Спосіб одержання азолових сполучень або їх кислотно аддітивних солей,їх простих або складних ефірів





Формула / Реферат
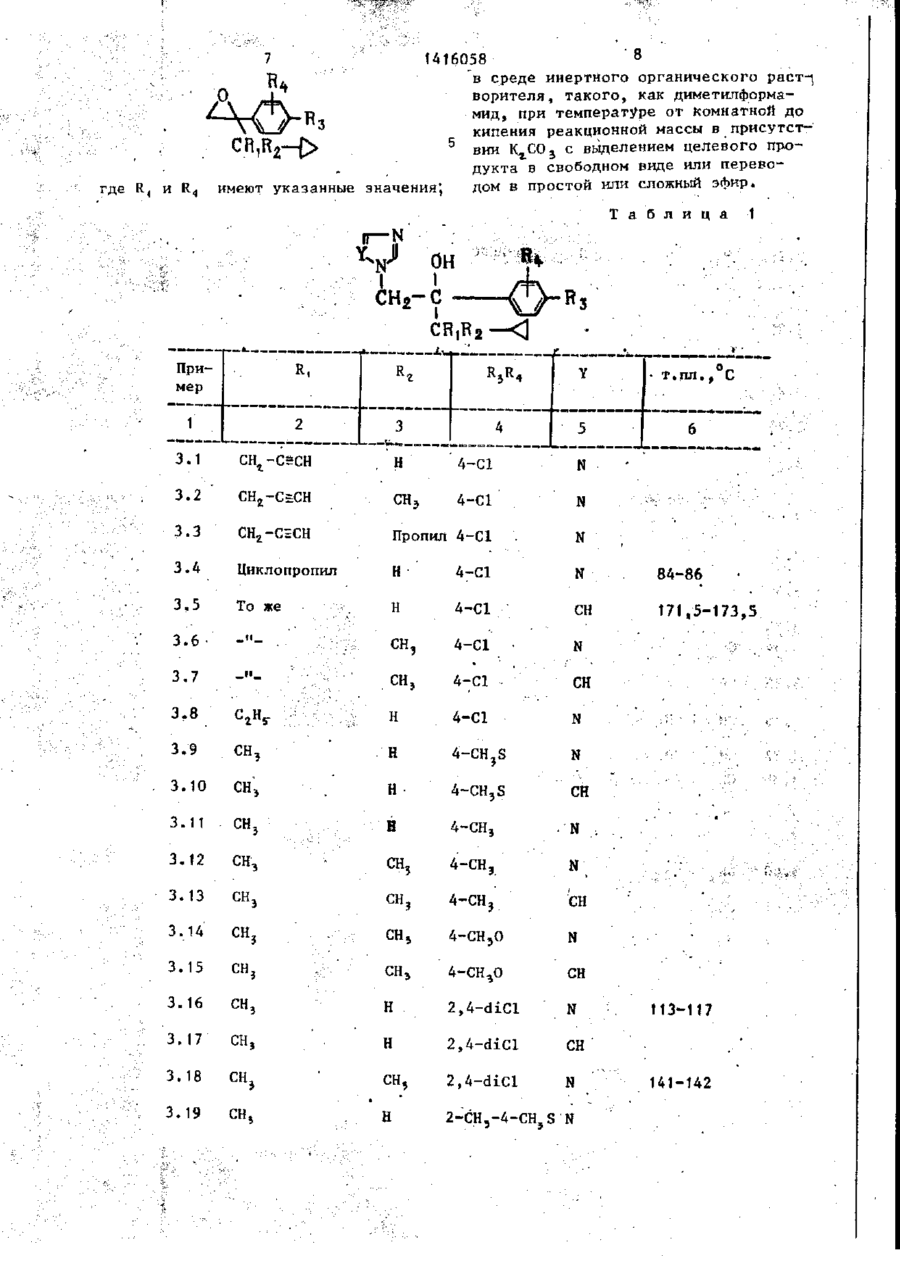
Способ получения азоловых соединений общей формулы
где R1 – С1-С5-алкил или циклопропил;
R2 - водород или С1-С5-алкил;
R1 и R2 вместе - СН2СН2;
R3 - галоген;
R4 - водород или галоген;
Y - ;
или их кислотно-аддитивных солей, их простых или сложных эфиров, отличающийся тем, что соединение общей формулы
где Y имеет указанные значения, подвергают взаимодействию с оксираном общей формулы
R1 R4 в среде имеют указанные значения; в среде инертного органического растворителя, такого, как диметилформамид, при температуре от комнатной до кипения реакционной массы в присутствии K2CO3 с выделением целевого продукта в свободном виде или переводом в простой или сложный эфир.
Текст
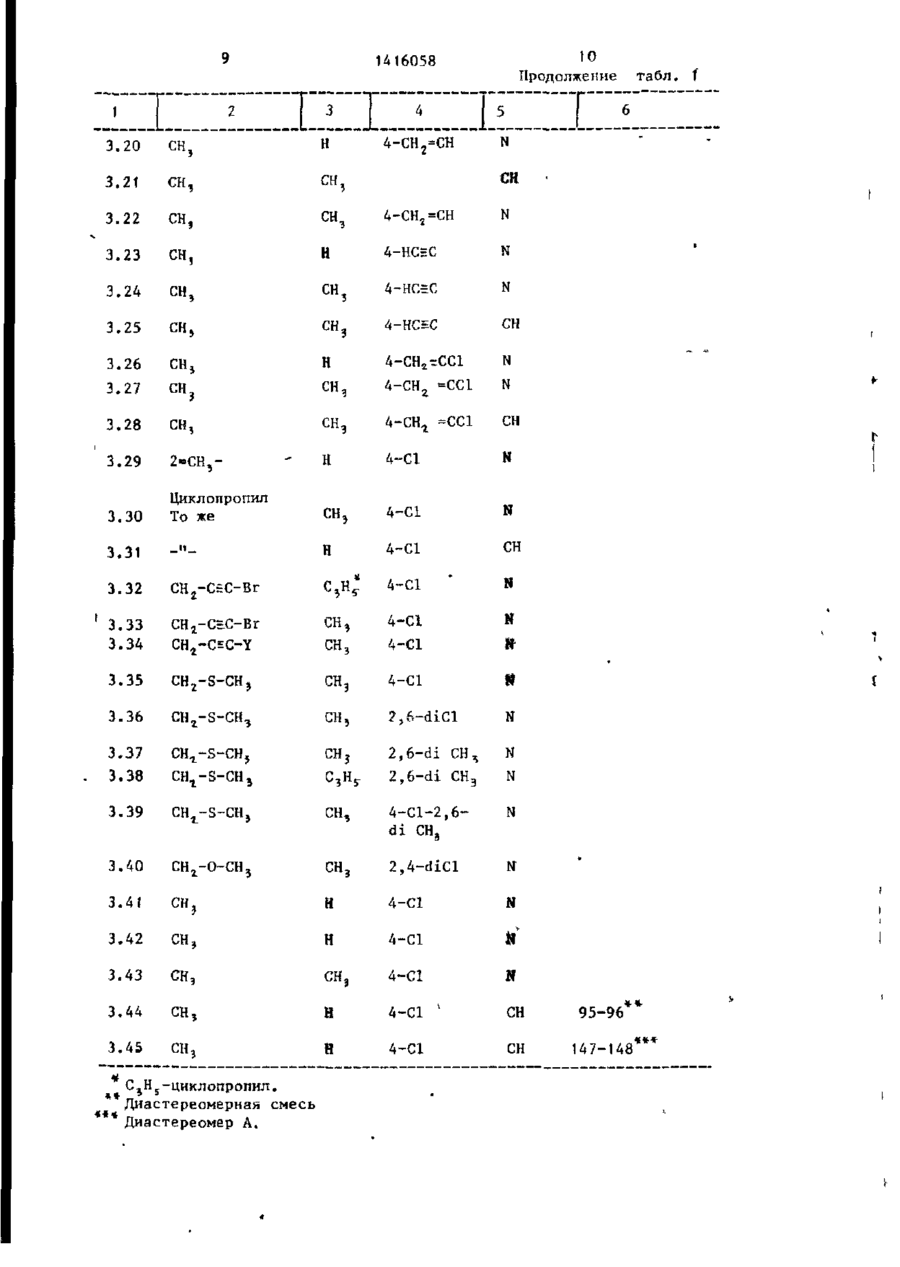
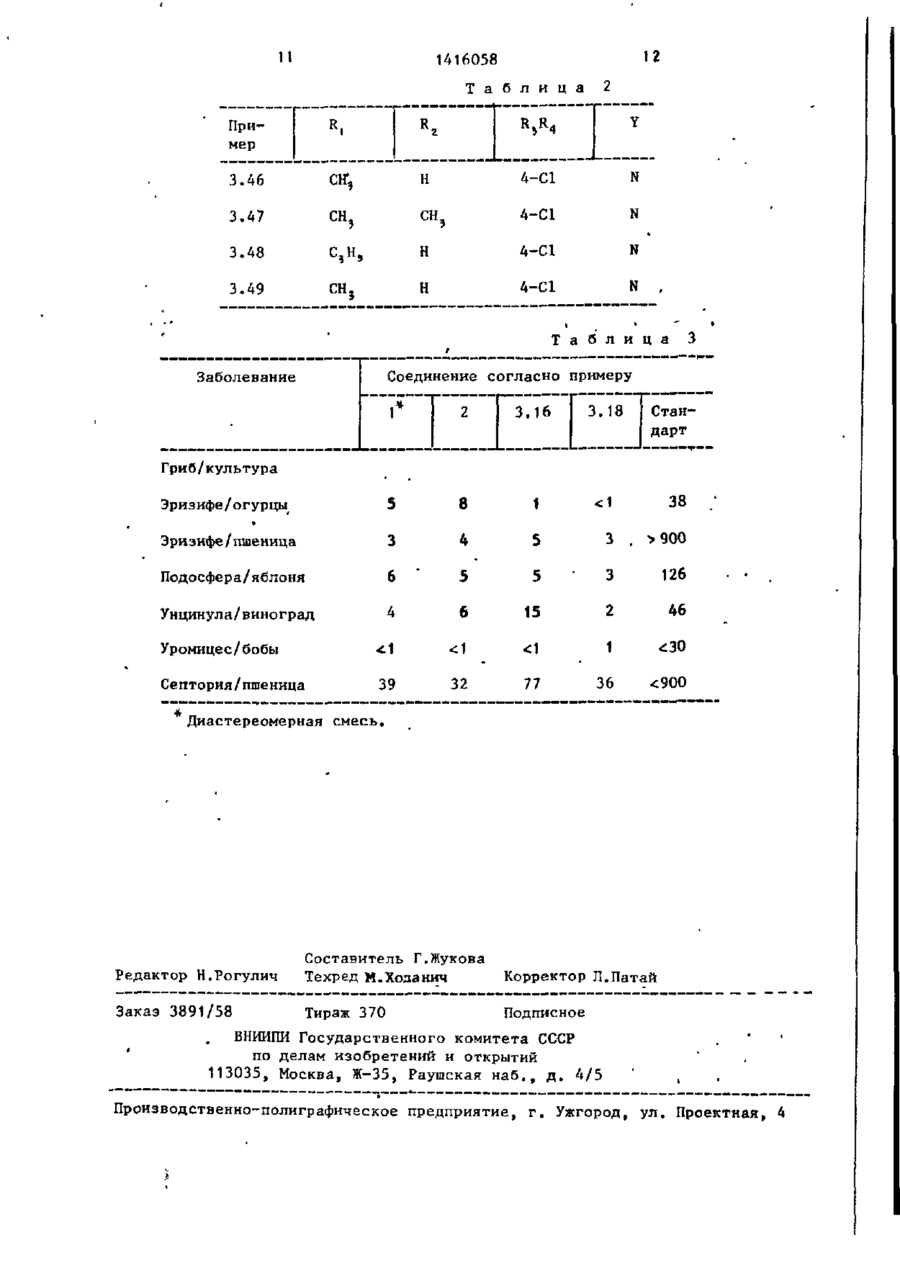
Изобретение касается азоловых соединений (АС) „в частности соединений общей формулыHC^N-CH^Y^-CHq-C(ОН) (CRT"** к CH 2 ~CH r CH 2 ;L -n=R 3 =C & - С^С^-алкил или циклопропші; - водород или С ( -С 5 -алкил; R, и R 2 вместе - ( С Н г ) 2 ; R 3 - галоген; R 4 - водород или галоген; Y -"СН >N, или их кислотно-аддитивных солей, их простых или сложных эфиров, которые обладают антимикотическими свойствами. Для повышения фун™ гицидной активности были получены новые АС. Их синтез ведут из соответствующего незамещенного азола и замещенного оксирана в среде органического инертного растворителя (дИ- * метилАюрмамид) при температуре от комнатной до температуры кипеййя реакционной массы в присутствии KgCOs с выделением АС в свободном виде или в виде простого или сложного эфира. Испытания АС показывают, что их активность выше активности стандарта - 1-циклоґексилметил~сі(п-метилфенил)~1Н-і, 2,4-триазол-1этанола против мучнистой росы винограда и хлебных злаков. 3 табл. 1\ \ 1 1416058 Изобретение относится к способу получения новых азоловых соединений,, 1 обладающих антимикотическими свойствами. Цель изобретения - способ получении новых соединений, обладающих более высокой фунгицидной активностью по сравнению со структурным аналогом, обладающим тем же видом активности. Все температуры показаны в градусах Цельсия. Значения Rf по силикагелю. П р и м е р 1. 2~(Хлорфенил)-3циклопропил-1-1Н-(1,2,4-триазоЛ-115 ил)-бутан-2-ол. , Стадия 1. 7,6 г 1-(4-хлорфенил)-2-циклопропилпропанона-1 растворяют в 120 мл сухого толуола, добавляют при окру20 жающей температуре до 28,6 г додецилдиметилсупьфонметилсульфата и суспензию перемешивают в течение 15 мин. К ней добавляют 6,3 г распыленного КОН и реакционную смесь пере- 25 мешивают в течение 18 ч при 35 С. Реакционную смесь охлаждают, выпивают на лед, и после добавления некоторого количества диэтилформамида экстрагируют диэтилэфиром. 30 Органические экстракты промывают трижды водой, а затем насыщенным водным раствором NaCl, высушивают на MgSO4 и испаряют в вакууме. Полученный маслянистый остаток содержит 2-(4-хлорфенил)-2-(1-циклопропилэтил)-оксирана (окись этилена) наряду с додецилметилсульфидом и додеценом-1. стериомерной смеси, бесцветными кристаллами с т.пл. 100-101°С. Значения Rf в тонкослойной хроматограмме (на пластине-тиз силикагеля с использованием этилацетата в качестве подвижной фазы) для диастереомера А 0,30, для диастерео^ера В 0,38. Путем повторной хроматографии на силикагеле с помощью диэтил-эфир/ е /ацетата (9 *: И и диэтил-эфир/этилацетат? 99:1 до 90:10 с последующей крист- ііизациєй из гексан/СН^СІ^ диастереомерную Смесь разделяют на чистые диастереомеры: a) диастереомер А т.пл,109-110°С; b) диастереомер В т.пл. 125-127°С; . с) Смесь 2,0 г моногидрата р-толуолсульфокислоты в 50 мл толуола концентрируют до объема 6 мл. К ней добавляют каплями с перемешиванием и при комнатной температуре раствор 2,8 г 2-(4-хлорфенил)-3-циклопропил1-(1Н-1,2,4-триазол-1-ил)-бутан-2-ол (диастереомерная смесь) в 35 мл толуола при абсолютной температуре. Дают реакционной смеси отстояться до кристаллизации. После добавления 20 мл диэтилэфира к образовавшемуся в толуоле кристаллизату смесь перемешивают в течение 30 мин, отфильтровывают, промывают диэтилэфиром и высушивают при 60 С в условиях глубокого вакуума, т.пл. 17О-171°С, Аналогично п. с получают следующие соли диастереомерной смеси титульного соединения: g) гидрогеноксалат, т.пл.180-182°CJ е) хлоргидрат, т.пл.19О-2ОО°С.# П р и м е р 2. 2-(4-Хлорфенил)-340 Стадия 2. Сырой оксирановый реакциклопропил-З-метил-1- (1Н-1. 2,4-триционный продукт из стадии 1 каплями азол-1-ил)-бутан-2-ол. добавляют к смеси из 4,2 г 1^2,4-три1-(4-Хлорфенил)-2-циклопропил-2азола и 15,4 кг К 2 С 0 3 в 80 мл сухого метил-пропан-1-од,ин реагирует аналодиметилформамида (ДМФ) при 90°С, и гично примеру 1 (стадия 1 и 2 ) . Очиэту смесь перемешивают при 90 С в тестку титульного соединения осущестчение 2 ч, После охлаждения реакционвляют путем кристаллизации из гексаную смесь выливают на лед и экстрагина с получением бесцветных кристалруют диэтилэфиром, промывают органилов с т.пл. 88-90 С (рацемат титульческие экстракты водой и насыщенным ного соединения). водным раствором NaCl, высушивают на 50 П р и м е р 3. Аналогично примеMgSO 4 и освобождают в вакууме от расру 1 (стадия 2) получают следующие творителя. Хроматография остатка на соединения (табл.1 и 2) путем реакснликагеле с помощью гексан/этилацеции азопа с желательным оксираном тата дает маслянистый бесцветный си(окись этилена). роп (диастереанерную смесь),которая 55 П р и м е р а 2-(4-Хлорфенкл)-2медленно кристаллизуется. Рекристал(1-циклопропил-циклопропил)-1-(1Нлизация кристаллизата из гексан-СН^СІ^ 1,2,4-триазол-1-ил)-этанол-2-ол. дает титульное соедиь че в виде диаСтадия 1, 3 141 6058 * Титульное соединение получают Суспензию 5,1 г 80% NaH в 50 мл аналогично примеру 5, за тем исклюабсолютного тетрагидрофурана (ТГФ) чением, что вместо СН 3 используют перемешивают под слоем азота. К ней аллилбромид, н что реакционную смесь каплями добавляют при комнатной темперемешивают в течение 18 ч при температуре 13,3 г диметилсульфоксида пературе не 20, а 70°С, т.пл. 58 (ДМСО) и после этого в течение 20 мин 60°С (белые кристаллы). 13,5 г 4-хлорфенил-(1-циклопропилциклопропил)-кетона в 50 мл абсолютП р и м е р 7. 2-(4~Хлорфенил)-3ного ТГФ, К полученной свежей суспен- 10 циклопропил-2-бензилокси-З-мєтилзии добавляют порциями 15,0 г иодтри1-(Н-1,2,4-триазол-1-ил)-бутан, метилсульфония. Суспензию перемешиТитульное соединение получают вают в течение 16 ч при комнатной аналогично примеру 6, за тем исклютемпературе и в течение 3 ч при 50 С, чением, что вместо аллилбромида нса затем охлаждают до 0-25 С. Каплями 15 попьзуют бензилбромид, т.пл. 130 добавляют воду, и экстрагируют реак132 С (белые -кристаллы). ционную смесь диэтилэфиром после за- ' П р и м е р е . 2-(4-Хлорфеншт)-3вершения экзотермической реакции. циклопропил-2~ацетокси-3-метил-1Органическую фазу трижды промыва(1Н-1,2 ,4--триазол-1 -ил) -бутан. ют водой и один раз водным раствором 20 Титульное соединение получают NaCl, просушивают на MgSO4 и испаряаналогично примеру 5, за тем исклюют в вакууме при 60 С. Остаток состочением, что используют ацетилхлорид' ит главным образом из 2-(4-хлорфенил)(вместо С Н Э ) и что реакционную смесь 2-(ї-циклопропил-циклопропил)-оксираперемешивают в течение 24 ч при 70°С. на. 25 Оно кристаллизуется из диэтилэфира, т.пл, 117-119 С (желтые кристаллы). Стадия 2. П р и м е р 9, ї-(4-Хлорфенил)-2Сырой оксиран (из стадии 1) реагициклопропил—пропанон-1. iрует с 1,2,4-триазолом аналогично по примеру 1 (стадия 2) и дает после хро15 г 4-хлорфенилциклопропилметилматографии на силикагеле и кристалли- 30 кетона, растворенного в 80 мл абсозации из гексана/СН г С1 2 чистое тилютного ДМФ, добавляют каплями к сустульное соединение, т.пл. 110—112°С ' пензии 2,6 г 80% NaH в 30 мл ДМФ под (рацематная форма). слоем N z и перемешивают смесь в течение 2 ч при 25-35°С. К ней добавП р и м е р 5. 2-(4-Хлорфенил)-3циклопропил-2-метокси~3-метил-1-(1Н- _ ляют каплями в течение 15 мин при комнатной температуре i и охлаждением 1,2,4-триазол-1 -ил),-бутан. ]593> г С Н 3 1 , смесь перемешивают в К суспензии 0,8 r N a H 8 0 % в 25 мл течение 15 мин при 25-30 С после доДМФ добавляют каплями при комнатной бавления холодной воды помещают в температуре раствор 7,64 г 2-(4-хлор4о эфир„ Органические экстракты промыфенил )-3-циклопропил-3-метил-1-(1Нвают водой и насыщенным водным раст1,2,4~триазол-1-ил)-бутан-2-ол в вором NaCl, высушивают на MgSO 4 , и 50 мл ДМФ. Реакционную смесь перемеиспаряют с целью получения сырого шивают при 40°С в течение 30 мин. титульного соединения, которое очиЗатем добавляют каплями при 50 С 3,76 г CHjI. Смесь перемешивают в тают посредством хроматографии на 4 5 течение 18 ч при 20 С, затем выливасиликагеле гексан/этилацетатрм 98;2, ют 1 л воды и экстрагируют с помощью 4-Хлорфенил~(циклопропилметил)С Н а С 1 2 . Органические фазы промывают кетон получают путем окисления по водой, высушивают на MgSO 4 и концентДжонсу соответствующего спирта порируют путем непрерывного мгновенно- ,-п средством СгО э в водном растворе го испарения. Затем получают титульH2SOfl /ацетон,, ное соединение путем хроматографии П р и м е р 10, 1-(4~Хлорфекил)осадка на силикагеле (подвижная фа2-(циклопропил-2-метил-пропанон-1)• за диэтилэфир: триэтиламин 10:2) в Поступают аналогично примеру 9, виде белых кристаллов, т.пл.87-89 С. используя 2,4 эквивалента NaH и 3 эквивалента СН 3 1 на эквивалент 4-хлорфенипциклопропилметнлкетона. Л р и м е р 6. 2-(4-Хлорфешіл)-3Титульное соединение хроматографи-' циклопропнл-2-аллилокси-3-ме7Ил-1руют на силикагеле с помощью фракции С1Н—1,2,4-триазол-1-ил)-бутан. гексана/этилацетата ( 9 9 : О 9 п ^ = = 15390. П р и м е р 11. 4-Хлорфенил-(1циклопропил-циклопролил)-кетон. Суспензию 4 г 80% NaH в 40 мл абсолютного ТГФ перемешивают под слоем N 2 . К ней добавляют каплями слабым противотоком в течение 40 мин 23,3 г 4-хлорфе нил-(циклопропилметил)-кетона в 250 мл абсолютного ТГФ. Затем медленно добавляют при 20° С 15,8 г фенилвинилсульфоксида с помощью шприца (экзотермическая реакция), и смесь перемешивают в течение 2,5 ч при 20-30 С, Получаемый промежуточный продукт (окись сернистого алкила) циклизуют до титульного соединения путем перемешивания в течение 18 ч противотоком. Реакционную смесь охлаждают до 0 (-5) С, затем каплями добавляют 200 мл воды и экстрагируют смесь диэтилэфиром. Органическую фазу трижды промывают водой и один раз насыщенным водным раствором NaCl, высушивают на MgSO^ и испаряют при 60 С в вакууме. 1416058 6 чивающая возможность контроля на 90% грибкового заболевания после применения путем разбрызгивания (табл. 3 ) . Контроль заболевания в полевых условиях. Фунгицидное действие соединения по примеру 1 оценено в полевых ус 10виях; 62 г/га обеспечивяпи лозмож10 ность контроля более "си на 90% порошковой ложной мучнистой росы вино- • града и контроль на 90% ржавчинного заражения хлебных злаков, а 2,6 г/га обеспьчивали возможность контроля на 15 99% порошковой ложной мучнистой росы на виноградниках. Другие оценки показывают фунгицидную активность соединения по примеру 1, а именно равноценное или лучшее 20 действие, чем пропиканозола, против порошковой ложной мучнистой росы на хлебных злаках и огурцах, а также против порошковой ложной мучнистой росы на яблонях и винограде против 25 вентурия на яблонях, соответственно лучше, чем триадимефон, например, 'против ржавчины на кофе. Чистое титульное соединение получают путем хроматографии осадка на 30 силикагеле гексан/эт^лацетатом (89:1), п2° - 15605. П р и м е р 12. Титульное соединение по примеру 9 можно также получать, взяв за исходное "4-хлор-бензил- -с , амид, путем реакции его с циклопропилметилкетоном в присутствии NaH, восстановления получаемого 1-(4-хлорфе нил)-1-циано-2-циклопропил-пропен1 с помощью Mg(CH 3 OH)NH 4 Cl в 1-(44 0 хлорфенил)-1-циано-2-циклопропилпро— паи с последующим окислением посредством 0 г цианового соединения в ш,е- ' лочных условиях в присутствии катализатора фазового переноса. В зависи- д о мости от ситуации (цена, окружающая среда и т.п.) способ этого примера может оказаться предпочтительным. Использование биологической активности - фунгицидного действия. 50 Результаты испытания в теплице. Приводимые результаты испытаний (согласно известным процедурам) показывают благоприятное фунгицидное действие предлагаемых соединений. « Стандартом является оС -циклогексилметил-оі~(р-метилфенил)~1Н-1 ,2,4-триазол-і-зтаноа. Результаты выражены в ЕС 90, т.е. концент, *ия, обеспе Ф о р м у л а и з о б р е т е н и я Способ получения азоловых соединений общей формулы где R - С,-С5-алкил или циклопропил; К2 - водород или С,-С 5 -алкил; R/p R вместе - СН 2 СН 2 ; ЇЦ - галоген; R^ - водород или галоген; Y - ^ С Н или' 2 N; или их кислотно-аддитивных солей, их простых или сложных эфиров, о т л и ч а ю щ и й с я тем, что соединение общей формулы где Y имеет указанные значения, подвергают взаимодействию с оксираном общей формулы •"V #-v;1-'-1 где R, и R 4 1416058 8 в среде инертного органического раст-^ ворителя, такого, как диметилформамид, при температуре от комнатной до кипения реакционной массы в присутствии K^COj с выделением целевого продукта в свободном виде или перевоимеют указанные значения; дом в простой или сложный эфир. Т а б л и ц а V 1 ОН I R сн2-с CR,R 2 —О j._ Пример Xud R, 1 R5R4 2 Y • т.пл., С 4 5 6 3 3.1 а 4-С1 N 3.2 сн э 4-С1 N 3.3 снг-с=сн Пропил 4-С1 3.4 Циклопропил Н 4-С1 N 84-86 3.5 То же н 4-С1 СН 171,5-173,5 3.6 СН, 4-С1 3.7 N • N • 3.9 СН снэ н н 3.10 СН\ н 4-CH3S 3.11 СН 3 н 4-СН3 3.12 СН 3 4-СН 3 N 3. 13 СН сн? сн3 4-СН 3 СН 3. 14 СН СН, 4-СН 3 О N 3. 15 СН CHj 4-СН3О СН 3. 16 СН н 2,4-diCl N 3.17 СН н 2,4-diCl СН 3.18 СН СН. 2,4-diCl N 3.19 СН, Н 2-СН,-4-СН S N 3.8 4-С1 СН 4-С1 N 4-CH ? S N СН • N 113-117 141-142 t 0 1416058 1 3.20 I 1> 1 • 2 H CH, 4-CH7=CH Продолжение 5 N T' 1 "" ..... CH 3.21 CH, CH, 3.22 CH, сн 3 4-CH2=CH N 3.23 CH, H 4-HCHC N 3.24 CH, CH, 4-HC=C N 3.25 CH, сн 3 4-HCEC CH 3.26 CHj H 4-CH 2 -CCl N 3.27 сн 3 4-CH 2 =CCl N 3.28 CH, сн 3 4-CH 2 =CC1 CH 3.29 2«CH 5 H 4-C1 N 3.30 Циклопропил To же CH, 4-C1 N 3.31 -» H 4-C1 CH 3.32 1 табл. f CH2-C=C-Br C3H^ 4-C1 N 3.33 3.34 CHj-CEC-Br CH, 4-C1 N ГЧІ CH 3 4-C1 N 3 сн 3 4-C1 N 3.36 CH2-S-CH? CH, 2,6-diCl N 3.37 CH-L~S-CHJ CH3 2,6-di CH, N 3.38 CHj-S-CH, 2,6-di CH3 N 3.39 CH.—S—CH» CH, 4-Cl-2,6di CH3 N 3.40 сн г -о-сн 5 сн 3 2,4-diCl N 3.41 % H 4-C1 N 3.42 сн3 H 4-C1 3.43 CH3 сн 3 4-C1 N 3.44 CH, H 4-C1 ^ CH 3.45 CH, H 4-C1 CH 3.35 PtP—V 2 С,H_-циклопропил. Диастереомерная смесь Диастереомер А. • 95-96** 147-148 11 12 1416058 Т а б л и ц а Пример R 2 R,R 4 z Y 3.46 Ctf, Н 4-С1 N 3.47 СН, СН, 4-С1 N 4-С1 N 4-С1 N с,н сн ? 3.48 3.49 н н Ї • і Т а б л и ц а 3 Соединение согласно примеру Заболевание 1* 2 Эризифе/огурцы 5 8 1 Эри зифе/пшеница 3 4 5 3 • >900 Подосфера/яблоня 6 5 5 3 126 Унцинула/виноград 4 6 15 2 46 Уромицес/бобы *1
ДивитисяДодаткова інформація
МПК / Мітки
МПК: C07D 249/08, A01N 43/50, C07D 521/00, C07D 233/60, A01N 43/653, A61P 31/04, A61K 31/415
Мітки: солей,їх, спосіб, аддітивних, складних, простих, сполучень, одержання, азолових, кислотної, ефірів
Код посилання
<a href="https://ua.patents.su/8-7048-sposib-oderzhannya-azolovikh-spoluchen-abo-kh-kislotno-additivnikh-solejjkh-prostikh-abo-skladnikh-efiriv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання азолових сполучень або їх кислотно аддітивних солей,їх простих або складних ефірів</a>
Спосіб одержання похідних цефалоспорину або їх складних ефірів, простих ефірів або солей, або їх гідратів, або гідратів їх складних ефірів, простих ефірів або солей

Номер патенту: 4199
Опубліковано: 27.12.1994
Автори: Марк Монтафон, Роланд Рейнер
МПК: C07D 501/00, A61K 31/545
Мітки: гідратів, похідних, солей, простих, спосіб, цефалоспорину, ефірів, складних, одержання
Формула / Реферат:
1. Способ получения производных цефалоспорина общей формулы І в которой X - 2,5-дигидро-6-оксо-2-метил-5-оксо-астриазин-З-илгруппа, которая находится в таутомерном равновесии с 1,2,5,6-тетрагидро-2-мстил-5,6-диоксо-астриазин-3-илгруппой, или 1,4,5,6-тетрагидро-4-метил-5,6-диоксо-астриазин-З-илгруппа, или их сложных эфиров, простых эфиров или солей, или их гидратов или гидратов их сложных эфиров,...
Спосіб одержання похідних простих арилфенілових ефірів або їх кислотно-аддитивних солей, або їх металевих комплексів

Номер патенту: 4203
Опубліковано: 27.12.1994
Автори: Адольф Губеле, Петер Ріблі
Мітки: кислотно-аддітивних, простих, комплексів, ефірів, металевих, спосіб, похідних, одержання, арилфенілових, солей
Формула / Реферат:
(57) Способ получения производных простых арилфениловых эфиров общей формулы г где У- -СН – или –N =;R1 и R2 - атом водорода или галогена или С, -С3 –алкил; Аг - незамещенный или однократно или двукратно замещенный атомом галогена, С, -С4 -алкилом, метоксигруппой, нитрогруппой и/или трехфтористым метилом фенил или нафтил;U и V - C1 - C3 – алкил или образуют один из следующих алкиленовых мостиков: где...
Спосіб одержання похідних 1н – 1, 2, 4 – триазолу або їх фармакологічно прийнятих кислотно-аддітивних солей

Номер патенту: 2709
Опубліковано: 26.12.1994
Автори: Лео Баккс, Жозеф Мостман, Ян Херес
Мітки: похідних, кислотно-аддітивних, солей, спосіб, фармакологічно, одержання, триазолу, прийнятих
Формула / Реферат:
(57) Способ получения производных 1Н—1,2,4-триазола общей формулыС3-С6-алкенил, арилалкил или арил-оксиалкил, в которых алкил с 1-6 атомами углерода, а арил выбран из группы, включающей фенил, нафтил или дифенил, который может быть замещен галогеном, или фенил, замещенный 1-3 заместителями, выбранными из группы, включающей галоген, С1-С6-алкил, или их фармакологически приемлемых кислотно-аддитивных солей,...
Спосіб отримання похідних теофілліну або їх кислотно-аддітивних фармакологічно припустимих солей

Номер патенту: 3512
Опубліковано: 27.12.1994
Автори: Еміль Мінкер, Ендре Палоші, Шандор Вірга, Габор Ковач, Іда Свобода, Юдіт Кун, Чаба Гьонци, Гергей Хейа, Пал Кіш, Дежьо Карбонітш, Тамаш Сютш, Маріа Сомор, Дьюла Шебештьєн
МПК: A61P 11/14, A61K 31/52, A61P 11/08, C07D 473/08, A61P 11/00, A61P 29/00, A61K 31/522
Мітки: отримання, солей, спосіб, припустимих, фармакологічно, теофілліну, кислотно-аддітивних, похідних
Формула / Реферат:
1. Способ получения производных теофиллина общей формулыгде А - прямой или разветвленный С1-С5-алкилен или группа СН2СН(ОН)СН2; R1- прямой или разветвленный алкил, галоидалкил или С1-С10-оксиалкил, С5-С6-циклоалкил, 2-этоксиэтил, С1-С6-карбонилалкил или аминоалкил формулы где n=0, 1, 2 или 3; R2 и R3- прямой или разветвленный С1-С4-алкил, или R2 и R3 вместе с общим атомом азота...
Спосіб одержання похідних 7-[2-/2-амінотіазоліл/2-оксиіміноацетамідо]-3-цефєм-4-карбонових кислот або їх складних ефірів, або їх солей з лужними металами

Номер патенту: 1749
Опубліковано: 25.10.1994
Автори: Такао Такая, Кіесі Цудзі, Хісасі Такасугі, Тосіюкі Тіба
Мітки: ефірів, одержання, лужними, спосіб, металами, кислот, складних, солей, похідних
Формула / Реферат:
Формула изобретенияСпособ получения производных 7-[2-(2-аминотиазолил)-2-оксиимино- ацетамидо ]-3-цефем-4-карбоновых кислот формулыв виде син-изомеров, где R1 - свободная аминогруппа или защищенная формильной или трифторацетильной группой аминогруппа;R2 - водород, C7-C8 -алкил, аллил, пропаргил, С5-С6-циклоалкил, карбоксиметил, этоксикарбонилметил или моно- или тризамещенный хлор или фтор этил;R3 -...
Попередній патент: Спосіб одержання похідних циклопропана
Наступний патент: Спосіб одержання фтороксігалосполучень
Випадковий патент: Багатокомпонентний дозатор сипучих матеріалів