Спосіб одержання високоочищеного вірусінактивованого препарату тромбіну
Номер патенту: 83940
Опубліковано: 26.08.2008
Автори: Кондрацький Богдан Олексійович, Новак Василь Леонідович, Магеровський Юрій Васильович, Брагінець Олена Григорівна




Формула / Реферат
Спосіб одержання високоочищеного вірусінактивованого препарату тромбіну передбачає виділення та очищення препарату тромбіну з утильної фракції III плазми крові за Коном, фракціонування поліетиленгліколем ПЕГ-115, осадження протромбінового комплексу на цитраті барію, активацію тромбіну тромбопластин-кальцієвою сумішшю, який відрізняється тим, що додатково включає сольвент-детергентну противірусну обробку та очистку від токсичних домішок агентів антивірусної обробки препарату тромбіну шляхом афінної хроматографії на Силохромі-Активний яскраво-голубий.
Текст
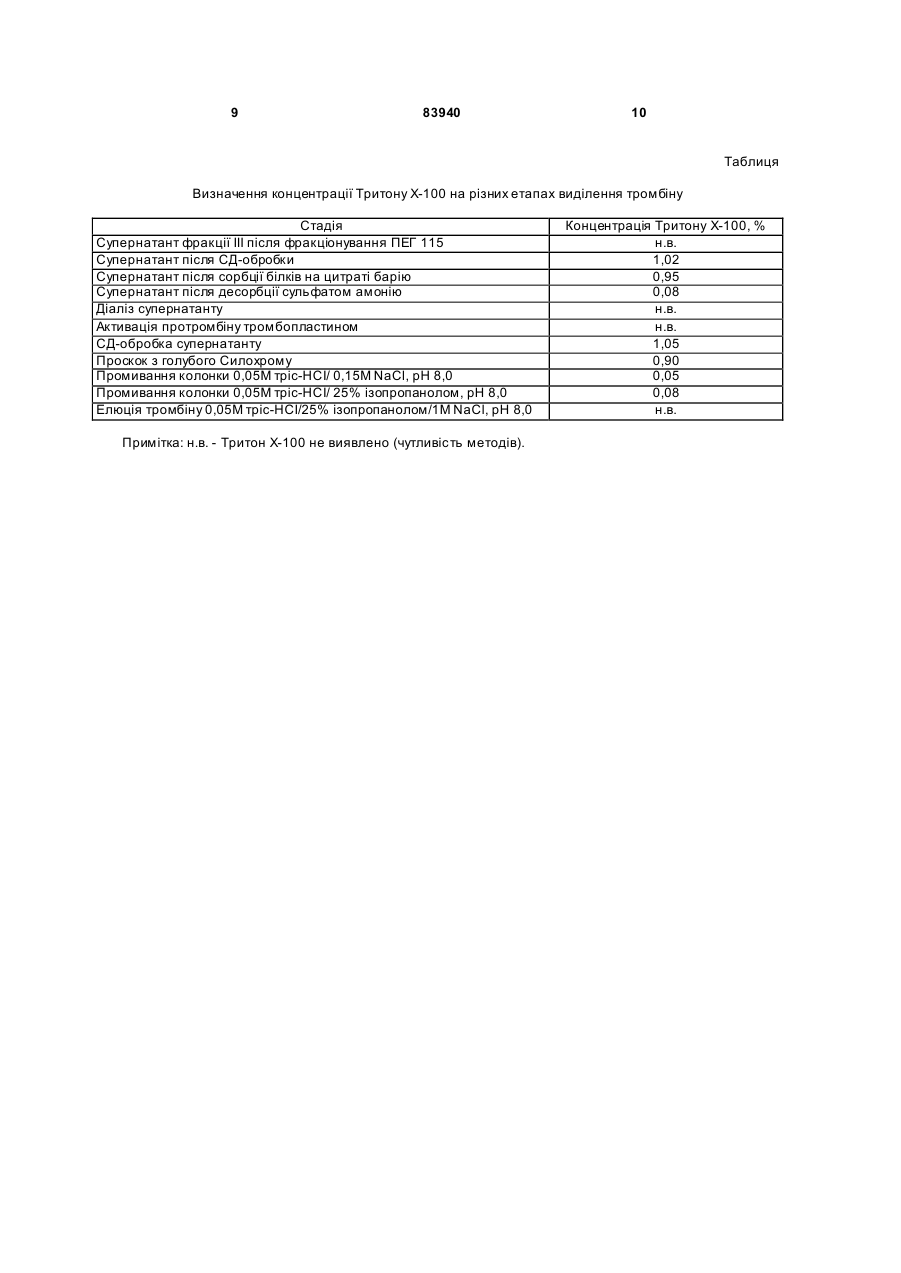
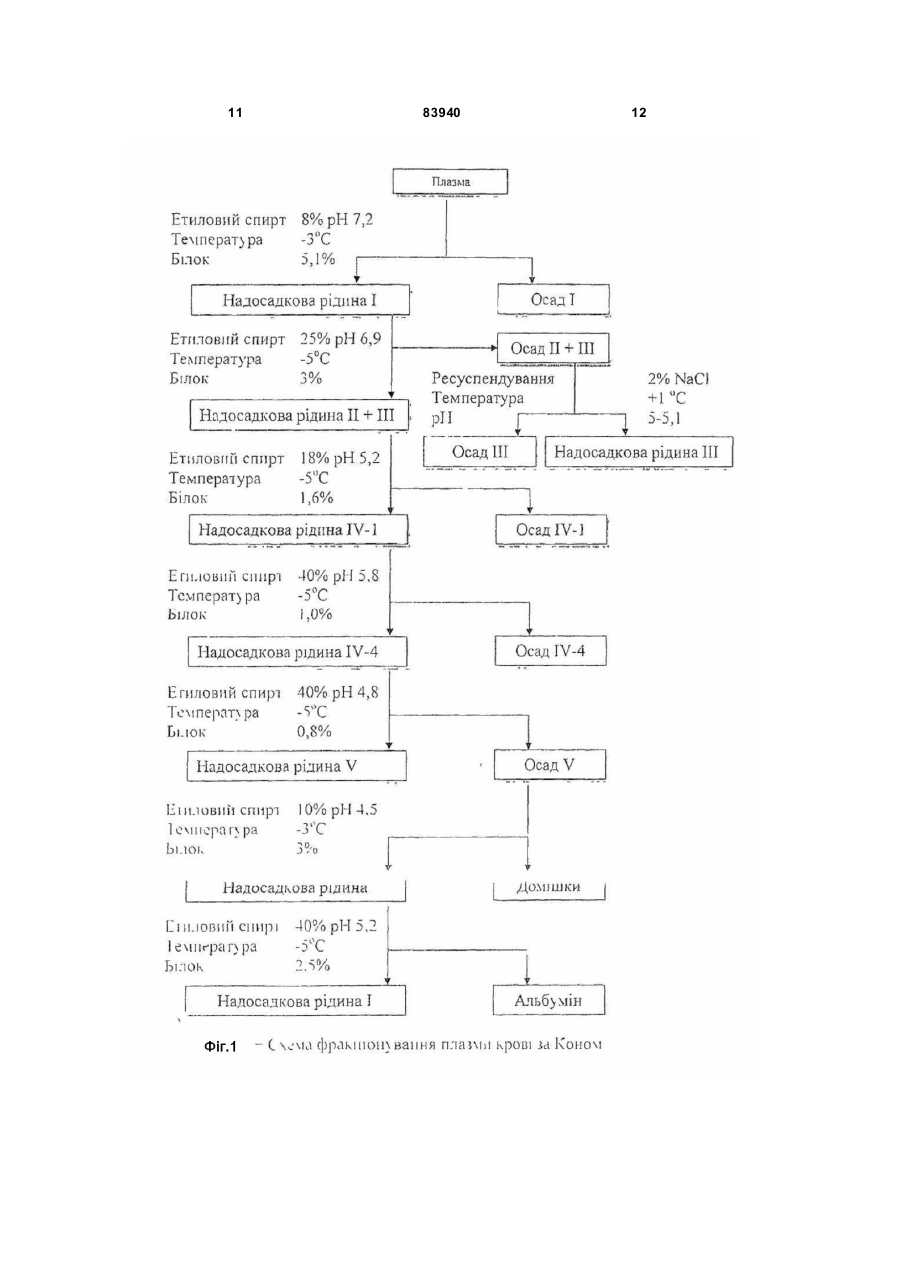
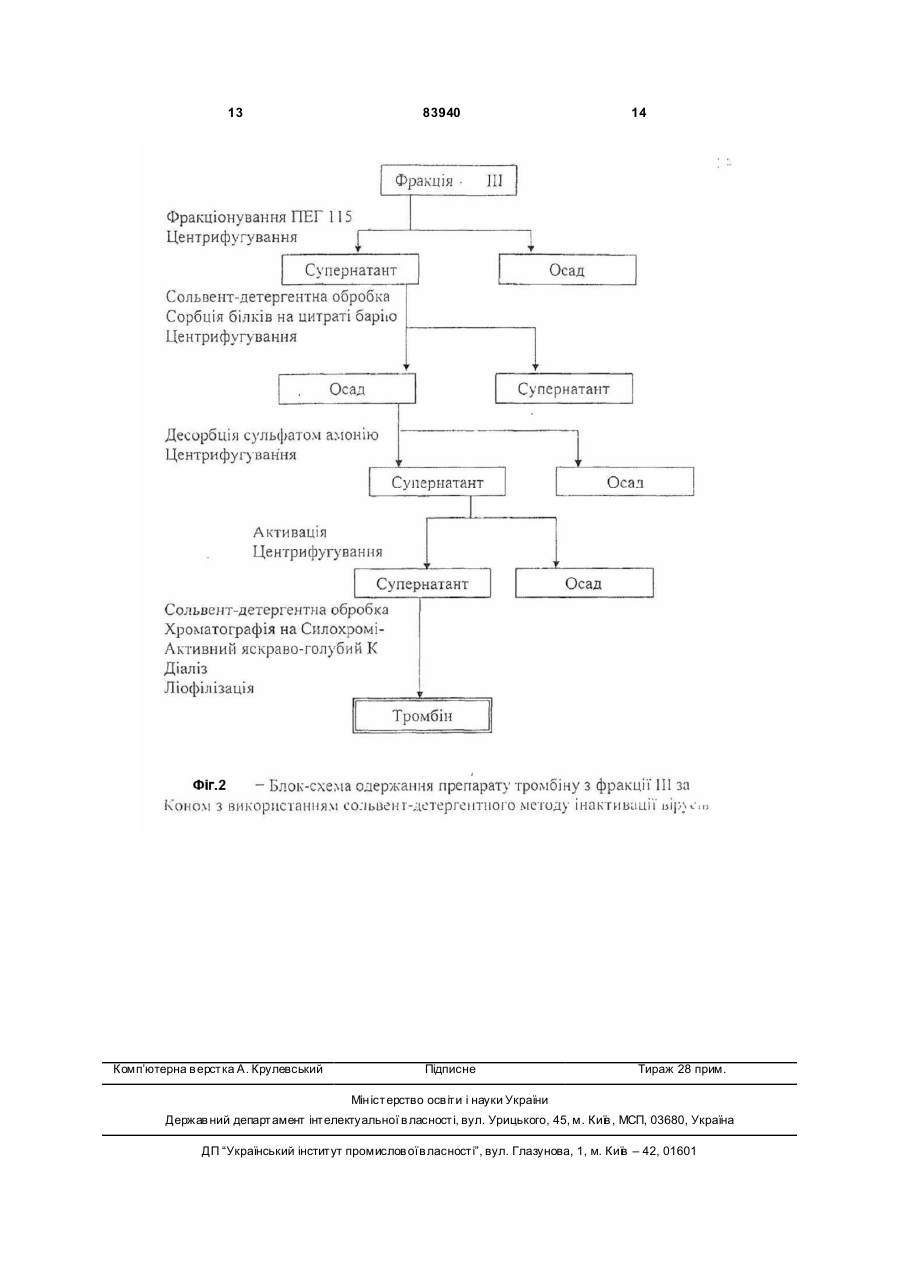
Спосіб одержання високоочищеного вірусінактивованого препарату тромбіну передбачає виділення та очищення препарату тромбіну з утильної фракції III плазми крові за Коном, фракціонування поліетиленгліколем ПЕГ-115, осадження протромбінового комплексу на цитраті барію, активацію тромбіну тромбопластин-кальцієвою сумішшю, який відрізняється тим, що додатково включає сольвент-детергентну противірусну обробку та очистку від токсичних домішок агентів антивірусної обробки препарату тромбіну шляхом афінної хроматографії на Силохромі-Активний яскравоголубий. Винахід належить до галузі клінічної і експериментальної коагулології, до способів отримання коагуляційних реагентів, зокрема до способів отримання високоочищеного тромбіну. Тромбін, протеолітичний фермент, який зумовлює зсідання крові (каталізує перетворення фібриногену у фібрин), і утворюється з неактивного протромбіну, який міститься у плазмі крові. Тромбін, як серинова протеїназа плазми крові (КФ 3.4.21.5), утворюється шляхом обмеженого протеолізу протромбіну за допомогою фактора Xa в присутності фактора Va, іонів кальцію і фосфоліпідів. Препарати тромбіну використовують для діагностичних коагуляційних досліджень (визначення тромбінового часу, антитромбіну III, фібриногену), а також для виробництва фібринового клею, який використовується у хірургії, отоларингології, стоматології, урології, онкології тощо. Технології виробництва тромбіну розроблені ще в 30-40-х роках XX століття і грунтувалися в осадженні неактивного зимогена фермента протромбіна буферно-сольовими розчинами [Препараты крови. Инструктивно-методические материа лы по контролю и производству / Под ред. СП. Буренкова. - М.: МЗ СССР, 1976. С.219-238] або у сорбції на сульфаті барію з наступною десорбцією і активацією тромбопластином. Отримані таким чином препарати тромбіну містили велику кількість баластних білків, характеризувалися поганою розчинністю і стабільністю у буферних розчинах. До кінця 80-х років основні концепції одержання білкових препаратів крові практично сформувалися. Технології їх очистки вдосконалились і поповнились такими принципово новими методами, як осадження поліетиленгліколем, гель-проникна-, іонообмінна, метало-хелатна, та афінна хроматографія тощо. Зараз найбільш ефективними методами одержання тромбіну є хроматографічні методи. Найчастіше в якості матриць для хроматографічних носіїв використовують декстран, агарозу, целюлозу, поліакриламід, полістирол, поліамід або комбіновані гелі. В останні 15-20 років основна увага приділялась в основному, методам одержання та збереження активності препаратів тромбіну. Але у зв'язку з широким застосуванням фібринового клею у медицині перше місце посідає вирішення (19) UA (11) 83940 (13) C2 (21) a200700172 (22) 05.01.2007 (24) 26.08.2008 (46) 26.08.2008, Бюл.№ 16, 2008 р. (72) МАГЕРОВСЬКИЙ ЮРІЙ ВАСИЛЬОВИЧ, UA, БРАГІНЕЦЬ ОЛЕНА ГРИГОРІВНА, UA, НОВАК ВАСИЛЬ ЛЕОНІДОВИЧ, UA, КОНДРАЦЬКИЙ БОГДАН ОЛЕКСІЙОВИЧ, UA (73) НАУКОВО-ВИРОБНИЧА ФІРМА "СІМКО", U A, ДЕРЖАВН А УСТАНОВА ІНСТИТУТ ПАТОЛОГІЇ КРОВІ ТА ТРАНСФУЗІЙНОЇ МЕДИЦИНИ, UA (56) US 5393666, 28.12.95. US 4820805, 11.04.89. EP 0366946 B1, 02.03.94. US 2006/0270014 A1, 30.11.06. RU 2144081 C1, 10.01.2000. Prince A.M., Horowitz В., Brotman В. Sterilization of 3 83940 проблеми противірусної безпеки тромбінових препаратів (СНІД, гепатити, цитомегаловірус і т.д.). Протягом останніх 10 років багатьма авторами та фірмами запатентовані різні методи одержання тромбіну та методи його вірусної інактивації. На сьогоднішній день традиційні методи фракцінування плазми за Коном вдало поєднують з сучасними хроматографічними методами білкових препаратів. Завдяки цьому в промислових масштабах одержують очищені препарати крові. Відомий спосіб одержання активного тромбіну [патент №5393666 US], суть якого полягає у активації протромбіну фосфоліпідним комплексом. Відомі також способи виділення та очищення тромбіну: 1. Виділення та очищення тромбіну з біологічних рідин із застосуванням гідрофобної хроматографії [патент №5094960 US], дозволяє одержати очищений препарат тромбіну; 2. Сорбент для виділення і очистки протеіназ і спосіб його отримання [A.C. №1300701 SU]; 2. Спосіб виділення і очистки протеолітичних ферментів [А.С. №1369291 SU]. Ці способи дозволяють виділити та очистити тромбін. Відомий спосіб сольвент-детергентної інактивації вірусів [патент №4820805 US]. Суть способу полягає у обробці біологічної рідини в розчині, що містить 1% три(н-бутил) фосфату і 1% ТритонуХ100, що дозволяє інактивувати вір уси з ліпідною оболонкою. Найбільш близьким по суті до способу, що заявляється є Спосіб сольвент-детергентної інактивації вірусів розроблений Нью-Йоркським Центром Крові та схвалений FDA у 1985 році [Prince A.M., Horowitz В., Brotman B. Sterilization of hepatitis and HTLV III viruses by exposure to Tri(n-butyl) phosphate and sodium cholate.// Lancet. - 1986. – V1. - P.706-710]. Цей спосіб поєднує у собі процес інактивації вірусів з процесами очищення і забезпечує високий рівень інактивації оболонкових вірусів (HIV, HBV, HCV, HGV, CMV), і високим виходом препаратів крові. Цей спосіб використовується багатьма потужними фірмами-виробниками і дозволяє інактивувати оболонкові віруси, забезпечуючи „м'які" умови обробки, які не впливають на лабільні фактори і є простим у застосуванні. Недоліками Способу сольвент-детергентної інактивації вірусів є те, що цей спосіб знижує рівень якості тромбіну (та інших препаратів крові), не забезпечує повної антивірусної безпеки та не виключає присутності у тромбіні сольвенту, детергенту та інши х токсичних агентів. В основу винаходу покладена технічна задача розробити спосіб одержання високоочищеного препарату тромбіну з утильної фракції III за Коном з використанням сольвентно-детергентного методу противірусної обробки та афінної хроматографії тромбіну на кремнеземних сорбентах. Технічна задача вирішується шляхом виділення та очищення препарату тромбіну з фракції III плазми крові за Коном, яка включає попереднє фракціонування поліетиленгліколем ПЕГ-115, осадження протромбінового комплексу на цитраті барію, активацію тромбіну тромбопластинкальцієвою сумішшю, сольвент-детергентну обробку та афінну хроматографію на Силохромі 4 Активний яскраво-голубий. При проведенні патентно-інформаційного пошуку заявником знайдено багато відомих фірм, які розробляють способи виділення та очистки тромбіну, і методи вірусної інактивації (США - 35 патентів, Японія - 5 патентів, Великобританія - 3 патентів). В Україні провідними установами по розробці способів виділення та очищення тромбіну є Інститут молекулярної біології та генетики AH України, Інститут органічної хімії AH України, Інститут біо хімії AH України. Для виділення тромбіну вони застосовують хроматографічні сорбенти на основі дорогих імпортних носіїв. При проведенні патентно-інформаційного пошуку заявником знайдено технічний результат Спосіб сольвент-детергентної інактивації вірусів розроблений Нью-Йоркським Центром Крові та схвалений FDA у 1985 році [Prince A.M., Horowitz В., Brotman B. Sterilization of hepatitis and HTLV III viruses by exposure to Tri(n-butyl) phosphate and sodium cholate.// Lancet. - 1986. - V1. - P.706-710], в якому є найбільша кількість суттєви х ознак, спільних із заявленим результатом. Цей спосіб включає хроматографічне виділення тромбіну та вірусінактактивацію. Однак, даних суттєви х ознак недостатньо для одержання очікуваного технічного результату. Те хнічних результатів, які містять сукупність всіх ознак, що повністю б співпадали із ознаками заявленого результату не виявлено. У джерелах патентної і науково-технічної інформації не знайдено відомостей про способи відділення шкідливих та токсичних сольвенту та детергенту (три(н-бутил) фосфа ту і тритону Х-100) від білків у поєднанні з одержанням білків у високоочищеному вигляді з використанням біоспецифічних сорбентів на основі силохрому. Таким чином заявником заявляється спосіб одержання високоочищеного вірусінактивованого препарату тромбіну, який включає використання утильної фракції III за Коном з використанням сольвент-детергентного методу противірусної обробки та афінної хроматографії на модифікованих макропористих кремнеземних сорбентах. Технікоекономічні показники способу, що заявляється: застосування хроматографічного носія на основі силохрому, чистота одержаного препарату (питома активність понад 1500 од.NIН/мг білка), спосіб активації протромбіну в тромбін (використання фосфоліпідного комплексу), включення в спосіб етапу інактивації оболонкових вірусів, видалення сольвенту і детергенту після вірусної інактивації. У процесі проведеного аналізу патентної і науково-технічної інформації встановлено, що заявлений винахід Спосіб одержання високоочищеного вірусінактивованого препарату тромбіну відповідає умовам патентоспроможності, він є новим, має винахідницький рівень і є промислово придатним. Фіг.1. На фігурі 1 відображені основні етапи фракціонування плазми крові за Коном на станціях переливання крові. Для отримання високо очищеного вірусінактивованого препарату тромбіну використовується Осад III (фракція C). Фіг.2. На фігурі 2 показано блок-систему одержання препарату тромбіну з фракції III за Коном з використанням сольвент-детергентного методу 5 83940 інактивації вірусів. Цей спосіб включає основні етапи: 1.Фракціонування полі етиленгліколем (ПЕГ 115), центрифугування; 2. Сольвентдетергентна обробка, сорбція білків на цитраті барію, центрифугування; 3. Активація тромбіну тромбопластиинкальцієвою сумішшю; 4. Сольвент-детергентна обробка; 5. Афінна хроматографія на Силохромі-Активний яскраво-голубий. Технологічна схема одержання високоочищеного вірусінактивованого препарату тромбіну з фракції III плазми крові за Коном включає попереднє фракціонування полі етиленгліколем, осадження протромбінового комплексу на цитраті барію, активацію тромбіну тромбопластикальцієвою сумішшю, сольвент-детергентну обробку та афінну хроматографію на Силохромі-Активний яскраво-голубий. Вихідною сировиною для одержання тромбіну є фракція III за Коном, яка не використовується і є утильною сировиною. Основні етапи фракціонування плазми крові за Коном на станціях переливання крові представлені на Фіг.1. При одержанні тромбіну на першому етапі виділяють фактори протромбінового комплексу з екстрактів фракції III. Для одержання протромбінового комплексу використовують сорбцію на цитраті барію з наступною десорбцією та активацією. При екстрагуванні білків на першому етапі необхідно видалити ліпопротеїни, присутність яких перешкоджає подальшому фракціонуванню. Для одержання протромбіну використовують модифікацію схеми [Human thrombin. Production, evaluation and properties of a-thrombin./ Fenton J.W., Campbell W.P., Stackrow A.B. et al.// J.Biol. Chem. 1977. - 252, N 11. - P.3587-3597] з використанням вітчизняного ПЕГ-115 замість зарубіжного ПЕГ600. Схема одержання протромбіну: 1. 2кг фракції III за Коном, що зберігалось за температури -40°C, витримується 2-3 години у 8л 0,15M NaCL з 0,03M тринатрій-цитратом, 50г/л ПЕГ-115, 0,02M тріс-буфером, pH 8,0. Суспендується за допомогою електричної мішалки 1,5-2 години. Далі центрифугується протягом 10хв на центрифузі РС-6 при 2500g. Осад відкидається; 2. додається краплями протягом 15-20хв охолоджений до +4°C хлорид барію, помішуючи 20хв. При цьому утворюється нерозчинний цитрат барію, на якому сорбуються фактори протромбінового комплексу. Центрифугували протягом 10хв при 2500g. Супернатант можна використовувати для виділення плазміногену. Осад промивається двома (2) л дистильованої води; 3. цитрат барію суспендується у об’ємі 600мл дистильованої води. Додається краплями протягом 15-20хв 3,3М сульфа т амонію з pH 8,0 при постійному перемішуванні. Центрифугується 20хв при 6000g; 4. діаліз проти 8л 0,15M хлориду натрію при +4-8°C протягом 12 годин. Діалізуючий розчин за цей час міняється двічі. Активація протромбіну в тромбін проводиться за схемою: 1. 100мл свіжозамороженої плазми розморожують, нагріваючи до +37°C, додають 0,25M хло 6 рид кальцію з 0,25M тріс-буферу, pH 7,4, інкубують 5-10хв до утворення згустка. Згусток видаляється центрифугуванням протягом 15хв при 3000g; 2. до супернатанту додається 10г тромбопластину з кадаверного мозку людини, перемішується 10-15хв на магнітній мішалці. Центрифугується 30хв при 6 000g; 3. осад суспендується у 100-120мл 0,25M хлорид кальцію з 0,25M тріс-HCL-буферу, pH 7,4, і додавали до діалізату факторів протромбінового комплексу з розрахунку 1 об'єм активаційної суміші на 6,5-7 об'ємів діалізату; 4. активацію проводять за температури +37°C протягом 2-3 годин, постійно перемішуючи розчин за допомогою мішалки. Активацію припиняють коли активність тромбіну виходить на плато. Розчин центрифугують протягом 45хв при 6000g, осад відкидається, а супернатант, що містить тромбін, використовується для подальшої роботи. Таким способом з 2кг фракції III за Коном одержують від 1 000 000 до 1 500 000 NIH од. тромбіну. В розчині активність тромбіну становить 1 0001 500 NIH од./мл. На цій стадії до неочищеного гомогенату тромбіну додавали Тритон Х-100 і три(н бутил)-фосфат до концентрації 1%. Перемішували протягом 30-60хв і використовували гомогенат для визначення сорбційних властивостей модифікованих кремнеземних сорбентів. На колонки з сорбентом розміром 1,2х6см наносили максимальну кількість тромбіну (до появи його у фракціях проскоку). Сольвент-детергентна інактивація вірусів: На активність тромбіну добавки тритону Х-100 і трибутилфосфату практично не впливають і в той же час основна їх кількість не зв'язується з сорбентами, а виходить у фракціях проскоку. Тромбін в цих умовах сорбується на модифікованих силохромах і не вимивається ні одним з де сорбуючих розчинів, за винятком 1M NaCl в присутності 25%ного пропанову-2, pH 8,0. Залишкові кількості Тритону Х-100 і трибутилфосфату повністю вимиваються 25%-ним розчином пропанову-2. Активність тромбіну в елюючи х розчинах становила 10002000 од. NIH/мг білка, а ємність по тромбіну 1500, 1900, 2000 і 10500 од. NIH/г сорбенту L-лізинглутарил-силохрому, L-лізин-діол-силохрому, nхлорбензил- і активного-яскраво-голубого Лсилохрому, відповідно. Синтезовані силохромні сорбенти мають ряд переваг перед іншими методами очищення з використанням сольвентдетергентної обробки, в яких для видалення детергентів використовують спеціальні сорбенти для їх сорбції. Оптимальний сорбент для сольвентдетергентної обробки (очистки тромбіну) є Силохром-голубий. На Фіг.2 представлена блок-схема одержання препарату тромбіну з фракції III за Коном з використанням сольвент-детергентного методу інактивації вірусів. Афінна хроматографія на Силохромі-Активний яскраво-голубий: Для визначення концентрації сольвента і детергента в процесі одержання препарату тромбіну необхідні чутливі методи їх кількісного визначення. Описані методи газової хроматографії [Enveloped 7 83940 virus inactivation by caprylate: a robust alternative to solvent-detergent treatment in plasms derived intermediates./ Korneeva M., Notta J., Lebing W., et al.// Biologicals. -2002. - V.30. - P. 153-162.], високоефективної зворотно-фазової хроматографії [Determination of X-100 in plasma-derived coagulation factor VIII and factor IX products by reversed-phase high-performance liquid chromatography./ Karlsson Q., Hinz A.C., Henriksson E., Winge S.// J Chromatography A. - 2002. - V.946, No.1-2. - P.163-168.]. Для визначення Тритону використовують його здатність поглинати світло при 275 та 223нм. Заявником було знято спектр поглинання 0,01% водного розчину Тритону X-100 в інтервалі довжини хвиль від 200 до 310нм. Дійсно в ультра фіолетовій області розчин Тритону Х-100 має два максимум поглинання - 223 та 275нм, причому 223нм пік значно вищий (абсолютний показник поглинання майже в 4 рази вищий). Оскільки в нашому випадку визначення концентрації Тритону Х-100 необхідно проводити в розчинах, що містять білки, для яких характерне поглинання в цьому діапазоні хвиль, постає необхідність попереднього їх видалення з розчину, що і досягається осадженням 10% три хлороцтовою кислотою (співвідношення проба: кислота як 1:1 по об'єму; центрифугування 30хв при 3000g). Для недопущення помилки внаслідок можливої інтерференції інших речовин вимірювання проводяться при довжинах хвиль (223 та 275нм) проти контролю, що містив 5% три хлороцтової кислоти. Визначення концентрації Тритону Х-100 в розчині проводили використовуючи калібровочні графіки для визначення концентрації Тритону Х-100 лінійні, похибка визначення не перевищує 5%. Чутливість визначення при 275нм - 0,0019%, а при 223нм - 0,00035% Тритону Х-100. Для визначення концентрації Тритону Х-100 використовували також метод [Knight J.Б., Anderson S., Rawle J.M. Chemical basis of the sulfophospho-vanillin reaction for estimation total serum lipids.// Clin.Chem. - 1972. - V.18, No.3. P.199-202.], принцип якого полягає в тому, що органічні сполуки, які містять ненасичені подвійні зв'язки, після гідролізу за температури 100°C у присутності сірчаної кислоти, реагують з ваніліном і фосфорною кислотою з утворенням ефірних сполук малинового кольору. В певних межах кількість Тритону Х-100 пропорційна поглинанню розчину 8 при довжині хвилі 500-560нм (максимум при 530нм). Хід визначення. До 0,5мл проби у скляній термостійкій пробірці обережно додають 2мл концентрованої сірчаної кислоти, що витримує пробу Сабаля. Вміст пробірки добре перемішують і поміщають на 10хв у киплячу баню. Після цього пробірку з пробою швидко охолоджують водою до кімнатної температури і додають 2мл 10мM розчину ваніліну в 5M ортофосфорній кислоті, перемішують, повторно охолоджують в холодній воді, і міряють поглинання в 1см кюветі при довжині хвилі 500-560нм проти контролю. Контроль обробляють аналогічно, але замість проби беруть відповідний буферний розчин. Для побудови калібрувальної кривої з 25%ного розчину Тритону Х-100 готують ряд розчинів Тритону Х-100 з кінцевими концентраціями 0,25%, 0,1%, 0,05%, 0,025%, 0,0125%. Відбирали по 0,5мл кожного розчину і обробляли, як вище зазначено. Одержані результати свідчать, що метод з використанням фосфорно-ванілінового реактиву дозволяє визначити не менше 125мкг/пробу Тритону Х-100, який використовували для СДінактивації препаратів крові. Обидва апробовані методи мають пряму кореляцію для визначення концентрації Тритону Х-100 (r=0,985). Для визначення залишкової кількості Тритону Х-100 в ліофілізованих препаратах їх попередньо розчиняли в мінімальній кількості дистильованої води. Наведені результати свідчать про те, що в концентрованих розчинах одержаних препаратів тромбіну Тритон Х-100 не визначається, тобто його можлива концентрація не перевищує 0,0019% (чутливість УФ методу). Одержані результати переконують в тому, що метод хроматографії з використанням макропористого Силохрому-Активного яскраво-голубого дозволяє одержувати препарати тромбіну вільні від токсичних домішок детергенту (тритону X-100). У таблиці наведені дані по визначенню Тритону Х-100 на різних стадіях отримання препаратів тромбіну. Застосування запропонованої схеми фракціонування плазми крові у поєднанні з методами антивірусної обробки та хроматографічних методів виділення та очищення дозволяє отримувати препарати тромбіну високого ступеня чистоти з достатнім виходом, безпечних у застосуванні стосовно можливої вірусної контамінації. 9 83940 10 Таблиця Визначення концентрації Тритону X-100 на різних етапах виділення тромбіну Стадія Супернатант фракції III після фракціонування ПЕГ 115 Супернатант після СД-обробки Супернатант після сорбції білків на цитраті барію Супернатант після десорбції сульфатом амонію Діаліз супернатанту Активація протромбіну тромбопластином СД-обробка супернатанту Проскок з голубого Силохрому Промивання колонки 0,05М тріс-НСІ/ 0,15М NaCl, pH 8,0 Промивання колонки 0,05М тріс-НСІ/ 25% ізопропанолом, pH 8,0 Елюція тромбіну 0,05М тріс-НСІ/25% ізопропанолом/1M NaCl, pH 8,0 Примітка: н.в. - Тритон Х-100 не виявлено (чутливість методів). Концентрація Тритону Х-100, % н.в. 1,02 0,95 0,08 н.в. н.в. 1,05 0,90 0,05 0,08 н.в. 11 83940 12 13 Комп’ютерна в ерстка А. Крулевський 83940 Підписне 14 Тираж 28 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for obtaining of highly purified thrombin virus inactivated preparation
Автори англійськоюMaherovskyi Yurii Vasyliovych, Brahinets Olena Hryhorivna, Novak Vasyl Leonidovych, Kondratskyi Bohdan Oleksiovych
Назва патенту російськоюСпособ получения высокоочищенного вирусинактивированного препарата тромбина
Автори російськоюМагеровский Юрий Васильевич, Брагинец Елена Григорьевна, Новак Василий Леонидович, Кондрацкий Богдан Алекссевич
МПК / Мітки
МПК: C07K 14/435, A61K 35/16, C12N 9/74, A61K 38/36
Мітки: тромбіну, високоочищеного, препарату, вірусінактивованого, одержання, спосіб
Код посилання
<a href="https://ua.patents.su/7-83940-sposib-oderzhannya-visokoochishhenogo-virusinaktivovanogo-preparatu-trombinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання високоочищеного вірусінактивованого препарату тромбіну</a>
Спосіб виділення високоочищеного стандарту тромбіну людини

Номер патенту: 55005
Опубліковано: 15.06.2005
Автори: Колодзейська Марія В'ячеславівна, Волков Георгій Леонідович, Макогоненко Євген Митрофанович
Мітки: високоочищеного, виділення, тромбіну, стандарту, спосіб, людини
Формула / Реферат:
Спосіб застосування поліоксидонію для лікування герпетичного стоматиту у дітей, що полягає у застосуванні традиційної базової терапії у поєднанні з новим імуномодулятором поліоксидонієм, який відрізняється тим, що поліоксидоній вводять сублінгвально із розрахунку 0,15 мг на 1 кг маси тіла дитини 1 раз на добу упродовж 5 днів.
Спосіб виділення та очищення тромбіну

Номер патенту: 24182
Опубліковано: 25.06.2007
Автори: Даниш Тарас Васильович, Вороняк Мирослав Іванович, Шурко Наталія Олегівна, Вус Марія Миронівна, Брагінець Олена Григорівна, Орлова Любов Володимирівна, Магеровський Юрій Васильович, Даниш Ольга Йосифівна
МПК: A61K 38/02, C12N 9/96
Мітки: очищення, спосіб, тромбіну, виділення
Формула / Реферат:
Спосіб виділення та очищення тромбіну з активованого концентрату протромбінового комплексу, який включає використання методу афінної хроматографії на макропористому кремнеземному носії, який відрізняється тим, що для підвищення ступеня очищення препарату, ефективності способу, а також для покращення аналітичних характеристик кінцевого продукту як ліганд використовують тріазиновий барвник Активний яскраво-голубий К.
Спосіб одержання високоочищеного a2-антиплазміну з плазми крові людини

Номер патенту: 53146
Опубліковано: 17.01.2005
Автори: Юсова Олена Іванівна, Волков Георгій Леонідович, Гриненко Тетяна Вікторівна, Задорожна Марина Борисівна, Макогоненко Євген Митрофанович
МПК: A61K 35/16, C07K 14/81
Мітки: плазми, крові, високоочищеного, одержання, людини, a2-антиплазміну, спосіб
Формула / Реферат:
Спосіб одержання високоочищеного α2-антиплазміну з плазми крові людини шляхом афінної хроматографії, який відрізняється тим, що афінну хроматографію проводять послідовно на лізин-сефарозі, К1-3-сефарозі (Tyr79-Val353, І форма) та К1-3-сефарозі (Tyr79-Val337, ІІ форма).
Композиція, яка містить інгібітор тромбіну, спосіб її одержання та застосування для профілактики або лікування тромбоемболії

Номер патенту: 75567
Опубліковано: 15.05.2006
Автори: Карлссон Крістер, Форсман Сіґбріт, Карлссон Магнус
МПК: A61K 9/22, A61K 38/55, A61K 47/36, A61P 7/02
Мітки: тромбоемболії, профілактики, спосіб, тромбіну, лікування, інгібітор, композиція, містить, яка, одержання, застосування
Формула / Реферат:
1. Пероральна композиція у твердій формі з негайним вивільненням, що містить (а) інгібітор тромбіну низької молекулярної маси на основі пептиду, який має рН-залежну розчинність та розмір частинок, менший за 300 мкм, і комбінацію мікрокристалічної целюлози та натрій-гліколяту крохмалю у кількості більше 35% (мас./мас.).2. Пероральна композиція за п. 1, яка відрізняється тим, що містить, необов’язково, цукор, дезінтегратор, зв'язуюче...
Спосіб одержання препарату лейкоцитів для лікування невиношуваності вагітності

Номер патенту: 67162
Опубліковано: 15.06.2004
Автори: Каднікова Наталія Георгіївна, Благовещенський Євген Вячеславович, Горюнова Галина Іванівна, Гуріна Тетяна Михайлівна, Марценюк Валентина Пилипівна
МПК: A61K 35/14
Мітки: спосіб, вагітності, одержання, невиношуваності, препарату, лікування, лейкоцитів
Формула / Реферат:
1. Спосіб одержання препарату лейкоцитів для лікування невиношуваності вагітності, що включає забір крові у чоловіка вагітної жінки, осадження еритроцитів, відділення плазми з лейкоцитами, центрифугування, видалення надосадової рідини і ресуспендування лейкоцитів, який відрізняється тим, що для осадження еритроцитів використовують плазмозамінник з молекулярною масою не менше 70000 Д, а ресуспендування здійснюють у кріозахисному розчині, який...
Попередній патент: Спосіб комплексного лікування хворих на рак яєчника
Наступний патент: Гербіцидний засіб та спосіб боротьби з небажаним ростом рослин
Випадковий патент: Спосіб приготування алкогольвмісного коктейлю