Похідні [2r, 3r (2’r, 3’r), 6r, 7s, 8s, 9r, 10r]-3-(2′, 3′-дигідроксипент-2-іл)-2,6,8,10,12-пентаметил-4, 13-діоксабіцикло[8,2,1]-тридец-12-єн-5-ону, спосіб їх отримання та фармацевтичний засіб, що стимулює дію шлунково-кишкового тракту







Текст
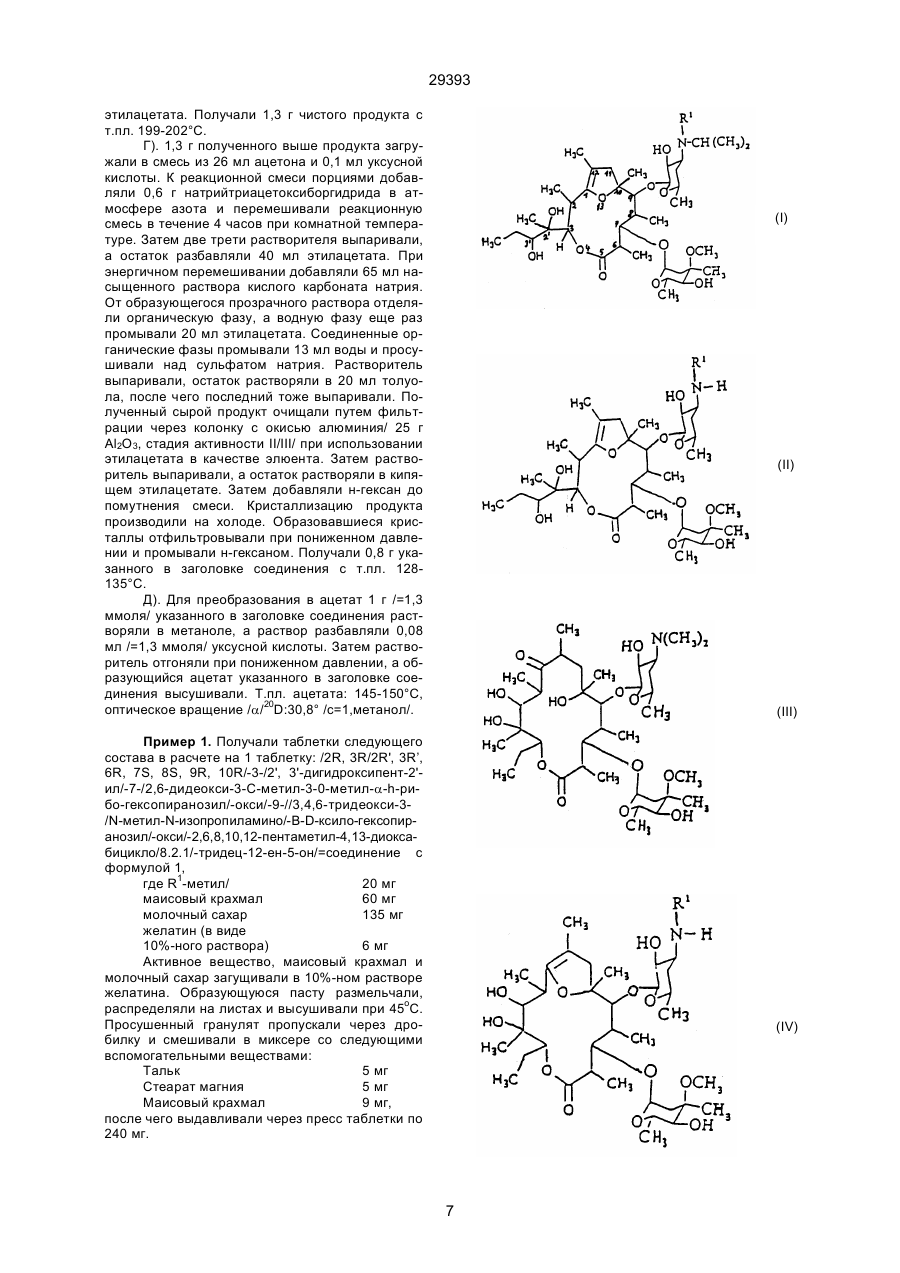
1. Производные [2R, 3R (2'R, 3'R), 6R, 7S, 8S, 9R, 10R]-3-(2', 3'-дигидроксипент-2 '-ил) -2, 6, 8, 10,12-пентаметил-4,13-диоксабицикло[8,2,1 ]-тридец-12-ен-5-она общей формулы (I): (І) (ІІ) 29393 где R1 -метил или водород, вводят остаток изопропила и, при необходимости, свободное соединение формулы (I) переводят в соль или соль соединения формулы (I) переводят в свободное соединение формулы (I). ____________________ Настоящее изобретение относится к новым N-замещенным /2R, 3R (2R', 3R'), 6R, 7S, 8S, 9R, 10R/-3-(2;3'-дигидрокcипeнт-2'-ил/-7-/(2,6-дидeoкcи-3-C-мeтил-3-0-d-h-рuбoгeксапиранозил/-окси/9-/(3,4,6-тридеокси-3-амино-В-D-ксилогексапиранозил/-окси/-2,6,8,10; 12-пентаметил-4,13-диоксабицикло/8,2,1/-тридец-12-ен-5-он-соединениям, с мотилинагонистическими свойствами к их солямпродуктам кислотного присоединения, а также к фармацевтическим средствам, содержащим указанные соединения и способы, а также промежуточным продуктам для их получения. Соединения согласно изобретению являются суженными в кольце N-дезметил-N-изопропил-производными эритромицина А. Как известно, антибиотик эритромицин А наряду с антибиотической активностью отличается нежелательным побочным действием, связанным с деятельностью желудочно-кишечного тракта, а именно ярко выраженным увеличением контракционной активности в области желудка и кишечника, сопровождающейся спазмами и иногда резкими болями в желудке и кишечнике, тошнотой, рвотой и поносом. Было предпринято множество попыток преобразовать эритромицин А таким образом, чтобы получить производные, в которых антибиотическая активность практически более не проявлялась, но сохранялся бы мотилитет желудочнокишечного тракта. Из Европейской патентной заявки 0 349 100 А2 известны фармацевтические средства, содержащие в качестве активного вещества гастропрокинетического характера сужений в кольце эритромицин-А-производный или его четвертичные соли, способные усиливать по холинергическому механизму мотилитет желудка. Поэтому в основу настоящего изобретения была положена заявка разработать новые суженные в кольце производные эритромицина А, не обладающие антибиотической активностью, но обладающие способностью оказывать благоприятное влияние на мотилитет желудочно-кишечного тракта. Было установлено, что новые суженные в кольце N-диметил-N-изопропил-производные эритромицина А обладают селективным мотилинагонистическими свойствами, стимулируя мотилитет желудочно-кишечного тракта благоприятным образом и обнаруживая в то же время действие, способное усиливать тонус нижнего сфинктера пищевода. На основе такого профиля активности вещества согласно изобретению могут быть использованы для лечения повреждений мотилитета, в желудочно-кишечном тракте, обнаруживая при этом хорошую совместимость. Настоящее изобретение относится к новым /2R, 3R(2R', 3R'), 6R, 7S, 8S, 9R, 10R/(-3-)2', З'-дигидроксипент-2'-ил)-2,6,8,10,12-пентаметил-4,13, диоксабицикло/8.2.1/-тридец-12-ен-5-он-производным с общей формулой I, (см. формулу I). где R1 - метил или водород, а также их стабильных и физиологически совместимых солей, продуктов присоединения кислот. Наиболее удачным в этом смысле можно считать, в частности, соединение с формулой I, где R1 - метил. Соединения формулы I могут быть получены известным способом тем, что в /2R, 3R (2R’ 3R')6R, 7S, 8S, 9R, 10R/-3-(2', 3'-дигидроксипент2'-ил)-2,6,8,10,12-пентаметил-4,13-диоксабицикло /8.2.1/тридец-12-ен-5-он-производные соединения общей формулы II, (см. формулу II) где R1 имеет указанное выше значение, вводят радикал изопропила или, по желанию, в полученное соединение с формулой I, где R1 - водород, вводят метил-радикал R1 или в полученное соединение формулы I, где R1 означает метил, отщепляют метиловый радикал R1, и, по желанию, свободное соединение формулы I переводят в их стабильные соли-продукты кислотного присоединения или, наоборот, переводят соли продуктов присоединения кислоты в свободные соединения формулы I. Для введения изопропилового радикала соединения формулы II можно алкилировать известным образом. Предпочтительно проводить 2 29393 алкилирование как восстановительное алкилирование путем взаимодействия соединений формулы II с ацетоном, при восстановительных условиях. Например, соединения с формулой II могут взаимодействовать с ацетоном в присутствии восстановителя, типа комплексного боргидридного соединения, такого как цианоборгидрид натрия, триацетоксиборгидрад натрия или боргидрид натрия. По желанию можно производить алкилирование, в частности, такого соединения с формулой II, где R1 - метил, также путем взаимодействия с изопропилгалогенидом, в частности изопропилйодидом, изопропилсульфатом или эфиром изопропил-сульфокислоты. Алкилирование целесообразно проводить в инертном органическом растворителе. Для восстановительного алкилирования, например, в качестве растворителя можно использовать избыток ацетона. В качестве растворителя могут быть использованы также циклические эфиры, такие как тетрагидрофуран или диоксан, ароматические углеводороды, например, толуол или низшие спирты. Алкилирование можно проводить в интервале температур от комнатной до температуры кипения растворителя. При алкилировании с применением изопропилпроизводного, например, изопропилгалогенида, такого как изопропилйодид, целесообразно проводить работу в присутствии основания, например, карбоната щелочного металла или третичного органического амина. Полученное соединение с формулой I, где R1 означает водород, можно по желанию, в последующем алкилировать по известному методу в соответствующее N-метил-соединение. Алкилирование можно проводить известным образом путем взаимодействия с метиленгалогенидом или как восстановительное алкилирование путем взаимодействия с формальдегидом при восстановительных условиях или можно, например, проводить в условиях, аналогичных проведению алкилирования соединений формулы II. Из соединений с формулой I, где R1 означает метил, можно по желанию отщепить метиловый радикал R1. Деметилирование можно проводить известным самим по себе способом путем обработки соединения галогеном, в частности йодом или бромом, в инертном растворителе в присутствии подходящего основания. В качестве оснований могут быть использованы, например, гидроокиси и соли щелочных металлов слабых органических кислот. Деметилирование проводят предпочтительно в слабо щелочной области преимущественно при рН 9, во избежание побочных реакций гидролиза. Соединения с формулой I можно выделять, используя известные сами по себе способы и очищать. Соли присоединенных кислот можно преобразовывать обычным образом в свободные основания с последующим их превращением, по желанию, известным образом в фармакологически совместимые соли присоединения кислот. Для устранения побочных реакций гидролиза является целесообразным для образования солей следует использовать только эквивалентные количества кислот. В качестве фармакологически приемлемых солей-продуктов присоединения кислоты соеди нений с формулой I могут быть использованы, например, их соли с неорганическими кислотами, например, угольной, галоидводородными кислотами, в частности, хлористоводородной кислотой, или с органическими кислотами, например, низшими алифатическими моно- или дикарбоновыми кислотами, такими как малеиновая, фумаровая, молочная, виннная или уксусная кислоты. Исходные соединения формулы II еще недостаточно хорошо описаны в литературе. В соответствии с изобретением соединения формулы II представляют собой весьма ценные промежуточные продукты для получения фармакологически активных соединений, например, соединений формулы I. Соединения с формулой II могут быть получены исходя из эритромицина А с формулой III при использовании известных методов. Так, например, эритромицин А по известному методу, в частности по методу, описанному в патентной заявке Германии 21 54 032, подвергается однократному или двукратному деметилированию путем взаимодействия с галогеном, предпочтительно, с йодом, в инертном растворителе в присутствии подходящего основания. В качестве оснований могут применяться, например, гидроокиси щелочных металлов, карбонаты и соли щелочных металлов слабых карбоновых кислот, такие как, например, ацетаты и пропионаты щелочных металлов. Предпочтительно вводить 1 до 5 эквивалентов галогена в перерасчете на количество подлежащего деметилированию соединения эритромицина. Количество оснований предпочтительно выбирать таким образом, чтобы гарантировать поддержание рН на уровне 5-9, чтобы устранить возможность побочных реакций гидролиза или алкоголиза. В качестве растворителей подходят метанол, циклические эфиры, такие как диоксан или тетрагидрофуран, диметилформамид или смеси указанных растворителей с водой. Деметилирование целесообразно проводить при температурах от комнатной до 50°С. Реакция требует облучения светом, например, светом с длиной волны свыше 290 нм от ртутной лампы с пониженным давлением, снабженной фильтром из кварцевого или огнеупорного стекла (например, марки "пирекс"). Реакция протекает с получением монодеметилированного или дидеметилированного продукта в зависимости, главным образом, от количества используемого галогена. При применении одного эквивалента гало 3 29393 гена получают предпочтительно монодеметилированный продукт, а при применении же двух и более эквивалентов галогена получают, дидеметилированный продукт. По желанию для получения дидеметилированного продукта можно исходить также из монодеметилированного продукта. Моно- и дидеметилированный эритромицин А можно также, используя известную методику, путем обработки кислотой в щадящих условиях преобразовывать в соответствующие моноили дидеметилированные 8,9-ангидроэритромицин-А-6,9-гемикетали с общей формулой IV Чтобы обеспечить регулярное переваривание принимаемой пищи, в здоровом состоянии совместно действует автономная нервная система и гормоны желудочно-кишечного тракта, которые обеспечивают упорядоченную деятельность по сокращению желудочно-кишечного тракта не только в случае приема пищи, но и, как говорится "на пустой желудок". Мотилин представляет собой известный пептидный гормон желудочно-кишечного тракта, стимулирующий его деятельность и координирующий состояние во всей желудочно-кишечной деятельности натощак, а также при приеме пищи. Соединения формулы I проявляют физиологическую активность, аналогичную действию мотилина, действуя в качестве агонистов для рецепторов мотилина. Так, соединения формулы I проявляют ярко выраженное стимулирующее действие желудочно-кишечной области и нижнего сфинктера пищевода. В частности, они способствуют быстрому опорожнению желудка, повышая на длительное время спокойный тонус сфинктера. Ввиду своей мотилиноподобной активности эти вещества могут быть использованы для лечения болезненных состояний, связанных с нарушениями функции мотилина в желудочнокишечном тракте и/или обратного выброса пищевой кашицы из желудка в пищевод. Так соединения формулы I например, показаны при парезе желудка, различнейших происхождений, нарушениях функции опорожнения желудка и /гастроэзофогальном рефлексе, дисперсии, аномалиях мотилитета толстой кишки / = усиленной перистальтике толстого кишечника, сокращению УПК/, а также постоперативных нарушениях мотилитета, например, кишечной непроходимости /илеусе/. Соединения формулы I, обладающие желудочно-кишечной активностью, можно испытывать в стандартных фармакологических опытах in vivo и in vitro. Описание методов испытаний. 1.Определение связывающей способности исследуемых веществ на рецепторах мотилина. Сродство соединений формулы I с рецепторами мотилина измеряли in vitro на фракции тканевого гомогената антруми (пещеры сосцевидного отростка) кролика. Целью исследования было определение степени вытеснения мотилина, меченного радиоактивным йодом, из мотилин-рецепторной связи испытываемым веществом. Изучение рецепторных связей проводили по одной из модификаций метода Бормана и сотр. /Regulatory Peptides, 15-/1986/, 143-153/. Для получения 125йод-маркированного мотилина проводилось йодирование мотилина по известному методу, например, по аналогии с методом Блоома и сотр., описанному /Scand J . Gastroenterol., II/ 1976/47-52/ с применением ферментативного способа при использовании лактопероксидазы. Для выделения применяемой при испытании фракции гомогена из ткани антрума кролика слизистую оболочку выделенной части антрума размельчали и гомогенизировали в 10-кратном объеме холодного гомогенизирующего буферного раствора /50 ммолей трис-НСl-буфер, 250 где R1 - водород или метил. Образование гемикеталя можно осуществлять например, путем обработки ледяной уксусной или разбавленной минеральной кислотой при температурах от комнатной до 50°С. В соединения формулы IV известным самим по себе образом можно проводить внутримолекулярную транслактонизацию по сужению 14членного лактонового кольца в молекуле эритромицина, преобразуя его в 12-членное лактоновое кольцо с получением соответствующих соединений формулы II. Для этой цели соединения формулы IV необходимо нагреть по известной методике в низшем спирте в присутствии основания, например, при температурах от 40оС до 70оС, предпочтительно до температуры кипения реакционной смеси. В качестве оснований могут быть использованы, в частности, карбонаты щелочных металлов, а также органические основания, такие как третичные амины, в частности низшие алкиламины. При таком сужении кольца конфигурация центров хиральности центров не изменяется. Новые соединения с формулой I или их физиологически совместимые соли присоединения кислот обладают интересными фармакологическими свойствами, в частности, способностью воздействовать на мотилин желудочно-кишечного тракта, стимулируя его деятельность. Антибиотическое действие в них отсутствует, однако они обладают высоким селективным средством к рецепторам мотилина, в то время как в эффективных дозах, обладающих агонистическим в отношении мотилина действием они практически не проявляют релевантного сродства с другими рецепторами желудочно-кишечного тракта, такими как, например, адреналин, ацетилхолин, гистамин, допамин или серотонин. 4 29393 ммолей сукрозы, 25 ммолей KCI, 10 ммолей Mg2CI2, рН 7,4/ при добавлении ингибиторов /1 ммоль йодацетамида, 1 мкмоль пепстатина, 0,1 г/л бактразина/ при использовании гомогенизатора в течение 15 секунд /1500 об/мин/. Затем гомогенизат центрифугировали в течение 15 минут в количестве 1000 г, полученный остаток четырежды промывали раствором буферного гомогенизированного раствора, после чего повторно суспендировали в 0,9%-ном растворе хлорида натрия/, в объеме, соответствующем 5-кратному весовому количеству антрума/. Полученную таким образом фракцию ткани, получившую название "мембрана заготовки", использовали для испытаний. Для опыта брали 200 мкл мембранной фракции /0,5 - 1 мг протеина/ в 400 мкл буферного раствора А /50 ммолей трис-HCl-буфера, 1,5% BSA, 10 ммолей MgCl2, pH 8,0/ разбавляли 100 мкл йодированного мотилина в буферном растворе Б/10 ммолей трис-НСl - буфер, 1% BSA, pH 8//конечная концентрация 50 рМ/ и инкубировали в течение 60 минут при 30°С. Реакцию останавливали при добавлении 3,2 мл холодного буферного раствора Б, после чего производили отделение связанного мотилина от несвязанного, используя для этого метод центрифугирования /1000 г, 15 минут/. Остаток, образующийся после центрифугирования в виде шелухи, промывали буферным раствором Б, после чего производили подсчет в гамма-счетчике. Исследование степени вытеснения проводилось при добавлении все возрастающих количеств исследуемого вещества в инкубационную среду. В качестве растворов исследуемых веществ использовали водные растворы, полученные при подходящем разбавлении 60 х 100-4 молярных водных штаммовых растворов. Трудно растворимые в воде исследуемые вещества предварительно растворялись в 60%-ном этаноле, и эти растворы разбавлялись таким количеством воды, чтобы концентрация этанола в испытуемом веществе /растворе/ не превышала 1.6 об.%. Исходя из данных, полученных при измерении, определяли ту концентрацию соответствующего исследуемого вещества как IC50, которая вызывала 50%-ное торможение специфической связи йодированной связи в рецепторах мотилина. Из этого рассчитывали соответствующее рIC50-значение. По указанному методу определяли величину pIC50 для вещества примера 1, составляющую 8.32. зок и рассматривался как мера опорожнения желудка. На данной тестовой модели соединение примера 1 обнаруживало ярко выраженное стимулирующее действие на скорость опорожнения желудка при дозе 0,46 мкмоль/кг. Время, необходимое для 50%-ного опорожнения желудка, составило в контрольной группе животных 46 минут, в то время как в группе, которой вводилось исследуемое вещество, оно снижалось до 27 минут. 3.Определение влияния веществ на тонус покоя сфинктера в опытах in vivo. Определение проводилось на тренированных, взрослых собаках породы "гончая" натощак, которым до начала проведения опыта вводилась фистула и вставлялся дуоденальный канал. Давление на нижнем конце сфинктера измерялось с помощью перфундированной системы катетеров с боковым отверстием, которая соединялась с приемником давления и самописцем. Катетер через фистулу эзофагуса вводился в желудок, а затем медленно вручную выводился из него /=манометрия протока/. При прохождении части катетера с боковым отверстием через зону повышенного давления на нижнем конце сфинктера регистрировался пик. По этому пику определялось давление в мм ртутного столба. Таким образом сначала в качестве контрольного значения определялось основное (базальное) давление сфинктера пищевода. Затем исследуемое вещество вводилось внутрь двенадцатиперстной кишки и через 15 минут проводились измерения давления на нижнем сфинктере пищевода. Интервалы между измерениями составили 2 минуты, продолжительность измерений составила 46 минут. Рассчитывалось повышение давления после введения исследуемого вещества в сравнении с установленным вначале базальным давлением. В этом опыте базальный тонус сфинктера пищевода при дозе 0,251 мкммоль на кг вещества из примера 1 более чем удваивался. Этот эффект сохранялся в течение всего хода испытаний, т.е. 45 минут. Вследствие своей активности в желудочно-кишечном тракте соединения с формулой 1 используются в гастроэнтерологии в качестве лекарств для крупных млекопитающихся, в частности для человека, для профилактики и лечения нарушения функции мотилина в желудочно-кишечной деятельности. Принимаемые дозы должны назначаться строго индивидуально и могут варьироваться с учетом состояния больного или формы применения лекарства. Например, парэнтерально вводимые препараты могут содержать меньше активного вещества, чем оральные. В основном, однако, для лечения крупных млекопитающих, в частности, человека, используются лекарственные формы с содержанием активного вещества от 5 до 200 мг на разовую дозу. Соединения формулы I, используемые в качестве лекарственных средств, принимаются вместе с обычными вспомогательными фармацевтическими веществами в галенических формах, таких как таблетки, капсулы, свечи или растворы. Эти галенические составы могут быть получены по известным методам с применением 2.Определение влияния вещества на скорость опорожнения желудка при опытах in vivo. Скорость опорожнения желудка определяли на собаках породы "гончая", которым перед опытом проводили операцию по наложению фистулы и имплантации дуоденального канала. Через 15 минут после дуоденального введения исследуемого вещества голодным взрослым собакам вводили через фистулу 285 г полужесткой калорийной пищи. Содержимое опорожняющегося желудка отбирали с интервалом в 15 минут через дуоденальный канал. Из выделенных количеств содержимого желудка рассчитывали тот отрезок времени, в течение которого происходило 50%ное опорожнение желудка. Этот временной отре 5 29393 обычных твердых носителей, таких как молочный сахар, крахмал, тальк или жидких разбавителей, например, воды, жирных масел или жидких парафинов, или же с применением обычных фармацевтических вспомогательных средств, таких как таблетообразующие средства, стимуляторы растворения или консервирующих средств. Приведенные ниже примеры призваны более подробно иллюстрировать предмет изобретения, однако перечнем примеров сущность представляемого изобретения не ограничивается. Пример 1. /2R, 3R/2R', 3R/6R, 7S, 8S, 9R, 10R/-3-/2', 3'-дигидроксипент-2'-ил/-7-//2,6-дидеокси-3-С-метил-3-0-метил-a-h-рибогексопиранозил/окси/-9-//3,4,6-тридеокси-3-/N-метил-N-изопропиламино/-В-D-ксилогексопиразонил/-окси/-2,6,8,10,12пентаметил-4,13-диоксабицикло/8.2.1/-тридец-12ен-5-он/=соединение с формулой 1, где R' – метил/. А). Получение N-дезметилэритромицина А. 20 г эритромицина А /=27,2 ммоля/ и 11,2 г/=136,2 ммоля/ ацетата натрия растворяли в 200 мл смеси из метанола и воды /852/. Раствор нагревали до 47°С, после чего добавляли 6,9 г/=136,2 ммоля/ йода. Путем добавления разбавленного раствора (водного) гидроокиси натрия поддерживали рН на уровне 8-9. Через 3 часа реакционную смесь с целью дальнейшей обработки сливали в смесь из 1 л воды и 20 мл раствора гидроокиси аммония. Проводили экстрагирование реакционной смеси, используя этиловый эфир уксусной кислоты, а органический экстракт промывали водой, содержащей гидроокись аммония, и концентрировали. Оставшийся после удаления растворителя сырой продукт перекристаллизовывали из смеси ацетона с раствором гидроокиси аммония, взятых в соотношении 50:3. Точка плавления 143-148°С. Б). Получение N-дезметил-8,9-ангидроэритромицин-А-6,9-гемикеталя/=соединения с формулой IV, R1-метил/. 21 г полученного в соответствии с п.А/ продукта растворяли в 110 мл ледяной уксусной кислоты и полученный раствор перемешивали в течение 1 часа при комнатной температуре. Затем реакционную смесь с целью обработки при ледяном охлаждении добавляли по каплям в 400 мл концентрированного раствора гидроокиси аммония. Реакционную смесь экстрагировали этиловым эфиром уксусной кислоты, органический экстракт промывали водой и растворитель отгоняли. Образующийся в качестве остатка сырой продукт перекристаллизовывали сначала из эфира, а затем из метанола. Получали 14 г чистого продукта с т.пл. 145°С. В). Получение /2R, 3R/2R', 3R'/, 6R, 7S, 8S, 9R, 10R/-3-/2', 3'-диrидpoкcипeнт2'-ил/-7-//2,6-дидeoкcи-3-C-мeтил-3-O-метил-a-hрибо-гексапиранозил/-окси/9-//3,4,6-тридеокси-3метиламино-В-D-ксило-гексапиранозил/-окси/-2, 6,8,10,12-пентаметил-4,13-диоксабицикло/-/8.2.1/тридец-12-ен-5-она/=соединение формулы II, где R1-метил/. 9,4 г (=13,4-ммоля) продукта, полученного п.п.Б), кипятили с 1,9 г (=13,4 ммоля) карбоната калия в метаноле в течение 2,5 часов до флегмы. С целью обработки реакционную смесь сгущали, разбавляли водой и экстрагировали этило вым эфиром уксусной кислоты. Оставшийся после отгонки растворителя сырой продукт перекристаллизировали из изопропанола. Получали 7,1 г чистого продукта с т.пл. 199-200°С и оптическим вращением /d/ 20D:-31,6° (с=1, метанол). Г). Получение указанного в заголовке соединения 2 г (=2,8 ммолей) продукта, полученного в соответствии с п.В), растворяли в метаноле и путем добавления разбавленного раствора соляной кислоты устанавливали рН на уровне 4. К раствору добавляли 2 г молекулярного сита/алюo минийсиликат кальция, диаметр пор 4 A /, избыток ацетона и 0,4 г/ = 6,4 ммоля/ натрийцианборгидрида. Реакционную смесь перемешивали в течение 12 часов. С целью обработки молекулярное сито отфильтровывали, фильтрат сгущали, разбавляли водой и экстрагировали этиловым эфиром уксусной кислоты. Образующийся после сгущения экстракта этилового эфира уксусной кислоты в качестве остатка сырой продукт очищали методом хроматографии на колонке через кизельгель/элюент-этиловый эфир уксусной кислоты (метанол 95:5). Получали 1,4 г указанного в заголовке соединения с точкой плавления 130134°С с оптическим вращением /a/20D:-32,8о. Пример 2. /2R,3R/2R',3R/, 6R, 7S, 8S, 9R,10R/-3-/2',3'-дигидроксипент-2'ил/-7-//2,6-дидеокси-3-С-метил-3-0-метил-a'-h-рибо-гексопиранозил/окси/-9-/3,4,6-тридеокси-3-/N-метил-N-изопропиламино/-В-D-ксило-гексопиразонил/-окси/-2,6,8,10, 12-пентаметил-4,13-диоксабицикло/8.2.1/-тридец12-ен-5-он/=соединение с формулой 1,R1-метил/. А) Получение N-дезметилэритромицина А 5 г эритромицина А и 4,7 г ацетата натрия (х 3 Н2О) растворяли в 200 мл смеси, состоящей из метанола и воды 8:2. К раствору добавляли 1,75 г йода, после чего реакционную смесь при комнатной температуре в течение 20 минут облучали кварцевой лампой. Затем половину растворителя выпаривали, а оставшуюся реакционную смесь сливали в смесь из 140 мл воды и 10 мл аммиака. Реакционную смесь трижды экстрагировали, используя каждый раз 20 мл метил-1-бутилового эфира. Эфирный экстракт отделяли и эфир частично выпаривали. Затем происходила кристаллизация реакционного продукта, который в дальнейшем перекристаллизовывали из ацетона. Получали 2 г N-дезметилэритромицина А. Б). Для получения N-дизметил-8,9ангидроэритромицин-А-6,9-гемикеталя /=соединение с формулой IV,R1 -метил/ 2 г полученного в соответствии с А) продукта обрабатывали по методике, приведенной в примере 1Б). Получали 2,3 г гемикеталя в качестве аморфного продукта. В). Для получения /2R, 3R/2R’, 3R'/, 6R, 7S, 8S, 9R, 10R/-3-/2',3'-дигидроксипент-2'-ил/-7-2,6дидеокси-3-С-метил-3-0-метил-a-h-рибо-гексопиранозил/-окси/-9-//3,4,6-тридеокси-3-метиламиноВ-D-ксило-гексопиранозил/-окси/-2,6,8,10,12-пента-метил-4,13-диоксабицикло/8,2,1/-тридец-12-ен5-она/=соединение с формулой II, где R1-метил/ 2,3 г полученного выше продукта обрабатывали в соответствии с методикой примера 1В. Полученный сырой продукт перекристаллизовывали из 6 29393 этилацетата. Получали 1,3 г чистого продукта с т.пл. 199-202°С. Г). 1,3 г полученного выше продукта загружали в смесь из 26 мл ацетона и 0,1 мл уксусной кислоты. К реакционной смеси порциями добавляли 0,6 г натрийтриацетоксиборгидрида в атмосфере азота и перемешивали реакционную смесь в течение 4 часов при комнатной температуре. Затем две трети растворителя выпаривали, а остаток разбавляли 40 мл этилацетата. При энергичном перемешивании добавляли 65 мл насыщенного раствора кислого карбоната натрия. От образующегося прозрачного раствора отделяли органическую фазу, а водную фазу еще раз промывали 20 мл этилацетата. Соединенные органические фазы промывали 13 мл воды и просушивали над сульфатом натрия. Растворитель выпаривали, остаток растворяли в 20 мл толуола, после чего последний тоже выпаривали. Полученный сырой продукт очищали путем фильтрации через колонку с окисью алюминия/ 25 г Аl2О3, стадия активности II/III/ при использовании этилацетата в качестве элюента. Затем растворитель выпаривали, а остаток растворяли в кипящем этилацетате. Затем добавляли н-гексан до помутнения смеси. Кристаллизацию продукта производили на холоде. Образовавшиеся кристаллы отфильтровывали при пониженном давлении и промывали н-гексаном. Получали 0,8 г указанного в заголовке соединения с т.пл. 128135°С. Д). Для преобразования в ацетат 1 г /=1,3 ммоля/ указанного в заголовке соединения растворяли в метаноле, а раствор разбавляли 0,08 мл /=1,3 ммоля/ уксусной кислоты. Затем растворитель отгоняли при пониженном давлении, а образующийся ацетат указанного в заголовке соединения высушивали. Т.пл. ацетата: 145-150°С, оптическое вращение /a/20D:30,8° /с=1,метанол/. (I) (II) (III) Пример 1. Получали таблетки следующего состава в расчете на 1 таблетку: /2R, 3R/2R', 3R’, 6R, 7S, 8S, 9R, 10R/-3-/2', 3'-дигидроксипент-2'ил/-7-/2,6-дидеокси-3-С-метил-3-0-метил-a-h-рибо-гексопиранозил/-окси/-9-//3,4,6-тридеокси-3/N-метил-N-изопропиламино/-В-D-ксило-гексопиранозил/-окси/-2,6,8,10,12-пентаметил-4,13-диоксабицикло/8.2.1/-тридец-12-ен-5-он/=соединение с формулой 1, 20 мг где R1-метил/ маисовый крахмал 60 мг молочный сахар 135 мг желатин (в виде 10%-ного раствора) 6 мг Активное вещество, маисовый крахмал и молочный сахар загущивали в 10%-ном растворе желатина. Образующуюся пасту размельчали, распределяли на листах и высушивали при 45оС. Просушенный гранулят пропускали через дробилку и смешивали в миксере со следующими вспомогательными веществами: Тальк 5 мг Стеарат магния 5 мг Маисовый крахмал 9 мг, после чего выдавливали через пресс таблетки по 240 мг. (IV) 7 29393 Отчет о проведении фармакологических опытов "определение минимальной токсической дозы" Мужским особям мышей весом от 20 до 25 г даются максимальные дозы в 300 мг/кг испытуемого вещества. В течение трех часов ведутся тщательные наблюдения за животными в части выявления симптомов токсичности. В течение 72 часов после применения регистрируются все дополнительные симптомы и смертные случаи. Побочные симптомы также наблюдаются и регистрируются. Если наблюдается смерть или сильные токсичные симптомы, то другим мышам дают значительно уменьшенные дозы до того момента, пока не исчезнут случаи появления токсичных симптомов. Самая низкая доза, которая вызывает смерть или появление сильных токсичных симптомов, указывается как минимальная токсичная доза. Вещество примера 1 при р.о. применении в дозе 300 мг/кг р.о. не показало токсичных симптомов. При применении i.p. при дозе в 100 мг/кг наблюдались легкие симптомы, такие как легкое ослабление тонуса конечностей и силы хватания. Пример 3. [2R,3R(2R',3R'), 6R, 7S, 8S, 9R, 10R]-3-(2',3'-дигидроксипент-2'-ил)-7-[(2,6-дидеокси-3-С-метил-3-0-метил-a-L-рибо-гексопиранозил)-окси]-9-(3,4,6-тридеокси-3-(N-метил-N-изопропиламино)-b-D-ксило-гексопиранозил)-окси)-2,6,8, 10,12-пентаметил-4,13-диоксабицикло[8.2.1]тридек12-ен-5-он- (= соединение формулы 1, R' - метил). 5 г [2R,3R(2R’R'), 6R, 7S, 8S, 9R, 10R]-3(2',3'-дигидроксипент-2'-ил)-7-[(2,6-дидеокси-3-Сметил-3-0-метил-a-L-рибо-гексопиранозил)-окси]9-[(3,4,6-тридеокси-3-метиламино-b-D-ксило-гексопиранозил)-окси]-2,6,8,10,12-пентаметил-4,13диоксабицикло-[8.2.1]тридек-12-ен-5-она (= соединение формулы 11, R' = метил, получение смотри пример 1C) были добавлены в смесь из 12 л диметилформамида и 1,4 мл диизопропилэтиламина ( = основание Гюнига). После этого к реакционной смеси по каплям добавляли 8,3 мл йодида изопропила, после чего смесь перемешивали в течение 3 часов при температуре 70°С. Для приготовления реакционная смесь упаривалась при пониженном давлении, а оставшийся осадок поглощается смесью из 50 мл сложного этилового эфира уксусной кислоты и 50 мл воды. Все еще нерастворившийся осадок переводился в раствор путем добавления небольшого количества 1n раствора едкого натра. Органическая фаза отделялась, а водная фаза еще два раза промывалась каждый раз 20 мл сложного этилового эфира уксусной кислоты. После этого объединенные органические фазы высушивались и упаривались над сульфатом натрия. Сырой продукт, оставшийся в виде остатка, многократно очищался с помощью хроматографии на колонке над силикагелем (растворитель дихлорметан/метанол 95:5). Получали 420 мг указанного в названии соединения с точкой плавления от 128 до 134оС, оптическое значение [a]D20: - 32,8° (с=1, метанол). Тираж 50 екз. Відкрите акціонерне товариство «Патент» Україна, 88000, м. Ужгород, вул. Гагаріна, 101 (03122) 3 – 72 – 89 (03122) 2 – 57 – 03 8
ДивитисяДодаткова інформація
Назва патенту англійськоюDerivatives of [2r, 3r (2'r, 3'r), 6r, 7s, 8s, 9r, 10r]-3-(2', 3'- dihydroxypent-2-yl)- 2,6,8,10,12-pentamethyl-4,13- dioxobicyclo [8,2,1]-tridec-12-ene-5-on, method for their production, pharmaceutical agent stimulating gastrointestinal tract activity
Автори англійськоюDagmar Khelte, Ulf Projshoff, Kristian Ehkkhout
Назва патенту російськоюПроизводные [2r,3r (2'r,3'r,), 6r,7s,8s,9r,10r] -3-(2',3'- дигидроксипент-2'-ил)-2,6,8,10,12-пентаметил -4,13- диоксабицикло [8,2,1]-тридец-12-ен-5-она, способ их получения, фармацевтическое средство, стимулирующее деятельность желудочно-кишечного тракта
Автори російськоюХельте Дагмар, Пройшофф Улф, Экхоут Кристиан
МПК / Мітки
МПК: C07H 17/08, A61K 31/70, A61P 1/14, A61K 31/7048, A61K 31/7042, A61P 43/00
Мітки: 3'-дигідроксипент-2-іл)-2,6,8,10,12-пентаметил-4, тракту, 10r]-3-(2, 2'r, засіб, отримання, спосіб, 3'r, стимулює, похідні, 13-діоксабіцикло[8,2,1]-тридец-12-єн-5-ону, дію, фармацевтичний, шлунково-кишкового
Код посилання
<a href="https://ua.patents.su/8-29393-pokhidni-2r-3r-2r-3r-6r-7s-8s-9r-10r-3-2-3-digidroksipent-2-il-2681012-pentametil-4-13-dioksabiciklo821-tridec-12-ehn-5-onu-sposib-kh-otrimannya-ta-farmacevtichnijj-zasib-shho-stim.html" target="_blank" rel="follow" title="База патентів України">Похідні [2r, 3r (2’r, 3’r), 6r, 7s, 8s, 9r, 10r]-3-(2′, 3′-дигідроксипент-2-іл)-2,6,8,10,12-пентаметил-4, 13-діоксабіцикло[8,2,1]-тридец-12-єн-5-ону, спосіб їх отримання та фармацевтичний засіб, що стимулює дію шлунково-кишкового тракту</a>
Засіб для лікування запальних захворювань стравоходу та запальних виразкових захворювань шлунково-кишкового тракту

Номер патенту: 15913
Опубліковано: 30.06.1997
Автори: Альфред Шмідт, Ханс-Юрген Упмейєр
МПК: A61K 31/80
Мітки: стравоходу, запальних, тракту, засіб, лікування, захворювань, виразкових, шлунково-кишкового
Формула / Реферат:
(57) 1. Применение диметилполисилоксана в качестве средства для лечения воспалительных заболеваний пищевода и воспалительных и язвенных заболеваний желудочно-кишечного тракта.2. Применение средства по п.1 г добавкой силикагеля.3. Применение средства по пп.1 и 2 с кинематической вязкостью в диапазоне 100-10000 мм2 с-1.
Фармацевтична композиція в твердій одиничній дозованій формі, придатній для орального застосування при розладах шлунково-кишкового тракту, яка містить сіль ранітидину і карбоксилату вісмуту та спосіб її одержання

Номер патенту: 27002
Опубліковано: 28.02.2000
Автори: ХЕППЕНСТОЛЛ Колін, СМІТ Норман, ДУГЛАС Стефен
МПК: A61K 33/00, A61K 31/34
Мітки: орального, придатній, дозованій, ранітидину, спосіб, сіль, шлунково-кишкового, композиція, форми, фармацевтична, розладах, карбоксилату, застосування, яка, вісмуту, твердий, одиничній, тракту, містить, одержання
Формула / Реферат:
1. Фармацевтическая композиция в твердой единичной дозированной форме, пригодная для орального применения при расстройствах желудочно-кишечного тракта, включающая в качестве активного начала комплекс N-[2-[[[5-[(диметиламино)метил]-2-фуранил]метил]тио]этил]-N'-метил-2-нитро-1,1-этендиамин:2-гидрокси-1,2,3-пропантрикарбоксилат-висмут(3+) или комплекс...
Спосіб реєстрації моторної активності шлунково-кишкового тракту

Номер патенту: 1216
Опубліковано: 30.12.1993
Автори: Саєнко Валерій Федосьович, Слабінський Валерій Володимирович, Дорош Анатолій Володимирович, Романішен Володимир Семенович
МПК: A61B 5/053
Мітки: реєстрації, тракту, шлунково-кишкового, спосіб, активності, моторної
Формула / Реферат:
Ф о р м у л а и з о б р е т е н и я Способ регистрации моторной активности желудочно-кишечного тракта путем введения в него измерительного зонда с последующим измерением моторной активности, о т л и ч а ю щ и й с я тем, что, с целью уменьшения травматичности способа измерения, измеряют электрическую емкость между конденсаторами, вмонтированными в зонд.
Спосіб лікування захворювань шлунково-кишкового тракту сільскогосподарських тварин

Номер патенту: 22459
Опубліковано: 03.03.1998
Автори: Гладишев Віталій Валентинович, Головкіна Лідія Василівна, Головкін Вячеслав Олександрович, Дунаєв Віктор Володимирович, Григор'єв Володимир Ніканорович, Візір Вадим Анатолійович
МПК: A61K 31/425
Мітки: спосіб, тварин, шлунково-кишкового, сільськогосподарських, захворювань, тракту, лікування
Формула / Реферат:
1. Способ лечения заболеваний желудочно-кишечного тракта сельскохозяйственных животных, включающий введение биологически активного вещества, отличающийся тем, что в качестве биологически активного вещества используют 2-меркаптобензтиазол из расчета 0,008-0,01 г на 1 кг массы животного на прием, а введение его осуществляют перорально 3 раза в день в пригодной для перорального введения лекарственной форме.2. Способ лечения заболеваний...
Спосіб санації ділянки шлунково-кишкового тракту, що використовується для пластики травного каналу

Номер патенту: 24858
Опубліковано: 06.10.1998
Автори: Слонецький Ігор Іванович, Бродовський Сергій Петрович, Слонецький Борис Іванович, Ватаман Віктор Миколайович, Фундюр Володимир Дмитрович, Карлійчук Олександр Оксентійович, Самохваленко Ігор Борисович, Кулачек Федір Григорович, Волянюк Петро Михайлович, Тутченко Микола Іванович, Вінніченко Ігор Олександрович
МПК: A61B 17/3209, A61K 33/14
Мітки: шлунково-кишкового, травного, спосіб, ділянки, пластики, використовується, каналу, санації, тракту
Формула / Реферат:
Способ санации участка желудочно-кишечного тракта, используемого для пластики пищеварительного канала, включающий мобилизацию его сегмента и пластическое замещение им одного из участков пищеварительного канала, отличающийся тем, что до начала осуществления пластического этапа операции мобилизированный сегмент трехкратно промывается от содержимого физиологическим раствором натрия хлорида, после чего его полость наполняется двухкратно с...
Наступний патент: Спосіб отримання речовини для лікування захворювань підшлункової залози
Випадковий патент: Сепаратор магнітно-гравітаційний стрічковий