Спосіб одержання рацемічних або оптично активних сполук
Номер патенту: 66359
Опубліковано: 17.05.2004
Автори: Баконіі Марія, Гаярі Анталь, Алаттіані Едіт, Чатаріне Нагі Маріанна, Мольнар Левентене





Формула / Реферат
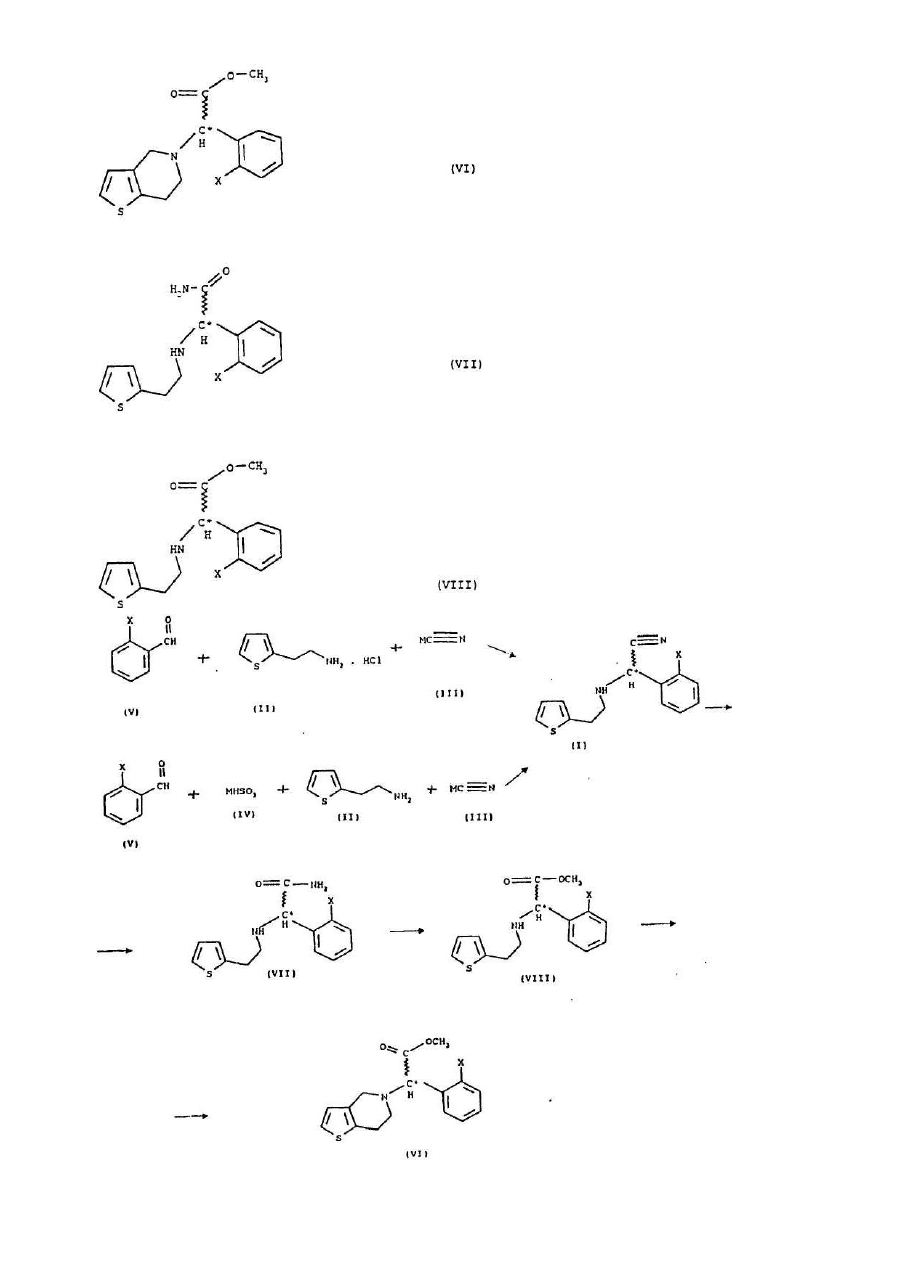
1. Спосіб одержання рацемічних або оптично активних сполук загальної формули (VI)
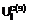 , (VI)
, (VI)
де Х означає атом галогену, або їх солей, який відрізняється тим, що рацемічну або оптично активну сполуку загальної формули (VIІ)
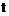 , (VII)
, (VII)
де Х означає атом галогену – перетворюють шляхом відомої реакції утворення естерів в рацемічну або оптично активну сполуку загальної формули (VIII)
 , (VIII)
, (VIII)
де Х означає атом галогену і, якщо бажано, отримані рацемічні сполуки загальної формули (VIII) розділяють на їх оптично активні ізомери, а потім шляхом замкнення циклу, який відомий з рівня техніки для сполук загальної формули (VIII), перетворюють в рацемічні та оптично активні сполуки загальної формули (VI), і, якщо бажано, рацемічні сполуки загальної формули (VI) розділяють на їх оптичні ізомери, і/або перетворюють на їх солі, і/або рацемічні або оптично активні сполуки виділяють з їх солей.
2. Спосіб за пунктом 1, який відрізняється тим, що сполуки загальної формули (VII), де значення Х є таким, як вказано в пункті 1, за допомогою метанолу в присутності метилгідросульфату перетворюють в сполуки загальної формули (VIII).
3. Спосіб за пунктом 1, який відрізняється тим, що перетворення відбувається при температурі від 50 °С до 150 °С.
4. Спосіб за пунктом 1, який відрізняється тим, що сполуки загальної формули (VII), де Х означає атом хлору - перетворюють у сполуку загальної формули (VIII), де Х означає атом хлору.
Текст
Винахід стосується нового способу виготовлення сполук з загальною формулою (VI), де X означає атом галогену. Відомо, що метил (2-галогенфеніл)-(6,7-дігидро-4Н-тієно[3,2-с]пірідин-5-іл) ацетати та їх солі можуть успішно застосовуватися при лікуванні, перш за все завдяки здатності гальмувати утворення агрегацій тромбоцитів та їх антитромбогенному ефекту. Особливо сприятливим представником цих сполук, що підпадають під загальну формулу (VI), де X означає хлорозаміщений атом, є правообертальний метил (+)-[(S)-(2-хлорфеніл)-(6,7-дигідро-4Н-тієно[3,2с]піридін-5-іл)ацетат бісульфат з міжнародною непатентованою назвою клопідогрель (clopidogrel) (Європейська заявка, публікація Nr.099802). Широкомасштабне одержання сполук з загальною формулою (VI) - де X означає атом галогену - раніше було можливим лише за допомогою похідних a-галогензаміщеної фенілоцтової кислоти, які здатні викликати сильну сльозоточивість та подразнення слизових оболонок, з якими важко обходитися під час технологічних операцій і які є несприятливими з точки зору здоров'я та довкілля (Європейська заявка, публікації Nr.099802, 0420706, 0466569). Окрім того, продуктивність відомих способів може вважатися скоріше незначною. Мета винаходу полягала в усуненні використання згаданих вище проміжних продуктів (таких, наприклад, як a-бром-(2-хлорфеніл)оцтова кислота та її метиловий ефір) та в значному підвищенні виходу сполук з загальною формулою (VI) в синтезі. Оскільки в синтезі згідно з даним винаходом кожний проміжний продукт є хіральним, при виготовленні оптично активного кінцевого продукту, такого, наприклад, як клопідогрель, відкривається можливість використання - починаючи з першої стадії - оптично активних сполук як проміжних продуктів. Економічна перевага цього способу, поряд з іншим, полягає в вилученні виробництва небажаного ізомеру. Було визначено, що при виготовленні сполук з загальною формулою (VI) шляхом, зображеним на схемі 1, можна уникнути використання небажаного проміжного продукту. І до того ж значно підвищити результативність синтезу. Об'єктом даного винаходу є третій розділ схеми 1, яка зображує реакцію. Оптично активні сполуки з загальною формулою (VI) виготовляються або з оптично активних сполук з загальною формулою (VII), або починаючи з оптично активного проміжного продукту, який одержують шляхом розділення проміжного продукту з загальною формулою (VII), або шляхом розділення рацемічних сполук з загальною формулою (VI). Згідно з винаходом рацемічна або оптично активна сполука з загальною формулою (VII) - де X означає атом галогену - перетворюється в рацемічну або оптично активну сполуку з загальною формулою (VIII) - де X означає атом галогену - і, при бажанні, одержану рацемічну сполуку з загальною формулою (VIII) розділяють на її оптично активні ізомери, а потім в замкнутому циклі за допомогою способів, що відомі самі по собі, сполуки з загальною формулою (VIII) перетворюють в рацемічну або оптично активну сполуку з загальною формулою (VI) і, при бажанні, рацемічні сполуки з загальною формулою (VI) розщеплюють на їх оптичні ізомери і/або їх перетворюють в їх солі, і/або рацемічну або оптично активну сполуку вивільняють з їх солей. Краще, коли сполуки з загальною формулою (VII) реагують з метанолом в присутності метил-сірчаної кислоти. До того ж, реакція може бути проведена під тиском, краще за все при 5-20бар. Найбільш сприятливі значення температури знаходяться між 50°С та 150° С. Метил-сірчану кислоту приготовляють в реакційній камері при зворотному потоці метанолу та сірчаної кислоти. Замкнутий цикл отримання сполук з загальною формулою (VIII) здійснюють способом, що відомий сам по собі. Розщеплення відомих рацемічних проміжних продуктів з загальною формулою (VIII) або рацемічних сполук з загальною формулою (VI) здійснюють способом розщеплення, що відомий сам по собі, приводячи до оптично активних сполук з загальною формулою (VI). Приготовлення вихідних сполук, використаних в даному винаході, пояснюється за допомогою прикладів. Вихідні матеріали, показані на схемі 1, можна придбати, а синтез сполуки з формулою (II) описаний, наприклад, у французькій заявці No 2608607. Подальші подробиці винаходу проілюстровані в наступних прикладах, які не обмежують обсяг винаходу. Приклад 1 [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрил 104г (1моль) бісульфату натрію розчиняють в суміші, що складається з 900мл води та 250мл етанолу, і до розчину додають 140,6г (1моль) о-хлорбензальдегіду. Через кілька хвилин аддукт бісульфату альдегіду осідає в формі білих кристалів, причому температура зростає до 40°С. Після розмішування, що триває 1 годину, до реактивної суміші додають 127,2г (1моль) 2-(2-тієніл)етиламіну, а потім розмішують протягом 2 годин при 50°С. За цей час кристалічний бісульфат альдегіду перетворюється в маслянисту речовину. Суміш охолоджують до кімнатної температури і додають до неї розчин з 49г (1моль) ціаніду натрію в 100мл води. Під час додавання температура реакційної суміші зростає до 40°С. Потім суміш розмішують при 60°С, поки реакція не завершиться (1 година). Після цього маслянисту органічну фазу екстрагують за допомогою 400мл 1,2-діхлоретану, промивають від ціаніду за допомогою 2x200мл води, а сліди 2-(2-тієніл)етиламіну усувають шляхом обробки з використанням 100мл 3%-ного розчину соляної кислоти. Дихлоретанову фазу просушують над безводним сульфатом натрію та випаровують в вакуумі. Залишок в вигляді масла, що швидко кристалізується, є кінцевим продуктом. Вага: 260г (94%), точка плавлення 40-41°С. Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 2 [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрил 9,8г (0,2моль) ціаніду натрію розчиняють в 70мл води і до розчину додають спочатку 32,8г (0,2моль) 2(2-тієніл)етиламін гідрохлориду, а потім протягом кількох хвилин - розчин 28,2г (0,2моль) охлорбензальдегіду в 30мл етанолу. Під час додавання температура суміші зростає до 45°С. Потім реакційну суміш розмішують при 60°С протягом 2 годин, після чого охолоджують до кімнатної температури та розріджують водою в кількості 50мл. Отриманий маслянистий продукт екстрагують за допомогою 100мл 1,2-діхлоретану, органічну фазу промивають від ціаніду за допомогою 2x50мл води, а сліди 2-(2тієніл)етиламіну усувають шляхом обробки з використанням 20мл 3%-ного розчину соляної кислоти. Залишок в вигляді масла, що швидко кристалізується, є кінцевим продуктом. Вага: 52г (94%), точка плавлення 40-41°С. Продукт було ідентифіковано тим же способом, що і в прикладі 1. Якість цього продукту ідентична якості продукту, отриманого згідно з прикладом 1. Приклад 3 [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрил хлоргідрат 276,7г (1моль) [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрилу, приготовленого як в прикладі 1 або 2, розчиняють в 600мл етанолу і до розчину додають 600мл 10%-ного водного розчину соляної кислоти. Через кілька хвилин осідають білі кристали, їх збирають, промивають за допомогою 60мл суміші з 10%-ої соляної кислоти та етанолу у співвідношенні 1:1, потім ацетоном, після чого просушують. Вага: 305г (94%), точка плавлення 153-154°С. Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 4 [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрил бромгідрат 13,8г (0,05моль) [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрилу, приготовленого як в прикладі 1 або 2, розчиняють в 30мл етанолу і до розчину додають 40мл 20%-ного водного розчину бромистого водню. Продукт, що осідає через кілька хвилин, збирають, промивають етиловим ацетатом, потім просушують. Вага: 14г (78,2%), точка плавлення 144-145°С. Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 5 [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетамід хлоргідрат В 1200мл метилового ацетату вводять 204г (5,6моль) хлороводню при 15-25°С, і до розчину додають 221,4г (0,8моль) [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрилу з формулою (І), приготовленого як в прикладі 1, та 48мл (1,2моль) метанолу, і цю суміш розмішують при 20-25°С протягом 6 годин. В ході реакції в формі білих кристалів випадає спочатку хлоргідрат первісного "нітрилу", а потім частково хлоргідрат остаточного "аміду кислоти". Кристали збирають шляхом фільтрації, промивають метилацетатом та просушують. Вага: 249г (94%), точка плавлення 231-232°С Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 6 [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетамід хлоргідрат В 700мл етилацетату при 0-10°С вводять 109,8г (3моль) газоподібного хлороводню, і до розчину додають 83г (0,3моль) [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрилу з формулою (І), приготовленого як в прикладі 1 або 2, та 15мл (0,37моль) метанолу, а потім цю суміш повільно, протягом 20 хвилин, нагрівають до 45-50°С. Після цього реакційну суміш перемішують при 40-50°С протягом 4 годин, кристалічний продукт відфільтровують при кімнатній температурі, промивають етилацетатом та просушують. Вага: 90,4г (91 5), точка плавлення 231-232°С. Якість цього продукту ідентична якості продукту, отриманого згідно з прикладом 5. Приклад 7 [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетамід 24,8г (0,075моль) [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетамід хлоргідрату, приготовленого як в прикладі 5 або 6, змішують з 170мл води, після чого при слабкому охолодженні додають 30мл 10%-ного розчину гідроокису натрію та 170мл 1,2-діхлоретану. Фази підлягають сепарації, водну фазу екстрагують за допомогою 2x20мл 1,2-діхлоретану, а комбіновану органічну плівку випаровують в вакуумі. Залишок: 22г, масло, що швидко кристалізується. Сирий продукт є рекристалізованим з 80мл ізопропілацетату, що дає 19,5г кристалічної основи з формулою (VII). Вихід: 88,2%, точка плавлення 90-92°С. Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 8 [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетамід бромгідрат 14,7г (0,05моль) [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетаміду, приготовленого як в прикладі 7, розчиняють в 150мл ацетону. До розчину додають 4мл 60%-ного водного розчину бромоводню, а білі кристали, що випали в осад, відфільтровують, промивають ацетоном та просушують. Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 9 Метил [2-(2-тієніл)етиламіно](2-хлорфеніл)хлоргідрат ацетату 21,5мл (0,4моль) 100%-ної сірчаної кислоти розчиняють при охолодженні в 100мл метанолу, розчин нагрівають зі зворотним холодильником на ? год., потім охолоджують до кімнатної температури, і додають до нього 33,1г (0,1моль) [2-(2-тієніл)етиламіно](2-хлорфеніл)хлоргідрат ацетаміду, приготовленого як в прикладі 5, потім цю суміш нагрівають зі зворотним холодильником протягом 10 годин. Після цього метанол відгонять в вакуумі, а до залишку додають 150мл 1,2-діхлоретану та 150мл води, добре переколочують і розділяють на дві фази. Водяний шар екстрагують за допомогою 2x30мл 1,2-діхлоретану, а комбіновані органічні шари промивають за допомогою 80мл 5%-ного розчину гідроокису натрію, а потім за допомогою 100мл води, висушують над безводним гідроокисом натрію та випаровують в вакуумі. Вага залишку: 28,5г. Маслянистий продукт, що є основою формули (VIII), розчиняють в 50мл ізопропілацетату, додають до нього 7,3мл (0,087моль) концентрованого розчину соляної кислоти, а суміш перемішують при кімнатній температурі протягом 1год. Продукт, що випав в осад, відфільтровують, промивають за допомогою 2x10мл ізопропілацетату та просушують. Вага: 28,4г (82%), точка плавлення: 177-178°С (літр 175°С). Продукт було ідентифіковано шляхом елементарних аналізів, за допомогою інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н, а також масс-спектрометрії та визначення точки плавлення. Приклад 10 Метил[2-(2-тієніл)етиламіно](2-хлорфеніл)хлоргідрат ацетату В 150мл метанолу вводять при охолодженні 8,5мл (0,15моль) 96%-ної сірчаної кислоти, а потім розчин нагрівають зі зворотним холодильником протягом ? год. Після охолодження до кімнатної температури 20г (0,0678моль) [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетаміду з загальною формулою (VII), приготовленого як в прикладі 7, додають до розчину, суміш поміщують в герметичний апарат (автоклав) і перемішують в ньому при 130°С протягом 5год., причому внутрішній тиск зростає до 13бар. Потім реакційну суміш охолоджують до кімнатної температури (тиск залишається на рівні 1-2бар), метанол усувають дистиляцією в вакуумі, до залишку додають 100мл ізопропілацетату та 100мл води, а рН суміші доводять до значення 7,5 шляхом додавання по краплям 60мл 10%-ного розчину гідроокису натрію в умовах охолодження та розмішування, причому температура суміші залишається кімнатною. Фази розділяють, органічну фазу розмішують з 60мл 3%-ного водного розчину малеїнової кислоти при 40-50°С протягом 10 хвилин, після чого розділяють дві фази. Після повторного екстрагування водного розчину малеїнової кислоти за допомогою 30мл ізопропілацетату органічні шари зв'язують, шляхом просушування усувають безводний сульфат натрію і концентрують до половини їх об'єму. При додаванні 5мл концентрованого розчину соляної кислоти осідає продукт в вигляді масла, який кристалізується через кілька хвилин. Його охолоджують до 0(+5)°С і через 2 години кристали збирають шляхом фільтрації, промивають невеликою кількістю ізопропілацетату та просушують. Вага: 19,4г (82,5%), точка плавлення 177-178°С. Якість цього продукту ідентична якості продукту, отриманого згідно з прикладом 9. Приклад 11 Метил[2-(2-тієніл)етиламіно](2-хлорфеніл)бромргідрат ацетату Процес відбувається, як описано в прикладі 9, отриманий метил[2-(2-тієніл)етиламіно](2-хлорфеніл) ацетат розчиняють в 50мл ізопропілацетату, до розчину додають 8мл 62%-ного водного розчину бромоводню, і цю суміш перемішують при кімнатній температурі протягом 1 години. За цей час відбувається кристалізація продукту. Кристали збирають, промивають за допомогою 2x10мл ізопропілацетату та просушують. Вага: 32,5г (83%), точка плавлення 164-165°С. Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 12 Метил(2-хлорфеніл)(6,7-дігідро-4Н-тієно[3,2-с]пірідін-5-іл)ацетат гідрохлорід гідрат До 28,4г (0,082моль) метил[2-(2-тієніл)етиламіно](2-хлорфеніл)хлоргідрат ацетату, приготовленого як в прикладі 9 або 10, додають 50мл 1,2-діхлоретану та розчин 7,5г (0,09моль) гідрокарбонату натрію в 100мл води. Суміш добре перемішують, фази розділяють, водну фазу вимивають за допомогою 2x30мл 1,2діхлоретану, комбіновані органічні шари просушують над безводним сульфатом натрію, а розчинник усувають у вакуумі. Залишок в вигляді 25 г речовини ( ацетатної основи) розчиняють в 90мл мурашиної кислоти, до розчину додають 4г (0,13моль) поліоксіметилену, і цю суміш перемішують при 50°С протягом 20 хвилин. Потім переважну більшість мурашиної кислоти усувають дистиляцією в вакуумі, залишок розчиняють в суміші з 100мл води та 100 мл1,2-діхлоретану, фази розділяють, водну фазу знову екстрагують за допомогою 30мл 1,2-діхлоретану, комбіновану органічну фазу добре розколочують разом з 100мл 5%-ного розчину гідрокарбонату натрію, фази розділяють, а органічну фазу просушують над безводним сульфатом натрію та випарюють в вакуумі. Залишок розчиняють в 45мл ацетону, і до розчину додають 6,5мл (0,077моль) концентрованого розчину соляної кислоти при 5-10°С в умовах охолодження. Відбувається повільна кристалізація продукту. Суміш перемішують протягом 1год. при 0-10°С, потім кристали відфільтровують, промивають за допомогою 2x10мл ацетону та просушують. Вага: 26,7г (теоретично: 30,8г). Вихід: 86,6%, точка плавлення 138-40°С (точка плавлення за літературою 130-140°С). Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н, а також визначення точки плавлення. Приклад 13 Лівооборотний [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрил хлоргідрат 10г (0,036моль) рацемічного [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрилу (І) розчиняють в 15мл ацетону, до розчину додають 10г (0,043моль) (1R)-(-)-камфора-10-сульфокислоти та 0,5мл (0,013моль) мурашиної кислоти, суміш нагрівають до 50-55°С, після чого охолоджують до кімнатної температури. Поступово випадає сіль, що формується між правооборотним енантіомером вихідного матеріалу та (1R)-(-)камфора-10-сульфокислотою, в легко забрудненій на вигляд формі. Кристали відділяють шляхом фільтрації. До початкової рідини додають 7мл метилацетату, що містить 10% хлороводню, або вводять розраховану кількість сухого газу хлороводню, кристалічний осад відфільтровують, промивають ацетоном та просушують. Вага: 2,5г [a]22D=-43° (с=1, метанол). Вихід: 43% від вмісту лівооборотного енантіомеру в первісному матеріалі. Після повторної кристалізації з етанолу: [a]22D=-48° (с=1, метанол). Точка плавлення: 151-152°С (розклад). Оптична чистота >98% (визначено шляхом дослідження ВЕРХ). Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 14 Правооборотний [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрил хлоргідрат Процес відбувається, як описано в попередньому прикладі, але в якості розчинної кислоти додають (ІS)-(+)-камфора-10-сульфокислоти. Продукт: вага 2,5г, [a]22D=+43° (с=1, метанол). Вихід: 43% від вмісту лівооборотного енантіомеру в первісному матеріалі. Після повторної кристалізації з етанолу: [a]22D=-48° (с=1, метанол). Точка плавлення: 151-152°С (розклад). Оптична чистота >98% (визначено шляхом дослідження ВЕРХ). Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 15 Правооборотний [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетамід 11,8г (0,037моль) лівооборотного [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетонітрил хлоргідрату розмішують в 100мл метилового ацетату і вводять 9,6г сухого газу хлороводню при кімнатній температурі. Після цього додають 3,6г (0,113моль) метанолу, і суміш помішують при кімнатній температурі, поки реакція не закінчиться через 6 годин. Кристалічний матеріал, що випав в осад і являє собою хлоргідратну сіль продукту, відфільтровують, розмішують у воді, нейтралізують бікарбонатом натрію при помішуванні. Осаджений білий кристалічний сирий продукт відфільтровують, просушують та шляхом повторної рекристалізації звільняють від метанолу. Вага: 5г, [a]22D==63° (с=1, метанол). Точка плавлення: 122-124°С. Вихід: 46%. Оптична чистота 97%. Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 16 Правооборотний [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетамід 38г (0,129моль) рацемічного [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетаміду розчиняють при 50°С в 380мл ізопропанолу, що містить 0-0,4, краще 0,2% води, і до цього розчину додають 50°С розчин 10,6г (0,071моль) L(+)-винної кислоти в 230мл ізопропанолу, що містить 0,04%, краще 0,2% води. Суміш розмішують протягом 30хв при 50°С. Утворюється густий білий осад. До суміші додають 3,4мл (0,09моль) мурашиної кислоти та помішують протягом 1 години при 50°С. Потім реакційну суміш охолоджують до кімнатної температури, помішують протягом ще однієї години, і тверду фазу відфільтровують. Осаджений матеріал являє собою сіль, що утворилася між лівооборотним енантіомером первісного матеріалу та L(+)винною кислотою в забрудненій на вигляд формі. Вага: 30г. Точка плавлення: 167-169°С, після кристалізації з етанолу. Початкову рідину випаровують в вакуумі. Залишок (29г) поглинають за допомогою 200мл води і 200мл 1,2-діхлоретану та нейтралізують при помішуванні за допомогою 16г (0,19моль) гідрокрбонату натрію. Фази розділяють, водяний шар промивають за допомоою 2х30мл 1,2-діхлоретану, комбінований органічний шар екстрагують за допомогою 50мл води, просушують над безводним сульфатом натрію та випаровують в вакуумі. Вага: 18г. Сирий продукт повторно кристалізують з 70мл етанолу, промивають невеликою кількістю етанолу та просушують. Вага: 12,6г. Точка плавлення: 122-124°С, [a]22D=+69° (с=1, метанол). Вихід: 66,3%від вмісту правооборотного енантіомеру в початковому матеріалі. Оптична чистота: 99-100%, зазвичай вище ніж 98% (визначено за допомогою ВЕРХ). Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. При концентрації фільтрату може бути регенеровано 4г рацемічного початкового матеріалу. Приклад 17 Правооборотний [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетамід 76г (0,257моль) рацемічного [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетаміду розчиняють при 50°С в 1200мл ізопропанолу, що містить 0,2% води, і до цього розчину додають 21,2г (0,141моль) L(+)-винної кислоти та 8,3г (0,18моль) мурашиної кислоти. Суміш перемішують при 50°С протягом 1год, причому утворюється густий білий осад. Потім реакційну суміш охолоджують до кімнатної температури протягом 1год, розмішують протягом наступних 2год, і тверду фазу відфільтровують. Осаджений матеріал являє собою сіль, що утворилася між лівооборотним енантіомером первісного матеріалу та L(+)-винною кислотою в забрудненій на вигляд формі. Вага: 57г. Точка плавлення: 167-169°С, після кристалізації з етанолу. Після фільтрації попереднього твердого матеріалу до фільтрату додають 5,2г (0,141моль) газу соляної кислоти, щоб хлоргідрат випав в осад з продукту. Отриманий білий кристалічний матеріал відфільтровують та просушують. Вага: 41,7г. Отримана сіль, на вигляд злегка забруднена, проглинається 100мл етанолу, потім 5,3г (0,13моль) гідроокису натрію, розчиненого в 70мл етанолу, додають до неї поступово, щоб виділити вільну основу. Отриманий продукт, що містить невелику кількість хлористого натрію, відфільтровують та промивають дистильованою водою. Після просушування його вага становить 27,7г, 73% вмісту правооборотного енантіомеру в початковому матеріалі. Точка плавлення: 122-124°С, [a]22D=+69° (с=1, метанол). Якщо етаноловий фільтрат випаровують в вакуумі, а залишок поглинається водою, то регенерується 9г рацемічного початкового матеріалу. Приклад 18 Правооборотний метил [2-(2-тієніл)етиламіно](2-хлорфеніл)хлоргідрат ацетату 40мл метанолу при охолодженні розчиняють 11,5мл (0,215моль) 100%-ої сірчаної кислоти, розчин нагрівають зі зворотним холодильником протягом 30хв, потім після охолодження до кімнатної температури додають 12,4г (0,042моль) правооборотного [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетаміду, і суміш нагрівають зі зворотним холодильником протягом 6-7год, поки не завершиться реакція. Метанол усувають дистиляцією в вакуумі, до залишку додають 75мл 1,2-діхлоретану та 75мл води, суміш добре розколочують, фази розділяють. Водяну фазу екстрагують за допомогою 2х20мл 1,2-діхлоретану, спільну органічну фазу екстрагують за допомогою 50мл 5%-ного розчину гідроокису натрію, а потім 50мл води, просушують над безводним сульфатом натрію. Просушений матеріал відфільтровують, після чого в розчин вводять 1,5г (0,041моль)сухого газу хлороводню в умовах охолодження. Осаджений кристалічний продукт відфільтровують, промивають 1,2-діхлоретаном та просушують. Вага: 12,1г, точка плавлення: 185-186°С (розпад), [a]22D=+107°. Вихід: 83%. Оптична чистота: загалом 99-100%. Продукт було ідентифіковано шляхом елементарних аналізів, інфрачервоного спектру та дослідження ядерно-магнітного резонансу ядра 1Н. Приклад 19 Правооборотний метил [2-(2-тієніл)етиламіно](2-хлорфеніл)ацетат, отриманий шляхом розщеплення рацемату а) 175г хлоргідратної солі сполуки з загальною формулою (VIII) - де X означає хлорозаміщений атом розчиняють в суміші, що складається з 0,75л діхлорметану та 0,25л води, і до розчину поступово додають 45г бікарбонату натрію. Після розмішування органічну фазу відділяють за допомогою декантації. Шляхом звичної процедури отримують аміноефір, який потім розчиняють в 850мл ацетону, і до розчину додають 87г (+)-камфора-10-сульфокислоти. Суміш витримують при кімнатній температурі протягом 12 годин, а потім отриманий осад сепарують. Таким чином отримують 146,5г камфорсульфанату, [a]22D=+51,7° (с=1, метанол). Камфорсульфанат суспендують в 700мл ацетону в умовах нагрівання зі зворотним холодильником, і для отримання повного розчину додають 300мл метилетилкетону. Суміш охолоджують до кімнатної температури. Отриманий осад сепарують і при кімнатній температурі обробляють 500мл ацетону та 300мл метилетилкетону. Так отримують 95г (+)-камфорсульфанату очікуваного продукту, точка плавлення: 95°С, [a]22D=+82° (с=1, метанол). б) 33,5г хлоргідратної солі сполуки з загальною формулою (VIII) - де X означає хлорозаміщений атом та 14,6г (+)-винної кислоти змішують в 500мл ізопропанолу, нагрівають до 50°С, потім витримують при кімнатній температурі. Отриманий осад сепарують та кристалізують 4 рази з ізопропанолу. Так одержують (+)-тартрат бажаного правообертового продукту, точка плавлення: 105°С. Питоме обертання аміну [a]22D=+99,76° (с=1, метанол). Приклад 20 Лівообертовий [2-(2-тієніл)етиламіно](2-хлорфеніл) складний метиловий ефір оцтової кислоти, отриманий шляхом розщеплення рацемату. 100г рацемату хлоргідрату сполуки з загальною формулою (VIII) - де X означає хлорозаміщений атом та 30г бікарбонату натрію змішують в 500мл діхлорметану та 200мл води. Після помішування органічну фазу відділяють за допомогою декантації. Залишок розчиняють в 800мл ацетону, і до цього розчину додають 53г (-)-камфора-10-сульфокислоти. Суміш витримують при кімнатній температурі протягом 12 годин, а отриманий осад сепарують та суспендують в 300мл ацетону. Нерозчинний твердий осад кристалізують з суміші, що складається з 600мл ацетону та 160мл метилетилкетону, щоб отримати 52,5г (-)камфорсульфонату бажаного продукту, точка плавлення: 95°С, [a]22D=-82° (c=1, метанол). Приклад 21 (+)-(3)-(2-хлорфеніл)(6,7-дігідро-4Н-тієно[3,2-с]пірідін-5-іл)хлоргідрат складного метилового ефіру оцтової кислоти 6г (0,017моль) правооборотного метилу [2-(2-тієніл)етиламіно](2-хлорфеніл)хлоргідрат ацетату суспендують в 6,7мл 38%-ного водного розчину формаліну і нагрівають до 60°С при помішуванні. Початковий матеріал розчиняють при 60°С, отриманий розчин розмішують при тій же температурі протягом 30хв, поки не завершиться реакція. Потім реакційну суміш розводять 100мл 1,2-діхлоретану та 150мл води, добре розколочують і фази сепарують. Водяну фазу екстрагують за допомогою 2x30мл 1,2-діхлоретану, спільну органічну фазу екстрагують за допомогою 100мл води, просушують над безводним сульфатом натрію, фільтрують та випаровують в вакуумі. Залишок в вигляді 6г матеріалу розчиняють в 30мл діетилового ефіру і при охолодженні суміші в розчин вводять 0,6г сухого газу хлороводню при кімнатній температурі. Осаджений кристалічний матеріал відфільтровують, промивають ефіром та просушують. Вага: 5,5г. Точка плавлення: 130-132°С, [a]22D=+60°. Вихід: 90,1%. Оптична чистота: 99% (визначено за допомогою HPLC). Приклад 22 а) (+)-(2-хлорфеніл)-(6,7-дігідро-4Н-тієно[3,2-с]пірідін-5-іл) сіль (-)-камфорсульфокислоти складного метилового ефіру оцтової кислоти 32г (0,0994моль) (2-хлорфеніл)-(6,7-дігідро-4Н-тієно[3,2-с]пірідін-5-іл) складного метилового ефіру оцтової кислоти розчиняють в 150мл ацетону і до розчину додають 9,95г (0,0397моль) лівообертового моногідрату 10-камфорсульфокислоти. Однорідну суміш витримують при кімнатній температурі. Після 48 годин з'являються кристали. Суміш концентрують до 50мл шляхом випаровування і залишають при кімнатній температурі на 24 години. Отримані кристали відфільтровують, промивають ацетоном та просушують. Потім їх знову розчиняють в дуже малій кількості (50мл) гарячого ацетону, і після охолодження кристали відфільтровують, промивають ацетоном та просушують. Так отримують означену в заголовку сполуку. Вихід: 88%. Точка плавлення: 165°С, [a]20D=+24° (с=1,68г/100мл; метанол). б) (+)-(2-хлорфеніл)-(6,7-дігідро-4Н-тієно[3,2-с]пірідін-5-іл) складний метиловий ефір оцтової кислоти До суспензії, виготовленої з 200г (+)-(2-хлорфеніл)-(6,7-дігідро-4Н-тієно[3,2-с]пірідін-5-іл) солі (-)камфорсульфокислоти складного метилового ефіру оцтової кислоти та 800мл діхлорметану додають 800мл розчину бісульфату натрію. Після розмішування органічну фазу відділяють за допомогою декантації, просушують над безводним сульфатом натрію, а розчинник усувають в вакуумі. (+)-(2-хлорфеніл)-(6,7дігідро-4Н-тієно[3,2-с]пірідін-5-іл) складний метиловий ефір оцтової кислот отримують як розчин в 800мл діхлорметану. Після розмішування органічну фазу відділяють за допомогою декантації, просушують над безводним сульфатом натрію, а розчинник усувають в вакуумі. (+)-(2-хлорфеніл)-(6,7-дігідро-4Н-тієно[3,2-с]пірідін-5-іл) складний метиловий ефір оцтової кислоти отримують в вигляді масла без кольору. с) (+)-(2-хлорфеніл)-(6,7-дігідро-4Н-тієно[3,2-с]пірідін-5-іл) бісульфат складного метилового ефіру оцтової кислоти Залишок, що отримано в попередньому прикладі, розчиняють в 500мл охолодженого льодом ацетону, і до розчину краплями додають 20,7мл концентрованої сірчаної кислоти (93,64%; густина 1,83). Отриманий осад сепарують шляхом фільтрації, промивають в 1000мл ацетону та висушують в вакуумі при 50°С. Так отримують 139г означеної в заголовку солі в вигляді білих кристалів. Точка плавлення: 184°С, [a]20D=+55,1° (с=1,891г/100мл; метанол).
ДивитисяДодаткова інформація
Назва патенту англійськоюA process for the preparation of pharmacologically active substance
Назва патенту російськоюСпособ изготовления фармакологически активного вещества
МПК / Мітки
МПК: C07D 495/04, A61K 31/4365, A61P 7/02
Мітки: активних, рацемічних, одержання, спосіб, сполук, оптично
Код посилання
<a href="https://ua.patents.su/6-66359-sposib-oderzhannya-racemichnikh-abo-optichno-aktivnikh-spoluk.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання рацемічних або оптично активних сполук</a>
Спосіб одержання оптично активних або рацемічних ефірів циклопропанкарбонових кислот

Номер патенту: 3145
Опубліковано: 26.12.1994
Автори: Габор Ковач, Маріанне Ловас, Іштван Секей, Рудольф Шоош, Йожеф Дукаі
МПК: C07C 255/39, C07C 67/00, C07C 69/743, C07B 53/00, C07B 31/00, C07C 253/00, C07D 307/935, C07D 209/49, A01N 53/00
Мітки: спосіб, циклопропанкарбонових, рацемічних, одержання, оптично, активних, кислот, ефірів
Формула / Реферат:
Способ получения оптически активных или рацемических эфиров циклопропанкарбоновых кислот общей формулыгде R и R— атом галогена или низшая алкилгруппа; R — группа формулыили бензильная группа; символ ~ означает оси/или a-положение; символ - означает b-положение, отличающийся тем, что, с целью повышения выхода целевого продукта и упрощения процесса, оптически активные или рацемические циклопропановые...
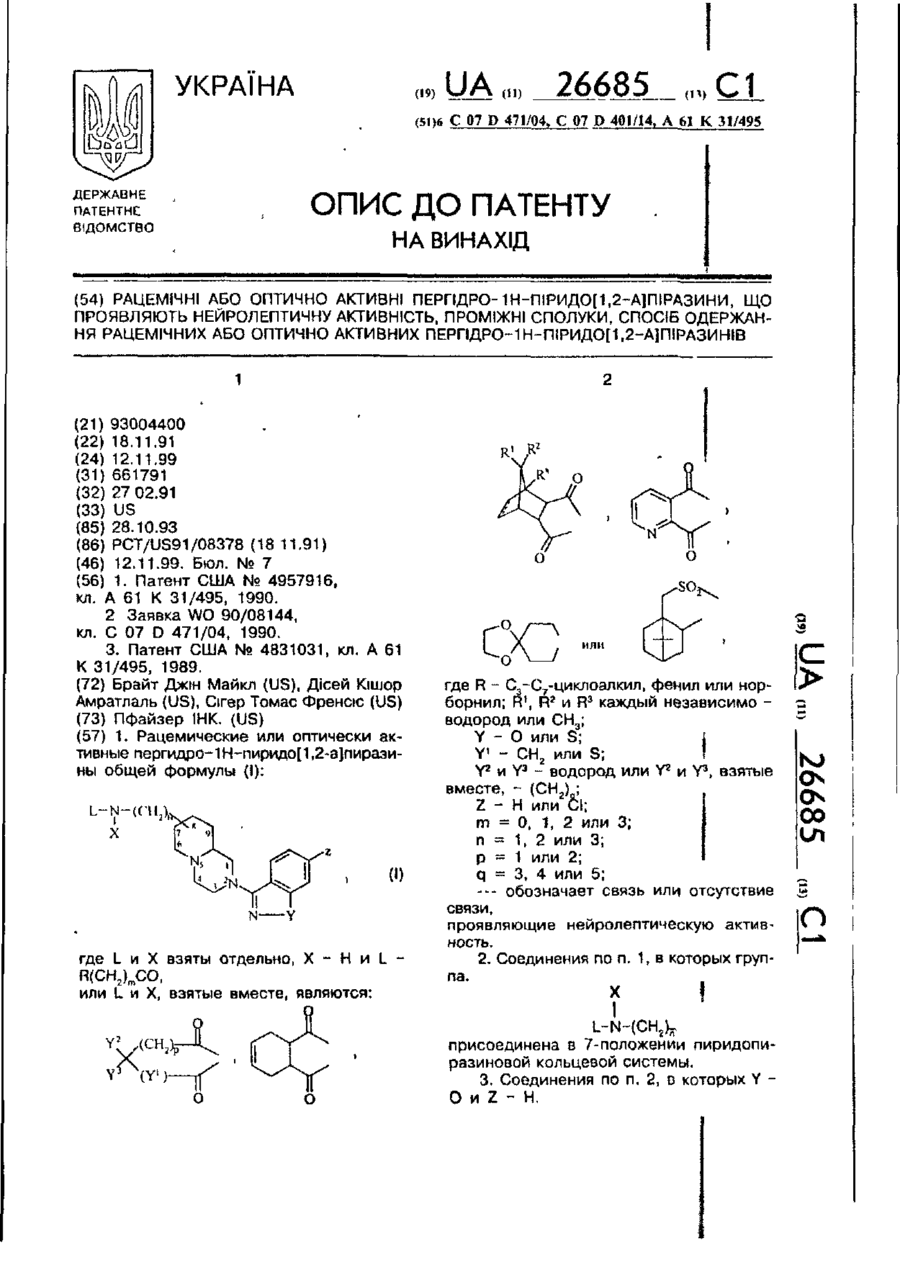
Рацемічні або оптично активні пергідро-1н-піридо[1,2-а]піразини, що проявляють нейролептичну активність, проміжні сполуки, спосіб одержання рацемічних або оптично активних пергідро-1н-піридо[1,2-а]піразинів
Номер патенту: 26685
Опубліковано: 12.11.1999
Автори: Сігер Томас Френсіс, Брайт Джін Майкл, Дісей Кішор Амратлаль
МПК: C07D 211/62, C07D 519/00, C07D 211/60, A61K 31/495, C07D 471/04, A61P 25/18
Мітки: одержання, активних, пергідро-1н-піридо[1,2-а]піразинів, активні, пергідро-1н-піридо[1,2-а]піразини, проявляють, сполуки, активність, проміжні, нейролептичну, рацемічні, спосіб, рацемічних, оптично
Формула / Реферат:
1. Рацемические или оптически активные пергидро-1Н-пиридо[1,2-a]пиразины общей формулы (I):где L и X взяты отдельно, X - H и L - R(CH2)mCO,или L и X, взятые вместе, являются:где R - C3-C7-циклоалкил, фенил или норборнил; R1, R2 и R3 каждый независимо - водород или CH3;Y - O или S;Y1 - CH2 или S;Y2 и Y3 - водород или Y2 и Y3, взятые вместе, - (CH2)q;Z - H или Cl;m =...
Спосіб одержання похідних оксіаміноебурнану або їх солей або оптично активних ізомерів

Номер патенту: 3539
Опубліковано: 27.12.1994
Автори: Тібор Кеве, Марія Газдаг, Дьордь Калауш, Чаба Сантаі, Лайош Сабо, Янош Шапі, Лайош Данчі
МПК: C07D 471/14, A61K 31/475, A61P 9/08, A61K 31/435, C07D 461/00
Мітки: спосіб, одержання, ізомерів, оптично, активних, оксіаміноебурнану, похідних, солей
Формула / Реферат:
1. Способ получения производных оксиаминоэбурнана формулы Iгде R1 и R2, С1-С4- алкил, или их солей, или их оптически активных изомеров, отличающийся тем, что соединение гексагидроиндолохинолизина общей формулы IIгде R2 имеет указанные значения; Х - кислотный остаток, подвергают взаимодействию с ди-эфиром метиленмалоновой кислоты общей формулы IIIгде R1 имеет указанные значення, в среде инертного...
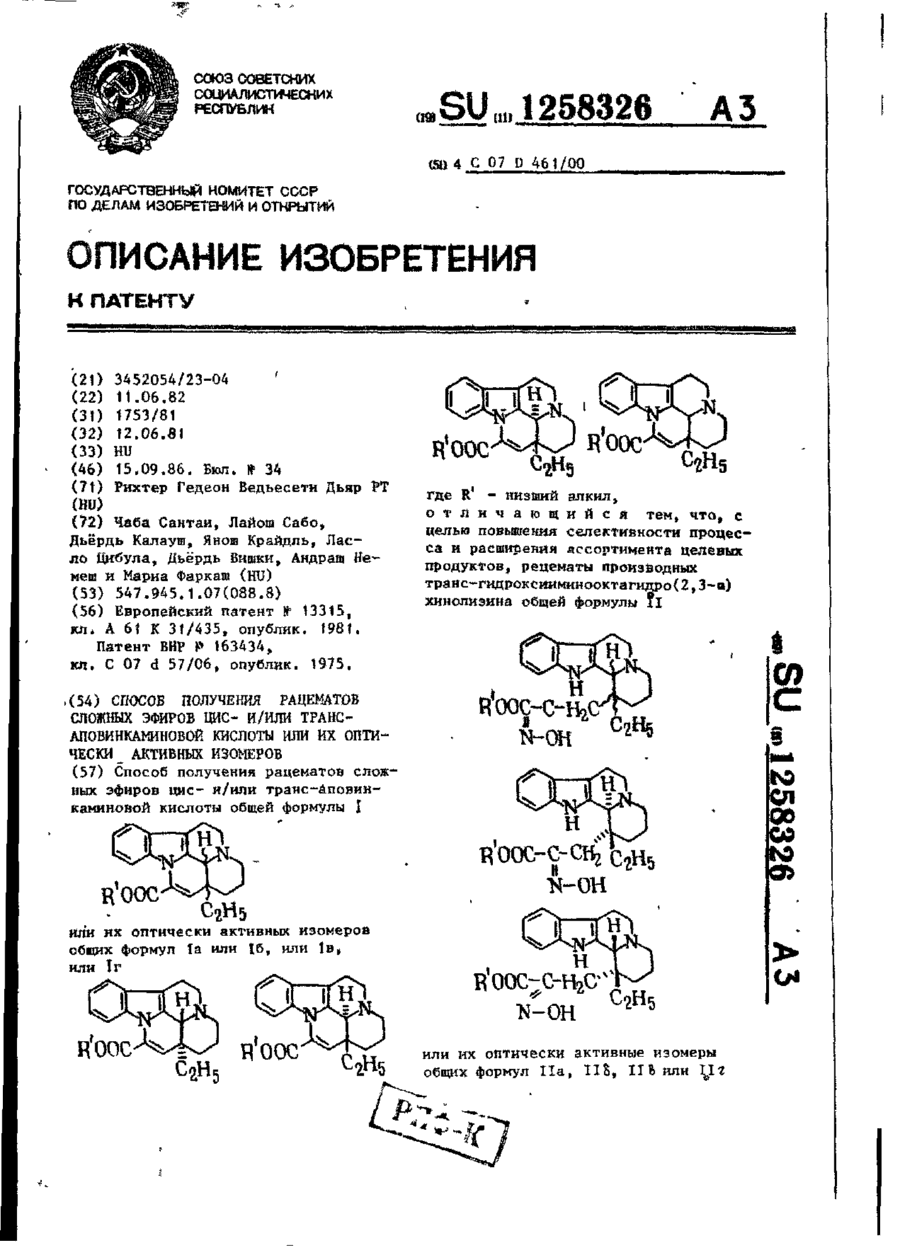
Спосіб одержання рацематів складних ефірів цисі/або трансаповінкамінової кислоти або їх оптично активних ізомерів
Номер патенту: 3472
Опубліковано: 27.12.1994
Автори: Ласло Цібула, Андраш Немеш, Чаба Сантаі, Янош Крайдль, Дьєрдь Вішкі, Лайош Сабо, Дьєрдь Калауш, Маріа Фаркаш
МПК: C07B 57/00, C07D 471/14, C07D 461/00
Мітки: активних, складних, одержання, оптично, спосіб, ефірів, кислоти, рацематів, ізомерів, трансаповінкамінової
Формула / Реферат:
Способ получения рацематов сложных эфиров цис- и/или трансаповинкаминовой кислоты общей формулы I или их оптически активных изомеров общих формул Iа или Iб, или Iв, или Iг где R' - низший алкил, отличающийся тем, что, с целью повышения селективности процесса и расширения ассортимента целевых продуктов, рацематы производных транс-гидроксииминооктагидро (2, 3-а) хинолизина общей формулы...
Мікрокапсула для біологічно активних сполук та спосіб її одержання

Номер патенту: 46025
Опубліковано: 15.05.2002
Автори: Шер Герберт Бенсон, Чен Дзін Лінг
МПК: B01J 13/02, A01N 25/22, A01N 25/04, A01N 25/30, A01N 25/28, B01J 13/06, B01J 13/04, A01N 53/00
Мітки: активних, одержання, сполук, біологічно, спосіб, мікрокапсула
Формула / Реферат:
1. Мікрокапсула для біологічно активних сполук, що містить органічну рідину, яка включає чутливий до ультрафіолетового світла біологічно активний матеріал та ефективну кількість засобу захисту від ультрафіолетового світла у вигляді часток, яка відрізняється тим, що засіб захисту від ультрафіолетового світла вибраний із групи, яка включає діоксид титану, оксид цинку та їх суміші, що суспендовані та ретельно дисперговані у рідини.2....
Попередній патент: Замковий пристрій
Наступний патент: Спосіб одержання масляної емульсії (варіанти), емульсія, препарат для годування риб
Випадковий патент: Фарш рибний з кашею з цільних зерен єсо