Спосіб одержання вінільних функціональних похідних гетероциклів
Номер патенту: 37721
Опубліковано: 10.12.2008
Автори: Сливка Марина Василівна, Сливка Михайло Васильович, Усенко Руслан Миколайович, Лендєл Василь Георгійович




Формула / Реферат
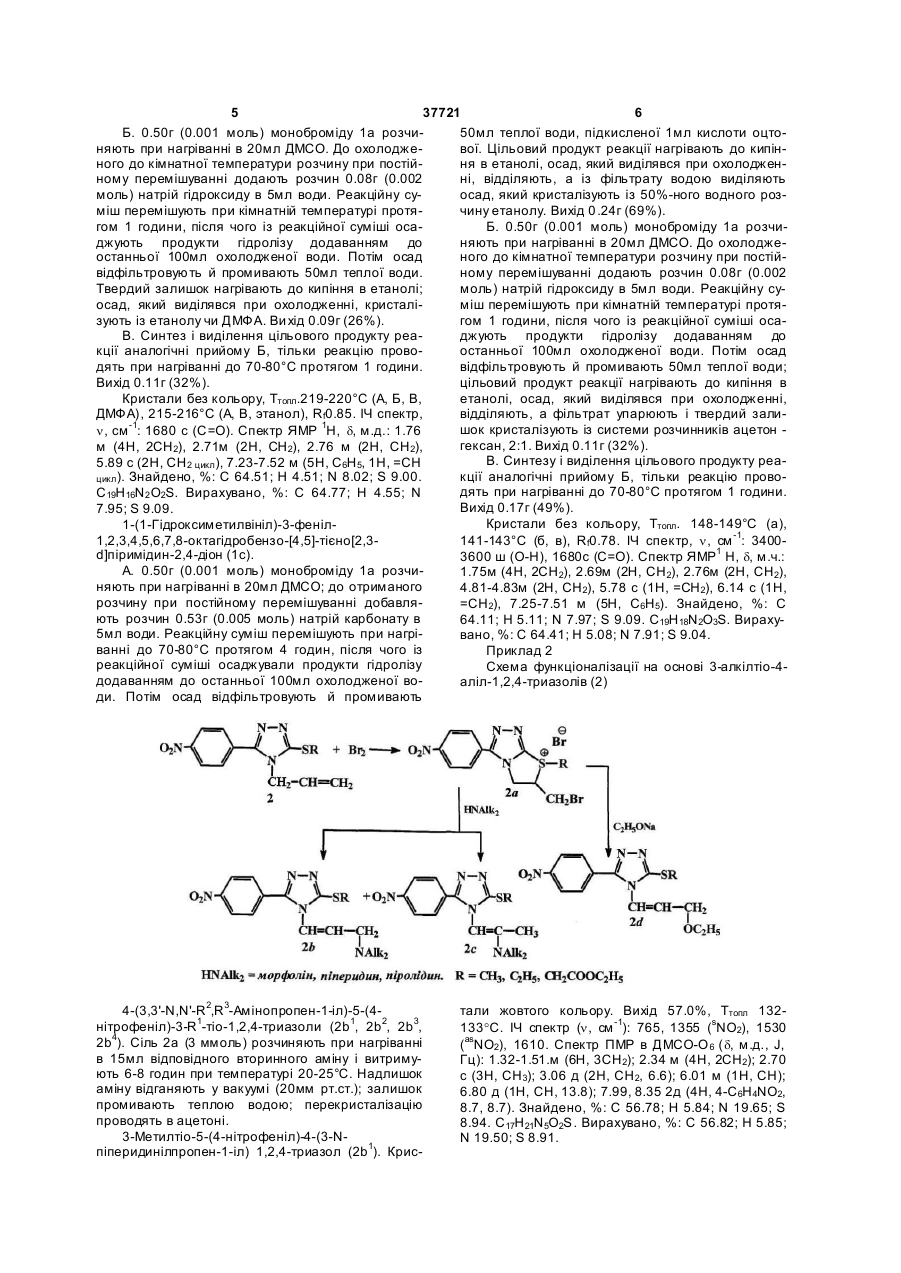
Спосіб одержання вінільних функціональних похідних гетероциклів, який включає взаємодію нуклеофільних реагентів із гетероциклічними системами, який відрізняється тим, що використовують штучно створений методом електрофільної галогеноциклізації реакційний центр в гетероциклічних системах, що супроводжується утворенням поліконденсованих гетероциклів з позитивно зарядженим гетероатомом, які містять атом гідрогену в ![]() -положенні до позитивно зарядженого гетероатома та до галогенометильної групи, одержують як приклад O,N-функціональні вінільні похідні тієно[2,3-d]піримідин-2,4-діону, 3-меркапто-1,2,4-триазолу та 1,2,4-триазол-3-ону.
-положенні до позитивно зарядженого гетероатома та до галогенометильної групи, одержують як приклад O,N-функціональні вінільні похідні тієно[2,3-d]піримідин-2,4-діону, 3-меркапто-1,2,4-триазолу та 1,2,4-триазол-3-ону.
Текст
Спосіб одержання вінільних функціональних похідних гетероциклів, який включає взаємодію нуклеофільних реагентів із гетероциклічними сис 3 37721 4 виходами, так як відомі методи функціоналізації кли з позитивно зарядженим гетероатомом, що гетероциклів є неприйнятними для їх синтезу. містять атом Гідрогену в a-положенні до позитивПоставлене завдання досягається таким чино зарядженого гетероатому та до галогенометином, що в способі одержання вінільних функціональної групи. Суть розробленого способу функціольних похідних гетероциклів, який включає взаєналізації гетероциклічних систем полягає в тому, модію нуклеофільних реагентів із що дія нуклеофільних реагентів різної природи гетероциклічними системами, згідно корисної мопризводить з одного боку до нуклеофільного заделі використовують штучно створений методом міщення атому галогену на фрагмент діючого нукелектрофільної галогеноциклізації реакційний леофілу (чи конкурентного нуклеофілу середовицентр в гетероциклічних системах, що супровоща, в якому проводять синтез); а з іншого боку джується утворенням поліконденсованих гетерозавдяки основності нуклеофілу відбувається еліміциклів з позитивно зарядженим гетероатомом, які нування атому Гідрогену в a-положенні до позитимістять атом Гідрогену в a-положенні до позитиввно зарядженого гетероатому, яке супроводжуєтьно зарядженого гетероатому та до галогенометися руйнуванням поліциклічної системи з льної групи, -одержують як приклад O,Nформуванням вихідного гетероциклу (1-3) та утвофункціональні вінільні похідні тієно[2,3-d]ренням подвійного зв'язку в a-положенні до фрагпіримідин-2,4-діону, 3-меркапто-1,2,4-триазолу та менту нуклеофілу та гетероциклу - тобто, відбува1,2,4-триазол-3-ону. ється утворення вінільних функціональних Модельним об'єктами для досліджень було похідних тієно[2,3-d]піримідин-2,4-діону та 1,2,4використано алільний етер тієно[2,3d]піримідинтриазолу. Причому, відмічається утворення вініль2,4-діону (1) (представник p-електронодефіцитних них функціональних похідних різного характеру, гетероциклічних систем) та похідні 3-алкілтіо-4залежно від природи нуклеофільного реагенту, аліл-1,2,4-триазолу (2), 3-алілтіо-4,5-дифенілумов синтезу та природи штучно створеного акти1,2,4-триазол (3) (представники pвного реакційного центру. електронодонорних гетероциклічних систем), які Склад синтезованих вінільних похідних гетеможна отримати майже з кількісними виходами роциклічних систем (1b,с; 2b,c,d; 3d) підтверджено методами, описаними в роботах [1-3]. Для штучноданими елементного аналізу; індивідуальність го створення активного реакційного центру (по методом тонкошарової хроматографії (ТШХ); буякому відбувається функціоналізація) - позитивно дову доведено методом інфрачервоної спектрозаряджених атомів Нітрогену та Суль фуру - було скопії (14) в таблетках калій броміду та методом обрано прийом електрофільної галогеногетероциспектроскопії протонного магнітного резонансу клізації алільних похідних (1-3), який характеризу(ПМР) високого розрішення (300МГц). ється простотою виконання, доступністю реактивів Приклад 1 та високими (іноді кількісними) виходами [4]. В Схема функціоналізації на основі алільного результаті отримують поліконденсовані гетероциетеру тієно[2,3-d]-піримідин-2,4-діону (1). 1-(2H-3-Оксетеніл)-3-феніл-1,2,3,4,5,6,7,8октагідробензо[4,5]тієно-[2,3-d]піримідин-2,4-діон (1b). А. 0.50г (0.001 моль) моноброміду 1а розчиняють при нагріванні в 20мл диметилсульфоксиду (ДМСО); до отриманого розчину при постійному перемішуванні добавляють розчин 0.53г (0.005 моль) натрій карбонату в 5мл води. Реакційну суміш перемішують при нагріванні до 70-80°С протя гом 4 годин, після чого із реакційної суміші осаджують продукти гідролізу додаванням до останньої 100мл охолодженої води. Потім осад відфільтровують й промивають 50мл теплої води, подкисленої 1мл кислоти оцтової. Цільовий продукт реакції нагрівають до кипіння в етанолі; осад, який виділявся при охолодженні, кристалізують із етанолу чи диметилформаміду (ДМФА). Ви хід 0.10г (29%). 5 37721 6 Б. 0.50г (0.001 моль) моноброміду 1а розчи50мл теплої води, підкисленої 1мл кислоти оцтоняють при нагріванні в 20мл ДМСО. До охолоджевої. Цільовий продукт реакції нагрівають до кипінного до кімнатної температури розчину при постійня в етанолі, осад, який виділявся при охолодженному перемішуванні додають розчин 0.08г (0.002 ні, відділяють, а із фільтрату водою виділяють моль) натрій гідроксиду в 5мл води. Реакційну суосад, який кристалізують із 50%-ного водного розміш перемішують при кімнатній температурі протячину етанолу. Вихід 0 .24г (69%). гом 1 години, після чого із реакційної суміші осаБ. 0.50г (0.001 моль) моноброміду 1a розчиджують продукти гідролізу додаванням до няють при нагріванні в 20мл ДМСО. До охолоджеостанньої 100мл охолодженої води. Потім осад ного до кімнатної температури розчину при постійвідфільтровують й промивають 50мл теплої води. ному перемішуванні додають розчин 0.08г (0.002 Твердий залишок нагрівають до кипіння в етанолі; моль) натрій гідроксиду в 5мл води. Реакційну суосад, який виділявся при охолодженні, кристаліміш перемішують при кімнатній температурі протязують із етанолу чи ДМФА. Ви хід 0.09г (26%). гом 1 години, після чого із реакційної суміші осаВ. Синтез і виділення цільового продукту реаджують продукти гідролізу додаванням до кції аналогічні прийому Б, тільки реакцію провоостанньої 100мл охолодженої води. Потім осад дять при нагріванні до 70-80°С протягом 1 години. відфільтровують й промивають 50мл теплої води; Вихід 0.11г (32%). цільовий продукт реакції нагрівають до кипіння в Кристали без кольору, Ттопл.219-220°С (А, Б, В, етанолі, осад, який виділявся при охолодженні, ДМФА), 215-216°С (А, В, этанол), Rf0.85. ІЧ спектр, відділяють, а фільтрат упарюють і твердий залишок кристалізують із системи розчинників ацетон n, см -1: 1680 с (С=О). Спектр ЯМР 1Н, d, м.д.: 1.76 гексан, 2:1. Вихід 0.11г (32%). м (4Н, 2СН2), 2.71м (2Н, СН2), 2.76 м (2Н, СН2), В. Синтезу і виділення цільового продукту реа5.89 с (2Н, СН2 цикл), 7.23-7.52 м (5Н, С6Н5, 1Н, =СН кції аналогічні прийому Б, тільки реакцію провоцикл ). Знайдено, %: С 64.51; Η 4.51; N 8.02; S 9.00. дять при нагріванні до 70-80°С протягом 1 години. C19H16N2 O2S. Вирахувано, %: С 64.77; Η 4.55; Ν Вихід 0.17г (49%). 7.95; S 9.09. Кристали без кольору, Ттопл. 148-149°С (а), 1-(1-Гідроксиметилвініл)-3-феніл1,2,3,4,5,6,7,8-октагідробензо-[4,5]-тієно[2,3141-143°С (б, в), Rf0.78. ІЧ спектр, n, см -1: 3400d]піримідин-2,4-діон (1с). 3600 ш (О-Н), 1680с (С=О). Спектр ЯМР1 Н, d, м.ч.: А. 0.50г (0.001 моль) моноброміду 1а розчи1.75м (4Н, 2СН2), 2.69м (2Н, СН2), 2.76м (2Н, СН2), няють при нагріванні в 20мл ДМСО; до отриманого 4.81-4.83м (2Н, СН2), 5.78 с (1Н, =СН2), 6.14 с (1Н, розчину при постійному перемішуванні добавля=СН2), 7.25-7.51 м (5Н, С6Н5). Знайдено, %: С ють розчин 0.53г (0.005 моль) натрій карбонату в 64.11; Η 5.11; Ν 7.97; S 9.09. C19H18N2O3S. Вираху5мл води. Реакційну суміш перемішують при нагрівано, %: С 64.41; Η 5.08; Ν 7.91; S 9.04. ванні до 70-80°С протягом 4 годин, після чого із Приклад 2 реакційної суміші осаджували продукти гідролізу Схема функціоналізації на основі 3-алкілтіо-4додаванням до останньої 100мл охолодженої воаліл-1,2,4-триазолів (2) ди. Потім осад відфільтровують й промивають 4-(3,3'-N,N'-R 2,R3-Амінопропен-1-іл)-5-(4нітрофеніл)-3-R1-тіо-1,2,4-триазоли (2b1, 2b2, 2b3, 2b4). Сіль 2a (3 ммоль) розчиняють при нагріванні в 15мл відповідного вторинного аміну і витримують 6-8 годин при температурі 20-25°С. Надлишок аміну відганяють у вакуумі (20мм рт.ст.); залишок промивають теплою водою; перекристалізацію проводять в ацетоні. 3-Метилтіо-5-(4-нітрофеніл)-4-(3-Nпіперидинілпропен-1-іл) 1,2,4-триазол (2b1). Крис тали жовтого кольору. Вихід 57.0%, Ттопл 132133°С. ІЧ спектр (n, см -1): 765, 1355 (sΝΟ2), 1530 (asNO2), 1610. Спектр ПМР в ДМСО-О 6 (d, м.д., J, Гц): 1.32-1.51.м (6Н, 3СН2); 2.34 м (4Н, 2СН2); 2.70 с (3Н, СН3); 3.06 д (2Н, СН2, 6.6); 6.01 м (1Н, СН); 6.80 д (1Н, СН, 13.8); 7.99, 8.35 2д (4Н, 4-C6H4NO2, 8.7, 8.7). Знайдено, %: С 56.78; Η 5.84; Ν 19.65; S 8.94. C17H21N5O2S. Вирахувано, %: С 56.82; Η 5.85; Ν 19.50; S 8.91. 7 37721 8 3-Метилтіо-4-(3-N-морфолінілпропен-1-іл)-5при температурі 20-25°С. Надлишок аміну відганя(4-нітрофеніл)-1,2,4-триазол (2b2). Кристали жовли у вакуумі (20мм рт.ст.); залишок промивали теплою водою і кип'ятили в гексані протягом 2 готого кольору. Вихід 61.0%, Ттопл 163-164°С 14 дин - фільтрат відділяли декантацією; із залишка спектр (n, см -1): 765, 1355 (sNO2), 1530 (asNO2), виділяли цільовий продукт 2с; перекристалізацію 1610. Спектр ПМР в ДМСО-D6 (d, м.д., J, Гц): 2.39 т проводили в ацетоні. (4Н, 2СН2, 4.5); 2.70 с (3Н, СН3); 3.12 д (2Н, СН2, Кристали жовтого кольору. Вихід 63.0 %, Ттопл 6.9); 3.56 т (4Н, 2СН2, 4.8); 6.03 м (1Н, СН); 6.84 д 144-145°С. 14 спектр (n, см -1): 760, 1350 (sNO2), (1Н, СН, 14.1); 7.99, 8.35 2д (4Н, 4-C6H4NO2, 8.7, 1530 (asNO2), 1610. Спектр ПМР в ДМСО-D6 (d, 8.7). Знайдено, %: С 53.22; Η 5.24; Ν 19.47; S 8.83. м.д., J, Гц): 1.45 с (3Н, СН3); 1.88 м (4Н, 2СН2); 2.64 C16H19N5 O3S. Вирахувано, %: С 53.19; Η 5.26; Ν с (3Н, СН3); 3.12-3.23 м (4Н, 2NCH2); 5.19 с (1H, 19.39; S 8.86. 5-(4-Нітрофеніл)-3-(N,N’СН); 8.17, 8.32 2д (4Н, 4-C6H4NO2, 8.7, 8.7). Знайдено, %: С 55.61; Η 5.50; Ν 20.37; S 9.33. піперидинілкарбамоїлометилтіо)-4-('3-NC16H19N5 O2S. Вирахувано, %: С 55.65; Η 5.51; Ν піперидинілпропен-1-іл)-1,2,4-триазол (2b3). Крис20.29; S 9.28. тали жовтого кольору. Вихід 61.0%, Ттопл 1584-(3-Етоксипропен-1-іл)-5-(4-нітрофеніл)-3-R159°С. ІЧ спектр (n, см -1): 760, 1350 (sNO2), 1530 тіо-1,2,4-триазоли (2d1, 2d2). До розчину відповід(asNO2), 1605, 1645. Спектр ПМР в ДМСО-D6 (d, ної солі 2а (1.5 ммоль) в абсолютному етанолі м.д., J, Гц): 1.32-1.61 м (12Н, 6СН2); 2.34 м (4Н, (18мл) добавляють 15% розчин етилату натрію 2СН2); 3.08 д (2Н, СН2, 6.6); 3.47 м (4Н, 2СН2); 4.38 (6мл); отриманий розчин через 2 години розбавс (2Н, SCH2); 6.02 м (1Н, СН); 6.85 д (1H, СН, 13.5); ляють трьохкратною кількістю води; потім прово8.00, 8.36 2д (4Н, 4-C6H4NO2, 8.7, 8.7). Знайдено, дять нейтралізацію оцтовою кислотою до рН=8. %: С 58.54; Η 6.33; Ν 18.02; S 6.67. C23H30N6O3S. Цільові продукти екстрагують бензеном, бензеноВирахувано, %: С 58.72; Η 6.38; Ν 17.87; S 6.81. вий екстракт сушать сульфа том натрію, бензен 3-(N,N'-Морфолінілкарбамоїлометилтіо)-4-(3відганяють у вакуумі (20мм рт.ст.), залишок переN-морфолінілпропен-1-іл)-5-(4-нітрофеніл)-1,2,4кристалізовують із метанолу і сушать в ексикаторі триазол (2b4). Кристали жовтого кольору. Вихід над хлоридом кальцію. 59.0 %, Ттопл 141-142°С. ІЧ спектр (n, см -1): 760, 4-(3-Етоксипропен-1-іл)-3-метилтіо-5-(41350 (sNO2), 1530 (asNO2), 1610, 1645. Спектр ПМР нітрофеніл)-1,2,4-триазол (2d1). Кристали жовтого в ДМСО-D6 (d, м.д., J, Гц): 2.40 т(4Η, 2СН2, 4.5); кольору. Вихід 52.1%, Ттопл 74-76°С. 14 спектр (n, 3.13 д (2Н, СН2, 6.0); 3.45-3.68 м (12Н, 6СН2); 4.38 см -1): 765, 1350 (sNO2), 1530 (asNO2), 1610, 1625, с (2Н, СН2); 6.05 м (1Н, СН); 6.89 д (1Н, СН, 13.5); 2820. Спектр ПМР в метанолі-D4 (d, м.д., J, Гц): 8.00, 8.36 2д (4Н, 4-C6H4NO2, 8.7, 8.7). Знайдено, 1.20 т (3Н, СН3, 7.4); 2.74 с (3Н, SCH3); 3.52 к (2Н, %: С 53.01; Η 5.44; Ν 17.91; S 6.68. C21H26N6O5S. ОСН2, 7.4); 4.15 д (2Н, СН2, 6.2); 6.16 м (1Н, СН); Вирахувано, %: С 53.16; Η 5.49; Ν 17.72; S 6.75. 6.78 д (1Н, СН, 16.0); 8.00, 8.38 2д (4Н, 4-C6H4NO2, 3-Метилтіо-5-(4-нітрофеніл)-4-(3-N9.8, 9.8). Знайдено, %: С 52.74; Η 4.91; Ν 17.77; S піролідинілпропен-1-іл)-1,2,4-триазол (2b5). Сіль 2а 10.23. C14H16N4O3S. Вирахувано, %: С 52.50; Η (3 ммоль) розчиняли при нагріванні в 15мл піролі5.00; Ν 17.50; S 10.00. дину і витримували 6 годин при температурі 203-Етилтіо-4-(3-етоксипропен-1-іл)-5-Г425°С. Надлишок аміну відганяли у вакуумі (20мм нітрофеніл)-1,2,4-триазол (2d2). Кристали жовтого рт.ст.); залишок промивали теплою водою і кип'я кольору. Вихід 47.9 %, Ттопл 58-59°С. ІЧ спектр (n, тили в гексані протягом 2 годин - фільтрат відділясм -1): 765, 1350 (sNO2), 1530 (asNO2), 1610, 1625, ли декантацією і при охолодженні останнього ви5 2820. Спектр ПМР в метанолі-D4 (d, м.д., J, Гц): кристалізовується цільовий продукт 2b ; 1.20 т (3Н, СН3, 7.4); 1.43 т (3Н, СН3, 7.4); 3.28 к перекристалізацію проводять в ацетоні. Кристали жовтого кольору. Вихід 12.0 %, Ттопл (2Н, SCH2, 7.4); 3.55 к (2Н, ОСН2, 7.4); 4.18 д (2Н, СН2, 6.2); 6.15 м (1Н, СН); 6.79 д (1Н, СН, 15.8); 118-120°С. ІЧ спектр (n, см -1): 765, 1355 (sNO2), 7.99, 8.37 2д (4Н, 4-C6H4NO2, 9.8, 9.8). Знайдено, 1530 (asNO2), 1610. Спектр ПМР в ДМСО-D6 (d, %: С 54.02; Η 5.33; Ν 16.94; S 9.73. C15H18N4О3S. м.д., J, Гц): 1.68 м (4Н, 2СН2); 2.48 м (4Н, 2СН2); Вирахувано, %: С 53.89; Η 5.39; Ν 16.77; S 9.58. 2.70 с (3Н, СН3); 3.20 д (2Н, СН2, 6.6); 6.08 м (1Н, Приклад 3 СН); 6.84 д (1Н, СН, 13.8); 8.01, 8.36 2д (4Н, 4Схема функціоналізації на основі 3-алілтіо-4.5C6H4NO2, 8.7, 8.7). Знайдено, %: С 55.63; Η 5.51; Ν дифеніл-1,2,4-триазо-лу(3) 20.31; S 9.29. C16H19N5O 2S. Вирахувано, %: С 55.65; Η 5.51; Ν 20.29; S 9.28. 3-Метилтіо-5-(4-нітрофеніл)-4-(2-Nпіролідинілпропен-1-іл)-1,2,4-триазол (2с). Сіль 2а (3 ммоль) розчиняли при нагріванні в 15мл відповідного вторинного аміну і витримували 6-8 годин 9 37721 4,5-дифеніл-2-(2-меркаптопропен-2-іл)-1,2,4триазол-3-ону дисульфід (3b). Сіль 3а (0.50г) перемішують в 5мл відповідного аміну при кімнатній температурі протягом двох годин. Перекристалізовують кінцевий продукт з ацетону. Кристали білого кольору. Вихід 76%, Ттопл=152153°С, Rf=0.87 (ізопропанол : гексан = 5:1). Спектр ПМР в ДМСО-D6 (d, м.ч., J, Гц): 4.24 с (2Н, СН2); 5.59 с, (1Н, =СН2транс); 6.02 с (1Н, =СН2цис); 7.377.57 м (10Н, 2С6Н5). Знайдено, %: N 13.82. C34Н28N6S 2O 2. Вирахувано, %: N 13.64. Таким чином, було розроблено прийом взаємодії штучно створеного активного реакційного центру з нуклеофільними реагентами, що забезпечило проведення функціоналізації гетероциклічних систем при звичайних умовах чи помірному нагріванні без використання важкодоступних реагентів та складного апаратурного оформлення. Використання запропонованого способу нуклеофільного розщеплення отриманих електрофільною галогеноциклізацією поліконденсованих гетероциклів з позитивно зарядженим гетероатомом забезпечує селективність утворення вінільних функціональних похідних, для синтезу яких відомі методи функціоналізації гетероциклів є неприйнятними. Спосіб є простим, не потребує використання складного апаратурного оформлення та важкодоступних реактивів; спосіб одназнач Комп’ютерна в ерстка В. Мацело 10 но діє як у випадку p-електронодефіцитних, так і pелектронодонорних гетероциклічних систем, тому може бути використаний як загальний для функціоналізації гетероциклічних систем любої природи. Спосіб може бути використаний у науководослідних хімічних та хіміко-фармацевтичних лабораторіях. Джерела інформації: 1. S. Ernst, C. Richter, A. Hobert, G.G. Mariam, K. Schulze. Synthesis of new 1,2,4-triazolines and 1,3,4-thiadiazolines from bithioureas. // J.Heterocycl.Chem. - 1995. - V.32.-№№1-2.-P.275281. 2. Мелик-Оганджанян Р.Г., Хачатрян В.Э., Гапоян A.C. Фуро-, тиено- и пир-роло[2,3а]пиримидины. // Успехи химии. - 1985. - т.54, №3. - С.450-478. 3. E.S.H.El Ashry, A.A. Kassem, Η. Abdel-Hamid, F.F. Louis, Sh. A. Khattab, and M.R. Aouad. Synthesis and Alkylation of 5-(3-Chlorobenzo[b]thien-2-yl)4H-l,2,4-triazole-3-thiol Under Classical and Microwave Conditions. // J.Heterocycl.Chem. - 2006. V.43. - №№11-12. - P.1427-1430. - Прототип. 4. M.M. Цицика, Хрипак СМ., Смоланка И.В. Бром- и иодциклизация некоторых 4-аллил-1,2,4триазолинтионов-3. // Укр.Хим.Журн. - 1976. - т.42, №8.-С.841-842. Підписне Тираж 28 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the preparation of vinyl functional derivatives of heterocycles
Автори англійськоюSlyvka Mykhailo Vasyliovych, Slyvka Maryna Vasylivna, Usenko Ruslan Mykolaiovych, Lendiel Vasyl Heorhiiovych
Назва патенту російськоюСпособ получения винильных функциональных производных гетероциклов
Автори російськоюСливка Михаил Васильевич, Сливка Марина Васильевна, Усенко Руслан Николаевич, Лендел Василий Георгиевич
МПК / Мітки
МПК: C07D 249/00, C07D 513/00, C07B 41/00, C07B 43/00
Мітки: гетероциклів, функціональних, спосіб, похідних, одержання, вінільних
Код посилання
<a href="https://ua.patents.su/5-37721-sposib-oderzhannya-vinilnikh-funkcionalnikh-pokhidnikh-geterocikliv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання вінільних функціональних похідних гетероциклів</a>
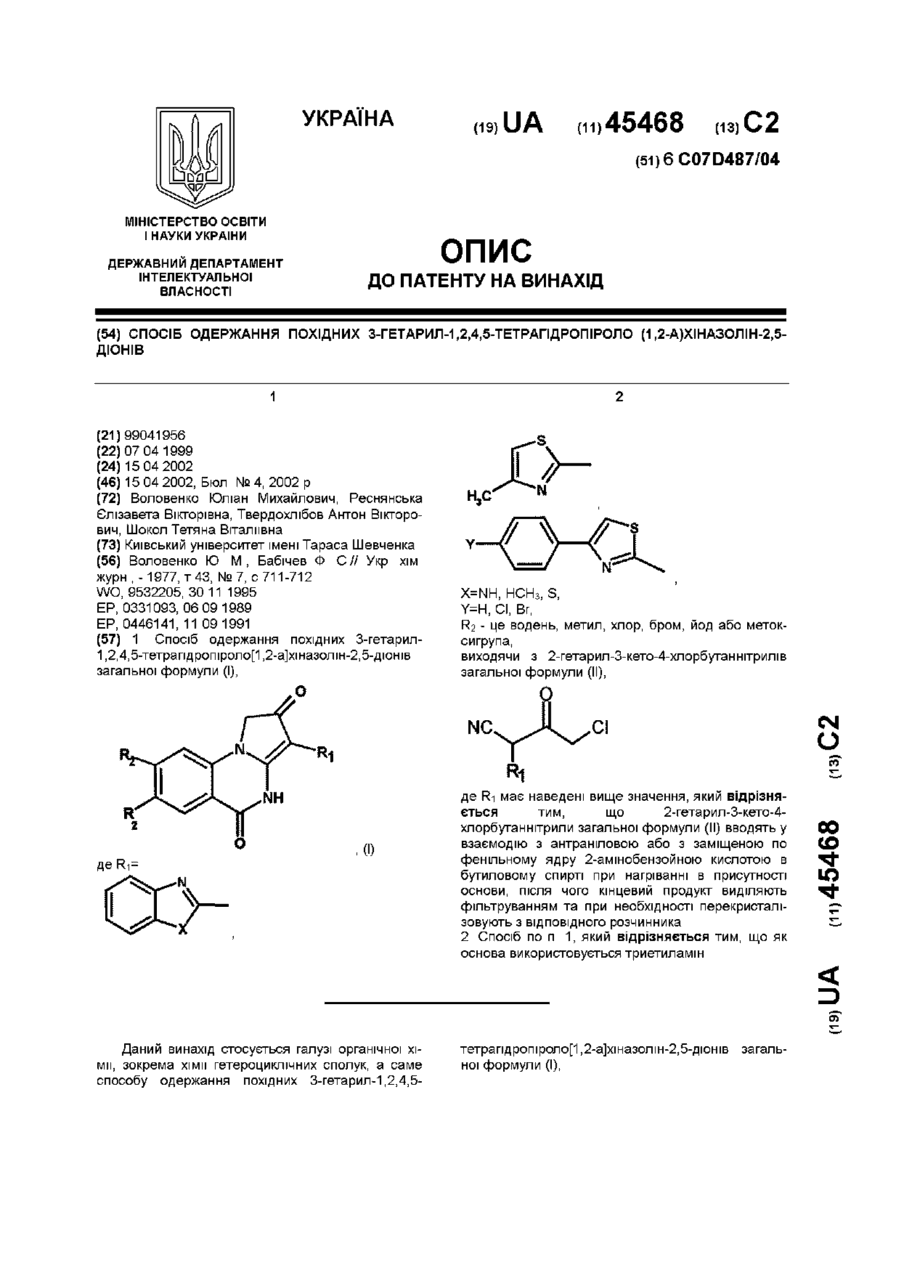
Спосіб одержання похідних 3-гетарил-1,2,4,5-тетрагідропіроло (1,2-а)хіназолін-2,5-діонів
Номер патенту: 45468
Опубліковано: 15.04.2002
Автори: Шокол Тетяна Віталіївна, Реснянська Єлізавета Вікторівна, Твердохлібов Антон Вікторович, Воловенко Юліан Михайлович
МПК: C07D 487/04, C07D 277/20, C07D 235/04
Мітки: спосіб, похідних, одержання, 3-гетарил-1,2,4,5-тетрагідропіроло, 1,2-а)хіназолін-2,5-діонів
Формула / Реферат:
1. Спосіб одержання похідних 3-гетарил-1,2,4,5-тетрагідропіроло[1,2-а]хіназолін-2,5-діонів загальної формули (І),, (І)де R1=,,,X=NH, НСН3, S,Y=H, Сl, Вr,R2 - це водень, метил, хлор, бром, йод або метоксигрупа,виходячи з 2-гетарил-3-кето-4-хлорбутаннітрилів загальної формули (ІІ),де R1 має наведені вище значення, який відрізняється тим, що...
Спосіб отримання ізомірних заміщених циклопропанкарбонових кислот або їх функціональних похідних

Номер патенту: 4992
Опубліковано: 28.12.1994
Автори: Жан-П'єр Демут, Жак Мартель, Жан Тесс'є
МПК: C07C 69/747, A61K 31/215, A01N 53/00, A61P 33/00, A23K 1/16, C07D 209/48, C07D 307/45, C07D 209/49, C07C 69/74
Мітки: спосіб, заміщених, функціональних, циклопропанкарбонових, кислот, ізомірних, похідних, отримання
Формула / Реферат:
1. Способ получения изомерных замещенных циклопропанкарбоновых кислот или их функциональных производных общей формулы 1 где Х1- атом водорода, фтора, хлора или брома; Х2-отличный или одинаковый с X1 атом фтора, хлора или брома;Х3- атом хлора, брома или йода, Z-гидроксил, галоген или сложноэфирная группа ОR, где R - либо радикал бензил, который может быть заменен одним или несколькими радикалами, выбираемыми из группы...
Спосіб одержання фенільних гетероциклів, придатних як інгібітори циклооксигенази-2

Номер патенту: 57029
Опубліковано: 16.06.2003
Автори: Тілльєр Річард Д., Десмонд Річард, Доллінг Ульф Х., Тшаєн Девід М., Фрей Лайза Ф.
МПК: C07D 307/58, C07D 307/38
Мітки: гетероциклів, спосіб, придатних, циклооксигенази-2, одержання, інгібітори, фенільних
Формула / Реферат:
1. Способ получения соединений формулы 11,где R2 является моно- или дизамещенным фенилом, где заместитель выбран из группы, включающей:(1) водород,(2) галоген,(3) C1-6-алкокси,(4) C1-6-алкилтио,(5) CN,(6) СF3 и(7) C1-6-алкил,R3 и R3' независимо выбраны из водорода и C1-4-алкила, который...
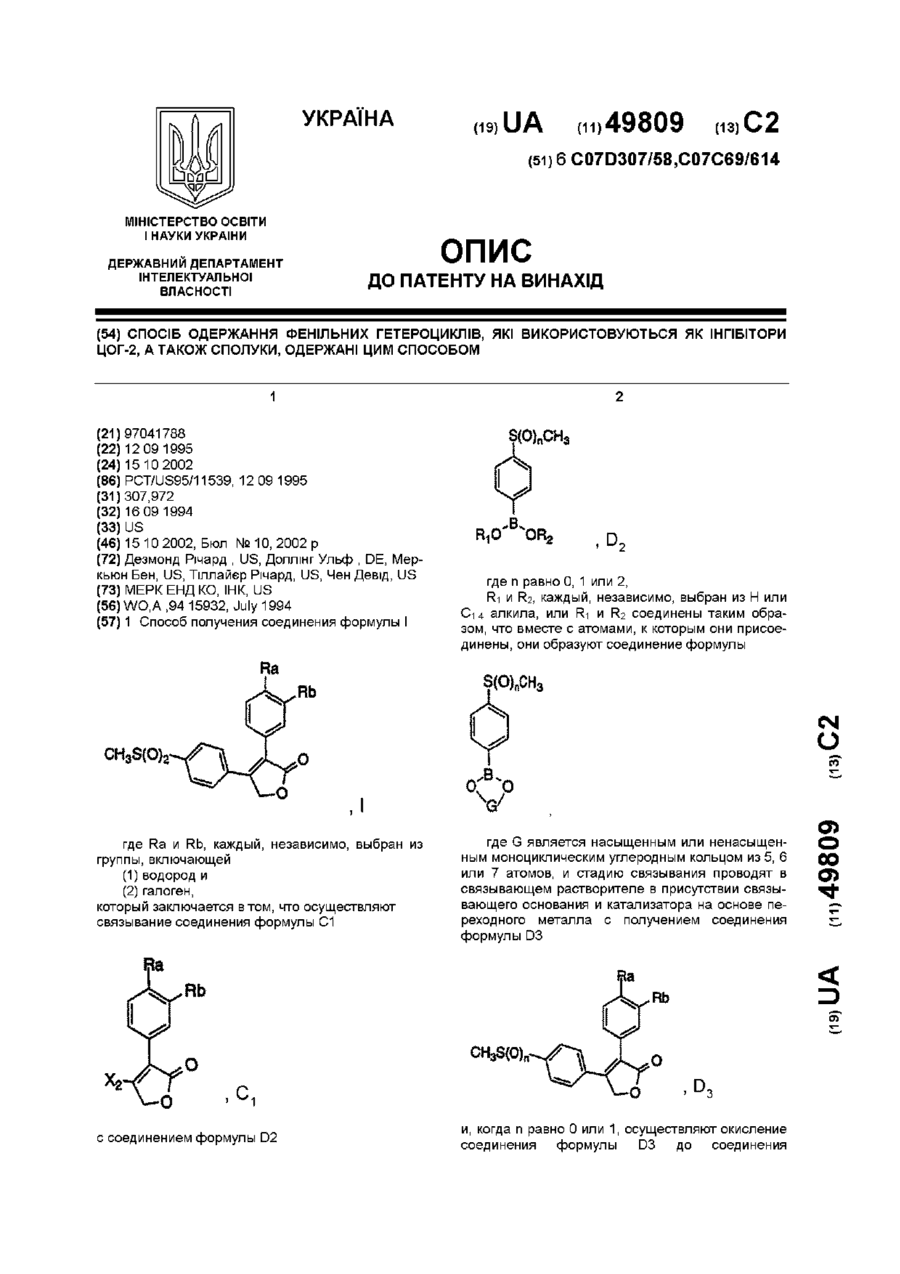
Спосіб одержання фенільних гетероциклів, які використовуються як інгібітори цог-2, а також сполуки, одержані цим способом
Номер патенту: 49809
Опубліковано: 15.10.2002
Автори: Доллінг Ульф, Дезмонд Річард, Чен Девід, Меркьюн Бен, Тіллайєр Річард
МПК: C07D 307/58, C07C 69/675
Мітки: інгібітори, одержані, спосіб, цог-2, сполуки, гетероциклів, способом, одержання, використовуються, цим, також, фенільних
Формула / Реферат:
1. Способ получения соединения формулы Iгде Ra и Rb, каждый, независимо, выбран из группы, включающей(1) водород и(2) галоген;который заключается в том, что осуществляют:связывание соединения формулы С1с соединением формулы D2где n равно 0, 1 или 2;R1 и R2, каждый, независимо, выбран из Н или C1-4 алкила, или R1 и R2 соединены таким образом, что вместе с атомами, к...
Спосіб одержання 4-нітро та/або 4-нітрозодифеніламіну або їх похідних, спосіб одержання 4-амінодифеніламіну або його заміщених похідних та спосіб одержання алкілованих р-фенілендіамінів або їх заміщених похідних

Номер патенту: 26851
Опубліковано: 29.12.1999
Автори: ЕЛЛМАН Джеймс Малькольм, ШТЕРН Майкл Кейт, БЕШКІН Джеймс Кін, РЕЙНС Роджер Керанен
МПК: C07C 211/54, C07C 209/36, C07C 209/38, C07C 209/60, C07C 307/00, C07C 213/00, C07C 211/56, C07C 211/64, C07C 229/54, C07C 217/92, C07C 209/26, C07B 61/00, C07C 209/02
Мітки: 4-нітрозодифеніламіну, 4-нітро, заміщених, 4-амінодифеніламіну, р-фенілендіамінів, одержання, спосіб, похідних, алкілованих
Текст:
...собой соединение, присутствующее в ходе реакции анилина или замещенных производных анилина и нитробензола в дополнение к соответствующему используемому основанию. Примеры подходящих осушителей включают, но не ограничиваются, безводный сульфат натрия, молекулярные сита, такие как сита типа 4А, 5А и 13Х, поступающие от Vion Carbide Corporation, хлорид кальция, дигидрат гидроксида тетраметиламмония, безводные основания, такие как КОН,...
Попередній патент: Прес-форма для рівноканального кутового пресування
Наступний патент: Спосіб приготування льонотрести
Випадковий патент: Спосіб оцінки ефективності використання епадолу при гіпотиреоїдній полінейропатії та енцефалопатії