрували 4 імпульси, масштабованих із коефіцієнтом 0,2. R s компенсацію використовували до рівня, що дозволяє надійну реєстрацію без загасання. Приготування та застосування сполуки: Різні концентрації тестованої сполуки послідовно застосовували у відношенні кожної з різних досліджуваних клітин. Рівень стійкого стану базового струму реєстрували протягом, щонайменше, 6 циклів вимірювань до використання першої концентрації тестованої сполуки. Тестовану сполуку розчиняли в DMSO з одержанням основного маткового розчину, що надалі розбавляли DMSO до одержання маткових розчинів, необхідних для більше низьких концентрацій. Перед початком кожного експерименту з цих маткових розчинів наготовлювали свіжі кінцеві розведення позаклітинним буфером шляхом розведення 1:1000. Дані аналізу: Максимальні амплітуди струму вимірювали через 3 мс після лінійної зміни до +40 мВ. Перед 7 UA 113641 C2 5 10 15 20 25 30 35 40 45 50 55 60 застосуванням наступної концентрації для базової лінії та кожної концентрації усереднювали значення максимальні значення струму в трьох останніх циклах. Залишкові струми (I/I 0) обчислювали для кожної клітини у вигляді частки дійсного середнього значення максимального струму та середнього значення максимального струму на базовій лінії. Результати представляли у вигляді відсотка (%) інгібування (1-I/I0)*100% при 10 мкМ. Метод аналізу фосфоліпідозу in vitro (метод аналізу 4) Фосфоліпідогенний потенціал тестованих сполук оцінювали з використанням гематопоетичної лінії клітин U937 людини. Принцип тесту полягав в аналізі вмісту фосфоліпідів шляхом фарбування клітин флуоресцентним барвником нільським червоним. 6 Клітини U937 висіювали в культуральні планшети в концентрації 0,5×10 клітин/мл у середовищі RPMI, що містить 10% FBS, 1% DMSO та 0,005% гентаміцину. Потім, клітини культивували у присутності або під час відсутності різних концентрацій тестованих сполук протягом 48 годин у стандартних культуральних умовах. Для відбору на аналіз клітини центрифугували при 130g протягом 4 хвилин й однократно промивали PBS. Потім, для вимірювання в нефіксованих клітинах наготовлювали 2×0,5 мл суспензії клітин (0,5 мл для вимірювання життєздатності з йодидом пропідія та 0,5 мл для вимірювання з нільським червоним). Клітини, що залишилися, фіксували 3,7% формальдегідом протягом 30 хвилин. Після наступного етапу центрифугування клітини ресуспендували з 1,3 мл робочого розчину нільського червоного (1 мкг/мл) й інкубували протягом 5 хвилин при к. т. Потім, суспензію клітин двічі промивали 3 мл PBS і центрифугували при 130g протягом 4 хвилин. Супернатант дискадрували, клітини ресуспендували в 0,5 мл PBS і зберігали для вимірювання методом проточної цитометрії. Для фарбування нільським червоним 0,5 мл зразків із нефіксованими клітинами, до зразка додавали 50 мкл готового до використання розчину нільського червоного (10 мкг/мл). Зразки витримували на льоді протягом 5 хвилин. Потім їх однократно промивали 4 мл PBS (4°C, 250g протягом 8 хвилин), а потім ресуспендували в 400 мкл PBS і зберігали для вимірювання методом проточної цитометрії. Для вимірювання життєздатності, до 0,5 мл суспензії з нефіксованими клітинами додавали 12,5 мкл готового до використання розчину PI (10 мкг/мл). Після 15 хвилин інкубації на льоді, у зразках проводили вимірювання методом проточної цитометрії з використанням проточного цитометра Coulter Epics XL/MCL. Життєздатність клітин у кожному зразку визначали шляхом вимірювання вмісту PI на каналі 2 методом проточної цитометрії (568/590 нм). Гейти для поділу живих і мертвих клітин на підставі розходжень у флуоресценції визначали на підставі аналізу в контрольних зразках із культуральним середовищем. На фосфоліпідоз аналізували лише зразки з життєздатністю клітин ≥ 90% у порівнянні з контрольними зразками. Для цього, методом проточної цитометрії на каналі 1 (504/541 нм) і на каналі 4 (660/680 нм) проводили вимірювання в кожному зразку з нільським червоним (зразки з нефіксованими та фіксованими клітинами). Для кожного каналу, відносну інтенсивність флуоресценції нільського червоного в тестованому зразку розраховували щодо контрольних зразків і виражали у відсотках щодо інтенсивності флуоресценції в контролі. Оцінку фосфоліпідогенного потенціалу та найменшої ефективної концентрації (FEC) тестованої сполуки проводили вручну на підставі значень інтенсивності флуоресценції на обох довжинах хвиль для фіксованих клітин, а також для нефіксованих клітин. Аналіз зниження A у головному мозку пацюків (метод аналізу 5) Ефективність сполук згідно з даним винаходом in vivo була продемонстрована методом аналізу зниження (редукції) вмісту A у головному мозку пацюків, і дані представлені в таблиці 3. Для демонстрації здатності сполук згідно з даним винаходом знижувати вміст амілоїдних пептидів A1-x у головному мозку використовували самців пацюків лінії Sprague-Dawley у віці від 5 до 6 тижнів. Сполуки вводили через пероральний зонд однократними дозами в 1% ® Полісорбаті-80 та 0,5% Натрозолі , зазначеними в таблиці 3. Тварин умертвляли через 3 години після введення дози, головний мозок витягали, розділяли на мозочок і ліву й праву півкулю та швидко заморожували в рідкому азоті. Півкулі головного мозку гомогенізували (5 обсягів за вагою) в 20 мМ Трис-HCl, pН 8,5, 0,2% Тритон-X100, з додаванням інгібіторів протеаз (cOmplete, Roche Applied Science) при 4°C з використанням скляного гомогенізатора Даунса. Гомогенат центрифугували при 120000g протягом 60 хвилин при 4°C, супернатант збирали й аналізували на вміст A1-x з використанням імуноаналізу з хемілюмінесцентною детекцією (Meso-Scale Discovery, Rockville, 8 UA 113641 C2 5 10 15 20 MD (MSD)). Оброблені стрептавідином 96-ямкові планшети (MSD) попередньо обробляли 5% розчином блокатора A (MSD) протягом 1 години при к. т. на орбітальному шейкері та чотири рази промивали фосфатним буферним сольовим розчином (PBS). Лунки попередньо покривали біотинільованими антитілами SIG-39155 (20 нг/лунку, Клон M3.2, специфічний для амінокислот 10-15 A гризунів) протягом 1 години при к. т. і чотири рази промивали PBS. Для аналізу вмісту A1-x, 25 мкл очищених лізатів головного мозку або A1-40 стандартів (8-500 пг/мл, із дворазовим збільшенням) інкубували протягом 1 години при к. т. при постійному перемішуванні. Лунки чотири рази промивали PBS, додавали 25 мкл антитіла, що виявляє (мічене сульфоміткою анти-A40-антитіло виробництва MSD), і інкубували протягом 1 години при к. т. Після4кратного промивання PBS додавали 150 мкл хемілюмінесцентного реагенту, що виявляє (Read Buffer T, MSD), і проводили зчитування з планшета на приладі MSD Sector Imager 6000. Калібровану криву апроксимували з використанням нелінійної регресійної моделі з чотирма параметрами, і обчислювали концентрації A1-x у кожній лунці, що містить очищені лізати головного мозку. Відсоток зниження A обчислювали на підставі відмінностей від середніх значень концентрації A, отриманих для головного мозку тварин, що піддавалися обробці тільки розчинником. У таблиці 1 представлені наступні властивості сполук згідно з даним винаходом: здатність інгібувати BACE1, вимірювана відповідно до методу аналізу 1; інгібуюча здатність у клітинах, вимірювана відповідно до методу аналізу 2; інгібування hERG, вимірюване відповідно до методу аналізу 3; і найменша ефективна концентрація (FEC) у моделі фосфоліпідозу, вимірювана відповідно до методу аналізу 4. Таблиця 1 Біологічні властивості сполук згідно з даним винаходом IC50 (нМ) IC50 (нМ) в Приклад BACE1 клітинах H4-APPwt (метод аналізу 1) (метод аналізу 2) 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 11 8 10 5 8 11 2 5 9 9 4 3 9 5 11 11 14 9 6 5 8 12 5 9 6 0,76 0,29 0,57 0,28 0,90 1,24 1,42 0,49 1,90 1,12 0,35 1,10 0,92 0,12 0,11 0,20 0,89 1,02 0,40 0,28 0,48 0,26 0,18 0,65 0,99 0,20 0,95 інгібування FEC у моделі фосфоліпідозу hERG (%) IC50 (мкМ) 10 мкМ (метод аналізу 4) (метод аналізу 3) 8 200 22 200 9 > 200 0 > 400 7 200 35 200 8 400 16 > 200 16 400 38 200 6 > 400 11 400 18 200 20 > 200 11 200 6 200 12 > 200 17 > 400 8 400 8 > 400 24 400 9 > 400 16 > 200 23 200 10 200 8 > 200 19 > 200 9 UA 113641 C2 Таблиця 1 Біологічні властивості сполук згідно з даним винаходом IC50 (нМ) IC50 (нМ) в Приклад BACE1 клітинах H4-APPwt (метод аналізу 1) (метод аналізу 2) 28 29 5 0,5 0,3 інгібування FEC у моделі фосфоліпідозу hERG (%) IC50 (мкМ) 10 мкМ (метод аналізу 4) (метод аналізу 3) 9 400 7 200 У таблиці 2 представлені дані, які демонструють, що певні сполуки згідно з даним винаходом характеризуються значно меншими значеннями IC50 у методі аналізу інгібування BACE1 (метод аналізу 1) і в методі аналізу в клітинах Abeta (метод аналізу 2) у порівнянні з певними сполуками порівняння, описаними у WO 2010/105179. Таблиця 2 Сполуки згідно з даним винаходом Приклад 5 Приклад 14 Порівняльні приклади Приклад 15 Приклад 281 у WO 2010/105179; BACE IC50 (метод аналізу 1) 35 нМ; метод аналізу в клітинах H4 (метод аналізу 2) 7,0 нМ; hERG (метод аналізу 3) 16% при 10 мкМ H2 N O N N N O O Приклад 7 Приклад 259 у WO 2010/105179; BACE IC50 (метод аналізу 1) 49 нМ; метод аналізу в клітинах H4 (метод аналізу 2) 16 нМ Приклад 8 Приклад 121 у WO 2010/105179; BACE IC50 (метод аналізу 1) 107 нМ; метод аналізу в клітинах H4 (метод аналізу 2) 16 нМ Приклад 11 10 UA 113641 C2 Таблиця 2 Сполуки згідно з даним винаходом Порівняльні приклади Приклад 18 Приклад 320 у WO 2010/105179 BACE IC50 (метод аналізу 1) 415 нМ; метод аналізу в клітинах H4 (метод аналізу 2) 16 нМ Приклад 12 Здатність сполук згідно з даним винаходом знижувати вміст A у головному мозку продемонстрували на пацюках, як описано в методі аналізу 5, і дані ефективності in vivo представлені в таблиці 3. 5 Таблиця 3 Приклад 2 4 6 8 9 12 13 14 15 17 18 20 21 22 23 26 28 29 10 15 20 Доза (мг/кг) 30 25 50 25 25 25 30 25 25 25 25 25 50 12,5 25 25 12,5 25 Зниження вмісту A ( %) 42 75 60 37 37 39 47 67 62 70 56 73 59 45 68 71 30 78 Спосіб лікування Даний винахід відноситься до сполук, які застосовні для лікування порушень або захворювань, що характеризуються підвищеними відкладеннями -амілоїду або рівнями амілоїду в суб'єкта, при яких інгібування активності ферменту -секретази (BACE1) терапевтично ефективно, включаючи без обмеження лікування, зменшення інтенсивності або профілактику нейродегенеративних порушень, порушень, що характеризуються зниженням когнітивних здатностей, когнітивних порушень, деменції та захворювань, що характеризуються формуванням відкладень -амілоїду або нейрофібрилярних клубків. Сполуки згідно з даним винаходом застосовні для лікування хвороби Альцгеймера, трисомії за 21 парою хромосом (синдром Дауна), спадкоємної церебральної геморрагії з амілоїдозом голандського типу (HCHWA-D), сенільної деменції, церебральної амілоїдної ангіопатії, дегенеративної деменції, деменцій змішаного судинного та дегенеративного походження, деменції, асоційованої з хворобою Паркінсона, деменції, асоційованої з прогресуючим супрануклеарним паралічем, деменції, асоційованої з корковою базальною дегенерацією, хвороби Альцгеймера з дифузійними тільцями Леві, сухої вікової макулярної дегенерації (AMD) 11 UA 113641 C2 5 10 15 20 25 30 35 40 45 50 55 60 та глаукоми. «Суха» форма AMD, також відома як «центральна географічна атрофія», виникає внаслідок атрофії пігментного епітеліального шару сітківки під нейросенсорною частиною сітківки, що викликає втрату зору внаслідок втрати фоторецепторів (паличок і колбочок) у центральній частині ока. На даний момент для такого стану не існує якого-небудь медичного або хірургічного лікування. На даний день доступні способи лікування (наприклад, запропоновані Національним Інститутом Ока) містять у собі використання вітамінних добавок із високими дозами антиоксидантів, лютеїну та зеаксантину, які можуть сповільнити прогресію сухої макулярної дегенерації. Глаукома являє собою захворювання, при якому тиск рідини всередині ока зростає, викликаючи необоротне ушкодження зорового нерва та втрату зору. Абета колокалізується з апоптотичними клітинами ганглія сітківки при експериментальній глаукомі й індукує виражений апоптоз клітин ганглія сітківки з дозовою та часовою залежностями. Відповідно, даний винахід відноситься до сполуки або її фармацевтично прийнятної солі для застосування як лікарський засіб. Крім того, даний винахід відноситься до застосування сполуки для лікування захворювання та/або стану, при якому інгібування активності ферменту -секретази (BACE1) терапевтично ефективно. Крім того, даний винахід відноситься до застосування сполуки для лікування нейродегенеративних порушень, порушень, що характеризуються зниженням когнітивних здатностей, когнітивних порушень, деменції та захворювань, що характеризуються формуванням відкладень -амілоїду або нейрофібрилярних клубків. Тому, даний винахід відноситься до застосування сполуки згідно з даним винаходом для лікування хвороби Альцгеймера, трисомії за 21 парою хромосом (синдром Дауна), спадкоємної церебральної геморрагії з амілоїдозом голандського типу (HCHWA-D), сенільної деменції, церебральної амілоїдної ангіопатії, дегенеративної деменції, деменцій змішаного судинного та дегенеративного походження, деменції, асоційованої з хворобою Паркінсона, деменції, асоційованої з прогресуючим супрануклеарним паралічем, деменції, асоційованої з корковою базальною дегенерацією, хвороби Альцгеймера з дифузійними тільцями Леві, сухої AMD і глаукоми. Даний винахід також відноситься до способу лікування порушення, пов'язаного з надмірною активністю BACE1 або асоційованого з нею, у пацієнта, що потребує цього, який включає в себе введення згаданому пацієнтові ефективної кількості розкритої сполуки або її фармацевтично прийнятної солі. Даний винахід також відноситься до способів інгібування активності BACE1 у пацієнта, що потребує цього, який включає в себе введення суб'єкту ефективної кількості, щонайменше, однієї розкритої сполуки або її фармацевтично прийнятної солі та/або приведення її рецептора в контакт із ним. Даний винахід також відноситься до способів зменшення інтенсивності відкладення -амілоїду в суб'єкта, що потребує цього, який включає в себе введення згаданому суб'єкту ефективної кількості, щонайменше, однієї розкритої сполуки або її фармацевтично прийнятної солі. Даний винахід включає в себе терапевтичний спосіб лікування або зменшення інтенсивності опосередкованого BACE1 порушення в суб'єкта, що потребує цього, який включає в себе введення суб'єкту, що потребує цього, ефективної кількості сполуки згідно з даним винаходом, описаної в даному документі, або її фармацевтично прийнятної солі або її композиції. Використовуваний у даному документі термін «суб'єкт» або «пацієнт» може бути використаний взаємозамінно й означає ссавця, що потребує лікування, наприклад свійських тварин (наприклад, собаки, кішки, та т.п.), сільськогосподарських тварин (наприклад, корови, свині, коні, вівці, кози та т.п.) і лабораторних тварин (наприклад, пацюки, миші, морські свинки та т.п.). Зазвичай, суб'єкт являє собою людину, що потребує лікування. Використовуваний у даному документі термін «проведення лікування» або «лікування» відноситься до одержання бажаного фармакологічного та/або фізіологічного ефекту. Такий ефект може бути профілактичним (тобто знижуючим імовірність розвитку порушення або захворювання) або терапевтичним, який включає досягнення, часткове або істотне, одного або декількох із наступних результатів: часткове або повне зменшення ступеня захворювання, порушення або синдрому; зменшення інтенсивності або поліпшення асоційованого з таким порушенням клінічного симптому або індикатора; або вповільнення, інгібування або зниження ймовірності прогресування захворювання, порушення або синдрому. Діапазон доз сполук згідно з даним винаходом, який застосовують протягом доби, зазвичай становить від 0,1 до 3000 мг, переважно від 1 до 2000 мг, більш переважно від 10 до 1000 мг, найбільше переважно, 50 або 500 мг. Кожна одиниця дозування може традиційно містити від 0,1 до 1000 мг, переважно від 25 до 250 мг. 12 UA 113641 C2 5 10 15 20 25 30 35 40 45 50 55 60 Конкретна фармацевтично ефективна кількість або терапевтична доза буде, безумовно, залежати від факторів, відомих фахівцям у даній області техніки, таких як вік і вага пацієнта, шлях введення та важкість захворювання. У будь-якому випадку, сполучення буде вводитися в дозуваннях і способом, які дозволять доставити фармацевтично ефективну кількість, виходячи з індивідуального стану пацієнта. Фармацевтичні композиції Підходящі препарати для введення сполук згідно з даним винаходом будуть очевидні фахівцям у даній області техніки та містять у собі, наприклад, таблетки, пігулки, капсули, супозиторії, льодяники, пастилки, розчини, сиропи, еліксири, саше, ін'єкційні препарати, інгаляційні препарати та порошки, і т.п. Вміст фармацевтично активної(их) сполуки(к) повинен бути в діапазоні від 0,1 до 95%, переважно від 5 до 90% від маси композиції в цілому. Підходящі таблетки можуть бути отримані, наприклад, шляхом змішування однієї або декількох сполук згідно з даним винаходом із відомими наповнювачами, наприклад, з інертними розріджувачами, носіями, розпушувачами, ад’ювантами, поверхнево-активними речовинами, зв'язувальними речовинами та/або змазками. Такі таблетки також можуть складатися з декількох шарів. Сполучена терапія Відповідно до одного варіанта здійснення, даний винахід включає в себе сполучену терапію для лікування або зменшення інтенсивності захворювання або порушення, описаного в даному документі. Сполучена терапія включає в себе введення сполучення, щонайменше, однієї сполуки згідно з даним винаходом із одним або декількома засобами, вибраними з групи, наприклад, яка складається з інгібіторів або модуляторів гамма-секретази; інгібіторів агрегації амілоїду, блокуючих формування Абета олігомерів або Абета фібрил (наприклад, ELND-005); нейропротективних і/або модифікуючих захворювання речовин, що діють прямо або опосередковано; антиоксидантів (наприклад вітамін E або гінколід); протизапальних речовин (наприклад, інгібітори Cox, NSAID, додатково або винятково мають властивості знижувати Абета); інгібіторів HMG-Co редуктази (статини); інгібіторів ацетилхолінестерази (наприклад, донепезил, ривастигмін, такрин, галантамін, такрин); антагоністів NMDA рецептора (наприклад, мемантин); агоністів AMPA рецептора; позитивних модуляторів AMPA рецептора, AMPAкінів, інгібіторів рецептора зворотного захоплення моноаміну, речовин, що модулюють концентрацію або вивільнення нейротрансміттерів; речовин, що індукують секрецію гормону росту (наприклад, ібутаморен мезилат і капроморелін); антагоністів або зворотних агоністів CB-1 рецептора; антибіотиків (наприклад, міноциклін або рифаміпіцин); інгібіторів PDE2, PDE4, PDE5, PDE9, PDE10, зворотних агоністів GABAA рецептора, антагоністів GABAA рецептора, агоністів або часткових агоністів або позитивних модуляторів альфа-4-бета-2-нікотинового рецептора, агоністів або часткових агоністів або позитивних модуляторів альфа-7-нікотинового рецептора; антагоністів гістаміну Н3, агоністів або часткових агоністів 5 HT-4, антагоністів 5HT-6, антагоністів альфа-2-адренорецептора, антагоністів кальцію, агоністів або часткових агоністів або позитивних модуляторів мускаринового М1 рецептора, антагоністів мускаринового М2 рецептора, антагоністів мускаринового М4 рецептора, позитивних модуляторів метаботропного рецептора глутамату 5, антидепресантів, таких як циталопрам, флуоксетин, пароксетин, сертралін і тразодон; анксіолітиків, таких як лоразепам й оксазепам; антипсихотиків, таких як арипіпразол, клозапін, галоперидол, оланзапін, кветіапін, рисперидон і зипрасидон, і інших речовин, які модулюють рецептори або ферменти таким чином, що ефективність та/або безпека сполук згідно з даним винаходом збільшується, і/або небажані побічні ефекти знижуються. Сполуки згідно з даним винаходом також можуть бути використані в сполученні з імунотерапіями (наприклад, активна імунізація Abeta або його частинами або пасивною імунізацією гуманізованими анти-Абета антитілами або нанотілами) для лікування вищезгаданих захворювань або станів. Сполучена терапія включає в себе спільне введення сполуки згідно з даним винаходом із одним або декількома іншими засобами, послідовне введення сполуки й одного або декількох інших засобів, введення композиції, що містить сполуку й одного або декількох інших засобів, або одночасне введення окремих композицій, що містять сполуку й один або декілька інших засобів. Експериментальний розділ Способи одержання сполук Сполуки згідно з даним винаходом можуть бути отримані зі застосуванням традиційних способів, у яких використовуються легкодоступні реагенти та вихідні речовини. Реагенти, використані при одержанні сполук згідно з даним винаходом, можуть бути або комерційно доступними, або можуть бути отримані за допомогою стандартних методик, описаних у 13 UA 113641 C2 5 10 15 20 25 30 літературі. Реакції з обробкою мікрохвилями проводять у реакторі CEM з використанням системи Discovery SP. У випадку надання даних ЯМР, спектри одержували на спектрометрі Varian-400 (400 Мгц), і наводили дані в м. д. убік слабкого поля відносно тетраметилсилану з вказівкою в круглих дужках номера протона, мультиплетностей та констант взаємодії, а також з посиланням на дейтерований розчинник. Сполуки очищали базовим методом препаративної HPLC, описаним нижче. Метод 1: Рухома фаза A: вода з 0.05% розчином аміаку; рухома фаза B: ACN; швидкість потоку: 25 мл/хв; детекція: УФ 220 нм/254 нм; колонка: Phenomenex Gemini C18 250×30 мм, 5 мкм; температура колонки: 30°C Час (хв) %A %B 0.0 68 32 12.00 38 62 12.20 0 100 13.5 0 100 13.7 90 10 Метод 2: Рухома фаза A: вода з 0.05% розчином аміаку; рухома фаза B: ACN; швидкість потоку: 25 мл/хв; детекція: УФ 220 нм/254 нм; колонка: Durashell C18 250×30 мм, 5 мкм; температура колонки: 30°C Час (хв) %A %B 0.0 67 33 12.00 47 53 12.20 0 100 13.5 0 100 13.7 90 10 Дані LC-MS одержували з використанням наступних умов проведення хроматографії: TM HPLC-система: Waters ACQUITY; колонка: Waters ACQUITY CSH C18 1.7 мкм Предколонка: Waters Assy. Frit, 0.2 мкм, 2.1 мм; температура колонки: 40°C Рухома фаза A: TFA/вода (1/1000, об./об.); рухома фаза B: TFA/ACN (1/1000, об./об.); швидкість потоку: 0.65 мл/хв; об’єм, що вводиться: 2 мкл; час зрівноважування: приблизно 1.5 хв. Схема градієнта: Час (хв) B% 0 10 0.8 90 1.20 90 1.21 10 Параметри мас-спектрометра: Мас-спектрометр: Waters SQD; іонізація: іонізація електророзпиленням (ESI) з детекцією позитивно заряджених іонів; режим сканування (100-1400 m/z кожні 0.2 с); напруга на ES капілярі: 3.5 кВ; напруга на ES конусі: 25 В; температура джерела: 120°C; температура десольватації: 500°C; потік газу-осушувача: азот у режимі 650 л/год; потік газу на конусі: азот в режимі 50 л/год На стадії 2 прикладу 10 і на стадіях 1 і 2 альтернативного синтезу проміжного продукту 38 використовували наступні умови проведення хроматографії й устаткування: Дані LC-MS одержували з використанням наступних умов проведення хроматографії: HPLC-система: Agilent 1100 Zorbax Eclipse XDB-C8, колонка: 2.1×50 мм температура 35°C колонки: A: мурашина Рухома фаза кислота/вода (1/1000, об./об.) B: мурашина кислота/ACN (1/1000, об./об.) 14 UA 113641 C2 Схема градієнта: Час (хв) 0 3 4.5 5.0 Швидкість потоку: Об’єм, що вводиться Час втримання: Час зрівноважування: Параметри масспектрометра: Масспектрометр: Іонізація: Режим Напруга на ES капілярі: Напруга на ES конусі: Температура джерела температура десольватації: потік газуосушувача: потік газу на конусі: B% 5 95 95 5 0.60 мл/хв : 2 мкл приблизно 1-4 хв приблизно 5 хв Agilent 77 іонізація електророзпиленням (ESI) у режимі детекції позитивно заряджених іонів сканування (100-800 m/z кожні 0.2 с) 3.5 кВ 25 В 120°C 500°C азот у режимі 650 л/год азот у режимі 50 л/год У прикладі 27 використовували наступні умови проведення хроматографії й устаткування: HPLC-система: Waters Alliance / DA- і MS-детектор Колонка: Waters XBridge C18, 4.6×30 мм, 3.5 мкм 5 Схема градієнта: час [хв] 0,0 1,6 1,85 1,9 10 15 % розчину [H2O, 0.1%TFA] 95 0 0 95 % розчину [метанол] 5 100 100 5 Потік [мл/хв] 4 4 4 4 Температура [°C] 60 60 60 60 Поділ методом SFC й охарактеризацію сполук проводили у відповідності з наступним способом. Метод A: Апарат: Thar SFC 80; колонка: AD 250 мм×30 мм, 5 мкм; Рухомі фази: A: надкритичний CO2, B: IPA (0.05% DEA), A/B =80/20 при 60 мл/хв; температура колонки: 38°C; тиск у форсунці: 100 бар; температура форсунки: 60°C; температура випарника: 20°C; температура тримера: 25°C; довжина хвилі: 220 нм. Метод B: Апарат: SFC MG2; колонка: OJ 250 мм×30 мм, 5 мкм; Рухомі фази: A: надкритичний CO2, B: MeOH (0.05% DEA), A/B =90/10 при 70 мл/хв; температура колонки: 38°C; тиск у форсунці: 100 бар; температура форсунки: 60°C; температура випарника: 20°C; температура тримера: 25°C; довжина хвилі: 220 нм. 15 UA 113641 C2 Наступні методики, розчинники та реагенти можуть позначатися наступними скороченнями: Скорочення Значення ACN ацетонітрил Boc трет-бутоксикарбоніл або t-бутоксикарбоніл сольовий розчин насичений водний NaCl DCM хлористий метилен DIEA діізопропілетиламін DMF диметилформамід DMSO диметилсульфоксид dppf 1,1-біс(дифенілфосфіно)фероцен EDCI гідрохлорид 1-(3-диметиламінопропіл)-3-етилкарбодііміду EtІ етил йодид Et етил Et2O етиловий ефір EtOAc етилацетат EtOH етанол HPLC високоефективна рідинна хроматографія LDA діізопропіламід літію MeOH метанол MeІ метил йодид Me метил Me2S диметилсульфід MsCl метансульфонілхлорид NaOMe метоксид натрію PdCl2dppf [1,1-біс(дифенілфосфіно)фероцен]дихлорпаладій (II) Pd2(dba)3 трис(дибензиліденацетон)дипаладій(0) PE петролейний ефір к. т. кімнатна температура SFC надкритична рідинна хроматографія t-BuOK трет-бутоксид калію t-BuLi трет-бутиллітій t-BuNH2-BH3 комплекс трет-бутиламін/боран t-BuOOH трет-бутилпероксид TFA трифтороцтова кислота TFAA ангідрид трифтороцтової кислоти THF тетрагідрофуран TLC тонкошарова хроматографія Ti(OEt)4 тетраетоксид титану Приклад 1 N H2N NC N N N O OCH3 5 10 Стадія 1: Синтез проміжного продукту 3 Суміш сполуки 1 (50,0 г, 236 ммоль) і метил акрилату (42,0 г, 472 ммоль) у безводному THF (900 мл) попередньо охолоджували при 0°C, протягом 30 хв рівними порціями додавали t-BuOK (31,8 г, 284 ммоль, 1,1 екв.), потім суміш нагрівали до к. т. протягом 1 год і перемішували протягом 40 хв при к. т. До цієї реакційної суміші додавали DMF (200 мл) і EtІ (74 г, 472 ммоль), і перемішували при к. т. протягом ночі. В умовах зниженого тиску видаляли THF. Залишок розбавляли H2O (300 мл) і екстрагували EtOAc, концентрували з одержанням неочищеної 16 UA 113641 C2 5 10 15 20 25 30 35 40 сполуки 2 (120,0 г). Цей продукт використовували як є на наступній стадії. Суміш сполуки 2 (120,0 г, 310 ммоль) і LiCl (130,0 г, 3100 ммоль) в DMSO (900 мл) нагрівали зі зворотним холодильником протягом ночі. Суміш гасили додаванням води (3 л) і екстрагували EtOAc (3×400 мл). Розділену органічну фазу сушили та концентрували в умовах зниженого тиску. Залишок очищали методом колоночної хроматографії на силікагелі (петролейний ефір/EtOAc = 20/1) з одержанням проміжного продукту 3 (15 г, 20%). 1 H ЯМР: (CDCl3): 7,91 (s, 1H), 7,74 (dd, J = 8,0 Гц, 1H), 7,41 (d, J = 8,0 Гц, 1H), 3,80 (s, 2H), 2,48-2,53 (m, 2H), 2,33-2,49 (m, 1H), 2,15-2,23 (m, 1H), 1,75-1,95 (m, 4H), 1,21-1,40 (m, 1H), 0,88 (t, J = 8,0 Гц, 3H). Стадія 2: Синтез проміжного продукту 5 До суміші THF (20 мл) і MeOH (5 мл) при -78°C додавали проміжний продукт 3 (6,0 г, 18,7 ммоль), NaBH4 (355 мг, 9,3 ммоль) і CeCl3·7H2O (70 мг, 0,19 ммоль). Суміш перемішували при 78°C протягом 20 хв, гасили додаванням нас. розчину NH4Cl (30 мл) і екстрагували EtOAc (4×400 мл). EtOAc фази поєднували та концентрували з одержанням неочищеної сполуки 4 (6,5 г, неочищ.). До суміші сполуки 4 (6,5 г, 20,0 ммоль) і NaН (3,2 г, 80,0 ммоль) в DMF (100 мл) при 0°C додавали MeІ (11,4 г, 80,0 ммоль). Суміш перемішували при к. т. протягом ночі. Суміш гасили додаванням H2O, екстрагували EtOAc, концентрували з одержанням неочищеного продукту, який очищали на колонці з силікагелем (елюент: петролейний ефір/етилацетат = 20/1 15/1) з одержанням проміжного продукту 5 (3,5 г, 56%). + LC-MS: tR = 1,315 хв, MS (ESI) m/z 339,1 [M+H] . 1 H ЯМР: (CDCl3): 7,88 (s, 1H), 7,69 (dd, J = 8,4, 2,0 Гц, 1H), 7,31 (d, J = 8,4 Гц, 1H), 3,39 (s, 3H), 2,97 (s, 2H), 2,88-2,94 (m, 1H), 2,21-2,26 (m, 1H), 1,81-1,87 (m, 1H), 1,70-1,78 (m, 1H), 1,401,59 (m, 4H), 1,12-1,39 (m, 2H), 0,88 (t, J = 8,0 Гц, 3H). Стадія 3: Синтез проміжного продукту 6A і 6B Суміш проміжного продукту 5 (3,5 г, 10,4 ммоль) і етоксиду титану (IV) (23,7 г, 104 ммоль) у безводному THF (40 мл) перемішували при к. т. протягом 1 год. Додавали (S)-N-третбутилсульфінамід (1,6 г, 11,6 ммоль), і перемішували отриману суміш при 80°C в атмосфері N2 протягом ночі. Потім, реакційну суміш охолоджували, додавали воду (400 мл) і фільтрували. Водний шар екстрагували EtOAc (3×400 мл). Розділені органічні фази поєднували, сушили та концентрували в умовах зниженого тиску. Залишок очищали методом колоночної хроматографії на силікагелі (петролейний ефір/EtOAc = 20/1) з одержанням проміжного продукту 6A (1,5 г, 33%) і 6B (1,5 г, 33%), елюйованих у зазначеному порядку. Стадія 4: Синтез проміжного продукту 7B До суміші етоксіетену (1,3 г, 17,0 ммоль) у безводному THF (20 мл) при -78°C в атмосфері N2 по краплях додавали t-BuLi (13,0 мл, 17,0 ммоль, 1,3 M у гексані), і перемішували протягом 20 хв. Потім, отриману суміш додатково перемішували при 0°C протягом 45 хв. До цієї суміші при 78°C по краплях додавали проміжний продукт 6B (1,5 г, 3,4 ммоль) у безводному THF (20 мл), і перемішували протягом 2,5 год. Реакційну суміш гасили додаванням нас. NH 4Cl (50 мл) і екстрагували EtOAc (3×300 мл). Органічні фази поєднували та концентрували з одержанням 17 UA 113641 C2 залишку, який очищали на колонці з силікагелем (петролейний ефір/EtOAc = 20/1) з одержанням проміжного продукту 7B (1,2 г, 69%). Стадія 5: Синтез проміжного продукту 8B 5 10 15 20 25 30 До DCM/MeOH (5/1, 20 мл) додавали проміжний продукт 7B (1,2 г, 2,4 ммоль), суміш охолоджували до -78°C, і барботували суміш озоном протягом 20 хв. Потім, суміш продували N 2 й обробляли Me2S при -78°C. Потім, реакційну суміш залишали нагріватися до к. т. і перемішували протягом 3 год. Розчинник видаляли в умовах вакууму, залишок очищали методом препаративної TLC (петролейний ефір/EtOAc = 3/1) з одержанням сполуки 8B (860 мг, 70%). + LC-MS: tR = 1,351 хв, MS (ESI) m/z 516,1 [M+H] . Стадія 6: Синтез проміжного продукту 9B До сполуки 8B (860 мг, 1,7 ммоль) в MeOH (10 мл) додавали 4 M розчин HCl у діоксані (2 мл). Отриману суміш перемішували протягом 30 хв. Розчинник видаляли в умовах зниженого тиску з одержанням неочищеної сполуки 9B (800 мг). Залишок використовували на наступній стадії без додаткового очищення. Стадія 7: Синтез проміжного продукту 10B Суспензію проміжного продукту 9B (500 мг, 1,9 ммоль), Zn(CN)2 (300 мг, 2,6 ммоль), Pd2(dba)3 (150 мг, 0,16 ммоль), dppf (160 мг, 0,32 ммоль) і Zn пилу (60 мг, 0,9 ммоль) в DMF (15 мл) нагрівали при 120°C протягом 3 год у мікрохвильовому реакторі CEM. Суміш концентрували в умовах вакууму, і очищали залишок на колонці з силікагелем (елюент: петролейний ефір/EtOAc = 20/1 8/1) з одержанням сполуки 10B (150 мг, 40%). Стадія 8: Синтез проміжного продукту 11B До DCM (10 мл), H2O (10 мл) і NaHCO3 (350 мг, 4,2 ммоль) додавали проміжний продукт 10B (150 мг, 0,42 ммоль). До цієї суміші при енергійному перемішуванні додавали тіофосген (100 мг, 0,84 ммоль), перемішували протягом 50 хв при к. т. й екстрагували DCM (3×40 мл). Органічний шар промивали сольовим розчином (2×40 мл), сушили, і видаляли розчинник в умовах зниженого тиску з одержанням неочищеної сполуки 11B (150 г, 93%), яку використовували на наступній стадії без додаткового очищення. Стадія 9: Синтез проміжного продукту 12B 18 UA 113641 C2 5 10 15 20 До суміші сполуки 11B (150 мг, 0,39 ммоль) в THF (5 мл) додавали 2-амінометилпіримідин (67 мг, 0,78 ммоль) і TEA (395 мг, 3,90 ммоль). Суміш перемішували протягом ночі при к. т. Реакційну суміш розбавляли водою й екстрагували EtOAc (30 мл). Залишок очищали методом колоночної хроматографії (петролейний ефір/етилацетат = 10/1) з одержанням 12B (100 мг, 70%). + LC-MS: tR = 1,204 хв, MS (ESI) m/z 462,2 [M+H] . Стадія 1: Синтез сполуки прикладу 1 До сполуки 12B (100 мг, 0,22 ммоль) в MeOH (10 мл) і NH4OH (3 мл) додавали t-Bu2H (1 мл). Після додавання, суміш перемішували при к. т. протягом 24 год. Суміш гасили додаванням насиченого розчину Na2S2O3 (0,5 мл). Залишок розподіляли між EtOAc (20 мл) і H2O (10 мл). Органічний шар розділяли та промивали сольовим розчином (10 мл), сушили, фільтрували та концентрували в умовах вакууму. Залишок очищали методом HPLC (спосіб 1) з одержанням сполуки прикладу 1 (14,60 мг, 15%). + LC-MS: tR = 0,933 хв, MS (ESI) m/z 445,2 [M+H] . 1 H ЯМР: (CD3OD): 8,74 (d, J = 5,2 Гц, 2H), 7,61 (dd, J = 7,6, 1,6 Гц, 1H), 7,52 (s, 1H), 7,45 (d, J = 8,0 Гц, 1H), 7,35 (t, J = 5,2 Гц, 1H), 4,94 (s, 2H), 3,38 (s, 3H), 3,17 (s, 2H), 2,80-2,87 (m, 1H), 2,082,13 (m, 1H), 1,90-1,94 (m, 1H), 1,38-1,85 (m, 2H), 1,22-1,39 (m, 3H), 1,12-1,18 (m, 2H), 0,76 (t, J = 8,0 Гц, 3H). Приклад 2 Зазначену сполуку синтезували з проміжного продукту 10B, отриманого у прикладі 1, як представлено нижче на схемі. 25 30 Стадія 1: Синтез проміжного продукту 13 До перемішаного розчину тіосечовини (23 г, 302 ммоль) в THF (5,0 л) в атмосфері аргону при 0°C додавали гідрид натрію (29,9 г, 755 ммоль, 60% у мінеральному маслі). Через 5 хв, баню з льодом видаляли, і перемішували реакційну суміш при кімнатній температурі протягом 10 хв. Суміш знову охолоджували до 0°C, додавали ди-трет-бутилдикарбонат (138 г, 635 ммоль), і після 30 хв перемішування при зазначеній температурі видаляли лазню з льодом. Отриману суспензію додатково перемішували протягом 2 год при к. т. Потім, реакційну суміш гасили додаванням насиченого водяного розчину NaHCO3 (500 мл). Реакційну суміш вливали у 19 UA 113641 C2 5 10 15 20 25 30 35 40 45 50 воду (5,0 л) і екстрагували EtOAc (3×2,0 л). Об'єднаний органічний шар сушили, фільтрували та концентрували в умовах вакууму з одержанням проміжного продукту 13 (80 г, 96%) у вигляді білої твердої речовини, яку використовували на наступній стадії без додаткового очищення. До суміші проміжного продукту 13 (4,14 г, 15,0 ммоль) і безводного THF (300 мл) при 0°C додавали NaН (60% у мінеральному маслі, 720 мг, 18,0 ммоль). Реакційну суміш перемішували при 0°C протягом 1 год, потім додавали TFAA (3,47 г/2,33 мл, 16,5 ммоль), і додатково продовжували перемішування протягом 1 год. Потім, додавали 4-(амінометил)тетрагідропіран (2,5 г, 16,5 ммоль) і Et3N (3,03 г/4,16 мл, 30,0 ммоль) у безводному THF (130 мл), і перемішували отриману реакційну суміш при к. т. протягом ночі. Для гасіння реакційної суміші додавали H 2O (150 мл), і екстрагували суміш EtOAc (3×200 мл). Об'єднані органічні шари сушили, і видаляли розчинник в умовах зниженого тиску. Залишок очищали методом флеш-хроматографії з одержанням сполуки 13 (3,54 г, 86%) у вигляді білої твердої речовини. + LCMS: tR = 0,973 хв; MS (ESI) m/z 219 [M-t-Bu] . Стадія 2: Синтез проміжного продукту 14 До суміші сполуки 10B (2,5 г, 7,0 ммоль) в 30 мл DMF додавали сполуку 13 (2,3 г, 8,4 ммоль), EDCI (2,5 г, 14,0 ммоль) і DIEA (1,7 г, 14,0 ммоль). Суміш перемішували при к. т. протягом ночі. Суміш екстрагували EtOAc (3×80 мл), промивали сольовим розчином (3×50 мл), сушили, і видаляли розчинник в умовах зниженого тиску. Залишок очищали методом колоночної хроматографії (петролейний ефір/етилацетат = 5/1) з одержанням сполуки 14 (2,7 г, 75%). + LC-MS: tR = 0,972 хв, MS (ESI) m/z 495,3 [M-t-Bu] . Стадія 3: Синтез сполуки прикладу 2 До суміші проміжного продукту 14 (2,7 г, 4,9 ммоль) в DCM (30 мл) додавали TFA (6 мл). Після додавання, суміш перемішували при к. т. протягом 1 год. Значення pН реакційної суміші коректували до 8,0-9,0 додаванням розчину NaHCO3. Органічний шар концентрували в умовах зниженого тиску. Залишок очищали методом колоночної хроматографії (петролейний ефір/етилацетат = 1/1) з одержанням сполуки прикладу 2 (1,83 г, 83%) у вигляді білої твердої речовини. + LC-MS: tR = 0,897 хв, MS (ESI) m/z 451,2 [M+H] . 1 H-ЯМР: (CD3OD): 7,66 (dd, J = 8,0, 1,6 Гц, 1H), 7,51 (d, J = 7,6 Гц, 1H), 7,33 (s, 1H), 3,923,98 (m, 2H), 3,37-3,43 (m, 7H), 3,20 (m, 2H), 2,78-2,83 (m, 1H), 2,16-2,20 (m, 1H), 1,87-2,03 (m, 1H), 1,71-1,77 (m, 1H), 1,58-1,62 (m, 1H), 1,51-1,54 (m, 2H), 1,28-1,37 (m, 7H), 1,09-1,10 (m, 1H), 0,76 (t, J = 7,6 Гц, 3H). Приклад 3 Синтез проміжного продукту 18 Стадія 1: Синтез проміжного продукту 16 До розчину метилмагнійброміду (14 мл, 42 ммоль, 3,0 M в Et 2O) при -30°C в атмосфері азоту додавали суміш сполуки 15 (2,0 г, 10,6 ммоль) у безводному THF (20 мл). Суміш перемішували при -30°C протягом 4 год, а потім гасили додаванням води (40 мл) і водн. HCl (50 мл, 1 M) при перемішуванні при 0°C. Суміш розділяли, і екстрагували водний шар EtOAc (2×50 мл). Об'єднані органічні шари промивали сольовим розчином (2×50 мл), сушили, фільтрували та концентрували в умовах вакууму з одержанням неочищеного проміжного продукту 16 (2,1 г, 100% неочищ.) у вигляді безбарвного масла, яке використовували безпосередньо на наступній стадії без очищення. 1 H ЯМР: (CDCl3): 4,97 (br, 1H), 3,10 (s, 2H), 2,17 (br, 1H), 1,44 (s, 9H), 1,20 (s, 6H). Стадія 2: Синтез проміжного продукту 17 До суміші проміжного продукту 16 (3,0 г, 15,9 ммоль, неочищ.) у безводному DCM (50 мл) при -78°C в атмосфері азоту додавали DAST (2,3 мл, 17,4 ммоль). Суміш перемішували при 78°C протягом 1 год і залишали нагріватися до к. т. протягом ночі. Потім, суміш охолоджували до 0°C, і гасили додаванням насиченого водяного розчину NaHCO 3 (30 мл) при повільному 20 UA 113641 C2 5 10 15 20 25 перемішуванні при 0°C. Суміш розділяли, і водний шар екстрагували DCM (2×20 мл). Об'єднані органічні шари промивали сольовим розчином (2×30 мл), сушили, фільтрували та концентрували в умовах вакууму з одержанням неочищеного проміжного продукту 17 (2,5 г, 76% неочищ.), який використовували безпосередньо на наступній стадії без очищення. 1 H ЯМР: (CDCl3): 4,82 (br, 1H), 3,30-3,35 (d, J = 6,0 Гц, 1H), 3,24-3,26 (d, J = 6,0 Гц, 1H), 1,44 (s, 9H), 1,37 (s, 3H), 1,35 (s, 3H). 19 F ЯМР: (CDCl3 400 MГц): -144,93. Стадія 3: Синтез проміжного продукту 18 До суміші проміжного продукту 17 (2,0 г, 10,5 ммоль, неочищ.) у безводному DCM (10 мл) при перемішуванні додавали HCl у діоксані (10 мл, 40 ммоль, 4 M у діоксані). Суміш перемішували при к. т. протягом 2 год, після чого концентрували розчинник в умовах вакууму. Залишок промивали сумішшю DCM/петролейний ефір (1/1, 3×10 мл), осад збирали та сушили в умовах вакууму з одержанням неочищеної сполуки 18 (1,1 г), яку використовували безпосередньо на наступній стадії без очищення. 1 H ЯМР: (CD3OD): 3,15-3,25 (d, J = 20,0 Гц, 2H), 1,51 (s, 3H), 1,48 (s, 3H). 19 F ЯМР: (CDCl3 400 MГц): -147,59. Сполука прикладу 3 Сполуку прикладу 3 синтезували з проміжного продукту 11B, отриманого у прикладі 1, слідуючи тим самим методикам, що й описані у прикладі 1, і використовуючи проміжний продукт 18 на стадії 9 прикладу 1. + LC-MS: tR = 1,12 хв, MS (ESI) m/z 427 [M+H] . 1 H-ЯМР: (CD3OD) 7,65 (dd, 1H, J = 8, 2 Гц), 7,51 (d, 1H, J = 8 Гц), 7,31 (s, 1H), 3,72 (dd, 2H, J = 22, 4 Гц), 3,37 (s, 3H), 3,20 (ap q, 2H, J = 16 Гц), 2,82 (m, 1H), 2,18 (m, 1H), 1,90 (m, 1H), 1,791,70 (m, 1H), 1,52-11,22 (m, 10H), 1,21-1,09 (m, 1H), 0,77 (t, 3H, J = 7 Гц). Приклад 4 Синтез проміжного продукту 25 30 35 Стадія 1: Синтез проміжного продукту 20 Суміш дигідро-2H-піран-4(3H)-ону (19, 50,0 г, 500 ммоль) і 2-хлорацетонітрилу (35,0 г, 350 ммоль) у трет-бутанолі (50 мл) перемішували протягом 30 хв. До цієї суміші протягом 40 хв додавали розчин t-BuOK (60 г, 550 ммоль) у трет-бутанолі (500 мл). Реакційну суміш перемішували при к. т. протягом 16 год. Суміш розбавляли водою та гасили додаванням 10% HCl. Реакційну суміш концентрували до однієї третьої первинного об’єму, і чотири рази екстрагували діетиловим ефіром. Об'єднані органічні шари промивали сольовим розчином, сушили над MgSO4, фільтрували та концентрували з одержанням проміжного продукту 20 (57 г), 21 UA 113641 C2 5 10 15 20 25 30 35 40 45 50 який використовували безпосередньо на наступній стадії без очищення. Стадія 2: Синтез проміжного продукту 21 Проміжний продукт 20 (57 г) змішували з дихлорметаном (200 мл) у сулії з поліпропілену. Сулію охолоджували до 0°C, і повільно додавали 70% фтороводень у піридині (50 мл). Суміш залишали нагріватися до кімнатної температури протягом ночі. Реакційну суміш розбавляли етилацетатом (500 мл) і вливали в насичений водний NaHCO3. Для нейтралізації суміші обережно, до виділення пухирців газу, використовували додаткову кількість твердого NaHCO 3. Органічний шар розділяли, і екстрагували водний шар етилацетатом (3×500 мл). Об'єднані органічні шари промивали 1% водним розчином HCl, сольовим розчином, сушили (MgSO4), фільтрували та концентрували з одержанням неочищеного проміжного продукту 21 (54 г), який використовували безпосередньо на наступній стадії без очищення. 1 H ЯМР: (CDCl3): 4,37 (m, 2H), 3,96-2,70 (m, 4H), 1,97 - 1,81 (m, 4H). Стадія 3: Синтез проміжного продукту 22 До суміші проміжного продукту 21 (54 г; 340 ммоль) в 2-пропанолі (1000 мол) і воді (250 мл) при 0°C додавали боргідрид натрію (20 г, 509 ммоль). Суміш перемішували та залишали нагріватися до к. т. протягом 3 год. Реакційну суміш гасили додаванням ацетону, і додатково перемішували протягом 1 год. Прозору рідину відокремлювали від твердої речовини шляхом декантування. Для промивання твердої речовини використовували додаткову кількість EtOAc, і декантували. Об'єднаний органічний розчин концентрували. Залишок очищали методом колоночної флеш-хроматографії на силікагелі, елюючи 5-20% EtOAc у гексанах, з одержанням проміжного продукту 22 (22 г, 40% за 3 стадії) у вигляді рідини. 1 H ЯМР: (CDCl3): : 3,82-3,77 (m, 4H), 3,72-3,52 (dd, J = 20,8, 6,4 Гц, 2H), 2,69(s, 1H), 1,82-1,60 (m, 4H). Стадія 4: Синтез проміжного продукту 23 До суміші проміжного продукту 22 (20 г, 150 ммоль) і триетиламіну (22,7 г, 225 ммоль) в DCM (200 мл) при 0°C додавали MsCl (25,8 г, 225 ммоль). Суміш перемішували при к. т. протягом 2 год, а потім додавали воду. Водний шар екстрагували DCM (2×200 мл). Розчинник сушили та видаляли з одержанням неочищеного проміжного продукту 23 (30 г, 100%), який використовували на наступній стадії без додаткового очищення. 1 H ЯМР: (CDCl3): : 4,22 (d, J = 20,0 Гц, 2H), 3,87-3,82 (m, 4H), 3,06(s, 3H), 1,88-1,68 (m, 4H). Стадія 5: Синтез проміжного продукту 24 До суміші проміжного продукту 23 (10 г, 47 ммоль) і DMF (150 мл) при 120°C додавали NaN3 (16 г, 250 ммоль) і NaHCO3 (9,3 мг, 100 ммоль). Суміш перемішували при 120°C протягом 20 год, реакційну суміш гасили додаванням води й екстрагували EtOAc (2×300 мл). Розчинник сушили та видаляли в умовах вакууму з одержанням неочищеного проміжного продукту 24 (8 г), який використовували на наступній стадії без додаткового очищення. Стадія 6: Синтез проміжного продукту 25 До суміші проміжного продукту 24 (8 г, 50 ммоль) в етилацетаті (100 мл) в атмосфері азоту додавали Pd/C (0,8 г, вміст 10%), з суміші відкачували повітря та заміняли його воднем (тричі). Кінцеву суміш перемішували протягом 24 год при кімнатній температурі та тиску 1 атм. водню. ® Каталізатор відфільтровували через шар Celite і промивали EtOAc (2×50 мл). Об'єднаний фільтрат концентрували в умовах зниженого тиску з одержанням проміжного продукту 25 (5,3 г, 80%). 1 H ЯМР: (CD3OD): 3,83-3,79 (m, 4H), 2,76-2,71 (d, J = 8,0 Гц, 2H), 1,83-1,65 (m, 4H). 19 F ЯМР: (CD3OD, 400 MГц) : -169,66. Сполука прикладу 4 Сполуку прикладу 4 синтезували з проміжного продукту 11B, слідуючи тій самій методиці, що й описана у прикладі 1, використовуючи на стадії 9 проміжний продукт 25 замість 2піримідилметанаміну. + LC-MS: tR = 0,98 хв, MS (ESI) m/z 469 [M+H] . 1 H-ЯМР: (CD3OD) 7,64 (d, 1H, J = 8 Гц), 7,50 (d, 1H, J = 8 Гц), 7,31 (s, 1H), 3,84-3,65 (m, 6H), 3,36 (s, 3H), 3,19 (ap q, 2H, J = 16 Гц), 2,81 (m, 1H), 2,17 (m, 1H), 1,89-1,66 (m, 6H), 1,50-1,37 (m, 3H), 1,34 (m, 2H), 1,20-1,11 (m, 1H), 0,76 (t, 3H, J = 8 Гц). 22 UA 113641 C2 Приклад 5 Стадія 4: Синтез проміжного продукту 7A 5 10 15 20 25 До суміші етоксіетену (1,3 г, 17,0 ммоль) у безводному THF (20 мл) при -78°C в атмосфері N2 по краплях додавали t-BuLi (13,0 мл, 17,0 ммоль, 1,3 M у гексані), і перемішували суміш протягом 20 хв. Потім, отриману суміш додатково перемішували при 0°C протягом 45 хв, додавали сполуку 6A (1,5 г, 3,4 ммоль) у безводному THF (20 мл), і перемішували протягом 2,5 год. Реакційну суміш гасили додаванням нас. NH 4Cl (50 мл) і екстрагували EtOAc (3×300 мл). Органічні фази поєднували та концентрували з одержанням неочищеного продукту. Продукт очищали на колонці з силікагелем (петролейний ефір/EtOAc= 20/1) з одержанням сполуки 7A (1,2 г, 69%), яку використовували як є на наступній стадії. Стадія 5: Синтез проміжного продукту 8A Суміш сполуки 7A (1,2 г, 2,4 ммоль) в DCM/MeOH = 5/1 (20 мл) охолоджували до -78°C, і барботували суміш озоном протягом 20 хв. Суміш продували N 2 й обробляли Me2S (5 мл) при 78°C, а потім залишали нагріватися до к. т. і перемішували протягом 3 год. Розчинник видаляли в умовах вакууму, залишок очищали методом препаративної TLC (петролейний ефір/EtOAc= 3/1) з одержанням сполуки 8A (860 мг, 70%). + LC-MS: tR = 1,333 хв; MS (ESI) m/z 516,1 [M+H] . Стадія 6: Синтез проміжного продукту 9A До суміші сполуки 8A (860 мг, 1,7 ммоль) в MeOH (10 мл) додавали 4 M розчин HCl у діоксані (2 мл). Отриману суміш перемішували протягом 30 хв при к. т. Розчинник видаляли в умовах зниженого тиску з одержанням неочищеної сполуки 9A (800 мг), яку використовували на наступній стадії без додаткового очищення. + LC-MS: tR = 0,976 хв; MS (ESI) m/z 361,1 [M+H] . Стадія 7: Синтез проміжного продукту 10A 23 UA 113641 C2 5 10 15 20 25 30 Суміш сполуки 9A (500 мг, 1,9 ммоль), Zn(CN)2 (300 мг, 2,6 ммоль), Pd2(dba)3 (150 мг, 0,16 ммоль), dppf (160 мг, 0,32 ммоль) і Zn пил (60 мг, 0,9 ммоль) в DMF (15 мл) нагрівали до 120°C протягом 3 год у мікрохвильовому реакторі CEM. Суміш концентрували в умовах вакууму, і очищали залишок на колонці з силікагелем (елюент: петролейний ефір/EtOAc = 20/1 8/1) з одержанням сполуки 10A (300 мг, 40%). + LC-MS: tR = 0,880; MS (ESI) m/z 308,1 [M+H] . Стадія 8: Синтез проміжного продукту 11A До суміші сполуки 10A (300 мг, 0,84 ммоль) в DCM (10 мл), H2O (10 мл) і NaHCO3 (655 мг, 8,4 ммоль) додавали тіофосген (180 мг, 1,68 ммоль). Суміш перемішували протягом 50 хв, потім екстрагували DCM (3×40 мл), промивали сольовим розчином (2×40 мл), сушили, і видаляли розчинник в умовах зниженого тиску з одержанням неочищеної сполуки 11A (300 г), яку використовували на наступній стадії без додаткового очищення. Стадія 9: Синтез проміжного продукту 12A До сполуки 11A (200 мг, 0,50 ммоль) в THF (10 мл) додавали R-(2амінометил)тетрагідрофуран (61 мг, 0,6 ммоль) і триетиламін (2 мл, 5,0 ммоль). Суміш перемішували при к. т. протягом ночі. Реакційну суміш розбавляли водою й екстрагували EtOAc (30 мл). Залишок очищали методом колоночної хроматографії (петролейний ефір/EtOAc = 10/1) з одержанням сполуки 12A (180 мг, 79%). Стадія 10: Синтез сполуки прикладу 5 До суміші проміжного продукту 12A (250 мг, 0,54 ммоль) в MeOH (10 мл) і NH4OH (3 мл) додавали розчин t-Bu2H (1 мл, 9M у гексані), і перемішували при к. т. протягом 24 год. Реакційну суміш гасили додаванням насиченого Na2S2O3 (0,5 мл). Залишок розподіляли між EtOAc (20 мл) і H2O (10 мл). Органічний шар розділяли та промивали сольовим розчином (10 мл), сушили, фільтрували та концентрували в умовах вакууму. Залишок очищали методом HPLC (спосіб 1) з одержанням сполуки прикладу 5 (89,10 мг, 52%). + LC-MS: tR = 0,971 хв, MS (ESI) m/z 437,2 [M+H] . 1 H ЯМР: (CD3OD): 7,60 (dd, J = 8,0, 1,6 Гц, 1H), 7,46 (d, J = 7,6 Гц, 1H), 7,28 (s, 1H), 4,08-4,01 (m, 1H), 3,63-3,90 (m, 4H), 3,33 (s, 3H), 3,09-3,20 (m, 2H), 2,74-2,79 (m, 1H), 1,80-2,06 (m, 5H), 1,65-1,78 (m, 1H), 1,55-1,64 (m, 2H), 1,29-1,35 (m, 3H), 1,07-1,29 (m, 1H), 0,89-0,96 (m, 1H), 0,85 (t, J = 7,6 Гц, 3H). Приклад 6 24 UA 113641 C2 Стадія 1: Синтез проміжного продукту 27 5 10 15 20 25 30 35 40 У висушену в печі колбу ємністю 3 л завантажували 6-бром-1-інданон (100 г, 473,8 ммоль), метилакрилат (86,4 г, 90 мл, 995 ммоль, 2,1 екв.) і безводний THF (800 мл), колбу занурювали в лазню з льодом у воді та перемішували. Спочатку обережно додавали tBuOK (0,5 г), а через 2 хв додавали другу порцію tBuOK (0,5 г). Охолодну лазню видаляли та додавали tBuOK (63 г), що залишився, рівними порціями протягом 20 хв (усього 64 г, 568,6 ммоль, 1,2 екв.). Суміш додатково перемішували протягом 2 год при к. т. До реакційної суміші додавали DMF (240 мл), а потім MeІ (134,6 г, 60 мл, 947,6 ммоль, 2,0 екв.), і додатково перемішували суміш протягом 2 год. Реакційну суміш гасили додаванням 10% розчину лимонної кислоти. Потім, для видалення більшої частини розчинника реакційну суміш концентрували в умовах зниженого тиску, після чого фільтрували. Осад на фільтрі промивали водою, а потім MeOH, з одержанням неочищеного проміжного продукту 26 (200 г), який використовували безпосередньо на наступній стадії. До розчину сполуки 26 (200 г, 547,6 ммоль, неочищ.) в THF/H2O (1,8 л/1,8 л) додавали LiOH·H2O (92 г, 2190 ммоль, 4,0 екв.). Суміш перемішували протягом 16 год при к. т., а потім 12 год при 70°C. Реакційну суміш концентрували в умовах зниженого тиску для видалення THF і фільтрували. Осад на фільтрі промивали H2O, а потім перемішували з MeOH (50 мл) протягом декількох хвилин, знову фільтрували та промивали додатковою кількістю MeOH (50 мл). Тверду речовину збирали з одержанням проміжного продукту 27 (75 г, 51,7%). Стадія 2: Синтез проміжного продукту 29 У трьохгорлу колбу в атмосфері азоту завантажували CeCl3·7H2O (1,2 г, 3,3 ммоль) і безводний MeOH (60 мл), і перемішували з одержанням прозорого розчину. В атмосфері азоту добавляли сполуку 27 (10,0 г, 32,6 ммоль) і безводний THF (240 мл), суміш охолоджували до 78°C. При -78°C в атмосфері азоту добавляли NaBH4 (0,4 г, 13,0 ммоль) при енергійному перемішуванні. Суміш перемішували при -78°C протягом 20 хв. Реакційну суміш гасили додаванням насиченого водного NH4Cl (100 мл) і H2O (200 мл) при перемішуванні при -78°C. Суміш залишали повільно нагріватися до температури навколишнього середовища. Суміш екстрагували EtOAc (3×150 мл). Об’єднані органічні шари промивали H 2O (2×200 мл), сольовим розчином (2×200 мл), сушили, фільтрували та концентрували в умовах вакууму, залишок очищали методом колоночної хроматографії на силікагелі, елюючи петролейним ефіром/EtOAc (20/1 3/1) з одержанням проміжного продукту 28 (7,5 г, 75%). + LC-MS: tR = 3,195 хв: MS (ESI) m/z 311,0 [M+H] . 1 H ЯМР: (CDCl3): 7,59 (s, 1H), 7,22-7,25 (d, J = 8,4 Гц, 1H), 7,08 (s, 1H), 6,88-6,91 (dd, J = 2,4, 8,4 Гц, 1H), 6,80-6,81 (d, J = 2,4 Гц, 1H), 5,84 (s, 1H), 4,87 (s, 2H), 4,31-4,36 (m, 2H), 3,50-3,55 (q, J = 6,8 Гц, 2H), 3,15-3,25 (m, 1H), 3,09-3,14 (d, J = 15,6 Гц, 1H), 3,00-3,06 (d, J = 15,2 Гц, 1H), 1,902,10 (m, 3H), 1,25-1,50 (m, 5H), 1,15-1,25 (t, J = 6,4 Гц, 3H). 25 UA 113641 C2 5 10 15 20 25 30 35 До суміші сполуки 28 (6,18 г, 20 ммоль) в DMF (20 мл) при 0°C додавали NaН (60% у мінеральному маслі, 0,96 г, 40 ммоль). Потім, суміш перемішували при 0°C протягом 2 год, потім до суміші додавали MeІ (3,5 мл), і перемішували протягом ночі. Суміш розбавляли EtOAc (40 мл) і H2O (40 мл), екстрагували EtOAc (2×60 мл). Об'єднані органічні фази сушили, і видаляли розчинник із одержанням проміжного продукту 29 (5,0 г). Стадія 2: Синтез проміжного продукту 30A і 30B До розчину проміжного продукту 29 (5,0 г, 15,3 ммоль) в THF (100 мл) додавали Ti(OEt) 4 (35,0 г, 153 ммоль). Після перемішування при к. т. протягом 1 год додавали (S)-N-третбутилсульфінамід (7,4 г, 61,2 ммоль). Реакційну суміш перемішували при нагріванні зі зворотним холодильником протягом ночі, і розподіляли суміш між H 2O (80 мл) і EtOAc (80 мл). Суміш фільтрували, і екстрагували фільтрат EtOAc (3×80 мл). Об'єднані органічні шари промивали сольовим розчином (50 мл), сушили та концентрували з одержанням залишку. Залишок очищали методом колоночної хроматографії на силікагелі (петролейний ефір/EtOAc = 20/1) з одержанням проміжного продукту 30A (1,6 г, 35%) і 30B (1,4 г, 33%), елюйованих у зазначеному порядку. Синтез сполуки прикладу 6 Проміжний продукт 30A додатково обробляли, як представлено на стадіях 4-10 прикладу 5. На стадії 9, замість R-(2-амінометил)тетрагідрофурану використали 2-амінометилпіримідин. + LC-MS: tR = 1,05 хв: MS (ESI) m/z 431,4 [M+H] . 1 H ЯМР: (CD3OD): 8,78 (d, J = 4,8 Гц, 2H), 7,76 (s, 1H), 7,75 (dd, J = 6,0, 1,6 Гц, 1H), 7,56 (d, J = 8,4 Гц, 1H) 7,44 (t, J = 5,2 Гц, 1H), 5,16 (m, 2H), 3,38 (s, 3H), 3,24 (m, 2H), 2,79 (m, 1H), 2,15 (m, 1H), 1,74 (m, 1H), 1,65 (d, J = 6,8 Гц, 1H), 1,39-1,57 (m, 4H), 0,99 (d, J = 6,4 Гц, 3H). Приклад 7 Зазначену сполуку синтезували відповідно до методики, описаної у прикладі 6. Проміжний продукт 30A додатково обробляли, як представлено на стадіях 4-10 прикладу 1. На стадії 9 використали (2-метоксі)етиламін, а потім проводили окислювання, як представлено на стадії 10, з одержанням сполуки прикладу 7. + LC-MS: tR = 1,08 хв, MS (ESI) m/z 397 [M+H] . 1 H ЯМР: (CD3OD) 7,74 (d, 1H, J = 8 Гц), 7,63 (d, 1H, J = 1 Гц), 7,57 (d, 1H, J = 8 Гц), 4,02-3,95 (m, 1H), 3,89-3,83 (m, 1H), 3,54 (m, 2H), 3,36 (s, 3H), 3,35 (s, 3H), 3,24 (ap q, 2H, J = 16 Гц)), 2,75 (m, 1H), 2,10 (m, 1H), 1,79 (dt, 1H, J = 13, 2 Гц), 1,56 (m, 1H), 1,41 (m, 3H), 1,14 (t, 1H, J = 13 Гц), 1,01 (d, 3H, J = 6 Гц). 26 UA 113641 C2 Приклад 8 5 10 15 20 25 30 Зазначену сполуку синтезували відповідно до методики, описаної у прикладі 6. (3Метилоксетан-3-іл)метанамін використали, як описано на стадії 9 прикладу 1, а потім проводили окислювання, як представлено на стадії 10, з одержанням сполуки прикладу 8. + LC-MS: tR = 0,930 хв, MS (ESI) m/z 423,0 [M+H] . 1 H ЯМР: (CD3OD): 7,66-7,64 (d, J = 7,2 Гц, 1H), 7,51-7,49 (d, J = 7,6 Гц, 1H), 7,34 (s, 1H), 4,72-4,67 (m, 2H), 4,29-4,25 (m, 2H), 3,74-3,59 (m, 2H), 3,37 (s, 3H), 3,25-3,14 (m, 2H), 2,74-2,67 (m, 1H), 2,08-2,03 (m, 1H), 1,80-1,53 (m, 3H), 1,30 (m, 5H), 1,08 (m, 1H), 0,90 (m, 3H). Приклад 9 Зазначену сполуку синтезували відповідно до методики, описаної у прикладі 6. 4(Амінометил)піримідин використали, як описано на стадії 9 прикладу 1, а потім проводили окислювання, як представлено на стадії 10 прикладу 6, з одержанням сполуки прикладу 9. + LC-MS: tR = 0,88 хв, MS (ESI) m/z 431,2 [M+H] . 1 H ЯМР: (CD3OD): 9,05 (s, 1H), 8,70-8,71 (d, J = 5,2 Гц, 1H), 7,60-7,62 (d, J = 7,6 Гц, 1H), 7,44-7,47 (m, 3H), 4,86 (s, 2H), 3,35 (s, 3H), 3,10-3,20 (q, 2H), 2,70-2,71 (m, 1H), 2,04-2,06 (m, 1H), 1,70 (m, 2H), 1,491 (m, 1H), 1,30-1,33 (m, 2H), 1,15-1,18 (m, 1H), 0,95-0,96 (d, J = 6,0 Гц, 3H). Приклад 10 Стадія 1: Синтез проміжного продукту 32 До суміші 6-броміндан-1-ону (100,00 г, 473,8 ммоль) у безводному THF (1 л) при 0°C додавали t-BuOK (58,5 г, 521,2 ммоль, 1,1 екв.), через 2 хв суміш нагрівали до к. т. і додатково перемішували протягом 10 хв, після чого однією порцією додавали метилметакрилат (49,8 г, 53,2 мл, 497,5 ммоль, 1,05 екв.). Через 2 год, до реакційної суміші додавали метилакрилат (49,0 г, 51,2 мл, 568,6 ммоль, 1,2 екв.). Через 3 год при к. т., до реакційної суміші додавали MeІ (101 г, 44,3 мл, 710,7 ммоль, 1,5 екв.), і перемішували її протягом 16 год. Додавали H2O (1 л), а потім LiOH·H2O (79,5 г, 1895,2 ммоль, 4,0 екв.), і перемішували суміш протягом 28 год при кімнатній температурі. В умовах зниженого тиску видаляли THF. Залишок розбавляли H 2O (1 л), 27 UA 113641 C2 фільтрували та промивали H2O до нейтральних значень фільтрату. Продукт промивали MeOH з одержанням 50 г проміжного продукту 32. Стадія 2: Синтез проміжного продукту 33 5 10 15 20 25 30 35 До суміші проміжного продукту 32 (60,0 г, 186,9 ммоль) і FeCl3 (33,0 г, 205,5 ммоль, 1,1 екв.) в THF (600 мл) при 0°C додавали NaBH3CN (29,4 г, 367,1 ммоль, 2,5 екв.). Суміш залишали нагріватися до кімнатної температури та перемішували протягом 1 год при к. т. Реакційну суміш гасили додаванням води, і видаляли THF в умовах вакууму. Суміш екстрагували DCM (3×200 мл). Об'єднані органічні фази промивали H2O і сольовим розчином, сушили та концентрували в умовах вакууму з одержанням неочищеного продукту, який очищали методом колоночної хроматографії на силікагелі з одержанням сполуки 33 (25,2 г, 42%) і 33A (12,0 г). + LC-MS: tR = 1,239 хв, MS (ESI) m/z 323,1 [M+H] . 1 H-ЯМР (CDCl3): : 7,889-7,894 (s, 1H), 7,671-7,696 (d, 1H), 7,311-7,332 (d, 1H), 3,605 (s, 1H), 2,981 (s, 2H), 1,769-1,797 (m, 4H), 1,072-1,082 (m, 2H), 1,019-1,056 (m, 6H). Стадія 2: Альтернативний синтез проміжного продукту 33 Суміш FeCl3 (6,0 г, 37,0 ммоль) з толуолом (60 мл) охолоджували до 0°C. Потім, до суміші додавали суміш сполуки 32 (11,9 г, 37,0 ммоль) в THF (48 мл). Суміш перемішували протягом 5 хв при 0°C, а потім охолоджували до -10°C. До реакційної суміші при -10°C по краплях додавали розчин t-BuNH2-BH3 (3,5 г, 40,7 ммоль) в THF (12 мл). Реакційну суміш перемішували приблизно при -10°C протягом 30 хв, гасили додаванням 6н водн. розчину HCl (10 мл), перемішували приблизно при 0°C протягом 30 хв, а потім залишали нагріватися до кімнатної температури. Суміш концентрували для видалення THF, і додавали толуол (60 мл). Водний шар видаляли, і промивали органічну фазу водою (3×60 мл). Органічну фазу концентрували до половини обсягу, нагрівали до 50°C з одержанням розчину, а потім охолоджували до 0°C протягом 1 год і витримували при 0°C протягом 1 год. Тверду речовину фільтрували та промивали холодним (0°C) толуолом (12 мл), і сушили в умовах вакууму з одержанням сполуки 33 (9,93 г, 83%). + LC-MS: tR = 2,36 хв, MS (ESI) m/z 323,0/325,0 [M+H] Стадія 3: Синтез проміжного продукту 34 До суміші сполуки 33 (20,0 г, 61,9 ммоль) з DMF (200 мл) при 0°C додавали NaН (5,0 г, 123,8 ммоль, 2,0 екв.). Потім, суміш перемішували протягом 15 хв при 0°C, і при 0°C додавали MeІ (17,6 г, 123,8 ммоль, 2,0 екв.). Потім, суміш нагрівали до к. т. і перемішували протягом 1,5 год при к. т. Суміш гасили додаванням H2O й екстрагували EtOAc. Об'єднані органічні фази промивали H2O і сольовим розчином, сушили, концентрували з одержанням неочищеного продукту, який очищали на колонці з силікагелем (елюент: петролейний ефір/EtOAc = 100/1 5/1) з одержанням проміжного продукту 34 (20 г, 96,2%). Стадія 4: Синтез проміжного продукту 35 28
ДивитисяДодаткова інформація
Назва патенту англійськоюInhibitors of beta-secretase
Автори англійськоюBukhtiyarov, Yuri, Cacatian, Salvacion, Dillard, Lawrence, Wayne, Dorner-Ciossek, Cornelia, Fuchs, Klaus, Jia, Lanqi, Lala, Deepak, S., Morales-Ramos, Angel, Rast, Georg, Reeves, Jonathan, Singh, Suresh, B., Venkatraman, Shankar, Xu, Zhenrong, Yuan, Jing, Zhao, Yi, Zheng, Yajun
Автори російськоюБухтияров Юрий, Какатиан Сальвасион, Диллард Лоуренс Уейн, Дорнер-Чосек Корнэлия, Фукс Клаус, Джиа Ланки, Лала Дипак С., Моралес-Рамос Энджел, Раст Георг, Ривз Джонатан, Сингх Сурэш Б., Венкатраман Шанкар, Сю Ченронг, Йуан Цзин, Чжао Йи, Чжен Яюн
МПК / Мітки
МПК: C07C 49/323, C07D 403/06, C07D 235/02, C07D 407/06, A61K 31/4184
Мітки: інгібітори, бета-секретази
Код посилання
<a href="https://ua.patents.su/47-113641-ingibitori-beta-sekretazi.html" target="_blank" rel="follow" title="База патентів України">Інгібітори бета-секретази</a>
Похідні 3,4-дигідропіроло[1,2-a]піразин-1-іламіну, придатні як інгібітори бета-секретази (bace)

Номер патенту: 109687
Опубліковано: 25.09.2015
Автори: Дельгадо-Хіменес Франциска, Трабанко-Суарес Андрес Авеліно
МПК: C07D 487/04, A61P 25/28, A61K 31/506, A61K 31/4985
Мітки: бета-секретази, інгібітори, bace, похідні, 3,4-дигідропіроло[1,2-a]піразин-1-іламіну, придатні
Формула / Реферат:
1. Сполука формули (І)або її таутомерна або стереоізомерна форма, де R1, R2, R3 незалежно вибрані з групи, що складається з водню, галогену, ціано, С1-3алкілу, моно- та полігалоген-С1-3алкілу та С3-6циклоалкілу; R4 вибраний із групи, що складається з водню, С1-3алкілу, метоксиметилу, С3-6циклоалкілу, моно- та...
Лактами як інгібітори бета-секретази

Номер патенту: 99787
Опубліковано: 25.09.2012
Автори: Хелал Крістофер Джон, Єфремов Іван Вікторовіч, Бродні Майкл Аарон, О'Нілл Брайан Томас
МПК: A61P 25/28, A61K 31/435, C07D 471/10
Мітки: лактами, інгібітори, бета-секретази
Формула / Реферат:
1. Сполука формули І:, Ів якій стереохімія формули І біля атома вуглецю, до якого приєднаний R2, і спіроциклічного атома вуглецю є абсолютною стереохімією;В означає алкіл, арил, гетероарил, циклоалкіл або гетероциклоалкіл, де В необов'язково заміщений 0-3 R3 групами;А незалежно означає арил, циклоалкіл, гетероциклоалкіл або...
Похідні 6,7-дигідропіразоло[1,5-a]піразин-4-іламіну, корисні як інгібітори бета-секретази (bace)

Номер патенту: 109800
Опубліковано: 12.10.2015
Автори: Гійсен Хенрікус Якобус Марія, Вега Раміро Хуан Антоніо, Дельгадо-Хіменес Франциска, Трабанко-Суарес Андрес Авеліно, ван Гоол Мішель Люк Марія
МПК: C07D 487/04, A61P 25/28, A61K 31/4985
Мітки: корисні, похідні, 6,7-дигідропіразоло[1,5-a]піразин-4-іламіну, інгібітори, bace, бета-секретази
Формула / Реферат:
1. Сполука формули (І)або її таутомерна або стереоізомерна форма, де R1 та R2 незалежно вибрані з групи, що складається з водню, галогену, ціано, С1-3алкілу, моно- та полігалоген-С1-3алкілу або С3-6циклоалкілу; R3 вибраний з групи, що складається з водню, С1-3алкілу, С3-6циклоалкілу, моно- та...
Дифторгексагідроциклопентаоксазиніли і дифторгексагідробензооксазиніли як інгібітори бета-секретази 1

Номер патенту: 113309
Опубліковано: 10.01.2017
Автор: Хільперт Ханс
МПК: C07D 265/12, C07D 413/12, A61K 31/536
Мітки: дифторгексагідроциклопентаоксазиніли, бета-секретази, інгібітори, дифторгексагідробензооксазиніли
Формула / Реферат:
1. Сполука формули І, IдеR1 вибраний з групи, що складається зі) арилу,іі) арилу, заміщеного 1-4 замісниками, незалежно вибраними з ціано, ціано-С1-6-алкілу, атома галогену, галоген-С1-6-алкокси, галоген-С1-6-алкілу, С1-6-алкокси, С1-6-алкоксі-С1-6-алкілу, С2-6-алкініл-С1-6-алкокси, С2-6-алкінілу і С1-6-алкілу,ііі) гетероарилу,...
Похідні 4,7-дигідропіразоло[1,5-a]піразин-6-іламіну, що використовуються як інгібітори бета-секретази (васе)

Номер патенту: 109663
Опубліковано: 25.09.2015
Автори: Трабанко-Суарез Андрес Авеліно, Тресадерн Ґері Джон, Делгадо-Джіменез Франціска
МПК: C07D 487/04, A61K 31/4985, A61P 25/28
Мітки: використовуються, інгібітори, похідні, бета-секретази, 4,7-дигідропіразоло[1,5-a]піразин-6-іламіну, васе
Формула / Реферат:
1. Сполука формули (І) (І)або її таутомерна або стереоізомерна форма, де R1 і R2 є воднем; R3 є метилом або циклопропілом; X1, X2, X3, X4 є CН та, необов’язково, Х1 та Х3 можуть бути СF; L є зв'язком або -N(R5)CO-, де R5 є воднем або С1-3алкілом; R6 є...
Попередній патент: Світна панель і стіна будівлі, що її містить
Наступний патент: Спосіб і система для відновлення сухим способом дрібних і понаддрібних частинок окисненої залізної руди, що містить магнітну роздільну установку
Випадковий патент: Двоярусний чотиримісний паркінг