Похідні 3(2н)піридазинону, що мають фармацевтичну активність, бронхолітичний, антиалергічний, антитромбоцитний засоби на їх основі
Номер патенту: 34481
Опубліковано: 15.03.2001
Автори: Саіто Акіра, Шікада Кен-іші, Хіротсука Мішуакі, Танікава Кеізо








Текст
1. Производные 3(2Н)пиридазинона общей формулы (I): 34481 атом галогена, аминогруппу, N-формильную группу или (С1-С4)алкилкарбониламиногруппу; остатком пиридила, остатком хинолила или группой формулы: где: R6 представляет собой (С1-С4)алкильную группу, причем эта алкильная группа может быть замещена фенильной группой, которая может быть замещена группой Y3, где Y3 представляет собой атом галогена, аминогруппу, N-формильную группу или (С1-С4)алкилкарбониламиногруппу; остатком пиридила, остатком хинолила или группой формулы: где: R9 представляет собой (С1-С4)алкильную группу, которая может быть замещена фенильной группой, которая может быть замещена атомом галогена, или их фармацевтически приемлемые соли. 4. Производные 3(2Н)пиридазинона по п. 1, в которых R4 и R5 вместе с соседним атомом азота образуют замещенное в положении 4 пиперидиновое кольцо формулы: где: R9 представляет собой (С1-С4)алкильную группу, которая может быть замещена фенильной группой, которая может быть замещена атомом галогена, или (с) R4 и R5 вместе с соседним атомом азота образуют замещенное в положении 4 пиперидиновое кольцо формулы: где: R11 представляет собой (С1-С4)алкильную группу, которая может быть замещена фенильной группой,или их фармацевтически приемлемые соли. 5. Производные 3(2Н)пиридазинона по любому из пп. 1-4 или их фармацевтически приемлемые соли, обладающие бронхолитическим, антиаллергическим, антитромбоцитным действием. 6. Бронхолитическое средство, содержащее производное 3(2Н)пиридазинона или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, отличающееся тем, что в качестве производного 3(2Н)пиридазинона содержит соединения общей формулы (I): где: R11 представляет собой (С1-С4)алкильную группу, которая может быть замещена фенильной группой, в количестве от 0,1 до 99,5 % по массе. 7. Антиаллергическое средство, содержащее производное 3(2Н)пиридазинона или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, отличающееся тем, что в качестве производного 3(2Н)пиридазинона содержит соединения общей формулы (I): где: R1 представляет собой атом водорода или (С1-С4)алкильную группу, каждый из R2 и R3 представляет собой атом водорода, Х представляет собой атом хлора или атом брома, Y1 представляет собой атом водорода, атом галогена, нитрогруппу или (С1-С4)алкоксильную группу, Y2 представляет собой (С1-С4)алкоксильную группу, А представляет собой (С1-С5)алкиленовую цепь, которая может быть замещена гидроксильной группой, В представляет собой карбонильную группу или метиленовую цепь, которая может быть замещена (С1-С4)алкильной группой, и (а) R4 представляет собой атом водорода и R5 представляет собой группу -Z-Ar, где Z – (С1С5)алкиленовая цепь и Ar – остаток пиридила, или (б) R4 и R5 вместе с соседним атомом азота образуют замещенное в положении 4 пиперазиновое кольцо формулы: где:R1 представляет собой атом водорода или (С1С4)алкильную группу, каждый из R2 и R3 представляет собой атом водорода, Х представляет собой атом хлора или атом брома, Y1 представляет собой атом водорода, атом галогена, нитрогруппу или (С1-С4)алкоксильную группу, Y2 представляет собой (С1-С4)алкоксильную группу, А представляет собой (С1-С5)алкиленовую цепь, которая может быть замещена гидроксильной группой, В представляет собой карбонильную группу или метиленовую цепь, которая может быть замещена (С1-С4)алкильной группой, и (а) R4 представляет собой атом водорода и R5 представляет собой группу -Z-Ar, где Z – (С1С5)алкиленовая цепь и Ar – остаток пиридила, или (б) R4 и R5 вместе с соседним атомом азота образуют замещенное в положении 4 пиперазиновое кольцо формулы: 2 34481 Y1 представляет собой атом водорода, атом галогена, нитрогруппу или (С1-С4)алкоксильную группу, Y2 представляет собой (С1-С4)алкоксильную группу, А представляет собой (С1-С5)алкиленовую цепь, которая может быть замещена гидроксильной группой, В представляет собой карбонильную группу или метиленовую цепь, которая может быть замещена (С1-С4)алкильной группой, и (а) R4 представляет собой атом водорода и R5 представляет собой группу -Z-Ar, где Z – (С1С5)алкиленовая цепь и Ar – остаток пиридила, или (б) R4 и R5 вместе с соседним атомом азота образуют замещенное в положении 4 пиперазиновое кольцо формулы: где: R6 представляет собой (С1-С4)алкильную группу, причем эта алкильная группа может быть замещена фенильной группой, которая может быть замещена группой Y3, где Y3 представляет собой атом галогена, аминогруппу, N-формильную группу или (С1-С4)алкилкарбониламиногруппу; остатком пиридила, остатком хинолила или группой формулы: где: R9 представляет собой (С1-С4)алкильную группу, которая может быть замещена фенильной группой, которая может быть замещена атомом галогена, или (с) R4 и R5 вместе с соседним атомом азота образуют замещенное в положении 4 пиперидиновое кольцо формулы: где: R6 представляет собой (С1-С4)алкильную группу, причем эта алкильная группа может быть замещена фенильной группой, которая может быть замещена группой Y3, где Y3 представляет собой атом галогена, аминогруппу, N-формильную группу или (С1-С4)алкилкарбониламиногруппу; остатком пиридила, остатком хинолила или группой формулы: где: R11 представляет собой (С1-С4)алкильную группу, которая может быть замещена фенильной группой, в количестве от 0,1 до 99,5 % по массе. 8. Антитромбоцитное средство, содержащее производное 3(2Н)пиридазинона или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, отличающееся тем, что в качестве производного 3(2Н)пиридазинона содержит соединения общей формулы (I): где: R9 представляет собой (С1-С4)алкильную группу, которая может быть замещена фенильной группой, которая может быть замещена атомом галогена, или (с) R4 и R5 вместе с соседним атомом азота образуют замещенное в положении 4 пиперидиновое кольцо формулы: где: R1 представляет собой атом водорода или (С1-С4)алкильную группу, каждый из R2 и R3 представляет собой атом водорода, Х представляет собой атом хлора или атом брома, где: R11 представляет собой (С1-С4)алкильную группу, которая может быть замещена фенильной группой, в количестве от 0,1 до 99,5 % по массе. _______________________ Настоящее изобретение относится к новым производным 3/2Н/-пиридазинона и их солям фармацевтического назначения, обладающим бронхолитическим, антиаллергическим и/или антитромбоцитным действием. 1) Область бронхолитических средств При лечении хронических обратимых, сопровождающихся закупориванием дыхательных путей респираторных заболеваний, например, бронхиальной астмы, бронхита и респираторного дистресс-синдрома взрослых, важным обстоятельством является ремиссия в дыхательных путях во время приступа. Именно для этого используются бронхолитические препараты. Большинство бронхолитических средств, применяемых в настоящее время в клинических целях, могут быть подразделены, с точки зрения общей классификации, на bстимуляторы, включая сальбутамол, и ксантиновые лекарственные средства, представленные теофиллином. Недостатком последних является ослабление действия при трудноизлечимых болезнях, а при лечении бронхиальной астмы отмечается ухудшение симптоматологии вследствие частого и продолжительного введения лекарст 3 34481 венных cредств (The New England Journal of Мedicine, vol 321, p. 1517-1527, 1989). С другой стороны, лекарственные препараты на основе теофиллина ограничены в употреблении, поскольку их безвредность ограничена узким диапазоном. 2) Область антиаллергических лекарств Предполагается, что в возникновении прямых аллергических заболеваний типа бронхиальной астмы, аллергического ринита, крапивницы и сенной лихорадки непосредственное участие принимают разнообразные химические медиаторы in vivo. Среди них одним из главных медиаторов является гистамин, и антигистаминные средства как противоаллергические препараты применяются уже очень давно. Однако многие противоаллергические лекарственные средства антигистаминного типа имеют побочное действие на центральную нервную систему, вызывающее, например, сонливость. Для лечения астмы было бы целесообразно, как с терапевтической, так и с экономической точек зрения, иметь лекарственное средство, которое обладало бы как антиаллергическим, так и бронхолитическим действием, однако в клиническом смысле такое лекарство еще не разработано. 3) Область антитромбоцитных препаратов Известно, что тромбоциты играют важную роль при образовании тромбов, что проявляется в форме их активизации в результате раздражения, адгезии к стенкам сосудов и агрегации. К числу заболеваний, вызываемых образованием тромбов, относятся, например, церебральный тромбоз, легочный тромбоз, инфаркт миокарда, стенокардия и закупорка периферических артерий, рассматриваемые как основные болезни, и все эти болезни требуют разработки эффективных лекарств. В качестве профилактического или терапевтического средства внимание обращается на антитромбоцитное средство, обладающее ингибиторным воздействием на агрегацию тромбоцитов. До настоящего времени широко исследовалось действие аспирина, а в последнее время в клинической практике появились такие препараты, как тиклопидин и цилостазол. Однако с точки зрения эффективности желательно более сильнодействующее лекарственное средство. Помимо упомянутых выше тромботических болезней, существует и множество других заболеваний, связанных с тромбоцитами. Примерами могут служить нефрит, метастаз раковых клеток и т.п., причем в последнее время были проведены разнообразные исследования, касающиеся профилактики и лечения этих болезней, осуществляемых в основном путем применения антитромбоцитных средств, действие которых направлено на управление функциями тромбоцитов ("Journal of Royal College of Physicians", Vol. 7, №. 1, р. 5-18, 1972; "Japan Clinics (Nihon Rinsho)", Vol. 4, №. 6, р. 130 -136,1988; Anticancer Research, Vol 6, р. 543548, 1986). ложенными в соответствии с настоящим изобретением, с соединениями, раскрытыми в опубликованных ссылочных документах. Соединения, в которых замещаемая бензиламиногруппа связана с положением 5 в кольце 3/2Н/-пиридазинона, и которые относительно аналогичны соединениям, предложенным в соответствии с настоящим изобретением, раскрыты в следующих ссылочных документах. (а) В опубликованной патентной заявке Японии № 41455/1994, EP186817B или патенте США 5098900 (упоминаемых ниже как ссылка (а)) раскрыты соединения, включающие производные 3/2Н/-пиридазинона, в которых положение 2 - низшая алкильная группа, положение 4 атом хлора или атом бора, положение 5 - бензиламиногруппа, имеющая бензольное кольцо, замещаемое заместителем, включающим w-аминоалкильную группу, w-карбамоилалкиленоксильную группу, -w-моно низшую алкиламинокарбонилалкиленоксильную группу и аминокарбонильную группу, и их фармацевтическое применение в качестве средства против медленно реагирующих субстанций аллергии (МРСА), а также их фармакологическое действие. (б) В опубликованной не подвергнутой экспертизе патентной заявке Японии № 030769/1987, EP201765B или патенте США 4892947 /упоминаемых ниже как ссылка /б// раскрыты соединения, включающие производные 3/2Н/-пиридазинона, в которых положение 2 - атом водорода, положение 4 - атом хлора или атом брома, положение 5 - бензиламиногруппа, имеющая бензольное кольцо, замещаемое заместителем, включающим алкоксильную группу, w-фенилалкиленоксильную группу и диалкиламиногруппу, а положение 6 - атом водорода, и их фармацевтическое применение в качестве средства против МРСА, а также их фармакологическое действие. (в) В опубликованной не подвергнутой экспертизе патентной заявке Японии № 301870/1988, ЕР275997В или патенте США 4978665 (упоминаемых ниже как ссылка (в)) раскрыты соединения, включающие производные 3/2Н/-пиридазинона, в которых положение 2 – атом водорода или низкая алкильная группа, положение 4 – атом хлора или атом бора, положение 5 – бензиламиногруппа, имеющая бензольное кольцо, замещаемое заместителем, включающим алкоксильную группу, wфенилалкиленоксильную группу и диалкиламиногруппу, а положение 6 – атом галогена, нитрогруппа, аминогруппа или алкоксильная группа, и их фармацевтическое применение в качестве средства против МРСА, а также их фармакологическое действие. (г) В опубликованной международной заявке WO 91/16314, ЕР482208А или патенте США 5202323 (упоминаемых ниже как ссылка (г)) раскрыты соединения, включающие производные 3/2Н/-пиридазинона, в которых положение 2 – атом водорода или низшая алкильная группа, положение 4 – атом хлора или атом бора, положение 5 – бензиламиногруппа, имеющая бензольное кольцо, замещаемое заместителем, включающим алкоксильную группу, w-фенилалкиленоксильную группу, где бензольное кольцо может быть замещено алкильной группой или атомом галогена, Ниже следует описание взаимосвязи между производными 5-w-/аминоалкиленокси- или wаминокарбонилалкиленокси-замещенная-бензиламино/-/2Н/-пиридазинона общей формулы (I) и их солями фармацевтического назначения, пред 4 34481 w-алкоксикарбонилалкиленоксильную группу и w-аминокарбонилалкиленоксильную группу, а положение 6 – алкиленоксильная группа, имеющая одну из различных функциональных групп в положении w, и их фармацевтическое применение в качестве антитромбоцитных, кардиотонических, сосудорасширяющих средств, и средств против МРСА, а также их фармакологическое действие. В результате обширных исследований авторами настоящего изобретения было установлено, что производные 3/2Н/-пиридазинона и их соли, приемлемые с фармацевтической точки зрения, которые предложены в соответствии с настоящим изобретением и которые отличаются от любого из соединений, раскрытых в упомянутых выше ссылочных документах (а)-(г), представляют собой превосходные соединения с точки зрения их применения в качестве сосудорасширяющих, антиаллергических и/или антитромбоцитных средств, особенно превосходное действие проявляется в результате их орального введения в организм, и их целесообразно применять в качестве активных ингредиентов профилактических и терапевтических лекарственных препаратов, например, при упомянутых выше респираторных болезнях, прямых аллергических заболеваниях и/или тромботических болезнях. Настоящее изобретение основано именно на этом открытии. Другими словами, в соответствии с настоящим изобретением предложены производное 3/2Н/-пиридазинона общей формулы (I) и его приемлемая с фармацевтической точки зрения соль, способ их получения и фармацевтический состав, содержащий их в качестве активного ингредиента: {где R6 – алкильная группа С1-4 (эта алкильная группа может быть замещена одним или более заместителями, выбранными из группы заместителей, состоящей из алкильной группы С1-4, фенильной группы, которая может быть замещена Y3 (где Y3 – атом водорода, атом галогена, алкильная группа С1-4, алкоксильная группа С1-4, аминогруппа, N-формильная группа или алкилкарбониламиногруппа С1-4), (где каждый из R7 и R8 – атом водорода, или R7 и R8 образуют совместно с атомами углерода, к которым они привязаны, бензольное кольцо, а каждый из А, B, C и D, которые независимы один от другого, – атом азота или атом углерода) и /где Y3 - как определено выше, а R9 - алкильная группа С1-4 или бензильная группа, которая может быть замещена алкильной группой С1-4, алкоксильной группой С1-4 или атомом галогена// или -COR10 /где R10 - атом водорода или алкильная группа С1-4, /} или 4-замещенное пиперидиновое кольцо формулы: {где R11 - алкильная группа С1-4 (эта алкильная группа может быть замещена одним или более заместителями, выбранными из группы заместителей, состоящей из фенильной группы, которая может быть замещена Y3 /где Y3 – как определено выше/, и гидроксильной группы)}. Ниже следует описание R1 , R2 , R3, R4, R5 , 1 2 А, B, X, Y и Y в соединении формулы (I), предложенном в соответствии с настоящим изобретением. К числу конкретных примеров каждого из R1 , R2 И R3 можно отнести атом водорода, метильную группу, этильную группу, n-пропильную группу, i-пропильную группу, n-бутильную группу, i-бутильную группу, втор-бутильную группу и t-бутильную группу. Для каждого из них предпочтительным является атом водорода. А - алкиленовая цепь, имеющая общее углеродное число от 1 до 5, которая может быть замещена гидроксильной группой или алкильной группой в любом произвольном положении и, например, представлять собой какую-либо разновидность связи, например, метиленовую группу, этиленовую группу, пропиленовую группу, бутиленовую группу или пентиленовую группу. Более предпочтительной является линейная алкиленовая группа, имеющая от 1 до 4 атомов углерода. (I) где каждый из R1, R2 и R3, которые независимы один от другого, – атом водорода или алкильная группа С1-4, X - атом хлора или атом брома, Y1 атом водорода, атом галогена, нитрогруппа, аминогруппа или алкоксильная группа С1-4, Y2 – атом водорода, атом галогена, гидроксильная группа, алкильная группа С1-4 или алкоксильная группа С1-4, А – алкиленовая цепь С1-5, которая может быть замещена гидроксильной группой, B – карбонильная группа или метиленовая цепь, которая может быть замещена алкильной группой C1-4, а каждый из R4 И R5, которые независимы один от другого, – алкильная группа С1-4, или R4 – атом водорода, а R5 – -Z-Ar (где Z – алкиленовая цепь С1-5, а Аг – ароматическое 6-членное кольцо, которое может содержать один или два атома азота), или R4 и R5 совместно образуют циклическую алкиленовую группу С2-6, или R4 и R 5 образуют совместно с соседним атомом азота 4-замещенное пиперазиновое кольцо формулы: 5 34481 нильная группа, которая может быть замещена Y3 (где Y3 – атом водорода, атом галогена, алкильная группа С1-4, алкоксильная группа С1-4, аминогруппа, N-формильная группа или алкилкарбониламиногруппа С1-4), B может быть карбонильной группой или разновидностью связи по метиленовой цепи, которая может быть замещена алкильной группой С1-4. X может быть атомом хлора или атомом брома. Y1 может быть, например, атомом водорода, атомом хлора, атомом брома, атомом иода, нитрогруппой, аминогруппой, метоксильной группой, этоксильной группой, n-пропоксильной группой, iпропоксильной группой, n-бутоксильной группой, iбутоксильной группой, втор-бутоксильной группой или t-бутоксильной группой. Y2 может быть, например, атомом водорода, атомом хлоpa, атомом брома, атомом иода, гидроксильной группой, метильной группой, этильной группой, n-пропильной группой, i-пропильной группой, n-бутильной группой, i-бутильной группой, втор-бутильной группой, t-бутильной группой, метоксильной группой, этоксильной группой, n-пропоксильной группой, i-пропоксильной группой, nбутоксильной группой, i-бутоксильной группой, втор-бутоксильной группой или t-бутоксильной группой. R4 и R5 - следующие: (1) каждый из них – алкильная группа С1-4, например, метильная группа, этильная группа, nпропильная группа, i-пропильная группа, n-бутильная группа, i-бутильная группа, втор-бутильная группа или t-бутильная группа; (2) R4 – атом водорода, а R5 – -Z-Ar (где Z – алкиленовая цепь С1-5, a Ar – ароматическое 6-членное кольцо, которое может содержать один или два атома азота). Ароматическое 6-членное кольцо содержит фенильную группу, 2-пиридильную группу, 3-пиридильную группу, 4-пиридильную группу, 3-пиридазинильную группу, 4-пиридазинильную группу, 2-пиримидинильную группу, 4пиримидинильную группу, 5-пиримидинильную группу и 2-пиразинильную группу; (3) R4 и R5 совместно образуют циклическую алкиленовую группу С2-6, а также совместно с атомом азота, к которому они привязаны, образуют азиридиновое кольцо, азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо или гомопиперидиновое кольцо; (4) R4 и R5 совместно с соседним атомом азота, к которому они привязаны, образуют 4-замещенное пиперазиновое кольцо формулы: (где каждый из R7 И R8 – атом водорода, или R7 И R8 совместно с атомами углерода, к которым они соответственно привязаны, образуют бензольное кольцо, а каждый из А, В, С и D, которые независимы один от другого, - атом азота или атом углерода) и (где Y3 – как определено выше, а R9 – алкильная группа С1-4 или бензильная группа, которая может быть замещена алкильной группой С1-4, алкоксильной группой С1-4 или атомом галогена на бензольном кольце). Число таких заместителей может составлять от одного и больше. К конкретным примерам R6 можно отнести бензильную группу, которая может иметь атом галогена, замещенный в любом произвольном положении о, m или р на бензольном кольце, a,a-дифенилметильную группу, пиридиметильную группу, которая может быть замещена в любом произвольном положении 2, 3 или 4, пиримидилметильную группу, пиразилметильную группу, пиридазилметильную группу, хинолилметильную группу, изохинолилметильную группу, хиноксалилметильную группу, хиназолилметильную группу, бензимидазолилметильную группу, имеющую бензильную группу, которая может быть замещена атомом галогена на бензольном кольце или алкильной группой С1-4 в положении N, и комбинацию таких ароматических колец, как a,a-фенилпиридилметильная группа, a,a-фенилпиримидилметильная группа, a,a-фенилпиразилметильная группа, a,aфенилпиридазилметильная группа, a,a-фенилхинолилметильная группа, a,a-фенилизохинолилметильная группа, a,a-фенилхиноксалилметильная группа или a,a-фенилхиназолилметильная группа. R11 - алкильная группа С1-4, причем эта алкильная группа может иметь заместители. Эти заместители подразделяются на два типа, а именно: на фенильную группу, которая может быть замещена Y3 (где Y3 - как определено выше), и гидроксильную группу. Одна из них или несколько могут быть замещены. К конкретным примерам R11 можно отнести бензильную группу, которая может иметь атом галогена, замещенный в любом произвольном положении о, m или р на бензольном кольце a,a-дифенилметильную группу и a,a,a-гидроксидифе или 4-замещенное пиперидиновое кольцо формулы: R6 – алкильная группа С1-4 или -COR10 (где R10 – атом водорода или алкильная группа С1-4). Алкильная группа С1-4 для R6 предпочтительно представляет собой метильную группу и может иметь заместитель. Таким заместителем может быть, например, алкильная группа C1-4, фе 6 34481 где R11 - алкильная группа С1-4 {эта алкильная группа может быть замещена одним или более заместителями, выбранными из группы заместителей, состоящей из фенильной группы, которая может быть замещена Y3 (где Y3 - как определено выше), и гидроксильной группы}. (3) Соединение, как определено в упомянутом выше пункте (2), где R4 и R5 совместно с соседним атомом азота, к которому они привязаны, образуют 4-замещенное пиперазиновое кольцо формулы: нилметильную группу. Предпочтительными примерами каждого из R4 и R5 являются 4-замещенный пиперазин-1-ил и 4-замещенный пиперидин-1ил, как это описано выше. В приведенном выше описании "n" означает "нормальный", "і" - "изо", "втор" - "вторичный", "t" "третичный", "о'' - "орто", "m" - "мета" и "р" - "пара". Среди соединений формулы (I), предложенных в соответствии с настоящим изобретением, в качестве предпочтительных могут быть упомянуты следующие соединения. (1) Соединение формулы (І), где каждый из R2 и R3 -атом водорода, а Y1 - атом водорода, атом галогена, нитрогруппа или алкоксильная группа С1-4. (2) Соединение формулы (I), как определено в вышеупомянутом пункте (1), где R4 и R5 совместно с соседним атомом азота, к которому они привязаны, образуют 4-замещенное пиперазиновое кольцо формулы: где R13 - метильная группа {эта метильная группа может быть замещена одним или более заместителями, выбранными из группы заместителей, состоящей из фенильной группы, которая может быть замещена Y3 (где Y3 - атом водорода, атом галогена, алкильная группа С1-4, алкоксильная группа С1-4, аминогруппа, N-формильная группа или алкилкарбониламиногруппа С1-4), где R12 – алкильная группа С1-4 {эта алкильная группа может быть замещена одним или более заместителями, выбранными из группы заместителей, состоящей из алкильной группы С1-4, фенильной группы, которая может быть замещена Y3 (где Y3 – атом водорода, атом галогена, алкильная группа С1-4, алкоксильная группа С1-4, аминогруппа, N-формильная группа или алкилкарбониламиногруппа С1-4), (где каждый из R7 и R8 - атом водорода, или R7 и R8 совместно с атомами углерода, к которым они привязаны, образуют бензольное кольцо, а каждый из А, в, с и D, которые независимы один от другого, - атом азота или атом углерода) и (где каждый из R7 и R8 - атом водорода, или R7 и R8 совместно с атомами углерода, к которым они привязаны, образуют бензольное кольцо, а каждый из А, в, с и D, которые независимы один от другого, - атом азота или атом углерода) и (где Y3 - как определено выше, a R9 - алкильная группа С1-4 или бензильная группа, которая может быть замещена алкильной группой С1-4, алкоксильной группой С1-4 или атомом галогена)} или -COR10 (где R10 - атом водорода или алкильная группа С1-4). (4) Соединение, как определено в упомянутом выше пункте (3), где Y2 - атом галогена или алкоксильная группа С1-4. (5) Соединение, как определено в упомянутом выше пункте (4), где R4 и R5 совместно с соседним атомом азота, к которому они привязаны, образуют 4-замещенное пиперазиновое кольцо формулы: (где Y3 - как определено выше, a R9 - алкильная группа С1-4 или бензильная группа, которая может быть замещена алкильной группой С1-4, алкоксильной группой С1-4 или атомом галогена на бензольном кольце)} или -COR10 (где R10 - атом водорода или алкильная группа С1-4), или 4-замещенное пиперидиновое кольцо формулы: где R14 – 7 34481 (где Y4 – атом водорода, атом галогена, аминогруппа, N-Формильная группа или алкилкарбониламиногруппа С1-4), лот (например, гидрохлориды, гидробромиды, сульфаты, гидросульфаты, нитраты, фосфаты, гидрофосфаты или дигидрофосфаты), соли органических кислот (например, формиаты, ацетаты, пропионаты, сукцинаты, малонаты, оксалаты, малеаты, фумараты, малаты, цитраты, тартраты, лактаты, глутаматы, аспартаты, пикраты или карбонаты) и соли сульфоновых кислот (например, метансульфонат, бензолсульфонат или толуолсульфонат). Эти соли могут быть приготовлены с использованием соответствующих известных способов. В таблице 1 приведены типичные примеры производных 3/2Н/-пиридазинона и его приемлемых с фармацевтической точки зрения солей, предложенных в соответствии с настоящим изобретением. Однако следует понимать, что настоящее изобретение ни в коей мере не ограничивается такими конкретными примерами. или (где R15 - бензильная группа, которая может быть замещена атомом галогена). Соединения формулы (I) включают оптические изомеры и стереоизомеры на основе 1-5 асимметрических атомов утлерода. Соединения формулы /I/, предложенные в соответствии с настоящим изобретением, могут быть превращены в приемлемые с фармацевтической точки зрения нетоксичные соли путем использования соответствующих кислот. Для целей настоящего изобретения соединения формулы /I/ могут быть использованы как в свободной форме, так и в форме солей, приемлемых с фармацевтической точки зрения. К солям такого типа могут быть отнесены, например, соли минеральных кис В таблице 1 "n" означает "нормальный", "i" – "изо", "t – "третичный", "Mе" – "метильная группа", "Et" – "этильная группа", "Pr" – "пропильная группа", "Bu'' – "бутильная группа" и "Ph" – "фенильная группа". В таблице 1 Q1 -Q42 – группы, представленные следующими формулами. 8 34481 Ниже следует описание способов получения соединений, предложенных в соответствии с настоящим изобретением. Производные 3/2Н/-пиридазинона формулы (I) и их приемлемые с фармацевтической точки зрения соли, предложенные в соответствии с настоящим изобретением, могут быть получены, например, способами, представленными следующими формулами реакций (1)-(7). Формула реакции (I) 9 34481 где R1, R2, R3, R4, R5, X, Y1, Y2, А и B – как определено выше. Способ получения в соответствии с формулой реакции (I) представляет собой способ, согласно которому для получения соединения формулы (I), предложенного в соответствии с настоящим изобретением, соединение 4,5-дигалоген3/2Н/-пиридазинона формулы (ІІ) и производное w -аминоалкиленокси- или w-аминокарбонилалкиленокси-замещенного бензиламина формулы (III) по выбору вводятся в реакцию в присутствии дегидрогалогенирующего агента в инертном растворителе. В приведенной выше формуле реакции (I) изомер положения соединения формулы (I), т.е. соединение формулы (IY), имеющее оксибензиламиногруппу, замещенную в положении 4: или всевозможная хроматография с использованием силикагеля. Во время реакции между соединением формулы (II) и соединением формулы (III) образуется хлористый водород или бромистый водород. Выход, как правило, можно улучшить путем введения в реакционную систему дегидрогалогенирующего агента, который захватывает такой галогеноводород. Может быть использован любой дегидрогалогенирующий агент при условии, что он не оказывает отрицательного влияния на реакцию и способен захватить галогеноводород. В качестве такого дегидрогалогенируюцего агента может быть использовано неорганическое основание, например, карбонат калия, карбонат натрия, гидрокарбонат калия или гидрокарбонат натрия, или органическое основание, например, N,N-диметиланилин, N,N-диэтилакилин, триметиламин, триэтил-амин, N,N-диметиламиноэтанол, N-метилморфолин, пиридин или 2,6-диметил-4-N,N-диметиламинопиридин. В альтернативном варианте в качестве дегидрогалогенирующего агента может быть использовано избыточное количество производного бензиламина формулы (III), взятого как исходный материал. Во многих случаях это приводит к улучшению выхода продукта. Температура реакции может, как правило, колебаться в пределах от 10°С до температуры кипения применяемого в реакции растворителя. Молярная концентрация исходных материалов может быть установлена произвольно. Однако производное бензиламина формулы (III) или его соль может быть использовано, как правило, в количестве от 1 до 10 моль, предпочтительно от 1,2 до 5 моль, на 1 моль производного 4,5-дигалоген-3/2Н/-пиридазинона формулы (ІІ). Производное 4,5-дигалоген-3/2Н/-пиридазинона формулы (ІІ) может быть получено, например, путем использования обычной органической реакции или следующего известного способа получения, а именно, либо в котором заместителем Y1 в положении 6 является атомом водорода и который соответствует способу, раскрытому в ссылочных документах (а) и (б), либо в котором заместителем Y является атом галогена, нитрогруппа, аминогруппа или алкоксильная группа и который соответствует способу, раскрытому в ссылочных документах (в). где R1 , R2, R3, R4, R5, X, Y1 , Y2 , А и B – как определено выше, образуется в виде побочного продукта. Процент получения соединений формул (I) и (IY) зависит в первую очередь от полярности применяемого растворителя. Более конкретно, если используется растворитель с высокой полярностью, процент получения соединения формулы (I), предложенного в соответствии с настоящим изобретением, стремится к повышению. Соответственно, в качестве растворителя, пригодного для эффективного получения соединения формулы (I), предложенного в соответствии с настоящим изобретением, при одновременном подавлении побочной реакции получения соединения формулы (IY) могут быть упомянуты растворители на основе простых эфиров (например, тетрагидрофуран или 1,4-диокcан), растворители на амидной основе (например, формамид, N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон), ацетонитрил, диметилсульфоксид, растворители на спиртовой основе (например, метанол, этанол или пропанол), растворители на основе органических аминов (например, пиридин, триэтиламин, N,N-диметиламиноэтанол или триэтаноламин) или вода, а также их смеси. Для выделения соединения формулы (I), предложенного в соответствии с настоящим изобретением, из упомянутой выше смеси соединения формулы (I) и соединения формулы (IY) и для его очистки могут быть использованы способы, известные в органическом синтезе, например, фракционированная перекристаллизация Производное w-аминоалкиленокси- или w-аминокарбонилалкиленокси-замещенного бензиламина формулы (ІІІ) или его соль в формуле реакции (I) может быть получено, например, способами в соответствии со следующими реакционными схемами (А)-(Д) путем использования способов, раскрытых в ссылочных документах (а). 10 34481 Схема (А) где hal - отщепляемая группа, например, атом хлора, атом брома, атом иода, метансульфонилоксильная группа или р-толуолсульфонилоксильная группа, R атом водорода, гидроксильная группа, алкильная группа C1-4 или алкоксильная группа С1-4, a R2, R3, R4, R5, Y2, А И В - как определено выше. Схема (Б) 11 34481 где T - аминозащитная группа, например, бензилоксикарбонильная группа, t-бутоксикарбонильная группа, формильная группа, ацетильная группа, бензоильная группа, метоксикарбонильная группа или этоксикарбонильная группа, а R2 , R3 , R4, R5, Y2, А, B, R и hal – как определено выше. Схема (В) где R9 – атом водорода или низшая алкильная группа, а R2, R3, R4, R5, Y2, А, T и hal – как определено выше. Схема (Г) 12 34481 где R10 – атом водорода или алкильная группа С1-4, hal' – отщепляемая группа в тех же границах, которые определены выше для hal в реакционной схеме (А), однако является заместителем, ко торый обладает такой же или меньшей способностыо к отщеплению, что и hal в конкретной комбинации, а R2, R3, R4, R5, Y2, А, T и hal - как определено выше. Схема (Д) где D - алкиленовая группа С1-4, а R2, R3,R4,R5,Y2 и hal - как определено выше. Реакционная схема (А) иллюстрирует способ, в котором в качестве исходного материала используется производное гидроксилкарбонила (IX) и в котором сначала соединение формулы (YIII) реагирует в направлении фенольного участка с целью образования соответствующей алкоксильной боковой цепи, а затем в результате восстановления карбонильный участок превращается в аминогруппу. Реакционная же схема /Б/ иллюст рирует способ получения, в котором этот порядок обратен порядку в схеме реакции (А). Реакционная схема (В) иллюстрирует способ, в котором в качестве исходного материала используется N-защищенное производное гидроксибензиламина формулы (X) как промежуточный продукт маршрута реакции в схеме (Б), и боковая цепь его фенольного участка постепенно расширяется, а из производного w-аминокарбонил-алкиленоксибензиламина формулы (IIIа) в результате его восста 13 34481 щее в Y2 и R4 или R5 атом галогена или бензильную группу, который или которая относительно неустойчив или неустойчива в условиях восстановления гидрогенизацией, среди производных бензиламина формулы (III). В то же время для получения вторичного амина, где R2 - алкильная группа C1-4, среди производных бензиламина формулы (III), в качестве RNH2 может быть использован первичный алкиламин формулы R2NH2, а затем в восстановлении производного имина, полученного в результате этой реакции конденсации, может быть использован не только восстановитель, описанный в отношении упомянутого выше способа получения первичного амина, но и более мягкий восстановитель, например, борогидрид натрия или цианоборогидрид натрия (NaCNBH3) может быть добавлен в качестве восстановителя, который можно применять соответствующим образом и в наиболее широком масштабе. Реакционная схема (Б) представляет собой маршрут реакции для получения бензиламина формулы (III) путем обратного осуществления ступеней реакции в реакционной схеме (А). Соответственно, конверсия карбонильной группы в аминометильную группу и реакция алкилирования фенольного участка могут быть осуществлены при соответствующих условиях реакции способа получения, описанного в отношении схемы (А). В соответствии с таким маршрутом процесс требует введения защитной группы для атома бензиламиноазота. В качестве защитной группы формулы T, подлежащей использованию в настоящем изобретении, можно задействовать широкий диапазон защитных групп для аминогрупп, которые широко применяются при обычном синтезе пептидов, например, бензилоксикарбонильная группа, t-бутоксикарбонильная группа, формильная группа, ацетильная группа, бензоильная группа, метоксикарбонильная группа и этоксикарбонильная группа. Не существует какого-либо строгого ограничения в выборе конкретной защитной группы из такого многочисленного ряда защитных групп. В некоторых случаях, однако, бывает необходимо выбрать конкретную защитную группу или условия для ее удаления в зависимости от видов заместителей Y2, B, R4 и R5. Так, например, для получения соединения, содержащего в Y2 или R4 и R5 атом галогена или бензильную группу в бензиламине (III) в некоторых случаях может возникнуть необходимость в правильном выборе заместителей и условий реакции с тем, чтобы реакция для удаления защитной группы могла быть проведена эффективно и избирательно даже с использованием способа, отличного от каталитической гидрогенизации. Для получения бензиламина формулы (ІІІ), где B – карбонильная цепь, во многих случаях предпочтительно использовать бензилоксикарбонильную группу или t-бутоксикарбонильную группу, поскольку именно они способствуют удалению защитной группы в условиях отсутствия гидролизации. Для указанных выше введения и удаления всевозможных защитных групп в качестве условий реакции могут применяться известные условия реакции. Реакционная схема (В) иллюстрирует способ, в котором при использовании в качестве исходного материала гидроксибензиламина форму новления образуется продукт формулы (ІІІ), имеющий восстановленный участок амидной связи формулы (IIIa). Реакционная схема (Г) иллюстрирует способ получения производного w-аминоалкиленоксибензиламина формулы (IIIс), содержащего разветвленную метиленовую цепь, в которой В замещен низшей алкильной группой, среди производных бензиламина формулы (ІІІ). Реакционная схема (Д) иллюстрирует способ получения соединения формулы (IIId), где А – метиленовая цепь, имеющая гидроксильную группу, среди производных бензиламина формулы (ІІІ). Используя покупной исходный материал или исходный материал на его основе, среди способов (А)–(Д) можно выбрать соответствующий условиям применения. Для проведения реакции производного гидроксикарбонила (IX) с (YIII) в схеме (А) широко могут применяться условия, используемые обычно для алкилирования фенолов. Как правило, эта реакция протекает относительно быстро, если использовать неорганическое основание, например, карбонат натрия, карбонат калия, гидроксид натрия, гидроксид калия, гидрокарбонат натрия или гидрокарбонат калия в кетоновом растворителе (например, ацетоне, метилэтилкетоне или диэтилкетоне), растворителе на амидной основе (формамид, N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон), растворителе на спиртовой основе (например, метанол, этанол или n-пропанол) или воде, а также в смеси указанных растворителей в условиях нагрева до температуры от 40 до 150 °С. Последующая реакция с целью конверсии карбонильной группы (формильная группа или кетоновая группа) в аминометильную группу осуществляется путем конденсации какого-либо амина формулы RNH2 с целью получения иминосоединения с последующим восстановлением последнего. При осуществлении данного способа это иминосоединение может не подвергаться выделению и может образовываться в реакционной системе, непрерывно подвергаясь в дальнейшем восстановлению. С точки зрения выхода или экономии применение данного способа целесообразно во многих случаях. Здесь получение первичного амина, где R2 – атом водорода, среди производных бензиламина формулы (III) может быть обеспечено путем использования в качестве RNH2 амина, например, аммиака, гидроксиламина или O-алкилгидроксиламина и восстановления имина, полученного таким образом. Для восстановления такого типа широко применяется реакция гидрогенизации, в которой в качестве катализатора используется скелетный никель (никель Ренея), палладиевая чернь и т.п. В данном случае, если полученное иминосоединение используется с О-алкилгидроксиламином, реакция может быть проведена с применением гидридов металлов, например, трифторацетоксиборогидрида натрия /NaBH3(OCOCF3)/ или бис-метоксиэтоксиалюмогидрида натрия /NaAlH2(OCH2CH2ОСН3)2/ (Chemical and Pharmaceutical Bulletin, vol. 26, р. 289.7- 2898, 1978). Упомянутый последним способ восстановления, предполагающий применение гидрида металла, целесообразно применять в тех случаях, когда необходимо получить соединение, содержа 14 34481 лы (X), защищенного защитной группой T, эфирная боковая цепь постепенно расширяется с образованием соединения формулы (IIIа), где B - карбонильная цепь, и соединения формулы (IIIb), где B - линейная метильная цепь, получаемая в результате восстановления карбонильного участка, среди бензиламинов формулы (III). В реакции для образования амидной связи на участке эфирной боковой цепи, где R9 - атом водорода, широко могут быть использованы способы, которые обычно применяются при синтезе пептидов. Если амин имеет относительно богатую нуклеофильную природу, можно использовать сложный эфир, где R9 низшая алкильная группа, и в этом случае обычно возможно применять условия нагрева в инертном растворителе. В качестве восстановителя, подлежащего применению для получения бензиламина формулы (IIIb), может быть упомянут восстановитель на основе гидрида металла, например, алюмогидрид лития. Алкилирование фенольного участка и реакция для удаления защитной группы на других стадиях могут быть осуществлены путем использования соответствующих реакций, приведенных в схемах (А) и (Б). Реакционная схема (Г) иллюстрирует способ получения производного аминоалкиленоксибензиламина формулы (ІІІс), где a-углерод аминогруппы на концевом участке фенольной боковой цепи представляет собой метиленовую цепь, замещенную линейным или низшим алкилом. Для введения аминосоставляющей могут быть использованы условия реакции, широко применяемые в реакции замещения алкиламина алкилгалогенидом. Реакционная схема (Д) предназначена для введения гидроксильной группы в фенольную боковую цепь формулы (IIId) и иллюстрирует способ, в котором в фенольную боковую цепь вводится эпоксильная группа в результате реакции с различными эпоксиалкилгалогенидными соединениями, а в результате реакции с различными аминами образуется соединение формулы (IIId). Формула реакции (2) где R1' - алкильная группа С1-4, hal – атом хлора, атом брома или атом иода, a R2,R3,R4,R5,X,Y1,Y2,А и B - как определено выше. Формула реакции (2) иллюстрирует способ получения замещенного в положении 2 продукта – пиридазинона формулы (І-b) как соединения, предложенного в соответствии с настоящим изобретением, путем введения в реакцию соединения формулы (1-а), которое представляет собой соединение формулы (I), предложенное в соответствии с настоящим изобретением, где положение 2 пиридазинона - атом водорода, с галогенопроизводным формулы R1' - hal. Для проведения этой реакции используется неорганическое основание, например, карбонат калия, карбонат натрия, карбонат лития, гидрокарбонат калия, гидрокарбонат натрия или гидрокарбонат лития, органическое основание, например, триэтиламин или три-n-пропиламин, или гидрид металла или металлоорганическое соединение, например, гидрид натрия или n-бутиллитий. В качестве растворителя для проведения этой реакции может быть использован кетоновый растворитель (например, ацетон, метилэтилкетон или диэтилкетон), растворитель на амидной основе (например, формамид, N,N-диметилформамид или N,N-диметилацетамид), растворитель на спиртовой основе (например, метанол или этанол), вода или их смесь. В тех случаях, когда используется гидрид металла, предпочтительно применять, как правило, растворитель на основе простых эфиров. В тех случаях, когда используется неорганическое или органическое основание, температура реакции может быть, как правило, установлена в пределах от 0°С до температуры кипения применяемого растворителя. В тех случаях, когда используется гидрид металла или металлоорганическое соединение, как правило, можно устанавливать температуру в пределах от –78 °С до 60 °С. Молярная концентрация исходных материалов может устанавливаться произвольно. Однако реакционноспособное производное формулы R1' – hal целесообразно использовать, как правило, в концентрации от 1 до 5 моль на 1 моль соединения формулы (I-а). 15 34481 Для выделения и очистки целевого продукта могут быть применены способы, обычные при проведении органического синтеза, например, перек ристаллизация, всевозможная хроматография с использованием силикагеля и дистилляция. Формула реакции (3) где R1,R2,R3,R4,R5,R9,X,Y1,Y2 и А - как определено выше. Формула реакции (3) иллюстрирует способ, в котором производное 5-/w-карбоксиалкиленокси/бензиламина или 5-/w-алкоксикарбонилалкиленокси/бензиламина формулы (Y) совместно с соединением амина формулы (YI) вводятся в реакцию конденсации посредством дегидратации или удаления спирта с целью получения соответствующего производного амида формулы (I-c). Для проведения реакции конденсации при R9 - атом водорода могут быть широко использованы способы конденсации, традиционно применяемые при синтезе пептидов. Так, например, могут быть использованы хлорангидридный или смешанные ангидридные методы, а также способы конденсации, в которых применяются такие конденсирующие агенты, как дициклогексилкарбодиимид, карбонилдиимидазол или N-гидроксисукцинимид, а наиболее приемлемый метод конденсации может быть выбран с учетом химической активности амина формулы (YI). Условия реакции могут быть аналогичны обычно применяемым в таких случаях. В том случае, когда реакция проводится с амином, обладающим богатой нуклеофильной природой, среди аминов формулы (YI), реакция конденсации может быть проведена даже в присутствии сложного эфира, где R9 - алкильная группа. В этом случае в качестве растворителя может быть использован любой растворитель без какихлибо конкретных ограничений, лишь бы данный растворитель был инертным в отношении реакции. Во многих случаях реакция может быть проведена и при отсутствии растворителя. Температура реакции может быть установлена в пределах от комнатной температуры до 200 °С, однако общепринятым считается проведение реакции такого типа при температуре от 50 до 150°С. Формула реакции (4) 16 34481 где R1, R2, R3, R4, R5, X,Y1, Y2, A, B и hal - как определено выше. Формула реакции (4) иллюстрирует способ получения соединения формулы (I), предложенного в соответствии с настоящим изобретением, путем введения в реакцию соединения формулы (YII) с галогенопроизводным формулы (YIII). ническое основание, например, триэтиламин или три-n-пропиламин. В качестве растворителя для этой реакции может быть использован кетоновый растворитель (например, ацетон, метилэтилкетон ИЛИ диэтилкетон), растворитель на амидной основе (например, формамид, N,N-диметилформамид или N,N-диметилацетамид), растворитель на спиртовой основе (например, метанол или этанол), вода или смесь на основе указанных растворителей. Температура реакции устанавливается, как правило, в диапазоне от 0°С до температуры кипения применяемого растворителя. Для проведения этой реакции, как правило, может быть использовано неорганическое основание, например, карбонат калия, карбонат натрия, карбонат лития, гидрокарбонат натрия, гидрокарбонат калия или гидроксид лития, или орга Формула реакции (5) где R1, R2, R3, R4, R5, R10, X, Y1, Y2, А и hal - как определено выше, а R7 – атом водорода или алкильная группа C1-4. Формула реакции (5) иллюстрирует способ получения производного амина формулы (I-d) как соединения, предложенного в соответствии с настоящим изобретением, путем введения в реакцию соединения формулы (IX), получаемого способом в соответствии с формулой реакции (4), с соединением амина формулы (YI). Данная реакция может быть проведена способом, аналогичным описанному в связи с формулой реакции (4). Формула реакции (6) 17 34481 где R2' – алкильная группа С1-4, a R1, R2, R3, R4, R5, X, Y1, Y2, A, B и hal – как определено выше. Формула реакции (6) иллюстрирует способ получения соединения, где R2 – алкильная группа С1-4, среди соединений, предложенных в соответствии с настоящим изобретением, путем введения в реакцию соединения формулы (I-е), которое является соединением формулы (I), предложенным в соответствии с настоящим изобретением, где R2 - атом водорода, с алкилгалогенидом формулы R2 –hal в присутствии основания. Для получения хороших результатов в качестве органического растворителя может быть использован растворитель на амидной основе, например, диметилформамид, растворитель на основе простых эфиров, например, тетрагидрофуран или диэтиловый эфир, или апротонный органический растворитель, например, n-гексан, бензол или толуол, а в качестве основания может быть использован гидрид металла, например, гидрид натрия, n-бутиллитий, диизопропиламид лития или амид натрия. При реакции в присутствии основания температура реакции может быть установлена в пределах от -78 °С до 10 °С, а в присутствии алкилгидрида - в пределах от -15 °С до 70 °С. Формула реакции (7) где R1,R2,R3,R4,R5,X,Y1,Y2,А,B и hal - как определено выше. Формула реакции (7) иллюстрирует способ получения соединения формулы (I), предложенного в соответствии с настоящим изобретением, путем введения в реакцию 3/2Н/-пиридазинона формулы (XI), имеющего группу -NНR2 в положении 5, с производным бензилгалогенида формулы (ХІІ) в присутствии основания. Условия реакции могут быть аналогичными описанным в связи с формулой реакции (6). Способ введения 3/2Н/-пиридазинонов формулы (I) или их приемлемых с фармацевтической точки зрения солей, предложенных в соответствии с настоящим изобретением, может представлять собой неоральный способ введения и использованием составов для инъекций (подкожных, внутривенных, внутримышечных или внутрибрюшинных), мазей, суппозиториев или аэрозолей или оральный способ введения в виде таблеток, капсул, гранул, пилюль, сиропов, растворов, эмульсий или суспензий. ветствии с настоящим изобретением, могут быть введены и другие соединения, обладающие фармакологической активностью. Соединение, предложенное в соответствии с настоящим изобретением, может быть приготовлено в форме, удобной для введения в организм, с применением известных способов, общепринятых в технологии приготовления фармацевтических составов. Более конкретно, таблетки, капсулы, гранулы или пилюли для орального введения могут быть приготовлены с использованием наполнителя, например, сахара, лактозы, глюкозы, крахмала ИЛИ маннита; связующего, например, сиропа, гуммиарабика, желатина, сорбитола, траганта, метилцеллюлозы или поливинилпирролидона; расщепителя, например, крахмала, карбоксилметилцеллюлозы или ее кальциевой соли, кристаллического целлюлозного порошка или полиэтиленгликоля; глянцеобразующего агента, например, талька, магния или стеарата кальция или кремнезема; или смазывающего вещества, например, лаурата натрия или глицерина. Инъекционные составы, растворы, эмульсии, суспензии, сиропы или аэрозоли могут быть приготовлены с использованием растворителя для активного ингредиента, например, воды, этилового спирта, изопропилового спирта, пропиленгликоля, 1,3-бутиленгликоля или полиэтиленгликоля; поверхностно-активного вещества, например, сорбитанового эфира жирной кислоты, полиоксиэтиленсорбитанового эфира жирной кисло Упомянутые выше фармакологические составы содержат соединение, предложенное в соответствии с настоящим изобретением, массовая доля которого составляет примерно от 0,1 до 99,5 %, предпочтительно от 0,5 до 95 %, от общей массы состава. В соединение, предложенное в соответствии с настоящим изобретением, или в состав, содержащий соединение, предложенное в соот 18 34481 ты, полиоксиэтиленового эфира жирной кислоты, полиоксиэтиленового эфира гидрогенизированного касторового масла или лецитина; суспендирующего агента, например, натриевой соли карбоксилметилцеллюлозы, производного целлюлозы, например, метилцеллюлозы, или натурального каучука, например, траганта или гуммиарабика; или консерванта, например, эфира параоксибензойной кислоты, бензальконхлорида или соли сорбиновой кислоты. Аналогичным образом могут быть приготовлены и суппозитории с использованием, например, полиэтиленгликоля, ланолина или кокосового масла. Лучший вариант осуществления изобретения Примеры (справочные примеры, примеры приготовления, примеры рецептуры и примеры испытаний) Ниже следует более подробное описание настоящего изобретения со ссылкой на примеры (справочные примеры, примеры приготовления, примеры рецептуры и примеры испытаний). Однако следует понимать, что настоящее изобретение ни в коей мере не ограничено данными конкретными примерами. В контрольных примерах, примерах приготовления или таблице 2 сокращения "ЯМР" и "МC" означают "спектр ядерного магнитного резонанса" и "масс-спектр" соответственно. Измерение ЯМР производилось в хлороформе с тяжелым водородом, если не оговорено иначе. В МC-данных в таблице 2 приведены лишь основные пики или пики характерных фрагментов. Cправочный пример 1 N-Бензилоксикарбонил-3-гидрокси-4-метоксибензиламин тилового эфира с образованием 95,11 г вышеуказанного соединения в виде белых кристаллов. ЯMP d : 7,34(s,5H), 6,79(s,3H), 5,78(s,1H), 5,12(br. s,2H), 4,25(d,2H), 3,84(s,3H). MC(m/z): 287(M+), 196, 152, 137, 91(100%). Справочный пример 2 N-Бутилоксикарбонил-3-гидрокси-4-метоксибензиламин Смесь, содержащая 150 г изованилина, 91 г гидроксида натрия, 89 г гидроксиламиносульфата, 500 мл этанола и 1300 мл воды, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение 1 часа, а затем охлаждена до 40 °С. Затем к ней было добавлено 91 г гидроксида натрия, после чего при внутренней температуре 3050 °С к ней постепенно было добавлено 150 г сплава Ренея. После этого смесь была подвергнута перемешиванию в течение 1 часа. Нерастворимые вещества были отфильтрованы и промыты 150 мл этанола и 150 мл воды. Фильтрат и промывочные растворы были объединены с последующей нейтрализацией концентрированной хлористоводородной кислотой в условиях охлаждения до достижения рH 8. Затем к смеси был добавлен 1 л ацетонитрила, после чего к ней по каплям в течение 1 часа при комнатной температуре было добавлено 215 г ди-t-бутилдикарбоната. После этого смесь была оставлена на ночь при постоянном перемешивании. Затем органический слой был промыт насыщенным водным раствором хлорида натрия и просушен над безводным сульфатом натрия. Затем был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (этилацетат : бензол =1:5), в результате чего было получено 126 г указанного выше соединения в виде маслянистого вещества. ЯМРd : 6,54- 6,85(m,3H), 6,14 - 6, 47(bs,1Н), 4,92-5,34(m,1H), 4,09(d,2H), 3,25(s,3H), 1,44(s,9H). MC(m/z): 153(M+ -100), 137(100 %). Смесь, содержащая 150 г изованилина, 93,2 г гидроксида натрия, 99 г гидроксиламиносульфата, 600 мл этанола и 1500 мл воды, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение 30 мин, а затем охлаждена до 40°С. Затем в течение 30 мин к ней было добавлено 93,2 г гидроксида натрия и 180 г сплава Ренея. После этого смесь была подвергнута перемешиванию в течение 1 часа. Нерастворимые вещества были отфильтрованы и промыты 100 мл этанола и 200 мл воды. Фильтрат и промывочные растворы были объединены с добавлением 53,6 г гидроксида натрия. Затем в условиях охлаждения льдом к смеси по каплям было добавлено 186 г бензилоксикарбонилхлорида. Полученная смесь была подвергнута перемешиванию в течение 4 часов. К этому реакционному раствору была добавлена хлористоводородная кислота до достижения рН 12, после чего раствор был подвергнут экстракции этилацетатом. Органический слой был промыт водой и насыщенным водным раствором хлорида натрия и просушен над безводным сульфатом натрия. Затем был отогнан растворитель. Полученный остаток был выкристаллизован из диэ Справочный пример 3 N-Бензилоксикарбонил-3-этоксикарбонилметилокси-4-метоксибензиламин Смесь, содержащая 20 г N-бензилоксикарбонил-3-гидрокси-4-метоксибензиламина, 17,43 г этилбромацетата, 14,43 г карбоната калия и 200 мл 2-бутанона, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение ночи. Затем смесь была охлаждена до комнатной температуры. После этого были отфильтрованы неорганические вещества, а фильтрат был под 19 34481 вергнут дистилляции в условиях пониженного давления. Полученный остаток был экстрагирован хлороформом, а органический слой был промыт водой и насыщенным водным раствором хлорида натрия, а затем просушен над безводным сульфатом натрия. Затем был отогнан растворитель. Полученный остаток был выкристаллизован из смеси диэтиловый эфир–n-гексан, в результате чего было получено 17,83 г указанного выше соединения в виде белых кристаллов. ЯМР d : 7,33(s,5H), 6,85(s,3H), 5,12(s,2H), 4,63(s,2H), 4,26(d,2H), 4,25(q,2H), 3,84(s,3H), 1,26(t,3H). MC(m/z): 373(M+), 282, 239(100 %), 210, 164, 136, 91. Аналогичным образом были приготовлены следующие соединения. N-Бензилоксикарбонил-3-этоксикарбонилпропокси-4- метоксибензиламин ЯМР d : 7,25 - 7,55(m,5H), 6,72 - 7,06(m,3Н), 5,14(s,2H), 3,71 - 4,52(m,10H), 1,90 - 2,80(m,4H), 1,24(t,3H). N-Бензилоксикарбонил-3-этоксикарбонилпентилокси-4-метоксибензиламин Справочный пример 4 N-Бензилоксикарбонил-3-карбоксиметилокси-4-метоксибензиламин Смесь, содержащая 2 г N-бензилоксикарбонил-3-гидрокси-4-метоксибензиламина, 20 мл диметилформамида, 1,4 г карбоната калия и 1,4 г эпибромогидрина, была подвергнута перемешиванию при 60 °С в течение ночи. После отгонки растворителя в условиях пониженного давления реакционная смесь была подвергнута экстракции этилацетатом. Полученный органический слой был последовательно промыт водным раствором карбоната калия и насыщенным водным раствором хлорида натрия, после чего просушен над безводным сульфатом натрия. Затем был отогнан растворитель, в результате чего было получено 2,6 г указанного выше соединения в виде маслянистого вещества. ЯМР d : 7,32(s,5H), 6,81(s,3H), 5,0-5,5(m,3H), 3,9-4,6(m,7H), 3,8(s,3H). MC(m/z): 343(M+), 252,208,19(100 %). Справочный пример 6 N-Бензилоксикарбонил-3-/4-метилпиперазин-1-ил/-карбонилметокси-4-метоксибензиламин Смесь, содержащая 5 г N-бензилоксикарбонил-3-карбоксиметилокси-4-метоксибензиламина, 1,67 г триэтиламина и 40 мл тетрагидрофурана, была подвергнута охлаждению льдом, после чего к ней по каплям было добавлено 1,79 г этилхлорформиата, растворенного в 10 мл тетрагидрофурана. Затем смесь была подвергнута перемешиванию в течение 2 часов. После этого к реакционному раствору было добавлено 1,65 г метилпиперазина, растворенного в 10 мл тетрагидрофурана, после чего смесь была подвергнута перемешиванию при комнатной температуре в течение 4,5 часов. Остаток был отфильтрован, а фильтрат подвергнут дистилляции в условиях пониженного давления. К полученному остатку была добавлена вода, после чего смесь была подвергнута экстракции хлороформом. Раствор экстракта был промыт водой и насыщенным водным раствором хлорида натрия и просушен над безводным сульфатом натрия. Затем был отогнан растворитель. Полученный остаток был выкристаллизован из смеси этилацетат – диэтиловый эфир-nгексан, в результате чего было получено 3,53 г указанного выше соединения в виде белых кристаллов. ЯМР d: 7,25(s,5H), 6,78(s,3H), 5,03(s,3H), 4,62(s,2H), 4,23(d,2H), 3,78(s,3H), 3,40 - 3,72(m, 4Н), 2,11-2,60(m, 7H). MC(m/z): 427(M+), 292, 235, 141, 91(100 %). Аналогичным образом были приготовлены следующие соединения. N-Бензилоксикарбонил-3-/4-(3-пиридилметил)-пиперазин-1-ил/карбонилметокси-4-метоксибензиламин MC(m/z): 504(M+), 92(100 %). N-Бензилоксикарбонил-3-/4-бензилпиперазин-1-ил/-карбонилметокси-4-метоксибензиламин ЯMP d : 7,15 - 7,43(m,10H), 6,7 - 6,92(m,3Н), 4,85 - 5,24(m,3H), 4,62(s,2Н), 4,22(d,2H), 3,4 3,96(m,9H), 2,25 - 2,7(m, 4Н). Смесь, содержащая 23,56 г N-бензилоксикарбонил-3-этоксикарбонилметилокси-4-метоксибензиламина, 7,29 г гидроксида натрия, 300 мл метанола и 30 мл воды, была подвергнута перемешиванию при 60oC в течение 1 часа. Реакционный раствор был затем нейтрализован путем добавления к нему хлористоводородной кислоты, а растворитель был отогнан в условиях пониженного давления. К полученному остатку была добавлена разбавленная хлористоводородная кислота, после чего смесь была подвергнута экстракции хлороформом. Слой экстракта был промыт водой и насыщенным водным раствором хлорида натрия, а затем просушен над безводным сульфатом натрия. После этого был отогнан растворитель. Полученный остаток был выкристаллизован из смеси диэтиловый эфир–n-гексан, в результате чего было получено 21,55 г указанного выше соединения в виде белых кристаллов. ЯМР d: 7,34(s,5H), 6,84(s,3H), 5,13(s,3H), 4,62( s, 2H), 4,25(d,2H), 3,83(s,3H). MC(m/z): 345(M+), 254, 210(100 %), 91. Аналогичным образом были приготовлены следующие соединения. N-Бензилоксикарбонил-3-карбоксипропилокси-4-метоксибензиламин N-Бензилоксикарбонил-3-карбоксипентилокси-4-метоксибензиламин Справочный пример 5 N-Бензилоксикарбонил-3-/2,3-эпоксипропилокси/-4-метоксибензиламин 20 34481 N-Бензилоксикарбонил-3-/4-(4-фторбензил)пиперазин-1-ил/карбонилметокси-4-метоксибензиламин ЯМР d: 6,60-7,50(m,12H), 5,0 - 5,5(m, 3H), 4,62(s,2H), 4,22(d,2H), 3,22-3,95(m,9H), 2,2 2,7(m,4H). N-Бензилоксикарбонил-3-/4-(3-пиридилметил)-пиперазин-1-ил/-карбонилпропокси-4-метоксибензиламин MC(m/z): 532(M+), 92(100 %). N-Бензилоксикарбонил-3-/4-бензилпиперазин-1-ил/-карбонилпропокси-4-метоксибензиламин ЯМР d : 7,0 - 7,40(m,10H), 6,60 - 6,90(m,3H), 5,50-5,51(m,3H), 3,22-4,37(m,13H), 2,0 - 2,68(m,8H). N-Бензилоксикарбонил-3-/4-бензилпиперазин-1-ил/-карбонилпентилокси-4-метоксибензиламин ЯМР d : 7,0-7,35(m,10H), 6,60 - 6,80(m, 3H), 5,0 - 5,50(m,3H), 3,20-4,32(m,13Н), 1,1 2,48(m,12Н). Справочный пример 7 N-Бензилоксикарбонил-3-/[4-(4-фторбензил)пиперазин-1-ил]b-гидроксипропилокси/-4-метоксибензиламин 3-/4-Метилпиперазин-1-ил/-карбонилметокси-4-метоксибензиламин Смесь, содержащая 3,26 г N-бензилоксикарбонил-3-/4-метилпиперазин-1-ил/-карбонилметокси-4-метоксибензиламина, 0,5 г 5 %-ной палладиевой черни и 70 мл этанола, была подвергнута перемешиванию при 60 °С в течение 6 часов в атмосфере водорода, а затем при комнатной температуре в течение ночи. Палладиевая чернь была затем отфильтрована, а фильтрат был затем отогнан в условиях пониженного давления, в результате чего было получено 2,45 г указанного выше соединения в виде масла, слегка окрашенного в коричневый цвет. ЯМР d: 6,88(s,3H), 4,74(s,2H), 3,50 - 4,10(m, 9H), 2,29-2,58(m,7H), 1,65(s,2H). MC(m/z): 293(М+), 152, 299, 70(100%). Аналогичным образом были приготовлены следующие соединения. 3-/4-(3-Пиридилметил)-пиперазин-1-ил/карбонилметокси-4-метоксибензиламин МC(m/z): 370(M+), 92(100 %). 3-/4-Бензилпиперазин-1-ил/-карбонилметокси-4-метоксибензиламин MC(m/z): 369 (М+), 91(100 %). 3-/4-(4-Фторбензил)-пиперазин-1-ил/карбонилметокси-4-метоксибензиламин MC(m/z): 387 (М+), 109(100 %). 3-/4-(3-Пиридилметил)-пиперазин-1-ил/карбонилпропокси-4-метоксибензиламин МС(m/z): 398 (M+), 92(100 %). 3-/4-Метилпиперазин-1-ил/-карбонилпропокси-4-метоксибензиламин MC(m/z): 321(М+), 99(100 %). 3-/4-Бензилпиперазин-1-ил/-карбонилпропокси-4-метоксибензиламин МС(m/z): 397(М+), 91(100 %). 3-/4-(4-Фторбензил)-пиперазин-1-ил)-1-оксо2-метилэтилокси/-4-метоксибензиламин МС(m/z): 401(M+), 109(100 %). 3-/4-Бензилпиперазин-1-ил/-карбонилпентилокси-4-метоксибензиламин МС(m/z): 425(M+), 91(100 %). Пример приготовления 1 4-Хлор-5-/3-(4-метилпиперазин-1-ил)-карбонилметокси-4-метоксибензиламино/-3/2H/-пиридазинон Смесь, содержащая 2,4 г N-бензилоксикарбонил-3-/2,3-эпоксипропилокси/-4-метоксибензиламина, 30 мл этанола и 1,4 г 4-фторбензилпиперазина, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение ночи. Затем смесь была охлаждена до комнатной температуры, после чего реакционный раствор был подвергнут концентрированию в условиях пониженного давления с последующей экстракцией хлороформом. Органический слой был промыт водным раствором карбоната калия и просушен над безводным сульфатом натрия. Затем в условиях пониженного давления был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (этилацетат : метанол = 19 : 1), в результате чего было получено 2,6 г указанного выше соединения. ЯМР d : 6,75 - 7,42(m,12Н), 5,0 - 5, 5(m, 3Н), 4,26(d,2H), 3,82-4,10(m,2H), 3,77(s,3H), 3,20 3,60(m,3Н), 2,20-2,85(m,10Н). MC(m/z): 537(M+), 207(100 %), 109. Аналогичным образом были приготовлены следующие соединения. N-Бензилоксикарбонил-3-/[4-(2-хинолилметил)-пиперазин-1-ил]-b-гидроксипропилокси/-4-метоксибензиламин ЯМР d: 7,03-8,12(m,11Н), 6,60 - 6,87(m,3H), 5,30-5,70(m,1H), 5,05(s,2H), 3,22-4,37(m,11H), 2,222,80(m,10H). N-Бензилоксикарбонил-3-/[4-(4-аминобензил)-пиперазин-1-ил] -b-гидроксипропилокси/-4-метоксибензиламин Смесь, содержащая 1,16 г 3-/4-метилпиперазин-1-ил/-карбонилметокси-4-метоксибензиламина, 0,5 г 4,5-дихлор-3/2Н/-пиридазинона, 0,46 г триэтиламина, 10 мл этанола и 10 мл воды, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение ночи. Затем в условиях пониженного давления был отогнан растворитель, а к остатку был добавлен водный раствор карбо ЯMР d: 6,45-7,41(m,12H), 5,40-6,78(m,1H), 5,04(s,2H), 3,50-4,38(m,11H), 3,30(s,2H), 2,10 2,80(m,8H). Справочный пример 8 21 34481 ната калия. Полученная смесь была подвергнута экстракции хлороформом. Затем раствор экстракта был промыт водой и насыщенным водным раствором хлорида натрия и просушен над безводным сульфатом натрия. После этого был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле и затем выкристаллизован из смеси хлороформ – диэтиловый эфир, в результате чего было получено 0,61 г указанного выше соединения в виде белых кристаллов. ЯМР d: 12,66(br. s,1H), 7,44(s,1H), 6,78(s,3H), 5,43(t,1H), 4,68(s,2H), 4,39(d,2H), 3,77(s,3H), 3,30 3,75(m,4H), 2,0 - 2,60(m,7H). MC(m/z): 421 (M+), 386, 140, 99, 70 (100 %). Справочный пример 9 4-Хлор-5-/3-карбоксиметилокси-4-метоксибензиламино/-3/2Н/-пиридазинон лученному остатку была добавлена вода, после чего смесь была подвергнута экстракции хлороформом. Раствор экстракта был промыт водой и насыщенным водным раствором хлорида натрия и просушен над безводным сульфатом натрия. Затем был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (элюент: хлороформ : метанол =9:1), в результате чего было получено 129 мг указанного выше соединения в виде твердого вещества белого цвета. ЯМР d : 8,35-8,58(m,2Н), 7,81 - 8,33(m,1H), 7,72(s,1H), 7,45-7,60(m,2H), 6,88(s,3H), 6,40 6,80(m,1H), 4,31-4,62(m,6Н), 3,75(s,3H). MC(m/z): 429(M+), 394, 298, 137, 121, 107, 92(100 %). Справочный пример 10 N-Бeнзилoкcикapбoнил-3-/3-xлopпpoпoкcи/4-мeтoкcибeнзиламин Смесь, содержащая 0,3 г 4-хлор-5-/3-(4-метилпиперазин-1-ил)-карбонилметокси-4-метоксибензиламино/-3/2Н/-пиридазинона, 2,0 г гидроксида калия, 10 мл этанола и 2 мл воды, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение ночи. Затем реакционный раствор был подвергнут нейтрализации водным раствором хлористоводородной кислоты. После этого в условиях пониженного давления был отогнан растворитель. К полученному остатку была добавлена вода, после чего смесь была подвергнута экстракции хлороформом. Раствор экстракта был промыт водой и насыщенным водным раствором хлорида натрия и затем просушен над безводным сульфатом натрия. Затем был отогнан растворитель, в результате чего было получено 212 мг указанного выше соединения в виде твердого вещества белого цвета. МC(m/z): 281(М+-CHCO2H), 246, 209, 159, 145(100 %), 116. Пример приготовления 2 4-Хлор-5-/3-(3-пиридилметиламинокарбонилметокси)-4-метоксибензиламино/-3/2Н/-пиридазинон Смесь, содержащая 20 г N-бензилоксикарбонил-3-гидрокси-4-метоксибензиламина, 14,43 г карбоната калия, 16,44 г бромхлорпропана и 200 мл 2-бутанона, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение 16 часов. Затем смесь была охлаждена до комнатной температуры. После этого были отфильтрованы неорганические вещества, а фильтрат был подвергнут дистилляции в условиях пониженного давления. Полученный остаток был подвергнут экстракции хлороформом, после чего органический слой был промыт водой и насыщенным водным раствором хлорида натрия и просушен над безводным сульфатом натрия. Полученный остаток был выкристаллизован из смеси диэтиловый эфир–n-гексан, в результате чего было получено 23,19 г указанного выше соединения в виде белых кристаллов. ЯMР d: 7,21(s,5H), 6,71(s,3H), 5,04(s,3H), 4,20(d,2H), 4,02(t,2H), 3,75(s,3H), 3,67(t,2Н), 1,942,47(m, 2Н). MC(m/z): 363(M+), 316, 273(100 %), 228, 152, 137, 125, 91. Аналогичным образом были приготовлены следующие соединения. N-Бензилоксикарбонил-3-/2-хлорэтокси/-4метоксибензиламин N-Бензилоксикарбонил-3-/2-диэтиламиноэтокси/-4-метоксибензиламин Cправочный пример 11 N-Бензилоксикарбонил-3-/3-(4-формилпиперазин-1-ил)пропокси/-4-метоксибензиламин Смесь, содержащая 200 мг 4-хлор-5-/3-карбоксиметилокси-4-метоксибензиламино/-3/2Н/-пиридазинона, 65 мг триэтиламина и 10 мл N,N-диметилформамида, была подвергнута охлаждению льдом, после чего к ней было добавлено 88 мг изобутилхлорформиата. При этой температуре смесь была подвергнута перемешиванию в течение 1 часа, после чего к ней было добавлено 140 мг 3-пиколиламина. Полученная смесь была оставлена на ночь в условиях постоянного перемешивания. Затем в условиях пониженного давления был отогнан растворитель. Затем к по Смесь, содержащая 23,1 г N-бензилоксикарбонил-3-/3-хлорпропокси/-4-метоксибензиламина, 8,7 г N-формилпиперазина, 13,16 г карбоната калия, 0,95 г иодида натрия и 300 мл N,N-диметилформамида, была подвергнута перемешиванию при 80°С в течение 16 часов. Затем смесь была 22 34481 охлаждена до комнатной температуры. После этого были отфильтрованы неорганические вещества, а фильтрат был подвергнут дистилляции в условиях пониженного давления. Полученный остаток был подвергнут экстракции хлороформом, а органический слой был промыт водой и насыщенным водным раствором хлорида натрия и просушен над безводным сульфатом натрия. Затем был отогнан растворитель, в результате чего было получено 30,67 г указанного выше соединения в виде масла, слегка окрашенного в коричневый цвет. ЯMР d: 7,97(s,1H), 7,32(s,5H), 6,81(s,3H), 5,36(brt,1H), 5,11(s,2H), 4,26(d,2H), 4,02(t,2H), 3,81(s,3H), 3,12-3,66(m,4H), 1,78-2,78 (m,8H). MC(m/z): 441(M+), 383, 306, 155(100 %), 128, 91. Аналогичным образом были приготовлены следующие содинения. N-Бензилоксикарбонил-3-/3-диэтиламинопропокси/-4-метоксибензиламин N-Бензилоксикарбонил-3-/2-(4-бензилпиперазин-1-ил) -этокси/-4-метоксибензиламин N-Бензилоксикарбонил-3-/2-[4-(4-хлорбензил)-пиперазин-1-ил]-этокси/-4-метоксибензиламин N-Бензилоксикарбонил-3-/2-[4-(4-фторбензил)-пиперазин-1-ил]-этокси/-4-метоксибензиламин N-Бензилоксикарбонил-3-/3-(4-бензилпиперазин-1-ил)-пропокси/-4-метоксибензиламин N-Бензилоксикарбонил-3-/3-(4-метилпиперазин-1-ил) - пропокси/-4-метоксибензиламин Cправочный пример 12 3-/3-(4-Формилпиперазин-1-ил)-пропокси-4метоксибензиламин 3-/2-[4-(4-Фторбензил)-пиперазин-1-ил]-этокси/-4-метоксибензиламин 3-/3-(4-Бензилпиперазин-1-ил)-пропокси/-4метоксибензиламин 3-/3-(4-Метилпиперазин-1-ил)-пропокси/-4метоксибензиламин Пример приготовления 3 4-Хлор-5-/3-[3-(4-формилпиперазин-1-ил)-пропокси]-4-метоксибензиламино/-3/2H-пиридазинон (Соединение № 50) Смесь, содержащая 11,58 г 3-/3-( 4-формилпиперазин-1-ил)-пропокси/-4-метоксибензиламина, 5,0 г 4,5-дихлор-3/2Н/-пиридазинона, 4,6 г триэтиламина, 50 мл n-пропанола и 50 мл воды, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение 14 часов. Затем в условиях пониженного давления был отогнан растворитель, а к полученному остатку был добавлен водный раствор карбоната калия, после чего смесь была подвергнута экстракции хлороформом. Органический слой был промыт водой и насыщенным водным раствором хлорида натрия и затем просушен над безводным сульфатом натрия. После этого был отогнан растворитель, а остаток был подвергнут очистке методом колоночной хроматографии на силикагеле, в результате чего было получено 6,21 г указанного выше соединения в виде твердого вещества, имеющего слегка желто-белую окраску. ЯМР d: 12,49(br.s,1H), 8,06(s,1H), 7,65(s,1H), 6,88(s,3H), 5,37(t,1H), 4,51(d,2H), 4,08(t,2H), 3,87(s,3H), 3,19- 3,74(m,4H), 2,30- 2,84m,6Н), 1,76 2,30(m, 2Н). Пример приготовления 4 4-Хлор-5-/3-[3-(4-этилпиперазин-1-ил)-пропокси]-4-метоксибензиламино/-3/2H/-пиридазинон Смесь, содержащая 30,4 г N-бензилоксикарбонил-3-/3-(4-формилпиперазин-1-ил)-пропокси/-4метоксибензиламина, 3,1 г 5 %-ной палладиевой черни и 300 мл этанола, была подвергнута перемешиванию при 60оС в течение 9 часов в атмосфере водорода. Затем палладиевая чернь была отфильтрована, после чего фильтрат был подвергнут дистилляции в условиях пониженного давления, в результате чего было получено 17,99 г указанного выше соединения в виде масла, слегка окрашенного в коричневый цвет. ЯМР d: 8,03(s,1H), 6,86(s,3H), 4,11(t,2H), 3,84(s,3H), 3,25-3,71 (m, 4H), 2,30 - 2,82(m, 4H), 1,82 - 2,30(m, 4H). MC(m/z): 307(M+), 292, 246, 171, 155, 125, 99(100 %). Аналогичным образом были приготовлены следующие соединения. 3-/2-Диэтиламиноэтокси/-4-метоксибензиламин 3-/3-Диэтиламинопропокси/-4-метоксибензиламин 3-/2-(4-Бензилпиперазин)-1-ил/-этокси-4-метоксибензилпиперазин 3-/2-[4-(4-Хлорбензил)-пиперазин-1-ил]-этокси/-4- метоксибензиламин Смесь, содержащая 1,0 г 4-хлор-5-/3-[3-( 4формилпиперазин-1-ил)-пропокси]-4-метоксибензиламано/-3/2Н/-пиридазинона, 0,62 г гидроксида калия, 7 мл этанола и 7 мл воды, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение 3,5 часов, после чего к ней было добавлено 0,32 г карбоната калия и 570 мг этилбромида. Полученная смесь была подвергнута перемешиванию при 60 °С в течение 4 часов. Затем в условиях пониженного давления был отогнан растворитель, а к полученному остатку была добавлена вода. Затем смесь была подвергнута экстракции хлороформом. Раствор экстракта был промыт водой и насыщенным водным раствором хлорида натрия и затем просушен над безводным сульфатом натрия. После этого был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле, в результате чего было получено 0,50 23 34481 вор был подвергнут дистилляции в условиях пониженного давления с последующей экстракцией хлороформом. Органический слой был промыт водным раствором карбоната калия и просушен над безводным сульфатом натрия. Затем в условиях пониженного давления был отогнан растворитель, в результате чего было получено 750 мг указанного выше соединения в виде маслянистого вещества. ЯМР d: 7,32-8,20(m,6Н), 4,01(s,2H), 3,20 3,90(m, 6H), 2,30-2,74(m,4Н). MC(m/z): 143 (M+-160). Аналогичным образом были приготовлены следующие соединения. 1-Хлорацетил-4-/4-хлорбензил/-пиперазин MC(m/z): 286(M+), 125(100 %). 1-Хлорацетил-4-/1-(4-фторбензил)-2-метилбензоимидазол/-пиперазин ЯMР d: 6,66-7,40(m,8H), 5,44(s,2H), 3,95(s,2H), 3,74(s,2H), 3,04-3,60(m,4H), 2,24 - 2,66(m,4H). 1-Хлорацетил-4-бензилпиперазин MC(m/z): 252(M+), 91(100 %). 1-Хлорацетил-4-бензилпиперидин MC(m/z): 251 (M+), 91(100 %). 1-Хлорацетил-4-/t-бутилоксикарбониламинобензил/-пиперазин MC(m/z): 368(M+), 150(100%). г указанного выше соединения в виде твердого вещества, слегка окрашенного в коричневый цвет. ЯМР d: 7,65(s,1H), 6,89(s,3H), 5,41(разрушено, 1H), 4,50(d,2H), 4,08(t,2H), 3,87(s,3H), 1,73 3,10(m,14H), 1,08(t,3H). MC(m/z): 435(M+), 365, 343, 206, 127(100 %), 99. Аналогичным образом было приготовлено следующее соединение. 4-Хлор-5-/3- [3-{4-(4-фторбензил)-пиперазин1-ил}-пропокси]-4-метоксибензиламино/-3/2Н/-пиридазинон MC(m/z): 515(M+), 109(100%). Пример приготовления 5 2-Этил-4-хлор-5-/3-[2-{4-(4-фторбензил)-пиперазин-1-ил}-этокси]-4-метоксибензиламино/3/2Н/-пиридазинон Смесь, содержащая 500 мг 4-хлор-5-/3-[2-{4(4-фторбензил )-пиперазин-1-ил]-этокси] -4-метоксибензиламино/-3/2Н/-пиридазинона, 130 мг этилбромида, 190 мг карбоната калия и 10 мл 2-бутанона, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение 5 часов. Затем были отфильтрованы неорганические вещества, после чего в условиях пониженного давления был отогнан растворитель. К полученному остатку была добавлена вода, и смесь была подвергнута затем экстракции хлороформом. Раствор экстракта был промыт водой и насыщенным водным раствором хлорида натрия и затем просушен над безводным сульфатом натрия. После этого был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (элюент: хлороформ : этанол = 19 : 1), в результате чего было получено 429 мг указанного выше соединения в виде бесцветного прозрачного клейкого вещества. ЯМР d: 7,47(s,1Н), 7,00-7,31(m,4H), 6,88(s,3H), 5,20(t,1H), 4,46(d,2H), 4,14(t,2H), 4,12(q,2H), 3,85(s,3H), 3,47(s,2H), 2,73(t,2H), 2,21 - 3,05(m,10Н), 1,32(t,3H). MC(m/z): 574(M+), 493, 273, 221, 192(100 %), 164, 111, 84. Справочный пример 13 1-Хлорацетил-4-/2-хинолилметил/-пиперазин Справочный пример 14 N-t-Бутилоксикарбонил-3-/4-(2-хинолилметилпиперазин)-1-ил/карбонилметокси-4-метоксибензиламин Смесь, содержащая 660 мг t-бутилоксикарбонил-3-гидрокси-4-метоксибензиламина, 10 мл диметилформамида, 750 мг 1-хлорацетил-4-/2-хинолилметил/-пиперазина и 510 мг карбоната калия, была подвергнута нагреву до 80оС и при этой температуре оставлена на ночь при постоянном перемешивании. Нерастворимые включения были отфильтрованы, после чего реакционный раствор был подвергнут дистилляции в условиях пониженного давления с последующей экстракцией хлороформом. Раствор экстракта был промыт водным раствором карбоната калия, после чего подвергнут очистке методом колоночной хроматографии на силикагеле (этилацетат : метанол = 19 : 1), в результате чего было получено 1,2 г указанного выше соединения в виде маслянистого вещества. ЯМР d: 7,32-8,03(m,6Н), 6,63-6,93(m,3H), 5,15-5,50(m,1H), 4,64(s,2H), 4,16(d,2H), 3,38 3,93(m,9H), 2,30-2,73(m,4H), 1,43(s,9H). MC(m/z): 520(М+), 144(100 %). Аналогичным образом были приготовлены следующие соединения. N-t-Бутилоксикарбонил-3-/4-(4-хлорбензил)пиперазин-1-ил/-карбонилметокси-4-метоксибензиламин MC(m/z): 503(М+), 125(100 %). N-t-Бутилоксикарбонил-3-/4-[1-(4-фторбензил)-2-метилбензоимидазол]-пиперазин-1-ил/-карбонилметокси-4-метоксибензиламин Раствор, содержащий 600 мг N-хинолилметилпиперазина и 20 мл сухого тетрагидрофурана, был охлажден до -60оС, после чего к нему по каплям в течение 10 мин был добавлен смешанный раствор, содержащий 330 мг ацетилхлорида и 5 мл сухого тетратидрофурана. Полученная смесь была подвергнута перемешиванию при –60 °С в течение 1 часа, после чего к ней было добавлено 10 мл воды. Затем смесь была подвергнута перемешиванию при комнатной температуре в течение 20 минут. Полученный реакционный раст 24 34481 ЯМР d: 6,10-7,35(m,11H), 5,45(s,2H), 4,80 5,17(m,1H), 4,10(s,2H), 4,15(d,2H), 3,76(s,ЗН), 3,70 (s,12H), 3,26-3,65(m,4H), 2,27- 2,65(m,4H). N-t-Бутилоксикарбонил-3-/4-бензилпиперидин-1-ил/-карбонилметокси-4-метоксибензиламин MC(m/z): 468(M+), 91(100 %). N-t-Бyтилоксикарбонил-3-/4-t-бутилоксикарбониламинобензилпиперазин-1-ил/-карбонилметокси-4-метоксибензиламин MC(m/z): 585(М+), 150(100%). Справочный пример 15 3-/4-(2-Хинолилметил) -пиперазин-1-ил/-карбонилметокси-4-метоксибензиламин Смесь, содержащая 2,4 г 3-/4-( 2-хинолилметил)-пиперазин-1-ил/-карбонилметокси-4-метоксибензиламина, 1 г 4,5-дихлор-6-этокси-3/2Н/-пиридазинона, 580 мг триэтиламина, 10 мл пропанола и 1,0 мл воды, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение ночи. Затем в условиях пониженного давления был отогнан растворитель, а остаток был подвергнут экстракции хлороформом. Органический слой был промыт водным раствором карбоната калия и затем просушен над безводным сульфатом натрия. После этого был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (этилацетат : метанол = 6:1® хлороформ : метанол = 12 : 1) и затем выкристаллизован из диэтилового эфира, в результате чего было получено 1,5 г указанного выше соединения в виде белых кристаллов. ЯМР d: 7,40-8,28(m,6H), 6,72-7,05(m,3H), 4,62-5,40(m, 5Н), 3,48-4,50(m,11H), 2,32 2,70(m,4H), 1,31(t,3H). MC(m/z): 592 (М+), 143(100 %). Справочный пример 16 1-Формил-4-/4-аминобензил/-пиперазин Смесь, содержащая 1,3 г t-бутилоксикарбонил-3-/4-(2-хинолилметил)-пиперазин-1-ил/-карбонилметокси-4-метоксибензиламина, 14 мл хлороформа и 2,8 г трифторуксусной кислоты, была подвергнута перемешиванию при комнатной температуре в течение суток. К полученному реакционному раствору было добавлено 50 мл хлороформа и 50 мл 0,5 н. раствора хлористоводородной кислоты, после чего смесь была подвергнута обратной экстракции. Водный слой был доведен до рН 12 водным раствором гидроксида натрия и подвергнут экстракции хлороформом. Органический слой был промыт водным раствором карбоната калия и затем просушен над безводным сульфатом натрия. После этого в условиях пониженного давления был отогнан растворитель, в результате чего было получено 850 мг указанного выше соединения в виде маслянистого вещества. ЯМР d: 7,39 - 8,20(m,6H), 6,72 - 7,0(m,3H), 4,7(s,2H), 3,40-4,00(m,11H), 2,32-2,70(m, 4H), 2,05(br. s,2H). MC(m/z): 420 (M+), 143(100 %). Аналогичным образом были приготовлены следующие соединения. 3-/4-(4-Хлорбензил)-пиперазин-1-ил/-карбонилметокси-4-метоксибензиламин MC(m/z): 403(M+), 125(100 %). 3-/3-[4-(4-Фторбензил)-пиперазин-1-ил]-2,2диметилпропокси/-4-метоксибензиламин МС(m/z): 429 (М+), 109(100 %). 3-/4-Бензилпиперазин-1-ил/-карбонилметокси-4-метоксибензиламин MC(m/z): 368 (М+),91(100 %). 3-/4-[1-(4-Фторбензил)-2-бензимидазолилметил]-пиперазин-1-ил/-карбонилметокси-4-метоксибензиламин MC(m/z): 517(M+), 109(100 %). Пример приготовления 6 4-Хлор-5-/3-[4-(2-хинолилметил)-пиперазин1-ил]-карбонилметокси-4-метоксибензиламино/-6этокси-3/2Н/-пиридазинон Смесь, содержащая 9 г 1-формил-4-/4-нитробензил/-пиперазина, 180 мл метанола и 14,6 г гексагидрата хлорида никеля, была охлаждена в ледяной бане, после чего к ней медленно было добавлено 4,6 г боргидрида натрия. Затем смесь была подвергнута перемешиванию при 0оС в течение 30 минут, а затем еще в течение 30 мин при комнатной температуре. Полученный реакционный раствор был подвергнут дистилляции в условиях пониженного давления, а остаток был растворен путем добавления к нему 200 мл 10 %-ной хлористоводородной кислоты и доведен до рН 10 28 %-ным водным раствором аммиака. Затем смесь была подвергнута экстракции этилацетатом. Раствор экстракта был промыт насыщенным водным раствором хлорида натрия и затем просушен над безводным сульфатом натрия. После этого в условиях пониженного давления был отогнан растворитель. Остаток был выкристаллизован из диэтилового эфира, в результате чего было получено 8,0 г указанного выше соединения в виде белых кристаллов. ЯМР d: 7,82(s,1H), 6,97(d,2H), 6,47(d,2H), 3,01-3,91(m,8Н), 2,11-2,48(m,4H). MC(m/z): 263(M+), 218(100 %). Справочный пример 17 1-Формил-4-/4-t-бутилоксикарбониламинобензил/-пиперазин Смесь, содержащая 4 г 1-формил-4-аминобензилпиперазина, 50 мл толуола и 4,8 г ди-t-бутилдикарбоната, была подвергнута дефлегмации в условиях нагрева в течение 5 часов. Получен 25 34481 ный реакционный раствор был подвергнут концентрации в условиях пониженного давления, а остаток очищен методом колоночной хроматографии на силикагеле (этилацетат : метанол = 9 : 1) и затем выкристаллизован из диэтилового эфира, в результате чего было получено 5,1 г указанного выше соединения в виде белых кристаллов. ЯМР d: 7,87(s,1Н), 6,97-7,42(m,5H), 3,15 3,65(m,6H), 2,15-2,57(m,4H), 1,45(s,9H). MC(m/z): 319 (M+), 106(100 %). Справочный пример 18 1-/4-t-Бутилоксикарбониламинобензил/-пиперазин цетат : метанол =9:1® хлороформ : метанол = 15 : :1) и затем выкристаллизован из диэтилового эфира, в результате чего было получено 1,6 г указанного выше соединения в виде белых кристаллов. ЯМР d: 6,55-7,15(m,7Н), 4,45-5,33(m,6H), 3,13-3,88(m,11H), 2,13 - 2,58(m,4H), 1,28(d,6H). MC(m/z): 465(M+-106), 430, 106(100 %). Пример приготовления 8 4-Хлор-5-/3-[4-(4-N-формилбензил)-пиперазин-1-ил]-карбонилметокси-4-метоксибензиламино/-6-изопропокси-3/2Н/-пиридазинон 4 г 1-формил-4-/-t-бутилоксикарбониламинобензил/-пиперазина было растворено в 50 мл метанола, после чего к полученному раствору был добавлен водный раствор, содержащий 1,5 г гидроксида натрия, растворенного в 10 мл воды. Затем полученная смесь была подвергнута нагреву до 60 °С и оставлена при этой температуре на 5 часов. Полученный реакционный раствор был подвергнут концентрации в условиях пониженного давления с последующей экстракцией хлороформом. Органический слой был промыт водным раствором карбоната калия и затем просушен над безводным сульфатом натрия. Затем в условиях пониженного давления был отогнан растворитель. Остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (хлороформ : метанол = 5 : 1) и затем выкристаллизован из диэтилового эфира, в результате чего было получено 3,2 г указанного выше соединения в виде белых кристаллов. ЯMP d: 7,0-7,7(m,5H), 3,38(s,2H), 2,60 3,12(m, 4H), 1,90-2,60(m,5H), 1,50(s,9H). MC(m/z): 291(M+), 206, 106(100 %). Пример приготовления 7 4-Хлор-5-/3-[4-(4-аминобензил)-пиперазин-1ил]-карбонилметокси-4-метоксибензиламино/-6-изопропокси-3/2Н/-пиридазинон 400 мг 4-хлор-5-/3-(4-аминобензил)-пиперазин-1-ил/-карбонилметокси-4-метоксибензиламино-6-изопропокси-3/2Н/-пиридазинона было растворено в 3 мл фенилформиата. Раствор был подвергнут перемешиванию в течение ночи при комнатной температуре. Затем полученный реакционный раствор был подвергнут дистилляции в условиях пониженного давления. После этого полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (хлороформ : метанол = 9 : 1) и затем выкристаллизован из диэтилового эфира, в результате чего было получено 380 мг указанного выше соединения в виде белых кристаллов. ЯМР d: 11,75(bг. s,1Н), 8,2-8,85(m,2Н), 6,75 7,62(m, 7Н), 4,58-5,30(m,6Н), 3,77(s,3H), 3,20 3,75(m,6H), 2,05-2,60(m,4H), 1,27(d,6H). MC(m/z): 464(M+-134), 137(100 %). Пример приготовления 9 4-Хлор-5-/3-[4-(4-N-ацетиламинобензил)-пиперазин-1-ил]-кapбoнилмeтoкcи-4-метoкcибeнзилaминo/-6-изoпpoпoкcи-3/2H/-пиридазинон 400 мг 4-хлор-5-/3-(4-аминобензил)-пиперазин-1-ил/-карбонилметокси-4-метоксибензиламино-6-изопропокси-3/2Н/-пиридазинона было растворено в 400 мл пиридина, и к полученному раствору было добавлено 220 мг уксусного ангидрида. Смесь была подвергнута перемешиванию при комнатной температуре в течение 2 часов. Растворитель был отогнан в условиях пониженного давления, а остаток подвергнут экстракции хлороформом. Органический слой был промыт водным раствором карбоната калия и просушен над безводным сульфатом натрия. Затем в условиях пониженного давления был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (хлороформ : метанол = 9 : 1) и затем выкристаллизован из диэтилового эфира, в результате чего было получено 340 мг указанного выше соединения в виде белых кристаллов. Смесь, содержащая 1,6 г 3-/4-(4-аминобензил)-пиперазин-1-ил/-карбонилметокси-4-метоксибензиламина, 770 мг 4,5-дихлор-6-изопропокси3/2Н/-пиридазинона, 460 мг триэтиламина и 20 мл метанола, была подвергнута дефлегмации в условиях нагрева и перемешивания в течение 2 суток. Затем в условиях пониженного давления был отогнан растворитель, а остаток был подвергнут экстракции хлороформом. Органический слой был промыт водным раствором карбоната калия и затем просушен над безводным сульфатом натрия. После этого был отогнан растворитель. Полученный остаток был подвергнут очистке методом колоночной хроматографии на силикагеле (этила 26 34481 ЯМР d: 11,84(br. s,1H), 8,24(br. s,1H), 6,63 7,52(m,8H), 4,52-5,30(m,6H), 3,30-3,92(m,9H), 2,0 2,62(m, 7Н), 1,25(d,6H). MC(m/z): 613(M+ +H), 466. Пример приготовления 10 Гидрохлорид 4-бром-5-/3-[2-{4-(4-хлорбензил)-пиперазин-1-ил}-этокси]-4-метоксибензиламино/-3/2Н/-пиридазинона /Соединение № 7/ Смесь, содержащая 163 мг 4-бром-5-/3-[2-{4(4-хлорбензил)-пиперазин-1-ил}-этокси]-4-метоксибензиламино/-3/2Н/-пиридазинона, 33 мг фумаровой кислоты и 4 мл хлороформа, была подвергнута перемешиванию при комнатной температуре в течение 3 часов. С целью кристаллизации в полученный реакционный раствор был введен диэтиловый эфир, в результате чего было получено 120 мг указанного выше соединения в виде белых кристаллов, имеющих температуру плавления от 178 до 185 °С. MC(m/z) : 562(М+–(СНСО2Н2), 482, 237, 223, 125(100 %), 91. Пример приготовления 12 Сульфат 4-бpом-5-/3-[2-{4-(4-хлорбензил) -пиперазин-1-ил}-этокси]-4-метоксибензиламино/3/2Н/-пиридазинона /Соединение № 9/ 5 мл хлороформа и 140 мг серной кислоты, была подвергнута перемешиванию при комнатной температуре в течение 3 часов. Полученный реакционный раствор был подвергнут дистилляции в условиях пониженного давления, а полученный остаток был выкристаллизован из смеси изопропиловый эфир - диэтиловый эфир, в результате чего было получено 800 мг указанного выше соединения в виде белых кристаллов, имеющих температуру плавления от 158 до 162°С. MC(m/z): 482(M+–Br–H2SO4), 238, 223(100%). 125. Соединения, приготовленные в соответствии с приведенными выше примерами приготовления, представлены в таблице 2. Структура этих соединений представлена в таблице 1 под соответствующими номерами. В крайней правой колонке таблицы 2 указан соответствующий номер примененного примера приготовления. Рецептурный пример 1 /Таблетки/ Соединение № 39 10 г Лактоза 20 г Крахмал 4г Крахмал для приготовления пастообразной массы 1г Стеарат магния 0,1 г Кальциевая соль карбоксиметилцеллюлозы 7г Всего 42,1 г Приведенные выше компоненты были смешаны обычным спoсобом и приготовлены в виде таблеток с сахарным покрытием, каждая из которых содержала 50 мг активного ингредиента. Pецептурный пример 2 /Капсулы/ Соединение № 43 10 г Лактоза 20 г Микрокристаллическая целлюлоза 10 г Стеарат магния 1г Всего 41 г Приведенные выше компоненты были смешаны обычным способом и помещены в желатиновые капсулы, в результате чего были получены капсулы, каждая из которых содержала 50 мг активного ингредиента. Pецептурный пример 3 /Мягкие капсулы/ Соединение № 7 10 г Кукурузное масло 35 г Всего 45 г Приведенные выше компоненты были смешаны и приготовлены обычным способом, в результате чего были получены мягкие капсулы. Рецептурный пример 4 (Мазь) Соединение № 25 1,0 г Оливковое масло 20 г Светлый вазелин 79 г Всего 100 г Приведенные выше компоненты были смешаны обычным способом с целью получения 1 %ной мази. Смесь, содержащая 700 мг 4-бpом-5-/3-[2-{4(4-хлорбензил)-пиперазин-1-ил}-этокси]-4-метоксибензиламино/-3/2Н/-пиридазинона, 5 мл метанола, Pецептурный пример 5 (Аэрозольная суспензия) (А) Соединение № 37 0,25 % Изопропилмиристат 0,10 % Этиловый спирт 26,40 % (Б) 60-40 %-ная смесь 1,2-дихлортетрафторэтана и 1-хлорпентафторэтана 73,25 % К смешанному раствору, содержащему 440 мг 4-бром-5-/3-[2-{4-(4-хлорбензил)-пиперазин-1ил}-этокси]-4-метокcибeнзилaмино/-3/2H/-пиpидaзинoнa и 5 мл хлороформа, был добавлен 10%ный раствор хлористоводородной кислоты в метаноле до достижения рН 2-3, после чего смесь была подвергнута перемешиванию при комнатной температуре в течение 2 часов. С целью кристаллизации полученного реакционного раствора в него был введен диэтиловый эфир, в результате чего было получено 465 мг указанного выше соединения в виде белых кристаллов, имеющих температуру плавления от 176 до 183 °С. MC(m/z): 562(М+–2HCl), 482, 238, 223(100 %), 203, 125, 91. Пример приготовления 11 Фумарат 4-бром-5-/3-[2-{4-(4-хлорбензил)пиперазин-1-ил}-этокси]-4-метоксибензиламино/3/2Н/-пиридазинона (Соединение № 8) × 27 34481 Приведенный выше состав (А) был смешан. Полученный в результате раствор был загружен в сосуд, оборудованный клапаном, через сопло которого был инжектирован диспергатор (Б) до достижения манометрического давления в сосуде порядка 2,46-2,81 мг/см2, в результате чего была получена аэрозольная суспензия. Примеры испытаний І. Бронхолитическое действие 1. Испытание in vitro Лекарственное средство: В 100 %-НОМ диметилсульфоксиде (DMSO, Wako Junyaku) был растворен образец лекарственного препарата для испытаний с последующим разбавлением до концентрации, пригодной для применения. В дистиллированной воде были растворены лейкотриен Д4, ультратонкий порошок (LTD4, Ultrafine) и изопротеренол (Isoproterenol, Sigma). В 100%-ном этаноле (EtOH, Komune Kagaku) был растворен индометацин (Indo , Sigma). В дистиллированной воде был растворен дигидрохлорид гистамина (His, Wako Junyaku), аминофиллин (АР, Sigma). Конечная объемная концентрация DMSO и EtOH в растворе не превышала 0,25 % и 0,1 % соответственно. Способ 1-1: Из предварительно обескровленной морской свинки массой 300-450 г был извлечена трахея. После удаления жира и соединительных тканей она была разделена на 2 - 3 спиральные полоски, каждая из которых имела ширину около 2 мм и содержала 4 отрезка тканей гладких мышц. Каждый из приготовленных таким образом образцов был подвешен в 8-миллилитровой ванне с модифицированным раствором Тироде, аэрированным газовой смесью 95 % О2 и 5 % СО2, при 37 °С и снабжен грузом массой 1 г. Релаксация мышцы регистрировалась самописцем (тип 3066, фирма Yokogawa Hokushin Electric) при помощи изотонического датчика (TD-112S, фирма Nihon Kohden). Состав модифицированного раствора Тироде был следующим (ммоль): NaCl 137, KCl 2,7, CaCl 2 1,8, MgCl 2 1,0, NaHCO3 20, NaH2PO4 0,32, глюкоза 11. концентрация, необходимая для достижения 50%ной релаксации (ЕС50, мкмоль). Результаты приведены в таблице 3-1. Способ 1-2: Применялся способ измерения, аналогичный способу 1-1. Образец был оставлен на 60-90 мин, после чего был подвергнут раздражению, приводящему к его релаксации, путем добавления изопротеренола (1 мкмоль). Затем образец был промыт, причем эта операция повторялась с интервалами 30-40 мин до тех пор, пока не была достигнута реакция постоянной релаксации. Затем с целью обеспечения релаксации образца был кумулятивно введен испытуемый лекарственный препарат. И, наконец, для достижения реакции максимальной релаксации был добавлен АР (1 ммоль). Результат выражался процентным отношением релаксации, достигаемой применением испытуемого лекарственного препарата, к релаксации, достигаемой применением АР, которая оценивалась как 100%-ная, после чего измерялась концентрация, необходимая для достижения 50%ной релаксации (EC50 мкмоль). Конечная объемная концентрация DMSO в растворе была установлена на уровне 0,2%. В качестве контрольного лекарственного средства использовался АР. Результаты приведены в таблице 3-2. 2. Испытание in vivo Действие при анафилактическом бронхостенозе, вызванном у пассивно сенсибилизированной морской свинки посредством эндогенно освобожденной МРСА За 1 – 2 дня до эксперимента была произведена пассивная сенсибилизация самцов морских свинок (350 - 450 г) путем внутривенной инъекции противоовальбуминной сыворотки кролика. Для измерения антиген-индуцированных анафилактических сужений просвета бронха, вызываемых посредством эндогенно освобожденной МРСА, использовался модифицированный метод Концетта и Росслера (Arch. Exp. Рath. Pharmak., 195, 71, 1940). Сенсибилизированные морские свинки были подвергнуты наркозу внутрибрюшинной инъекцией уретана (1,5 г/кг). Правая яремная вена была катетеризована для введения всех необходимых агентов, а трахея была снабжена трахеотомической канюлей для регистрации общего сопротивления дыханию. Mорские свинки были подключены к небольшому аппарату искусственной вентиляции легких для животных (модель SN-480-7, фирма Shinano), установленному на частоту 50 вдохов в минуту с объемом дыхания 4,5 мл. Изменения в сопротивлении дыханию регистрировалось датчиком давления (модель ТР-602Т, фирма Nihon Kohden), подключенным к Т-трубке, установленной на трахеотомической канюле. Процент максимального сужения просвета бронха определялся полным перекрытием трахеи. После хирургической подготовки и перед введением провокационной пробы овальбумина (0,2 мг/кг) животным были предварительно введены индометацин (2 мг/кг, 10 мин), пириламин (2 мг/кг, 6 мин) и пропранолол (0,1 мг/кг, 5 мин). Все испытуемые соединения вводились орально за 2 часа до введения провокационной пробы овальбумина. Подавление (%) эффекта сужения просвета бронха определялось следующим образом: подавление, % = (1,0 - % Образец был оставлен на 50 - 60 мин и подвергнут раздражению, приводящему к сокращению мышцы, дигидрохлоридом гистамина (100 мкмоль). После того, как промежуток времени между раздражением и ответной реакцией стал постоянным, образец был промыт и оставлен на 2030 мин. После этого к нему был добавлен индометацин (5 мкмоль), и через 30-минутный период инкубации образец был подвергнут раздражению, приводящему к сокращению мышцы, путем добавления LTD4 (30 нмоль). После того, как промежуток времени между раздражением и ответной реакцией стал постоянным, был кумулятивно введен образец лекарственного препарата для испытаний. И, наконец, для достижения реакции максимальной релаксации был введен АР (1 ммоль). Результат выражался процентным отношением релаксации, достигаемой применением образца лекарственного препарата для испытаний, к релаксации, достигаемой применением АР, которая оценивалась как 100%-ная, после чего измерялась 28 34481 нут центрифугированию при 2000 х g в течение 10 мин с целью получения плазмы с низким содержанием тромбоцитов (ПНСТ). Измерения производились путем разбавления ПВСТ в ПНСТ до 333000/мм3. ПВСТ и ПНСТ были помещены в кювету, после чего был отрегулирован диапазон измерений прозрачности, достигающий 0% в отношении ПВСТ и 100% в отношении ПНСТ. После этого к ПВСТ был добавлен образец испытуемого лекарственного препарата, растворенного в 100%ном диметилсульфоксиде (DMSO), при этом конечная концентрация DMSO составила 0,25 %. После 2-минутного периода инкубации при 37°С на скорости 900 оборотов в минуту был добавлен препарат, вызывающий агрегацию тромбоцитов, с целью получения кривой агрегации. Антитромбоцитное действие образца испытуемого лекарственного препарата выражалось концентрацией (IC50, мкмоль), при которой происходило 50%-ное подавление агрегации тромбоцитов в контрольном образце. Аденозиндифосфат, применяемый в качестве препарата, вызывающего агрегацию тромбоцитов, использовался в минимальной концентрации (5 - 10 мкмоль), что позволяло достигнуть максимальной агрегации. Измерение агрегации тромбоцитов производилось с использованием изотопного индикатора NBS НЕМА TRACER 601. Результаты приведены в таблице 5. Промышленная применимость максимального сужения просвета бронха в испытуемой группе/% максимального сужения просвета бронха в контрольной группе) х 100. Максимальное сужение просвета бронха составляло 62±6 % (среднее±среднеквадратическая ошибка; n = 6), а число подопытных животных составляло 5-6. Коэффициент подавления при дозе испытуемого соединения 30 мг/кг представлен в таблице 3-3. II. Антиаллергическое действие Тест на связывание с радиоактивным пириламином - 3Н (тест на связывание Н1-гистаминорецепторов). Испытания проводились в соответствии с методом Чанга и др. (J. Neurochem., 32, 1653 (1979)). К суспензии коровьевого мозжечка и 50 ммоль фосфатного буферного раствора (рН 7,5) был добавлен меченный тритием пириламин, после чего смесь была оставлена на отстой при 25°С в течение 30 мин. Затем в условиях разреженной атмосферы смесь была быстро профильтрована через стекловолоконную фильтровальную бумагу, после чего была замерена радиоактивность на поверхности фильтровальной бумаги. Коэффициент подавления в отношении H1-рецептора при концентрации испытуемого соединения, равной 10 мкмоль, определялся в соответствии со следующим равенством. Коэффициент подавления, % = {1– (степень связывания в присутствии лекарственного препарата – степень неспецифического связывания / /суммарная степень связывания – степень неспецифического связывания)} х 100, где суммарной степенью связывания является радиоактивность связывания пириламина3 Н при отсутствии испытуемого соединения, а степенью неспецифического связывания является радиоактивность связывания пириламина-3Н в присутствии мкмоль трипролизина. Результаты приведены в таблице 4. 3. АНтитромбоцитное действие Антитромбоцитное действие у кроликов В шприц, содержащий 0,1 объема 3,8 %-ного цитрата натрия, была взята кровь из брюшной артерии самцов японского белого кролика (масса: 1,8-2,5 кг). Полученная таким образом кровь была подвергнута центрифугированию при 200 х g в течение 7 мин при комнатной температуре с целью получения плазмы с высоким содержанием тромбоцитов (ПВСТ). Кроме того, остаток был подверг Как свидетельствуют приведенные выше результаты измерений, соединения, предложенные в соответствии с настоящим изобретением, обладают превосходными бронхолитическим действием, антиаллергическим действием и антитромбоцитным действием. Соединения, предложенные в соответствии с настоящим изобретением, проявляют высокую фармакологическую активность даже при оральном способе введения в организм.Таким образом, они могут быть использованы в качестве профилактических и терапевтических лекарственных препаратов, пригодных для лечения аллергических заболеваний, сопровождающихся реакциями немедленного типа, например, бронхиальной астмы, аллергического ринита, крапивницы и сенной лихорадки, различных заболеваний, сопровождающихся воспалительными процессами, например, ревматического артрита и спинномозгового артрита, ишемических заболеваний, например, стенокардии и инфаркта миокарда, а также различных тромбозных заболеваний. 29 34481 Таблица 1 30
ДивитисяДодаткова інформація
Назва патенту англійською3(2h)-pyridazinone derivatives with pharmaceutical activity, broncholytic, antiallergenic, antiplatelet agents based thereon
Автори англійськоюTanikawa Keizo, Saito Akira, Hirotsuka Mitsuaki, Shikada Ken-Ichi
Назва патенту російськоюПроизводные 3(2н)пиридазинона, имеющие фармацевтическую активность, бронхолитические, антиаллергические, антитромбоцитные средства на их основе
Автори російськоюТаникава Кеизо, Саито Акира, Хиротсука Мишуаки, Шикада Кен-иши
МПК / Мітки
МПК: C07D 237/20, C07D 401/12
Мітки: активність, бронхолітичний, основі, похідні, 3(2н)піридазинону, антиалергічний, фармацевтичну, засоби, антитромбоцитний, мають
Код посилання
<a href="https://ua.patents.su/55-34481-pokhidni-32npiridazinonu-shho-mayut-farmacevtichnu-aktivnist-bronkholitichnijj-antialergichnijj-antitrombocitnijj-zasobi-na-kh-osnovi.html" target="_blank" rel="follow" title="База патентів України">Похідні 3(2н)піридазинону, що мають фармацевтичну активність, бронхолітичний, антиалергічний, антитромбоцитний засоби на їх основі</a>
Похідні 3(2н)піридазинону або їх фармацевтично прийнятні солі, що мають протитромботичну, позитивну інотропну, судинорозширювальну дію й антиалергічну активність та фармацевтична композиція на їх основі

Номер патенту: 27031
Опубліковано: 28.02.2000
Автори: Кен-Іті Сікада, Ріозо Сакода, Акіра Саіто, Кейзо Танікава, Набутомо Тсурузое, Такасі Матсумото
МПК: C07D 401/12, C07D 237/22, C07D 405/12
Мітки: солі, прийнятні, антиалергічну, судинорозширювальну, фармацевтично, мають, протитромботичну, похідні, основі, 3(2н)піридазинону, дію, інотропну, фармацевтична, позитивну, активність, композиція
Формула / Реферат:
1. Производные 3(2Н)пиридазинона общей формулы (І):где R1=водород, алкильная группа С1-С4, группа общей формулы -(CH2)nCO2R5, где n=1, 2 или 3; R5= водород или алкильная группа С1-С4;R2= группа формулы A1-Y1, где А1= алкилен С1-С12 с прямой или разветвленной цепью; Y1= группа CO2R5, фенокси, тиенил, пиридил, цианогруппа, группа общей формулы CONR7R8, где R5 имеет указанные выше значения, R7 и R8 независимо - водород,...
Похідні 3-циклоалкілпропен-2-аміду, їх таутомерні форми та їх солі, що мають протизапальну активність, спосіб їх одержання та фармацевтична композиція на їх основі
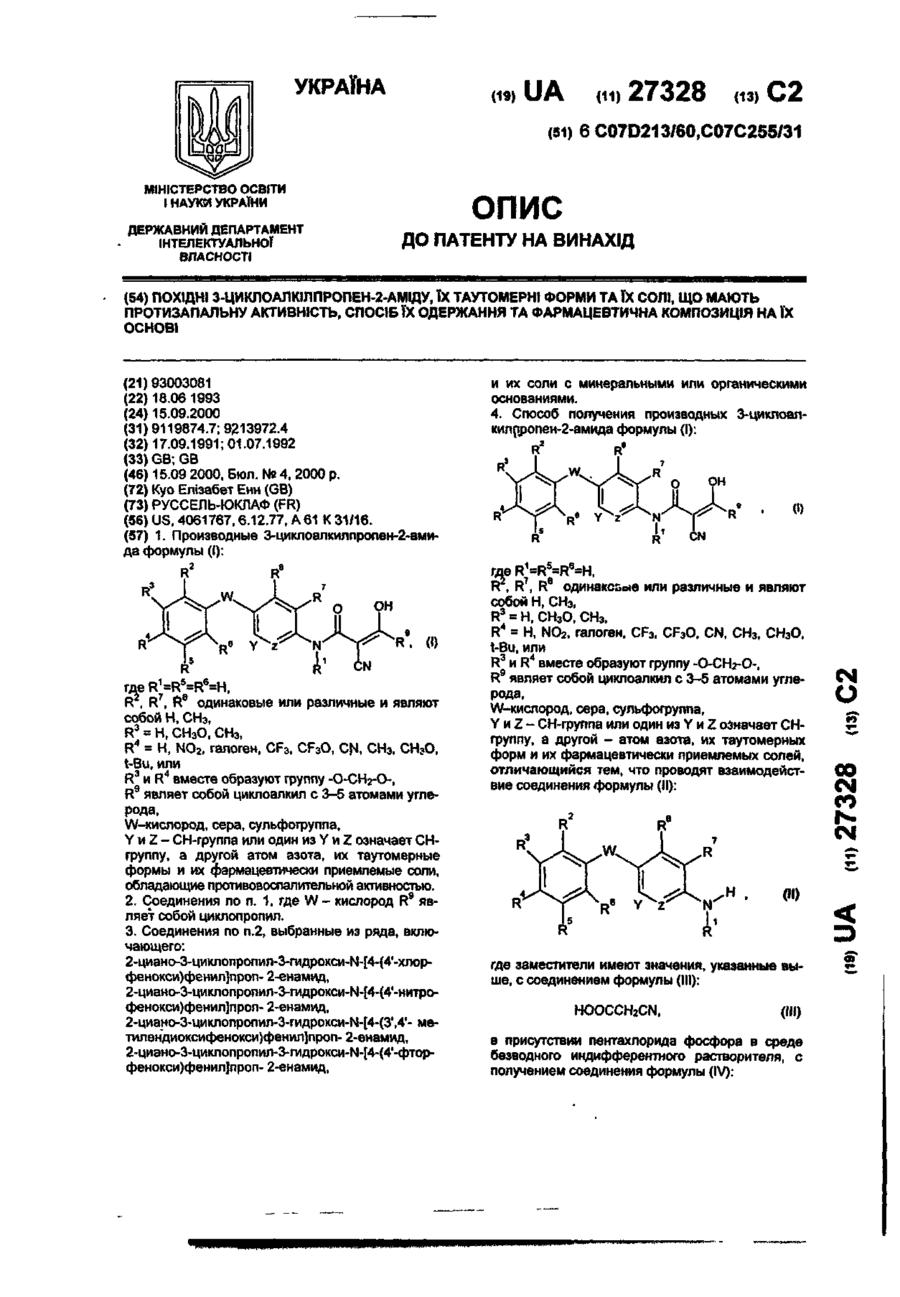
Номер патенту: 27328
Опубліковано: 15.09.2000
Автор: Куо Елізабет Енн
МПК: C07C 255/31
Мітки: 3-циклоалкілпропен-2-аміду, похідні, спосіб, активність, основі, одержання, композиція, солі, фармацевтична, таутомерні, мають, форми, протизапальну
Текст:
...фармакологическими свойствами. В частности, они проявляют противовоспалительное действие Они ингибируют, с одной стороны, воспалительные явления, вызванные раздражающими агентами, а с другой стороны, тормозят реакции запаздывающей суперчувствительности, препятствуя активации иммунных клеток специфическим антигеном. Эти свойства иллюстрированы ниже в опытной части. Эти свойства позволяют использовать новые производные...
Амідинові похідні бензолу, що мають гіпоглікемічну активність, та їх фармацевтично прийнятні солі
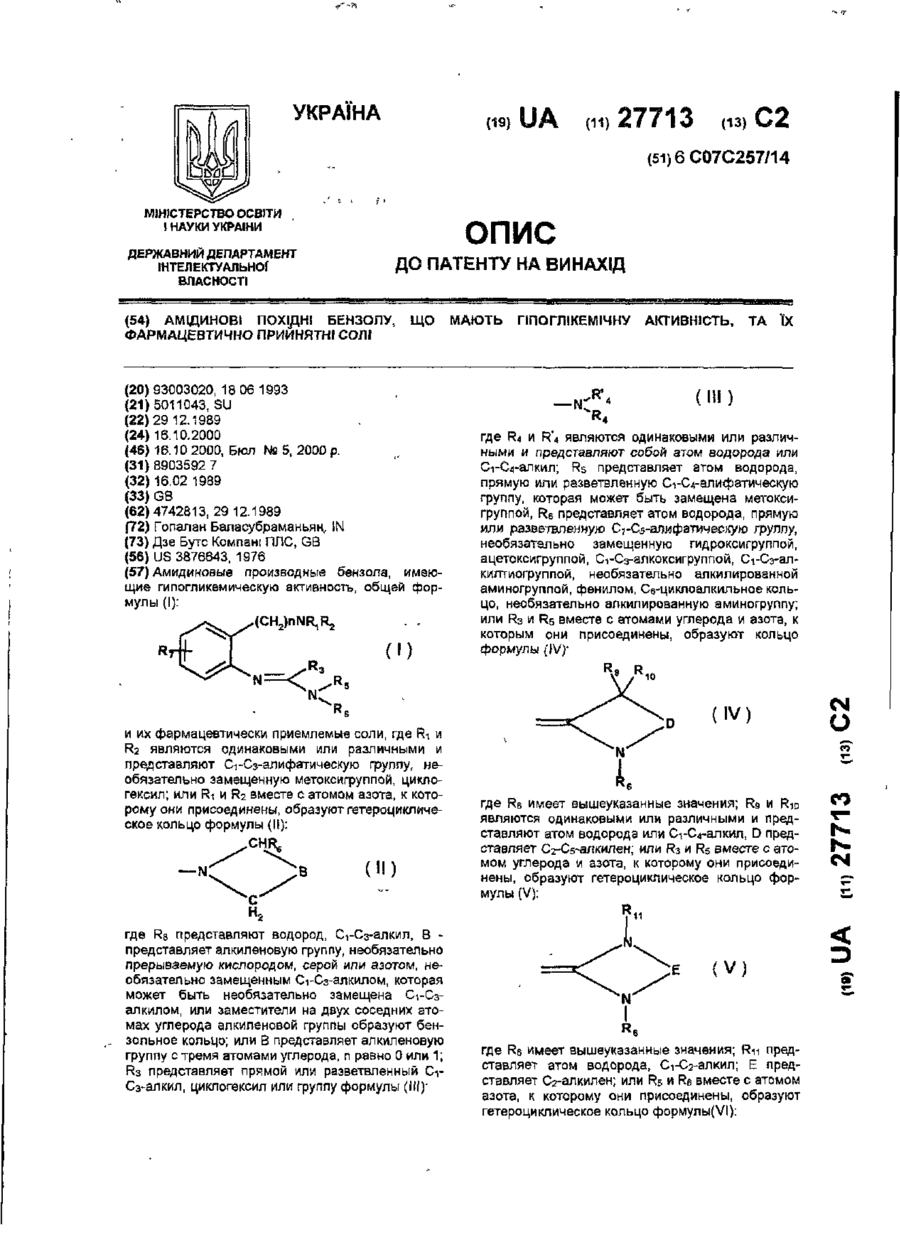
Номер патенту: 27713
Опубліковано: 16.10.2000
Автор: Гопалан Баласубраманьян
МПК: A61K 31/445, C07D 295/135, C07C 257/00
Мітки: активність, похідні, солі, фармацевтично, гіпоглікемічну, мають, амідинові, прийнятні, бензолу
Формула / Реферат:
Амидиновые производные бензола, имеющие гипогликемическую активность, общей формулы (I):и их фармацевтически приемлемые соли, где R-1 и R2 являются одинаковыми или различными и представляют С1-С3-апифатическую группу, необязательно замещенную метоксигруппой, циклогексил; или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо формулы (II):где R6 представляют водород,...
Похідні піримідину, що мають активність антагоністів ангіотензину іі, спосіб їх отримання та фармацевтична композиція

Номер патенту: 27748
Опубліковано: 16.10.2000
Автори: Бажлі Жак Фрамрос, Нікайдо Маделен, Еллінгбой Джон Ватсон
МПК: C07D 487/04, C07D 471/04
Мітки: похідні, фармацевтична, мають, спосіб, активність, композиція, антагоністів, ангіотензину, піримідину, отримання, іі
Текст:
...Результаты анализа из расчеты на формулу 3 или 30 мг/кг (объем вводимой суспензии составлял 1 мл/кг). Через 15, 30, 60, 90, 120, 150, 180, 210 и 240 минут после введения испытуемого соединения определяли среднее артериальное давление и частоту сердечных сокращений. Так, например, соединение, в соответствии с примером 1, при введении его внутрикожно в количестве 3 мг/кг снижало зависимость кровяного давления от А II в среднем на 41 % в...
Похідні оксазолу, які мають гіпоглікемічну і/чи гіпохолестеринемічну активність, та інтермедіати для іх отримання.

Номер патенту: 27417
Опубліковано: 15.09.2000
Автор: Кларк Девід Е.
МПК: C07D 417/06, C07D 417/10, A61K 31/425, C07D 263/32, A61P 3/08, A61P 3/06, C07F 7/18, C07B 57/00
Мітки: інтермедіати, активність, мають, похідні, оксазолу, отримання, гіпоглікемічну, гіпохолестеринемічну
Текст:
Попередній патент: Спосіб виготовлення будівельних виробів
Наступний патент: Вимірник комплексного коефіцієнта відбиття
Випадковий патент: Причіпний коток