Спосіб одержання азациклопептидів (варіанти)
Номер патенту: 44763
Опубліковано: 15.03.2002
Автори: Хьюджес Девід Л., Белік Кевін М., Блек Регіна М., Бендер Дін Р., Леонард Уільям




Формула / Реферат
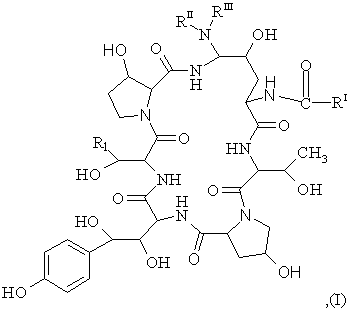
1. Спосіб одержання азациклопептидів формули (І)
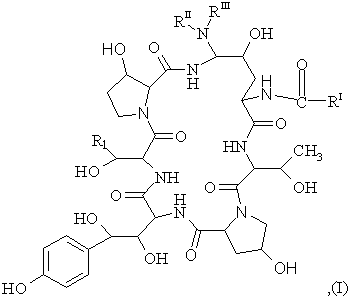
(SEQ ID No. l)
де
R1 становить собою СН2СН2NН2 або CH2CONH2;
RI становить собою (С9-С21)-алкіл, (С9-С21)-алкеніл, (С1-С10)-алкоксифеніл, (С1-С10)-алкоксинафтил або (С1-С10)-алкокситерфеніл;
RII становить собою Н, (С1-С4)-алкіл, (С3-С4)-алкеніл, (СН2)2-4OН або (CH2)2-4NRIVRV;
RIII становить собою Н, (С1-С4)-алкіл, (С3-С4)-алкеніл, (СН2)2-4OН, (CH2)2-4NRIVRV або
RIIта RIII разом становлять (СН2)4, (СН2)5, (СН2)2O(СН2)2 або (CH2)2NH(CH2)2;
RIV становить собою Н або (С1-С4)-алкіл;
RV становить собою Н або (С1-С4)-алкіл; або їх фармацевтичнo пристосованих кислотно-аддитивних солей, що складається з наступних стадій:
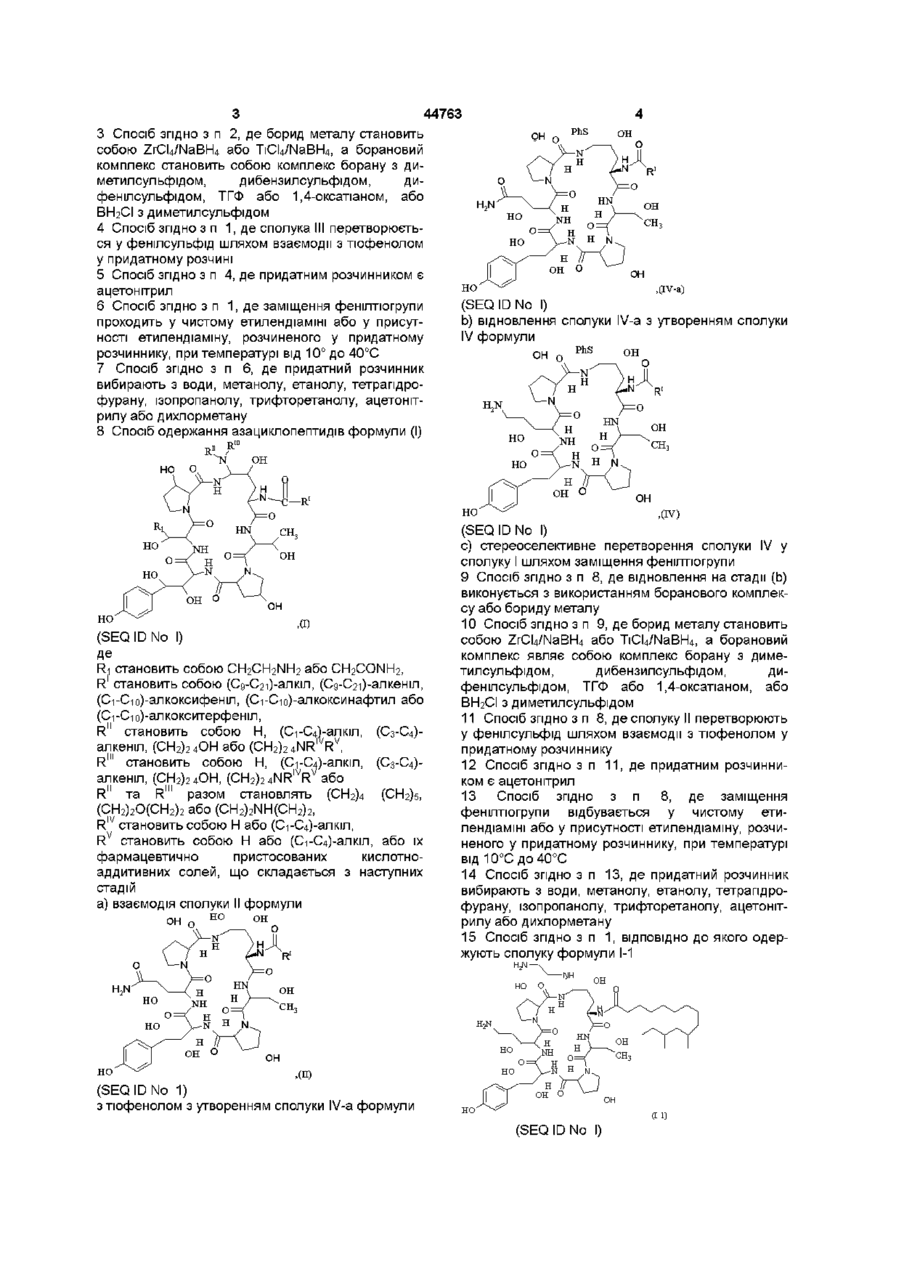
а) відновлення сполуки II формули
(SEQ ID No. l)
з утворенням сполуки III формули
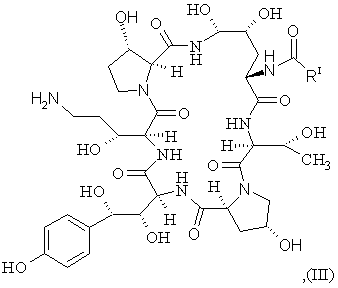
(SEQ ID No. l)
b) перетворення сполуки III з утворенням сполуки IV формули
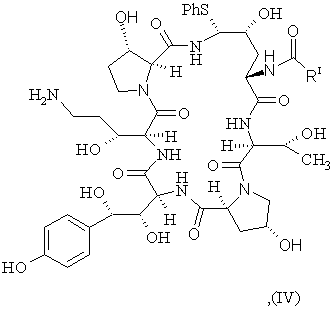
(SEQ ID No. l)
с) стереоселективне перетворення сполуки IV у сполуку І шляхом заміщення фенілтіогрупи.
2. Спосіб згідно з п. 1, де відновлення на стаії (а) виконують з використанням боранового комплексу або бориду металу.
3. Спосіб згідно з п. 2, де борид металу становить собою ZrCl4/NaBH4 або ТіСl4/NаВН4, а борановий комплекс становить собою комплекс борану з диметилсульфідом, дибензилсульфідом, дифенілсульфідом, ТГФ або 1,4-оксатіаном, або BH2Cl з диметилсульфідом.
4. Спосіб згідно з п. 1, де сполука ІІІ перетворюється у фенілсульфід шляхом взаємодії з тіофенолом у придатному розчині.
5. Спосіб згідно з п. 4, де придатним розчинником є ацетонітрил.
6. Спосіб згідно з п. 1, де заміщення фенілтіогрупи проходить у чистому етилендіаміні або у присутності етилендіаміну, розчиненого у придатному розчиннику, при температурі від 10° до 40°С.
7. Спосіб згідно з п. 6, де придатний розчинник вибирають з води, метанолу, етанолу, тетрагідрофурану, ізопропанолу, трифторетанолу, ацетонітрилу або дихлорметану.
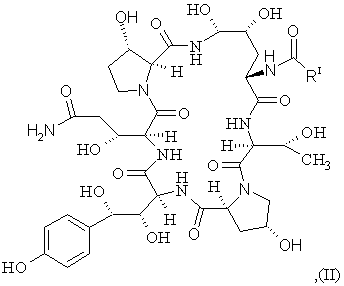
8. Спосіб одержання азациклопептидів формули (І)
(SEQ ID No. l)
де
R1 становить собою СН2СН2NН2 або CH2CONH2;
RI становить собою (С9-С21)-алкіл, (С9-С21)-алкеніл, (С1-С10)-алкоксифеніл, (С1-С10)-алкоксинафтил або (С1-С10)-алкокситерфеніл;
RII становить собою Н, (С1-С4)-алкіл, (С3-С4)-алкеніл, (СН2)2-4OН або (CH2)2-4NRIVRV;
RIII становить собою Н, (С1-С4)-алкіл, (С3-С4)-алкеніл, (СН2)2-4OН, (CH2)2-4NRIVRV або
RII та RIII разом становлять (СН2)4, (CН2)5, (СН2)2O(СН2)2 або (CH2)2NН(CH2)2;
RIV становить собою Н або (С1-С4)-алкіл;
RV становить собою Н або (С1-С4)-алкіл; або їх фармацевтичнo пристосованих кислотно-аддитивних солей, що складається з наступних стадій:
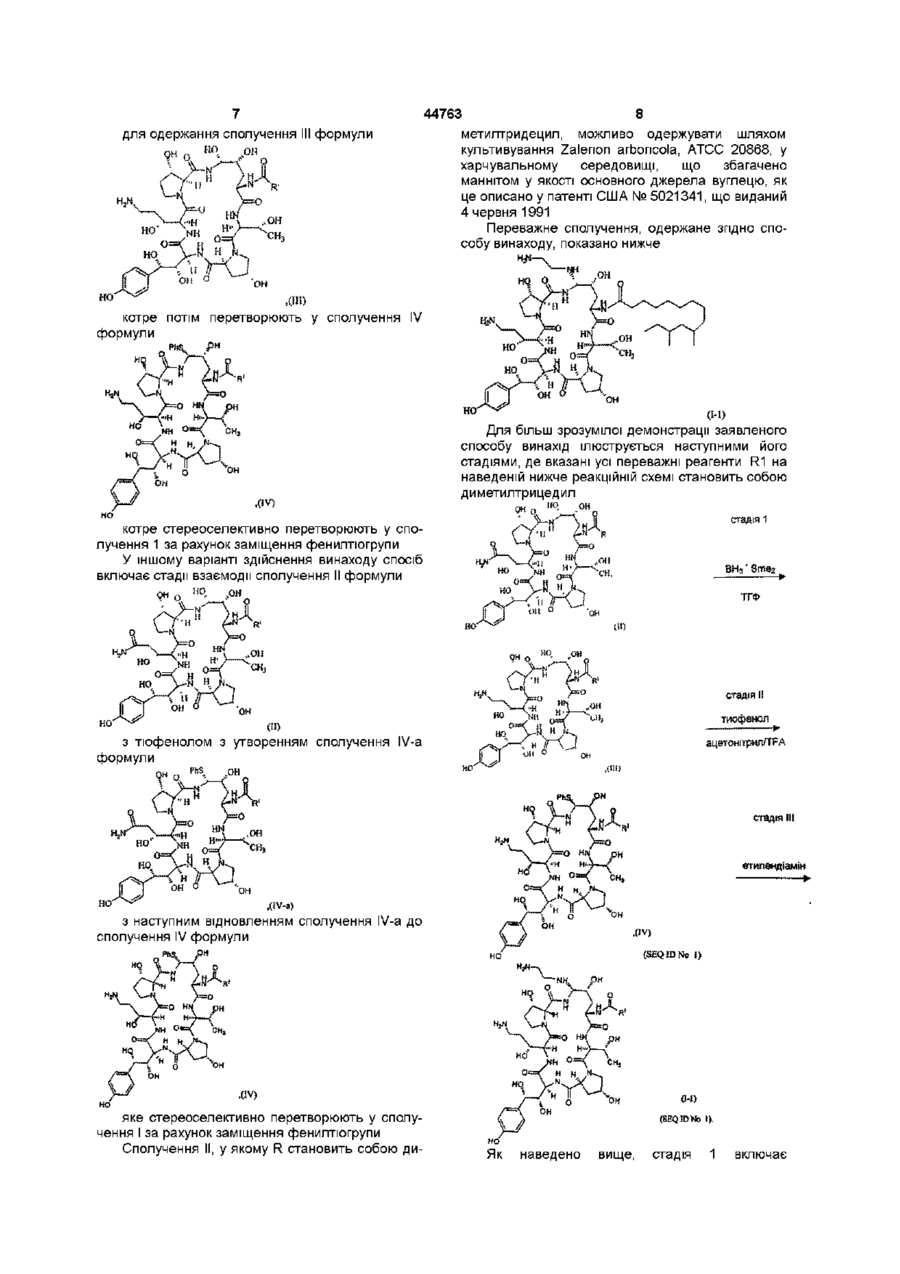
а) взаємодія сполуки II формули
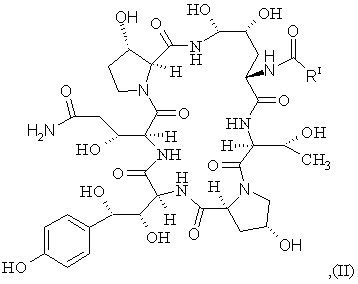
(SEQ ID No. 1)
з тіофенолом з утворенням сполуки IV-a формули
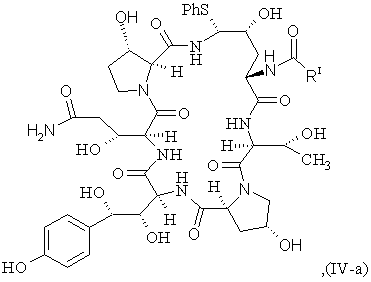
(SEQ ID No. l)
b) відновлення сполуки IV-a з утворенням сполуки IV формули
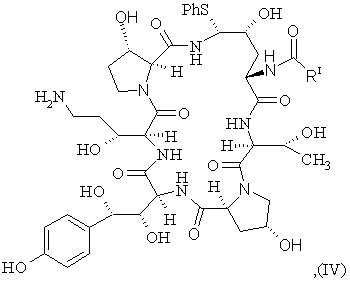
(SEQ ID No. l)
с) стереоселективне перетворення сполуки IV у сполуку І шляхом заміщення фенілтіогрупи.
9. Спосіб згідно з п. 8, де відновлення на стадії (b) виконується з використанням боранового комплексу або бориду металу.
10. Спосіб згідно з п. 9, де борид металу становить собою ZrCl4/NaBH4 або ТіСl4/NаВН4, а борановий комплекс являє собою комплекс борану з диметилсульфідом, дибензилсульфідом, дифенілсульфідом, ТГФ або 1,4-оксатіаном, або ВН2Сl з диметилсульфідом.
11. Спосіб згідно з п. 8, де сполуку II перетворюють у фенілсульфід шляхом взаємодії з тіофенолом у придатному розчиннику.
12. Спосіб згідно з п. 11, де придатним розчинником є ацетонітрил.
13. Спосіб згідно з п. 8, де заміщення фенілтіогрупи відбувається у чистому етилендіаміні або у присутності етилендіаміну, розчиненого у придатному розчиннику, при температурі від 10°С до 40°С.
14. Спосіб згідно з п. 13, де придатний розчинник вибирають з води, метанолу, етанолу, тетрагідрофурану, ізопропанолу, трифторетанолу, ацетонітрилу або дихлорметану.
15. Спосіб згідно з п. 1, відповідно до якого одержують сполуку формули І-1
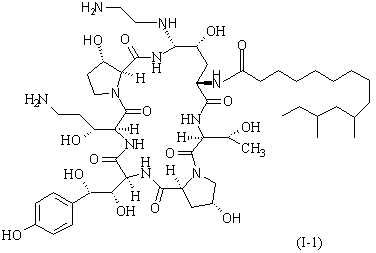
(SEQ ID No. l).
Текст
1 Спосіб одержання азациклопептидів формули (І) но но H-,N но (SEQ ID No з утворенням сполуки III формули но он о -N' н Н он H,N ^-N R . N R1 но (SEQ ID No Де Ri становить собою CH2CH2NH2 або CH2CONH2, R1 становить собою (Сд-С2і)-алкіл, (С9-С21)-алкеніл, (Сі-Сю)-алкоксифеніл, (Сі-Сю)-алкоксинафтил або (Сі-Сю)-алкокситерфеніл, R11 становить собою Н, (С1-С4)-алкіл, (C3-C4)lV v алкеніл, (СН2)2 4ОН або (CH 2 ) 2 4NR IVR V , 1 R " становить собою Н, (С1-С4)-алкіл, (C3-C4)алкеніл, (СН2)2 4ОН, (CH2)2 4NRIVRV або R" та R1" разом становлять (СН2)4 (СН2)5, (СН2)2О(СН2)2 або (CH2)2NH(CH2)2, RIV становить собою Н або (С1-С4)-алкіл, Rv становить собою Н або (С1-С4)-алкіл, або їх фармацевтично пристосованих кислотноаддитивних солей, що складається з наступних стадій а) відновлення сполуки II формули о (SEQ ID No b) перетворення сполуки III з утворенням сполуки IV формули PhS ОН о -N' HJST \ .'. н н но .(TV) (SEQIDNo I) с) стереоселективне перетворення сполуки IV у сполуку І шляхом заміщення фенілтюгрупи 2 Спосіб згідно з п 1, де відновлення на стам (а) виконують з використанням боранового комплексу або бориду металу со (О 44763 PhS 3 Спосіб згідно з п 2, де борид металу становить собою ZrCI4/NaBH4 або TiCI4/NaBH4, а борановий комплекс становить собою комплекс борану з диметилсульфідом, дибензилсульфідом, дифенілсульфідом, ТГФ або 1,4-оксатіаном, або ВНгСІ з диметилсульфідом 4 Спосіб згідно з п 1, де сполука III перетворюється у фенілсульфід шляхом взаємодії з тіофенолом у придатному розчині 5 Спосіб згідно з п 4, де придатним розчинником є НО ацетонітрил (SEQ ID No 6 Спосіб згідно з п 1, де заміщення фенілтюгрупи b) відновлення сполуки IV-a з утворенням сполуки проходить у чистому етилендіаміні або у присутності етилендіаміну, розчиненого у придатному IV формули PhS ОН розчиннику, при температурі від 10° до 40°С о 7 Спосіб згідно з п 6, де придатний розчинник вибирають з води, метанолу, етанолу, тетрапдрофурану, ізопропанолу, трифторетанолу, ацетонітНД рилу або дихлорметану 8 Спосіб одержання азациклопептидів формули (І) R но in ОН НО но (SEQ ID No Де Ri становить собою CH 2 CH 2 NH 2 або CH 2 CONH 2 , R1 становить собою (Сд-С2і)-алкіл, (Cg-C2i)-алкеніл, (Сі-Сю)-алкоксифеніл, (Сі-Сю)-алкоксинафтил або (Сі-Сю)-алкокситерфеніл, R11 становить собою Н, (Сі-С4)-алкіл, (Сз-С4)алкеніл, (СН 2 ) 2 4 ОН або (CH 2 ) 2 4 NR | V R V , R1" становить собою Н, (С1-С4)-алкіл, (Сз-С4)алкеніл, (СН 2 ) 2 4 ОН, (CH 2 ) 2 4 NR I V R V або R та R разом становлять (СН2)4 (СН2)5, (СН 2 ) 2 О(СН 2 ) 2 або (CH 2 ) 2 NH(CH 2 ) 2 , RIV становить собою Н або (Сі-С4)-алкіл, v R становить собою Н або (Сі-С4)-алкіл, або їх фармацевтично пристосованих кислотноаддитивних солей, що складається з наступних стадій а) взаємодія сполуки формули но он он о (SEQ ID No с) стереоселективне перетворення сполуки IV у сполуку І шляхом заміщення фенілтюгрупи 9 Спосіб згідно з п 8, де відновлення на стадії (Ь) виконується з використанням боранового комплексу або бориду металу 10 Спосіб згідно з п 9, де борид металу становить собою ZrCI4/NaBH4 або TiCI4/NaBH4, а борановий комплекс являє собою комплекс борану з диметилсульфідом, дибензилсульфідом, дифенілсульфідом, ТГФ або 1,4-оксатіаном, або ВН2СІ з диметилсульфідом 11 Спосіб згідно з п 8, де сполуку II перетворюють у фенілсульфід шляхом взаємодії з тіофенолом у придатному розчиннику 12 Спосіб згідно з п 11, де придатним розчинником є ацетонітрил 13 Спосіб згідно з п 8, де заміщення фенілтюгрупи відбувається у чистому етилендіаміні або у присутності етилендіаміну, розчиненого у придатному розчиннику, при температурі від 10°С до40°С 14 Спосіб згідно з п 13, де придатний розчинник вибирають з води, метанолу, етанолу, тетрапдрофурану, ізопропанолу, трифторетанолу, ацетонітрилу або дихлорметану 15 Спосіб згідно з п 1, ВІДПОВІДНО до якого одержують сполуку формули 1-1 (SEQ ID No 1) зтіофенолом з утворенням сполуки IV-a формули (SEQ ID No 44763 Данний винахід стосується удосконаленого способу одержання деяких азациклогексапептидів, що описані у патенті США №5378804,який виданий З січня 1995 р Попередній синтез цих сполучень передбачав проведення п'яти стадій та не мав достатньої стереоселективності або високого виходу ВІДОМІ способи відновлення початкових амідів, такі як гідрування, застосування гідриду металу та електрохімічне відновлення, потребують форсованих режимів, несумісних з іншими амідами та функціональними групами в ряді пневмокандинів Недоліком цих способів відновлення є відсутність хемоселективності серед різних заміщених амідів Новий спосіб, описаний у даній заявці, виключає дві стадії та забезпечує, в результаті, більш високий вихід та більш легкий синтез аналогів вказаних сполучень Стислий виклад суті винаходу Даний винахід стосується способу одержання азациклогексапептидів формули Де R1 становить собою CH2CH2NH2 або CH2CONH2, RI становить собою (С9-С21)-алкіл, (С9-С21)алкенил, (СІ-СЮ)-алкоксифенил, (С1-С10)алкоксинафтил або (СІ-СЮ)-алкокситерфенил, RII становить собою Н, (С1-С4)-алкіл, (СЗ-С4)алкенил, (СН2)2-4 ОН або (СН2)2-4 NRIVRV, RIII становить собою Н, (С1-С4)-алкіл, (СЗ-С4)алкенил, (СН2)2-4 ОН або (СН2)2-4 NRIVRV або RII та RUN узяті разом, становлять (СН2)4, (СН2)5, (СН2)2-О(СН2)2 або (СН2)2 NH(CH2)2, RIV становить собою Н або (С1-С4)-алкіл, Rv становить собою Н або (С1-С4)-алкіл, або їх фармацевтичне пристосованих кислотноаддитивних солей Встановлено, що сполучення, одержані заявленим способом, корисні для лікування грибкових інфекцій та для лікування та попередження інфекцій, що викликані Pneumocyctis саппм, котрі часто виявляються у пацієнтів з послабленим імунітетом, наприклад, у пацієнтів, що потерпають від СНІДу Ретельний опис винаходу Даний винахід стосується способу одержання сполучень формули (1) шляхом стереоселективного, високо продуктивного синтезу, котрий виключає дві стадії з попереднього способу їх одер жання Згідно ВСЬОГО описання та докладаємої формули винаходу дана хімічна формула або назва буде охоплювати всі оптичні та стереїзоміри, а також їх рацемічні суміші, коли такі ізоміри та суміші існують Термін "алкіл" припускає ЛІНІЙНІ, розгалужені або ЦИКЛІЧНІ вуглеводні групи, наприклад, метил, етил, н-пропіл, ізопропіл, н-бутил, пентил, гексил, гептил, циклолентил, циклогексил, циклогексилметил та подібні групи Термін "циклоалкіл" стосується типу алкіла, що містить від 3 до 15 атомів вуглецю, без подвійних зв'язків, ЯКІ чергуються або резонують, між атомами вуглецю Термін "алкенил" стосується груп, таких як, наприклад, винил, 1-пропен-2-іл, 1-бутен-4-іл, 2бутен-4-іл, 1-пентен-5-іл, та подібних груп Термін "алкокси" стосується ЛІНІЙНИХ або розгалужених оксиалкільних груп, таких як, наприклад, метокси, етокси, бутокси, гептокси, додецилокси, та подібних груп Сполучення даного винаходу, як правило, одержують у вигляді сумішів стереоізомірних форм, в яких звичайно переважає одна з них, причому спеціаліст може знайти умови для одержання переважно необхідного ізоміру Сполучення переважної стереїзомірної форми, що позначається у даному винаході як "нормальна" форма, є сполученнями, в яких група в положенні "C-5-orn" знаходиться нижче площини у вказаній позиції Позначення "ері" використовується для тих сполучень, у яких група у положенні "C-5-orn" знаходиться вище цієї площини Положення "C-5-orn" визначається як вуглець у положенні 5, 4-пдроксиорнітінового компоненту Фармацевтичне пристосованими солями, що підходять в якості солей приєднання кислот, є солі таких кислот, як хлористоводнева, бромістоводнева, фосфорна, сірчана, малеїнова, лимонна, оцтова, винна, янтарна, щавлева, яблучна, глутамшова та подібні кислоти, та включають солі інших кислот, родинні з фармацевтичне пристосованими солями, перерахованими у Journal of Pharmaceutical Science, 66 2 (1977) У переважному варіанті здійснення винаходу спосіб даного винаходу включає стадії відновлення сполучення II формули для одержання сполучення III формули 9н0. «° НА 8 44763 метилтридецил, можливо одержувати шляхом культивування Zalenon arboncola, ATCC 20868, у харчувальному середовищі, що збагачено маннітом у якості основного джерела вуглецю, як це описано у патенті США № 5021341, що виданий 4 червня 1991 Переважне сполучення, одержане згідно способу винаходу, показано нижче и о ,01» котре потім перетворюють у сполучення IV формули PhS,. Для більш зрозумілої демонстрації заявленого способу винахід ілюструється наступними його стадіями, де вказані усі переважні реагенти R1 на наведеній нижче реакційній схемі становить собою диметилтрицедил котре стереоселективно перетворюють у сполучення 1 за рахунок заміщення фенилтіогрупи У іншому варіанті здійснення винаходу спосіб включає стадії взаємодії сполучення II формули он 0 Щ .он ВНз ТГФ стадія li тиофеноп !. ацетонітрип/TFA з тюфенолом з утворенням сполучення IV-a формули PhS, OH 9 й о, стадія lit з наступним відновленням сполучення IV-a до сполучення IV формули н ^ \ .(IV) {SEQIDNo f) ґ ч HO ) = o HN \ -. / \ (W) яке стереоселективно перетворюють у сполу чення І за рахунок заміщення фенилтіогрупи Сполучення II, у якому R становить собою ди (SEQIDNo !). Як наведено вище, стадія 1 включає 44763 10 ізопропанол, тетрапдрофуран, трифторетанол, діхлоретан або ацетонітрил Даний винахід детально описується у наступних прикладах, у яких всі КІЛЬКІСНІ вирази у вигляді частин, а також співвідношення та відсотки є ваговими, якщо на це немає інших вказівок У наведеному прикладі R1 станосить собою діметилтридецил Приклад 1 a) Синтез та відокремлення сполуки III від сполуки II Сполука II (15,9г, з чистотою 89% по площі піка хроматограми, 3,4ваг% води, 0,0128моль) додають до сухого ТГФ (0,64л), та суспензію висушують до вмісту води менше 10мол% шляхом стікання флегми через шар молекулярних сит ЗА Додають додаткову КІЛЬКІСТЬ сухого ТГФ для відновлення початкового об'єму суміші, та суспензію охолоджують до температури менше 4°С на бані лід/вода/метанол Додають протягом десяти хвилин чистий BH3'SMe2 (10,91г, 0,144моль), та реакційну суміш Стадія II включає реакцію сполучення III з витримують при 0 - 4°С Розвиток реакції контротюфенолом в ацетонитрілі та трифтороцтовій килюють методом ВЕЖХ до співвідношення вихідної слоті (ТФК) з утворенням вміщуючого фенилсульречовини та кінцевого продукту 1 1, що показує фид проміжного сполучення Очікується, що будьна закінчення часу витримування реакційної суміші яка кислота помірної сили дасть проміжне сполу(3,5 години) Через 4 години суміш охолоджують чення з хорошим виходом Можна використовувадо -12°С та повільно гасять 2N НСІ (0,036л) Цей ти ІНШІ сульфіди, такі як 4-метокситюфенол, 2розчин розбавляють водою до 1,14л Вихід сполумеркапто-1-метилімідазол та 2ки III за аналізом складає 6,60г (47%) меркаптобензимідазол Сполучення III екстрагують шляхом внесення розбавленого реакційного розПогашений розчин розбавляють до 4л та зачину у колонку для хроматографії зі зворотною вантажують у колонку для хроматографії середньфазою С-18 з наступним елюїруванням метаноого тиску з адсорбентом LiChroprep RP-C18 (158г) лом Після завантаження колонку промивають 1,2л води, та амін елюїрують 1,9л суміші води з ацетоКІЛЬКІСТЬ використовуємої ТФК є критичним нитрілом (1 4 по об'єму), а потім 0,38л суміші ацефактором для ступеня заміщення, так само, як і тонітрилу з водою (1 3 по об'єму) для наступного утворення небажаного сульфіду в гомотирозиновій частині циклічного пептиду ВиБагаті продуктом фракції (більш 80% площі явлено, що 5-25% ТФК у ацетонітрилі дає найкрапика) поєднують та розбавляють водою 1 7,3 по щий вихід, а для часу необоротного процесу переоб'єму (всього 1,70л) Цю суміш завантажують у важає інтервал вміщуючий ТФК 7 -15% таку ж колонку, що описана вище, та колонку промивають 0,57л води Необхідну сполуку елюїрують Не виявлено, що КІЛЬКІСТЬ води відносно 0,57л метанолу Збагачені фракції (більш 85% вихідної речовини у реакційній суміші істотно площі пику) поєднують та концентрують шляхом впливає на вихід випаровування на роторному випарувальнику та в КІЛЬКІСТЬ тюфенолу, що використовується на умовах статичного високого вакуума, та одержуцій стадії, також є критичним фактором для виходу ють 6,81г (з чистотою 87ваг%, 6,8ваг% води), що кінцевого продукту Найкращий вихід забезпечумістить 5,92г пдрохлоріду сполуки III (де R1 є диють 3 - 5 еквівалентів цього сполучення метилтридецилом) з виходом у чистому вигляді Встановлено, що переважними умовами для 43% утворення сульфіду є 5 еквівалентів тюфенолу у 10% ТФК ацетонітрилі при 0°С За таких умов b) Одержання фенилсульфідної сполуки IV після екстракції на твердій фазі досягають виходу Сполука III (5,80г по аналізу, 0,00533моль) за65-70% вантажують у 0,23л сухого ацетонітрилу та охолоджують до -5°С, після чого додають тюфенол Стадія 3, що складається з заміщення фе(3,10г, 0,028моль) Додають протягом 20 хвилин нилтюгрупи, здійснюється в обхід попереднього ТФК (36г, 24,5мл, 0,318моль), щоб підтримати шляху, котрий проходив через утворення температуру реакційної суміші нижче 0°С Репроміжного сульфону акційну суміш витримують при температурі від Фенилсульфід реагує у нерозбавленому ети10°С до 0°С до тих пір, поки аналіз за допомогою лендіаміні (1 3) при температурі навколишнього ВЕЖХ не покаже менше 3% площі вихідної речосередовища з одержанням сполуки 1 -1 з виходом вини (3,75 годин) У цей час повільно (1 година) 95% Реакція проходить при температурі від 10° С додають охолоджену воду (0,56л), причому редо 40° С протягом 0,5 - 6,0 годин Переважно, щоб акційну суміш охолоджують таким чином, щоб и реакція проходила при кімнатній температурі протемпература залишалась нижче 5°С Вихід по тягом приблизно 1,5 години аналізу а- та р-фенилсульфідного аддукту у виРеакцію також можна проводити з використангляді солі трифтороцетової кислоти складає 4,82г ням етилендіаміну, розчиненого у придатному (71 %) розчині, такому як вода, метанол, етанол, відновлення аміду (сполучення II) до аміну з використанням боранового комплексу, наприклад, борану з тетрапдрофураном 0~ГФ), діметилсульфідом, діфенилсульфідом, дібензилсульфідом, 1,4-оксатіаном, або ВН2СІ з діметилсульфідом, або бориду металу, такого як ZrCI4/NaBH4 або TiCI4NaBH4 у ТФГ або іншому придатному розчиннику Відновлення також можна здійснити, використовуючи бориди тітану або цирконія або боранові комплекси з аміаком, діметиламіном, пірідином або піперазином Переважними відновлювачами є боранові комплекси з тетрапдрофураном (ТГФ), діметилсульфідом, діфенилсульфідом, дібензилсульфідом, 1,4оксатіаном, або ВН2СІ з діметилсульфідом, або борид металу, такий як ZrCI4/NaBH4 або TiCL4/NaBH4, у ТГФ або іншому придатному розчиннику Амід, ЩО не зазнав перетворення при такому відновленні, ВІДДІЛЯЮТЬ, використовуючи хроматографію зі зворотною фазою 11 44763 12 Цей розчин завантажують на таку ж колонку, мл/0,5мл, ВІДПОВІДНО) Сирий залишок на фільтрі що описана у стадії а), та колонку промивають висушують у струмі азоту, т а одержують 1,91г водою (0,57л), потім адсорбовані органічні сполуки (1,75г по аналізу, витягання 88%) діацетату сполуелюїрують метанолом (0,50л) Збагачені фракції ки 1-1 концентрують за допомогою випарювання на роСПИСОК ПОСЛІДОВНОСТЕЙ торному випарювальнику та в умовах статичного (1) ЗАГАЛЬНА ІНФОРМАЦІЯ високого вакууму Одержують 7,20г (з чистотою (1) ЗАЯВИТЕЛЬ Belyk, Kevin M Bender, Dean R 57% ваг, 5,1 ваг % води) сирої солі трифтороцетоBlack, Regma M Hughes, David L Leonard, William воі кислоти фенилсульфіду у вигляді аморфної (II) НАЗВА ВИНАХОДУ Спосіб одержання пінистої твердої речовини Скоригована стадія деяких азациклогексапептидів виділення дає 4,10г (61%) фенилсульфіду у ви(III) ЧИСЛО ПОСЛІДОВНОСТЕЙ 1 гляді суміші а-та р-амшальних діастереомірів 93 (IV) АДРЕСА ДЛЯ КОРЕСПОНДЕНЦІЇ 7 (A) А Д Р Е С А Т Elliott Korsen c) Перетворення фенилсульфіду у діамін (Б) ВУЛИЦЯ Р О Box 2000, 126 Е Lincoln Ave (сполука 1-1) (B) МІСТО Rahway Сирий трифторметансульфонат (8,4г сирого, (Г) ШТАТ NJ чистотою 57% ваг, 0,00377 моль) додають до ети(Д) КРАЇНА С Ш А лендіаміну (24мл) при перемішуванні при темпе(Е) ZIP 07065 ратурі навколишнього середовища Розчин, що (V) ФОРМА КОМПЬЮТЕРНОГО Р А Х У В А Н Н Я одержується в результаті, перемішують протягом (A) ТИП С Е Р Е Д О В И Щ А LBCRTNF 1,5 години для завершення заміщення, потім до(Б) К О М П Ь Ю Т Е Р сумісний з системою IBM дають метанол (40мл), а після нього оцетову киPC слоту (45мл), підтримуючи температуру нижче (B) ОПЕРАЦІЙНА СИСТЕМА PC-DOS/MS25°С шляхом охолодження на льодяній бані та DOS одержуючи у результаті густу суспензію Додають (Г) ПРОГРАМНЕ ЗАБЕЗПЕЧЕННЯ Patentm воду (160мл) для розчинення суспензії, та водний Release # шар екстрагують шляхом обережного встряхуван1 0, версія # 1 25 ня з гексаном (75мл) Гексановий шар знову екст(VI) ВІДОМОСТІ ПРО П О П Е Р Е Д Н Ю ЗАЯВКУ рагують водою (40мл), поєднаний водний шар (A) НОМЕР ЗАЯВКИ фільтрують через лійку з фільтром з плавленого (Б) ДАТА РЕЄСТРАЦІЇ скла середньої пористості, а потім очищують за (B) КЛАССИФіКАЦІЯ допомогою препаративної ВЕЖХ, використовуючи (VIII) ВІДОМОСТІ ПРО АГЕНТА/ПОВІРНИКА колонку С-18 діаметром 50мм, з використанням у (A) ІМ'Я Korsen, Elliott якості елюенту суміш з 22% ацетонітрилу та 78% (Б) Реєстраційний номер 2705 0,15%-і водної оцетової кислоти Збагачену (B) НОМЕР СПРАВИ/РЕЄСТРУ 19536 фракцію люфілізують, та одержують 4,2г (з чисто(IX) ТЕЛЕКОМУНІКАЦІЙНА ІНФОРМАЦІЯ (А) тою 85% ваг) сполуки 1-1 у вигляді діацетату з ТЕЛЕФОН 908-594-5493 (Б) ТЕЛЕФАКС 908-594виходом 78% на стадії одержання у чистому виді 4720 d) Кристалізація сполуки 1-1 (2) ІНФОРМАЦІЯ О НАСТУП № 1 Тверду речовину (2,3г) розчиняють в етанолі (I) ХАРАКТЕРИСТИКИ ПОСЛІДОВНОСТІ (25мл), і потім додають воду (2,7мл) Розчин про(A) Д О В Ж И Н А 6 амінокислот пускають через лійку з фільтром з плавленого (Б) ТИП амінокислота скла для видалення сторонніх речовин До одер(B) ТИП Л А Н Ц Ю Ж Е Н О С Т І невідомий жаного фільтрату додають оцтову кислоту (Г) циклічна (0,14мл), а потім послідовно (1,75 години) додають (II) ТИП МОЛЕКУЛИ пептид етилацетат (14мл) У розчин вносять затравку, та (III) ГІПОТЕТИЧНІСТЬ нема затравочний шар залишають для визрівання на 1 (IV) НЕАНТИЗМІСТОВА годину Додають протягом 5 годин решту етилаце(XI) О П И С А Н Н Я ПОСЛІДОВНОСТІ НАСТУП тату (32мл), та залишають для визрівання ще на 1 №1 годину Тверду кристалічну речовину збирають на Хаа Thr X a a Хаа Хаа Х а а ЛІЙЦІ з фільтром з плавленого скла та промивають 15 розчином етанолу з етилацетатом і водою (6мл/9 Д П «Український інститут промислової власності» (Укрпатент) вул Сім'ї Хохлових, 15, м Київ, 04119, Україна (044) 456 - 20 - 90
ДивитисяДодаткова інформація
МПК / Мітки
МПК: C07K 1/06, A61P 31/12, C07K 1/107, A61P 31/04, A61B 17/06, A61P 37/04, A61K 38/00, A61B 17/04, C07K 7/56
Мітки: варіанти, спосіб, одержання, азациклопептидів
Код посилання
<a href="https://ua.patents.su/6-44763-sposib-oderzhannya-azaciklopeptidiv-varianti.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання азациклопептидів (варіанти)</a>
Виділена молекула нуклеїнової кислоти (варіанти), химерний рослинний ген (варіанти), трансформуючий рослину вектор (варіанти), рослинна клітина (варіанти), спосіб одержання рослини, що проявляє стійкість до гер

Номер патенту: 44217
Опубліковано: 15.02.2002
Автори: Лебрюн Мішель, Фрейссіне Жорж, Бог Моллі, Томас Террі
МПК: C07K 14/415, C12N 9/10, C12N 15/09, C12N 15/82, A01H 1/00
Мітки: рослинна, трансформуючий, нуклеїнової, молекула, рослинній, проявляє, одержання, гер, стійкість, химерний, кислоти, ген, спосіб, варіанти, вектор, виділена, клітина, рослини, рослину
Формула / Реферат:
1. Выделенная молекула нуклеиновой кислоты, содержащая регуляторный элемент гена гелиантинина, где указанный регуляторный элемент управляет семя-специфичной генной экспрессией в растении и включает нукдеотиды 2304-2401 SEQ ID NO: 1: 2. Выделенная молекула нуклеиновой кислоты, содержащая регуляторный элемент гена гелиантинина, где указанный регуляторный элемент управляет корень-специфичной генной экспрессией в...
Спосіб одержання гомологічно рекомбінантної клітини (варіанти), плазміда для використання у способі (варіанти), штам гомологічно рекомбінантних клітин фібросаркоми людини (варіанти), спосіб одержання еритропоет

Номер патенту: 34493
Опубліковано: 15.03.2001
Автори: Треко Дуглас А., Хартлейн Мішель В., Селден Річард Ф.
МПК: C12N 1/15, C12P 21/00, A61K 38/22, A61K 38/28, A61K 48/00, A61K 38/00, C12N 5/10, A61K 38/27, C07K 14/505, A61K 38/43, C12N 15/85, C12N 15/90, C12N 15/67, C07H 21/04, C12N 15/09, A61P 43/00, A61K 39/395, A61K 31/70, A61K 38/21
Мітки: клітин, еритропоет, одержання, штам, рекомбінантних, фібросаркоми, людини, варіанти, клітині, спосіб, гомологічної, використання, способи, плазміда, рекомбінантної
Текст:
...ватывающий положения нуклеотидое HUMGHCSA (Genbank Entry) 3787-5432 (положе ния двух сайтов EcoRI, которые образуют фраг мент удобного размера для клонирования или диагностического расщепления включающих в себя этот фрагмент субклонов), амплифицируют при помощи праймеров ПЦР, сконструированных согласно анализу последовательности HUMGHCSA в этом районе Этот район прости рается от середины интрона 1 гена NhGH до по ложения против хода...
Комплекс аналога інсуліну та протаміну, фармацевтична композиція для парентерального введення (варіанти), спосіб її одержання (варіанти), спосіб одержання кристалів комплексу lysb28prob29-людського інсуліну та

Номер патенту: 34468
Опубліковано: 15.03.2001
Автор: де Феліппіс Майкл Розаріо
МПК: A61K 33/30, A61P 5/48, A61K 38/28, A61K 38/16, A61P 3/10, A61P 3/08
Мітки: інсуліну, аналога, кристалів, протаміну, композиція, спосіб, одержання, варіанти, введення, фармацевтична, комплекс, парентерального, комплексу, lysb28prob29-людського
Текст:
...приблизительно одинаковы. Однако, наиболее важно, что настоящая композиция повышает более быстро и остается стабильной в течение более длительного периода, чем инсулин-NPH. Это различие достаточно неожиданно с точки зрения быстродействующего профиля мономерного аналога. Особенно, предпочтительная композиция инсулиновый аналог-протамин, LysB28ProB29-человеческий инсулин-NPD, включает: LysB28ProB29человеческий инсулин, от 0,27 до 0,32 мг...
Стимулятор росту збудника туберкульозу “рідин” (варіанти), живильне середовище для виділення збудника туберкульозу (варіанти), спосіб одержання живильного середовища (варіанти), спосіб виділення збудника туберк

Номер патенту: 43467
Опубліковано: 17.12.2001
Автори: Багрій Петро Іванович, Власенко Володимир Васильович
МПК: C12N 1/38, C12Q 1/04, C12N 1/20, C12R 1/32, C12N 1/02
Мітки: живильного, стимулятор, рідин, туберкульозу, виділення, туберк, одержання, спосіб, живильне, середовище, збудника, росту, варіанти, середовища
Формула / Реферат:
1. Стимулятор росту збудника туберкульозу, що містить хімічні сполуки, до складу яких входять елементи: натрій, кисень, водень, вуглець, який відрізняється тим, що характеризується наступним складом інгредієнтів, мас. %: сахароза х/ч 2,0-5,0 спирт ректифікований 0,1-0,4 кальцій хлористий 0,1-0,15 натрій двовуглекислий ...
Фармацевтична композиція, яка містить мофетил мікофеноляту, для орального введення ( варіанти ) та спосіб її одержання ( варіанти )

Номер патенту: 39962
Опубліковано: 16.07.2001
Автори: Лідгейт Дебора М., Гу Ліо, Хедж Сейі Г., Уанг-Кеслер Лі-Хуа, Йоші Бінду
МПК: A61K 31/5377, A61P 31/12, A61K 9/10, A61P 35/00, A61P 17/06, A61K 31/365
Мітки: яка, варіанти, містить, мофетил, мікофеноляту, введення, орального, спосіб, одержання, композиція, фармацевтична
Формула / Реферат:
1 .Фармацевтическая композиция в виде жидкой суспензии, пригодной для орального введения, включающая мофетил микофенолята или микофенольную кислоту, отличающаяся тем, что она дополнительно содержит суспендирующий и/или повышающий вязкость агент, подслащивающие вещества, ароматизатор, буфер до рН 5,0-7,0 и дистиллированную воду, при следующем соотношении компонентов, % (мас./об): мофетил микофенолята или микофенольная...
Попередній патент: Спосіб ранньої діагностики абсцесу селезінки
Наступний патент: Спосіб отримання металевого порошку
Випадковий патент: Форма для виготовлення ніздрюватобетонних виробів