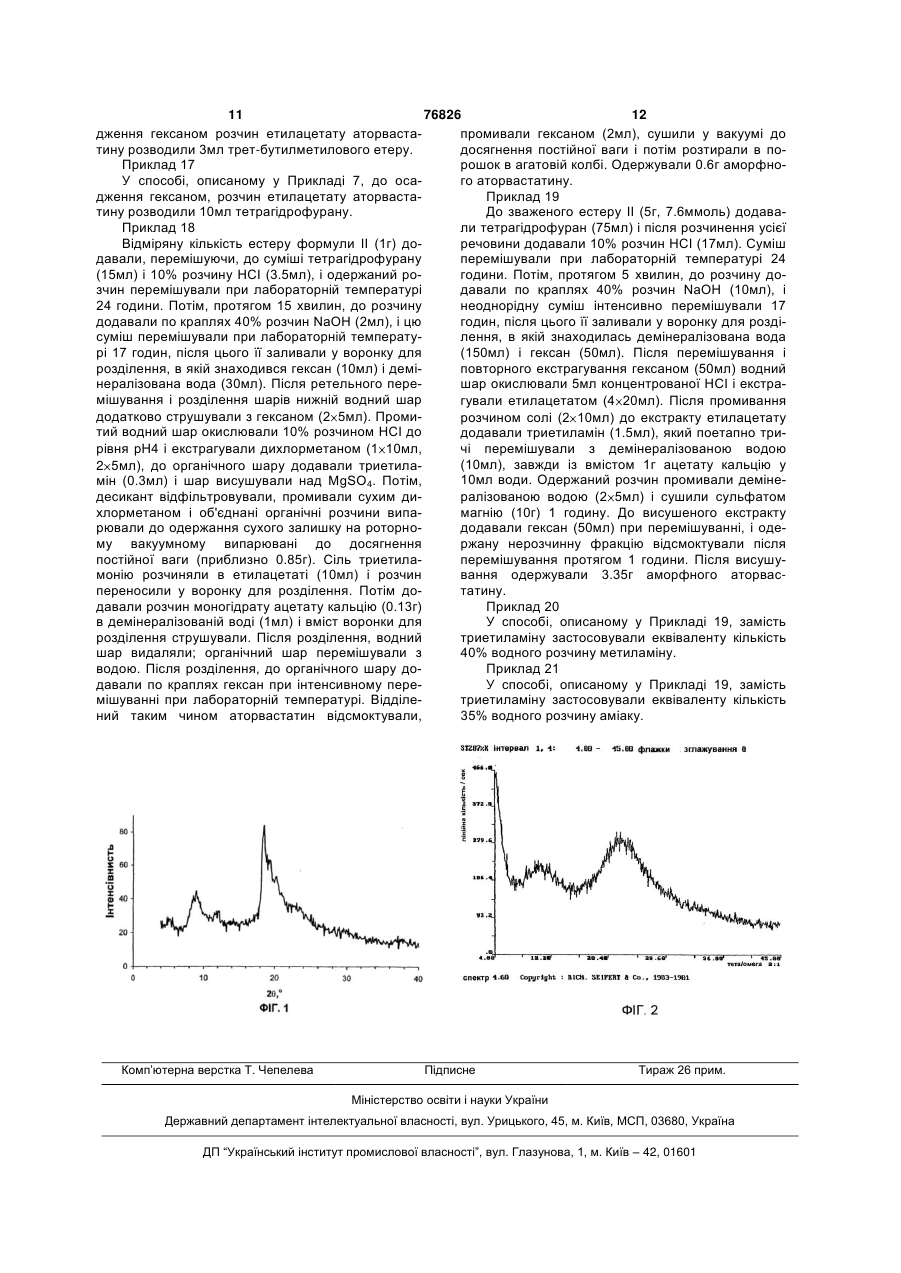
Спосіб одержання аморфної форми гемікальцієвої солі (3r,5r) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти (аторвастатину)





Формула / Реферат
1. Спосіб одержання аморфної форми гемікальцієвої солі (3R,5R) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти формули І
 , (I)
, (I)
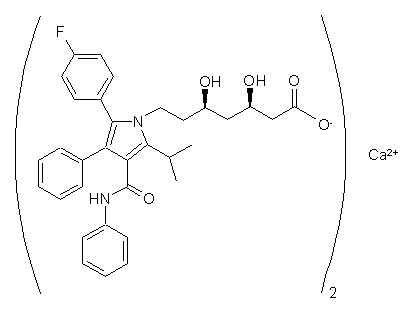
який відрізняється тим, що (3R,5R) трет-бутил (6-{2-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-етил)-2,2-диметил-[1,3]діоксан-4-іл)-ацетату формули II
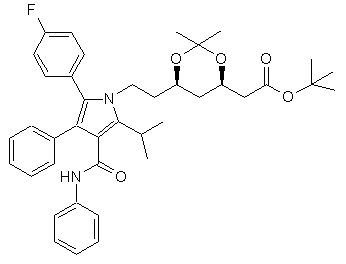 (II)
(II)
спочатку перетворюють у процесі кислотного гідролізу із наступним лужним гідролізом у сіль (3R,5R) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти з лужним металом, яку потім екстрагують в органічному розчиннику, вибраному з групи естерів, утворених C1-C5 кислотами зі C1-C5 спиртами, та у якому кислоту, після очищення, перетворюють шляхом обробки кальцієвою сіллю або гідроксидом кальцію, або алкоголятом C1-C5 кальцію у гемікальцієву сіль, яку осаджують вуглеводнем C5-C12 або діалкіловим етером формули R1OR2, де кожен з R1 і R2 є C1-C5 алкільною групою.
2. Спосіб згідно з пунктом 1, який відрізняється тим, що розчин солі (3R,5R) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти з катіоном лужного металу, одержаний із сполуки формули II, який містить надлишок хлориду і гідроксиду відповідного катіону, підкислюють і екстрагують з одержанням розчину (3R,5R) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти, який потім перетворюють, шляхом реакції з гідроксидом кальцію або алкоголятом кальцію, у розчин гемікальцієвої солі.
3. Спосіб згідно з принаймні одним з пунктів 1 і 2, який відрізняється тим, що розчин (3R,5R) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти у відповідному естері, утвореному C1-C5 кислотою зі C1-C5 спиртом, перетворюють у сіль лужного металу.
4. Спосіб згідно з принаймні одним з пунктів 1-3, який відрізняється тим, що гемікальцієву сіль осаджують з розчину етилацетату.
5. Спосіб згідно з принаймні одним з пунктів 1-4, який відрізняється тим, що осадження проводять шляхом додавання неполярного розчинника із групи, яка складається з пентану, гексану, гептану або циклогексану, або діалкілового етеру формули R1OR2, де кожен з R1 і R2 є C1-C5 алкільною групою, до розчину гемікальцієвої солі (3R,5R) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідроксигептанової кислоти у відповідному розчиннику.
6. Спосіб згідно з принаймні одним з пунктів 1-5, який відрізняється тим, що розчин гемікальцієвої солі (3R,5R) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти в естері, утвореному C1-C5 кислотою зі C1-C5 спиртом, додають до неполярного розчинника із групи, яка складається з пентану, гексану, гептану або циклогексану, або діалкілового етеру формули R1OR2, де кожен з R1 і R2 є C1-C5 алкільною групою.
7. Спосіб згідно з принаймні одним із пунктів 1-6, який відрізняється тим, що розчин гемікальцієвої солі (3R,SR) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти у відповідному естері розводять, до осадження, 5-95%, переважно 30-70%, придатним співрозчинником.
8. Спосіб згідно з пунктом 7, який відрізняється тим, що толуол, т-бутилметиловий етер або тетрагідрофуран використовують як співрозчинник.
Текст
1. Спосіб одержання аморфної форми гемікальцієвої солі (3R,5R) 7-[3-феніл-4-фенілкарбамоїл2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5дигідрогептанової кислоти формули І 2 (19) 1 3 76826 4 ізопропілпірол-1-іл]-3,5-дигідроксигептанової кис7. Спосіб згідно з принаймні одним із пунктів 1-6, який відрізняється тим, що розчин гемікальцієвої лоти у відповідному розчиннику. 6. Спосіб згідно з принаймні одним з пунктів 1-5, солі (3R,SR) 7-[3-феніл-4-фенілкарбамоїл-2-(4який відрізняється тим, що розчин гемікальцієвої фторфеніл)-5-ізопропілпірол-1-іл]-3,5солі (3R,5R) 7-[3-феніл-4-фенілкарбамоїл-2-(4дигідрогептанової кислоти у відповідному естері фторфеніл)-5-ізопропілпірол-1-іл]-3,5розводять, до осадження, 5-95%, переважно 30дигідрогептанової кислоти в естері, утвореному 70%, придатним співрозчинником. 8. Спосіб згідно з пунктом 7, який відрізняється C1-C5 кислотою зі C1-C5 спиртом, додають до неполярного розчинника із групи, яка складається з тим, що толуол, т-бутилметиловий етер або тетрапентану, гексану, гептану або циклогексану, або гідрофуран використовують як співрозчинник. діалкілового етеру формули R1OR2, де кожен з R1 і R2 є C1-C5 алкільною групою. Винахід стосується нового способу одержання аморфної форми гемі-кальцієвої солі (3R,5R) 7-[3феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5ізопропіл-пірол-1-іл]-3,5-дигідрогептанової кислоти, відомою під непатентованою назвою аторвастатин. Даний лікарський засіб є важливим представником медикаментів, що знижують рівень ліпідів та холестерину у крові. Аторвастатин (формула І) одержують відповідно до опублікованих патентів [патенти США №№4,681,893; 5,003,080; 5,097,045; 5,103,024; 5,124,482; 5,149,837; 5,155,251; 5,216,174; 5,245,047; 5,248,793; 5,273,995; 5,397,792; 5,342,952] звичайно із натрієвої солі (3R,5R) 7-[3феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5ізопропіл-пірол-1-іл]-3,5-дигідрогептанової кислоти і придатної, розчинної у воді солі кальцію, переважно із ацетату або хлориду кальцію. Початкову натрієву сіль (3R,5R) 7-[3-феніл-4фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти одержують із зазначеної кислоти, яку звичайно одержують з (3R,5R) трет-бутил (6-{2-[3-феніл-4фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-етил}-2,2-диметил-[1,3]диоксан-4-іл)ацетату (формула II). Цю основну проміжну сполуку перетворюють у натрієву сіль відповідної кислоти шляхом змішування спочатку із соляною кислотою і потім із великим надлишком гідроксиду натрію, який, однак, супроводжується великою кількістю надлишку гідроксиду, а також хлориду натрію. Окисленням із наступним екстрагуванням одержують розчин відповідної кислоти (формула III) без будь-яких неорганічних домішок. Одержану таким чином кислоту потім перетворюють у відповідний лактон (формула IV), який очищують кристалізацією. Очищений лактон потім перетворюють у натрієву сіль шляхом перемішуванням із еквівалентом гідроксиду натрію; у наступній стадії надлишок не може використовуватись, якщо він утворює, із сіллю кальцію, гідроксид кальцію, який не може бути повністю видалений із речовини у наступних стадіях процесу згідно із вищезазначеними патентами. Також, якщо використовується еквівалент гідроксиду, реакція займає багато часу і має контролюватись за допомогою ВЕРХ. Ще одним недоліком даного винаходу є втрата приблизно 20% виходу. Залежно від способу втілення, аторвастатин одержують у деяких його кристалічних формах, або у аморфній формі. В первинних патентах [наприклад, патентах №№4,681,893 і 5,273,995] форма речовини, одержаної відповідно до цих патентів, не зазначається. В наступних патентах [патенти США №№5,969,156 і 6,121,461], в яких описуються кристалічні форми аторвастатину, припускається, що речовина, одержана відповідно до первинних патентів, була у аморфній формі. [В патенті ЕР 839,132], в якому описується новий спосіб одержання аморфної форми аторвастатину шляхом розчинення аторвастатину в кристалічній формі І у розчиннику, який не гідроксилює (у патентах вказується тетрагідрофуран та суміш тетрагідрофурану та толуолу у якості прикладів таких розчинників), з наступним висушуванням, знову зазначається, що у перших патентах одержували аторвастатин у аморфній формі, але методом, який важко відтворити. Із нашого досвіду випливає, що аторвастатин, одержаний згідно із попередніми патентами [США 4,681,893, 5,298,627 і 5,273,995], не є абсолютно аморфним, і результати рентгенографічного аналізу свідчать про присутність кристалічних компонентів (див. Фіг.1). [В первинному патенті 4,681,893] також описується можливість очищення невідповідної речовини шляхом її розчинення в етилацетаті, фільтрації через суперцел, і осадження розчину гексаном при температурі 50°С. Патент на ім'я Ranbaxy Laboratories [WO 00/71116 А1] описує перетворення кристалічної форми аторвастатину у розчиннику, який не гідроксилює, та осадження одержаного розчину неполярним вуглеводневим розчинником. Той же самий принцип описаний у патенті на ім'я 5 76826 6 Lek [WO 01/42209 А1], в якому описується перетдо одного із вищезазначених неполярних розчинворення кристалічної форми аторвастатину у амоників. На жодній із вищенаведених стадій цього рфну шляхом її розчинення у різних розчинниках, процесу, гемі-кальцієву сіль не виділяють у твервключаючи розчинники, які не гідроксилюють, і дому стані і, тому, права на жоден з патентів, якинижчі спирти, із наступним осадженням таких розми охороняються кристалічні форми цієї речовини, чинів розчинами, в яких аторвастатин є нерозчинабо способи перетворення кристалічного аторвасним. Знову, такі розчинники мають широке визнататину у аморфний аторваститин, не поручення і, окрім неполярних вуглеводневих шуються. розчинників, включають аліфатичні етери. Розчин гемі-кальцієвої солі аторвастатину у Предметом даного винаходу є опис нового попридатному розчиннику, переважно у естері, утвокращеного способу одержання аморфної форми реному С1-С5 кислотою і С1-С5 спиртом, більш пегемі-кальцієвої солі (3R,5R) 7-[3-феніл-4реважно у етилацетаті, можна одержувати з розфенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілчину відповідної вільної кислоти, одержаної за пірол-1-іл]-3,5-дигідрогептанової кислоти, якому не допомогою описаного вище способу із проміжної будуть притаманні недоліки вищезазначених спосполуки II. Цей розчин перетворюють у розчин собів. гемі-кальцієвої солі шляхом змішування із водним На перевагах аморфного атовастатину для розчином гідроксиду кальцію, суспензією гідроксидеяких способів застосування було вже наголошеду кальцію в меншій кількості води, або, необов'яно у вищезазначеному [патенті ЕР 839,132]. Предзково, розчином алкоголяту кальцію С1-С5 у приметом даного винаходу є новий спосіб одержання датному розчиннику, переважно у відповідному аморфної форми гемі-кальцієвої солі (3R,5R) 7-[3спирті. Також можливо використати розчин однієї феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5із вищенаведений солей аторвастатину у придатізопропіл-пірол-1-іл]-3,5-дигідрогептанової кислоному розчиннику, переважно у етилацетаті, і перети, який полягає у перетворенні (3R,5R) 7-[3творити його у розчин гемі-кальцієвої солі аторвафеніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5статину шляхом перемішування із придатною ізопропіл-пірол-1-іл]-3,5-дигідрогептанової кислорозчинною у воді сіллю кальцію, переважно із ацети, або її солі, яка містить катіон М+, який є або татом кальцію. Надлишок використаних солей мокатіоном лужного металу, або катіоном амонію жна легко видалити шляхом промивання органічформули RnN(+)H(4-n), де R є нижчим алкілом С1-С5, ного розчину гемі-кальцієвої солі аторвастатину і n може досягати значень у проміжку 0-3, без відводою, або певними водними розчинами, наприділення сполуки у формі гемікальцієвої солі, або клад розчином солі і потім водою. В залежності від іншої солі, кислоти або лактону із розчину шляхом системи осадження і способу осадження, розчин обробки кальцієвої солі гідроксидом кальцію, або гемі-кальцієвої солі аторвастатину потім викорисалкоголятом кальцію С1-С5, з одержанням гемітовується у наступній стадії без висушування, або кальцієвої солі, після чого її осаджують вуглеводвін може бути попередньо висушений відповідним нем С5-С12, або диалкіловим етром формули десикантом, переважно сульфатом натрію, кальR1OR2, де кожен з R1 і R2 є С1-С5алкільною групою, цію або магнію. з утворенням твердої аморфної фази. Придатні розчинні у воді солі аторвастатину Весь вищевикладений спосіб ґрунтується на можуть бути переважно одержані із розчину відпонесподіваному відкритті, що в етилацетаті та у відної вільної кислоти відповідно до описаного деяких інших відповідних розчинниках не тільки вище способу із проміжної сполуки формули II. вільна кислота, яка відповідає аторвастатину, але і Розчин потім перетворюють у відповідну сіль шляряд її солей, наприклад сіль натрію, сіль калію, хом додавання розчину гідроксиду лужного металу солі амонію, які походять від аміаку, первинних, в воді, шляхом додавання розчину аміаку у воді, вторинних та третинних амінів, а також безпосеабо розчину відповідного аміну у воді з наступним редньо гемі-кальцієва сіль є легко розчинними. перемішуванням. Переважно, використовуються Аморфну форму аторвастатину переважно такі аміни як триетиламін, які є рідкими при нородержують безпосередньо осадженням розчину мальній температурі і які можуть додаватися до гемі-кальцієвої солі аторвастатину у придатному розчину відповідної вільної кислоти без викорисрозчиннику, переважно у естері, утвореному С1-С5 тання розчинника. Одержані таким чином розчини кислотою і С1-С5 спиртом, причому застосування солей можуть одразу використовуватись без будьетилацетату є особливо бажаним, неполярним якого очищення для перетворення у розчин гемірозчинником, переважно пентаном, гексаном, гепкальцієвої солі аторвастатину як було описано таном або циклогептаном, необов'язково, диалківище. Розчини деяких солей аторвастатину також ловим етером R1OR2, де кожний з R1 і R2 є С1можна випарювати на цій стадії і очищувати шляС5алкільною групою, переважно діетиловим етехом кристалізації в придатних розчинниках. Розчин ром, диізопропіловим етером або т-бутилнатрієвої або кальцієвої солі аторвастатину можна метиловим етером. Для покращення чистоти пропереважно одержувати також одразу із розчину дукту, було доведено доцільність розведення розпісля проведення реакції конверсії проміжної спочину аторвастатину в етилацетаті, до його випалуки формули II. На останній стадії одержують діння в осад, додатковим розчинником, наприклад розчин, який містить відповідний хлорид лужного толуолом, трет-бутилметиловим етером або тераметалу, натрієву або кальцієву сіль аторвастатину, гідрофураном, у кількості 5-95%, переважно 30а також надлишок використаного гідроксиду луж70%. Аморфну форму також можна одержувати ного металу. Було несподівано виявлено, що пооберненим способом, при якому розчин гемівторним екстрагуванням цього розчину відповідкальцієвої солі у придатному розчиннику додають ним розчинником, переважно естером, утвореним 7 76826 8 С1-С5 кислотою і С1-С5 спиртом, більш переважно шування, органічний шар видаляли і водний шар етилацетатом, майже всю кількість відповідної екстрагували сумішшю гексану (40мл) і тетрагідсолі можна перевести в органічну фазу, яка може рофурану (10мл). Після повного розділення, водбути використана надалі як було описано вище. ний шар екстрагували етилацетатом (1 40мл, Рентгенограма аторвастатину, одержаного 3 20мл). Після випарювання одержували 4г екстспособом, описаним у цьому винаході, свідчить ракту натрієвої солі аторвастатину. Після перекрипро повністю аморфну структуру (див. Фіг.2). сталізації з етанолу сіль розчиняли в етилацетаті Винахід також ілюструється доданими графі(100мл) і розчин етилацетату поступово тричі ками та наступним прикладами. Приклади, в яких струшували з демінералізованою водою (5мл), наведено переважні альтернативні шляхи одерзавжди із вмістом 1г ацетату кальцію у 5мл води. жання аторвастатину згідно з даним винаходом, Отриманий розчин етилацетату промивали демімають лише ілюстративний характер і ніяким чинералізованою водою (2 5мл) і, після просушуном не обмежують об'єм цього винаходу. вання сульфатом магнію, його концентрували на На Фіг.1 наведена рентгенограма аторваставакуумному випарювачі до об'єму 30мл. Після фітину, одержаного способом, описаним [у патенті льтрації, прозорий розчин додавали по краплях до США No.5,298,627]. гексану (300мл) при інтенсивному перемішуванні На Фіг.2 наведена рентгенограма аторвастапротягом 5 хвилин, потім суміш перемішували тину, одержаного способом, одержаним згідно з протягом наступних 20 хвилин, нерозчинні фракції даним винаходом. відсмоктували, промивали гексаном (20мл) і сушиПриклади ли у вакуумі при лабораторній температурі. ОдерПриклад 1 жували 3.1г аморфного аторвастатину. До зваженого естеру формули II (5г, 7.6ммоль) Приклад 4 додавали тетрагідрофуран (75мл) і, після розчиДо зваженого естеру формули II (5г, 7.6ммоль) нення усієї речовини, додавали 10% НСІ (17мл). додавали тетрагідрофуран (75мл) і, після розчиСуміш перемішували при лабораторній темперанення усієї речовини, додавали 10% розчин НСІ турі протягом 6 годин. До одержаного розчину до(17мл). Суміш перемішували при лабораторній давали розчин 40% NaOH (10мл) протягом 5 хвитемпературі 24 години. До розчину потім додавали лин для того, щоб температура не перевищувала 40% розчин NaOH (10мл) протягом 5 хвилин і не35°С, і неоднорідну суміш інтенсивно струшували однорідну суміш інтенсивно перемішували 17 гопротягом 15 годин і, після цього, заливали у вородин і, після цього, заливали у воронку для роздінку для розділення, в якій знаходилась демінералення із демінералізованою водою (150мл) і лізована вода (150мл) і гексан (50мл). Після стругексаном (50мл). Водний шар окислювали 10% шування, органічний шар видаляли і водний шар розчином НСІ до рівня рН3 і екстрагували етилаекстрагували сумішшю гексану (40мл) і тетрагідцетатом (4 20мл). Одержаний екстракт перемішурофурану (10мл). Після повного розділення, водвали із сумішшю гідроксиду кальцію (0.87г) в деміний шар екстрагували етилацетатом (1 40мл, нералізованій воді (15мл) 20 хвилин. Потім водний 4 20мл). Екстракт етилацетату потім поетапно шар відділяли, екстракт промивали демінералізострушували тричі з демінералізованою водою ваною водою (2 5мл) і сушили сульфатом магнію (5мл), завжди із вмістом 1г ацетату кальцію у 5мл (10г) 1 годину. До висушеного екстракту додавали води. Одержаний екстракт етилацетату промивали гексан (160мл) при перемішуванні і через 1 годину демінералізованою водою (2 5мл), концентрували одержані нерозчинні фракції відсмоктували. Після на вакуумному випарювачі до об'єму 20мл. Після висушування одержували 3.3г аморфного аторвафільтрації прозорий розчин додавали по краплях статину. до гексану (200мл) при інтенсивному перемішуПриклад 5 ванні протягом 5 хвилин, і потім суміш перемішуУ способі, описаному у Прикладі 4, замість гідвали протягом наступних 20 хвилин, нерозчинні роксиду кальцію використовували розчин метанофракції відсмоктували, промивали гексаном (20мл) лату кальцію в метанолі для перетворення кислоі сушили у вакуумі при лабораторній температурі. ти аторвастатину у відповідну кальцієву сіль, і Одержували 4.1 г аморфного аторвастатину. одержували таку ж кількість аморфного аторвасПриклад 2 татину після проведення процесу, описаного у У способі, описаному у Прикладі 1, замість гідПрикладі 4. роксиду натрію для гідролізу естеру використовуПриклад 6 вали гідроксид калію, таким чином одержували Відміряну кількість естеру формули II (8.7г, калійну сіль аторвастатину, яку далі оброблювали 13.3ммоль) додавали, при перемішуванні, до кругвідповідно до методики, описаної у Прикладі 1. лої колби, яка містила суміш тетрагідрофурану Приклад 3 (100мл) і 10% розчину НСІ (30мл, 0.082моль) і До зваженого естеру формули II (5г, 7.6ммоль) одержаний розчин перемішували при лаборатордодавали тетрагідрофуран (75мл), і після розчиній температурі 24 години. Потім, протягом 15 нення усієї речовини додавали 10% розчин НСІ хвилин, до розчину додавали по краплях 30% роз(17мл). Суміш перемішували при лабораторній чин NaOH (24мл, 7.2г, 0.18моль); реакційна суміш температурі протягом 24 годин. До розчину потім нагрівалась і ставала мутною. Цю суміш переміпротягом 5 хвилин додавали 40% розчин NaOH шували при лабораторній температурі протягом 15 (10мл) і неоднорідну суміш інтенсивно перемішугодин і, після цього, її заливали у воронку для розвали 17 годин і, після цього, заливали у воронку ділення, яка містила гексан (100мл) і демінералідля розділення, в якій знаходилась демінералізозовану воду (300мл). Після ретельного перемішувана вода (150мл) і гексан (50мл). Після перемівання і розділення шарів нижній водний шар 9 76826 10 умному випарювачі після перемішування при лаперемішували додатково з гексаном (2 50мл). бораторній температурі 0.5 годин. Одержану тверПромитий водний шар окислювали 10% розчином ду піну перемішували з сухим диетилетером НСІ до рівня рН4 і екстрагували дихлорметаном (25мл) і знову випарювали до одержання сухого (1 100мл, 2 50мл, 2 25мл), органічний шар прозалишку (приблизно 0.9г). Сіль триетиламонію мивали насиченим розчином солі (2 25мл) і суширозчиняли в етилацетаті (10мл) і розчин переноли CaSO4 (25г) протягом ночі. Потім десикант відсили до воронки для розділення. Потім туди додафільтровували, промивали сухим дихлорметаном вали розчин моногідрату ацетату кальцію (0.13г) в (50мл) і до одержаного розчину додавали розчин демінералізованій воді (1мл) і вміст воронки для триетиламіну (3мл, 21.5ммоль) в сухому дихлоррозділення перемішували. Після розділення водметані (20мл), розчин випарювали до одержання ний шар видаляли, органічний шар перетрушували сухого залишку на роторному вакуумному випарюз водою. Після розділення, у органічний шар при вачі після перемішування при лабораторній темінтенсивному перемішуванні додавали по краплях пературі протягом 0.5 годин. Одержану тверду піну пентан при лабораторній температурі. Після дода(приблизно 10-11г) перемішували з сухим дихлорвання по краплях приблизно 3мл пентану розчин метаном (25мл) і знову випарювали до одержання ставав помітно мутним, і суміш перемішували при сухого залишку (приблизно 9г). Цю процедуру полабораторній температурі 15 хвилин. Потім, додавторювали ще раз з одержанням 8.34г жовтуватої ткові 7.5мл пентану швидко додавали по краплях, твердої піни солі триетиламонію (12.6ммоль, 95%). суміш перемішували прилабораторній температуСіль триетиламонію розчиняли в етилацетаті рі 1 годину і потім залишали на ніч. Аторвастатин, (50мл) і розчин переносили в воронку для роздіякий таким чином відділяли, відсмоктували, пролення. Потім додавали розчин моногідрату ацетамивали пентаном (2мл), сушили на повітрі до доту кальцію (1.2г, 6.8ммоль) в демінералізованій сягнення постійної ваги і потім розтирали в пороводі (10мл), вміст воронки для розділення струшушок в агатовій колбі. Одержували 0.6г аморфного вали. Після розділення водний шар видаляли, ораторвастатину. ганічний шар струшували з водою (2 10мл) і суПриклад 8 шили сульфатом магнію. Після розділення, У способі, описаному у Прикладі 7, замість органічний шар розводили толуолом (50мл) і, при етилового ететру використовували етилацетат для інтенсивному перемішуванні при лабораторній виділення кислоти і одержували таку ж кількість температурі, додавали по краплях гексан (25мл). аморфного аторвастатину. Потім, температура суміші піднялась до 50°С і Приклад 9 суміш перемішували при цій температурі поки не У способі, описаному у Прикладі 7, замість пещезло раніш утворене помутніння. При інтенсивнтану використовували гексан для осадження, і ному перемішуванні, температурі дали мимоволі одержували таку ж кількість аморфного аторвасопуститись до 30°С (знову з'явилось деяке помуттатину. ніння) і, при цій температурі додавали по краплях Приклад 10 гексан (100мл) без подальшого нагрівання. Потім, У способі, описаному у Прикладі 7, замість песуміш перемішували при лабораторній температунтану використовували гептан для осадження, і рі протягом наступних 10 хвилин, кількість гексану, одержували таку ж кількість аморфного аторвасяка залишилась (100мл), додавали по краплях і татину. суміш перемішували при лабораторній температуПриклад 11 рі 1 годину. Відділений таким чином аторвастатин У способі, описаному у Прикладі 7, замість певідсмоктували, промивали гексаном (25мл), сушинтану використовували циклогексан для осадженли у вакуумі до досягнення постійної ваги і потім ня, і одержували таку ж кількість аморфного аторрозтирали в порошок в агатовій колбі. Одержували вастатину. 5.6г аморфного аторвастатину. Приклад 12 Приклад 7 У способі, описаному у Прикладі 7, замість пеВідміряну кількість естеру формули II (1г) донтану використовували диетиловий етер для осадавали, перемішуючи, до суміші тетрагідрофурану дження, і одержували таку ж кількість аморфного (20мл) і 10% розчину НСІ (3.5мл) і одержаний розаторвастатину. чин перемішували при лабораторній температурі Приклад 13 10 годин. Потім, протягом 15 хвилин, до розчину У способі, описаному у Прикладі 7, замість педодавали по краплях 40% розчин NaOH (2мл) і цю нтану використовували диізопропіловий етер для суміш перемішували при лабораторній температуосадження, і одержували таку ж кількість аморфрі 15 годин, після цього, її заливали у воронку для ного аторвастатину. розділення, в якій знаходився гексан (10мл) і деміПриклад 14 нералізована вода (30мл). Після ретельного переУ способі, описаному у Прикладі 7, замість пемішування і розділення шарів нижній водний шар нтану використовували трет-бутилметиловий етер додатково перемішували з гексаном (2 5мл). Продля осадження, і одержували таку ж кількість амомитий водний шар окислювали 10% розчином НСІ рфного аторвастатину. до рівня рН=4 і екстрагували диетилетером Приклад 15 (2 10мл, 2 5мл), органічний шар промивали насиУ способі, описаному у Прикладі 7, до осаченим розчином солі (2 5мл) і сушили Na2SO4. дження гексаном розчин етилацетату аторвастаПотім десикант відфільтровували, промивали сутину розводили 5мл толуолу. хим диетиловим етером і до одержаного розчину Приклад 16 додавали триетиламін (0.3мл), розчин випарювали У способі, описаному у Прикладі 7, до осадо одержання сухого залишку на роторному ваку 11 76826 12 дження гексаном розчин етилацетату аторвастапромивали гексаном (2мл), сушили у вакуумі до тину розводили 3мл трет-бутилметилового етеру. досягнення постійної ваги і потім розтирали в поПриклад 17 рошок в агатовій колбі. Одержували 0.6г аморфноУ способі, описаному у Прикладі 7, до осаго аторвастатину. дження гексаном, розчин етилацетату аторвастаПриклад 19 тину розводили 10мл тетрагідрофурану. До зваженого естеру II (5г, 7.6ммоль) додаваПриклад 18 ли тетрагідрофуран (75мл) і після розчинення усієї Відміряну кількість естеру формули II (1г) доречовини додавали 10% розчин НСІ (17мл). Суміш давали, перемішуючи, до суміші тетрагідрофурану перемішували при лабораторній температурі 24 (15мл) і 10% розчину НСІ (3.5мл), і одержаний рогодини. Потім, протягом 5хвилин, до розчину дозчин перемішували при лабораторній температурі давали по краплях 40% розчин NaOH (10мл), і 24 години. Потім, протягом 15 хвилин, до розчину неоднорідну суміш інтенсивно перемішували 17 додавали по краплях 40% розчин NaOH (2мл), і цю годин, після цього її заливали у воронку для роздісуміш перемішували при лабораторній температулення, в якій знаходилась демінералізована вода рі 17 годин, після цього її заливали у воронку для (150мл) і гексан (50мл). Після перемішування і розділення, в якій знаходився гексан (10мл) і деміповторного екстрагування гексаном (50мл) водний нералізована вода (30мл). Після ретельного перешар окислювали 5мл концентрованої НСІ і екстрамішування і розділення шарів нижній водний шар гували етилацетатом (4 20мл). Після промивання додатково струшували з гексаном (2 5мл). Промирозчином солі (2 10мл) до екстракту етилацетату тий водний шар окислювали 10% розчином НСІ до додавали триетиламін (1.5мл), який поетапно трирівня рН4 і екстрагували дихлорметаном (1 10мл, чі перемішували з демінералізованою водою (10мл), завжди із вмістом 1г ацетату кальцію у 2 5мл), до органічного шару додавали триетила10мл води. Одержаний розчин промивали демінемін (0.3мл) і шар висушували над MgSO4. Потім, десикант відфільтровували, промивали сухим диралізованою водою (2 5мл) і сушили сульфатом хлорметаном і об'єднані органічні розчини випамагнію (10г) 1 годину. До висушеного екстракту рювали до одержання сухого залишку на роторнододавали гексан (50мл) при перемішуванні, і одему вакуумному випарювані до досягнення ржану нерозчинну фракцію відсмоктували після постійної ваги (приблизно 0.85г). Сіль триетилаперемішування протягом 1 години. Після висушумонію розчиняли в етилацетаті (10мл) і розчин вання одержували 3.35г аморфного аторваспереносили у воронку для розділення. Потім дотатину. давали розчин моногідрату ацетату кальцію (0.13г) Приклад 20 в демінералізованій воді (1мл) і вміст воронки для У способі, описаному у Прикладі 19, замість розділення струшували. Після розділення, водний триетиламіну застосовували еквіваленту кількість шар видаляли; органічний шар перемішували з 40% водного розчину метиламіну. водою. Після розділення, до органічного шару доПриклад 21 давали по краплях гексан при інтенсивному переУ способі, описаному у Прикладі 19, замість мішуванні при лабораторній температурі. Відділетриетиламіну застосовували еквіваленту кількість ний таким чином аторвастатин відсмоктували, 35% водного розчину аміаку. Комп’ютерна верстка Т. Чепелева Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod of manufacturing amorphous form of hemi-calcium salt of (3r,5r) 7-[3-phenyl-4-phenylcarbamoyl-2-(4-fluorophenyl)-5-iso-propyl-pyrrol-1-yl]-3,5-dihydroxyheptanoic acid (atorvastatin)
Автори англійськоюRadl Stanislav, Stach Jan
Назва патенту російськоюСпособ получения аморфной формы гемикальциевой соли (3r,5r) 7-[3-фенил-4-фенилкарбамоил-2-(4-фторфенил)-5-изопропилпиррол-1-ил]-3,5-дигидрогептановой кислоты (аторвастатина)
Автори російськоюРадл Станислав, Стах Ян
МПК / Мітки
МПК: C07D 207/416, C07D 207/337
Мітки: спосіб, солі, одержання, 3r,5r, гемікальцієвої, аторвастатину, форми, аморфної, 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової, кислоти
Код посилання
<a href="https://ua.patents.su/6-76826-sposib-oderzhannya-amorfno-formi-gemikalciehvo-soli-3r5r-7-3-fenil-4-fenilkarbamol-2-4-ftorfenil-5-izopropilpirol-1-il-35-digidrogeptanovo-kisloti-atorvastatinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання аморфної форми гемікальцієвої солі (3r,5r) 7-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-ізопропілпірол-1-іл]-3,5-дигідрогептанової кислоти (аторвастатину)</a>
Спосіб одержання аморфної [r-(rr)]-2-(4-фторфеніл)-b, d-дигідрокси-5-(1-метилетил)-3-феніл-4-[(феніламіно)карбоніл]-1н-пірол-1-гептанової кислоти (аторвастатин) у вигляді солей кальцію (2:1)

Номер патенту: 50743
Опубліковано: 15.11.2002
Автори: Швайс Дітер, Лін Мін
МПК: A61K 31/00, A61P 3/00, A61P 3/06, A61K 31/40, C07D 207/34
Мітки: одержання, 2:1, d-дигідрокси-5-(1-метилетил)-3-феніл-4-[(феніламіно)карбоніл]-1н-пірол-1-гептанової, солей, кислоти, кальцію, аморфної, аторвастатин, r-(rr)]-2-(4-фторфеніл)-b, спосіб, вигляді
Формула / Реферат:
1. Спосіб одержання аморфного аторвастатину і його гідратів, що полягає в розчиненні аторвастатину кристалічної форми І у середовищі негідроксильного розчинника і подальшому видаленні розчинника.2. Спосіб за п.1, де негідроксильний розчинник обраний із групи, що включає тетрагідрофуран і суміші тетрагідрофурану і толуолу.3. Спосіб за п.2, де розчинник являє собою суміш тетрагідрофурану і толуолу.4. Спосіб за п.1, де...
Кристалічні форми i, ii, iv кислої кальцієвої солі [r-(rr)]-2-(4-фторфеніл)-b, d-дигідрокси-5-(1-метилетил)-3-феніл-4-[(феніламіно)карбоніл]-1н-пірол-1-гептанової кислоти (аторвастатин) та фармацевтична композиція на їх основі

Номер патенту: 51661
Опубліковано: 16.12.2002
Автори: Бріггс Крістофер А., Ічікава Шігеру, Вейд Роберт А., ХАРАСАВА Кікуко, Мінохара Казуо, Дженнінгс Рекс Ален, Накагава Шінсуке
МПК: C07C 51/06, A61K 31/40, C07D 207/34, A61P 3/06
Мітки: основі, кальцієвої, d-дигідрокси-5-(1-метилетил)-3-феніл-4-[(феніламіно)карбоніл]-1н-пірол-1-гептанової, фармацевтична, аторвастатин, композиція, кислоти, кристалічні, кислої, r-(rr)]-2-(4-фторфеніл)-b, форми, солі
Формула / Реферат:
1. Кристалічна форма І аторвастатину або його гідрату, для якої значення для основного піку дифракції рентгенівського випромінювання на порошку, виміряного з використанням випромінювання , становить 21,6.2. Кристалічна форма І аторвастатину або його гідрату згідно з пунктом 1, що характеризується...
Кристалічна форма кальцієвої солі [r-(r,r)]-2-(4-фторфеніл)-b, d-дигідрокси-5-(1-метилетил)-3-феніл-4-[(феніламіно)карбоніл]-1н-пірол-1-гептанової кислоти (2:1)(аторвастатин) (варіанти)

Номер патенту: 74075
Опубліковано: 17.10.2005
Автори: Берн Стівен Роберт, Влахова Петінка Іванова, Лі Женг Джан, Кшизаняк Джозеф Френсіс, Гушерст Карен Сью, Парк Аері, Котс Девід Ендрю, Моррісон Генрі Грант, II
МПК: A61P 19/10, A61K 31/40, A61P 3/06, A61P 25/28, C07D 207/34
Мітки: варіанти, r-(rr)]-2-(4-фторфеніл)-b, кислоти, кристалічна, 2:1)(аторвастатин, кальцієвої, форма, d-дигідрокси-5-(1-метилетил)-3-феніл-4-[(феніламіно)карбоніл]-1н-пірол-1-гептанової, солі
Формула / Реферат:
1. Кристалічна Форма V аторвастатину або її гідрат, що має порошкову дифрактограму, яка містить наступні значення , виміряні із використанням випромінювання : 4,9 (широкий), 6,0, 7,0, 8,0 (широкий), 8,6, 9,9, 16,6, 19,0 і 21,1.2. Кристалічна Форма VI аторвастатину або її гідрат, що має порошкову...
Кристалічна форма ііі кислої кальцієвої солі [r-(r r)]-2-(4-фторфеніл)-b, d-дигідрокси-5-(1-метилетил)-3-феніл-4-[(феніламіно)карбоніл]-1н-пірол-1-енантової кислоти (аторвастатин) та фармацевтична композиція на її основі

Номер патенту: 44796
Опубліковано: 15.03.2002
Автор: Маккензі Анн Т.
МПК: C07D 207/34, A61P 3/14, A61K 31/40, A61P 3/06, A61P 43/00
Мітки: основі, композиція, солі, r)]-2-(4-фторфеніл)-b, кислоти, фармацевтична, форма, кальцієвої, d-дигідрокси-5-(1-метилетил)-3-феніл-4-[(феніламіно)карбоніл]-1н-пірол-1-енантової, кристалічна, ііі, аторвастатин, r-(r, кислої
Формула / Реферат:
1. Кристалічна форма III аторвастатину або його гідрату, що має основний пік дифракції рентгенівських променів на порошку, виміряний з використанням випромінювання CuKa, при значенні 2q = 8,5.2. Кристалічна форма III аторвастатину або його гідрату згідно з п. 1, що також має такі значення 2q, виміряні з використанням випромінювання CuKa: 16,6; 20,0; 20,3 та 24,4.3. Кристалічна форма III аторвастатину або його гідрату згідно з п....
Спосіб одержання лікарської форми дигідрату динатрієвої солі метиленбісфосфонової кислоти

Номер патенту: 60372
Опубліковано: 15.10.2003
Автори: Сяркевич Олег Романович, Жебровська Філя Іванівна, Борщевська Марина Іллінічна, Лозинський Мирон Онуфрієвич, Жилеєв Володимир Тимофійович, Комісаренко Сергій Васильович, Чуйко Олексій Леонідович, Шарикіна Надія Іванівна, Борисевич Анатолій Миколайович
МПК: A61P 35/00, A61K 31/663
Мітки: солі, форми, лікарської, кислоти, одержання, спосіб, динатрієвої, дигідрату, метиленбісфосфонової
Формула / Реферат:
1. Спосіб одержання лікарської форми дигідрату динатрієвої солі метиленбісфосфонової кислоти шляхом взаємодії триізопропілфосфіту з дибромметаном при температурі 145-179°С протягом 10 годин для одержання тетраізопропілового ефіру метиленбісфосфонової кислоти з наступною відгонкою легко киплячих побічних речовин, гідролізом залишку 35%-ною соляною кислотою для утворення метиленбісфосфонової кислоти, виділенням метиленбісфосфонової кислоти...