Спосіб одержання 2′,3′-дидегідро-2′,3′-дидезоксипохідних уридину та тимінрибозиду при створенні анти-віл-активних препаратів
Номер патенту: 30564
Опубліковано: 15.11.2000
Автори: Усенко Любов Семенівна, Костина Валентина Григорівна, Шаламай Анатолій Севастьянович



Формула / Реферат
Спосіб одержання 2',3'-дидегідро-2',3'-дидез-оксипохідних уридину та тимінрибозиду при створенні анти-ВІЛ активних препаратів, полягає в деоксигенувані та вінілізації вуглеводного фрагменту піримідинових нуклеозидів, відбувається при його йодувані в умовах реакції Арбузова і відрізняється тим, що утворення проміжних квазифосфонієвих синтонів, O2, 2'-, 2'-йодпохідних та 2',3'-дийодиду 5'-0-захищених нуклеозидів проводять одностадійно без їх виділення і очистки, контролюючи хроматографічно тільки повноту послідовних перетворень.
Текст
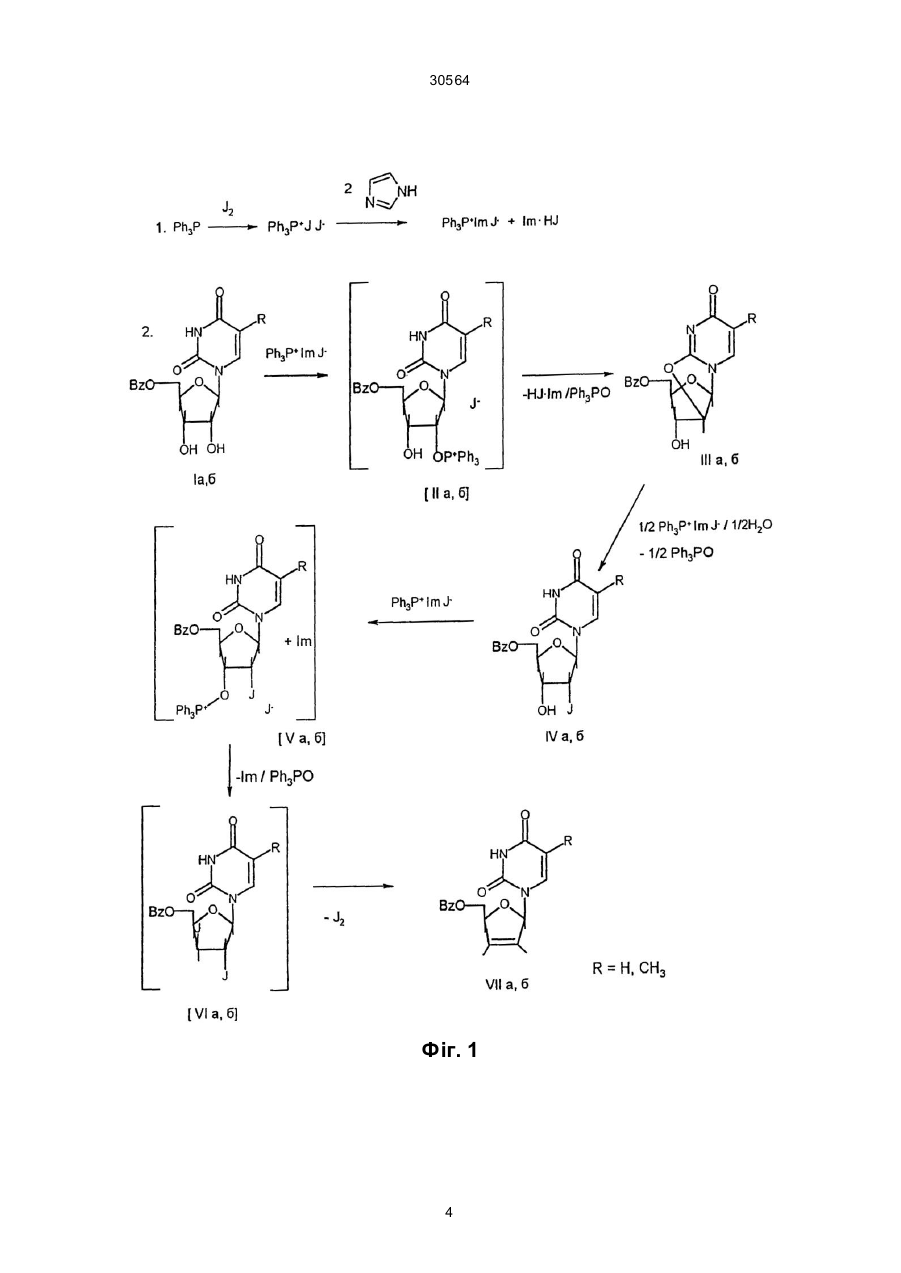
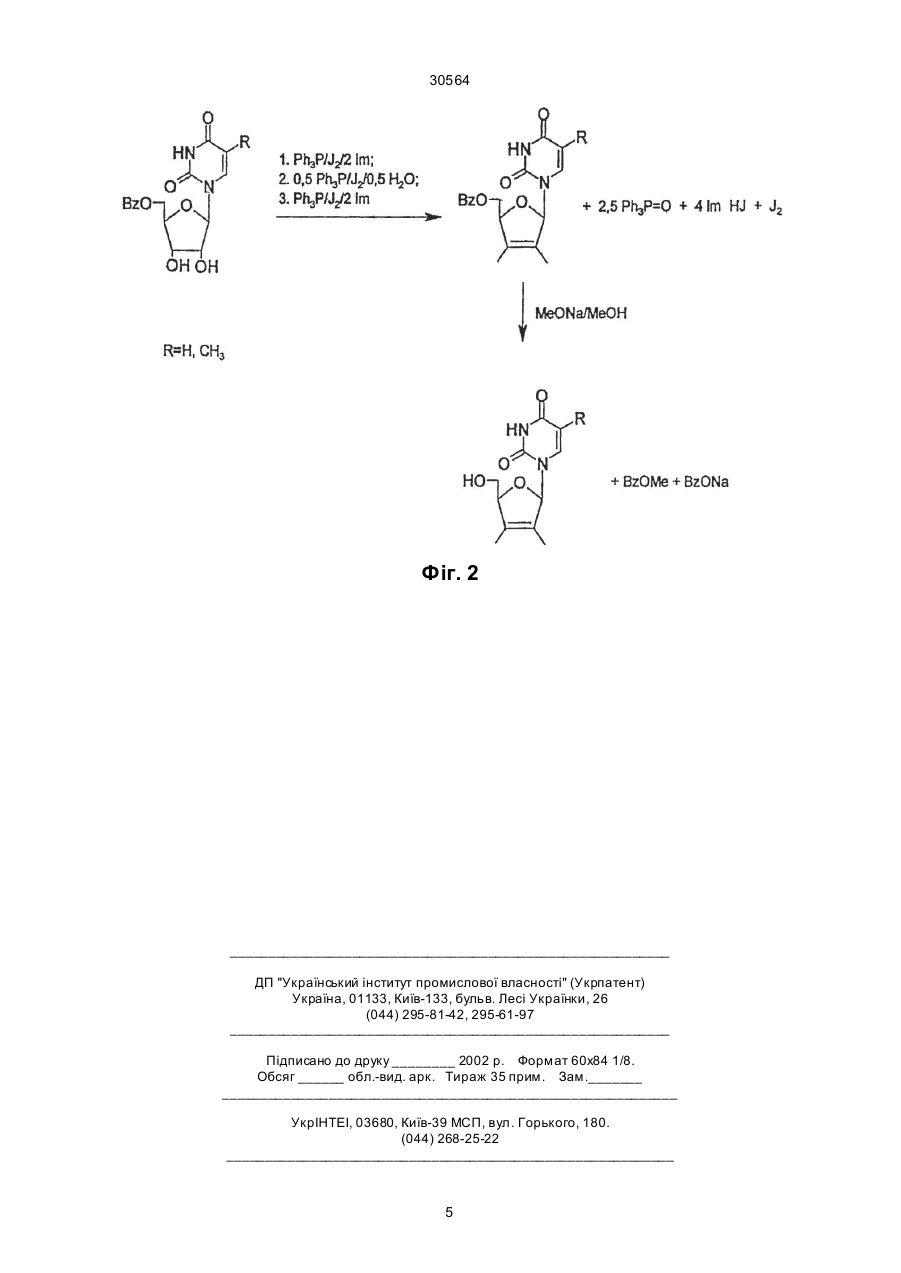
Спосіб одержання 2',3'-дидегідро-2',3'-дидезоксипохідних уридину та тимінрибозиду при ство 30564 Запропонований спосіб одержання 2',3'-дидегідронуклеозидів з постадійним введенням еквівалентних кількостей трифенілфосфіну, імідазолу та йоду і без виділення проміжних синтонів подано на загальній схемі (фіг. 2). Техніко-економічні показники запропонованого способу одержання 2',3'-дидегідронуклеозидів також значно кращі від наведених в прототипі. Насамперед, вихідні 5'-0-ацил-(бензоїл- чи ацетил-) похідні уридину та тимінрибозиду одержують в умовах стандартного методу селективного блокування гідроксилів нуклеозидів. Для проведення цих реакцій використовуються доступні - ацетон, бензоїлхлорид чи оцтовий ангідрид та мурашина кислота. Всі інші реактиви - уридин, трифенілфосфін, імідазол та йод також доступні та дешеві. Тимінрибозид одержують в умовах відомого методу одностадійної "силільної конденсації" гетероциклу з ацильованим цукром. Слід відмітити, що при масштабному виробництві можна регенерувати майже весь імідазол та 75% йоду. Важливим є те, що після 5'-0-деацилювання цільові 2',3'-дідегідронуклеозиди очищають від побічного трифенілфосфіноксиду шляхом екстрагування. Винахід пояснюється такими прикладами. Приклад 1. Синтез 2',3'-дидегідро-2',3'-дидезоксиуридину До попередньо висушених у вакуумі суміші 5,28 г (0,015 моль) 5'-0-бензоїлуридину та 6,91 г (0,0258 моль) трифенілфосфіну в 40 мл сухого діоксану додають 6,35 г (0,025 моль) йоду. Суміш перемішують протягом 15 хв і додають розчин 3,62 г (0,0532 моль) імідазолу в 70 мл сухого ацетонітрилу. Реакційну масу видержують при 5055°С протягом 7-10 год, контролюючи за допомогою тонкошарової хроматографії (ТШХ) повноту перетворення вихідного нуклеозиду в 5'-0-бензоїлО2,2'-циклоуридин. Потім розчинник випаровують у вакуумі досуха, залишок співупарюють з діоксаном і розчиняють в 100 мл сухого діоксану, додаючи 2,39 г (0,009 моль) трифенілфосфіну та 2,29 г (0,009 моль) йоду. До утвореної суспензії додають 5 мл діоксану, що містить 0,108 г (0,006 моль) води. Реакційну суміш перемішують при 20°С 2-3 год до повного перетворення О2,2'-циклонуклеозиду в 2'-йодпохідну (контроль ТШХ) і потім додають 4,43 г (0,0165 моль) трифенілфосфіну, 2,055 г (0,0806 моль) йоду та 2,245 г (0,033 моль) імідазолу. Масу перемішують при 20°С протягом 1012 год до повного перетворення 2'-йодпохідної в 2',3'-дидегідронуклеозид (контроль ТШХ). В кінці реакції відфільтровують 5,6 г йодгідрату імідазолу. До фільтрату додають 140 мл води, 0,5 г Na2SО3 і 100 мл хлороформу, перемішують, органічний шар відділяють і екстрагування хлороформом повторюють. Об'єднаний органічний екстракт сушать безводним Na2SO4, фільтрують через шар силікагелю (висота 3 см, діаметр фільтру 4 см), випарують досуха у вакуумі. Маслоподібну масу, що містить 2',3'-дидегідро-2',3'-дидезоксиуридин та трифенілфосфіноксид розчиняють в 160 мл 0,14 М розчину метилату натрія в сухому метанолі. Реакційну суміш при 20°С витримують 16 год, для нейтралізації додають катіоніт КУ-2х8, (Н+) і потім його відфільтровують. Фільтрат випарують у вакуумі. Залишок розчиняють в 100 мл хлороформу і екстрагують водою (50 мл ´ 3 рази), водний розчин ви парують досуха. Одержаний залишок кристаліують з суміші бензол-етанол (9:1). Одержують 2,07 г (62%) 2',3'-дидегідро-2',3'-дидезоксиуридину. Температура топлення 153-155°С з розкладом (літ. т.т. 155-156°С з розкладом). ТШХ R f 0,30 в системі розчинників бензол-етанол 9:1. УФ-спектр у воді: lмакс. 261 нм, e 10240. ПМР-спектр в ДМСО-d6: d (J, Гц); 11,20 с (1Н, Н-3), 7,72 д (1Н, J6-5=7,8, Н-6), 6,84 м (1Н, Н-1'), 6,38 дт (1Н, J3'-2' =6,1, Н-3'), 5,92 д (1Н, J2'-3' =6,1, Н2'), 5,52 д(1Н, J5-6=7,8, H-5), 4,88 м (1Н, 0Н), 4,73 м (1Н, Н-4'), 3,62 м (2Н, Н-5', Н-5''). Приклад 2. Синтез 2',3'-дидегідро-3'-дезокситимідину До попередньо висушених у вакуумі суміші 1,0 г (2,76 ммоль) 5'-0-бензоїлтимінрибозиду та 1,26 г (1,68 ммоль) трифенілфосфіну в 8 мл сухого диоксану додають 1,2 г (4,68 ммоль) йоду. Суміш перемішують протягом 15 хв і до утвореної жовтої суспензії додають розчин 0,65 г (9,62 ммоль) імідазолу в 10 мл сухого ацетонітрилу. Реакційну суміш при перемішуванні і температурі 50-55°С витримують протягом 7-10 годин до повного перетворення вихідного нуклеозиду в 5'-0-бензоїл-О2, 2'-циклотимінрибозид (контроль ТШХ). Потім розчин випаровують в вакуумі досуха, співупарують з діоксаном, залишок розчиняють в 20 мл диоксану і додають 0,55 г (2,10 ммоль) трифенілфосфіну та 0,55 г (2,11 ммоль) йоду. До утвореної суспензії додають 1 мл діоксану, що містить 0,024 г (1,38 ммоль) води. Реакційну суміш перемішують при 20°С протягом 2-3 год до повного перетворення циклонуклеозиду в 2'-йодтимідин (контроль ТШХ). Потім додають 0,57 г (3,31 ммоль) трифенілфосфіну, 0,21 г (0 ,83 ммоль) йоду і 0,38 г (5,5 ммоль) імідазолу. Масу перемішують при 20°С протягом 10-12 год до повного перетворення 2'йодпохідної в 2',3'-дидегідронуклеозид (контроль ТШХ). Після закінчення реакції осад йодгідрату імідазолу (біля 1,2 г) відфільтровують, до фільтрату додають 20 мл води, 0,1 г Na2SO3, 20 мл хлороформу, перемішують органічний шар відділяють і повторюють екстракцію водного шару. Об'єднані органічні екстракти сушать над безводним Na2SO4, фільтрують через шар силікагелю висотою 2 см і упарюють в вакуумі досуха. Залишок розчиняють в 30 мл 0,14 М розчину метилату натрія в сухому метанолі, розчин витримують 16 год при 20°С і в кінці нейтралізують смолою КУ 2x8 (Н'), яку потім відфільтровують. Фільтрат випарують в вакуумі, залишок розчиняють в 25 мл хлороформу. Розчин екстрагують водою (20 мл ´ 3 рази), водний екстракт випарюють в вакуумі досуха. Одержаний залишок кристалізують. Вихід 2',3'-дидегідро-3'-дезокситимідину 0,44 г (68%). Температура топлення 163-165°С (літ. [3] т.т. 163-166°С). ТШХ Rf 0,32 в системі розчинників бензол-етанол 9:1. УФ-спектр у воді: lмакс.=265 нм, e 9 680. ПМР-спектр в ДМСО-d6: d (J, Гц); 11,29 с (1Н, Н-3), 7,68 с (1Н, Н-6), 6,80 д (1Н, J1'-2' =1,2, Н-1'), 6,36 д (1Н, J3'-2' =5,9, Н-3'), 5,88 дд (1Н, J2'-1' =1,2, J2'-3' =4,7, H-2'), 5,00 м (1Н, 0Н), 4,74 с (1Н, Н-4'), 3,60 м (2Н, Н-5', Н-5''), 1,72 д (3Н, JCH-H=1,2, СН3). В підсумок слід відмітити, що наведений спосіб одержання 2',3'-дидегідронуклеозидів відрізня 2 30564 ється від відомих і наведеного в прототипі меншою кількістю стадій синтезу, простотою технологічних операцій його проведення і виділення кінцевого продукту, утилізації і регенерації побічних сполук і найбільш важливим показником є високі виходи D4U та D4T. Джерела інформації 1. Stavudine (Zerit). Original monograph // Drugs Fut. - 1996. - V. 21, № 10. - P. 1084-1086. 2. McDonald F.E., Gleason M.M. Assymmetric syntheses of stavudine (d4T) and cordycepin by cycloisamerization of alkynyl alcohols to endocyclic and ethers // Angew. Chem. Int. Ed Engl. - 1995. - V. 34. № 3. - P. 350-352. 3. Lipshutz B.H., Ste vens K.L., Lowe R.F. A novel route to the anti-HlV-nucleosides d4T. //Tetrahedron Lett. - 1996. - V. 36. № 16. - P. 2711-2713. 4. Horwitz J.P., Chua J., Klundt I.L., Da Rooge M.A., Noel M. The introduction of unsaturation in to the carbohydrate of a pyrimidine nucleoside via a 2',3' - anhydro bond. // J. Amer. Chem. Soc. - 1964. V. 86, №5. - P. 1896-1897. 5. Mansuri M.M., Starrett J.E., Ghazzouli I. et al. l-(2,3-Dideoxy-b-D-glycero-pent-2-enofuranosyl) thymine. A h yghly potent selective anti-HIV agent. // J. Med. Chem. - 1989. - V. 32, № 2. - P. 461 - 466. 3 30564 Фіг. 1 4 30564 Фіг. 2 __________________________________________________________ ДП "Український інститут промислової власності" (Укрпатент) Україна, 01133, Київ-133, бульв. Лесі Українки, 26 (044) 295-81-42, 295-61-97 __________________________________________________________ Підписано до друку ________ 2002 р. Формат 60х84 1/8. Обсяг ______ обл.-вид. арк. Тираж 35 прим. Зам._______ ____________________________________________________________ УкрІНТЕІ, 03680, Київ-39 МСП, вул. Горького, 180. (044) 268-25-22 ___________________________________________________________ 5
ДивитисяДодаткова інформація
Назва патенту англійськоюA process for preparation of 2',3'-didehydro-2',3'-didesoxyderivatives of uridine and thymine riboside in creating anti-hiv-active preparations
Автори англійськоюShalamai Anatolii Sevastianovych, Kostina Valrntyna Hryhorivna, Usenko Liubov Semenivna
Назва патенту російськоюСпособ получения 2',3'-дидегидро-2',3'-дидезоксипроизводных уридина и тиминрибозида при образовании анти-вич-активных препаратов
Автори російськоюШаламай Анатолий Севастьянович, Костина Валентина Григорьевна, Усенко Любовь Семеновна
МПК / Мітки
МПК: C07H 19/067
Мітки: препаратів, 2',3'-дидегідро-2',3'-дидезоксипохідних, створенні, одержання, спосіб, уридину, тимінрибозиду, анти-віл-активних
Код посилання
<a href="https://ua.patents.su/5-30564-sposib-oderzhannya-23-didegidro-23-didezoksipokhidnikh-uridinu-ta-timinribozidu-pri-stvorenni-anti-vil-aktivnikh-preparativ.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання 2′,3′-дидегідро-2′,3′-дидезоксипохідних уридину та тимінрибозиду при створенні анти-віл-активних препаратів</a>
Спосіб одержання поверхнево-активних речовин на основі природних тригліцеридів

Номер патенту: 29956
Опубліковано: 15.11.2000
Автори: Щербина Фелікс Федорович, Прокоф'єв Вадим Павлович, Кухар Валерій Павлович, Танчук Юлій Володимирович, Сакун Валентин Андрійович
МПК: C07C 241/00, C07C 69/00, C07C 243/00
Мітки: основі, поверхнево-активних, тригліцеридів, спосіб, одержання, речовин, природних
Формула / Реферат:
1. Спосіб одержання поверхневоактивних речовин на основі природних тригліцеридів загальної формули,де R=H, СН3, СН2ОН; х=110; у=0,1; z=0, 1, 2, 3; q=7, 11 - кількість структурних фрагментів ненасичених та насичених жирних кислот, що входять до складу рослинних олій, що передбачає використання тригліцеридів в реакції ацилювання органічних азотистих основ, який...
Спосіб одержання рацематів складних ефірів цисі/або трансаповінкамінової кислоти або їх оптично активних ізомерів
Номер патенту: 3472
Опубліковано: 27.12.1994
Автори: Янош Крайдль, Андраш Немеш, Чаба Сантаі, Лайош Сабо, Ласло Цібула, Дьєрдь Вішкі, Дьєрдь Калауш, Маріа Фаркаш
МПК: C07D 471/14, C07B 57/00, C07D 461/00
Мітки: оптично, складних, кислоти, одержання, ізомерів, рацематів, активних, спосіб, трансаповінкамінової, ефірів
Формула / Реферат:
Способ получения рацематов сложных эфиров цис- и/или трансаповинкаминовой кислоты общей формулы I или их оптически активных изомеров общих формул Iа или Iб, или Iв, или Iг где R' - низший алкил, отличающийся тем, что, с целью повышения селективности процесса и расширения ассортимента целевых продуктов, рацематы производных транс-гидроксииминооктагидро (2, 3-а) хинолизина общей формулы...
Спосіб одержання препаратів з торфу

Номер патенту: 9564
Опубліковано: 30.09.1996
Автори: Шеріна Наталія Миколаївна, Запорожченко Ольга Михайлівна, Лотош Тамара Дмитрівна, Іванов Валерій Іванович, Сотнікова Олена Петрівна, Тимошенко Ярослава Григорівна, Соловйова Віра Петрівна
МПК: A61K 35/10
Мітки: спосіб, одержання, препаратів, торфу
Формула / Реферат:
Способ получения препаратов из торфа, включающий отгон летучих соединений из торфа, отличающийся тем, что до отгона торфа его подвергают охлаждению и облучению ультрафиолетом, после чего к отгону добавляют пиридоксин, а из оставшейся массы получают гуминат, с последующим биотестированием готовой продукции.
Спосіб одержання препаратів з плаценти людини

Номер патенту: 20274
Опубліковано: 15.07.1997
Автори: Соловйова Віра Петрівна, Лотош Тамара Дмитрівна, Тимошенко Ярослава Григорівна, Абрамова Анна Борисівна
МПК: A61K 35/50
Мітки: плаценти, спосіб, одержання, препаратів, людини
Формула / Реферат:
Способ получения препаратов из плаценты человека, включающий предварительную консер- . вацию плацентарной ткани при пониженной температуре, измельчение ее и экстрагирование дистиллированной водой, отличающийся тем, что производится экстрагирование апирогенной водой гомогенизированной ткани плаценты при соотношении сырья и воды 1:4, получая взвесь, половина объема которой разливают в ампулы и автоклави-руют, вторую половину фильтруют,...
Спосіб одержання основи препаратів інтерферонів

Номер патенту: 10462
Опубліковано: 25.12.1996
Автори: Лазарєв Олексій Павлович, Співак Микола Якович, Собко Анатолій Іванович, Купчинський Леонід Георгійович, Карась Олег Якович
МПК: A61K 38/21
Мітки: основі, інтерферонів, одержання, спосіб, препаратів
Формула / Реферат:
1. Способ получения основы препаратов интерферонов, включающий культивирование клеток в питательной среде, п райминг, индукцию, выделение целевого продукта, отличающийся тем, что в качестве индуктора используют ротавирус или продукт его обработки, а выделение осуществляют, концентрируя культуральную среду индуцированных клеток на полых волокнах.2. Способ по п.1, отличающийся тем, что ротавирус дополнительно обрабатывают фенолом и...
Попередній патент: Споруда для біологічної очистки води
Наступний патент: Спосіб визначення об’єму загиблих тканин при опіках iv ступеню
Випадковий патент: Спосіб вимірювання внутрішньочеревного тиску