Макроциклічні деазаоксипурини для лікування вірусних інфекцій
Номер патенту: 115588
Опубліковано: 27.11.2017
Автори: Бонфанті Жан-Франсуа, Арну Ерік П'єр Александр, Фортен Жером Мішель Клод, Дублє Фредерік Марк Моріс, Рабуассон П'єр Жан-Марі Бернар, Мюллер Філіпп





Формула / Реферат
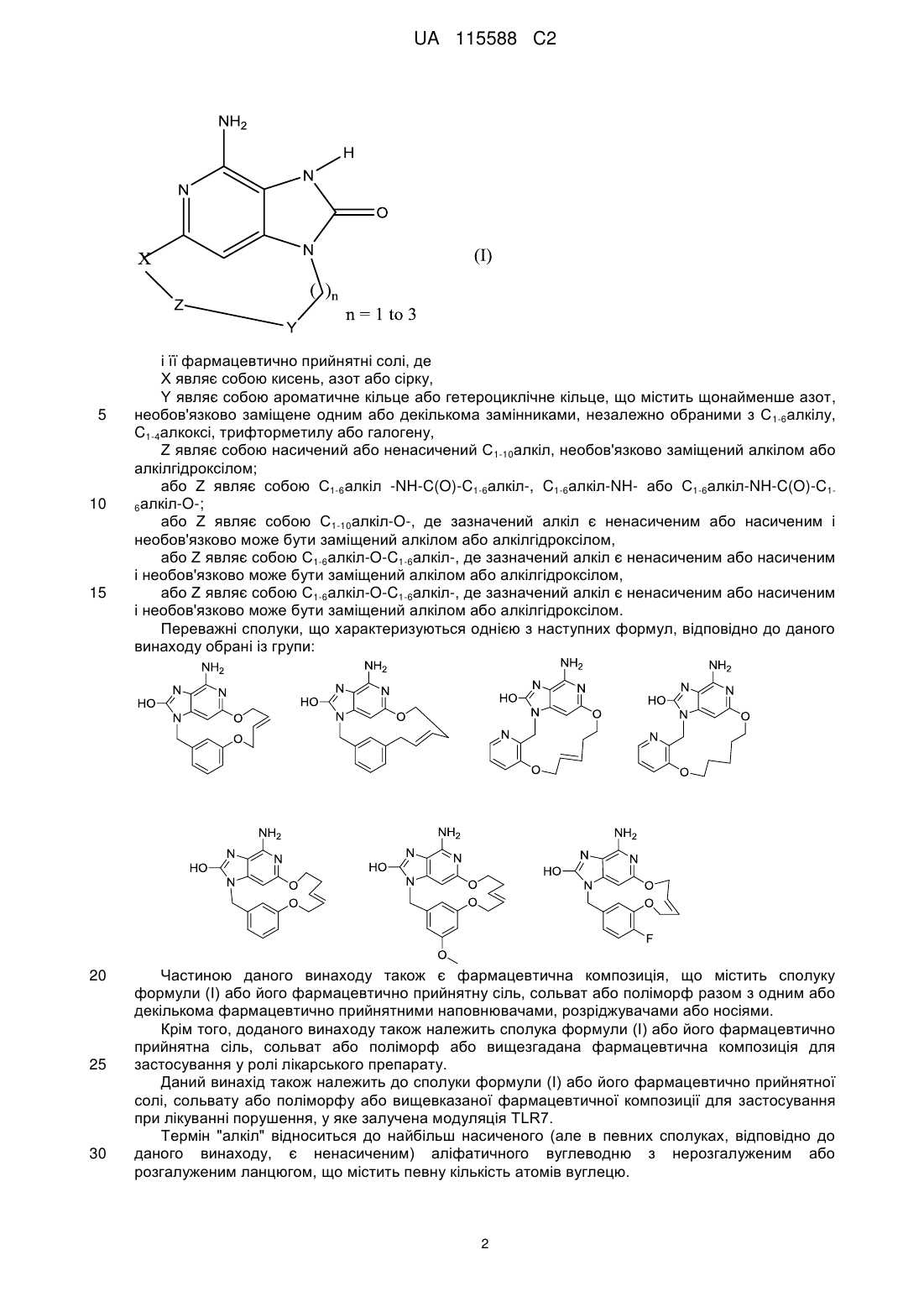
1. Сполука формули (І):
 , (I)
, (I)
або її фармацевтично прийнятні солі, де
X являє собою кисень, азот або сірку,
Υ являє собою ароматичне кільце або гетероциклічне кільце, що містить щонайменше азот, необов'язково заміщене одним або декількома замісниками, незалежно вибраними з С1-6алкілу, С1-4алкокси, трифторметилу або галогену,
Ζ являє собою насичений або ненасичений С1-10алкіл, необов'язково заміщений алкілом або алкілгідроксилом;
або Ζ являє собою С1-6алкіл-NН-С(О)-С1-6алкіл-, С1-6алкіл-NH- або С1-6алкіл-NН-С(О)-С1-6алкіл-О-;
або Ζ являє собою С1-10алкіл-О-, де вказаний алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом,
або Ζ являє собою С1-6алкіл-О-С1-6алкіл-, де вказаний алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом,
або Ζ являє собою С1-6алкіл-О-С1-6алкіл-О-, де вказаний алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом.
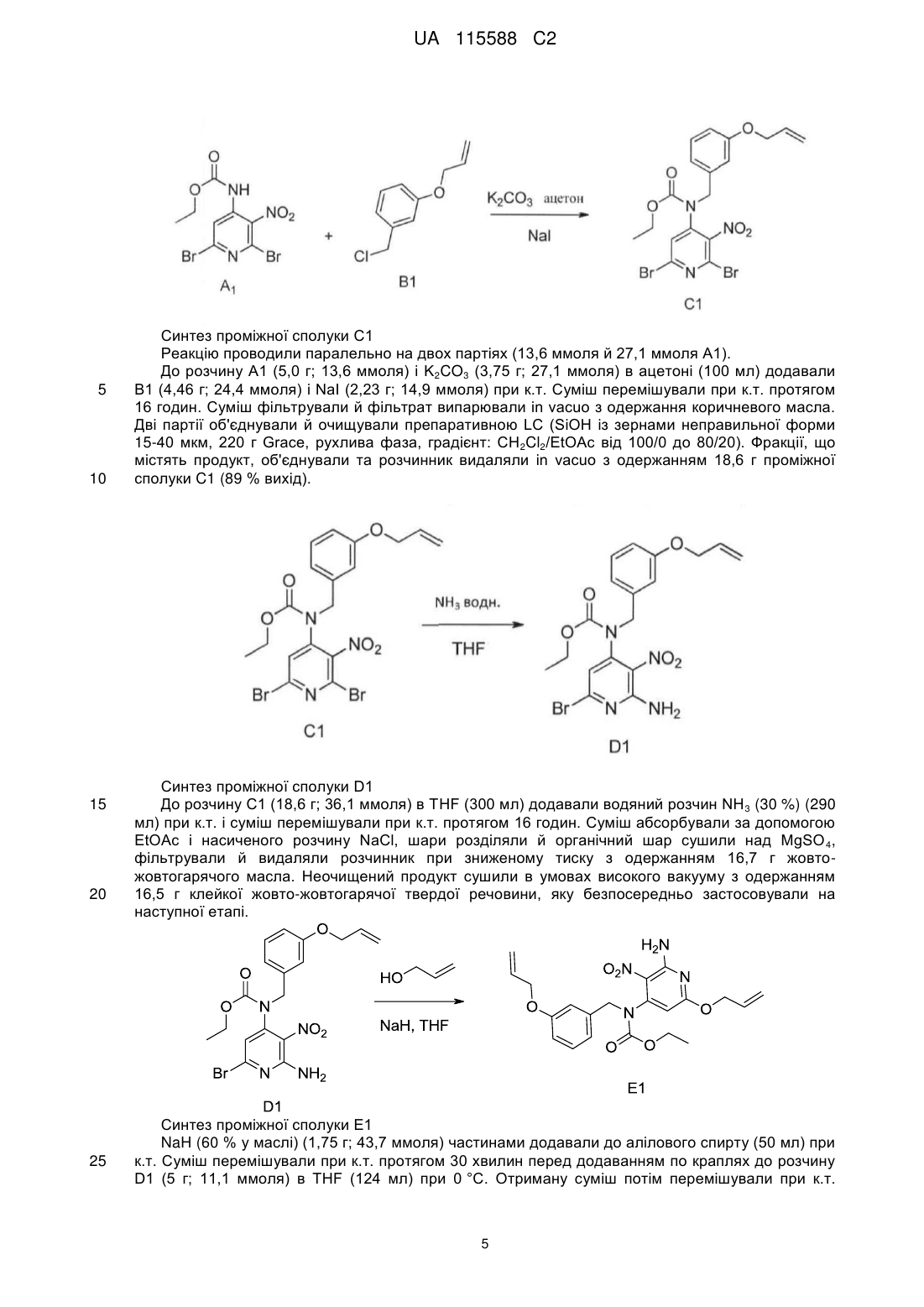
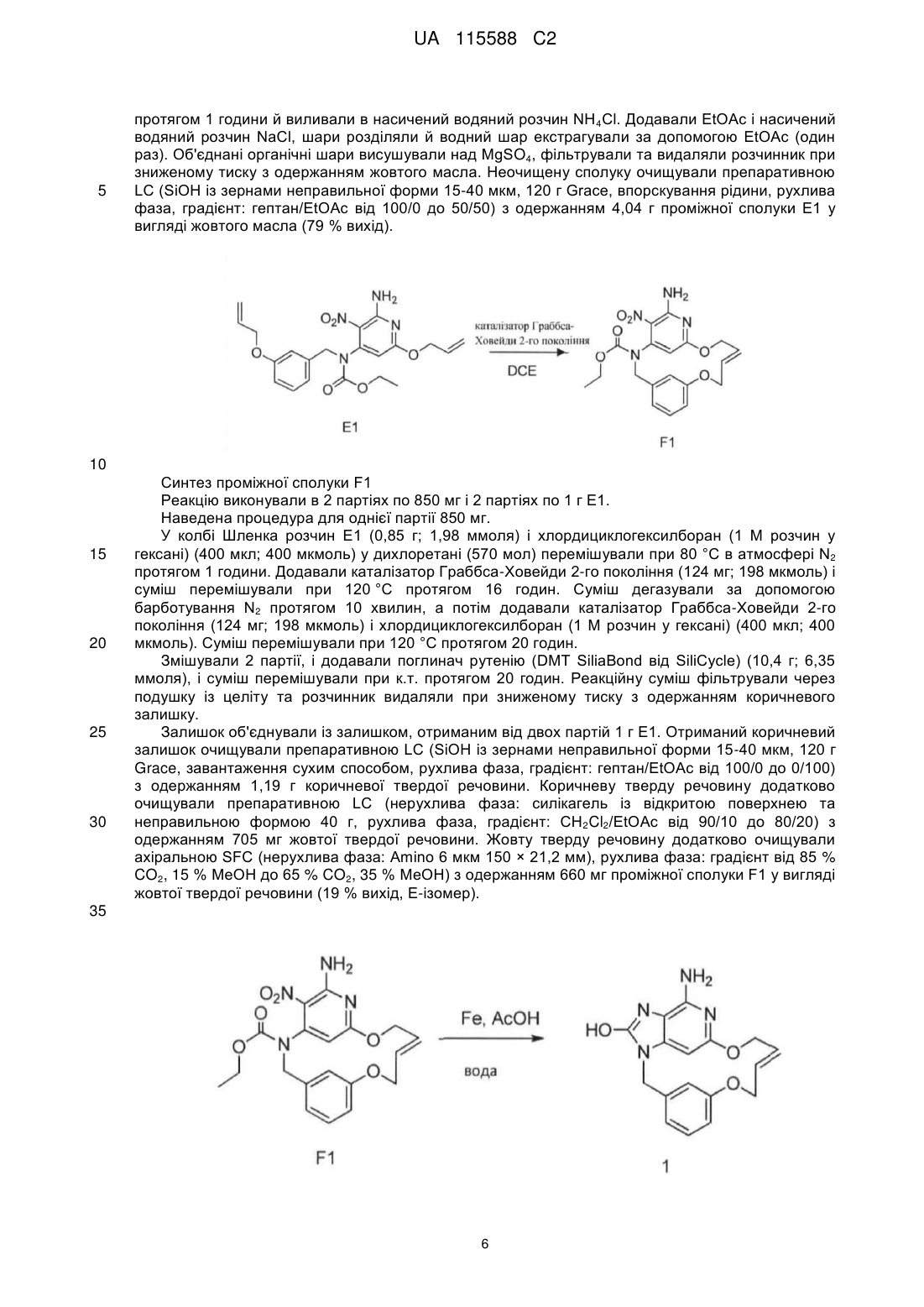
2. Сполука за п. 1, що представлена однією з наступних формул, вибраних із групи:
 ,
, 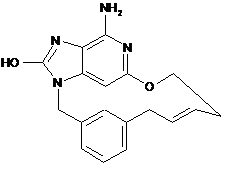 ,
, 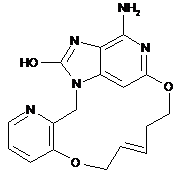 ,
, 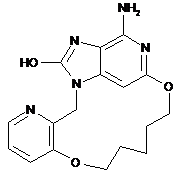 ,
, 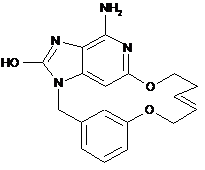 ,
, 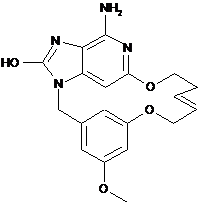 ,
, 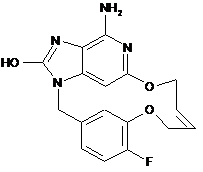 .
.
3. Сполука формули (І) за п. 1 або п. 2 або її фармацевтично прийнятна сіль для застосування як лікарського препарату.
4. Сполука формули (І) за п. 1 або п. 2 або її фармацевтично прийнятна сіль для застосування при лікуванні порушення, у яке залучена модуляція TLR7.
5. Фармацевтична композиція, що містить сполуку формули (І) за п. 1 або п. 2 або її фармацевтично прийнятну сіль разом з одним або декількома фармацевтично прийнятними наповнювачами, розріджувачами або носіями.
6. Фармацевтична композиція за п. 5 для застосування як лікарського препарату.
7. Фармацевтична композиція за п. 5 для застосування при лікуванні порушення, у яке залучена модуляція TLR7.
Текст
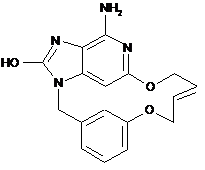
Реферат: Даний винахід стосується похідних макроциклічних деазаоксипуринів, способів їх одержання, фармацевтичних композицій і їх застосування при лікуванні вірусних інфекцій. UA 115588 C2 (12) UA 115588 C2 UA 115588 C2 5 10 15 20 25 30 35 40 45 Макроциклічні деаза-оксипурини для лікування вірусних інфекцій Даний винахід належить до похідних макроциклічних деаза-оксипуринів, способів їх одержання, фармацевтичних композицій і їх застосування при лікуванні вірусних інфекцій. Даний винахід належить до застосування похідних макроциклічних деаза-оксипуринів у лікуванні вірусних інфекцій, імунних або запальних порушень, у яке залучені модуляція або агонізм толл-подібних рецепторів (TLR). Толл-подібні рецептори представляють собою основні трансмембранні білки, що характеризуються позаклітиною ділянкою, багатою на лейцин і цитоплазматичним розширенням, яке містить консервативну ділянку. Вроджена імунна система може розпізнавати патогенасоційовані молекулярні патерни за допомогою даних TLR, що експресуються на клітинній поверхні певних типів імунних клітин. При розпізнаванні чужорідних патогенів активується вироблення цитокінів і підвищується експресія ко-стимулюючих молекул на фагоцитах. Це призводить до модуляції поведінки Т-клітин. Встановлено, що більшість видів ссавців мають від десяти до п'ятнадцяти типів толлподібних рецепторів. У загальній складності у людей і мишей виявили тринадцять TLR (під назвою TLR1-TLR13), а у інших видів ссавців виявили еквівалентні форми багатьох з них. Проте, еквіваленти певних TLR, виявлені у людей, не присутні у всіх ссавців. Наприклад, ген, що кодує білок, аналогічний TLR10 у людей, є присутнім у мишей, але, виявляється, що у певний момент у минулому він був пошкоджений ретровірусом. З іншого боку, у мишей експресуються TLR 11, 12 і 13, жоден з яких не представлений у людини. В інших ссавців можуть експресуватися TLR, які не виявлені у людини. Інші види, що не є ссавцями, можуть мати TLR, відмінні від таких у ссавців, доказом цьому служить TLR14, виявлений у риби фугу роду Takifugu. Це може ускладнити процедуру використання експериментальних тварин як моделей вродженого імунітету людини. Для детального огляду толл-подібних рецепторів див. наступні журнальні статті. Hoffmann, J.A., Nature, 426, p33-38, 2003; Akira, S., Takeda, K., and Kaisho, T., Annual Rev. Immunology, 21, p335-376, 2003; Ulevitch, R. J., Nature Reviews: Immunology, 4, p512-520, 2004. Раніше були описані сполуки, що проявляють активність по відношенню до толл-подібних рецепторів, такі як похідні пурину в WO 2006/117670, похідні аденіну в WO 98/01448 і WO 99/28321 і піримідини в WO 2009/067081. Проте, існує гостра потреба в нових модуляторах толл-подібних рецепторів, що володіють кращою селективністю, більш високою ефективністю, більш високою метаболічною стабільністю, а також поліпшеним профілем безпеки у порівнянні зі сполуками з відомого рівня техніки. При лікуванні певних вірусних інфекцій можуть регулярно вводитися ін'єкції інтерферону (IFN-альфа), як у випадку з вірусом гепатиту C (HCV). Для більшої інформації див. Fried et. al. Peginterferon-alfa plus ribavirin for chronic hepatitis C virus infection, N Engl J Med 2002; 347: 97582. Низькомолекулярні індуктори IFN, доступні для перорального застосування, пропонують потенційні переваги у вигляді зниженої іммуногенності й зручності введення. Таким чином, нові індуктори IFN представляють собою потенційно ефективний новий клас лікарських засобів для лікування вірусних інфекцій. Приклад низькомолекулярного індуктора IFN, що володіє противірусним ефектом, див. в De Clercq, E.; Descamps, J.; De Somer, P. Science 1978, 200, 563565. IFN-альфа також уводять у комбінації з іншими лікарськими засобами при лікуванні певних типів раку (Eur. J. Cancer 46, 2849–57, і Cancer Res. 1992, 52, 1056). Агоністи TLR 7/8 також становлять інтерес як вакцинні ад'юванти завдяки своїй здатності індукувати яскраво виражену Th1 реакцію (Hum. Vaccines 2010, 6, 322-335, і Hum. Vaccines 2009, 5, 381-394). Відповідно до даного винаходу представлена сполука формули (I) 1 UA 115588 C2 5 10 15 20 25 30 і її фармацевтично прийнятні солі, де X являє собою кисень, азот або сірку, Y являє собою ароматичне кільце або гетероциклічне кільце, що містить щонайменше азот, необов'язково заміщене одним або декількома замінниками, незалежно обраними з C 1-6алкілу, C1-4алкоксі, трифторметилу або галогену, Z являє собою насичений або ненасичений C1-10алкіл, необов'язково заміщений алкілом або алкілгідроксілом; або Z являє собою C1-6алкіл -NH-C(O)-C1-6алкіл-, C1-6алкіл-NH- або C1-6алкіл-NH-C(O)-C16алкіл-O-; або Z являє собою C1-10алкіл-O-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксілом, або Z являє собою C1-6алкіл-O-C1-6алкіл-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксілом, або Z являє собою C1-6алкіл-O-C1-6алкіл-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксілом. Переважні сполуки, що характеризуються однією з наступних формул, відповідно до даного винаходу обрані із групи: Частиною даного винаходу також є фармацевтична композиція, що містить сполуку формули (I) або його фармацевтично прийнятну сіль, сольват або поліморф разом з одним або декількома фармацевтично прийнятними наповнювачами, розріджувачами або носіями. Крім того, доданого винаходу також належить сполука формули (I) або його фармацевтично прийнятна сіль, сольват або поліморф або вищезгадана фармацевтична композиція для застосування у ролі лікарського препарату. Даний винахід також належить до сполуки формули (I) або його фармацевтично прийнятної солі, сольвату або поліморфу або вищевказаної фармацевтичної композиції для застосування при лікуванні порушення, у яке залучена модуляція TLR7. Термін "алкіл" відноситься до найбільш насиченого (але в певних сполуках, відповідно до даного винаходу, є ненасиченим) аліфатичного вуглеводню з нерозгалуженим або розгалуженим ланцюгом, що містить певну кількість атомів вуглецю. 2 UA 115588 C2 5 10 15 20 25 30 35 40 45 50 55 60 Термін "галоген" відноситься до фтору, хлору, брому або йоду. Термін "алкоксі" відноситься до алкільной групи (ланцюги з атомів вуглецю і водню), зв'язаної одинарним зв'язком з киснем, як, наприклад, метоксігрупа або етоксігрупа. Фармацевтично прийнятні солі сполук формули (I) включають їх солі приєднання кислоти й солі приєднання основи. Відповідні солі приєднання кислоти утворюються з кислот, які утворюють нетоксичні солі. Відповідні основні солі утворюються з основ, які утворюють нетоксичні солі. Сполуки відповідно до даного винаходу також можуть існувати в несольватованій і сольватованій формах. Термін "сольват" використовується в даному документі для опису молекулярного комплексу, що містить сполуки відповідно до даного винаходу й одну або кілька молекул фармацевтично прийнятного розчинника, наприклад, етанолу. Термін "поліморф" відноситься до здатності сполуки даного винаходу існувати в більш ніж одній формі або кристалічній структурі. Сполуки даного винаходу можуть бути присутні у формі так званого "таутомера(ів)», яка належить до ізомерів органічних сполук, які легко піддаються взаємоперетворенню шляхом хімічної реакції, яка називається таутомеризацією. Ця реакція призводить до формальної міграції атома водню або протону, яка супроводжується зрушенням одинарного та суміжного подвійного зв'язку. Сполуки даного винаходу можна вводити у вигляді кристалічних або аморфних продуктів. Вони можуть бути отримані, наприклад, у вигляді твердої пресованої маси, порошків або плівок за допомогою таких способів, як осадження, кристалізація, ліофільне сушіння, сушіння розпиленням або сушіння випарюванням. Їх можна вводити окремо або в комбінації з одним або декількома іншими сполуками даного винаходу або в комбінації з одним або декількома іншими лікарськими засобами. Як правило, їх вводитимуть у вигляді лікарської форми в комбінації з одним або декількома фармацевтично прийнятними наповнювачами. Термін "наповнювач" використовується в даному документі для опису будь-якого інгредієнта, відмінного від сполуки (сполук) даного винаходу. Вибір наповнювача більшою мірою залежить від таких факторів, як конкретний спосіб введення, вплив наповнювача на розчинність і стабільність і природа лікарської форми. Сполуки даного винаходу або будь-яка їхня підгрупа можуть бути виражені в різних фармацевтичних формах для цілей введення. В якості відповідних композицій можуть бути згадані всі композиції, звичайно використовувані для системного введення лікарських засобів. Для одержання фармацевтичних композицій даного винаходу ефективна кількість конкретної сполуки, необов'язково у формі солі приєднання, об'єднана в однорідну суміш із фармацевтично прийнятним носієм в якості активного інгредієнта, при цьому носій може приймати широку різноманітність форм залежно від форми препарату, необхідного для введення. Дані фармацевтичні композиції переважно представлені у вигляді одиничної лікарської форми, що підходить, наприклад, для перорального, ректального або черезшкірного введення. Наприклад, при одержанні композицій у вигляді пероральної лікарської форми можна використовувати будь-яке загальноприйняте фармацевтичне середовище, таке як, наприклад, вода, гліколі, масла, спирти і т.п., у випадку пероральних рідких препаратів, таких як суспензії, сиропи, еліксири, емульсії та розчини; або тверді носії, такі як крохмалі, цукри, каолін, розріджувачі, що змазують речовини, зв'язувальні речовини, розпушувачі й т.п., у випадку порошків, пігулок, капсул і таблеток. Завдяки простоті їх уведення, таблетки та капсули являють собою найбільш вигідні пероральні форми стандартних доз, у випадку яких, очевидно, що застосовують тверді фармацевтичні носії. Також включені препарати у твердій формі, які можуть бути перетворені безпосередньо перед застосуванням у препарати в рідких формах. У композиціях, що підходять для черезшкірного введення, носій необов'язково включає засіб, що підвищує проникність, і/або підходящий змочувальний засіб, необов'язково в комбінації з відповідними добавками будь-якої природи в мінімальних пропорціях, при цьому добавки не виявляють значного шкідливого впливу на шкіру. Зазначені добавки можуть полегшувати введення в шкіру й/або можуть бути корисними при одержанні необхідних композицій. Дані композиції можна вводити різними шляхами, наприклад, у формі трансдермального пластиру, у формі крапкового нанесення, у формі мазі. Сполуки відповідно до даного винаходу можна також уводити за допомогою інгаляції або інсуфляції за допомогою способів і складів, застосовуваних у даній області для введення таким шляхом. Таким чином, в основному сполуки відповідно до даного винаходу можна вводити в легені у формі розчину, суспензії або сухого порошку. Особливо вигідним являється складання вищевказаних фармацевтичних композицій у вигляді стандартної лікарської форми для простоти введення й рівномірного дозування. Стандартна лікарська форма, застосовувана в даному документі, належить до фізично окремих 3 UA 115588 C2 5 10 15 20 одиниць, що підходять у якості одиничних доз, при цьому кожна одиниця містить попередньо встановлену кількість активного інгредієнта, розраховане для одержання необхідного терапевтичного ефекту, у комбінації з необхідним фармацевтичним носієм. Прикладами таких стандартних лікарських форм є таблетки (у тому числі ділені або покриті оболонкою), капсули, пігулки, пакетики з порошком, облатки, суппозиторії, ін'єкційні розчини або суспензії й т.п., а також їх окреме різноманіття. Фахівці в області лікування інфекційних захворювань зможуть визначити ефективну кількість, виходячи з результатів тестів, представлених далі в даному документі. У цілому, вважається, що ефективна добова кількість буде становити від 0,01 до 50 мг/кг маси тіла, більш переважно від 0,1 до 10 мг/кг маси тіла. Може бути доцільним введення необхідної дози у вигляді двох, трьох, чотирьох або більших частин дози через відповідні інтервали протягом дня. Зазначені частини дози можуть бути виражені у вигляді одиничних лікарських форм, наприклад, що містять від 1 до 1000 мг і, зокрема, від 5 до 200 мг активного інгредієнта на одиницю лікарської форми. Точне дозування та частота введення залежать від конкретної використовуваної сполуки формули (I), конкретного стану, що підлягає лікуванню, тяжкості стану, що підлягає лікуванню, віку, ваги та загального фізичного стану конкретного пацієнта, а також іншого медикаментозного лікування, яке може одержувати індивідуум, і що добре відомо фахівцям у даній області. Більш того, очевидно, що ефективна кількість може бути зменшена або збільшена залежно від реакції суб'єкта, над яким проводиться лікування й/або залежно від оцінки лікаря, який призначає сполуки, відповідно до даного винаходу. Таким чином, вищезгадані діапазони ефективної кількості є лише рекомендаціями та не призначені для обмеження обсягу або застосування даного винаходу. 25 30 Синтез проміжної сполуки B1 SOCl2 (80 мл; 1,11 моля) краплями додавали до суміші G1 (19,1 г; 111 ммоль) в CH 2Cl2 (230 мл) при к.т. Суміш перемішували при к.т. протягом 16 годин. Розчинник випарювали, а залишок солюбілізували в CH2Cl2 і обробляли насиченим водяним розчином NaHCO 3 до основного pH. Шари розділяли та водний шар екстрагували за допомогою CH 2Cl2 (двічі). Об'єднані органічні шари сушили над MgSO4, фільтрували та концентрували у вакуумі з одержанням 20,1 г коричневого масла. Неочищену сполуку використовували на наступному етапі без додаткового очищення. 35 4 UA 115588 C2 5 10 15 20 25 Синтез проміжної сполуки C1 Реакцію проводили паралельно на двох партіях (13,6 ммоля й 27,1 ммоля A1). До розчину A1 (5,0 г; 13,6 ммоля) і K2CO3 (3,75 г; 27,1 ммоля) в ацетоні (100 мл) додавали B1 (4,46 г; 24,4 ммоля) і NaI (2,23 г; 14,9 ммоля) прик.т. Суміш перемішували при к.т. протягом 16 годин. Суміш фільтрували й фільтрат випарювали in vacuo з одержання коричневого масла. Дві партії об'єднували й очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 220 г Grace, рухлива фаза, градієнт: CH2Cl2/EtOAc від 100/0 до 80/20). Фракції, що містять продукт, об'єднували та розчинник видаляли in vacuo з одержанням 18,6 г проміжної сполуки C1 (89 % вихід). Синтез проміжної сполуки D1 До розчину C1 (18,6 г; 36,1 ммоля) в THF (300 мл) додавали водяний розчин NH 3 (30 %) (290 мл) при к.т. і суміш перемішували при к.т. протягом 16 годин. Суміш абсорбували за допомогою EtOAc і насиченого розчину NaCl, шари розділяли й органічний шар сушили над MgSO 4, фільтрували й видаляли розчинник при зниженому тиску з одержанням 16,7 г жовтожовтогарячого масла. Неочищений продукт сушили в умовах високого вакууму з одержанням 16,5 г клейкої жовто-жовтогарячої твердої речовини, яку безпосередньо застосовували на наступної етапі. Синтез проміжної сполуки Е1 NaH (60 % у маслі) (1,75 г; 43,7 ммоля) частинами додавали до алілового спирту (50 мл) при к.т. Суміш перемішували при к.т. протягом 30 хвилин перед додаванням по краплях до розчину D1 (5 г; 11,1 ммоля) в THF (124 мл) при 0 °C. Отриману суміш потім перемішували при к.т. 5 UA 115588 C2 5 протягом 1 години й виливали в насичений водяний розчин NH 4Cl. Додавали EtOAc і насичений водяний розчин NaCl, шари розділяли й водний шар екстрагували за допомогою EtOAc (один раз). Об'єднані органічні шари висушували над MgSO4, фільтрували та видаляли розчинник при зниженому тиску з одержанням жовтого масла. Неочищену сполуку очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 120 г Grace, впорскування рідини, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 50/50) з одержанням 4,04 г проміжної сполуки E1 у вигляді жовтого масла (79 % вихід). 10 15 20 25 30 Синтез проміжної сполуки F1 Реакцію виконували в 2 партіях по 850 мг і 2 партіях по 1 г E1. Наведена процедура для однієї партії 850 мг. У колбі Шленка розчин E1 (0,85 г; 1,98 ммоля) і хлордициклогексилборан (1 M розчин у гексані) (400 мкл; 400 мкмоль) у дихлоретані (570 мол) перемішували при 80 °C в атмосфері N2 протягом 1 години. Додавали каталізатор Граббса-Ховейди 2-го покоління (124 мг; 198 мкмоль) і суміш перемішували при 120 °C протягом 16 годин. Суміш дегазували за допомогою барботування N2 протягом 10 хвилин, а потім додавали каталізатор Граббса-Ховейди 2-го покоління (124 мг; 198 мкмоль) і хлордициклогексилборан (1 M розчин у гексані) (400 мкл; 400 мкмоль). Суміш перемішували при 120 °C протягом 20 годин. Змішували 2 партії, і додавали поглинач рутенію (DMT SiliaBond від SiliCycle) (10,4 г; 6,35 ммоля), і суміш перемішували при к.т. протягом 20 годин. Реакційну суміш фільтрували через подушку із целіту та розчинник видаляли при зниженому тиску з одержанням коричневого залишку. Залишок об'єднували із залишком, отриманим від двох партій 1 г E1. Отриманий коричневий залишок очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 120 г Grace, завантаження сухим способом, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 0/100) з одержанням 1,19 г коричневої твердої речовини. Коричневу тверду речовину додатково очищували препаративною LC (нерухлива фаза: силікагель із відкритою поверхнею та неправильною формою 40 г, рухлива фаза, градієнт: CH2Cl2/EtOAc від 90/10 до 80/20) з одержанням 705 мг жовтої твердої речовини. Жовту тверду речовину додатково очищували ахіральною SFC (нерухлива фаза: Amino 6 мкм 150 × 21,2 мм), рухлива фаза: градієнт від 85 % CO2, 15 % MeOH до 65 % CO2, 35 % MeOH) з одержанням 660 мг проміжної сполуки F1 у вигляді жовтої твердої речовини (19 % вихід, E-ізомер). 35 6 UA 115588 C2 5 10 15 20 Синтез кінцевої сполуки 1 Суміш F1 (570 мг; 1,42 ммоля) і заліза (795 мг; 14,2 ммоля) в AcOH (21 мл) і води (4,2 мл) перемішували при 50 °C протягом 2 годин. Суміш концентрували насухо. Додавали DMF, суміш обробляли ультразвуком, нагрівали та фільтрували через подушку із целіту, а целіт промивали використовуючи гарячий DMF. Поглинач заліза (імідазол SiliaBond від SiliCycle) (25,4 г; 29,5 ммоля) додавали до фільтрату та суміш перемішували при к.т. протягом 16 годин. Суміш фільтрували через целіт, целіт промивали за допомогою DMF і фільтрат концентрували in vacuo з одержанням 620 мг коричневої твердої речовини. Неочищену речовину очищували препаративною LC (SiOH із зернами неправильної форми, 15-40 мкм, 30 г Merck, рухлива фаза, градієнт: CH2Cl2/MeOH/NH3водн. від 98/2/0,2 до 85/15/1,5) з одержанням 360 мг кінцевої сполуки 1 у вигляді брудно-білої твердої речовини (75 % вихід). Альтернативний синтез проміжної сполуки C1 При 0 °C діізопропілазодикарбоксилат (DIAD) (3,0 мл, 15,0 ммоля) краплями додавали до суміші A1 (3,70 г, 10,028 ммоля), G1 (1,98 г, 12,0 ммоля) і PPh 3 (3,94 г, 15,0 ммоля) в THF (70 мл). Суміш перемішували при к.т. протягом 12 годин. Додавали EtOAc і воду. Шари декантували. Органічний шар промивали водою, сушили над MgSO 4, фільтрували та розчинник випарювали. Неочищену речовину очищували препаративною LC (SiOH із зернами неправильної форми 20-45 мкм 450 г Matrex), рухлива фаза (85 % гептан, 15 % AcOEt) з одержанням 4,5 г проміжної сполуки C1 (87 % вихід). Загальна схема одержання кінцевих продуктів: спосіб 2 25 30 35 Синтез проміжної сполуки H1 Каталізатор Вілкінсона (44 мг; 47,5 мкмоля) додавали до розчину F1 (190 мг; 475 мкмоль) в THF/MeOH (50/50) (50 мл), продутому шляхом барботування N 2 протягом 15 хвилин. Суміш гідрогенізовували (8 бар) при к.т. протягом 16 годин. Суміш продували протягом 15 хвилин і додатково додавали каталізатор Вілкінсона (44 мг; 47,5 мкмоль). Реакційну суміш гідрогенізовували (8 бар) при к.т. протягом 4 годин. Суміш концентрували in vacuo з одержанням коричневого масла. Масло очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 12 г Grace, завантаження сухим способом, рухлива фаза, градієнт: CH2Cl2/EtOAc від 100/0 до 80/20) з одержанням 150 мг проміжної сполуки H1 у вигляді жовтої твердої речовини (79 % вихід). 7 UA 115588 C2 5 10 Синтез проміжної сполуки J1 I1 (5,9 г; 35,6 ммоля) додавали до розчину A1 (7,3 г; 19,8 ммоля), K 2CO3 (5,5 г; 39,6 ммоля) і NaI (3,3 г; 21,8 ммоля) в ацетоні (145 мл). Суміш перемішували при к.т. протягом 20 годин. Суміш фільтрували через подушку із целіту та фільтрат випарювали in vacuo з одержанням жовтогарячої твердої речовини. Залишок абсорбували в CH 2Cl2. Осад фільтрували та фільтрат концентрували in vacuo з одержанням 13 г жовтого масла. Неочищену сполуку очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 300 г Interchim, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 80/20). Фракції, що містять продукт, об'єднували та розчинник видаляли in vacuo з одержанням 7,1 г проміжної сполуки J1 (72 % вихід) у вигляді жовтого масла. 15 Синтез проміжної сполуки K1 8 UA 115588 C2 У колбі Шленка розчин J1 (7,1 г; 14,2 ммоля) в THF (130 мл) і водяний розчин NH3 (30 %) (130 мл) перемішували при к.т. протягом 16 годин. Суміш абсорбували за допомогою EtOAc і насиченого водяного розчину NaCl, шари розділяли. Органічний шар висушували над MgSO 4, фільтрували та концентрували in vacuo з одержанням 6,4 г жовтого масла (100 % вихід). 5 10 15 Синтез проміжної сполуки L1 NaH (2,2 г; 54,2 ммоля) додавали частинами при к.т. і в атмосфері N 2 до 3-бутен-1-олу (76 мл). Суміш перемішували при к.т. протягом 30 хвилин перед її додаванням по краплях при температурі 0 °C у розчин K1 (5,9 г; 13,6 ммоля) в THF (150 мл). Отриману суміш перемішували при 0 °C протягом 1 години. Суміш виливали у водний насичений розчин NH 4Cl. Додавали EtOAc і насичений водяний розчин NaCl, шари розділяли. Органічні шари сушили над MgSO 4, фільтрували та концентрували in vacuo з одержанням жовтого залишку, який азеотропно відганяли використовуючи толуол (однократно) з одержанням 6,6 г жовтого масла. Неочищену сполуку очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 220 г Grace, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 50/50). Фракції, що містили продукт, об'єднували та розчинник видаляли in vacuo з одержанням 4,46 г проміжної сполуки L1 (77 % вихід) у вигляді жовтого масла. 20 25 30 35 Синтез проміжної сполуки M1 Реакцію виконували в 2 партіях. Типова процедура для однієї партії. Розчин L1 (2,45 г; 5,75 ммоля) у сухому CH 2Cl2 (1,7 л) дегазували за допомогою барботування N2 протягом 15 хвилин. Додавали каталізатор Граббса 2-го покоління (488 мг; 574 мкмоль) і суміш перемішували при к.т. протягом 72 години. Додавали DMT SiliaBond (7,66 г; 4,59 ммоля) і суміш перемішували при к.т. протягом 16 годин. Об'єднували 2 партії та фільтрували через целіт. Фільтрат концентрували in vacuo з одержанням чорної твердої речовини. Неочищену сполуку очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 150 г Merck, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 50/50). Фракції, що містили продукт, об'єднували та розчинник видаляли in vacuo з одержанням 230 мг фракції 1 і 2,3 г фракції 2. Фракцію 2 попередньо очищували препаративною LC (нерухлива фаза: SiOH із зернами неправильної форми 40 мкм 120 г, рухлива фаза: гептан/CH 2Cl2/MeOH 55/43/2). Виділену сполуку об'єднували із фракцією 1 і очищували ахіральною SFC (нерухлива фаза: Chiralpak IC 5 мкм 250 × 20 мм, рухлива фаза: 70 % CO2, 30 % iPrOH) з одержанням 1,51 г проміжної сполуки M1 (33 % вихід, ізомер E) у вигляді жовтої твердої речовини. 9 UA 115588 C2 5 10 15 Синтез кінцевої сполуки 3 Залізо (631 мг; 11,3 ммоля) додавали до розчину M1 (750 мг; 1,88 ммоля) в AcOH (150 мл) і води (25 мл). Суміш перемішували при 80 °C протягом 16 годин. Додавали залізо (315 мг; 5,65 ммоля) і суміш перемішували при 80 °C протягом 2 годин. Додавали залізо (315 мг; 5,65 ммоля) і суміш перемішували при 80 °C протягом 4 годин. Додавали залізо (315 мг; 5,65 ммоля) і сумішперемішували при 80 °C протягом 16 годин. Суміш концентрували насухо. Додавали DMF, суміш фільтрували через целіт і целіт промивали використовуючи гарячий DMF. Імідазол SiliaBond (48,7 г; 56,5 ммоля) додавали до фільтрату та суміш перемішували при к.т. протягом 16 годин. Суміш фільтрували через целіт, целіт промивали за допомогою DMF і фільтрат концентрували in vacuo. Неочищену сполуку очищували препаративною LC (SiOH із зернами неправильної форми, 15-40 мкм, 25 г Merck, рухлива фаза, градієнт: від CH2Cl2/MeOH/NH3водн. від 98/2/0,2 до 85/15/1,5) з одержанням 2 фракцій. Фракцію 1 абсорбували за допомогою EtOH і фільтрували з одержанням фракції 3, а фракцію 2 абсорбували за допомогою MeCN і фільтрували з одержанням фракції 4. Фракції 3 і 4 об'єднували в EtOH, фільтрували та сушили in vacuo з одержанням 199 мг кінцевої сполуки 3 (33 % вихід). 20 25 30 35 40 Синтез проміжної сполуки N1 Каталізатор Вілкінсона (46 мг; 50,2 мкмоля) додавали до розчину M1 (200 мг; 502 мкмоль) в THF/MeOH (50/50) (50 мл), продутому шляхом барботування N 2 протягом 15 хвилин. Суміш гідрогенізували (7 бар) при к.т. протягом 20 годин. Суміш продували шляхом барботування N 2 протягом 15 хвилин, також додавали каталізатор Вілкінсона (46 мг; 50,2 мкмоля) і реакційну суміш гідрогенізували (7 бар) при к.т. протягом 16 годин. Реакційну суміш концентрували in vacuo з одержанням зеленого масла. Масло очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 25 г Merck, завантаження сухим способом, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 70/30) з одержанням 130 мг проміжної сполуки N1 у вигляді жовтої твердої речовини (66 % вихід). Синтез проміжної сполуки O1 У реакторі високого тиску N1 (110 мг; 275 мкмоль) гідрогенізували в EtOH (5 мл) з Pd/C (10 %) (30 мг; 28,5 мкмоля) у якості каталізатора при 40 °C (3 бару) протягом 6 годин. Каталізатор видаляли шляхом фільтрації через целіт, а целіт промивали за допомогою EtOH і фільтрат випарювали під вакуумом з одержанням 100 мг жовтого залишку (98 % вихід). Проміжну сполуку О1 застосовували на наступному етапі без додаткового очищення. Синтез кінцевої сполуки 4 У запаяній пробірці O1 (100 мг; 270 мкмоль) у чистій оцтовій кислоті (5 мл) перемішували при к.т. протягом 90 хвилин. Розчинник видаляли при зниженому тиску з одержанням жовтого залишку. Залишок абсорбували за допомогою CH 2Cl2 і видаляли розчинник при зниженому тиску (двічі) з одержанням 87 мг жовто-зеленої твердої речовини. Тверду речовину азеотропно відганяли за допомогою толуолу (чотири рази), а потім розтирали в порошок і обробляли ультразвуком в Et2O. Суміш відфільтровували (склоподібна фрита n°5) з одержанням 75 мг 10 UA 115588 C2 кінцевої сполуки 4 (77 % вихід, ацетатна сіль). 5 10 Синтез проміжної сполуки Q1 До розчину A1 (3,52 г; 9,54 ммоля) і K2CO3 (2,64 г; 19,1 ммоля) в ацетоні (80 мл) додавали P1 (1,93 г; 10,5 ммоля) і NaI (1,57 г; 10,5 ммоля) при к.т. Суміш перемішували при к.т. протягом 16 годин, потім додавали P1 (1,5 г; 8,17 ммоля) і суміш перемішували при к.т. протягом 24 годин. Реакційну суміш фільтрували через подушку із целіту та фільтрат випарювали in vacuo з одержанням чорного залишку. Залишок очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 80 г Grace, завантаження сухим способом, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 50/50) з одержанням 3,28 г проміжної сполуки Q1 у вигляді жовто-гарячого масла (67 % вихід). 15 20 Синтез проміжної сполуки R1 У колбі Шленка до розчину Q1 (3,28 г; 6,36 ммоля) в THF (52 мл) додавали водяний розчин NH3 (30 %) (52 мл) при к.т. Суміш перемішували при к.т. протягом 26 годин і додатково додавали водяний розчин NH3 (10 мл) і суміш перемішували при к.т. протягом 4 годин. Суміш 11 UA 115588 C2 абсорбували за допомогою EtOAc і насиченого водяного розчину NaCl, шари розділяли й органічний шар сушили над MgSO4, фільтрували та розчинник видаляли при зниженому тиску з одержанням 2,74 г проміжної сполуки R1 у вигляді жовтого масла (87 % вихід). 5 10 15 20 25 30 35 Синтез проміжної сполуки S1 NaH (60 % у маслі) (888 мг; 22,2 ммоля) частинами додавали до 3-бутен-1-олу (30 мл; 354 ммоль) при к.т. Суміш перемішували при к.т. протягом 30 хвилин перед додаванням по краплях до розчину R1 (2,74 г; 5,63 ммоля) в THF (62 мл) при 0 °C. Отриману суміш перемішували при к.т. протягом 1 години та виливали в насичений водяний розчин NH 4Cl. Додавали EtOAc і насичений водяний розчин NaCl, шари розділяли й водний шар екстрагували за допомогою EtOAc (один раз). Об'єднані органічні шари висушували над MgSO 4, фільтрували та видаляли розчинник при зниженому тиску з одержанням жовтого масла. Масло очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 80 г Grace, завантаження сухим способом, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 20/80) з одержанням 1,06 г проміжної сполуки S1 у вигляді жовтого залишку (42 % вихід). Синтез проміжної сполуки T1 Реакцію виконували у двох партіях по 480 мг проміжної сполуки S1. Далі навена процедура для однієї партії. У колбі Шленка розчин S1 (480 мг; 1,08 ммоля) і хлордициклогексилборан (1 M у гексані) (216 мкл; 216 мкмоля) у сухому дихлоретані (300 мл) перемішували при 80 °C і в атмосфері N2 протягом 1 години. Додавали каталізатор Граббса-Ховейди 2-го покоління (68 мг; 108 мкмоль) і суміш перемішували при 120 °C протягом 2 годин. Змішували дві партії, додавали DMT SiliaBond (2,84 г; 1,73 ммоля) і суміш перемішували при к.т. протягом 20 годин. Суміш фільтрували через подушку із целіту, целіт промивали за допомогою EtOAc і фільтрат випарювали in vacuo з одержанням коричневої твердої речовини. Коричневу тверду речовину очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 40 г Grace, завантаження сухим способом, рухлива фаза, градієнт: CH 2Cl2/EtOAc від 100/0 до 20/80) з одержанням 610 мг жовтого залишку (суміш E- і Z-ізомерів, проміжна сполука U1). 310 мг проміжної сполуки U1 очищували зворотно-фазовою хроматографією (нерухлива фаза: Nucleodur-Sphinx rp 5 мкм 21 × 150 мм, рухлива фаза: градієнт від 70 % мурашиної кислоти 0,1 %, 30 % Mecn до 0 % мурашиної кислоти 0,1 %, 100 % MeCN) з одержанням 195 мг проміжної сполуки T1 (E-ізомер) у вигляді жовтої твердої речовини (22 % вихід). 12 UA 115588 C2 5 10 15 Синтез кінцевої сполуки 5 Суміш T1 (160 мг; 385 мкмоль) і заліза (129 мг; 2,31 ммоля) в оцтовій кислоті (21 мл) і воді (2,4 мл) перемішували при 80 °C протягом 7 годин. Додатково додавали залізо (129 мг; 2,31 ммоля) і суміш перемішували при 80 °C протягом 16 годин. Додатково додавали залізо (129 мг; 2,31 ммоля) і суміш перемішували при 80 °C протягом 3 годин. Суміш концентрували in vacuo з одержанням залишку. Залишок розбавляли в DMF і фільтрували через подушку із целіту. Імідазол SiliaBond (12,7 г; 14,7 ммоля) додавали до фільтрату та суміш перемішували при к.т. протягом 16 годин. Суміш фільтрували через подушку із целіту та фільтрат випарювали in vacuo з одержанням коричневої твердої речовини. Коричневу тверду речовину очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 12 г Grace, завантаження сухим способом, рухлива фаза, градієнт: CH2Cl2/MeOH/NH3водн. від 97/3/0,3 до 80/20/2) з одержанням 65 мг брудно-білої твердої речовини. Тверду речовину очищували зворотнофазовою хроматографією (нерухлива фаза: X-Bridge-C18 5 мкм 30*150 мм, рухлива фаза, градієнт: H2O (0,5 % NH4CO3)/MeOH від 70/30 до 0/100) з одержанням 43 мг кінцевої сполуки 5 у вигляді білої твердої речовини (31 % вихід, E-ізомер). Загальна схема одержання кінцевих продуктів: спосіб 6 20 25 30 35 40 Синтез проміжної сполуки V1 Каталізатор Вілкінсона (58 мг; 62,6 мкмоль) додавали до розчину U1 (Z/E суміш, 260 мг; 626 мкмоль) в THF/MeOH (50/50) (66 мл), продутому шляхом барботування N2 протягом 15 хвилин. Суміш гідрогенізували (7 бар) при к.т. протягом 16 годин. Додатково додавали каталізатор Вілкінсона (58 мг; 62,6 мкмоля) і суміш гідрогенізували (7 бар) при к.т. протягом 6 годин. Реакційну суміш концентрували in vacuo з одержанням коричневої твердої речовини. Тверду речовину очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 25 г Merck, завантаження сухим способом, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 50/50) з одержанням 250 мг проміжної сполуки V1 у вигляді жовтого масла (54 % вихід). Синтез кінцевої сполуки 6 Суміш V1 (238 мг; 359 мкмоль) і заліза (120 мг; 2,16 ммоля) в оцтовій кислоті (20 мл) і воді (2,2 мл) перемішували при 80 °C протягом 6 годин. Додатково додавали залізо (120 мг; 2,16 ммоля) і суміш перемішували при 80 °C протягом 20 годин. Додатково додавали залізо (120 мг; 2,16 ммоля) і суміш перемішували при 80 °C протягом 5 годин. Суміш концентрували in vacuo з одержанням залишку. Залишок розбавляли в DMF і фільтрували через подушку із целіту. Імідазол SiliaBond (11,1 г; 12,9 ммоля) додавали до фільтрату та суміш перемішували при к.т. протягом 16 годин. Суміш фільтрували через подушку із целіту та фільтрат випарювали in vacuo з одержанням коричневої твердої речовини. Тверду речовину очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 12 г Grace, завантаження сухим способом, 13 UA 115588 C2 5 10 15 рухлива фаза, градієнт: CH2Cl2/MeOH/NH3водн. від 97/3/0,3 до 80/20/2) з одержанням 32 мг брудно-білої твердої речовини. Тверду речовину абсорбували за допомогою води, розтирали в порошок і обробляли ультразвуком. Отриману суспензію відфільтровували (склоподібна фрита n°5) і промивали за допомогою Et2O (двічі) з одержанням 19 мг кінцевої сполуки 6 у вигляді брудно-білої твердої речовини (15 % вихід). Синтез проміжної сполуки W1 NaH (60 % у маслі) (2,1 г; 52,1 ммоля) частинами додавали до 1-олу (74 мл) при к.т. Суміш перемішували при к.т. протягом 30 хвилин перед додаванням по краплях до розчину D1 (5,97 г; 13,2 ммоля) в THF (150 мл) при 0 °C. Отриману суміш потім перемішували при к.т. протягом 2 годин 30 хвилин і виливали в насичений водяний розчин NH 4Cl. Додавали EtOAc і насичений водяний розчин NaCl, шари розділяли й водний шар екстрагували за допомогою EtOAc (один раз). Об'єднані органічні шари висушували над MgSO4, фільтрували та видаляли розчинник при зниженому тиску з одержанням 6,77 г жовтого масла. Неочищену речовину очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 120 г Grace, упорскування рідини, рухлива фаза, градієнт: гептан/EtOAc від 100/0 до 50/50) з одержанням 5,12 г проміжної сполуки W1 у вигляді жовтого масла (83 % вихід). 20 14 UA 115588 C2 5 10 15 20 25 30 35 Синтез проміжної сполуки X1 До розчину W1 (3 г; 6,78 ммоля) в CH2Cl2 (1,3 л), дегазованому шляхом барботування N2 протягом 15 хвилин, додавали каталізатор Граббса 2-го покоління (578 мг; 678 мкмоль) при к.т. Розчин перемішували при к.т. протягом 20 годин. Додавали DMT SiliaBond (8,89 г; 5,42 ммоля) і суміш перемішували при к.т. протягом 20 годин. Реакційну суміш фільтрували через подушку із целіту та розчинник видаляли при зниженому тиску з одержанням коричневого залишку, який об'єднували з іншою партією (у розмірі 0,226 ммоля). Об'єднаний залишок абсорбували за допомогою MeOH, обробляли ультразвуком і нагрівали з одержанням осаду, який відфільтровували з одержанням 3,2 г коричневої твердої речовини. Неочищену речовину очищували препаративною LC (SiOH із зернами неправильної форми, 15-40 мкм, 220 г Grace, впорскування рідини, рухлива фаза, градієнт: CH2Cl2/EtOAc від 100/0 до 50/50) з одержанням 1,7 г фракції 1 у вигляді ясно-коричневої твердої речовини. Фракцію 1 абсорбували за допомогою MeOH, обробляли ультразвуком і нагрівали з одержанням осаду, який відфільтровували з одержанням 820 мг фракції 2 у вигляді ясно-коричневої твердої речовини. Фільтрат концентрували in vacuo з одержанням 590 мг фракції 3 у вигляді коричневого залишку (неочищений продукт X1). Фракцію 2 очищували препаративною LC (нерухлива фаза: сферичні гранули оксиду кремнію без покриття 5 мкм 150 × 30,0 мм, рухлива фаза, градієнт: гептан/EtOAc від 85/15 до 0/100) з одержанням 435 мг проміжної сполуки X1 у вигляді жовтої твердої речовини (E-ізомер, 15 % вихід). Фракцію 3 очищували з іншою партією. Синтез кінцевої сполуки 10 Суміш X1 (430 мг; 1,04 ммоля) і заліза (579 мг; 10,4 ммоля) в оцтовій кислоті (43 мол) і воді (3 мл) перемішували при 50 °C протягом 4 годин. Суміш концентрували насухо. Додавали DMF. Суміш обробляли ультразвуком, нагрівали та фільтрували через подушку із целіту, а целіт промивали використовуючи гарячий DMF. Імідазол SiliaBond (17,9 г; 20,8 ммоля) додавали до фільтрату та суміш перемішували при к.т. протягом 16 годин. Суміш фільтрували через целіт, целіт промивали за допомогою DMF і фільтрат концентрували in vacuo з одержанням 670 мг неочищеної сполуки. Неочищену речовину очищували препаративною LC (SiOH із зернами неправильної форми, 15-40 мкм, 25 г Merck, рухлива фаза, градієнт: CH 2Cl2/MeOH/NH3водн. від 98/2/0,2 до 85/15/1,5) з одержанням брудно-білої твердої речовини. Тверду речовину сушили при 40 °C при зниженому тиску протягом 20 годин з одержанням 295 мг кінцевої сполуки 10 у вигляді брудно-білої твердої речовини (84 % вихід). Загальна схема одержання кінцевих продуктів: спосіб 8 40 Синтез проміжної сполуки Y1 Каталізатор Вілкінсона (103 мг; 111 мкмоль) додавали до розчину X1 (230 мг; 0,555 ммоля) в 15 UA 115588 C2 5 10 15 20 25 THF/MeOH (50/50) (40 мл), продутому шляхом барботування N 2 протягом 15 хвилин. Суміш гідрогенізували (8 бар) при к.т. протягом 24 годин. Реакційну суміш концентрували in vacuo з одержанням коричневого залишку. Тверду речовину очищували препаративною LC (SiOH із зернами неправильної форми, 15-40 мкм, 12 г Grace, завантаження сухим способом, рухлива фаза, градієнт: CH2Cl2/EtOAc від 100/0 до 90/10) з одержанням 55 мг проміжної сполуки Y1 у вигляді жовтого залишку (24 % вихід). Синтез кінцевої сполуки 14 Суміш Y1 (55 мг; 0,132 ммоля) і заліза (74 мг; 1,32 ммоля) в оцтовій кислоті (5,5 мол) і воді (0,4 мл) перемішували при 50 °C протягом 20 годин. Додавали ще заліза (37 мг; 0,66 ммоля) і суміш перемішували при 50 °C протягом 3 годин. Додавали ще заліза (37 мг; 0,66 ммоля) і суміш перемішували при 50 °C протягом 20 годин. Суміш фільтрували через подушку із целіту, а целіт промивали оцтовою кислотою. Додавали ще заліза (74 мг; 1,32 ммоля) до фільтрату та суміш перемішували при 50 °C протягом 88 годин. Додавали ще заліза (74 мг; 1,32 ммоля) до фільтрату та суміш перемішували при 80 °C протягом 24 годин. Циклізацію не здійснювали. Суміш концентрували in vacuo з одержанням коричневої твердої речовини. TiCl3 (8,60 мл; 10,0 ммоля) краплями додавали до розчину коричневої твердої речовини в C (19 мл). Суміш перемішували при к.т. протягом ночі. Суміш підлуговували шляхом додавання порошку K2CO3 при 0 °C. Отриману суміш фільтрували через подушку із целіту та целіт промивали за допомогою розчину AcOEt/MeOH (8:2). Фільтрат концентрували in vacuo. Неочищену тверду речовину очищували препаративною LC (SiOH із зернами неправильної форми, 15-40 мкм, 10 г Merck, завантаження сухим способом, рухлива фаза, градієнт: CH2Cl2/MeOH/NH3водн. від 98/2/0,2 до 85/15/1,5). Фракції, що містили продукт, об'єднували та розчинник видаляли in vacuo з одержанням 20 мг кінцевої сполуки 14 (12 % вихід) у вигляді брудно-білої твердої речовини. Загальна схема одержання кінцевих продуктів: спосіб 9 16 UA 115588 C2 5 Синтез проміжної сполуки A2 Метансульфонілхлорид (8,4 мл; 108 ммоль) додавали до розчину Z1 (14 г; 72,1 ммоля), NEt 3 (20 мл; 144 ммоля) і LiCl (4,6 г; 108 ммоль) у сухому CH 2Cl2 (980 мол). Суміш перемішували при к.т. протягом 1 години 30 хвилин. Додавали воду та шари розділяли. Органічний шар промивали водою (один раз), сушили над MgSO4, фільтрували та концентрували in vacuo з одержанням 18,8 г A2 (96 %) у вигляді зеленого масла. 17 UA 115588 C2 5 10 15 Синтез проміжної сполуки B2 Проміжну сполуку B2 одержували за допомогою процедури, описаної для проміжної сполуки C1 (78 % вихід у вигляді жовтого масла). Синтез проміжної сполуки C2 Проміжну сполуку C2 одержували за допомогою процедури, описаної для проміжної сполуки D1 (кількісний вихід у вигляді жовтого масла). Синтез проміжної сполуки D2 Проміжну сполуку D2 одержували за допомогою процедури, описаної для проміжної сполуки W1 (64 % вихід у вигляді жовтої твердої речовини). 18 UA 115588 C2 5 10 Синтез проміжної сполуки E2 Розчин D2 (1 г; 2,12 ммоля) у сухому CH2Cl2 (400 мл) дегазували за допомогою барботування N2 протягом 15 хвилин. Додавали каталізатор Граббса 2-го покоління (181 мг; 212 мкмоль) і суміш перемішували при к.т. протягом 16 годин. Додавали DMT SiliaBond (2,78 г; 1,69 ммоля) і суміш перемішували при к.т. протягом 16 годин. Суміш фільтрували через подушку із целіту, а фільтрат концентрували in vacuo з одержанням 1,11 г коричневого масла. Неочищену речовину очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 50 г, Merck, рухлива фаза, градієнт: CH2Cl2/EtOAc від 100/0 до 90/10). Фракції, що містили продукт, об'єднували та розчинник видаляли in vacuo з одержанням 386 мг проміжної сполуки E2 (41 %, ізомер E (96,2 %) плюс ізомер Z (3,8 %)) у вигляді жовтої піни. 15 20 25 30 Синтез кінцевої сполуки 15 Залізо (291 мг, 5,21 ммоля) додавали до розчину Е2 (386 мг, 0,869 ммоля) в оцтовій кислоті (36 мл) і воді (3 мл). Суміш перемішували при 80 °C протягом 6 годин. Додавали залізо (146 мг; 2,61 ммоля) і суміш перемішували при 80 °C протягом 16 годин. Знову додавали залізо (146 мг; 2,61 ммоля) і суміш перемішували при 80 °C протягом 5 годин. Суміш концентрували насухо. Додавали DMF, суміш фільтрували через целіт і целіт промивали використовуючи DMF. Імідазол SiliaBond (18 г; 20,9 ммоля) додавали до фільтрату та суміш перемішували при к.т. протягом 72 годин. Суміш фільтрували через целіт, целіт промивали за допомогою DMF і фільтрат концентрували in vacuo з одержанням 428 мг коричневої твердої речовини. Тверду речовину абсорбували в CH3CN, що призводило до його випадання в осад. Осад фільтрували з одержанням 267 мг коричневої твердої речовини. Тверду речовину очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 10 г Merck, завантаження сухим способом, рухлива фаза, градієнт: CH2Cl2/MeOH/NH3водн. від 95/5/0,5 до 85/15/1,5). Фракції, що містять продукт, об'єднували та розчинник видаляли in vacuo з одержанням 124 мг брудно-білої твердої речовини. Тверду речовину очищували зворотно-фазовою хроматографією (нерухлива фаза: Sunfire-C18 5 мкм 19 × 150 мм, рухлива фаза, градієнт: CH3CN/H2O (мурашина кислота 0,1 %) від 5/95 до 50/50) з одержанням 72 мг кінцевої сполуки 15 (23 % вихід) у вигляді білої твердої речовини. Загальна схема одержання кінцевих продуктів: спосіб 10 35 19 UA 115588 C2 5 10 15 Синтез проміжної сполуки F2 Проміжну сполуку F2 одержували за допомогою процедур, описаних для проміжної сполуки F1 (E-ізомер). Синтез кінцевої сполуки 16 При к.т. TiCl3 (12,3 мл; 14,341 ммоля) краплями додавали до суміші F2 (300 мг; 0,717 ммоля) в THF (30 мл). Суміш перемішували при к.т. протягом 2 годин. Суміш прохолоджували до 0 °C і підлуговували за допомогою порошку K2CO3. Отриману в результаті мутну суміш фільтрували через подушку із целіту, а целіт промивали за допомогою розчину AcOEt/CH 3OH 8/2. Фільтрат частково випарювали з одержанням 175 мг кінцевої сполуки через 16 після фільтрації білої твердої речовини й сушіння при зниженому тиску при 85 °C (71 % вихід). Синтез кінцевої сполуки 17 Одержували хлористоводневу сіль із 10 экв. 4 Н HCl у діоксані, яку додавали в суспензію сполуки 16 (100 мг; 0,292 ммоля) в CH3OH (10 мл). Осад перемішували протягом 3 годин, фільтрували та сушили у вакуумі при 90 °C протягом ночі. Проводили солюбілізацію твердої речовини в MeOH/CH2Cl2 50/50, додавали CH3CN і випарювали розчинник до одержання осаду білої твердої речовини, який фільтрували та сушили у вакуумі при 90 °C з одержанням 47 мг кінцевої сполуки 17 у вигляді солі HCl (0,93 HCl, 0,51 H2O; 42 % вихід). 20 20 UA 115588 C2 5 Синтез проміжної сполуки H2 При -20 °C у потоці N2 додавали G2 (22,0 г; 72,04 ммоля) в THF (100 мл) краплями до суспензії LiAlH4 (3,28 г; 86,45 ммоля) в THF (120 мл). Суміш перемішували при 0 °C протягом 1 години. 3,5 мл води додавали по краплях з наступним додаванням 3,5 мл NaOH, 3 N, і 10 мл води. Отриману суміш фільтрували через подушку із целіту, а целіт промивали за допомогою EtOAc. Фільтрат концентрували при зниженому тиску з одержанням 19 г проміжної сполуки H2 у вигляді жовтого масла (кількісний вихід). 21 UA 115588 C2 5 10 15 20 Синтез проміжної сполуки I2 При 0 °C діізопропілазодікарбоксілат (4,0 мл; 20,32 ммоля) краплями додавали до суміші А1 (5,0 г; 13,55 ммоля), H2 (4,28 г; 16,26 ммоля) і PPh3 (5,33 г; 20,327 ммоля) в THF (100 мл). Суміш перемішували при к.т. протягом 12 годин. Додавали EtOAc і воду. Шари декантували. Органічний шар промивали водою, сушили над MgSO4, фільтрували та розчинник випарювали. Неочищену речовину розчиняли в гептані/EtOAc 80/20, осад відфільтровували (в основному POPh3) і фільтрат очищували за допомогою хроматографії. Очищення проводили за допомогою флеш-хроматографії через силікагель (15-40 мкм, 220 г, гептан/EtOAc 80/20). Чисті фракції збирали й випарювали насухо з одержанням 8,2 г проміжної сполуки I2 (99 % вихід). Синтез проміжної сполуки J2 I2 (8,2 г; 13,349 ммоля) перемішували в NH4OH (100 мл) і THF (100 мл) при к.т. протягом 24 годин. Суміш наполовину випарювали при зниженому тиску. Залишок абсорбували за допомогою EtOAc. Органічний шар промивали водою, сушили над MgSO 4, фільтрували та розчинник випарювали з одержанням 8,15 г проміжної сполуки J2 (кількісний вихід). Неочищену речовину використовували безпосередньо на наступному етапі. 22 UA 115588 C2 5 10 15 20 25 Синтез проміжної сполуки K2 У потоці N2 додавали NaH (60 % у маслі) (1,15 г; 28,64 ммоля) частинами до алілового спирту (35 мл) при к.т. Суміш перемішували при к.т. протягом 30 хвилин перед додаванням по краплях до розчину J2 (4,0 г; 7,26 ммоля) в THF (80 мл) при 0 °C. Отриману суміш потім перемішували при к.т. протягом 2 годин 30 хвилин і виливали в насичений розчин NH4Cl. Додавали EtOAc і насичений водяний розчин NaCl, шари розділяли й водний шар екстрагували за допомогою EtOAc (один раз). Об'єднані органічні шари висушували над MgSO 4, фільтрували й видаляли розчинник при зниженому тиску з одержанням 4,7 г жовтого масла. Виконували очищення флеш-хроматографією на силікагелі (15-40 мкм, 80 г, CH2Cl2/гептан 65/35). Чисті фракції збирали й випарювали насухо з одержанням 2,65 г проміжної сполуки K2 (69 % вихід). Синтез проміжної сполуки L2 Перед здійсненням реакції дихлоретан дегазували шляхом барботування N 2. У пробірці Шленка розчин K2 (1,3 г; 2,464 ммоля) і хлордициклогексилборан (1 M у гексані) (493 мкл; 0,493 ммоля) у дихлоретані (600 мл) перемішували при 80 °C в атмосфері N2 протягом 1 години. Додавали каталізатор Граббса-Ховейди 2-го покоління (609 мг; 0,493 ммоля) і суміш перемішували при 120 °C протягом 16 годин. Додавали DMT SiliaBond (2,98 г; 1,82 ммоля) і суміш перемішували при к.т. протягом 16 годин. Реакційну суміш фільтрували через целіт і фільтрат випарювали in vacuo з одержанням 1,6 г, які об'єднували з іншою реакційною сумішшю (у розмірі 2,46 ммоля) перед очищенням (загальна вага до очищення 3,2 g). Виконували очищення флеш-хроматографією на силікагелі (15-40 мкм, 80 г, CH2Cl2/CH3OH: 99,5/0,5). Чисті фракції збирали й випарювали насухо з одержанням 0,99 г F1 (E/Z суміш передбачуваної сполуки, 40 % вихід) і 0,65 г F2 (вихідний матеріал K2). F1 додатково очищували ахіральною SFC (нерухлива фаза: NH 2 5 мкм 150*30 мм), рухлива фаза: 92 % CO2, 8 % MeOH) з одержанням 664 мг проміжної сполуки L2 (E-ізомер, 27 % вихід). 30 35 Синтез проміжної сполуки M2 Залізо (1,45 г, 26,025 ммоля) додавали до суміші L2 (0,65 г, 1,301 ммоля) в оцтовій кислоті (15 мл) і воді (1,5 мл). Суміш перемішували при 50 °C протягом 3 годин, а потім фільтрували через целіт за допомогою CH2Cl2/MeOH. Фільтрат концентрували при зниженому тиску. Сполуки очищували за допомогою флеш-хроматографії через силікагелеву колонку (15-40 мкм; 80 г, елюент CH2Cl2/CH3OH/NH4OH 96/4/0,5) з одержанням 640 мг. Проводили друге очищення за допомогою флеш-хроматографії через силікагель (15-40 мкм, 40 г, CH2Cl2/CH3OH/NH4OH: 97/3/0,2). Чисті фракції збирали й випарювали насухо з одержанням 240 мг проміжної сполуки М2 (38 % вихід). 23 UA 115588 C2 5 10 15 20 25 Синтез кінцевої сполуки 18 При 0 °C додавали CF3CO2H (0,455 мл) краплями до суміші M2 (100 мг, 0,236 ммоля) в CH2Cl2 (1 мл). Суміш перемішували при к.т. протягом ночі, а потім підлуговували 10 % розчином K2CO3 у воді. Осад відфільтровували, промивали водою та CH 3CN і наприкінці сушили у вакуумі з одержанням 35 мг кінцевої сполуки 18 (E-ізомер, 46 % вихід). Загальна схема одержання кінцевих продуктів: спосіб 12 Синтез проміжної сполуки N2 Суміш M2 (140 мг, 0,331 ммоля) в THF/CH3OH (50/50) (30 мл) гідрогенізували під тиском 10 бар з каталізатором Вілкінсона (61,2 мг, 0,0661 ммоля) протягом 72 годин. Додавали DMT SiliaBond (441 мг, 0,264 ммоля) і суміш перемішували при к.т. протягом 18 годин. Суміш фільтрували через подушку із целіту, а целіт промивали за допомогою CH 2Cl2/CH3OH 95/5. Фільтрат концентрували при зниженому тиску. Виконували очищення флеш-хроматографією на силікагелі (15-40 мкм, 10 г, CH2Cl2/CH3OH/NH4OH: 97/3/0,1). Чисті фракції збирали й випарювали насухо з одержанням 62 мг проміжної сполуки N2 (44 % вихід), яку використовували без додаткового очищення на наступному етапі. Синтез кінцевої сполуки 23 При 0 °C додавали CF3CO2H (0,281 мл, 3,643 ммоля) краплями до суміші N2 (62 мг, 0,146 ммоля) в CH2Cl2 (1 мл). Суміш перемішували при к.т. протягом ночі. Суміш підлуговували 10 % розчином K2CO3 у воді. Суміш двічі екстрагували за допомогою CH 2Cl2 і CH3OH (80/20). Органічний шар сушили над MgSO4, фільтрували та розчинник випарювали. Неочищену речовину абсорбували за допомогою DMF, додавали 2 г SiO 2, 60-200 мкм, і отриману суспензію випарювали насухо. Даний залишок вносили на хроматографічну колонку (для відкладання твердої речовини). Виконували очищення флеш-хроматографією на силікагелі (15-40 мкм, 25 г, CH2Cl2/CH3OH/NH4OH: 95/5/0,5). Чисті фракції збирали й випарювали насухо з одержанням 20 мг. Фракцію абсорбували за допомогою CH 3CN, осад відфільтровували й сушили у вакуумі з одержанням 18 мг кінцевої сполуки 23 (38 % вихід). 30 Синтез проміжної сполуки O2 Проміжну сполуку О2 одержували за допомогою процедур, описаних для проміжної сполуки X1 (E-ізомер). 24 UA 115588 C2 5 10 Синтез кінцевої сполуки 21 При к.т. додавали TiCl3 (51,5 мл; 60,128 ммоля) краплями до суміші O2 (1,3 г; 3,006 ммоля) в THF (130 мл). Суміш перемішували при к.т. протягом 2 годин. Суміш прохолоджували до 0 °C, а потім підлуговували за допомогою порошку K 2CO3. Отриману в результаті мутну суміш фільтрували через подушку із целіту, а целіт промивали за допомогою розчину AcOEt/CH 3OH 8/2. Фільтрат частково випарювали з одержанням 380 мг кінцевої сполуки 21 (35 % вихід) після фільтрації білої твердої речовини й сушіння у вакуумі при 85 °C. Синтез кінцевої сполуки 22 Сполука 21 (118 мг, 0,331 ммоля) в CH3OH (2 мл) плюс CH3CN (2 мл) прохолоджували до 10 °C. HCl (6M в ізопропанолі) (0,16 мл, 0,993 ммоля) краплями додавали й суміш перемішували при к.т. протягом 1 години. Осад відфільтровували, промивали за допомогою Et 2O і сушили у вакуумі з одержанням 109 мг кінцевої сполуки 22 у вигляді солі HCl (0,76 HCl 0,81 H 2O, 83 % вихід). 15 20 25 30 35 Синтез проміжної сполуки P2 Суміш O2 (320 мг; 0,74 ммоля) каталізатора Вілкінсона (137 мг; 0,148 ммоля) в THF/CH 3OH (50/50) (45 мл) гідрогенізували під тиском 10 бар при к.т. протягом 20 годин. Розчинник випарювали у вакуумі. Неочищену речовину очищували за допомогою флеш-хроматографії через силікагелеву колонку (15-40 мкм; 24 г) у гептані/AcOEt, 80/20, з одержанням 310 мг проміжної сполуки P2 (96 % вихід). Синтез кінцевої сполуки 19 При к.т. додавали TiCl3 (9,5 мл; 11,049 ммоля) краплями до суміші Р2 (0,24 г; 0,552 ммоля) в THF (25 мл). Суміш перемішували при к.т. протягом 2 годин. Суміш прохолоджували до 0 °C, а потім підлуговували за допомогою порошку K2CO3. Отриману в результаті мутну суміш фільтрували через подушку із целіту, а целіт промивали за допомогою розчину AcOEt/CH 3OH 8/2. Фільтрат частково випарювали з одержанням 100 мг кінцевої сполуки 19 (50 % вихід) після фільтрації білої твердої речовини й сушіння у вакуумі при 85 °C. Синтез кінцевої сполуки 20 Сполуку 19 (58 мг; 0,162 ммоля) в CH3OH (2 мл) плюс CH3CN (4 мл) охолоджували до 5 °C. Краплями додавали HCl (6 M в ізопропанолі) (81 мкл; 0,486 ммоля) і суміш перемішували при к.т. протягом 1 години. Осад відфільтровували, промивали діізопропіленефіром і сушили у вакуумі при 90 °C з одержанням 57 мг кінцевої сполуки 20 у вигляді солі HCl (0,88 HCl 0,04 H 2O, 89 % вихід). 25 UA 115588 C2 5 Синтез проміжної сполуки R2 При 0 °C діізопропілазодікарбоксілат (3,8 мл; 19,107 ммоля) краплями додавали до суміші А1 (4,7 г; 12,738 ммоля), Q2 (2,27 г; 12,738 ммоля) і PPh3 (5 г; 19,107 ммоля) в THF (100 мл). Суміш перемішували при к.т. протягом 12 годин. Додавали EtOAc і воду. Шари декантували. Органічний шар промивали водою, сушили над MgSO4, фільтрували та розчинник випарювали. Неочищену речовину очищували за допомогою колонкової хроматографії через силікагель (1540 мкм; 220 г) у гептані/AcOEt, 85/15, з одержанням 5,3 г проміжної сполуки R2 (79 % вихід). 10 26 UA 115588 C2 5 10 15 20 25 30 Синтез проміжної сполуки S2 R2 (5,3 г; 10,015 ммоля) вмішували в THF (80 мл) і NH4OH (80 мл) при к.т. протягом 24 годин. Суміш концентрували при зниженому тиску. Залишок абсорбували за допомогою CH 2Cl2, відфільтровували осад (неорганічний) іконцентрували фільтрат при зниженому тиску. Неочищену речовину очищували за допомогою колонкової хроматографії через силікагель (1540 мкм; 220 г) у гептані/AcOEt, 85/15, з одержанням 3,65 г проміжної сполуки S2 (78 % вихід). Синтез проміжної сполуки T2 NaH (1,35 г; 33,88 ммоля) частинами додавали до алілового спирту (41 мл) при к.т. Суміш перемішували при к.т. протягом 30 хвилин перед додаванням по краплях до розчину S2 (4 г; 8,597 ммоля) в THF (100 мл) при 0 °C. Отриману суміш потім перемішували при к.т. протягом 2 годин 30 хвилин і виливали в насичений водяний розчин NH 4Cl. Додавали EtOAc і насичений водяний розчин NaCl, шари розділяли й водний шар екстрагували за допомогою EtOAc (один раз). Об'єднані органічні шари висушували над MgSO 4, фільтрували й видаляли розчинник при зниженому тиску з одержанням жовтого масла. Неочищену речовину очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм, 120 г Grace, впорскування рідини, рухлива фаза, градієнт: гептан/EtOAc 85/15) з одержанням 3,2 г проміжної сполуки T2 у вигляді жовтого масла (84 % вихід). Синтез проміжної сполуки U2 Розчин T2 (1 г; 2,26 ммоля) і хлордициклогексилборану (1 M у гексані) (904 мкл; 904,013 мкмоля) у сухому дихлоретані (540 мл) перемішували при 80 °C і в атмосфері N2 протягом 1 години. Суміш дегазували шляхом барботування N2 протягом 15 хвилин, додавали каталізатор Граббса-Ховейди 2-го покоління (141,6 мг; 226 мкмоль), суміш ще раз дегазували шляхом барботування N2 протягом 15 хвилин, а потім перемішували при 120 °C протягом 16 годин. Ще раз додавали 0,25 екв. каталізатора та суміш перемішували при 120 °C протягом 16 годин. 27 UA 115588 C2 5 10 15 Додавали DMT SiliaBond (5,9 г; 3,616 ммоля) і суміш перемішували при к.т. протягом 16 годин. Суміш фільтрували через подушку із целіту та фільтрат концентрували у вакуумі з одержанням чорного масла. Неочищену сполуку очищували препаративною LC (SiOH із зернами неправильної форми 15-40 мкм 80 г, Merck, рухлива фаза: CH 2Cl2/AcOEt 97/3). Фракції, що містили продукт, об'єднували та розчинник видаляли у вакуумі з одержанням 335 мг проміжної сполуки U2 (E-ізомер, 36 % вихід). Синтез кінцевої сполуки 25 Залізо (0,45 г, 8,084 ммоля) додавали до суміші U2 (0,335 г, 0,808 ммоля) в оцтовій кислоті (24 мл) плюс вода (5 мл). Суміш енергійно перемішували при 50 °C протягом 5 годин. Додавали CH2Cl2 і фільтрували реакційну суміш через подушку із целіту, а потім промивали оцтовою кислотою. Розчинник видаляли при зниженому тиску. Неочищену речовину очищували хроматографією на силікагелевій колонці (SiO2 15-40 мкм, 25 г) в CH2Cl2/CH3OH/NH4OH 96/4/0,5 з одержанням 154 мг кінцевої сполуки 25 (56 % вихід). Сполуку кристалізували в CH 3OH, фільтрували та сушили у вакуумі при 90 °C з одержанням 70 мг (25 % вихід). 28
ДивитисяДодаткова інформація
Назва патенту англійськоюMacrocyclic deaza-purinones for the treatment of viral infections
Автори англійськоюBonfanti, Jean-Francois, Fortin, Jerome Michel Claude, Muller, Philippe, Doublet, Frederic Marc Maurice, Raboisson, Pierre Jean-Marie Bernard, Arnoult, Eric Pierre Alexandre
Автори російськоюБонфанти Жан-Франсуа, Фортен Жэром Мишель Клод, Мюллер Филипп, Дубле Фрэдэрик Марк Морис, Рабуассон Пьер Жан-Мари Бернар, Арну Эрик Пьер Александр
МПК / Мітки
МПК: A61K 31/522, C07D 473/00
Мітки: деазаоксипурини, лікування, макроциклічні, інфекцій, вірусних
Код посилання
<a href="https://ua.patents.su/56-115588-makrociklichni-deazaoksipurini-dlya-likuvannya-virusnikh-infekcijj.html" target="_blank" rel="follow" title="База патентів України">Макроциклічні деазаоксипурини для лікування вірусних інфекцій</a>
Макроциклічні пурини для лікування вірусних інфекцій

Номер патенту: 113651
Опубліковано: 27.02.2017
Автори: Мюллер Філіпп, Рабуассон П'єр Жан-Марі Бернар, Дубле Фредерік Марк Моріс, Бонфанті Жан-Франсуа, Фортен Жером Мішель Клод, Арну Ерік П'єр Александр
МПК: A61K 31/52, C07D 498/16, C07D 487/16
Мітки: лікування, пурини, інфекцій, вірусних, макроциклічні
Формула / Реферат:
1. Сполука, що характеризується формулою (І), (I)де n=1-3,або її фармацевтично прийнятні солі, де X являє собою кисень, азот, сірку або,Y являє собою ароматичне кільце або гетероциклічне кільце, що містить щонайменше азот, необов'язково заміщене одним або декількома...
Похідні піперидинопіримідину для лікування вірусних інфекцій

Номер патенту: 112668
Опубліковано: 10.10.2016
Автори: Даубі Хамлічі Мурад, Йонкерс Тім Хьюго Марія, МакГован Девід Крейг, Рабуассон П'єр Жан-Марі Бернар
МПК: C07D 471/04, A61K 31/519
Мітки: піперидинопіримідину, похідні, лікування, вірусних, інфекцій
Формула / Реферат:
1. Сполука формули (І) (I)або її фармацевтично прийнятна сіль, деА вибраний з групи, що складається з СН2 і NCOR2 в будь-якій стереохімічній конфігурації,В вибраний з групи, що складається з СН2 і NCOR4 в будь-якій стереохімічній конфігурації,за умови, що, якщо А являє собою NCOR2, тоді В не являє собою NCOR4,X вибраний з СН2 в...
Похідні піримідину для лікування вірусних інфекцій

Номер патенту: 113956
Опубліковано: 10.04.2017
Автори: Ембрехтс Вернер, Ласт Стефан Джульєн, Мк Гован Давід, Ребойсон П'єр Жан-Марія Бернард, Пітерс Серж Марія Алойсіс, Влач Яромір, Джонкерс Тім Хьюго Марія
МПК: C07D 405/12, C07D 403/12, C07D 401/12, C07D 239/48, C07D 413/12, C07D 471/04, A61K 31/505, A61K 31/506
Мітки: лікування, інфекцій, піримідину, вірусних, похідні
Формула / Реферат:
1. Сполука формули (І)або її фармацевтично прийнятна сіль, таутомер (таутомери) або сольват, деR1 являє собою водень, С1-4алкіл, циклопропіл або С1-6алкокси, галоген, гідроксил, трифторметил,R2 являє собою С1-8алкіл, (С1-4)алкоксі-(С1-4)алкіл, С3-7циклоалкіл, С4-7гетероцикл, ароматичний, біциклічний гетероцикл, арилалкіл, гетероарил,...
Алкілпіримідинові похідні для лікування вірусних інфекцій та подальших захворювань

Номер патенту: 115558
Опубліковано: 27.11.2017
Автори: Йонкерс Тім Хьюго Марія, МакГован Девід Крейг, Рабуассон П'єр Жан-Марі Бернар
МПК: A61K 31/505, C07D 401/06, C07D 239/48, A61K 31/506, C07D 409/06
Мітки: захворювань, вірусних, лікування, інфекцій, подальших, похідні, алкілпіримідинові
Формула / Реферат:
1. Сполука формули (І)або її фармацевтично прийнятна сіль, деR1 являє собою водень, піридиніл, тіофеніл, хінолініл або феніл, необов'язково заміщені однією або двома С1-6алкоксигрупами;R2 являє собою С1-6алкіл;за умови, що N-(2-аміно-5-фенетилпіримідин-4-іл)-N-пентиламін виключений.2. Сполука за п. 1, де R1 являє собою водень, і де...
Похідні 1,2,4-триазину для лікування вірусних інфекцій

Номер патенту: 114644
Опубліковано: 10.07.2017
Автори: Мак Ґован Дейвід Ґрейґ, Оаро Крістоф, Жюбо Філіп, ійємон Жером Еміль Жорж, Жамбю Венсан, Лєваше Венсан, Бонфанті Жан-Франсуа
МПК: C07D 253/07, C07D 401/12, A61K 31/53
Мітки: лікування, 1,2,4-триазину, вірусних, похідні, інфекцій
Формула / Реферат:
1. Сполука формули (І)або її фармацевтично прийнятна сіль або таутомер(и), де:R1 являє собою С1-6алкіл, арилалкіл або гетероарилалкіл, кожний з яких необов'язково заміщено одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, С1-6алкілу, ді-(С1-6)алкіламіно, С1-6алкіламіно, С1-6алкокси, С3-6циклоалкілу, карбонової...